 Iveta Nedvedova1
Iveta Nedvedova1 David Kolar1
David Kolar1 Jan Neckar2Martin Kalous3Michal Pravenec2Jan Šilhavý2Vlasta Korenkova4Frantisek Kolar2
Jan Neckar2Martin Kalous3Michal Pravenec2Jan Šilhavý2Vlasta Korenkova4Frantisek Kolar2 Jitka M. Zurmanova1*
Jitka M. Zurmanova1*- 1Department of Physiology, Faculty of Science, Charles University, Prague, Czechia
- 2Institute of Physiology, Czech Academy of Sciences, Prague, Czechia
- 3Department of Cell Biology, Faculty of Science, Charles University, Prague, Czechia
- 4Institute of Biotechnology, Czech Academy of Sciences, Prague, Czechia
Adaptation to continuous normobaric hypoxia (CNH) protects the heart against acute ischemia/reperfusion injury. Recently, we have demonstrated the infarct size-limiting effect of CNH also in hearts of spontaneously hypertensive rats (SHR) and in conplastic SHR-mtBN strain characterized by the selective replacement of the mitochondrial genome of SHR with that of more ischemia-resistant Brown Norway rats. Importantly, cardioprotective effect of CNH was more pronounced in SHR-mtBN than in SHR. Thus, here we aimed to identify candidate genes which may contribute to this difference between the strains. Rats were adapted to CNH (FiO2 0.1) for 3 weeks or kept at room air as normoxic controls. Screening of 45 transcripts was performed in left ventricles using Biomark Chip. Significant differences between the groups were analyzed by univariate analysis (ANOVA) and the genes contributing to the differences between the strains unmasked by CNH were identified by multivariate analyses (PCA, SOM). ANOVA with Bonferroni correction revealed that transcripts differently affected by CNH in SHR and SHR-mtBN belong predominantly to lipid metabolism and antioxidant defense. PCA divided four experimental groups into two main clusters corresponding to chronically hypoxic and normoxic groups, and differences between the strains were more pronounced after CNH. Subsequently, the following 14 candidate transcripts were selected by PCA, and confirmed by SOM analyses, that can contribute to the strain differences in cardioprotective phenotype afforded by CNH: Alkaline ceramidase 2 (Acer2), Fatty acid translocase (Cd36), Aconitase 1 (Aco1), Peroxisome proliferator activated receptor gamma (Pparg), Hemoxygenase 2 (Hmox2), Phospholipase A2 group IIA (Ppla2g2a), Dynamin-related protein (Drp), Protein kinase C epsilon (Pkce), Hexokinase 2 (Hk2), Sphingomyelin synthase 2 (Sgms2), Caspase 3 (Casp3), Mitofussin 1 (Mfn1), Phospholipase A2 group V (Pla2g5), and Catalase (Cat). Our data suggest that the stronger cardioprotective phenotype of conplastic SHR-mtBN strain afforded by CNH is associated with either preventing the drop or increasing the expression of transcripts related to energy metabolism, antioxidant response and mitochondrial dynamics.
Introduction
Hypertension represents one of the major risk factors for the development of ischemic heart disease (1). The most extensively studied experimental model of essential hypertension is an inbred strain of spontaneously hypertensive rats (SHR). This strain develops hypertension during the 10–15th postnatal week (2). SHR are also frequently used for metabolic syndrome studies due to the development of insulin resistance, hyperinsulinemia, glucose intolerance, and hypertriglyceridemia (3, 4). Moreover, SHR manifested an increased sensitivity to ischemia/reperfusion (I/R) injury when compared to Wistar-Kyoto strain (WKY) (5, 6).
It has been repeatedly shown that cardiac resistance to I/R injury is tightly related to mitochondrial status and energy homeostasis of cardiomyocytes (7). In compliance with this knowledge a unique model of the SHR-mtBN conplastic strain was developed by selective replacement of the mitochondrial genome of the SHR by the mitochondrial genome of normotensive, more ischemia resistant, Brown Norway rat strain (8). Recently, we have shown that the infarct size-limiting effect afforded by adaptation to continuous normobaric hypoxia (CNH; inspired oxygen fraction 0.1) was more pronounced in SHR-mtBN than in progenitor SHR and correlated with the decreased sensitivity to mitochondrial permeability transition pore (mPTP) opening in both strains (9). Indeed, affection of mitochondria determines the cell fate during early phases of reperfusion by mPTP opening (10) as the result of reactive oxygen species (ROS) overproduction and calcium overload [reviewed in (11)]. Likewise, we have showed that mitochondrial antioxidants play an important role in cardioprotective phenotype induced by CNH in normotensive rats (12). Beside that both glucose and lipid metabolism signaling pathways have been shown to contribute to cardioprotective phenotype of chronically hypoxic hearts (13, 14).
Based on the above-mentioned data, we aimed first, to detect genes with significantly different mRNA expressions in progenitor SHR and conplastic SHR-mtBN under normoxic and chronically hypoxic conditions using univariant analyses; and second, to identify candidate genes responsible for the differences between SHR and SHR-mtBN after adaptation to CNH using multivariant analyses. These potential candidates may elucidate the relationship between CNH-afforded protection and mitochondrial genome.
Materials and Methods
Animals
Inbred progenitor SHR and its conplastic strain SHR-mtBN (SHR harboring mitochondrial genome from Brown Norway rats) were used in present study. Both strains were exposed to CNH (inspired oxygen fraction 0.1) for 3 weeks and control groups were kept at normoxic conditions. The rats were housed at a 12/12-h light/dark cycle and fed by standard diet ad libitum and free access to the water. The animals were handled in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th edition, revised 2011). The experimental protocol was approved by the Animal Care and Use Committee of the Institute of Physiology of the Czech Academy of Sciences.
