 Aradhana Rani
Aradhana Rani Justin Stebbing2
Justin Stebbing2 Georgios Giamas
Georgios Giamas John Murphy
John Murphy- 1School of Life Sciences, University of Westminster, London, United Kingdom
- 2Department of Surgery and Cancer, Imperial College London, London, United Kingdom
- 3Department of Biochemistry and Biomedicine, School of Life Sciences, University of Sussex, Brighton, United Kingdom
The importance and role of the estrogen receptor (ER) pathway has been well-documented in both breast cancer (BC) development and progression. The treatment of choice in women with metastatic breast cancer (MBC) is classically divided into a variety of endocrine therapies, 3 of the most common being: selective estrogen receptor modulators (SERM), aromatase inhibitors (AI) and selective estrogen receptor down-regulators (SERD). In a proportion of patients, resistance develops to endocrine therapy due to a sophisticated and at times redundant interference, at the molecular level between the ER and growth factor. The progression to endocrine resistance is considered to be a gradual, step-wise process. Several mechanisms have been proposed but thus far none of them can be defined as the complete explanation behind the phenomenon of endocrine resistance. Although multiple cellular, molecular and immune mechanisms have been and are being extensively studied, their individual roles are often poorly understood. In this review, we summarize current progress in our understanding of ER biology and the molecular mechanisms that predispose and determine endocrine resistance in breast cancer patients.
Introduction
The complex association of endocrine ablation and breast cancer (BC) was discovered in 1896 when Beatson revealed that oophorectomy on an advanced cancer patient led to a pronounced and marked response (1). The estrogen receptor (ER) was first discovered in 1958 by EV Jensen and subsequent studies showed that estrogen (E2) is implicated in BC pathogenesis and nurtures the surge in ER expressing BC cells (2, 3). E2 modulates its activity through its two ERs: ERα (ERα) and β (ERβ) (4, 5). ERα was cloned in 1985 by Pierre Chambon's group and ERβ was cloned in 1996 by Jan-Ake Gustafsson's group (6, 7).
However, it is the ERα which is expressed predominantly in breast tumors, and considered the most suitable target for hormonal therapy (8). Consequently, most treatments are directed at reducing the levels of E2 or inhibiting the E2-mediated signaling. Endocrine therapy has been prevalent ever since the discovery of endocrine cancers and it is, to date, one of the most effective treatments in ER-positive (ER+) BC. At least six distinct therapeutic modalities dictate endocrine therapy, namely the selective ER modulators (SERMs), the selective ER down-regulators (SERDs), aromatase inhibitors (AIs), mTORC1 inhibitors in combination with aromatase inhibitors, and cyclin dependent kinases 4 and 6 (CDK4 and CDK6) inhibitors in combination with AIs and CDK4/CDK6 inhibitors in combination with SERDs (9).
Tamoxifen, a SERM that acts by blocking the ER, has been ubiquitously used over the last four decades as the treatment of choice in pre-menopausal BC patients, although in the adjuvant setting, it was the drug of choice in post-menopausal women with HR+ BC (10, 11). AIs have been shown to be effective in post-menopausal women and might be effective in patients resistant to SERMs (12–14). With the discovery of endocrine therapies, the population of BC cells have been evolving too and overcoming endocrine resistance is a major cause of concern.
ER signaling is a complex cascade of events and the function of ER activity is dependent on the microenvironment within the cell at the molecular level, i.e., other signaling molecules, coactivators, corepressors, and genomic sequence of consensus binding sites. These are some of the determinants leading to the inefficiency of endocrine therapy and are associated with integral or assumed endocrine resistance. The cellular and molecular milieu can determine the transformation from an E2-dependent to E2-independent cluster of proteins, which subsequently go on to regulate genes in the absence of the E2-ER signaling.
In the Immediate Preoperative Anastrozole, Tamoxifen, or Combined with Tamoxifen (IMPACT) neoadjuvant trial, BC cell proliferation was measured by Ki67 immunohistochemistry (IHC) and it was demonstrated that almost 40% of patients presented with a high correlation of a 5-years recurrence free survival (RFS) upon short term anti-E2 therapy (15). In this study, the AI anastrozole was more effective in suppressing Ki67 expression. The larger adjuvant Arimidex, Tamoxifen Alone or in Combination (ATAC) and Breast International Group (BIG) trials also reported that the set of patients exhibiting significant inhibition of cell proliferation upon endocrine therapy had a larger number of tumor cells that were hormone sensitive/dependent (11, 16). Whole exome sequencing of operable ER+ patient tumors after estrogen deprivation by an AI, has the potential to identify useful biomarkers and suitable therapy in a subset of BC patients (17). Large genomic datasets have been generated from ER+ BC patients revealing a catalog of somatic mutations within genes and each of these datasets have provided a wealth of information (18, 19). A recent study identified association with clinical variables and MAP3K1 and TP53 mutations were predictors of good and poor response, respectively (19). These findings were in line with observations in the METABRIC dataset (20).
An in-depth characterization of tumors through large integrated genomic landscape studies on metastatic breast cancer (MBC) patients has provided valuable insights into a few of the genomic drivers, the role of heterogenic genomic architecture of cells within the tumor, the cellular and molecular determinants that define response to endocrine therapy along with identified novel biomarkers and therapies (9, 21, 22). These studies have demonstrated a central clonal hub at the primary tumor site and acquired mutations and drivers that promotes metastasis (21). One such study identified the SWI-SNF and JAK2-STAT3 pathways as potential therapeutic targets (21). Another of the recent studies identified at least four separate clusters of cells: 1. A cluster of tumorous cells possessing ERα mutations 2. A cluster of cells harboring mutations within the RAS/RAF/MEK/MAPK pathway 3. A cluster of cells with mutations in a variety of transcriptional factors and 4. A cluster of cells with unknown mechanisms (9). Although ERα mutations are common in a small proportion of endocrine resistant BC patients, the mechanistic details of endocrine resistance in the remaining patients remains elusive and has been discussed in further details below. Clinico-genomic factors as well as the role of the immune system and its associated tumor microenvironment (TME) provide a strong rationale for stratifying therapeutic approaches in endocrine resistant BC patients.
The immune system, both innate and adaptive play a pivotal role in BC. The microenvironment and the soluble mediators involved, including cytokines, dictate to a large extent, the ability of immune cells to either subdue the proliferating cancer cells or support the growth and metastatic capacity of BC cells. Lymphocyte infiltration has been considered in a recent study and this has reinforced the importance of the immune system in ER+ BC (23). While the authors did not observe any prognostic significance on the scores for immune cell abundance accounting for tumor infiltrating lymphocytes (TILs) in histology sections, they did observe prognostic significance for the immune scores based on spatial heterogeneity of the TILs and an increase in spatial clustering across late recurrent ER+ cancer.
A thorough study of the molecular structure, components and cascade of events leading to endocrine resistance will highlight suitable schemes and strategies for therapeutic targeting. Illustrated in Figure 1 are the various factors leading endocrine sensitive cells toward reduced endocrine sensitivity/endocrine resistance. This review discusses in detail the various intricate and sophisticated web of pathways involved in ER signaling and highlights schemes for treatment.
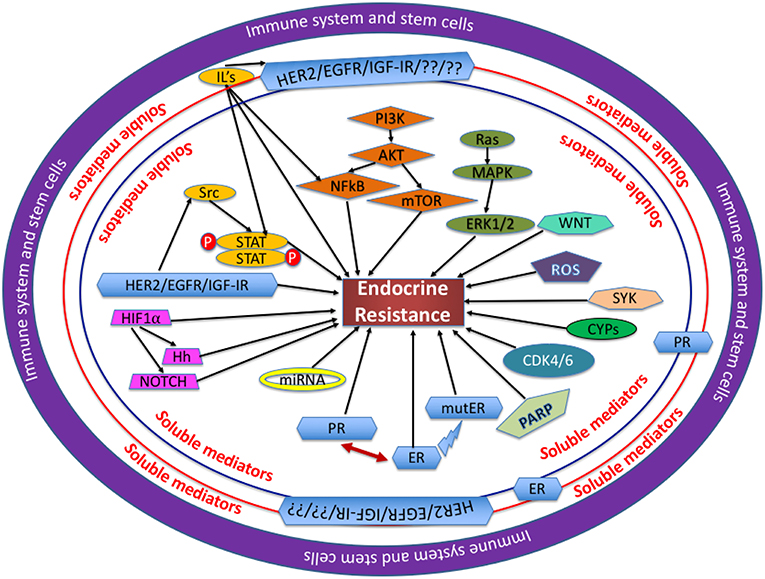
Figure 1. Mechanisms involved in endocrine resistance: Several proteins, soluble mediators and transcription factors are assimilated and function cohesively in a complex network, with each entity playing a unique role through the regulation of its own cascade of events as mentioned in each section of this review. In the opinion of the authors, the immune system and the stem cells are at the center of dysregulation observed in the proteins and pathways involved. The soluble mediators, like hormones, cytokines, and chemokines all play a crucial role in BC cells becoming endocrine resistant. IL, interleukin; ROS, reactive oxygen species; mutER, mutations in ERα.
1. Estrogen Receptor and Signaling
ER is a nuclear receptor and belongs to the steroid-thyroid-retinoid receptor superfamily (6). The human ER is comprised of two subdivisions: ERα and ERβ, located on chromosomes 6 and 14 at 6q21.5 and 14q23.2, respectively (7, 24). In some tissues, like the uterus, mammary gland, testis, pituitary, liver, kidney, heart, and skeletal muscle, ERα appears to dominate, whereas in the ovary and prostate, ERβ is highly expressed. The role of ERα in breast malignancy is well-documented, while the function of ERβ remains ambiguous (8). It is understood that ERβ has contrasting effects to ERα, and inhibits the stimulatory effects of E2 on cell proliferation. Studies have shown that downregulation of ERβ contributes to tumor progression and the chances of survival increase with an increase in the expression of ERβ (25, 26).
Both receptor subtypes share similarity at the protein level and are comprised of domains denoted as A to F (Figure 2) (27, 28). However, there is only limited homology between the two receptors, with 95% in the DNA binding domain (DBD), 50% in the ligand binding domain (LBD) and 15% in the N terminal domain of the two receptors (29). The ERα and ERβ contains two transactivation domains, the activation function 1 (AF1) and activation function 2 (AF2), with the N-terminal AB domain (containing the AF1), responsible for constitutive ER activity (30). The DNA-binding domain (DBD) or C domain, binds to the palindromic sequence, GGTCAnnnTGACC and leads to dimerization of the ER (28). The D domain/hinge domain leads to nuclear transport, while the E domain/Ligand-Binding Domain (LBD), is responsible for ligand binding (31–33). Located at the carboxy terminus is the F domain/AF2, which is responsible for E2 dependent activation of the ER. This region modulates the functions of the AF1 and AF2 domains (34). Consistent with previous reports on the role of polymorphisms in the ERα and ERβ genes on endocrine resistance, recent large scale projects, like The Cancer Genome Atlas (TCGA), have indicated similar results, in that ERα mutations were present in only 0.5% of BC patient samples and ERα amplification in 2.6% (35).
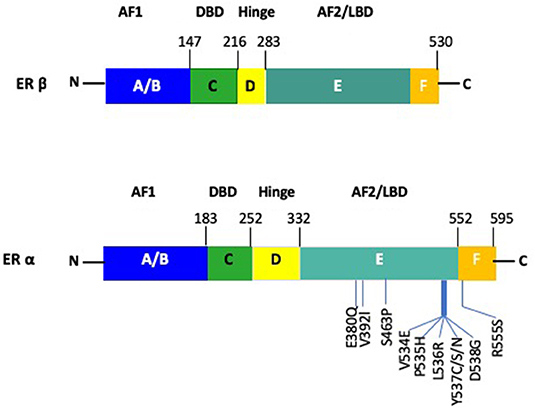
Figure 2. Schematic figure of the structure of ERα and ERβ. The AF1 site is located at the N-terminus A/B domain. The DBD and dimerization site is present within the C domain. The nuclear localization signal is contained in the D domain. The E/F domain contains the AF2 site as well as the ligand binding domain along with a dimerization site. Notable mutations within ERα are depicted for the ERα gene.
Upon binding of E2 to ER, a series of successive triggers, results in the translocation of chaperone proteins from the ERα, receptor dimerization, phosphorylation and subsequent binding of ER as a dimer to consensus binding sites on the DNA, known as E2 response elements (EREs). The EREs are located in the 5′ untranslated region (UTR) of genes, although it is well-known that ER can bind to intronic as well as distal regions of the transcription start site of a gene and that ERα binds within the enhancer rather than the promoter regions and also facilitates transcription through long distance interactions by DNA looping (36–38). ER signaling can be triggered by nuclear and non-nuclear mechanisms. At the molecular level, ER regulates the expression of genes by both canonical and non-canonical pathways, also known as “nuclear-initiated steroid signaling (NISS)” pathways (15). The consensus palindromic ERE was initially defined as 5′-GGTCAnnnTGACC-3′, from the X. laevis vitellogenin gene, although the ERE in humans ranges from 3 to 5 nucleotides between the penta half sites (GGTCA(n)3−5TGACC (39, 40). When ER binds to the ERE on the DNA, it leads to gene transcription of target genes, regulated by synergistic activity of AF2 and AF1. Additional co-activator (Co-A), specificity protein 1 (SP1) and activator protein 1 (AP1) are recruited to the ER/DNA complex and can regulate cellular function by upregulating or downregulating gene transcription (41, 42) (Figure 2). Essentially, the activity of the ER is modulated by post-translational modifications which include, phosphory/acety/palmitoy/sumoy-lations and ubiquitination (Table 1). ERα is phosphorylated at Ser118, 104, 106, and Tyr537, acetylated at Lys266, 268, 299, 302, and 303, palmitoylated at Cys447, sumoylated at Lys 299, 302, and 303 and ubiquitinated at Leu 429 and Ala 430 among few others. In the last decade, studies have shown that a proportion of target genes are regulated using a more complex machinery, where more than one ERE-consensus sequence and/or non-consensus ERE sites are present in the promoter region (47).
