 Martina Cusan†‡Giorgia Mungo†Mara De Marco Zompit†‡Ilenia Segatto
Martina Cusan†‡Giorgia Mungo†Mara De Marco Zompit†‡Ilenia Segatto Barbara Belletti
Barbara Belletti Gustavo Baldassarre*
Gustavo Baldassarre*- Division of Molecular Oncology, CRO of Aviano, IRCCS, National Cancer Institute, Aviano, Italy
The CDKN1B gene encodes for the p27Kip1 protein, firstly characterized as a cyclin dependent kinase (CDK)-inhibitor. Germline CDKN1B pathogenic variants have been described in hereditary tumors, such as multiple endocrine neoplasia (MEN)-like syndromes and familial prostate cancer. Despite its central role in tumor progression, for a long time it has been proposed that CDKN1B was very rarely somatically mutated in human cancer and that its expression levels were almost exclusively regulated at post-transcriptional level. Yet, the advent of massive parallel sequencing has partially subverted this general understanding demonstrating that, at least in some types of cancer, CDKN1B is mutated in a significant percentage of analyzed samples. Recent works have demonstrated that CDKN1B can be genetically inactivated and this occurs particularly in sporadic luminal breast cancer, prostate cancer and small intestine neuroendocrine tumors. However, a clear picture of the extent and significance of CDKN1B mutations in human malignances is still lacking. To fill this gap, we interrogated the COSMIC, ICGC, cBioPortal, and TRANSFAC data portals and current literature in PubMed, and reviewed the mutational spectrum of CDKN1B in human cancers, interpreting the possible impact of these mutations on p27Kip1 protein function and tumor onset and progression.
Introduction
The CDKN1B gene encodes for the p27Kip1 protein (hereafter p27), firstly characterized as an inhibitor of cell cycle progression for its ability to bind and regulate a broad range of cyclin-CDK (cyclin-dependent kinase) complexes (1). CDKN1B belongs to a family of CDK inhibitor (CKI) genes that also comprises CDKN1A (encoding for p21Waf1) and CDKN1C (encoding for p57Kip2). The three CKI proteins share a region of high homology at their N-terminal portion, encompassing the cyclin- and the CDK-binding domains (2, 3). This region confers the ability to bind and inhibit, although with different stoichiometry, all cyclin/CDKs complexes, eventually controlling progression through the cell cycle. Each member of the family has specific functions and, in this context, p27 has been more prominently indicated as a sensor of external stimuli in driving the decision of the cell to enter or not the cell cycle and, eventually, divide (4). However, depicting p27 only as a cell cycle regulating protein is an old representation of its cellular functions. Many studies have clearly demonstrated that p27 has the ability to interact with many different proteins and represents a target for many signal transduction pathways, thereby accomplishing a number of previously unexpected and so-called non-canonical functions (5) (Figure 1).
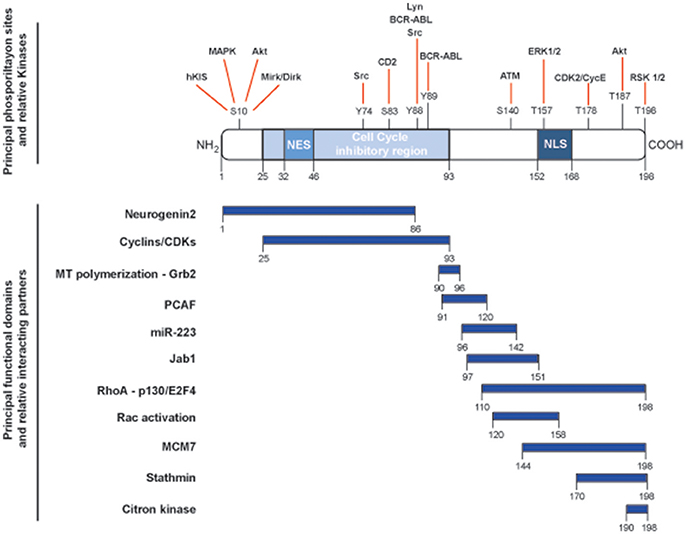
Figure 1. Schematic representation of the main functional domains and phosphorylation sites of p27. p27 protein is composed by 198 amino acids and contains a nuclear exportation signal (NES) at the N-terminus and a nuclear localization signal (NLS) at the C-terminus. Key phosphorylation sites and corresponding kinases are depicted in the upper part of the figure and linked with red lines. The cell cycle inhibitory region is comprised between amino acids 25and 93 and is necessary for the binding to cyclin/CDK complexes. Known functional domains and relative interacting protein/microRNA are reported below and highlighted by blue rectangles.
A central role in the determination of p27 interactions and functions is played by its subcellular localization. p27 shuttles from the nucleus to the cytoplasm, and vice versa, depending from the phase of the cell cycle and from the activation of specific signaling. Different functions can be played by p27 when localized in different subcellular compartment, including regulation of transcription, cytoskeleton organization, autophagy, cytokinesis, eventually impacting on cell proliferation, survival, differentiation, motility, and invasion (5).
These different activities of p27 are highly interconnected and it has been proven difficult to separate these functions only on the basis of specific interactions and/or subcellular localization, although this aspect has been a matter of intense study in the last decade. For instance, we reported that the cytoplasmic interaction of p27 C-terminal domain with the microtubule-destabilizing protein stathmin interferes with microtubule dynamics and regulates cellular migration (6). More recently, the study of p27 and stathmin double knock-out mice unveiled that the cytoplasmic interaction of p27 and stathmin also regulates cell cycle progression in a CDK-independent manner, acting on the H-Ras/MAPK pathway (7–9). Similarly, nuclear localization of p27 has been reported to be necessary for its tumor suppressive function in mice and it has been linked to the control of cell cycle progression via the binding with cyclins/CDK complexes (10). However, nuclear p27 localization is also necessary for embryonic stem cell differentiation and cell reprogramming into induced pluripotent stem cells (iPSCs), via the transcriptional repression of the SOX2 gene (11).
The fact that p27 plays many different functions and binds many different partners has stimulated the hypothesis that it could act either as oncogene or tumor suppressor gene, in a context-dependent or interaction-dependent manner (12). However this hypothesis has never been recapitulated in human tumors yet. Also the possibility that specific post-translational modifications, such as phosphorylation, ubiquitination or SUMOylation, could directly impact on p27 functions and/or on its interactions with other proteins is a scientific issue that might merit more investigations.