Tissue Preparation
All rats were killed by cervical dislocation in their environment, i.e., normoxic groups in room air and hypoxic groups in hypoxic chamber. The hearts were immediately excised and washed in ice-cold saline. Samples of left ventricle were rapidly frozen in liquid nitrogen and stored in −80°C until use.
RNA Isolation and Chip Analyses
Total RNA isolation and reverse transcription was performed as described previously (12), with a slight modification. Briefly, RNA was isolated using RNAzol reagent (Sigma Aldrich) according to manufacturer's instructions. The purity of isolated RNA was tested on Agilent 2100. One microgram of total RNA was loaded to the reverse transcription and the PCR reaction was performed as described previously using RevertAidTM H Minus First Strand cDNA Synthesis Kit with oligo(dT) primers (Fermentas). Gene-specific primers were designed using the Universal Probe Library Assay Design Center. The specific forward and reverse primer sequences are summarized in Supplement Table 1. At first, the samples for gene expression profiling were pre-amplified with 48 primers in 18 cycles with the following temperature profile: activation polymerase (95°C/3 min); amplification, 18 cycles of denaturation (95°C/15 s), and annealing (59°C/4 min) using iQ Supermix (Bio-Rad) and 2 μl cDNA (diluted on 10 ng input RNA). Subsequently, Biomark analysis were performed with following temperature profile: polymerase activation (95°C/3 min); amplification 30 cycles of denaturation (96°C/5 min), and annealing (60°C/20 s). Priming and pipetting were performed according to the manufacturer's instructions.
Statistical Analysis
The quality of the quantification cycles (Cq) data of 48 mRNA transcripts from 4 experimental groups (SHR and SHR-mtBN under normoxic and hypoxic conditions; n = 5) obtained from high-throughput qPCR instrument Biomark HD (Fluidigm) was checked by Fluidigm Real-Time PCR Analysis software (Fluidigm). The Cq data were basically processed by two approaches. First, the univariant analyses, based on the p-values, were used to reveal significant differences (p < 0.05) between four experimental groups within each mRNA transcript by ANOVA followed by Tukey's Multiple Comparison Posttest with Bonferroni correction using GenEx Enterprise (MultiD, SE) and GraphPad Prism software. Second, the multivariate principal component analysis analyses (PCA), based on the p-value and fold change of the gene, used auto scaled Cq data to reveal candidate genes responsible for diverse effects of CNH in SHR and SHR-mtBN. PCA is a powerful tool for reducing the dimensionality of a large data set in an unbiased way to identify clustering behavior. Subsequently, the Self-organizing maps (SOM) analysis was used to proof clustering obtained by PCA.
Results
The 48 mRNA transcripts were analyzed as representatives of six groups of interest as follows: lipid metabolism, glucose metabolism, apoptosis, mitochondria, antioxidant defense and cell signaling (see Supplement Table 1). NormFinder analysis selected ribosomal protein S18 with SD equal to 0.056, as the best reference gene from three candidates including hypoxanthine phosphoribosyltransferase 1 (Hprt; SD = 0.23) and beta-2-mikroglobulin (B2m; SD = 0.28).
Univariate Analysis