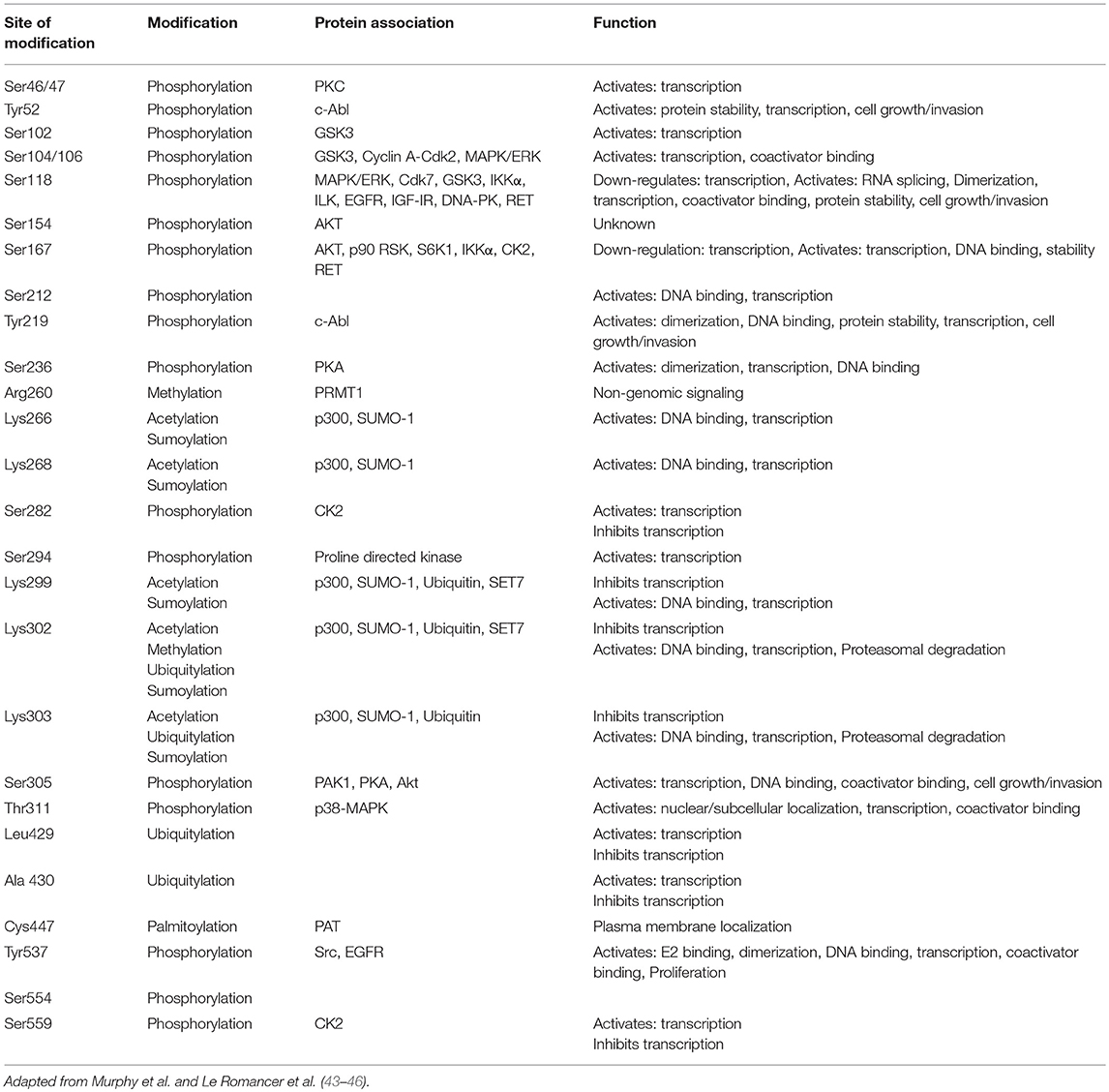
Table 1. Post-translational modifications in ERα.
Both the AF1 and AF2 domains are crucial, but the AF2 activation serves as the binding region for coactivators and corepressors (48, 49). The predominant steroid receptor coactivator (SRC) are ERAP-160, RIP-140, SRC-1, CBP, p300, TIF2, and AIB1 (49), while the corepressor is SMRT (50). A recent study identified Oct4 as a cofactor associated with ERα to promote tamoxifen resistance (51). In this study, enhanced expression of Oct4 led to a tamoxifen dependent increase in the proliferative potential of the tumor. This is important in the context of endocrine resistance, since the array of molecules mobilized by the ER is dependent on the organization of the E2 mediated ER-ERE complex (52).
In contrast, the AF1 domain is not dependent on a ligand and regulates gene transcription even in an ERα deletion mutant (53). In the ligand independent mechanism, activation of AF1 by phosphorylated ER at Ser104, Ser106, Ser118, Ser167, and Ser305 is mediated by crosslink and crosstalk between MAPK, PKA, PI3K/AKT and cyclin-dependent protein kinase 2/7 (Cdk2/7) pathways (54–58) (Figure 3A). The two prominent pathways mobilizing this cascade are those of the EGFR and IGFR1 (59). In response to E2, activation of AF1 leads to a concomitant action on the AF2 assisted by a complex of transcription factors to bring about a concerted effect on the ER mediated transcription (60, 61). These studies go on to highlight how redundancy can play a crucial role between signaling pathways and lead to an activation of the ER followed by subsequent activation, transcription and translation of ER regulated genes under conditions with inadequate E2 (62). This mechanism might lead to resistance against various endocrine therapies.
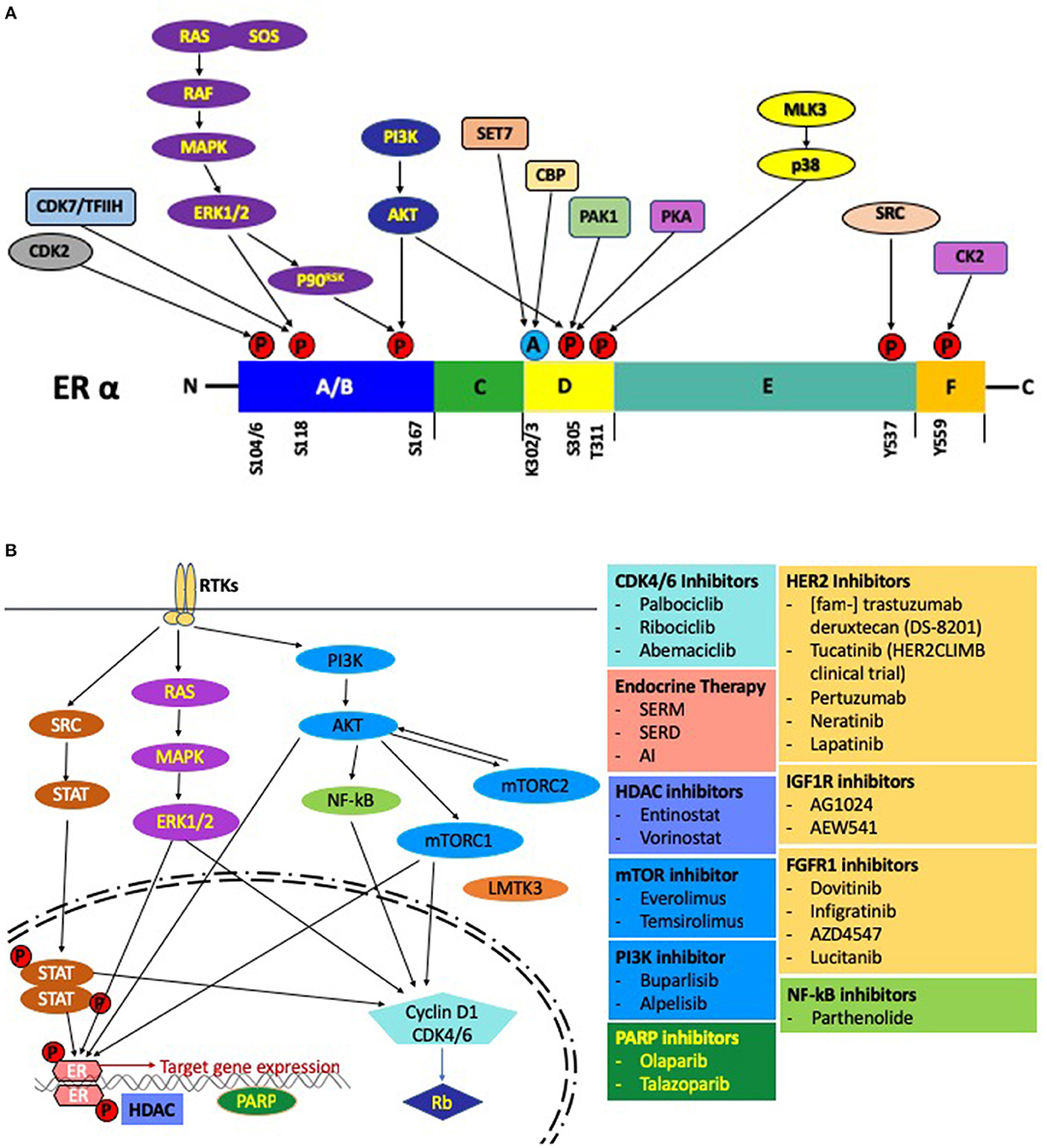
Figure 3. (A) Post-translational modifications of the ER. Activation of the growth factor receptor tyrosine kinases leads to phosphorylation of the ER through the RAS-MAPK and PI3K-AKT pathways. Several other pathways, including the CDK2 complex, CDK7/TFIIH complex can also phosphorylate the ER. Sensitivity to E2 is modulated by acetylation of the ER by src (CREB-binding protein). Pictorially represented above are the domains of the ER with phosphorylation/acetylation sites and the protein kinases mediating these modifications. (B) Regulation by E2 at the molecular level: A schematic representation of the pathways and the associated small molecule inhibitors involved in endocrine resistance.
In the non-classical nuclear mechanism of ER signaling, the functions are mediated by activation of membrane bound ER, that occurs within minutes by activating protein kinase cascades. This rapid non-nuclear mechanism is also referred to as membrane-initiated steroid signaling (MISS) pathway (63). The responses are mediated via ER associated with caveolar rafts, which collaborate as dimers precisely with signaling adaptor proteins (64, 65).
E2 mediated responses are also conducted through a novel ER GPR30 (renamed GPER), a G protein-coupled receptor (66, 67). Since one in four ER+ patients do not respond to antioestrogens, GPER provides another possible mechanism of signaling, bypassing the E2-ER signaling mode (68, 69). Recent evidence suggests an association of GPER expression with resistance to endocrine therapy (70–72). GPER is unique in that it activates a whole new cascade of pathways distinct from those activated by the classical E2-ERα pathway (72). A fine crosstalk between GPER and HER-2 is considered to be an important pathway in the progression of breast cancer upon activation via E2 (73, 74). The GPER serves as an alternative E2 receptor since this pathway may be used by ER negative BC patients to drive HER-2 dependent proliferation (75). Thus, the interactions between classical ER and GPER is important to reveal the complex crosstalk and activation of non-nuclear mechanisms of E2 (76, 77).
Ligand-independent pathways are activated upon phosphorylation of ER by MAPK/ERK or PI3K/AKT (55, 78). Recent studies have shown a role for isoforms of ERα in the non-nuclear E2 signaling pathways (79, 80). The various pathways are an interplay of events and act in tandem. Thus, inhibition of one pathway or mode of E2 signaling does not completely abolish oestrogenic or ER regulatory actions. There is crosstalk between both nuclear and non-nuclear pathways and these effects are coactive and interdependent.
1a. Loss of ERα
Downregulation of ER is hypothesized as a cause of acquired resistance to tamoxifen, although it is not downregulated in approximately a quarter of tumors with acquired resistance to tamoxifen and a fifth of the tamoxifen resistant cancers will go on to be responsive to treatment with an AI or a SERM (e.g., Fulvestrant) (81, 82). Endocrine therapy is effective in ER + BCs where they are dependent on ER activation to exert their effects on growth and differentiation. Thus, it is the expression of ER, which determines response to endocrine therapy, and a lack of the ER is the principal cause of de novo resistance to endocrine therapy.
Cyclical and continuous methylation/demethylation of CpG dinucleotides is a prominent characteristic of ER and its target genes (83). A small proportion of BCs presenting with non-existent ERα gene expression have an intrinsic gain in CpG site methylation (84, 85). Another theory suggests that an increase in the deacetylation of histones, which limits transcription by condensing the nucleosome structure, could be a cause of non-existent ER transcription (85). Inhibition of the histone deacetylase, HDAC, revives ER transcription in BC cell lines which do not express the receptor (86). It is understood that a combination treatment with histone deacetylase inhibitor (HDACi) and DNA methyltransferase-1 (DNMT1) inhibitor will re-establish sensitivity to SERMs in BC cells not expressing the receptor (87). Thus, inhibitors to HDAC and DNMT1, by interfering with the epigenetic changes, could prove to be promising anticancer drugs in a small proportion of BCs presenting with a loss of the ER.
Epigenetic mechanisms play an important role in DNA methylation, chromatin modification and miRNA regulation. Recently, reports suggest that despite dysregulated signaling through the ER by SERMs and inadequate expression of the receptor, the target genes affected promote specific phenotypic changes in BC subtypes (88). Unsurprisingly, epigenetic mechanisms in the form of histone deacetylase inhibitors or demethylation agents are now being used in anti-cancer therapy (89–91). The ER and HDAC pathway crosstalk lead to changes in the activity and expression of ER and p21Waf1/Cip1, affecting cell proliferation, differentiation and survival (92). Hanahan and Weinberg have reviewed the importance of the loss of genomic methylation as well as hyper/hypo methylation of genes, which are involved in cell signaling, proliferation, or apoptosis and are thought to favor cancer progression (93).
Clinical studies have been done on endocrine resistant MBC patients using HDACi in combination with an endocrine agent. In one recent Phase II study done by Munster et al., an HDACi (vorinostat) was used in combination with tamoxifen or tamoxifen alone and it was noted that in the combination arm the objective response rate (ORR) was 19% and the clinical benefit rate (CBR) was 40%, thereby demonstrating that the combination is effective in overcoming endocrine resistance (94). Another Phase II study was done in patients treated with an AI, who had locally recurrent or ER+ metastatic breast cancer (MBC). Here, they were given either exemestane, an AI, with or without entinostat (a benzamide HDACi) and the combination arm again showed a benefit in terms of the progression-free survival (PFS) and overall survival when compared with the patients on exemestane alone (95). The HDAC inhibitor entinostat is being studied in BC patients on an AI (ClinicalTrials.gov Identifier: NCT02820961) (Figure 3B). Thus, HDACi's appear to be have an important role in reversing endocrine resistance.
1b. Mutated ERα
Mutations of the ER gene play a crucial role in the effectiveness of anti BC drugs. Although such mutations have not been detected in primary breast tumors, Fuqua et al. have detailed on the occurrence of an ERα variant with an A-to-G bp transition, which introduces a Lysine-to-Arginine substitution at residue 303 in almost a third of hyperplastic breast lesions (96). Although mutations in tamoxifen treated metastatic ER + BC patients present as ERα mutations, the majority are in the ERα LBD region, leading to constitutive activation of the ER (97–100). A point mutation, leading to a defective ER-ERE complex due to an alteration in the LBD was observed in tamoxifen resistant MCF7 cells (101, 102). A recent study identified hot spot mutations, like the Tyr537Ser, Tyr537Asn, and Asp538Gly (Y537S, Y537N, D538G) mutants within the LBD which favored the agonist structure of the ERα receptor, driving E2 independent transcriptional activity and proliferation of cancerous cells, leading to endocrine resistance (97). Whole genome RNA profiling in ER+/HER-2 negative BC patients revealed mutations/aberrations within the LBD domain of the ERα gene (17). The Y537S, D538G mutants had been previously identified and defined, while the V422del aberration is consistently observed in ER+ MBC (17). Thus, ERα mutations are more prevalent in MBC patients treated with AIs and occur in ~25–30% of these patients (103). The most prevalent mutations present in these patients are the D538G, Y537S, Y537N, Y537C, and E380Q. A recent study has indicated that constitutive transcriptional activity of the D538G mutation in ER, leads to overexpression which in turn leads to enhanced proliferation, thereby conferring resistance to tamoxifen (99).