In this context, it is interesting to note that p27 is an intrinsically disordered protein (IDP) that adopts an extended conformation upon binding to cyclin A/CDK2 complex at the N-terminus while the C-terminus retains the characteristics of intrinsically disordered region (IDR) (13, 14). The presence of IDRs in a protein greatly weakens the old postulate that protein sequence determines protein structure and protein structure determines protein function (13). Indeed, in an interesting review of the literature, Babu proposed that IDRs increase the functional versatility of proteins and may contribute to human disease (13). The presence of IDRs in a protein allows for the promiscuous interaction with a large number of “partners” and/or facilitates protein function regulation via diverse upstream pathways and post-translational modifications.
Thanks to their conformational flexibility, IDPs are therefore excellent substrates to encode and decode information via post-translational modifications (13). This hypothesis is in line with the current knowledge on p27 protein functions and interactions (Figure 1) and supports the possibility that p27 is very well suited to perform signaling and regulatory functions in multiple contexts.
The CDKN1B gene was the first gene described as haplo-insufficient for tumor suppression, meaning that it did not follow the Knudson's “two-hit” theory. Animals lacking only one copy of Cdkn1b already displayed a tumor-prone phenotype, with increased tumor frequency and decreased latency when challenged with different carcinogens and spontaneously developed pituitary tumors with a mild penetrance, late in life (15). The complete ablation of Cdkn1b led to 20–30% increased body size respect to wild-type littermates, and predisposed to spontaneous pituitary adenomas and multiple organ hyperplasia (15, 16). Importantly, the phenotype observed in mice is fairly well recapitulated also in humans affected by the syndrome of multiple endocrine neoplasia (MEN) that carry germline CDKN1B mutations (17).
These molecular and preclinical evidences have acquired particular clinical interest in the last few years, when mutations of CDKN1B gene were unexpectedly identified as driver mutations in selected types of human cancers. Deletion, methylation or mutations of CDKN1B gene have been considered very rare for a long period of time. Consistently, loss of p27 in tumors has been always ascribed to an accelerated proteolysis (18, 19). However, the advent of massive parallel sequencing that allowed to analyze the cancer genome at very high sensitivity has subverted this notion and indicated that CDKN1B is mutated at a relatively high frequency in prostate cancer (PC) (20), small intestine neuroendocrine tumors (SI-NET) (21) and, especially, in luminal breast cancer (LBC) (22).
It is well established that, although all cancers carry many somatic mutations in their genomes, only a subset of those, known as driver mutations, confers clonal selective advantage to cancer cells and are implicated in oncogenesis. These mutations are opposed to the remaining ones that are referred to as passenger mutations (23). Mutations targeting CDKN1B have been considered driver in the above-mentioned sporadic tumors, as well as in the MEN familial syndromes.
In this review we aim to gather all reported mutations of CDKN1B in cancer and all related biological consequences, focusing our attention on human malignancies where pathogenic variants of CDKN1B are thought to give a selective clonal advantage and particularly discussing the significance of the mutations occurring in the IDR of the protein.
CDKN1B Mutational Status in Human Malignancies
In order to have a comprehensive overview of the mutational status of p27 in all human malignancies, we interrogated the COSMIC, ICGC, cBioPortal, and TRANSFAC data portals and the current literature in PubMed, and then annotated all pathogenic mutations described for CDKN1B (Table 1, Supplementary Table 1). From our analysis we excluded the known polymorphism V109G that in some articles was reported as a mutation.
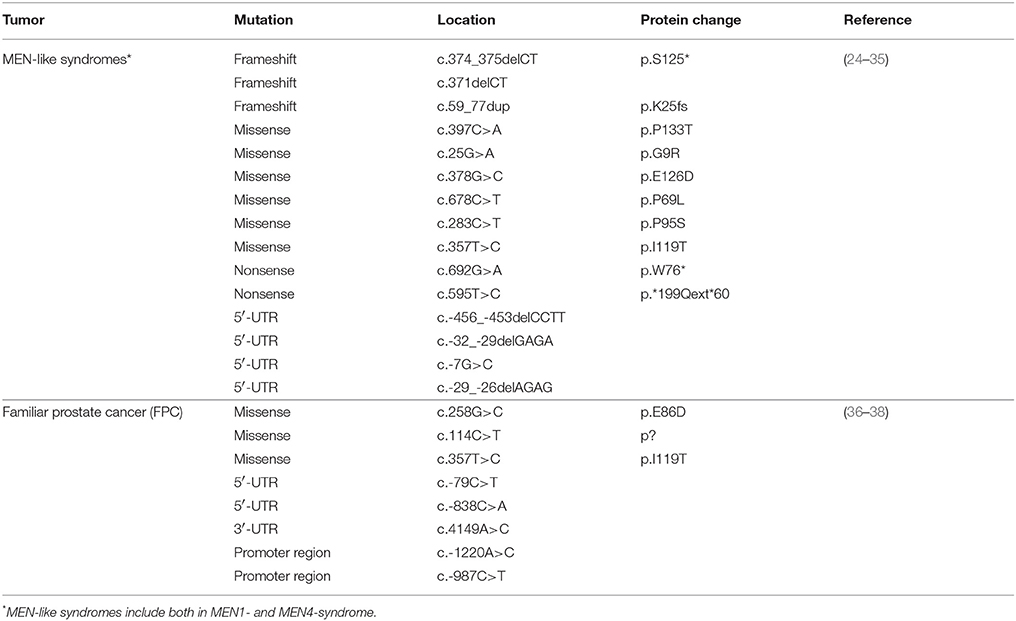
Table 1. CDKN1B mutations in hereditary syndromes.
Among sporadic cancers, CDKN1B mutations were present in a significant proportion of analyzed PC, LBC, and SI-NET. We will therefore describe more in detail the mutations and their possible significance in these tumor types. Among neoplastic hereditary syndromes, we will focus our attention on MEN1 and MEN4, a MEN1-like syndrome, and on familial prostate cancer (FPC), since these are the two types of hereditary syndromes in which variants of CDKN1B have been reported to be more frequent and of higher pathological significance.
An intriguing observation was that we consistently noticed a different distribution and type of mutations in the CDKN1B gene, between hereditary syndromes and sporadic cancers. While in sporadic tumors somatic CDKN1B mutations are mostly located in the protein coding sequence (CDS), CDKN1B germline variants are fairly distributed along the whole sequence, both in the CDS and in untranslated regions (UTRs) (Table 1, Supplementary Table 1). Moreover, in hereditary syndromes most of germline mutations affecting the CDS are missense mutations, which result in a single amino acid change. Conversely, in sporadic cancers frameshift or nonsense mutations are largely predominant (Figure 2A). These mutations often result in the production of truncated forms of p27 protein lacking the C-terminus, which encodes for the p27 IDR.