The univariant analyses (with Bonferroni correction) revealed significant differences predominantly in lipid metabolism and mRNA transcripts related to oxidative stress (see Figure 1). The mRNAs related to glucose metabolism remained mostly unchanged, except for pyruvate dehydrogenase kinase 3 (Pdk3) and pyruvate dehydrogenase phosphatase (Pdp2). Adaptation to CNH increased expression of Pdk and decreased expression of Pdp2 compared to normoxic groups similarly in both SHR and SHR-mtBN strains.
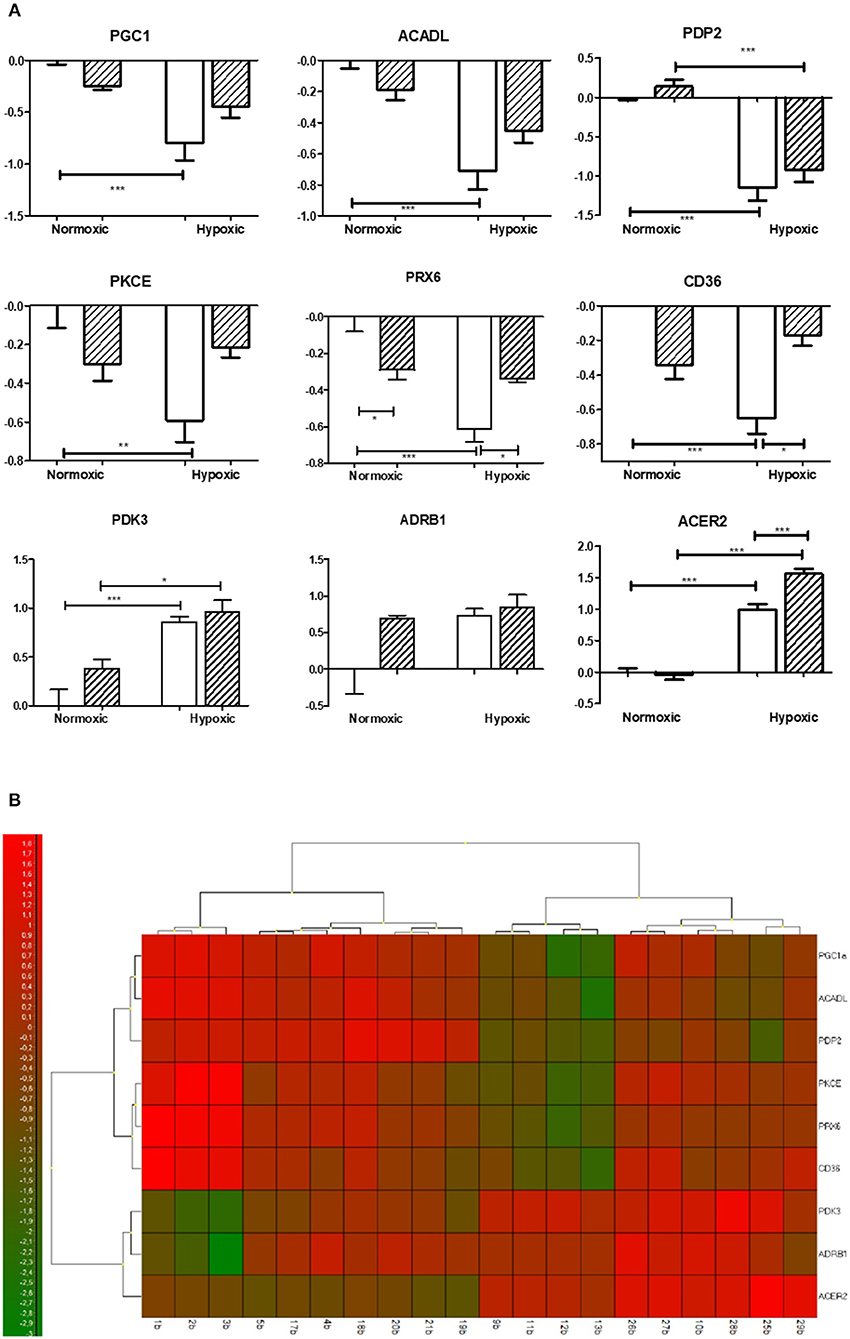
Figure 1. Effect of chronic continuous normobaric hypoxia on mRNA relative amount in the left ventricles of spontaneously hypertensive rats (SHR, empty bars) and its conplastic strain receiving mitochondria from normotensive Brown Norway rats (SHR-mtBN, hatched bars). Graphs showing genes with significant differences revealed by univariate analyses (ANOVA with Bonferroni correction) from 48 analyzed transcripts by Biomark Chip (A) and Heat map of all transcripts analyzed (B). Values are mean ± SEM, (n = 5), p-value * ≤ 0.05, ** ≤ 0.01, *** ≤ 0.001.
Regarding lipid metabolism, we observed a significant decrease in the expression of rate limiting enzyme of β-oxidation acyl-CoA dehydrogenase (Acadl) with a concomitant decline of fatty acid transporter (Cd36) in SHR but not in SHR-mtBN after CNH. In contrast, CNH increased the expression of secretory phospholipases Pla2g2a and Pla2g5a in conplastic SHR-mtBN compared to its normoxic counterpart. Interestingly, unlike Pla2g2a, Pla2g4a transcript level was lower in SHR-mtBN group than in SHR under normoxic conditions. Moreover, alkaline ceramidase 2 (Acer2) expression increased after CNH in both strains, but the effect was more pronounced in SHR-mtBN than in SHR.
Among the genes related to oxidative stress, CNH increased the expression of monoaminooxidase A (MaoA) and hemoxygenase 1 (Hmox1) in both strains. However, Hmox1 was lower in SHR-mtBN than in SHR under normoxia. The level of mRNA transcript of peroxiredoxin 6 (Prx6) was also lower in normoxic SHR-mtBN than in SHR. CNH led to a significant drop of Prx6 in SHR but not in SHR-mtBN, resulting in higher level in the later group.
Whereas we did not observe any changes in the level of transcripts related to apoptosis and mitochondria, the expression of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (Pgc1) markedly decreased in chronically hypoxic SHR but not in corresponding SHR-mtBN. Protein kinase C epsilon (Pkce) transcript level was lower in normoxic SHR than in SHR-mtBN and protein kinase C delta (Pkcd) significantly increased after CNH in SHR-mtBN only.
Multivariate Analysis
The basic PCA analysis of the whole set of 48 mRNA transcripts divided four experimental groups into two different clusters according to the experimental conditions, i.e., normoxia vs. hypoxia (Figure 2). Moreover, under hypoxic conditions the differences between strains became more pronounced.
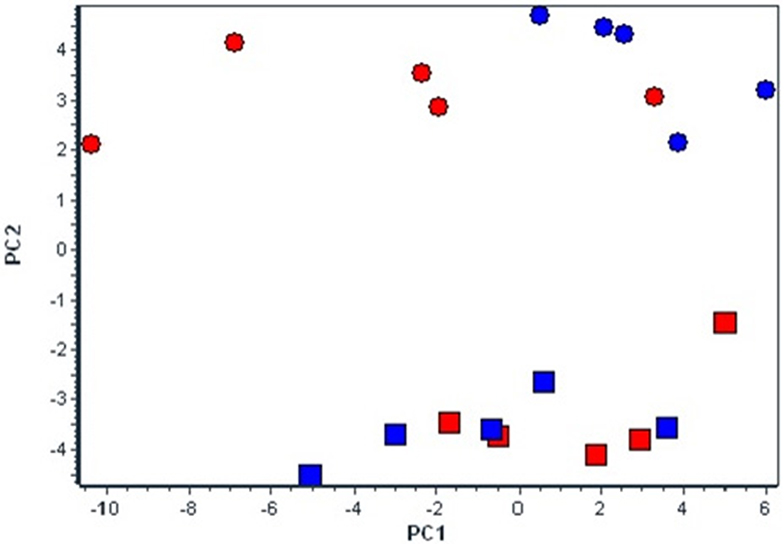
Figure 2. Effect of chronic continuous normobaric hypoxia on mRNA relative amount in spontaneously hypertensive rats (SHR, red) and its conplastic strain receiving mitochondria from normotensive Brown Norway rats (SHR-mtBN, blue) assessed by the multivariant principal component analysis (PCA) of all 48 genes. The diagram demonstrates clusters of selected candidate genes by PCA responsible for diverse effects of CNH in SHR and SHR-mtBN under normoxic (rectangle) and CNH conditions (circle).