Another point mutation in ER (nucleotide A908G), leads to an enhanced response upon activation by E2 and has been associated with invasive BCs (96). Such loss of regulation could contribute to the development of endocrine resistance as has been reported by Ellis et al., where they identified an ERα/YAP1 fusion gene and defined its association with endocrine-resistant BCs (104). In the last few years several studies have been done to provide a complete set of mutations that could cause BC although in primary tumors, no mutation has been identified in the ER (97, 98, 105). In the metastatic scenario, several ER mutations have been identified by at least three other studies (97–99). Basically, several studies using next generation sequencing and liquid biopsies in cohorts of clinical trials since 2013, led to an interest in the high prevalence of ERα mutations in ER+ MBC patients with prior AI treatment (100, 106, 107). Mutations at the D538G and/or the Y537S, E380Q, Y537N, and Y537C sites on the ERα gene, have been studied as part of the BOLERO-2, SoFEA as well as the PALOMA-3 trial and a few others (106, 108). The results from these studies demonstrate the possible use of mutations at these Y537S, Y537N, Y537C, D538G, and E380Q sites on the ERα gene as marker to screen for endocrine therapy resistant BC (109). The Y537S and D538G mutant forms of ERα have distinct cistromes when compared to the E2-stimulated WT ER (ERαWT) form and have been demonstrated to drive endocrine resistance and metastasis (110). A recent study on a small molecule SERD, AZD9496, demonstrated that it binds to and downregulates the D538G/Y537S/Y537N/Y537C containing ERα proteins in vitro, leading to an appreciable inhibition in the rate of tumor progression (111). AZD9496 was more effective than fulvestrant in suppressing the growth of tumors driven by ERαWT and ERαMUT (Y537S) (112). The drug is also well-tolerated in ER+/HER2 negative advanced BC as demonstrated in a phase I clinical trial (113). Some recent SERDs are being developed to target ER in both their wild-type (ERαWT) and mutant forms (ERαMUT). The purpose of this study was to evaluate the efficacy of a novel orally bioavailable SERD, elacestrant (RAD1901), in preclinical models of ER+ BC. Elacestrant (RAD1901) is one such SERD that inhibits cell proliferation in ER+ BC cell lines and is being studied as part of clinical trials in ER+/HER-2 negative advanced BC where partial response as an effective SERD was demonstrated in heavily pre-treated ER+/ER mutant MBC and patients (ClinicalTrials.gov Identifier: NCT03778931 and NCT02338349) (114, 115). A structurally and chemically unique SERD, GDC-0927, induces tumor regression in ER+ MBC patients including those with ERα mutations (ClinicalTrials.gov Identifier: NCT02316509) (116, 117). Essentially, a range of SERDs are being developed with differential activities as ER antagonists to combat the clinical effectiveness of fulvestrant due to poor bioavailability. A recent study evaluated three recently developed ER ligands, GDC-0810, AZD9496 and GDC-0927 along with fulvestrant and it was GDC-0927 and fulvestrant that showed enhanced transcriptional suppression of the ER (118). A new class of drug, the selective estrogen receptor covalent antagonist (SERCA) with H3B-5942 being identified as an ERα antagonists that inactivates both the ERαWT and ERαMUT forms (119).
Thus, present studies to-date demonstrate that ER mutations are rare in primary tumors but appear to be reasonably frequent in the progression to endocrine resistance and can be used as biomarkers for prognosis/prediction of response to endocrine therapy along with promoting the development of therapeutic strategies. Evaluating and studying a variety of potent ER antagonists will promote the development of clinically effective SERDs (118).
2. Progesterone Receptor (PR) and Signaling
In BC, the Progesterone Receptor (PR) also plays an important role and its signaling has been at the center of various targeted therapies, including the selective progesterone receptor modulators (SPRM) (120). The PR is regulated by the ER and is required for mammary gland development. Progesterone binds to the PR, which is followed by receptor dimerization and translocation to the nucleus where it binds to the progesterone response elements (PRE) within the promoter/enhancer regions of PR target genes, thereby leading to either the upregulation or downregulation of those genes. Apart from binding to the PRE in its target genes, it also binds as a complex of transcription factors (PR in complex with AP1 and SP1) to regulate genes devoid of canonical PRE binding sites (121, 122). Indeed, a study of PR mediated regulation of oncogenic genes in PR regulated BC models has yielded information on its varied actions (123).
The PR exists as two isoforms, the PR-A and PR-B with the PR gene regulated by a complex of ERα-AP1-SP1 at an ERE motif half site on the PR gene (124, 125). Although normal mammary epithelial cells express separate receptors (ER and PR) on designated cells, the receptors are co-expressed in oncogenic cells (126). A dominant pathway takes over the functions of another during targeted therapy, in one such instance, a loss of the PR during endocrine therapy leads to the cancer becoming more aggressive. These patients have a poor survival outcome (127). Evidence from various studies, reflect the importance of the PR, in that, ER+ BC tumors presenting with PR-negative status have a poorer outcome than those with PR-positive (PR+) ones (128). A loss in PR leads to activation and upregulation of the PI3K pathway (129). A recent study demonstrated that the PR-B in a complex with ER and PELP1 promoted the regulation of E2 dependent ERα target genes associated with BC cell proliferation and tamoxifen resistance (130). In another study by Mohammed et al., PR was shown to be complexed with the known ERα co-factors, NRIP1, GATA3, and TLE3, upon stimulation with progesterone. They also concluded that activation of the PR led to the formation of an ERα-PR complex (131).
3. Receptor Tyrosine Kinases (RTK)
Receptor Tyrosine Kinases (RTK) are a family of receptors that are activated upon binding their respective ligands, which are mainly the growth factors, hormones or cytokines. The most prominent RTK's are the family of epidermal growth factor receptors (EGFR), Insulin-like growth factor-I receptor (IGF-IR), hepatocyte growth factor receptor (HGFR), vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), anaplastic lymphoma kinase (ALK), ROS protooncogene 1 (ROS1) and receptor like tyrosine kinase (RYK). An upregulation of RTKs is observed in breast cancer and is indicative of poor prognosis (132, 133). Upon binding to their respective ligands, the prominent pathways activated are the mitogen activated protein kinase (MAPK), janus kinase (JAK)/signal transducer and activator of transcription (STAT) and phosphoinositide (PI3K)/AKT pathways. A multitude of evidence suggests that inhibitors of RTK's can reverse therapeutic resistance in metastatic breast cancers (134) (Figure 3B).
3a. EGF/EGFR/HER2 Signaling
The ErbB family of growth factor receptors includes epidermal growth factor receptor (EGFR) (also called ErbB1/HER1, ErbB2/HER2, ErbB3/HER3, and ErbB4/HER4). Upon activation by ligands, such as EGF, which induces dimerization and autophosphorylation, signaling proceeds through various pathways including the MAPK-ERK and PI3K-AKT pathways (Figure 3B) (135). EGFR, HER2, HER3, and HER4 are all implicated to varying degrees in BC with their relative involvement in the order HER2-EGFR-HER3-HER4. HER2 expression is reported in 20–30% of BCs and HER3 is up-regulated in 10% of BCs, and its common association with HER2 makes its specific role difficult to determine (136).
With the ever increasing evidence for a role of ERα in signaling via EGFR, an association with DNA-synthesis has been observed in MCF7 cells, whereby an E2 antagonist led to a reduction in the phosphorylation of the EGFR, followed by reduced DNA synthesis and cytoskeleton formation through a Src mediated pathway (137). The presence of alternative forms of ERα (66-KD): ERα36 (36-KD) and ERα46 (46-KD), which are mainly located in the cytoplasm and plasma membrane, play additional roles and ERα36 has also been associated with tamoxifen resistance (138).
GPER is activated by E2 and it represents a transmembrane receptor that modulates E2 actions (139). The activation of EGFRs by E2 mainly occurs via the membrane- bound GPER1 (76). E2 increases phosphorylated MAPK in SKBR3 cells which express the membrane bound GPER1, while phosphorylation remains unchanged in the MDA-MB-231 which do not express GPER1. However, transfecting the protein into MDA-MB-231 leads to activation by E2 (140). These studies show that ER works in concert with the ErbB family of proteins to foster and assist cancer cell proliferation. With GPR30 being a hindrance in the EGFR signaling pathway and attenuating the inhibition of MAP kinases, combination therapy with tamoxifen and GPER inhibitors could lead to novel therapeutic options (70).
Several retrospective clinical studies have shown the significance of growth factor signaling in both de novo as well as assumed endocrine resistance. While it was previously hypothesized that a loss in the expression of ER could lead to tamoxifen resistance, evidence suggests that only three quarter of tumors express the ER (82). Upregulation of growth factor signaling in MCF7 cells, as well as HER2 in patients treated with tamoxifen, suggests a redundancy in the operation and thus the use of combination therapy may be beneficial (82). Data from studies imply that tumors switch from HER2 to ER and vice versa as the preferred signaling pathway, with therapy toward HER2 leading to an activation of the ER pathway and vice-versa (82, 141). The dependence of the two pathways on each other is highlighted in MBC patients who have been treated with an AI or the ER downregulated with fulvestrant and have progressed with trastuzumab or lapatinib. Thus, combination therapy with inhibition of the ER and HER2 axis has proven to show a benefit in dual ER-HER2 positive (HER2+) patients as demonstrated in a phase III study (TAnDEM: trastuzumab in dual HER2+/ER+ MBC). Here, patients treated with trastuzumab plus anastrozole, an AI, showed a benefit in terms of the PFS when compared with the women on anastrozole alone (142). Recently, a study targeted the extracellular domain of HER2, which is responsible for HER2 homo- and heterodimerization. Consequently, disrupting the 16aa sequence within the extracellular domain of HER2 (HER2-ECD-Δ451-466), led to a reduction in the activity of HER2, and switched off signal transduction mediators, such as ERK1/2 and PI3K/AKT (143).
Signal transduction inhibitors are crucial mediators in overcoming endocrine resistant BC. It was reported in 2000 that inhibition of HER2 and MAPK in HER2 overexpressing tamoxifen resistant MCF7 cells reinstated the interdependence of ER with nuclear receptor corepressor (N-CoR), thereby enhancing the action of tamoxifen and abrogating the antioestrogen resistance (144). The findings above are supported by clinical studies where tumors over expressing HER2 and AIB1 had a worse outcome with tamoxifen (145). In a recent study done on AI treated patients, it was evidenced that AIB1 played an important role in regulating selective ER transcriptional activity and promoting tumor recurrence (146).
Lapatinib, a dual inhibitor of EGFR and HER2 was used to study its role in prototypes of HER2+ BC cell lines with assumed endocrine resistance, where it restores endocrine sensitivity (147). In a phase III study, a combined treatment with letrozole, an aromatase inhibitor, and lapatinib or letrozole alone, showed a benefit in the combination arm of the study with a significantly higher PFS in MBC with ER-negative/HER2+ tumors (148).
Clinical studies are being done using EGFR inhibitors in isolation or a combination therapy in order to address endocrine resistance. A phase II study using tamoxifen and the EGFR inhibitor gefitinib or gefitinib alone, showed no significant enhancement in PFS in the endocrine naïve or group treated with an AI (149). In a separate Phase II study on ER+ MBC, the combination treatment of anastrozole and gefitinib showed an increase in the PFS over patients treated with anastrozole alone (150).
Acquired HER2 mutations lead to endocrine resistance in a proportion of patients with ER+ MBC. Tumors driven by a HER2 amplified mechanism are known to be resistant to endocrine therapy (142, 148, 151). A whole genome landscape study identified hotspot mutations in HER2 (D769Y, L755S, and S310Y) which were more common in acquired endocrine resistant tumors (9). Mutations within the HER2 gene lead to E2 independence and resistance to the first line therapy, which includes tamoxifen, fulvestrant and palbociclib, a CDK4/6 inhibitor in BC patients. In these patients an ER-directed drug in combination with neratinib [a pan-HER tyrosine kinase inhibitor (TKI)] was an effective therapy (152). Mutations within the HER2 gene are more common in the ER+ MBC setting and HER2 mutations favor dimerization with HER3 (ERBB3) (153). This study also noted that inhibition of HER2 and the ER pathways is required for therapy in ER+/HER2 mutant BCs (153). Neratinib has been approved by the FDA for treatment in the adjuvant setting of BC patients with early stage HER2 amplified disease, based on the ExteNET trial (154, 155). In the phase III ExteNET study, the neratinib arm of treatment showed a benefit in the disease-free survival (DFS) when compared to the placebo arm in early stage HER2+ BC (156).
3b. IGF-IR
Insulin-like growth factor-I receptor (IGF-IR) is a protein expressed as a trans-membrane tyrosine kinase and is activated by the stimulatory hormone/ligands insulin-like growth factor-I and -II (IGF-I, IGF-II), resulting in proliferation as well as anti-apoptosis (157). The IGF-I system is implicated in the pathogenesis of BC (158). There are also several lines of evidence that dysregulation of the IGF-I system and enhanced IGF-IR activation are involved in resistance to endocrine therapy and that IGFs play a paracrine/autocrine role in promoting tumor growth in situ during tumor progression, depending on the tissue of origin (159).
Upon binding of ligand to its receptor, the IGF-IR, it leads to phosphorylation of insulin receptor substrate-1 and−2 (IRS1, IRS2), which promotes growth by signaling through the MAPK/ERK, PI3K/AKT, and JAK/STAT pathways. Elevated plasma concentrations of IGF-I have been linked to a higher risk for BC (160). Genetic polymorphisms in genes encoding IGF-I have been reported, and may contribute to an increased risk for BC (161, 162). IGF-IR activation leads to phosphorylation of the MAPK-AKT and subsequent activation/phosphorylation ERα (56).