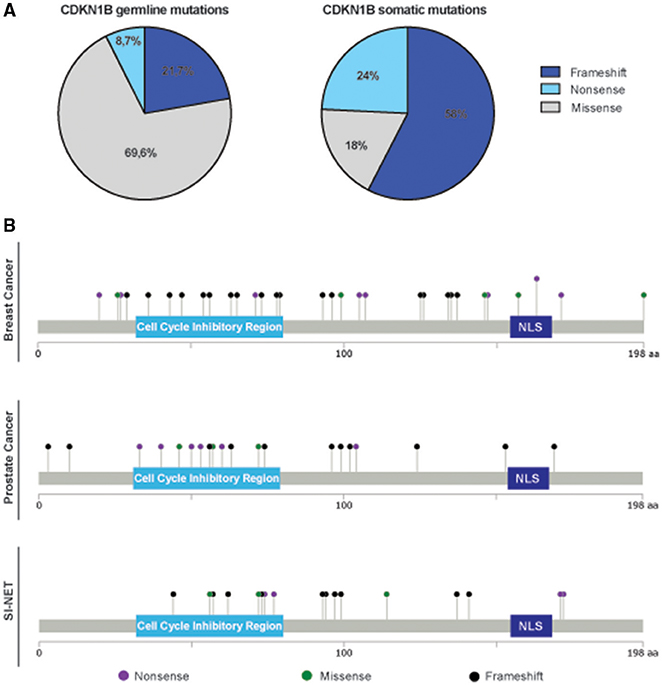
Figure 2. Distribution of germline and somatic CDKN1B mutations. (A) Pie graphs report the distribution of germline mutations in hereditary syndromes (left) and somatic ones in sporadic cancers (right). The percentage of each type of pathogenic variant (frameshift, including both insertion and deletions, missense, and nonsense) is reported. (B) Distribution of mutations identified in breast cancer (top), prostate cancer (middle), and SI-NET (bottom) are depicted along the sequence of p27 protein.
Hereditary Syndromes
Multiple Endocrine Neoplasia Syndromes
Multiple endocrine neoplasia type 1 (MEN1) is a rare syndrome characterized by occurrence of parathyroid, pancreatic, and pituitary tumors. MEN1 syndrome can be either inherited, as an autosomal-dominant disorder, or developed during life as a sporadic event. Although MEN1 syndrome is generally due to mutations in the MEN1 gene (39), 5–10% of MEN1 patients display mutations in different genes (17, 40).
After the identification of a novel p27 mutation (p27 p.177fs) in a rat model of MEN syndrome, Pellegata and colleagues reported the discovery of a germline nonsense mutation (p.W76*) in heterozygosis in a patient suspected for MEN1 syndrome, but negative for MEN1 mutations (33, 41). This mutation led to a truncation of the resulting p27 protein that lost its nuclear localization signal (NLS) and was thereby retained in the cytoplasm where it could not fully exert its cell cycle inhibitory function. These studies, followed by the discovery of other MEN patients carrying CDKN1B mutation (see below), allowed the recognition of a novel human form of multiple endocrine neoplasia that includes MEN1-related tumors and that was named MEN4 (33).
Among the pathogenic variants reported in MEN4 families (Table 1), the vast majority are missense mutations located in the coding sequence. Some of described mutations have been functionally studied in in vitro models and are known to result in altered functionality of p27. For instance, it has been established that the nucleotide substitutions c.678 C>T (p.P69L) and c.283 C>T (p.P95S) disrupt p27 binding to GRB2 and CDK2, respectively, while the in frame deletions c.374_375delCT, the c.59_77dup, and 371delCT affect p27 nuclear localization (35). However, in some cases no clear alteration in p27 functions could be observed as a consequence of germline mutations identified in MEN4 families. This is, for example, the case of the c.397C>A (p.P133T) missense mutations that, using in silico analyses, have been predicted to be well tolerated in a physiological context (42). It is however intriguing to consider that both these mutations create new possible phosphorylation sites in the unstructured region, IDR of the protein, suggesting that an alteration of the binding to specific partners and/or of the engagement in non-canonical pathways could be occurring in the presence of these mutations. These are however only suggestive hypotheses that will need formal experimental demonstration to assess their validity.
Interestingly, there is also a group of germline CDKN1B mutations affecting p27 expression by altering the UTRs. For instance, the deletions c.-456_-453delCCTT and c.-32_29delGAGA lead to an impairment of the mRNA ribosome entry, on one hand lengthening the uORF (upstream Open Reading Frame), on the other disrupting the mRNA secondary structure (29, 31). Considering the notion that IDRs-containing proteins preferentially undergo on-site synthesis to ensures specific localization of the protein, we can well hypothesize that these mutations, altering uORFs or disrupting mRNA secondary structure, may alter p27 mRNA abundance, localization and asymmetric translation, eventually impacting on protein function and/or interaction.
Furthermore, it is interesting to note that also in the absence of p27 mutations, MEN1-3 syndromes might ultimately rely on altered p27 regulation and/or expression. In fact, MEN1 gene directly regulates CDKN1B transcription and TGFβ pathway (43–45), the latter eventually strongly impinging on p27 nuclear functions (46, 47). MEN2-3 syndromes are characterized by activating mutation of the RET proto-oncogene (33), that also controls p27 expression and functions (48, 49). Thus, together these data suggest that altered p27 functions, due to specific signaling alterations or to direct genetic inactivation, could be a common feature of MEN syndromes. Whether this role of CDKN1B in MEN syndromes is due to the peculiar functions exerted by p27 in these tissues or to the interaction between p27 and specific signaling transduction pathway(s) necessary for MEN onset, it is an issue that remains to be fully clarified.
Familial Prostate Cancer
Several lines of evidence from clinical and experimental studies demonstrate that there is a linkage between the chromosomal location of the CDKN1B gene (12p13) and prostate cancer susceptibility in a considerable number of FPC (36). To test whether CDKN1B alteration contributes to an increased risk of prostate cancer, Chang and colleagues systematically sequenced 96 probands of families with prostate carcinoma. They identified a total of 10 germline CDKN1B variants, equally distributed in the ~800 bp promoter region, in the three exons and in the exon-intron junctions. Consulting the TRANSFAC database, the nucleotide changes c.-1220T>G, c.-987C>T, and c.-838C>A alter the binding sequences for transcription factors such as Pbx-1a, SP1, and E2F, respectively and could therefore affect the transcription of p27. The c.-79C>T was found to perturb one CpG site in a CpG island, known to be close to the transcription start site of a gene. The missense mutation p.E86D falls in the cell cycle inhibitory region and, as a consequence, could alter the binding of p27 protein with the cyclin/CDK complexes, although this hypothesis has not been experimentally confirmed. Among the other missense mutations described by the authors, they also propose that the p.I119T might alter the interaction between p27 and its negative regulator JAB1, which binds p27 between the amino acid residues 97 and 151 and is necessary for its cytoplasmic shuttling from the nucleus. Of note, they also identified the substitutions c.-79C>T and c.258G>C that fall in potentially exonic splice enhancer elements area36.