Subsequent PCA (Figure 3) aimed to analyse the diverse effects of hypoxia on SHR and SHR-mtBN strains, and selected most important candidate genes which might contribute to the strain differences in cardiopotective phenotype afforded by CNH according to increasing p-value as follows: Acer2 (0.0013), Cd36 (0.0021), Aco1 (0.0057), Pparg (0.0061), Hmox2 (0.0093), Ppla2g2a (0.0097), Drp (0.0115), Pkce (0.0145), Hk2 (0.0155), Sgms2 (0.0166), Casp3 (0.0398), Mfn1 (0.0491), Pla2g5 (0.0611), Cat (0.0633). As seen above the PCA also selected two transcripts in which the p-value exceeded 0.05 level (Pla2g5a and Cat) however the influence of fold change became more important for selection of that genes by PCA. Further SOM analysis confirmed the clustering by PCA, except one sample.
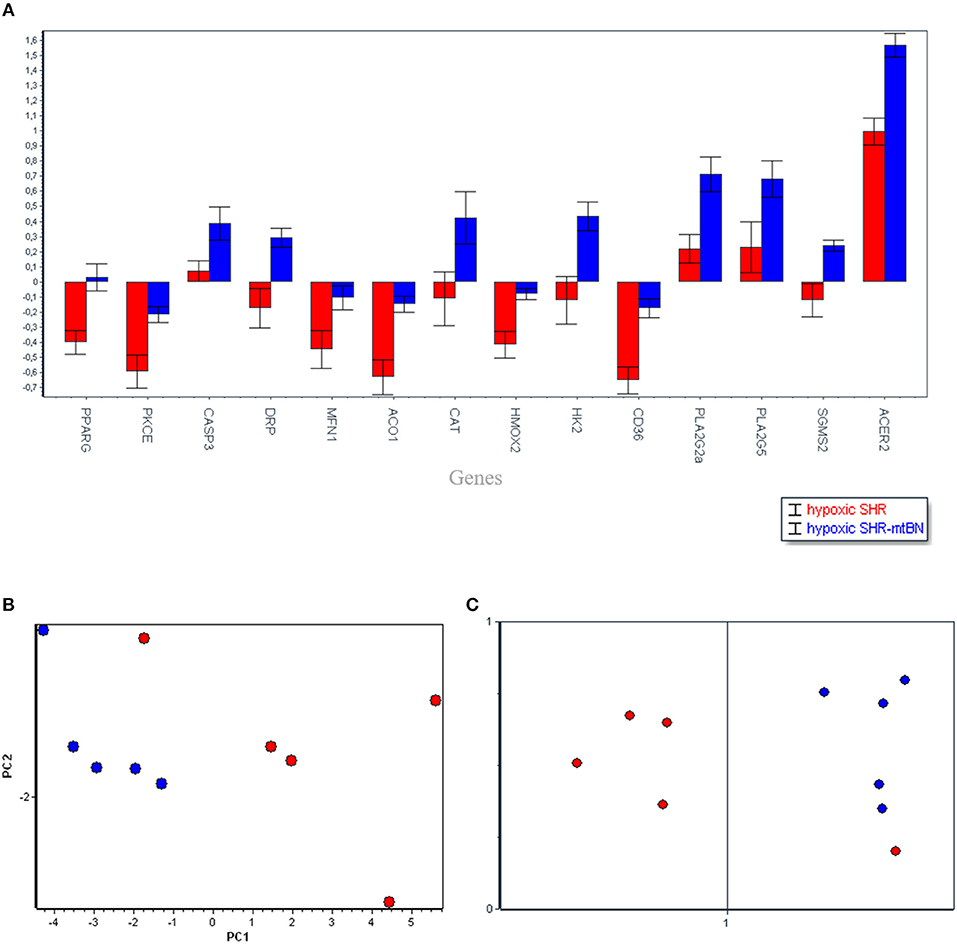
Figure 3. Selected candidate genes (14), responsible for the differences between spontaneously hypertensive rats (SHR, red) and its conplastic strain receiving mitochondria from normotensive Brown Norway rats (SHR-mtBN, blue) unmasked by adaptation to chronic continuous normobaric hypoxia (CNH) (A). The distribution of selected genes assessed by principal component analysis (PCA) based on the p-value and fold change of the genes (B); and confirmed by Self-organizing maps (SOM) (C). Normoxic conditions (rectangle) and CNH conditions (circle).
Discussion
Recently we demostrated that CNH adaptation of SHR and conplastic SHR-mtBN induced cardioprotective phenotype in both strains which was more pronounced in SHR-mtBN (9). In the present study we aimed to find out candidate genes which might contribute to the improved cardioprotective phenotype of hypertensive SHR-mtBN strain after CNH. The univariant analyses revealed predominant role of lipid metabolism and antioxidant system in the effect of hypoxia. Beside that, attenuation of pyruvate transport into the mitochondria may suggest increasing contribution of anaerobic glycolysis in hypertensive heart of both strains. Interestingly, multivariant PCA and SOM analysis has identified 14 candidates out of 48 genes with predominant role of lipid metabolism.
The overview of the obtained data from both analyses (Figures 1, 3) shows that mitochondrial genome replacement led to two different alterations of SHR-mtBN response to CNH: it either prevented drop of transcripts level induced by CNH or significantly increased upregulation of certain transcripts by CNH. The former effect was related to fatty acid transport and β-oxidation (Cd36, Acadl), antioxidants (Prx6, Hmox2, Aco1), signaling (PKCe, Pgc1, Pparg) and mitochondrial genes (Mfn1, Pdp2). However, the latter effect was observed in most transcripts related to lipid metabolism (Ppla2g2a, Pla2g5, Sgms2 Acer2) and transcripts which all might be functionally related to mitochondria (Casp3, Drp, Cat and Hk2).