Interdependence between IGF-IR and ERα in BC prototypes is well-studied, and is implicated as a mechanism of antioestrogen resistance (163, 164). A study in EGFR-positive tamoxifen-resistant variants of MCF7 (TAMR) cells, demonstrated reduced expression of IGF-IR protein levels when compared to their wild-type MCF7 cells. However, the phosphorylated IGF-IR protein levels were equivalent in the two cell lines under basal growth conditions, this was due to an increase in the IGF2 expression, which activated both IGF-IR and EGFR (158). A study by Creighton et al., identified a set of genes that were either upregulated or downregulated by IGF-I which represent hyperactive pathways and hormone independence (165). Basically, the IGF-I signature obtained from MCF7 cells presented with a similar observation in clinical BC patients. The profiled tumors exhibiting the IGF-I signature correlated with poor prognosis and was indicative of a poor outcome. In another study, patients with tamoxifen-resistant tumors with higher IGF1 and ERα expression developed tamoxifen resistance after a longer period of time, and tamoxifen-resistant tumors had lower IGF1 and ERα expression compared to tamoxifen-sensitive tumors (166). A separate study using a kinome wide siRNA screen demonstrated a role for the combined inhibition of IGF-IR and the insulin receptor (InsR), where the dual tyrosine kinase inhibitor OSI-906 (inhibitor of InsR and IGF-IR) in combination with fulvestrant inhibited the growth of hormone independent tumors when compared to either drug alone (167). The IGF-IR-specific inhibitors (like AG1024 and AEW541) or an IGF2 neutralizing antibody inhibited basal IGF-IR, c-SRC, AKT and EGFR phosphorylation, and significantly reduced tamoxifen-resistant basal cell growth. Interestingly, AEW541 also inhibited insulin and IGF2-stimulated effects in tamoxifen-resistant cells (168).
3c. FGFR Signaling
Binding of FGF ligands to the FGFR receptor promotes receptor dimerization followed by activation of the kinase domain and subsequent activation of the PI3K/AKT, RAS/RAF/MEK1/2-ERK, phospholipase Cγ (PLCγ), and STATs (169). Amplification of the genes located at the 11q12-14 chromosomal loci, which includes FGFR1 and FGF ligands 3, 4, and 19 occurs in more than 10% of BC patients, depending on the specific type of BC (170, 171). A third of patients with FRFR1 amplified tumors also exhibit amplification of oncogenes like CCND1, FGF3, FGF4, and FGF19 (172, 173). FRGR1 expression and signaling via FGFR1 is essential to survival of FGFR1 amplified BC cell lines (174). Apart from gene amplifications, enhanced expression of its ligand and mutations have been verified in the FGFRs from BC patients, which points to multifactorial mechanisms involved in the FGF/FGFR pathway (175). Dysregulated regulation of FGF/FGFR signaling and its control in oncogenic processes is poorly understood, although evidence suggests that aberrant FGFR1 signaling mediates endocrine resistance through the PI3K and MAPK pathways (176). A small molecule inhibitor dovitinib (TKI258) led to antitumor activity in FRFR1 amplified MDA-MB-134 and FGFR2 amplified SUM52 BC cell lines (177). Interestingly, the FGFR1 amplified cell lines MDA-MB-134 and SUM44 were resistant to treatment with tamoxifen (176). Activating mutations of the FGFR gene have been shown to have oncogenic potential and driving resistance to endocrine therapy (178, 179).
Amplification of FGFR1 correlated with short overall survival rates (OSR) in HR+ BC (180). Consistent with these findings, FGFR amplifications are associated with poor prognosis and endocrine resistance in HR+ BC (180–182). Resistance to endocrine therapy upon aberrant FGF/FGFR pathway amplifications/signaling, offers a strong rationale to study the role of FGFR inhibitors. FGFR TKIs offer one the best approaches in targeting tumors with ER+/HER2 negative/FGFR amplified status and some of these are in initial phase clinical trials. Some of the selective pan-FGFR inhibitors are BGJ398 (infigratinib), JNJ-42756493, Debio1347, TAS-120, AZD4547, ARQ087, and BAY1163877 (183). The role of BGJ398 was evaluated and initial reports suggest a promising role in breast cancer progression and metastasis to the lung (184). AZD4547 has shown promise as an anti-tumor drug in combination with an AI (anastrozole/letrozole) in the RADICAL trial (ClinicalTrials.gov Identifier: NCT01791985) (185).
Dovitinib, a non-selective TKI (TKI258) was assessed in a phase II study (ClinicalTrials.gov Identifier: NCT01528345) in combination with fulvestrant in HR+/HER2-negative MBC patients (186). The study was terminated due to low numbers of enrolled patients, although the combination arm of treatment showed promising clinical activity in the FGF pathway-amplified subgroup. Another non-selective TKI, lucitanib (E-3810) is in a phase II trial (FINESSE: ClinicalTrials.gov Identifier: NCT02053636) as monotherapy in ER+, FGFR1-amplified/FGFR1-non-amplified MBC (187). Whole exome sequencing revealed amplification of FGFR1, CCND1, and FGF3/4/19, all being associated with resistance to letrozole (17). The same study also noted that combined treatment of ER+ FGFR1/CCND1 co-amplified CAMA1 BC cells with palbociclib, and/or the FGFR1 inhibitor, lucitanib, lead to a reversal in antioestrogen resistance (17). A recent study demonstrated amplification of the FGFR gene which led to aberrant FGFR signaling and thus resistance to therapy with an anti-ER drug and a CDK4/6 inhibitor (188). In line with these findings, a phase Ib trial (ClinicalTrials.gov Identifier: NCT03238196) with erdafitinib in combination with palbociclib and fulvestrant is ongoing in ER+/HER2 negative/FGFR amplified MBC.
With regard to mutational studies, FGFR mutations are infrequent in BC, although mutations in the FGFR gene have been identified in BC with unknown functional roles (178, 189). Genome wide association studies have identified FGFR2 as one of the loci associated with BC (190). According to another GWAS study, the four genomic locations associated with BC were rs1219648, rs2420946, rs11200014, and rs2981579, all of which were located on intron 2 of the FGFR2 gene (191). Genetic fusions of the FGFR1-3 gene also represent a small proportion of aberrations and a causative agent in BC (178, 192, 193). These studies suggest a prominent role for FGFR1 amplifications and that all future studies and trials could focus their therapeutic strategy at targeting and inhibiting FGFR1.
3d. SYK
The spleen tyrosine kinase (SYK) protein tyrosine kinases are comprised of SYK and ZAP-70, which contain two SH2 domains and C-terminal kinase domains interrupted by two interdomains A and B (194, 195). SYK is primarily expressed by a variety of hematopoietic cells ranging from B cells, mast cells, neutrophils to macrophages and functions in proliferation, differentiation and adhesion (196, 197). Here, activation of the specific receptor (B cell receptor) promotes phosphorylation of immune-receptor tyrosine-based activation motifs (ITAMs) and recruitment, autophosphorylation of SYK (198). SYK subsequently modulates its actions by activating several downstream effectors like RAS-RAF-MEK-ERK (199). SYK functions as a tumor suppressor in BC with a reduction in SYK expression being associated with poor prognosis and metastasis (200, 201). In samples from patients, SYK expression is lost as the tumor progresses from ductal carcinoma in situ (DCIS) to invasive breast cancer (202, 203). However, in various other solid cancers it has tumor promoting activity depending on the association of the cancer with inflammation (204). With quite a few SYK inhibitors in clinical trials for other cancers, its role in ER+ BC associated with inflammation could be studied (205–208).
4. Cell Cycle Regulators
BC sensitivity to endocrine treatment is impacted by the activity of both positive and negative cell cycle regulators (209). Studies have shown that overexpression of positive cell cycle regulators like c-MYC, cyclins E1 and D, contribute to the development of endocrine resistance by activating CDKs (210, 211). Investigators have also demonstrated that aberrant c-MYC expression can contribute to endocrine resistance by altering the regulation of p21WAF1/Cip1 (210, 212). Consistent with the activity of CDK, downregulation or loss of function of the two G1 checkpoint CDK inhibitors, p21 and p27 (CDKN1A and CDKN1B), is associated with endocrine resistance (213, 214). A separate study surmised a central role for E2F proteins, in that unliganded ER plays a role in the E2 deprived growth of long term E2 deprived BC cells by regulating a transcriptional machinery (215). The same study also identified CDK4, an activator of E2F transcription as a modulator for E2 independent BC cell proliferation and inhibition of CDK4 led to an inhibition in the growth of fulvestrant-sensitive and -insensitive ER+ cell lines in the absence of E2 (215). These findings supported the development of CDK4 inhibitors as possible therapeutics for BC.
The cyclin D-CDK4/6-INK4-retinoblastoma (Rb) pathway plays a pivotal role in the proliferation of normal breast epithelial and cancer cells. The CDKs are crucial cyclin dependent drivers in cell cycle, cell division and thus cancer. Their importance in the cycle is well-known, with the most common being: G0 (quiescence), G1-phase (pre-DNA synthesis), G1/S-phase, S-phase(DNA synthesis), and M-phase (cell division) cyclins (216). There are typically four different CDK's (CDK1, 2, 4, and 6), dependent on the cyclins D (D1, D2, D3) (217–219). Nascent DNA synthesis occurs during the S phase of the cell cycle at which the cyclin D1-CDK4/6 complex serves as the enzyme that catalyzes the phosphorylation of Rb (retinoblastoma) protein and dictates DNA replication. Coupling of cyclin D to CDK4/6 is regulated by the INK4, Cip, and Kip group of proteins. CYCLIN D1 activates the CDK4/6 essential for mediating RB-induced cell cycle progression at the G1/S checkpoint (220, 221). In the TCGA network studies, CYCLIN D1 is amplified in 58% of luminal B BCs with 25% showing a gain in CDK4. However, CYCLIN D1 was amplified in only 29% of luminal A tumors with only 14% showing a CDK4 gain (35). In another study, luminal B-type BCs resistant to endocrine therapy were identified by a unique gene signature indicating a loss of RB protein (222). The E2F4 transcriptional cascade is suppressed upon CD4/6 inhibition in hormone independent ER+ BC cells and ER+ BC patients, thereby supporting the benefit of adjuvant CDK4/6 inhibition in ER+ patients (223).
Thus, Cyclin D1 and CDK4/6 inhibitors represent strategies to overcome endocrine resistant BCs and potent CDK4/6 inhibitors have become extensively available in the last decade. A recent study revealed aberrant FGFR signaling as the mechanism causing resistance to combinatorial therapy by anti-ER's with CDK4/6 inhibitors (188).
Preclinical studies have confirmed the usefulness of CDK4/6 inhibitors specific to a particular molecular subtype of BC (224, 225). Three FDA approved, novel CDK4/6 inhibitors used in treatment of ER+ MBC are palbociclib (PD0332991, Pfizer), ribociclib (LEE011, Novartis and Astex), and abemaciclib (LY835219, Eli Lilly) (226–228). In BC cell lines exhibiting the luminal ER+ BC subtype, proliferation was inhibited by palbociclib and ribociclib (225). The BC cell lines exhibiting non-luminal or the basal subtypes, presented with resistance to palbociclib (225). The clinical trials involving the three prominent CDK4/6 inhibitors: PALOMA for palbociclib, MONALEESA for ribociclib and MONARCH for abemaciclib; were studied for efficacy in MBC patients. CDK4/6 inhibitors are used both as a single agent and as a combination therapy. With endocrine resistance being an obstacle toward effective therapy, the addition of a CDK4/6 inhibitor to endocrine therapy as a combination therapy presents with prolonged PFS and has recently been included as a first line therapy in advanced HR+ BC (226, 228–230) (Figure 3B).
4a. Palbociclib (PD0332991)
In the largest clinical validation of a CD4/6 inhibitor, palbociclib was seen as a breakthrough therapeutic drug. In the phase II PALOMA-1 trial on ER+/HER2 negative advanced BC patients, significant prolongation of median PFS was demonstrated in the combination arm (palbociclib with letrozole) when compared to letrozole alone (231, 232). Again, the PALOMA-2 phase III study, validated on ER+/HER2 negative MBC patients reported significant benefit in PFS in the combination arm when compared to letrozole alone (226). In the PALOMA-3 phase III trial, a benefit in the PFS was demonstrated for women with ER + MBC in the combination arm (palbociclib with fulvestrant) when compared to fulvestrant alone (233, 234). The PALOMA-4 trial (ClinicalTrials.gov Identifier: NCT02297438) is ongoing as a first line treatment. A summary of the three PALOMA studies, pointed to palbociclib plus letrozole as the therapeutic first choice in ER + MBC, followed by palbociclib plus fulvestrant as another therapy of choice in ER + MBC, previously on an aromatase inhibitor. The Food and Drug Administration (FDA) granted approval for palbociclib to be used as a first line therapy in combination with an AI or letrozole in post-menopausal women with HR+/HER2 negative advanced or MBC. It has also been approved as a combination therapy with fulvestrant in the treatment of HR+/HER2-negative advanced or MBC in women with disease progression following endocrine therapy.
4b. Ribociclib (LEE011)
Ribociclib is another selective and reversible CDK4/6 inhibitor approved by the FDA. In the phase II MONALEESA-1 study on HR+/HER2-negative BC patients, a decrease in the expression of the cell proliferation marker Ki-67 was observed in the combination arm (letrozole with ribociclib) and single agent arm (letrozole alone) (235). The phase III MONALEESA-2 trial was a crucial trial focused as a first line treatment in untreated post-menopausal women with HR+/HER2-negative recurrent or MBC. The results from this trial presented with an extension in the PFS for the cohort of patients treated with ribociclib plus letrozole when compared with the placebo group of letrozole alone (228). In the subsequent phase III MONALEESA-7 trial, appreciable benefits were observed in HR+/HER2-negative advanced BC patients. Here too, the combination arm (ribociclib with tamoxifen/AI and goserelin) showed a 2-fold enhancement in the PFS when compared with the placebo arm (236, 237). Based on the results from the MONALEESA trials, the FDA approved ribociclib in combination with an AI as a first line therapy in pre/perimenopausal women with HR+/HER2-negative advanced or MBC. It was previously approved as a first line therapy or following disease progression on endocrine therapy in combination with fulvestrant or an AI in post-menopausal women with HR+/HER2-negative advanced or MBC.
4c. Abemaciclib (LY2835219)
Abemaciclib is another CD4/6 inhibitor, although it has higher selectivity and inhibitory effect for CDK4 than for CDK6 (238–240). In the phase II MONARCH-1 trial, abemaciclib showed enhanced clinical benefit as a single agent in refractory HR+/HER2-negative MBC following multiple prior therapies (241). The phase III MONARCH-2 study conducted on advanced BC patients, demonstrated an extension in the PFS in the combination arm (abemaciclib with fulvestrant) over the placebo arm (fulvestrant alone) (242). Again, the phase III MONARCH-3 study, of abemaciclib (CDK4/6 inhibitor) plus an AI (letrozole or anastrozole) compared to letrozole/anastrozole alone on HR+/HER2-negative MBC has shown promise as an initial therapy with significantly improved PFS (227, 243). Based on the results from the MONARCH trials, the FDA approved abemaciclib as a combination therapy with an AI in post-menopausal women with HR+/HER2-negative advanced or MBC, as a combination therapy with fulvestrant in women with HR+, HER2-negative advanced or MBC with disease progression following endocrine therapy and as a monotherapy for women and men with HR+/HER2-negative advanced or MBC with progression following endocrine therapy and prior chemotherapy.