To sum up the results on germline CDKN1B mutations described so far, we can draw some interesting conclusions. First of all, homozygous germline loss of CDKN1B has never been found, suggesting that this possibility could be not compatible with life in humans. Second, germinal CDKN1B mutations principally determine an unbalance of the fine-tuned protein expression and/or localization, suggesting that small variation of p27 could be sufficient to promote transformation, at least in parathyroid, anterior pituitary adrenals, kidneys, and reproductive organs (MEN4 syndrome) or in prostate (FPC). This observation is substantially in line with the haploinsufficient role as tumor suppressor gene, ascribed to p27 many years ago, by the study of genetically modified mouse models.
Somatic Cancers
As mentioned above, prostate cancer (PC), luminal breast cancer (LBC), and small intestine neuroendocrine tumors (SI-NET) are the cancer subtypes in which CDKN1B mutations have been identified as driver genetic lesions in a significant percentage of cases (Supplementary Table 1). Common characteristic of these tumors is that they rely on hormones for their growth. Since these patients are typically treated with radio- and endocrine-therapies and can develop resistance to these first-line treatments and a role for p27 in the response to both radiation- and endocrine-therapy has been proposed, it will be important to ascertain whether CDKN1B mutational status in these tumors could act not only as genetic driver but also as predictor of therapy response.
CDKN1B Mutations in Breast Cancer
The downregulation of p27 protein was first described in breast cancer (BC), where it had significance of poor prognosis (50, 51). These data were then confirmed by several subsequent studies that, interestingly, identified high p27 expression not only as a marker of good prognosis but also as an independent predictor of responsiveness to hormonal therapy (52).
Even though the first attempts to identify CDKN1B mutation in BC were unsuccessful (53), more recent efforts by whole genome sequencing revealed that mutations affecting CDKN1B are present and can represent driver genetic lesions in the estrogen receptor (ER)-positive (LBC) subtype. The mutations identified in LBC are reported in Figure 2B. Ellis and colleagues sequenced 77 biopsies from LBC patients enrolled in two clinical trials testing aromatase inhibitors in neoadjuvant setting, to identify driver genetic lesions involved in drug response (22). Aromatase inhibitors are widely used to treat hormone-sensitive BC in the adjuvant- and in first line metastatic-setting (relapse of the disease after Tamoxifen treatment) (54), to prevent the biosynthesis of estrogens from androgens. The authors describe CDKN1B as one of the most significantly mutated gene in this cohort and, interestingly, several of the reported mutations resided in the C-terminal portion of p27 (p.E171*, p.K134fs*, p.P137fs*, p.Q163*; p.T198A) (22). In the same time, another group also sequenced the coding exons of p27 in 100 primary LBC tumors, reaching very similar conclusions and confirming two CDKN1B truncating mutations. Interestingly, for the first time this work reported the discovery of biallelic inactivation of CDKN1B, a feature very commonly found in recessive tumor suppressor genes (55). Some of the mutations described were at the N-terminus and were very likely affecting the cyclin/CDK binding domain of p27 (e.g., p.T42I) and/or determined a substantial loss of the protein functions (e.g., p.C29fs*12; p.E71*, and p.K73fs).
Among the different mutations described in LBC, there are two that have been extensively characterized in experimental models, namely the p.T198A and p.E171* [(6, 8, 47, 56–59)]. The p.E171* mutation results in the loss of 28 C-terminal amino acids, which are necessary for the interaction with stathmin. This interaction can impinge on mechanisms regulating cancer cell migration and also vesicular trafficking and receptor recycling [(6, 8, 57–59)]. The residue threonine 198 is important for p27 stability and to control cell motility and, accordingly, both these processes are altered in cells expressing the p.T198A mutant (47, 56, 59). It is interesting to note that both these mutations impinge on the ability of p27 to interact with the microtubule destabilizing protein stathmin (6, 59), suggesting that these interaction could have a role in tumor onset and/or progression. Intriguingly, both stathmin and p27 are IDPs and are fully regulated by post-transcriptional modifications. Their interaction seems to have a prominent role in the regulation of microtubule dynamics, eventually affecting several cellular functions such as proliferation, motility, and invasion.
Overall, these data indicate that, in primary LBC, CDKN1B mutations are mostly located in the C-terminal portion of p27, possibly contributing to alteration of protein localization and stability that, in turn, may profoundly affect cancer cell proliferation and motility.
Very recently, Yates and colleagues reported a deeper characterization of driver mutations by sequencing all coding exons of 365 cancer genes, in 756 primary tumors and in 227 samples from either distant or loco regional relapses. Then, they split the primary and relapse cohorts with respect to ER status. Only 15, out of the 365 genes analyzed, were identified as significantly mutated in primary tumors and/or recurrent/metastatic diseases. Among those, CDKN1B resulted significantly mutated in both ER-positive and -negative primary tumors, but not in metastatic/recurrent BC samples, overall confirming the pathological relevance of CDKN1B in BC and putting a particular accent on its involvement more during early steps of tumor initiation rather than during cancer progression (60).
CDKN1B Mutations in Prostate Cancer
PC is the most common malignancy in men worldwide. The standard therapy for PC is represented by radiotherapy coupled with surgical or medical castration. However, more than half of patients develop resistance to therapy and develop metastatic castration-resistant PC (CRPC) (61). Although several new anti-tumoral drugs are currently under clinical development, including compounds targeting the androgen receptor (AR) signaling axis, no therapy is currently curative for CRPC and no biological biomarker is currently available to predict patients' response (62). Notably, it has been demonstrated that p27 constrains PC growth in mice and the FOXA1 transcription factor, known to modulate AR-driven transcription, also induces the expression of CDKN1B (20). Recent genome wide studies provided an exhaustive overview of mutations occurring in local, metastatic or lethal PC [(20, 63, 64)]. In these reports, several genes whose products regulate AR function are frequently mutated or altered, both in primary and metastatic therapy-resistant tumors, suggesting that deregulation of the AR pathway could be an early event during prostate tumorigenesis (61). Barbieri et al. performed exome sequencing followed by paired-end, massive parallel sequencing of 112 primary prostate adenocarcinomas and matched normal samples, providing a cross section of PC genomes at diagnosis before treatment. CDKN1B has been identified as one of the most frequently somatically altered genes, mutated in three and deleted in sixteen tumor samples. The authors described one new missense mutation, p.E46G, which resides in the nuclear exportation signal of p27, and two new frameshift mutation, p.R169fs* and p.R152fs*, both located in the nuclear localization signal of p27, leading to loss of the IDR region and likely protein displacement in the cytoplasm (63).