Glucose and Lipid Metabolism
In the present study we observed CNH-induced expression of Hk2 in SHR-mtBN in line with our recent studies which showed increased activity of Akt signaling pathway including hexokinase (HK) and glucose transporters (GLUTs) in SHR and SHR-mtBN as well as in normotensive rats (13–15). Chronic hypoxia-induced shift in substrate utilization from fatty acids to glucose has been already reported (16, 17). However, we describe a novel mechanism which include an essential peroxisomal role in β-oxidation support utilizing the enhanced glycolytic flux (unpublished data) that also may correspond with present data. Indeed, beside an increased glucose-utilizing capacity of CNH myocardium, we suggest that the capability of glucose oxidative metabolism is attenuated in both hypertensive strains due to upregulation of Pdk3 and downregulation of Pdp2.
Mitochondrial ability to process long-chain fatty acids (LCFAs) might be reduced in SHR after CNH due to downregulation of Acadl, while in conplastic SHR-mtBN this effect was suppressed. Similarly, Cd36 level dropped after CNH suggesting that FA influx mediated by CD36 can be decreased in SHR. In contrast, mRNA level of both secretoric PLA2 (sPLA2; Pla2g2a, gene and its paralog Pla2g5a) increased in SHR-mtBN hearts adapted to CNH and this change can be assumed as cardioprotective. However, the role of sPLA2 in myocardial susceptibility to I/R injury is rather controversial. Although the Pla2g2a gene deletion has been shown to correlate with infarct size expansion (18), increased plasma concentration of sPLA2 is considered as a marker of I/R injury in human (19, 20). Moreover, inhibition of sPLA2 activity during early phase of reperfusion decreased the extent of injury and increased viability of cardiomyocytes (18, 21). Beside the secretoric Pla2g2a and Pla2g5a, myocardium express cytosolic PLA2 (cPLA2) which has been shown to be activated by CNH in normotensive rats (22). These isoforms cooperate in producing cardioprotective eikosanoid prostagladin E2 (23–25).
Acer2 was more expressed after CNH in SHR-mtBN, which may suggest anti-apoptotic action associated with an improved energy balance and stronger infarct size-limiting effect shown previously (9). ACER2 is a Golgi complex associated alkaline ceramidase catalyzing the conversion of ceramide, with preferences to long- and very long-chain (VLC) unsaturated ceramides, to sphingosine, which in turn is phosphorylated to sfingosine-1-phosphate (S1P) (26, 27). S1P-activated signaling pathway leads to cell survival in opposite to ceramide which is associated with apoptosis (28).
Cell Signaling
Pkce markedly declined only in progenitor SHR after CNH showing an interesting difference between strains which was unmasked by long-lasting hypoxic exposure. PKCε has been shown to play a role roles in myocardial ischemic tolerance and cardioprotection induced by CNH (29). Proposed mitochondrial targets of PKCε include components of electron transport chain (30) and mPTP pore where PKCε is supposed to form a complex with various proteins such as hexokinase2, adenine nucleotide translocase and voltage-dependent anion channel and inhibit the pathological function of the pore (31). Moreover, murine hearts with constitutively active PKCε were shown to preserve mitochondrial electron transfer coupling with maintained mitochondrial membrane potential and decreased cytochrome c release induced by reperfusion (32).
Similarly as Pkce transcript, Pparg and Pgc1 decline after CNH were also prevented by mitochondrial replacement in SHR-mtBN. These data suggest possible preservation of mitochondrial biogenesis (33) in SHR-mtBN as PGC-1 may also indirectly control the expression of mitochondrial DNA (mtDNA) transcription via increased expression of mitochondrial transcription factor A (Tfam) 1 (NRF-1) (34, 35). PGC-1α may stimulate its own transcription by co-activation: PPARγ binding to the PGC-1α promoter (36). Activation of PPARγ promotes glucose uptake and triglyceride synthesis in adipose tissue and the resulting reduction in circulating glucose and FA may directly modulate cardiac PPARα and PPARδ activities (37, 38). PPARγ null mice showed a downregulation of manganese superoxide dismutase transcript and protein levels within the cardiomyocytes indicating an essential role of PPARγ in the antioxidant defense (39). Possible interplay between PPAR and PKC cannot be excluded (40, 41).
Antioxidant Defense
Mitochondrial genome replacement also prevented the decreased gene expression of Hmox2, Aco1 and Prx6 induced by CNH in SHR and simultaneously increased Cat mRNA transcript. These changes may improve antioxidant defense of SHRmtBN compared to SHR. Importantly, CNH increased inducible Hmox1 transcript in both SHR strains. Accordingly, in non-hypertrophied left ventricles of normotensive rats, we observed CNH-induced increase of Hmox1, whereas upregulation of Hmox2 and Aco1 was observed after cardioprotective regimen (8 h/day, 3 weeks) of intermittent normobaric hypoxia (12). Constitutively expressed HMOX2 possesses common antioxidant features (42) and Hmox2 deletion led to phenotype characterized by increased pro-inflammatory and oxidative markers (43). Increased expression of Cat in SHR-mtBN may contribute to H2O2 detoxifying in the myocardium within the peroxisomes. However, rat cardiac mitochondria were shown to contain CAT as well (44). CAT overexpressing transgenic mice are resistant to myocardial I/R injury (45).
PRX6 is primarily a member of cytosolic peroxiredoxins with two important functions (i) antioxidant defense and (ii) phospholipid homeostasis because of its PLA2 activity (46). Beside that PRX6 was shown to play an important role in the pathophysiology of type 2 diabetes (T2DM). The deficiency of Prx6(−/−) impaired insulin signaling in mice, leading to reduction of muscle glycogen uptake (47). SHR-mtBN have a lower skeletal muscle glycogen than SHR (8) in accordance with lower expression of Prx6. However, in the present study SHR responded to CNH by significant drop of Prx6 transcript which was prevented in conplastic strain. These findings are in agreement with our previous data (8) demonstrating that selective replacement of the mitochondrial genome of the SHR with the mitochondrial genome of the BN rat influences several major metabolic risk factors of type 2 diabetes.