The impressive and exceptional results from the CDK4/6 inhibitor trials have led researchers to study its efficacy and suitability in several other trials using the CDK4/6 inhibitor as a single agent and in early stage BC (244, 245). The essence of a role for CDK4/6 inhibitors stems from the need for novel agents to supplement endocrine therapy in high risk ER+ disease. Several other trials like the PEARL, NeoPalAna, neoMONARCH, FELINE, CORALLEEN, PELOPS, KENDO (ClinicalTrials.gov Identifier: NCT03227328), and SONIA (ClinicalTrials.gov Identifier: NCT03425838) trials, all are focused toward evaluating the efficacy of CDK4/6 in the advanced ER+ BC and in other BC subtypes (245).
4d. Combination of CDK4/6 With PI3K/AKT/mTOR
The remarkable results observed with CDK4/6 inhibitors has been very well-received and approved by the FDA. However, a large number of ER+ BC patients continue to experience relapse and recurrence (246). There is a complex crosstalk between ER+ BC, constitutive PI3K activation and the CDK4/6 pathway. This provides a strong rationale for the combined targeting of both the CDK4/6 and PI3K pathways in effective control of the tumor progression (247–249). Studies have suggested that inhibition of the either the CDK4/6 or the PI3K/AKT pathway, delays the onset of endocrine resistance (248, 250). Combinatorial inhibition of both the PI3K and CDK4/6 pathways overcomes the resistance due to single agent inhibition by CDK4/6 by downregulating cyclin D1 and arresting cell cycle progression (248, 249). Several combination therapy trials are underway to identify the best strategy to overcome endocrine resistance in ER+ BC. Trials that combine the CDK4/6 inhibitors with various PI3K/AKT/mTOR inhibitors (ClinicalTrials.gov Identifiers: NCT03128619, NCT03006172, NCT02684032, NCT02732119, NCT02871791, NCT02599714) are ongoing, with the aim to inhibit tumor growth and prevent relapse. The triplet inhibitor trial (ClinicalTrials.gov Identifier: NCT02088684) with ribociclib, fulvestrant and BKM 120 (buparlisib, PI3K-pan class I-inhibitor) or BYL719 (alpelisib, PI3K-alpha specific class I inhibitor) in HR+/HER2-negative advanced or MBC is one study aimed to study its efficacy and suitability as a mode of treatment in these patients.
4e. Combination of CDK4/6 With Immune Checkpoint Inhibitors
CDK4/6 inhibitors act as cell cycle checkpoint inhibitors and induce cell cycle arrest, senescence and immunogenicity in the TME. Studies have demonstrated an enhancement in the anti-tumor response upon treatment with CDK4/6 inhibitors due to an enhancement in interferon production, reduced T-regs and enhanced T cell activation (251, 252). Addition of immune checkpoint inhibitors (PD-1and CTLA-4) to CDK4/6 inhibitors holds promise by inhibiting the growth and proliferation of cancerous cells and, with this study as the background, a clinical trial (JPCE) has focused on studying the suitability and efficacy of using abemaciclib in combination with pembrolizumab in HR+/HER-2 negative metastatic BC patients (253). Another study is recruiting to study the response of fulvestrant and pembrolizumab as a combination therapy (ClinicalTrials.gov Identifier: NCT03393845) in HR+/HER-2 negative advanced or metastatic BC patients.
4f. CDK4/6 Associated Response and Fusion Genes
Although CDK4/6 inhibitor are effective in controlling tumor growth, a fraction of the cells acquire resistance to CDK4/6 inhibitors. Upon mechanistic dissection, a role for the FAT1 and RB1 proteins were identified, wherein, a loss of function in FAT1 was observed with a concomitant increase in CDK6 which was attributed to enhanced binding of YAP and TAZ on the CDK6 promoter and regulation of the Hippo pathway and associated factors within the pathway (254).
High throughput RNA sequencing (RNA-seq) methods have led to the identification of ERα gene fusions with some instances of more than four ERα coding exons fused in frame or out of frame (17, 104, 255). A number of ERα gene fusions have been identified in endocrine resistant and ER+ MBC, some of the common ones being the ERα-e6 > DAB2, ERα-e6 > GYG1 and ERα-e6 > YAP1 structures (104, 256). Li et al. in 2013, identified an in-frame ERα fusion protein consisting of ERα exons 1–6 and the c-terminal YAP1 sequence (ERα-e6 > YAP1) that functioned as a driver of endocrine resistance and E2 independent proliferation (104). A recent study by the same group identified at least two fusion genes (ERα-e6 > YAP1 and ERα–e6 > PCDH11X) in advanced ER+ BC and palbociclib was observed to inhibit the fusion gene driven T47D growth under hormone deprived conditions in a dose-dependent manner (257). This observation was consistent with previous reports of palbociclib being able to suppress the growth of pRb expressing tumors with ERα mutations under E2-deprived conditions (258).
A genetic landscape sequencing study on circulating tumor DNA of samples from the PALOMA-3 study demonstrated that acquired mutations in the RB1, PIK3CA, and ERα genes emerged in the treatment arm of fulvestrant and palbociclib (259). In this study too, ERα Y537S was one of the driver mutations that promoted resistance to fulvestrant and clonal evolution is surmised as the principle behind resistance to therapy (259).
5. Other Transcription Factors
5a. PI3K/AKT/mTOR
The PI3K/AKT pathway is mainly activated downstream of RTKs, which can phosphorylate several targets, including NFκB, BAD, IKK, p27, FOXO1, GSK-3β, MDM2, and mTOR, with the mTOR complex being the best described target of AKT signaling (260). The tumor suppressor gene phosphatase and tensin homolog (PTEN) is a negative regulator of mTOR pathway (261). PTEN may be downregulated through several mechanisms, including mutations, loss of heterozygosity, methylation, aberrant expression of regulatory microRNA, protein instability and activated mTOR signaling is also associated with Cowden's syndrome (PTEN mutations) (262).
PI3K is comprised of several isoforms of the regulatory (p85) and a catalytic subunit (p110). Mutations are frequently identified in BC within the PIK3CA gene, encoding the p110α catalytic subunit (263). A large number of the PIK3CA mutations occur within the kinase (H1047R) and helical domains (E542K and E545K) of p110α subunit (264, 265).
The PI3K/AKT and E2 signaling crosstalk has been studied extensively in BC. As previously mentioned, activated AKT can phosphorylate ERα at serine-167 in the AF-1 domain and increase ERα-dependent transcription (56). Activation of the PI3K pathway in breast tumors is associated with reduced ERα levels and endocrine resistance (266). Signaling via GPER is another mode of signaling utilized by E2 activated PI3K (p110α subunit) which leads to inactivation of FOXO3a, thereby promoting progression of BC (74). GPER antagonists in combination with a SERM/SERD therapy could serve as an effective therapeutic strategy in GPER positive/ER+ BC (74). The above studies have also shown the importance of a crosstalk between the PI3K and ER pathway in antioestrogen resistance and suggested that combining a PI3K inhibitor with an ER down regulator is more effectual than either of them alone (267). Thus, constitutive activation of GFR signaling pathways leads to reduced ERα levels and subsequent endocrine resistance. In contrast, inhibition of the pathways leads to sensitization in response to antioestrogens and other oncogenic pathways are activated upon E2 deprivation or inhibition of ER in the endocrine resistant scenario (267).
Two other interdependent factors, mTORC1 and mTORC2 are part of a positive feedback mechanism belonging to the PI3K pathway, which have independent regulatory mechanisms and exert their effects through distinct targets and mechanisms (268). Upon inhibition of mTORC1, the alternate complex mTORC2 leads to an activation of the PI3K pathway, which indicated that inhibition of one arm of the pathway (via inhibitor), led to activation of another arm and may not be sufficient to produce a clinical benefit (269).
Clinical studies have validated the association between activation of the PI3K and de novo/assumed resistance to endocrine therapy. Several trials have been done where inhibitors of PI3K pathway have been combined with endocrine therapy. In a neoadjuvant study by Baselga et al., treatment with letrozole and everolimus (mTOR inhibitor), a more pronounced reduction in tumor cell proliferation and improved clinical response was observed when compared with letrozole alone in patients with early-stage ER+ BC (270). In another Phase III combination study, also called the BOLERO-2, patients with ER+ advanced or MBC, were either given everolimus and exemestane (an AI), or exemestane and placebo. The combination of everolimus and exemestane was found to have a median PFS that was significantly much superior to the exemestane only arm (271). Due to highly successful rates of PFS, this combination therapy of exemestane and everolimus has been approved on selected advanced patients of ER+ BC in the USA and Europe. A recent study has produced a systematic approach and an effective rationale behind the use of an mTORC1 inhibitor (RAD001) in combination with neratinib and tamoxifen/fulvestrant to target BC patients who relapse due to endocrine resistance, for example patients treated with RAD001 and an AI (272). This study demonstrated that triple blockade with RAD001, neratinib and tamoxifen/fulvestrant was highly effective in the long term E2 deprived BC cell lines and was well-tolerated in a xenograft model (272).
The BOLERO-2 study also demonstrated that patients who has the D538G and Y537S mutation within the ERα gene presented with aggressive disease and a shorter OS. When considering the benefit based on mutation site, it was more pronounced in the D538G group due to a larger number of patients. In the TAMRAD phase II trial, patients with ER+ MBC becoming resistant to AI treatment were given tamoxifen and everolimus or tamoxifen alone. Here too, patients in the combination arm showed an improved clinical benefit rate, time to progression, as well as an overall survival compared to patients treated with tamoxifen alone. This study stratified the patients based on primary and secondary endocrine therapy resistance and interestingly, patients with relapse after 6 months of AI treatment presented with an improvement in the PFS when compared to those patients that relapsed during adjuvant AI before 6 months on the treatment (273). Thus, further studies are warranted where a proportion of patients could benefit from the combined mTOR/ER targeting approach based on specific mutations within the ERα gene.
In the phase 3 BELLE-2 trial, patients with ER+/HER2 negative, locally advanced or MBC who had progressed on or after AI treatment and had received at least one line of chemotherapy treatment, were given the pan-PI3K inhibitor buparlisib plus fulvestrant or fulvestrant alone and it was observed that the combination arm of the treatment showed improvement in the PFS in these patients, although the toxicity observed warrants the use of other more α specific PI3K inhibitors (274). With the focus on PIK3CA, PIK3CB, and PIK3CD mutant tumors, some of the more recent isozyme-specific inhibitors target the p110α, p110β, and p110δ isoforms of PI3K: BYL719 (alpelisib) and MLN1117 for p110α and GDC-0032 (taselisib) for 110β-sparing inhibitors (275). In the phase III SOLAR-1 study (ClinicalTrials.gov Identifier: NCT02437318), post-menopausal women with HR+/HER-2 negative advanced BC were given either alpelisib/placebo (a PI3Kα specific inhibitor) in combination with fulvestrant and a significant extension in the PFS was reported in patients with PIK3CA mutations in the alpelisib arm of the study (276).
5b. PARP
Poly (ADP-ribose) polymerase (PARP) is a family of nuclear proteins which plays a vital role in the recognition and repair of endogenously/exogenously induced DNA damage (277, 278). PARP like BRCA1 and BRCA2 acts by repairing DNA and PARP inhibitors act by disrupting the DNA repair mechanism and increasing genomic instability (279, 280). Basically, it is the synthetic cellular/DNA lethality caused due to BRCA1/2 mutation along with PARP inhibition that leads to a block in the DNA repair pathway and subsequent death/apoptosis of the BRCA mutated/deficient cells (281, 282). The two PARP inhibitors approved by the FDA are olaparib and talazoparib, with talazoparib having a higher potency than olaparib due to a mechanism of action called DNA trapping (283, 284). The PARP inhibitor olaparib has shown promise as a drug of choice in germline BRCA-mutated, ER+/HER-2 negative MBC patients who have been treated with a prior endocrine therapy or been considered inappropriate for endocrine treatment (285). The decision was based on the results from the OlympiAD (ClinicalTrials.gov Identifier: NCT02000622) study conducted on MBC patients with a germline BRCA1/2 mutation where there was an OS benefit among patients who had not received chemotherapy for MBC (285). Based on the EMBRACA study (ClinicalTrials.gov Identifier: NCT01945775) on BRCA mutated subjects with locally advanced and/or MBC, talazoparib provided benefit in the PFS over physician's choice of chemotherapy (286). The use of PARP inhibitors holds promise in BRCA1/2 mutated ER+ MBC patients in combination with other appropriate therapies. Another viewpoint is that activation of CDK4/6 when in complex with other cyclins leads to the phosphorylation of RB1 which blocks binding the binding of RB1 and repressing E2F regulated genes (287). PARP1 protein is reduced by the recruitment of E2F1-RB1-HDAC1-EZH2-SWI/SNF complex in hematopoietic stem cells and monocytes (288). Reduction/loss of PARP1 leads to an impairment in the 8-oxoguanine glycosylase (OGG1)-dependent DNA repair mechanism and an arrest at the G1 phase in PARP1 overexpressing cells, leads to the formation of a dense complex with OGG1 (PARP1-OGG1 complex) (289). In oxidatively stressed cells, CDK4/6 inhibitors also act by repressing PARP and causing an aberrant DNA repair mechanism (289). There is a functional complex between CDK4/6, RB1, PARP1, and OGG1 (289). However, with tumors eventually acquiring resistance to PARP inhibitors, the therapeutic advantage/possibility of combining CDK4/6 inhibitors and PARP inhibitors offers hope(290).
5c. MAPK/ERK
Activation of the RAS/RAF/MEK1/2-ERK pathway leads to phosphorylation of ERα at serine 118, resulting in cell proliferation (291, 292). The RAS/MAPK/ERK pathway increases sensitivity of ERα to low concentration of E2 leading to endocrine resistance (144, 293, 294). Tamoxifen resistant (TAMR) BC cells show increased levels of activated MAPK and ERα (295). McGlynn et al., have shown an association between increased expression of activated Raf-1, pRaf (ser338), MAPK and a greater risk of relapse following treatment with tamoxifen in clinical samples (296). ERα is activated following phosphorylation by MAPK at serine 118 in the AF1 domain (297) and previous studies have demonstrated that inhibiting the MAPK/ERK pathway leads to an upregulation of ERα and subsequent re-sensitization to tamoxifen (298). Another study has shown that tamoxifen resistance is mediated through CDK10 suppression by activation of the MAPK/ERK1/2 pathway, thereby overcoming the dependence upon ERα signaling (299). The active RAS-GTP to converted to inactive RAS-GDP through NF1, a large transcriptional protein, which acts as a tumor suppressor (300).