Robinson et al. conducted a systematic study of 150 metastatic pre-treated CRPCs to determine the landscape of somatic genomic alterations in this cohort. All alterations described in CDKN1B gene in this study, mainly frameshift mutations, were potentially driver and their overall incidence in the cohort reached the 4%. One frameshift variant was intronic, the others were the p.KGAC96fs and the p.P94fs, both likely leading to expression of a loss-of-function phenotype (65).
Grasso et al. reported the exome sequencing of 50 lethal-, heavily treated-, metastatic-CRPCs, obtained at autopsy of the patients. The authors describe a new frameshift mutation, the p.P101fs*, but this somatic variant did not result as driver in this cohort of clinical samples (66).
Beltran et al. investigated the mutational pattern of 45 formalin-fixed paraffin-embedded specimens from patients with localized PC, metastatic hormone-naïve PC and CRPC, but they did not find any considerable alteration in CDKN1B gene (62). However, given the heterogeneity and the small number of cases per cohort, it is conceivable that the presence of CDKN1B pathogenic variants could be underestimated in this study.
Altogether, mutations or deletions in CDKN1B have been often recognized as driver genetic lesions in primary and metastatic CPRCs, whereas their frequency in lethal metastatic CPRCs is not high enough to be considered clonal events. In support to this view, Baca et al. analyzed the clonal evolution of PC examining large genomic somatic rearrangements in 55 primary prostate adenocarcinoma and 2 metastatic Neuro Endocrine Prostate Cancer (NEPC). This study led to the conclusion that in PC large translocations and deletions arise in a highly interdependent manner. The authors name the emergence of these large genomic alterations as “chromoplexy” and suggest that chromoplexy disrupts multiple cancer genes in a coordinate manner, eventually leading to PC progression. Relevant to the topic of our review, Baca et al. reach the conclusion that CDKN1B inactivation by chromoplexy occurs frequently as a subclonal event and, therefore, could be linked with PC progression, possibly leading to genomic instability, proliferation and/or evasion of apoptosis (67).
Finally, the recently published atlas of metastatic tumors, collected from 10,336 patients (www.cbioportal.org/study?id=msk_impact_2017#) has reported the presence of CDKN1B mutations in 84 cases (somatic mutation rate = 0.7%). Altered allele frequency in these samples varied from 5 to 90%. Again, mutation of CDKN1B in metastatic tumors was restricted to some specific cancer types, including PC and BC. Among these, 5/500 (1%) PCs were found mutated and mutations included 1 missense, 1 nonsense, and 3 frameshift, at a frequency of 30–40%, overall confirming the driver role of CDKN1B mutation in, at least, a subset of PCs.
CDKN1B Mutations in Small Intestine-Neuroendocrine Tumors (SI-NET)
SI-NETs are rare tumors that derive from enterochromaffin cells of the neuroendocrine system of the gut. These cells contain a large amount of our body storage of serotonin. Clear information about how these tumors develop is limited (68), but recent studies tried to shed light on the genomic landscape of SI-NETs. Banck and colleagues have first analyzed 48 SI-NETs by massive parallel exome sequencing, both in tumors and in their normal tissue counterpart. They identified mutations in different genes implicated in cancer-related pathways, although none of them was consistently altered in a significant proportion of analyzed samples (69). However, in a subsequent study, Francis et al. profiled 55 tumors from 50 individuals, using a combination of whole-exome and whole-genome sequencing. The only gene identified as significantly mutated by this analysis was CDKN1B. In total, small insertion and deletions were found in the 10% of cases (Figure 2B). To confirm these data, they sequenced CDKN1B with a 800-fold mean coverage samples from two independent cohorts of SI-NETs, the one already sequenced by Banck et al. (48 samples) and another comprising 81 SI-NETs (21). Recurrent insertions and deletions leading to frameshift were identified in CDKN1B, in 14 out of 180 samples evaluated (8% of the individuals analyzed). In addition, hemizygous deletions, encompassing CDKN1B gene, were detected in 7 out of 50 patients. Altogether, the work from Francis et al. not only confirmed the previous findings but also found new heterozygous frameshift mutations in CDKN1B, strengthening the notion that this gene acts as a haploinsufficient tumor-suppressor in SI-NETs. These data were further confirmed in a larger study comprising 362 samples from 200 SI-NET patients. This work also led to the detection of CDKN1B mutations in 8% of the patients, making of CDKN1B the most frequently mutated gene in SI-NETs (70). However, it has to be highlighted that the expression of p27 protein did not correlate with CDKN1B mutational status and no clear difference in the clinical characteristics between CDKN1B mutated and CDKN1B wild type tumor carriers were found.
The works on SI-NETs give some interesting new insights on the possible role(s) and significance of CDKN1B mutations in human cancer. First, some CDKN1B mutations could be revealed only when NGS was carried out at high coverage (i.e., 800X) (21). If this was due to the subclonal nature of some CDKN1B mutations will be probably matter of future investigation. As for now, we can speculate that it may be worth using the same approach in LBC and PC, where total frequency reported for CDKN1B mutations is currently at 2%, but this percentage results from a whole-exome sequencing approach with relatively low coverage.
Second, the works on SI-NET confirm once again that CDKN1B acts as haploinsufficient tumor-suppressor, as already observed in different mouse models. Whether the same applies to other and more frequent cancers, such as LBC and PC, is something that certainly needs to be further investigated in the future.
Conclusions
Our review of the literature and available datasets reporting the mutations of CDKN1B gene in human cancers indicates that these events are generally rare in sporadic cancer, with the exception of LBC, PC, and SI-NET. However, also in these tumors the frequency of CDKN1B mutation is below the 10% of the cases, partially supporting the old evidence that p27 expression is mainly deregulated at post-transcriptional level in human cancer.
A second relevant observation is that mutations encompassing CDKN1B gene are mainly subclonal. This has been proven in PC when large genomic rearrangements were studied (67).
The most intriguing observation is that ~80% of mutated tumor samples display nonsense or frameshift mutations at the C-terminus of the protein, leading to the formation of a truncated protein. This evidence has, in our opinion, two potential readings: the first is that the IDR of p27 plays relevant tumor suppressive activities in sporadic human cancer; the second is that the cytoplasmic displacement of p27, due to the loss of its nuclear localization signal (located in the C-terminus of the protein) is sufficient to drive tumor progression. Yet, this latter hypothesis is weakened by the notion that many functions of p27 have been ascribed to cytosolic interactions with other proteins, mediated by the C-terminal part of the protein. It is to note that, at least in PC and in LBC, CDKN1B mutations are less frequent in more aggressive/recurrent than in primary tumors, suggesting that the presence of these mutations could be linked to a more favorable scenario with respect to the one having a complete loss of function of p27 protein.