Mitochondrial and Apoptosis Associated Genes
Mitochondrial biogenesis involves morphological changes in mitochondrial reticulum such as fusion and fission. Dynamin related protein coding by Drp1 gene belongs to fission proteins, while Mfn1 gene coding dynamin-related fusion protein mitofusin, which is involved in internal membrane fusion. Increased expression of both genes in SHR-mtBN after CNH suggests more pronounced mitochondrial dynamics in this strain. Beside that, Drp1 has been shown to participate in peroxisomal fission process (48–50) which can also reflect the improved metabolic interplay between mitochondria and peroxisomes in SHR-mtBN after CNH.
Traditionally, CASP3 is seen as the activator of apoptosis. Its activation starts the apoptotic cascade in pressure overload-induced heart failure (51). On the other hand, defective Akt activation in Casp3 KO mice was accompanied by impaired cell survival, increased apoptosis in stressed organs with marked lapse in their physiological functions (52). An increased level of Casp3 mRNA in the present study suggests an increase in the mitochondrial domain of the central apoptotic effector molecule in the form of precursor caspase 3 in SHR-mtBN after CNH with the possibility of rapid response to stress stimuli (53).
In conclusion, the PCA analysis of Cq data clearly selected 14 important genes out of 48 ones that can be considered to contribute to the differences observed in myocardial ischemic tolerance between chronically hypoxic progenitor SHR and conplastic SHR-mtBN strains. Most of them belong to glucose and lipid metabolism and were unmasked by adaptation to the cardioprotective regimen of CNH. The data also suggest an improvement in certain antioxidant, metabolic, and anti-inflammatory aspects induced by the replacement of SHR mitochondrial genome with that of normotensive BN rats.
Author Contributions
IN performed experiments, analyzed data, interpreted data, and drafted and edited manuscript. DK performed experiments and edited manuscript. JN contributed to experimental design and the hypoxic model. MK performed experiments. MP and JŠ contributed the rat conplastic strain donor, and edited the manuscript. VK performed experiments and data analysis. FK contributed to the experimental model of hypoxia, data interpretation, and edited the manuscript. JZ contributed to experimental design, performed experiments, interpreted data, drafted and edited the manuscript.
Funding
This work was supported by the Charles University Grant Agency (1214214), Charles University Institutional Project SVV260-434/2018, the Grant Agency of the Czech Republic (13-10267, 14-36804G, 17-07748S), the Ministry of Education, Youth and Sports of the Czech Republic (SVV-260313/2016), the European Regional Development Fund (project no. CZ.1.05/4.1.00/16.0347) and the state budget of the Czech Republic.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2018.00809/full#supplementary-material
References
1. Franklin SS, Wong ND. Hypertension and cardiovascular disease: Contributions of the Framingham Heart Study. Glob Heart (2013) 8:49–57. doi: 10.1016/j.gheart.2012.12.004
2. Okamoto K, Aoki K. Development of a strain of spontaneously hypertensive rats. Jpn Circ J. (1963) 27:282–93. doi: 10.1253/jcj.27.282
3. Hajri T, Ibrahimi A, Coburn CT, Knapp FFJ, Kurtz T, Pravenec M, et al. Defective fatty acid uptake in the spontaneously hypertensive rat is a primary determinant of altered glucose metabolism, hyperinsulinemia, and myocardial hypertrophy. J Biol Chem. (2001) 276:23661–6. doi: 10.1074/jbc.M100942200
4. Reaven GM, Chang H. Relationship between blood pressure, plasma insulin and triglyceride concentration, and insulin action in spontaneous hypertensive and Wistar-Kyoto rats. Am J Hypertens. (1991) 4:34–8. doi: 10.1093/ajh/4.1.34
5. Penna C, Tullio F, Moro F, Folino A, Merlino A, Pagliaro P. Effects of a protocol of ischemic postconditioning and/or captopril in hearts of normotensive and hypertensive rats. Basic Res Cardiol. (2010) 105:181–92. doi: 10.1007/s00395-009-0075-6
6. Besík J, Szárszoi O, Kunes J, Netuka I, Malý J, Kolár F, et al. Tolerance to acute ischemia in adult male and female spontaneously hypertensive rats. Physiol Res. (2007) 56:267–74.
7. Garlid KD, Costa ADT, Quinlan CL, Pierre SV. Cardioprotective signalling to mitochondria. J. Mol. Cell. Cardiol. (2009) 46:858–66. doi: 10.1016/j.yjmcc.2008.11.019.Cardioprotective
8. Pravenec M, Hyakukoku M, Houstek J, Zidek V, Landa V, Mlejnek P, et al. Direct linkage of mitochondrial genome variation to risk factors for type 2 diabetes in conplastic strains. Genome Res. (2007) 17:1319–26. doi: 10.1101/gr.6548207
9. Neckar J, Svatonova A, Weissova R, Drahota Z, Zajickova P, Brabcova I, et al. Selective replacement of mitochondrial DNA increases cardioprotective effect of chronic continuous hypoxia in spontaneously hypertensive rats. Clin Sci. (2017) 131:865–81. doi: 10.1042/CS20170083