A negative regulator of the RAS pathway, NF1 is associated with an increased risk of BC (301, 302). Independent Component Analysis (ICA) elucidated the association of NF1 and NFAT to clinical outcomes in BC (303). A lack of NF1 type 1 expression in the aggressive endocrine resistant MDA-MB-231 BC cell line was associated with an increase in phosphorylated MAPK and activated RAS (304). NF1 mutations have been identified in luminal or ER+/HER2-negative, HER2-enriched and triple negative BC (35, 300, 305). NF1 was one among the compendium of genes whose silencing led to tamoxifen resistance in MCF7 cells (306). A recent report revealed mutually exclusive hotspot mutations in NF1, ERBB2 and ERα that were acquired after endocrine therapy and play a role in endocrine resistance (9). Since the RAS/RAF pathway is induced by both ERBB2 (gain-of function) and NF1 (loss-of function), an in-depth analysis of the pathway alternations before and after endocrine therapy, revealed hotspot mutations in KRAS, HRAS, BRAF, and MAP2K1 (MEK1). Oncogenic mutations were present in more than one of the RAS/RAF pathway effectors that did not present with an ERα mutation in the post-hormonal HR+/HER2-negative tumors (254). Additionally, targeting the RAS/RAF pathway with an ERK inhibitor SCH772984 re-sensitized MCF7-EGFR cells to fulvestrant (9). Targeted sequencing was performed to study the prognostic effects of somatic mutations and it was reported that NF1 frame-shift nonsense (FS/NS) mutations has adverse effects on prognosis (19). A recent study identified an enrichment in NF1 alterations in metastatic Invasive Lobular Carcinoma (mILC) and a role for NF1 in endocrine resistance (307). The RAS/RAF pathway along with NF1 are therefore involved in crosstalk between ERα and RTK's, and are associated with tamoxifen resistance (297).
5d. c-SRC/Kinase
c-SRC, a non-receptor tyrosine kinase is associated with the progression of many human cancers, including BCs (308). Src-1 (NCOA1) serves as a master regulator, a transcriptional hub that complexes with AP1, NF-κB, p300/CBP and other co-activators to activate the ER and PR (309, 310). Basically, SRC-1 interacts with AIB1, ETS2 and HOX11 to define its association in BC progression and endocrine resistance (311, 312).
Studies have shown an activation and upregulation of SRC associated with acquiring tamoxifen resistance in ER+ cell lines (313). SRC is involved in various signal transduction pathways including ER and HER2/EGFR (314). Elevated levels of c-SRC may be due to overexpression of growth factors (315, 316). c-SRC is involved in various oncogenic signaling pathways including growth, invasion and metastases (317, 318). The interaction between ER and SRC is well-established and they form complexes with an array of proteins including PI3K, FAK, PELP, and MNAR leading to the activation of pAKT and pMAPK demonstrating the role that c-SRC has in endocrine resistance (318–323). A recent study demonstrated the effect of TGF-β, where it promoted epithelial mesenchymal transition (EMT) and expression of the EGFR and IGF-IR. These RTKs (EGFR and IGF-IR) formed complexes with ERα and SRC, thereby supporting endocrine resistance in BC (324). Furthermore, c-SRC phosphorylates the ER and has been shown to regulate ER localization (325, 326). Upon treatment with tamoxifen, c-SRC activity is increased and this subsequently amplifies the extent of invasion and motility in BC cells (313). Moreover, since c-SRC is essential in modulating tamoxifen resistance, and blocking its activity reverses tamoxifen resistance (327). c-SRC inhibitor could be exploited as a combinatorial therapeutic drug in the treatment of human BC.
5e. STATs
The STAT-family members (STAT1-4, STAT5a, STAT5b, and STAT6) represent a family of transcription factors involved in cell proliferation, differentiation, survival and apoptosis. They have all been shown to be expressed in BC cell lines, but only STATs 1, 3, 5a, and 5b are expressed in BC tissues (328, 329). It is well-understood that the STAT proteins mediate cell proliferation and survival by regulating and influencing the activities of several other transcription factors and associated pathways. It has been shown that the STAT3 and STAT5 signaling pathways are integrally involved in endocrine resistance and more so in the growth factor-stimulated cases. Also, there is functional redundancy between the two STAT proteins, where one protein overcomes the loss/gain in function due to the others gain/loss in function (330, 331). The activation of STAT3 and STAT5 pathways are downstream of the EGFR and c-SRC pathways, which present as being overexpressed in tamoxifen resistant tumors. SRC-1 also interacts with STAT1, STAT3, STAT5 and STAT6, and although STAT3 and STAT5 are considered as tumorigenic transcription factors, STAT1 is a tumor suppressor (309, 332–334). It has also been demonstrated that ER+ BC cells treated with tamoxifen, results in decreased phosphorylation of STAT3 at serine 727, suggesting an association between tamoxifen sensitivity and decreased STAT3 transcriptional activity (335). Yamashita et al. in 2006 had shown that STAT5 is a strong prognostic molecular marker in ER+ BC. In their study, they investigated the expression of STAT3 and STAT5 in more than 500 BC tissues by immunohistochemical techniques and observed that in ER+ patients with STAT5 positive tumors there was significantly increased overall survival, thereby suggesting that expression of STAT5 is helpful in selecting patients who could possibly benefit from endocrine therapy (336). Recently, a role for increased STAT1 signaling in endocrine resistance was reported in studies that identified STAT1 as a potential mediator of endocrine resistance/sensitivity in BC and an appropriate STAT1 inhibitor could serve as a therapy in endocrine resistant BC (331, 337).
5f. NF-kB
The NFκB family of transcription factors is comprised of five members (RelA/p65, RelB, cRel, NFκB1/p50, and NFκB2/p52), all of which play an important role in cellular homeostasis (338). Accumulating evidence in support of NFκB activation in cancerous stem cells has led researchers to focus on various genetic signatures (339). It not only creates a microenvironment suitable for stem cell survival, but also its invasiveness and metastatic capability. A line of evidence supports the notion that NFκB and CXCR4 helps to maintain the stemness and promotes migration of cancerous cells (340). Increased expression of p50/NFκB1, p52/NFκB2, and c-Rel was detected in breast tumors compared to adjacent normal tissue (341). Activated NFκB activity has been detected in hormone resistant BC cells and RelA/p65 expression was upregulated (342). A separate study demonstrated that upon inhibition of NFκB and thus also the RelA/p65 by parthenolide (an NFκB inhibitor), sensitivity to tamoxifen was restored in resistant MCF7 cell lines, along with decreasing BCL2 expression, which can be reversed by a caspase 8 (CASP8) specific inhibitor (343). Inflammatory molecules leading to endocrine resistance occurs through kinases that regulate ERα directly or through ERα and RelA/p65/NFκB complexes at ERE enhancer sites to either upregulate or downregulate respective genes (344).
A previous study showed that the expression and DNA binding of NFκB1/p50 and NFκB2/p52 were enhanced in LCC1 estrogen-independent, TAM-sensitive BC cells compared to MCF-7 estrogen-dependent cells, which further demonstrated a role for NFκB in the pathway to endocrine resistance (345). Gionet et al. reported that NFκB1/p50 binds to the ERα on EREs and inhibiting NFκB increased the expression of E2 responsive genes (346).
A separate study suggested that NFκB induces breast cancer progression by stimulating IL-6 and IL-8 (347, 348). Another study reported that FOXA1 led to suppressed IL6 expression by disrupting the binding of NFκB to the IL-6 promoter and thus concluded that reduced FOXA1 expression leads to cancer stem cell like properties in tamoxifen resistant cells through the preferential binding of NFκB to the IL-6 promoter and upregulating IL-6 expression (349).
5g. LMTK3
Lemur tyrosine kinase 3 (LMTK3) is a serine-threonine-tyrosine protein kinase involved in various cancers. Aberrant expression of the gene and polymorphisms within the gene serve as suitable biomarkers in cancer progression (350, 351). LMTK3 is expressed in both ER+ and ER-negative BCs and a kinome screen identified it as an ERα regulator (352). In an elaborate experimental setup, a role for LMTK3 in endocrine resistance was confirmed (353). Future studies could focus on its use as a valuable biomarker and therapeutic target.
6. Hypoxia Inducible Factor
The intra-tumoral pressure of oxygen (O2) serves as an important indicator of the possibility of tumor metastasis (354). In BC, hypoxia is induced due to reduced oxygen levels becoming available at the site of the tumor due to their distance from functionally viable blood vessels, and it in this simmering tumor conducive and expansive microenvironment that activation of its main effector, hypoxia-inducible factors (HIF) takes place. The HIF family of transcription factors are comprised of at least three factors: HIF1, HIF2, and HIF3, each of which exists as a heterodimer, with an O2 inducible α and a constitutively expressed β subunit (355). Hypoxia associated HIF1α is quite commonly coupled with errors in DNA replication including homologous replication and high mutation rates along with changes in gene expression which is mainly due to epigenetic changes regulated by histone demethylases.
Mounting evidence suggests that a large proportion of the HIF1α targets are also targets of ERα and that the HIF1α gene is endowed with the presence of an ERE within its genomic architecture and is regulated by ERα (356). The transcription factor HIF1α is regulated by ERα and that increased HIF1α expression confers endocrine resistance to ERα+ cancer cells. Clinical studies have thus shown that HIF1α is associated with endocrine resistant BC (356). One of the common target genes for both the ERα and HIF1α is KDM4B/JMJD2B, which is a H3K9me3/me2, H3K36me2/me3, H1.4K26m2/me3 histone demethylase and its genomic landscape contains binding sites for both HIF1α and ERα (356). It has also been demonstrated that both KDM4A and KDM4B form a complex with ERα and lead to the regulation of ERα target genes and an upregulation of both the genes has been observed in both ERα+ and ERα- cells (357). Another histone demethylase of the KDM4 family: KDM4C, promotes BC cells proliferation and metastasis by serving as a transcriptional activator of HIF1α (358). HIF1α promotes angiogenesis within tumors via regulation of its target gene VEGF, thereby leading to BC cell proliferation and metastasis. Hypoxia is the principal regulator of VEGF expression, as it is a direct transcriptional target of both HIF1α and HIF2α (359).
Accumulating evidence from preclinical and clinical studies have demonstrated a role for HIF1α in endocrine resistance. Endocrine resistance was observed in MCF7 cells transfected and stably expressing HIF1α, and targeting it with zoledronic acid led to endocrine sensitivity (360). Signaling via the GPER-HER2-ERK-cFOS pathway triggers HIF1α dependent VEGF activation and BC progression (73). An enhancement in the resistance of BC cells to tamoxifen and fulvestrant was observed in ERα+ cancer cells transfected with HIF1α (356). A recent study reported that HIF1α and p44/42MAPK play an important role in endocrine resistance and may serve as appropriate therapeutic targets for endocrine resistant patients treated with an AI (361, 362).
7. Stem Cell Population and Transcription Factors
Cancerous/tumorous stem cells give rise to tumors/cancers and define their ability to proliferate and metastasize. They encompass the ability to regenerate or self-renew and initiate cancer progression, proliferation, migration and metastasis. The stemness of these cells are correlated with poor prognosis and endocrine resistance. A few of the biomarkers that define these subpopulations are CD44, CD24, CD133, and ALDH1 along with others (363).
A recent study identified FOXA1 as a prominent factor for ERα activity in breast cancer (364–366). Induction of FOXA1 triggers a cascade of events leading to endocrine resistance, one of the most prominent being the induction of IL-8 by the FOXA1 expressing ER+ tumors (364). Another study identified the FOXM1 motif bound strongly to the ERα DNA in endocrine resistant cells by expansion of BC stem cells and could be a promising target aimed toward BC therapy (367). RUNX1 has been identified as a BC stemness repressor by repressing ZEB1 expression (368). RUNX2 is another stemness gene and a separate study identified that RUNX2 and ERα interact with each other in an ER+ endocrine resistant BC cell line to mediate EMT and metastasis, and supports a role for RUNX1 in endocrine resistance (367). Interestingly the same study also highlighted the upregulation of SOX9 in tamoxifen resistant clinical samples. RUNX1 is a tumor suppressor gene that has recently been identified to have point mutations in ER+ BC (369). Xue et al. identified FXYD3 as a target of the E2-ER pathway and it is upregulated in the ER+ BC stem cells (370). They also went on to validate its importance in ER+ BC cells. Crucially, the study identified a direct regulation of the FXYD3 by SOX9.
There are possibly many transcription factors and associated pathways that may transform a normal stem cell to a cancerous stem cell, although two of the well-defined ones are: NOTCH and Hedgehog (Hh). The NF-kB pathway has been described elsewhere.
7a. NOTCH
The 4 NOTCH transmembrane receptors: NOTCH 1–4, interact with multiple ligands (Delta-like 1, Delta-like 3, Delta-like 4, Jagged 1, and Jagged 2), resulting in cleavage of the intracellular domain by γ-secretase. The intracellular domain then translocates to the nucleus where it binds with co-activators to regulate transcription of target genes and thereby plays an important role in endocrine resistance (371). NOTCH signaling has been shown to play a critical role in normal human mammary development and also regulation of cancer stem cells in both invasive carcinoma of the breast as well as ductal carcinoma in situ (372, 373). Augmenting the activity of NOTCH4 in BC cells led to an increase in the expression of BC stem cell biomarkers and the higher expression of NOTCH in these cells, were evident in both the basal and luminal cell types (374). Another study went on to delineate the role of NOTCH1 in inhibiting tumor progression in BC, whereby inhibition of NOTCH1 resulted in tumor regression and prevention of recurrence in ~67% of the tumors studied (375). It has recently been demonstrated that NOTCH pathway is hyperactivated in resistant BC cells and can be abrogated by blocking the NOTCH pathway (376). Previously, it was shown that E2 inhibits NOTCH activity by affecting the NOTCH receptor cellular localization and that tamoxifen and raloxifene blocks this effect, thereby activating the NOTCH pathway. Inhibiting the NOTCH signaling pathway with γ-secretase inhibitors was more effective when used with tamoxifen (377). Recently, it has been reported that Nicastrin, an essential subunit of gamma secretase, and NOTCH4 are key molecules involved in resistance to endocrine therapy and that the gamma secretase inhibitor (GSI) PF03084014 and anti-Nicastrin MAbs can possibly reverse and potentially re-sensitize endocrine resistant BCs (378).
In BC, SOX2 was identified as a factor responsible for the reprogramming of BC cells into BC stem cells and its enhanced expression was associated with the HER2+ BC subtype (379). SOX2 is activated by the NOTCH signaling pathway and Oct4 localizes β-catenin within the nucleus and thus a role for the NOTCH (described later) and WNT pathways in defining the stemness of BC cells (379, 380). The two pathways interact among themselves, regulating each other's functions and HES1, a NOTCH target gene, is regulated by the WNT signaling pathway (381). WNT signaling has been reported to play an important role in BC stem cells (382).