Furthermore, it is well known that p27 expression can be profoundly regulated by the expression of microRNAs, particularly miR-221 and miR-222. Therefore, it will be important to take into account that not only mutations occurring in 5′ UTR and in CDS, but also those in the 3′ UTR (for instance identified in one case of FPC) could contribute to alter the levels of p27 protein expression, eventually affecting cell transformation. Notably, while CDKN1B mRNA spans 2,535 base pair (bp), CDKN1B CDS only covers 596 bp, suggesting that regulatory regions present in the 5′ UTR (571 bp) and, especially, in the 3′ UTR (1,368 bp) could have a functional relevance. Moreover, we recently showed that p27 protein directly binds selected microRNAs eventually altering the proliferation of normal and cancer cells under specific culture condition. In particular, we identified an interaction between p27 and miR-223 that regulated cell proliferation/cell cycle arrest after achievement of cell-cell contact (71). Interestingly, we also verified that CDKN1B mutations identified in LBC could affect the ability of p27 to bind miR-223 (71).
Altogether, from the review of existing literature it is clear that many new insights, previously not hypothesized, on CDKN1B role and implications in cancer have been disclosed by the recent advent of next generation sequencing technologies. Nevertheless, many observations suggest that the interplay between CDKN1B mutations and p27 protein expression and function is far to be completely clarified and certainly merits further investigation, in particular with regard to the reciprocal modulation with non coding RNAs.
More experimental work will be necessary to fully clarify the role of p27 in tumor suppression and the significance of its genetic or functional deregulation in familial and sporadic human cancers, especially in those tumors driven by hormonal factors. Combining genetic and functional studies and using the most appropriate model systems will be mandatory to reach this important goal and establish at which extent each specific mutation identified does effectively impact on cancer initiation, progression, and/or on treatment response.
Author Contributions
MC, GM, MD, GB contributed to the revision of literature. MC, GM, MD, IS, BB, and GB wrote the paper. All authors read, corrected and approved the paper. MC, GM, MD equally contributed to this work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
We like to thank all members of the S.C.I.C.C. lab for critical discussions and comments on the paper. This work was supported by the Associazione Italiana Ricerca sul Cancro (AIRC) to BB (IG 20061) and to GB (IG 16865); by CRO Intramural Research Grant (5X1000_2016_MdS) to IS; by L.R. 17/2014-Regione FVG (TuMaGiDo) to GB; by Associazione Italiana Ricerca sul Cancro (AIRC fellowships) to IS (#18171).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2018.00393/full#supplementary-material
References
1. Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. (1994) 8:9–22. doi: 10.1101/gad.8.1.9
2. Nakanishi M, Robetorye RS, Adami GR, Pereira-Smith OM, Smith JR. Identification of the active region of the DNA synthesis inhibitory gene p21Sdi1/CIP1/WAF1. EMBO J. (1995) 14:555–63.
3. Russo AA, Jeffrey PD, Patten AK, Massagué J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inibitor bound to the cyclin A–Cdk2 complex. Nature (1996) 382:325–31. doi: 10.1038/382325a0
4. Belletti B, Nicoloso M, Schiappacassi M, Chimienti E, Berton S, Lovat F, et al. p27kip1 functional regulation in human cancer: a potential target for therapeutic designs. Curr. Med. Chem. (2005) 12:1589–605. doi: 10.2174/0929867054367149
5. Sharma SS, Pledger WJ. The non-canonical functions of p27 Kip1 in normal and tumor biology. Cell Cycle (2016) 15:1189–201. doi: 10.1080/15384101.2016.1157238
6. Baldassarre G, Belletti B, Nicoloso MS, Schiappacassi M, Vecchione A, Spessotto P, et al. p27Kip1-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell (2005) 7:51–63. doi: 10.1016/j.ccr.2004.11.025
7. Berton S, Pellizzari I, Fabris L, D'Andrea S, Segatto I, Canzonieri V, et al. Genetic characterization of p27 kip1 and stathmin in controlling cell proliferation in vivo. Cell Cycle (2014) 13:3100–11. doi: 10.4161/15384101.2014.949512
8. Fabris L, Berton S, Pellizzari I, Segatto I, D'Andrea S, Armenia J, et al. p27 kip1 controls H-Ras/MAPK activation and cell cycle entry via modulation of MT stability. Proc. Natl. Acad. Sci. U.S.A. (2015) 112:13916–21. doi: 10.1073/pnas.1508514112
9. Pellizzari I, Fabris L, Berton S, Segatto I, Citron F, D'Andrea S, et al. (2016). p27kip1 expression limits H-Ras-driven transformation and tumorigenesis by both canonical and non-canonical mechanisms. Oncotarget 7:64560–74. doi: 10.18632/oncotarget.11656
10. Besson A. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. (2006) 20:47–64. doi: 10.1101/gad.1384406
11. Li H, Collado M, Villasante A, Matheu A, Lynch CJ, Cañamero M, et al. p27Kip1 Directly represses Sox2 during embryonic stem cell differentiation. Cell Stem Cell (2012) 11:845–52. doi: 10.1016/j.stem.2012.09.014
12. Larrea MD, Wander SA, Slingerland J. p27 as Jekyll and Hyde: regulation of cell cycle and cell motility. Cell Cycle (2009) 8:3455–61. doi: 10.4161/cc.8.21.9789
13. Babu MM. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem Soc Trans. (2016) 44:1185–200. doi: 10.1042/BST20160172
14. Das RK, Huang Y, Phillips AH, Kriwacki RW, Pappu RV. Cryptic sequence features within the disordered protein p27 Kip1 regulate cell cycle signaling. Proc Natl Acad Sci USA. (2016) 113:5616–21. doi: 10.1073/pnas.1516277113
15. Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature (1998) 396:177–80. doi: 10.1038/24179
16. Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, et al. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell (1996) 85:707–20. doi: 10.1016/S0092-8674(00)81237-4
17. Lee M, Pellegata NS. Multiple endocrine neoplasia type 4. In: Stratakis CA editor. Frontiers of Hormone Research. Basel: S. KARGER AG (2013). p. 63–78.