10. Halestrap AP, Richardson AP. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol. (2015) 78:129–41. doi: 10.1016/j.yjmcc.2014.08.018
11. Halestrap AP. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem Soc Trans. (2010) 38:841–60. doi: 10.1042/BST0380841
12. Kasparova D, Neckar J, Dabrowska L, Novotny J, Mraz J, Kolar F, et al. Cardioprotective and non-protective regimens of chronic hypoxia diversely affect the myocardial antioxidant systems. Physiol Genom. (2015) 47:612–20. doi: 10.1152/physiolgenomics.00058.2015
13. Kolar D, Gresikova M, Waskova-Arnostova P, Elsnicova B, Kohutova J, Hornikova D, et al. Adaptation to chronic continuous hypoxia potentiates Akt/HK2 anti-apoptotic pathway during brief myocardial ischemia/reperfusion insult. Mol Cell Biochem. (2017) 432:99–108. doi: 10.1007/s11010-017-3001-5
14. Nedvedova I, Kolar D, Elsnicova B, Hornikova D, Novotny J, Kalous M, et al. Mitochondrial genome modulates myocardial Akt/GLUT/HK salvage pathway in spontaneously hypertensive rats adapted to chronic hypoxia. Physiol Genomics. (2018) 50:532–41. doi: 10.1152/physiolgenomics.00040.2017
15. Waskova-Arnostova P, Kasparova D, Elsnicova B, Novotny J, Neckar J, Kolar F, et al. Chronic hypoxia enhances expression and activity of mitochondrial creatine kinase and hexokinase in the rat ventricular myocardium. Cell Physiol Biochem. (2014) 33:310–20. doi: 10.1159/000356671
16. Essop MF. Cardiac metabolic adaptations in response to chronic hypoxia. J Physiol. (2007) 584:715–26. doi: 10.1113/jphysiol.2007.143511
17. Rumsey WL, Abbott B, Bertelsen D, Mallamaci M, Nelson D, Erecinska M, et al. Adaptation to hypoxia alters energy metabolism in rat heart. AM J Physiol. (1999) 276(1 Pt 2):71–80. doi: 10.1152/ajpheart.1999.276.1.H71
18. Krijnen PA, Meischl C, Nijmeijer R, Visser CA, Hack CE, Niessen HW. Inhibition of sPLA2-IIA, C-reactive protein or complement: new therapy for patients with acute myocardial infarction? Cardiovasc. Hematol. Disord. Targets (2006) 6:111–21. doi: 10.2174/187152906777441830
19. Dutour A, Achard V, Sell H, Naour N, Collart F, Gaborit B, et al. Secretory type II phospholipase A2 is produced and secreted by epicardial adipose tissue and overexpressed in patients with coronary artery disease. J Clin Endocrinol Metab. (2010) 95:963–7. doi: 10.1210/jc.2009-1222
20. Nijmeijer R, Lagrand WK, Baidoshvili A, Lubbers YTP, Hermens WT, Meijer CJLM, et al. Secretory type II phospholipase A2 binds to ischemic myocardium during myocardial infarction in humans. Cardiovasc Res. (2002) 53:138–46. doi: 10.1016/S0008-6363(01)00480-1
21. Van Dijk A, Krijnen PAJ, Vermond RA, Pronk A, Spreeuwenberg M, Visser FC, et al. Inhibition of type 2A secretory phospholipase A2 reduces death of cardiomyocytes in acute myocardial infarction. Apoptosis (2009) 14:753–63. doi: 10.1007/s10495-009-0350-x
22. Alanova P, Chytilova A, Neckar J, Hrdlicka J, Micova P, Holzerova K, et al. Myocardial ischemic tolerance in rats subjected to endurance exercise training during adaptation to chronic hypoxia. J Appl Physiol. (2017) 122:1452–61. doi: 10.1152/japplphysiol.00671.2016
23. Cupillard L, Koumanov K, Mattei MG, Lazdunski M, Lambeau G. Cloning, chromosomal mapping, and expression of a novel human secretory phospholipase A2. J Biol Chem. (1997) 272:15745–52. doi: 10.1074/jbc.272.25.15745
24. Ishikawa Y, Komiyama K, Masuda S, Murakami M, Akasaka Y, Ito K, et al. Expression of type V secretory phospholipase A in myocardial remodelling after infarction. Histopathology (2005) 47:257–67. doi: 10.1111/j.1365-2559.2005.02227.x
25. Shinohara H, Balboa MA, Johnson CA, Balsinde J, Dennis EA. Regulation of delayed prostaglandin production in activated P388D1 macrophages by group IV cytosolic and group V secretory phospholipase A2s. J Biol Chem. (1999) 274:12263–8. doi: 10.1074/jbc.274.18.12263
26. Sun W, Jin J, Xu R, Hu W, Szulc ZM, Bielawski J, et al. Substrate specificity, membrane topology, and activity regulation of human alkaline ceramidase 2 (ACER2). J Biol Chem. (2010) 285:8995–9007. doi: 10.1074/jbc.M109.069203
27. Xu R, Jin J, Hu W, Sun W, Bielawski J, Szulc Z, et al. Golgi alkaline ceramidase regulates cell proliferation and survival by controlling levels of sphingosine and S1P. FASEB J. (2006) 20:1813–25. doi: 10.1096/fj.05-5689com
28. Van Brocklyn JR, Williams JB. The control of the balance between ceramide and sphingosine-1-phosphate by sphingosine kinase: oxidative stress and the seesaw of cell survival and death. Comp Biochem Physiol B Biochem Mol Biol. (2012) 163:26–36. doi: 10.1016/j.cbpb.2012.05.006
29. Holzerova K, Hlavackova M, Zurmanova J, Borchert G, Neckar J, Kolar F, et al. Involvement of PKCepsilon in cardioprotection induced by adaptation to chronic continuous hypoxia. Physiol Res. (2015) 64:191–201.