With STAT3 and STAT5 being constitutively activated in cancers, they have also been shown to defy each other's physiological actions with differential effects (383, 384). That NOTCH activated STAT3 pathway plays an important role in endocrine resistance, was verified in an endocrine resistant model of MCF7 cells (TAMR-MCF7 cells), whereby constitutive activation of STAT3 was observed and inhibition of the NOTCH signaling pathway by a NOTCH inhibitor, DAPT led to a concomitant reduction in the level of activated STAT3 observed in this model of endocrine resistant MCF7 cells (385).
7b. Hedgehog (Hh) Signaling
The Hedgehog (Hh) signaling pathway has been studied extensively in the past as an essential pathway dictating the initiation, progression and metastasis of cancers, although more recently the focus on this pathway is toward it being at essential crossroads as part of the BC stem cell circuit of pathways (386). This pathway is mainly activated in tamoxifen-resistant tumors and it is activated by the PI3K/AKT pathway (387). A recent study suggested the combined therapy targeting both the PI3K/AKT pathway along with the Hh pathway to tackle endocrine resistance in BC (388). Although the stem cell biomarker CD133 has not been clearly defined, it is a moiety that binds cholesterol and has a prominent role in Hedgehog (Hh) signaling (363). The Hedgehog signaling pathway is regulated by the E2 related receptor β and affect the downstream targets (389).
8. Other Factors and Proteins
There are several other transcription factors and proteins that determine cancer progression in the face of ongoing endocrine therapies, some known and many unknown. The EMT factors, Twist and Snail, have been implicated in endocrine resistance, with Twist downregulating ERα by epigenetically regulating the promoter of ERα (390–393). There are several other proteins and molecules that play a major role in endocrine resistance, including the NOTCH and WNT pathways described earlier. All these proteins and transcription factors function as individual molecules working together for/against the homeostasis of the human cells, to determine the fate of cancer cells.
Metabolic and oxidative stress are a few of the other factors affecting endocrine resistance. The mechanisms are elucidated in detail below for each of these factors. Recent publications have demonstrated that homeostasis in cell metabolism and cell proliferation is mediated by a few other transcription factors and pathways as well. Another mechanism for endocrine resistance that has been suggested relates to the ability of the metabolic enzymes to deliver the active compound. Some studies have reported lower concentrations of BC drugs at the site of the tumor when compared to levels in the plasma and an association with poor outcomes (394). Alternatively, a recent study demonstrated that an elevated concentration of tamoxifen metabolites at the site of the cancer, led to agonist effects (395).
8a. Oxidative Stress
Oxidative stress is caused due to an imperfect polarization of reactive oxygen species (ROS) vs. antioxidants. Although this holds true under all disease states that progress toward chronic disorders, it stands out as a cause of major concern in cancers, where there is excess cellular mass burden. Thus, it becomes imperative to also understand the role of toxicity to varied drug dosage/regimens and the associated oxidative stress, in other words: the oxidative stress created due to a sudden imbalance in the microenvironment of the cancer. The polyunsaturated fatty acids present in every cell are metabolized to malondialdehyde (MDA) by the ROS whereas nucleic acids (DNA) targeted by ROS are metabolized to 8-oxo-7, 8-dihydro-2′-deoxyguanosine (8-oxodG) (396, 397). A recent study, demonstrated the possibility of using concentrations of urinary 8-oxodG as a marker to define early stage breast cancer patients (398). Again, a study of serum levels of 8-oxodG, indicated an association with ductal carcinoma (399). Glutathione is another antioxidant that exists as reduced glutathione (GSH) and oxidized glutathione (GSSG), the ratio of which (GSH: GSSG) has been inferred as a marker of oxidative stress (400, 401).
A study revealed that E2 led to an increase in ROS production and an upregulation in the expression of genes involved in oxidative stress, e.g., hemeoxygenase 1 (HMOX1). An association with c-SRC phosphorylation was observed in this experimental setting and it was attributed to the oxidative stress induced by E2 (402).
In luminal B breast cancer, the loss of sirtuin proteins, particularly SIRT3, promotes tumorous phenotypes dictated by atypically regulated protein acetylation and thus cells are exposed to oxidative stress. One of the analysis from the loss of SIRT3 showed its association with the development of endocrine resistance in the luminal B BC (403). Tamoxifen builds up in tumors as part of the daily dosage regimen, and leads to an increase in tamoxifen induced oxidative stress. This effect leads to an activation of the protein, Nuclear factor-erythroid 2-related factor-2 (Nrf2) which subsequently activates the anti-oxidant response element (ARE). Thus, Nrf2 and ARE levels could serve as oxidative stress biomarkers in tamoxifen treated BC patients (404). These markers could also be studied for their role in endocrine resistant BC and also provide a rationale to study the role of oxidative stress.
8b. Drug Metabolism
The therapeutic drugs used in ER+ BC are administered at a certain dosage between fixed intervals to achieve optimal dosage and efficacy which is detected as plasma half-life. The plasma half-life is dictated by the metabolic rate and subsequent elimination of the drug from the plasma. Among the popular drugs studied in ER+ BC, tamoxifen is among one of the most widely used choice of available treatments and is metabolized to its active forms 4-hydroxytamoxifen (4-OH tamoxifen) and N-hydoxy desmethyl tamoxifen (N-OH desmethyl tamoxifen), which is further metabolized to endoxifen in the liver through the polymorphic cytochrome P450 enzymes (CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A4, CYP3A5), the most effective metabolic enzyme being CYP2D6 (405, 406). Interestingly, the potency of its active metabolite endoxifen is far more effective than tamoxifen in its native form. The active metabolites are degraded for excretion by the UDP-glucuronosyltransferases (UGTs) and sulfotransferase (SULTs) enzymes (407). The genotypic information for the CYPs, UGTs, and SULTs has been surmised as one of the molecular mechanisms that define endocrine resistance.
Differences in the expression and associated mutations in each enzyme (CYPs, UGTs, or SULT's) of the tamoxifen metabolic pathway, would help define the effectiveness of the drug in the individual, leading to the well-known concept of personalized medicine. Studies have demonstrated a role of the genotype (expression profiles and/or polymorphisms in the tamoxifen metabolic genes) along with co-intake of other drugs that could lead to altered effectiveness and/or plasma concentrations and associated half-life. One of the crucial CYP genes, CYP2D6, has several polymorphisms within its gene, and each have been associated with either an increase or decrease in enzyme activity. Each variant defines the metabolic rate of tamoxifen, denoted as either extensive (EM), intermediate (IM), or poor metabolizers (PM) (408, 409). Studies have reported altered plasma concentrations which correlate to polymorphisms in CYP2D6 (406). One of the first studies that investigated the polymorphisms in CYP2D6 and SULT1A1 in 226 BC patients on tamoxifen, presented with differences in recurrence of the disease based on the homozygosity/heterozygosity of the CYP2D6*4 and SULT1A1*1 alleles (410).
In another study conducted by a Korean group (Asian cohort), a polymorphism at CYP2D6*10/*10 correlated with lower plasma concentrations of the tamoxifen metabolites (411). The variation between activities of the CYP2D6 gene due to allelic variants across various ethnicities becomes imperative when translating preclinical data into clinical use for dosage and regimen (409, 412). Along with the genotype and associated polymorphisms, the enzymes metabolize other drugs too, and thus the effectiveness of tamoxifen is dependent on the intake of tamoxifen and any other drug taken simultaneously, for example paroxetine (406). In a separate study, it was noted that upregulation of the CYP3A4 enzyme played a crucial role in tamoxifen resistant BC cells and this was attributed partially to the 11,12-epoxyeicosatrienoic (11-12-EET) acid pathway and thus drugs targeting the CYP3A4/11,12-EET pathway may serve as suitable therapeutic drugs (413). A phase I trial conducted using Z-endoxifen as a treatment of choice rather than tamoxifen was performed to bypass the metabolism of the drug and thus differential effects due to the CYP and other enzymes, showed promising anti-tumor effects unaffected by toxicity (414).
9. Immune System
The complicated crosstalk between the cancer cells and immune cells at the site of the tumor has led researchers to harness the unique capability of the immune system to destroy and at times shrink the size of the tumor. In BC as in other cancers, increased levels of immune cells and soluble mediators like cytokines and some chemokines, predict poor prognosis. It has been reported that tumor associated macrophages (TAM) are associated with low survival rates and that the TAMs exhibit features that promote angiogenesis, migration, EMT and metastasis by suppressing anti-cancer immunity (415, 416). Interestingly, the levels of interleulin-1β (IL-1β) and tumor-necrosis factor α (TNFα) are higher in metastatic ER+ BC patients, where these cytokines activate the NF-κB and lead to endocrine resistance (417, 418). Antibodies and molecules directed against transcription factors upstream of NF-κB in the NF-κB pathway has been observed to restore sensitivity in cell line models of endocrine resistant BC (419, 420). IL-1β dependent activation of the ER, due to phosphorylation at the serine 305 locus of the protein by IKKβ leads to growth and proliferation of BC cells in the absence of E2, thus providing evidence for a prominent role for cytokines in BC (421).
Although in BC it is the negligible immunogenicity of the tumors that limits the effectiveness of immunotherapies like checkpoint inhibitors, yet the immune system plays an essential role in the development and branching of ductal and luminal epithelial differentiation (422, 423). According to some researchers, the immune system appears to play a very prominent role in the triple negative BC setting (ER-negative, PR-negative, and HER2-negative), and has a limited role in the endocrine sensitive setting (ER+ BC) (424, 425).
Thus, although there is a role of both the innate and adaptive immune cells in BC and immune mechanisms have been more intensely studied in ER negative patients, recent studies have focused on studies suggesting a role for the adaptive arm of the immune system in ER+ patients, namely the TILs (22). In post-menopausal women with ER + BC receiving neoadjuvant AI, anastrozole, an inflammatory gene expression signature was observed in baseline samples (426). The cluster of genes expressed were suggestive of infiltrating immune cells and these are associated with poor response to endocrine therapy. The most recent study focused on identifying a prognostic value for immune scores and associating it to the relapse in ER + BC patients receiving neoadjuvant AI, anastrozole or tamoxifen (23). The study reveals the importance of spatial heterogeneity of TILs and a crucial role for immune memory of tumor immune cells in ER + patients.
In the ER+, PR+, and HER2+ setting, it is the therapy based on monoclonal antibodies against HER2 that activate the antibody-dependent cytotoxic killing ability of natural killer (NK) cells through the adaptive arm of the immune system (427). hTERT is also capable of eliciting an immune response and it has been reported that there was an abundance of hTERT peptide 1540 specific CD8+ T cells in some cancers and in the metastatic BC setting, patients immunized with the peptide presented with increased TILs and necrosis of some tumorigenic areas (428).
Checkpoint inhibitors, anti-CTLA4 and anti-PD1 have been reported to elicit anti-cancer responses, although studies in the ER+ luminal BC subtype, demonstrate that due to the low titer of TILs in these patients, it is not the therapy of choice (422). In a study aimed at tumor rejection in HER+ metastatic BC patients, trastuzumab was given in combination with anti-PD1 (PANACEA study) and it was reported that the objective response rate in the PDL1+ BC patients was 15.2% and 0% in the PDL1-negative arm of the study (422). The myeloid derived suppressor cells (MDSCs) are another set of myeloid derived cells that have revealed their importance in promoting resistance to therapies (429). Therapies involving the immune system hold promise for future efforts to overcome BC endocrine resistance.
10. miRNA and Extracellular Vesicles
MicroRNAs (miRNA's) are small (~22-nucleotide) regulatory non-coding RNA molecules and miRNA deregulation was first reported in 2005 in BC (430). Investigators have reported that miRNAs can regulate the network/cascade of signal transduction pathways associated with endocrine resistance by several mechanisms: upregulating drug efflux transporters and anti-apoptotic proteins, promoting EMT and forming cancer stem cells (431).
Aberrant expression of specific miRNAs has been implicated in the development of tamoxifen resistance. Gene expression profiling has identified differential expression of miRNA expression profiles between tamoxifen resistant and sensitive BC cell lines (431–433). A miRNA library recently identified a plethora of miRNAs involved in TAM sensitivity in MCF7 cells (434). A previous study demonstrated that overexpression of miR-101, led to TAM resistance in MCF7 cells grown in an E2 free medium, mediated by the activation of AKT (435). The miR-519a regulates tumor suppressor genes within the PI3K and cell cycle proteins conferred endocrine resistance (436). The miR-451a, promotes endocrine resistance through a reduction in autophagosomes (437). Basically, The PI3K/AKT/mTOR pathway plays a crucial role in metastasis and endocrine resistance and overexpression of miR-451a enhances sensitivity to tamoxifen by repressing 14-3-3ζ expression along with reduced p-AKT and p-mTOR and an increase in the ERα expression. miR221/222 targets p27/Kip1, which is a cell cycle inhibitor in TAMR BC cells and promotes proliferation of cells even in the absence of E2 (432). Interestingly, expressing miR-320a in TAMR BC cells, leads to re-sensitization through downregulation of the c-MYC and CYCLIN D1 (438). Again, an increase of miR-873 in TAMR BC cells led to a restoration in the tamoxifen sensitivity through CD3 (439). Another study, highlighted the importance of the miRNA-375, in TAMR BC cells, where re-expression of miR-375, led to a reversal in tamoxifen resistance and associated EMT like behavior in these cells (440). BC stem cells also activate the HIF1α pathway during hypoxic conditions (441).
Extracellular vesicles (EVs) derived from cancer cells are similar to lipid vesicles, and contain oncogenic material in the form of overexpressed oncogenic genes/proteins/mediators/nucleic acids/non-coding RNA's and metabolic enzymes. They typically home into specific recipient cells where they can trigger a separate cascade of molecules and associated pathological response (442, 443). An analysis of the contents within the EV could shed light on their role in endocrine resistance and metastasis. EVs have been implicated in transmitting tamoxifen resistance from the TAM resistant cells to the TAM sensitive cells via the EVs containing miR-221/222 (434). Cancer biomarkers identified include HER2 and HLA-G, which is associated with circulating tumor cells and promotes proliferation, therapy resistance and metastasis (444). The translational capacity of miRNAs and EVs hold promise toward identifying therapies to combat endocrine resistance.