18. Loda M, Cukor B, Tam SW, Lavin P, Fiorentino M, Draetta GF, et al. Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat Med. (1997) 3:231–4. doi: 10.1038/nm0297-231
19. Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, et al. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science (1995) 269:682–5. doi: 10.1126/science.7624798
20. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat J.-P., et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. (2012) 44:685–9. doi: 10.1038/ng.2279
21. Francis JM, Kiezun A, Ramos AH, Serra S, Pedamallu CS, Qian ZR, et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat Genet. (2013) 45:1483–6. doi: 10.1038/ng.2821
22. Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, et al. (2012). Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486:353–60. doi: 10.1038/nature11143
23. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature (2009) 458:719–24. doi: 10.1038/nature07943
24. Costa-Guda J, Soong C.-P., Parekh VI, Agarwal SK, Arnold A. Germline and somatic mutations in cyclin-dependent kinase inhibitor genes CDKN1A, CDKN2B, and CDKN2C in sporadic parathyroid adenomas. Horm Cancer (2013) 4:301–7. doi: 10.1007/s12672-013-0147-9
25. Dénes J, Swords F, Rattenberry E, Stals K, Owens M, Cranston T, et al. Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: results from a large patient cohort. J Clin Endocrinol Metab. (2015) 100:E531–41. doi: 10.1210/jc.2014-3399
26. Elston MS, Meyer-Rochow GY, Dray M, Swarbrick M, Conaglen JV. Early onset primary hyperparathyroidism associated with a novel germline mutation in CDKN1B. Case Rep Endocrinol. (2015) 2015:1–4. doi: 10.1155/2015/510985
27. Georgitsi M, Raitila A, Karhu A, van der Luijt RB, Aalfs CM, Sane T, et al. Germline CDKN1B /p27 Kip1 mutation in multiple endocrine neoplasia. J Clin Endocrinol Metab. (2007) 92:3321–5. doi: 10.1210/jc.2006-2843
28. Lauter KB, Arnold A. Mutational analysis of CDKN1B, a candidate tumor-suppressor gene, in refractory secondary/tertiary hyperparathyroidism. Kidney Int. (2008) 73:1137–40. doi: 10.1038/ki.2008.28
29. Malanga D, De Gisi S, Riccardi M, Scrima M, De Marco C, Robledo M, et al. Functional characterization of a rare germline mutation in the gene encoding the cyclin-dependent kinase inhibitor p27Kip1 (CDKN1B) in a Spanish patient with multiple endocrine neoplasia-like phenotype. Eur J Endocrinol. (2012) 166:551–60. doi: 10.1530/EJE-11-0929
30. Molatore S, Marinoni I, Lee M, Pulz E, Ambrosio MR, Uberti EC, et al. A novel germline CDKN1B mutation causing multiple endocrine tumors: clinical, genetic and functional characterization. Hum Mutat. (2010) 31:E1825–35. doi: 10.1002/humu.21354
31. Occhi G, Regazzo D, Trivellin G, Boaretto F, Ciato D, Bobisse S, et al. A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PLoS Genet. (2013) 9:e1003350. doi: 10.1371/journal.pgen.1003350
32. Pardi E, Mariotti S, Pellegata NS, Benfini K, Borsari S, Saponaro F, et al. Functional characterization of a CDKN1B mutation in a Sardinian kindred with multiple endocrine neoplasia type 4. Endocr Connect. (2014) 4:1–8. doi: 10.1530/EC-14-0116
33. Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Hofler H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA. (2006) 103:15558–63. doi: 10.1073/pnas.0603877103
34. Sambugaro S, Di Ruvo M, Ambrosio MR, Pellegata NS, Bellio M, Guerra A, et al. Early onset acromegaly associated with a novel deletion in CDKN1B 5′UTR region. Endocrine (2015) 49:58–64. doi: 10.1007/s12020-015-0540-y
35. Tonelli F, Giudici F, Giusti F, Marini F, Cianferotti L, Nesi G, et al. A heterozygous frameshift mutation in exon 1 of CDKN1B gene in a patient affected by MEN4 syndrome. Eur J Endocrinol. (2014) 171, K7–17. doi: 10.1530/EJE-14-0080
36. Chang B, Zheng SL, Isaacs SD, Wiley KE, Turner A, Li G, et al. A polymorphism in the CDKN1B gene is associated with increased risk of hereditary prostate cancer. Cancer Res. (2004) 64:1997–9. doi: 10.1158/0008-5472.CAN-03-2340
37. Kluth M, Ahrary R, Hube-Magg C, Ahmed M, Volta H, Schwemin C, et al. Genomic deletion of chromosome 12p is an independent prognostic marker in prostate cancer. Oncotarget (2015) 6:27966–79. doi: 10.18632/oncotarget.4626
38. Xu J, Langefeld CD, Zheng SL, Gillanders EM, Chang B, Isaacs SD, et al. Interaction effect of PTEN and CDKN1B chromosomal regions on prostate cancer linkage. Hum Genet. (2004) 115:255–62. doi: 10.1007/s00439-004-1144-4
39. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1). Best Pract Res Clin Endocrinol Metab. (2010) 24:355–70. doi: 10.1016/j.beem.2010.07.003
40. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol. (2014) 386:2–15. doi: 10.1016/j.mce.2013.08.002
41. Fritz A, Walch A, Piotrowska K, Rosemann M, Schäffer E, Weber K, et al. Recessive transmission of a multiple endocrine neoplasia syndrome in the rat. Cancer Res. (2002) 62:3048–51.
42. Bugalho MJ, Domingues R. (2016). Uncommon association of cerebral meningioma, parathyroid adenoma and papillary thyroid carcinoma in a patient harbouring a rare germline variant in the CDKN1B gene. BMJ Case Rep. 2016:bcr2015213934. doi: 10.1136/bcr-2015-213934
43. Dreijerink KMA, Mulder KW, Winkler GS, Höppener JWM, Lips CJM, Timmers HTM. Menin links estrogen receptor activation to histone H3K4 trimethylation. Cancer Res. (2006) 66:4929–35. doi: 10.1158/0008-5472.CAN-05-4461
44. Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, et al. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci USA. (2005) 102:14659–64. doi: 10.1073/pnas.0503484102
45. Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci USA. (2005) 102:749–54. doi: 10.1073/pnas.0408836102
46. Depoortere F, Pirson I, Bartek J, Dumont JE, Roger PP. Transforming growth factor beta 1 selectively inhibits the cyclic AMP-dependent proliferation of primary thyroid epithelial cells by preventing the association of cyclin D3-cdk4 with nuclear p27kip1. Mol Biol Cell (2000) 11:1061–76. doi: 10.1091/mbc.11.3.1061
47. Lovisa S, Citro S, Sonego M, Dall'Acqua A, Ranzuglia V, Berton S, et al. SUMOylation regulates p27 Kip1 stability and localization in response to TGFβ. J Mol Cell Biol. (2016) 8:17–30. doi: 10.1093/jmcb/mjv056
48. Baldassarre G, Bruni P, Boccia A, Salvatore G, Melillo RM, Motti ML, et al. Glial cell line-derived neurotrophic factor induces proliferative inhibition of NT2/D1 cells through RET-mediated up-regulation of the cyclin-dependent kinase inhibitor p27(kip1). Oncogene (2002) 21:1739–49. doi: 10.1038/sj.onc.1205226
49. Vitagliano D, Carlomagno F, Motti ML, Viglietto G, Nikiforov YE, Nikiforova MN, et al. Regulation of p27Kip1 protein levels contributes to mitogenic effects of the RET/PTC kinase in thyroid carcinoma cells. Cancer Res. (2004) 64:3823–9. doi: 10.1158/0008-5472.CAN-03-3918
50. Catzavelos C, Bhattacharya N, Ung YC, Wilson JA, Roncari L, Sandhu C, et al. Decreased levels of the cell-cycle inhibitor p27Kip1 protein: prognostic implications in primary breast cancer. Nat Med. (1997) 3:227–30. doi: 10.1038/nm0297-227
51. Porter PL, Malone KE, Heagerty PJ, Alexander GM, Gatti LA, Firpo EJ, et al. Expression of cell-cycle regulators p27Kip1 and cyclin E, alone and in combination, correlate with survival in young breast cancer patients. Nat Med. (1997) 3:222–5.