30. Guo D, Nguyen T, Ogbi M, Tawfik H, Ma G, Yu Q, et al. Protein kinase C-epsilon coimmunoprecipitates with cytochrome oxidase subunit IV and is associated with improved cytochrome-c oxidase activity and cardioprotection. Am J Physiol Heart Circ Physiol. (2007) 293:H2219–30. doi: 10.1152/ajpheart.01306.2006
31. Budas G, Costa HMJ, Ferreira JCB, Teixeira da Silva Ferreira A, Perales J, Krieger JE, et al. Identification of epsilonPKC targets during cardiac ischemic injury. Circ J. (2012) 76:1476–85. doi: 10.1253/circj.CJ-11-1360
32. Baines CP, Zhang J, Wang G-W, Zheng Y-T, Xiu JX, Cardwell EM, et al. Mitochondrial PKC? and MAPK Form Signaling Modules in the Murine Heart Enhanced Mitochondrial PKC?-MAPK Interactions and Differential MAPK Activation in PKC?-Induced Cardioprotection. Circulation (2002) 90:390–7. doi: 10.1161/01.RES.0000012702.90501.8D
33. Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science (2003) 299:896–9. doi: 10.1126/science.1079368
34. Choi YS, Kim S, Kyu Lee H, Lee K-U, Pak YK. In vitro methylation of nuclear respiratory factor-1 binding site suppresses the promoter activity of mitochondrial transcription factor A. Biochem Biophys Res Commun. (2004) 314:118–22. doi: 10.1016/j.bbrc.2003.12.065
35. Virbasius JV, Scarpulla RC. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc Natl Acad Sci USA. (1994) 91:1309–13. doi: 10.1073/pnas.91.4.1309
36. Hondares E, Mora O, Yubero P, Rodriguez de la Concepcion M, Iglesias R, Giralt M, et al. Thiazolidinediones and rexinoids induce peroxisome proliferator-activated receptor-coactivator (PGC)-1alpha gene transcription: an autoregulatory loop controls PGC-1alpha expression in adipocytes via peroxisome proliferator-activated receptor-gamma coactivation. Endocrinology (2006) 147:2829–38. doi: 10.1210/en.2006-0070
37. Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res. (2004) 95:568–78. doi: 10.1161/01.RES.0000141774.29937.e3
38. Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. (2003) 24:78–90. doi: 10.1210/er.2002-0012
39. Ding G, Fu M, Qin Q, Lewis W, Kim HW, Fukai T, et al. Cardiac peroxisome proliferator-activated receptor gamma is essential in protecting cardiomyocytes from oxidative damage. Cardiovasc Res. (2007) 76:269–79. doi: 10.1016/j.cardiores.2007.06.027
40. Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta (2007) 1771:952–60. doi: 10.1016/j.bbalip.2007.04.018
41. Tsao W-C, Wu H-M, Chi K-H, Chang Y-H, Lin W-W. Proteasome inhibitors induce peroxisome proliferator-activated receptor transactivation through RXR accumulation and a protein kinase C-dependent pathway. Exp Cell Res. (2005) 304:234–43. doi: 10.1016/j.yexcr.2004.11.004
42. Williams SEJ, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, et al. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science (2004) 306:2093–7. doi: 10.1126/science.1105010
43. Ayer A, Zarjou A, Agarwal A, Stocker R. Heme oxygenases in cardiovascular health and disease. Physiol Rev. (2016) 96:1449–508. doi: 10.1152/physrev.00003.2016
44. Nohl H, Hegner D. Evidence for the existence of catalase in the matrix space ofrat-heart mitochondria. FEBS Lett. (1978) 89:126–30. doi: 10.1016/0014-5793(78)80537-7
45. Li G, Chen Y, Saari JT, Kang YJ. Catalase-overexpressing transgenic mouse heart is resistant to ischemia-reperfusion injury. Am J Physiol. (1997) 273:H1090–5. doi: 10.1152/ajpheart.1997.273.3.H1090
46. Fisher AB. Peroxiredoxin 6: a bifunctional enzyme with glutathione peroxidase and phospholipase A 2 activities. Antioxid Redox Signal. (2011) 15:831–44. doi: 10.1089/ars.2010.3412
47. Pacifici F, Arriga R, Sorice GP, Capuani B, Scioli MG, Pastore D, et al. Peroxiredoxin 6, a novel player in the pathogenesis of diabetes. Diabetes (2014) 63:3210–20. doi: 10.2337/db14-0144
48. Koch A, Thiemann M, Grabenbauer M, Yoon Y, McNiven MA, Schrader M. Dynamin-like protein 1 is involved in peroxisomal fission. J Biol Chem. (2003) 278:8597–605. doi: 10.1074/jbc.M211761200
49. Koch A, Yoon Y, Bonekamp NA, McNiven MA, Schrader M. A role for Fis1 in both mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell (2005) 16:5077–86. doi: 10.1091/mbc.E05-02-0159
50. Schrader M, Bonekamp NA, Islinger M. Fission and proliferation of peroxisomes. Biochim Biophys Acta (2012) 1822:1343–57. doi: 10.1016/j.bbadis.2011.12.014
51. Moorjani N, Ahmad M, Catarino P, Brittin R, Trabzuni D, Al-Mohanna F, et al. Activation of apoptotic caspase cascade during the transition to pressure overload-induced heart failure. J Am Coll Cardiol. (2006) 48:1451–8. doi: 10.1016/j.jacc.2006.05.065
52. Khalil H, Peltzer N, Walicki J, Yang J-Y, Dubuis G, Gardiol N, et al. Caspase-3 protects stressed organs against cell death. Mol Cell Biol. (2012) 32:4523–33. doi: 10.1128/MCB.00774-12
Keywords: SHR, conplastic strain, SHR-mtBN, left ventricle, hypoxia, metabolism
Citation: Nedvedova I, Kolar D, Neckar J, Kalous M, Pravenec M, Šilhavý J, Korenkova V, Kolar F and Zurmanova JM (2019) Cardioprotective Regimen of Adaptation to Chronic Hypoxia Diversely Alters Myocardial Gene Expression in SHR and SHR-mtBN Conplastic Rat Strains. Front. Endocrinol. 9:809. doi: 10.3389/fendo.2018.00809
Received: 25 July 2018; Accepted: 24 December 2018;
Published: 22 January 2019.
Edited by:
Slava Berger, Johns Hopkins Medicine, United StatesReviewed by:
Luciano Drager, University of São Paulo, BrazilZhichao Feng, Albert Einstein College of Medicine, United States
Copyright © 2019 Nedvedova, Kolar, Neckar, Kalous, Pravenec, Šilhavý, Korenkova, Kolar and Zurmanova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jitka M. Zurmanova, aml0a2EuenVybWFub3ZhQG5hdHVyLmN1bmkuY3o=