52. Guan X, Wang Y, Xie R, Chen L, Bai J, Lu J, et al. p27Kip1 as a prognostic factor in breast cancer: a systematic review and meta-analysis. J Cell Mol Med. (2010) 14:944–53. doi: 10.1111/j.1582-4934.2009.00730.x
53. Ferrando AA, Balbín M, Pendás AM, Vizoso F, Velasco G, López-Otín C. Mutational analysis of the human cyclin-dependent kinase inhibitor p27kip1 in primary breast carcinomas. Hum Genet. (1996) 97:91–4. doi: 10.1007/BF00218840
54. Fabian CJ. The what, why and how of aromatase inhibitors: hormonal agents for treatment and prevention of breast cancer: hormonal agents for treatment and prevention of breast cancer. Int J Clin Pract. (2007) 61:2051–63. doi: 10.1111/j.1742-1241.2007.01587.x
55. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, et al. (2012). The landscape of cancer genes and mutational processes in breast cancer. Nature 486:400–4. doi: 10.1038/nature11017
56. Kossatz U, Vervoorts J, Nickeleit I, Sundberg HA, Arthur JSC, Manns MP, et al. C-terminal phosphorylation controls the stability and function of p27kip1. EMBO J. (2006) 25:5159–70. doi: 10.1038/sj.emboj.7601388
57. Berton S, Belletti B, Wolf K, Canzonieri V, Lovat F, Vecchione A, et al. The tumor suppressor functions of p27(kip1) include control of the mesenchymal/amoeboid transition. Mol Cell Biol. (2009) 29:5031–45. doi: 10.1128/MCB.00144-09
58. Belletti B, Pellizzari I, Berton S, Fabris L, Wolf K, Lovat F, et al. p27kip1 controls cell morphology and motility by regulating microtubule-dependent lipid raft recycling. Mol Cell Biol. (2010) 30:2229–40. doi: 10.1128/MCB.00723-09
59. Schiappacassi M, Lovisa S, Lovat F, Fabris L, Colombatti A, Belletti B, et al. Role of T198 modification in the regulation of p27Kip1 protein stability and function. PLoS ONE (2011) 6:e17673. doi: 10.1371/journal.pone.0017673
60. Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell (2017) 32:169–184.e7. doi: 10.1016/j.ccell.2017.07.005
61. Hieronymus H, Sawyers CL. Traversing the genomic landscape of prostate cancer from diagnosis to death. Nat Genet. (2012) 44:613–4. doi: 10.1038/ng.2301
62. Beltran H, Tomlins S, Aparicio A, Arora V, Rickman D, Ayala G, et al. Aggressive variants of castration-resistant prostate cancer. Clin Cancer Res. (2014) 20:2846–50. doi: 10.1158/1078-0432.CCR-13-3309
63. Barbieri CE, Bangma CH, Bjartell A, Catto JWF, Culig Z, Grönberg H, et al. The mutational landscape of prostate cancer. Eur Urol. (2013) 64:567–76. doi: 10.1016/j.eururo.2013.05.029
64. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell (2010) 18:11–22. doi: 10.1016/j.ccr.2010.05.026
65. Robinson D, Van Allen EM, Wu Y.-M., Schultz N, Lonigro RJ, Mosquera J.-M., et al. Integrative clinical genomics of advanced prostate cancer. Cell (2015) 161:1215–28. doi: 10.1016/j.cell.2015.05.001
66. Grasso CS, Wu Y.-M., Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature (2012) 487:239–43. doi: 10.1038/nature11125
67. Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated evolution of prostate cancer genomes. Cell (2013) 153:666–77. doi: 10.1016/j.cell.2013.03.021
68. Neklason DW, VanDerslice J, Curtin K, Cannon-Albright LA. Evidence for a heritable contribution to neuroendocrine tumors of the small intestine. Endocr Relat Cancer (2016) 23:93–100. doi: 10.1530/ERC-15-0442
69. Banck MS, Beutler AS. Advances in small bowel neuroendocrine neoplasia: Curr Opin Gastroenterol. (2014) 30:163–7. doi: 10.1097/MOG.0000000000000043
70. Crona J, Gustavsson T, Norlén O, Edfeldt K, Åkerström T, Westin G, et al. Somatic mutations and genetic heterogeneity at the CDKN1B locus in small intestinal neuroendocrine tumors. Ann Surg Oncol. (2015) 22:1428–35. doi: 10.1245/s10434-014-4351-9
Keywords: p27Kip1 protein, CDKN1B gene, somatic mutation, germinal mutation, driver mutation, passenger mutation, Intrinsically disordered protein/region
Citation: Cusan M, Mungo G, De Marco Zompit M, Segatto I, Belletti B and Baldassarre G (2018) Landscape of CDKN1B Mutations in Luminal Breast Cancer and Other Hormone-Driven Human Tumors. Front. Endocrinol. 9:393. doi: 10.3389/fendo.2018.00393
Received: 28 March 2018; Accepted: 25 June 2018;
Published: 17 July 2018.
Edited by:
Antonio Brunetti, Università degli Studi Magna Græcia di Catanzaro, ItalyReviewed by:
Eva Surmacz, Temple University, United StatesJudy Sue Crabtree, LSU Health Sciences Center New Orleans, United States
Matteo Fassan, Università degli Studi di Padova, Italy
Copyright © 2018 Cusan, Mungo, De Marco Zompit, Segatto, Belletti and Baldassarre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gustavo Baldassarre, Z2JhbGRhc3NhcnJlQGNyby5pdA==
†These authors have contributed equally to this work.
‡Present Address: Martina Cusan, Department of Systems Biology, NCI Comprehensive Cancer Center, Beckman Research Institute, City of Hope, Monrovia, CA, United States
Mara De Marco Zompit, Department of Gynecology, University of Zurich, USZ, Schlieren, Switzerland