Conclusion
The molecular mechanisms underlying resistance to endocrine therapy have been much studied and these studies have provided novel insights and yielded potentially new therapeutic strategies that may overcome endocrine resistance in BC. While an improvement in the quality of life and survival of women with BC has increased with the discovery of novel ER targeted therapies, endocrine resistance still remains at large. The ER signaling pathway represents a complex cascade of events with several regulators and comprehensive crosstalk with and between pathways, thereby resulting in the emergence of endocrine resistance. The authors view stem cells and the immune system as a focus toward all future studies on overcoming endocrine resistance in BC and other solid cancers. The soluble microenvironment around the BC, which consists of hormones, growth factors, cytokines, ROS, and various other types of metabolic compounds determine the effectiveness of the immune system in eliminating or shrinking the size of tumors. The upcoming efforts should now be to study potential biomarkers for definitive use in the clinic in greater detail. It is well-understood that results that appear promising in cell lines will not entirely translate into clinically reproducible results, thereby rendering clinical validation as a necessary step in evaluating novel therapeutic strategies. Advances in integrative approaches examining the transcriptomic and proteomic profiles in combination with robust bioinformatics support studying the differences between normal and resistant tumor tissue from a large dataset of patients will help tease out the differences/similarities between the two datasets and furthering our understanding. A focused study of the immune cell infiltrates and tumor spatial architecture around the site of the cancer, polymorphisms within metabolic enzymes and oxidative stress milieu are also aspects that will help clinicians in treatment decisions. This approach will help identify novel mediators, molecules and pathways of interest thereby resulting in tailor-made treatment for different sets of patients with similar clinical symptoms and molecular profiles.
Author Contributions
All authors contributed to the text and design of the review and approved the final manuscript.
Conflict of Interest Statement
JS sat on SABs for Celltrion, Singapore Biotech, Vor Biopharma, TLC Biopharmaceuticals and Benevolent AI, has consulted with Lansdowne partners, Vitruvian and Social Impact Capital and he Chairs the Board of Directors for BB Biotech Healthcare Trust and Xerion Healthcare.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Lesley-Ann Martin who had helped with discussions and Aidan M. Murphy who proof read and edited the review for better readability.
References
1. Beatson GT. On the treatment of inoperable cases of carcinoma of the mamma: suggestions for a new method of treatment, with illustrative cases. Trans Med Chir Soc Edinb. (1896) 15:153–79.
2. Key T, Appleby P, Barnes I, Reeves G, Endogenous G, Endogenous H, Breast Cancer Collaborative G. Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J Natl Cancer Inst. (2002) 94:606–16. doi: 10.1093/jnci/94.8.606
3. Engelsman E, Korsten CB, Persijn JP, Cleton FJ. Human breast cancer and estrogen receptor. Arch Chir Neerland. (1973) 25:393–7.
4. Pedram A, Razandi M, Blumberg B, Levin ER. Membrane and nuclear estrogen receptor alpha collaborate to suppress adipogenesis but not triglyceride content. FASEB J. (2016) 30:230–40. doi: 10.1096/fj.15-274878
5. Flesher JW, Gupta GN, Jacobson HI, Jensen EV. Fate of steroid estrogens in target tissues. Fed Proc. (1960) 19:170.
6. Weatherman RV, Fletterick RJ, Scanlan TS. Nuclear-receptor ligands and ligand-binding domains. Annu Rev Biochem. (1999) 68:559–81. doi: 10.1146/annurev.biochem.68.1.559
7. Walter P, Green S, Greene G, Krust A, Bornert JM, Jeltsch JM, et al. Cloning of the human estrogen receptor cDNA. Proc Natl Acad Sci USA. (1985) 82:7889–93. doi: 10.1073/pnas.82.23.7889
8. Anderson WF, Chatterjee N, Ershler WB, Brawley OW. Estrogen receptor breast cancer phenotypes in the surveillance, epidemiology, and end results database. Breast Cancer Res Treat. (2002) 76:27–36. doi: 10.1007/978-1-4615-1173-1
9. Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell. (2018) 34:427–38.e6. doi: 10.1016/j.ccell.2018.08.008
10. Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. (2003) 2:205–13. doi: 10.1038/nrd1031
11. Baum M, Budzar AU, Cuzick J, Forbes J, Houghton JH, Klijn JG, et al. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. Lancet. (2002) 359:2131–9. doi: 10.1016/S0140-6736(02)09088-8
12. Strasser-Weippl K, Goss PE. Advances in adjuvant hormonal therapy for postmenopausal women. J Clin Oncol. (2005) 23:1751–9. doi: 10.1200/JCO.2005.11.038
13. Arimidex TAoiCTG, Forbes JF, Cuzick J, Buzdar A, Howell A, Tobias JS, et al. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 100-month analysis of the ATAC trial. Lancet Oncol. (2008) 9:45–53. doi: 10.1016/S1470-2045(07)70385-6
14. Ma CX, Reinert T, Chmielewska I, Ellis MJ. Mechanisms of aromatase inhibitor resistance. Nat Rev Cancer. (2015) 15:261–75. doi: 10.1038/nrc3920
15. Dowsett M, Smith IE, Ebbs SR, Dixon JM, Skene A, A'Hern R, et al. Prognostic value of Ki67 expression after short-term presurgical endocrine therapy for primary breast cancer. J Natl Cancer Inst. (2007) 99:167–70. doi: 10.1093/jnci/djk020
16. Thürlimann B, Keshaviah A, Coates AS, Mouridsen H, Mauriac L, Forbes JF, et al. A comparison of letrozole and tamoxifen in postmenopausal women with early breast cancer. N Engl J Med. (2005) 353:2747–57. doi: 10.1056/NEJMoa052258
17. Giltnane JM, Hutchinson KE, Stricker TP, Formisano L, Young CD, Estrada MV, et al. Genomic profiling of ER. Sci Transl Med. (2017) 9:eaai7993. doi: 10.1126/scitranslmed.aai7993
18. Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. (2012) 486:405–9. doi: 10.1038/nature11154
19. Griffith OL, Spies NC, Anurag M, Griffith M, Luo J, Tu D, et al. The prognostic effects of somatic mutations in ER-positive breast cancer. Nat Commun. (2018) 9:3476. doi: 10.1038/s41467-018-05914-x
20. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. (2012) 486:346–52. doi: 10.1038/nature10983
21. Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell. (2017) 32:169–84.e7. doi: 10.1016/j.ccell.2017.07.005
22. Savas P, Salgado R, Denkert C, Sotiriou C, Darcy PK, Smyth MJ, et al. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat Rev Clin Oncol. (2016) 13:228–41. doi: 10.1038/nrclinonc.2015.215
23. Heindl A, Sestak I, Naidoo K, Cuzick J, Dowsett M, Yuan Y. Relevance of spatial heterogeneity of immune infiltration for predicting risk of recurrence after endocrine therapy of ER+ breast cancer. J Natl Cancer Inst. (2018) 110:166–75. doi: 10.1093/jnci/djx137
24. Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. (1996) 93:5925–30. doi: 10.1073/pnas.93.12.5925
25. Roger P, Sahla ME, Makela S, Gustafsson JA, Baldet P, Rochefort H. Decreased expression of estrogen receptor beta protein in proliferative preinvasive mammary tumors. Cancer Res. (2001) 61:2537–41.
26. Mann S, Laucirica R, Carlson N, Younes PS, Ali N, Younes A, et al. Estrogen receptor beta expression in invasive breast cancer. Hum Pathol. (2001) 32:113–8. doi: 10.1053/hupa.2001.21506
27. Evans RM. The steroid and thyroid hormone receptor superfamily. Science. (1988) 240:889–95. doi: 10.1126/science.3283939
28. Kumar V, Green S, Stack G, Berry M, Jin JR, Chambon P. Functional domains of the human estrogen receptor. Cell. (1987) 51:941–51. doi: 10.1016/0092-8674(87)90581-2
29. Kumar R, Zakharov MN, Khan SH, Miki R, Jang H, Toraldo G, et al. The dynamic structure of the estrogen receptor. J Amino Acids. (2011) 2011:812540. doi: 10.4061/2011/812540
30. Tora L, White J, Brou C, Tasset D, Webster N, Scheer E, et al. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell. (1989) 59:477–87. doi: 10.1016/0092-8674(89)90031-7
31. Norris JD, Fan D, Kerner SA, McDonnell DP. Identification of a third autonomous activation domain within the human estrogen receptor. Mol Endocrinol. (1997) 11:747–54. doi: 10.1210/mend.11.6.0008
32. Picard D, Kumar V, Chambon P, Yamamoto KR. Signal transduction by steroid hormones: nuclear localization is differentially regulated in estrogen and glucocorticoid receptors. Cell Regulat. (1990) 1:291–9. doi: 10.1091/mbc.1.3.291
33. Berry M, Metzger D, Chambon P. Role of the two activating domains of the oestrogen receptor in the cell-type and promoter-context dependent agonistic activity of the anti-oestrogen 4-hydroxytamoxifen. EMBO J. (1990) 9:2811–8. doi: 10.1002/j.1460-2075.1990.tb07469.x
34. Osborne CK, Schiff R, Fuqua SA, Shou J. Estrogen receptor: current understanding of its activation and modulation. Clin Cancer Res. (2001) 7(12 Suppl.):4338s−42s; discussion 411s−412s.
35. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. (2012) 490:61–70. doi: 10.1038/nature11412
36. Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. (2006) 38:1289–97. doi: 10.1038/ng1901
37. Lin CY, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M, et al. Whole-genome cartography of estrogen receptor alpha binding sites. PLoS Genet. (2007) 3:e87. doi: 10.1371/journal.pgen.0030087
38. Wang Q, Carroll JS, Brown M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol Cell. (2005) 19:631–42. doi: 10.1016/j.molcel.2005.07.018
39. Klein-Hitpass L, Ryffel GU, Heitlinger E, Cato AC. A 13 bp palindrome is a functional estrogen responsive element and interacts specifically with estrogen receptor. Nucleic Acids Res. (1988) 16:647–63. doi: 10.1093/nar/16.2.647
40. Ascenzi P, Bocedi A, Marino M. Structure-function relationship of estrogen receptor alpha and beta: impact on human health. Mol Aspects Med. (2006) 27:299–402. doi: 10.1016/j.mam.2006.07.001
41. Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, et al. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. (1997) 277:1508–10. doi: 10.1126/science.277.5331.1508
42. Gaub MP, Bellard M, Scheuer I, Chambon P, Sassone-Corsi P. Activation of the ovalbumin gene by the estrogen receptor involves the fos-jun complex. Cell. (1990) 63:1267–76. doi: 10.1016/0092-8674(90)90422-B
43. Murphy LC, Seekallu SV, Watson PH. Clinical significance of estrogen receptor phosphorylation. Endocr Relat Cancer. (2011) 18:R1–14. doi: 10.1677/ERC-10-0070
44. Le Romancer M, Poulard C, Cohen P, Sentis S, Renoir JM, Corbo L. Cracking the estrogen receptor's posttranslational code in breast tumors. Endocr Rev. (2011) 32:597–622. doi: 10.1210/er.2010-0016
45. Williams CC, Basu A, El-Gharbawy A, Carrier LM, Smith CL, Rowan BG. Identification of four novel phosphorylation sites in estrogen receptor alpha: impact on receptor-dependent gene expression and phosphorylation by protein kinase CK2. BMC Biochem. (2009) 10:36. doi: 10.1186/1471-2091-10-36
46. Pesiri V, Di Muzio E, Polticelli F, Acconcia F. Selective binding of estrogen receptor α to ubiquitin chains. IUBMB Life. (2016) 68:569–77. doi: 10.1002/iub.1514
47. Gruber CJ, Gruber DM, Gruber IM, Wieser F, Huber JC. Anatomy of the estrogen response element. Trends Endocrinol Metab. (2004) 15:73–8. doi: 10.1016/j.tem.2004.01.008
48. Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. (1997) 389:753–8. doi: 10.1038/39645
49. McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. (1999) 20:321–44. doi: 10.1210/er.20.3.321
50. Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. (1995) 377:454–7. doi: 10.1038/377454a0
51. Bhatt S, Stender JD, Joshi S, Wu G, Katzenellenbogen BS. OCT-4: a novel estrogen receptor-alpha collaborator that promotes tamoxifen resistance in breast cancer cells. Oncogene. (2016) 35:5722–34. doi: 10.1038/onc.2016.105
52. Yi P, Driscoll MD, Huang J, Bhagat S, Hilf R, Bambara RA, et al. The effects of estrogen-responsive element- and ligand-induced structural changes on the recruitment of cofactors and transcriptional responses by ER alpha and ER beta. Mol Endocrinol. (2002) 16:674–93. doi: 10.1210/mend.16.4.0810
53. Lees JA, Fawell SE, Parker MG. Identification of two transactivation domains in the mouse oestrogen receptor. Nucleic Acids Res. (1989) 17:5477–88. doi: 10.1093/nar/17.14.5477
54. Thomas RS, Sarwar N, Phoenix F, Coombes RC, Ali S. Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is important for estrogen receptor-alpha activity. J Mol Endocrinol. (2008) 40:173–84. doi: 10.1677/JME-07-0165
55. Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. (1995) 270:1491–4. doi: 10.1126/science.270.5241.1491
56. Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem. (2001) 276:9817–24. doi: 10.1074/jbc.M010840200
57. Bostner J, Skoog L, Fornander T, Nordenskjold B, Stal O. Estrogen receptor-alpha phosphorylation at serine 305, nuclear p21-activated kinase 1 expression, and response to tamoxifen in postmenopausal breast cancer. Clin Cancer Res. (2010) 16:1624–33. doi: 10.1158/1078-0432.CCR-09-1733
58. Rogatsky I, Trowbridge JM, Garabedian MJ. Potentiation of human estrogen receptor alpha transcriptional activation through phosphorylation of serines 104 and 106 by the cyclin A-CDK2 complex. J Biol Chem. (1999) 274:22296–302. doi: 10.1074/jbc.274.32.22296
59. Schiff R, Massarweh S, Shou J, Osborne CK. Breast cancer endocrine resistance: how growth factor signaling and estrogen receptor coregulators modulate response. Clin Cancer Res. (2003) 9(1 Pt 2):447S−54S.
60. Kobayashi Y, Kitamoto T, Masuhiro Y, Watanabe M, Kase T, Metzger D, et al. p300 mediates functional synergism between AF-1 and AF-2 of estrogen receptor alpha and beta by interacting directly with the N-terminal A/B domains. J Biol Chem. (2000) 275:15645–51. doi: 10.1074/jbc.M000042200
61. McInerney EM, Tsai MJ, O'Malley BW, Katzenellenbogen BS. Analysis of estrogen receptor transcriptional enhancement by a nuclear hormone receptor coactivator. Proc Natl Acad Sci USA. (1996) 93:10069–73. doi: 10.1073/pnas.93.19.10069
62. Weigel NL, Zhang Y. Ligand-independent activation of steroid hormone receptors. J Mol Med. (1998) 76:469–79. doi: 10.1007/s001090050241
63. Razandi M, Pedram A, Park ST, Levin ER. Proximal events in signaling by plasma membrane estrogen receptors. J Biol Chem. (2003) 278:2701–12. doi: 10.1074/jbc.M205692200
64. Wong CW, McNally C, Nickbarg E, Komm BS, Cheskis BJ. Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc Natl Acad Sci USA. (2002) 99:14783–8. doi: 10.1073/pnas.192569699
65. Schiff R, Massarweh SA, Shou J, Bharwani L, Mohsin SK, Osborne CK. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin Cancer Res. (2004) 10(1 Pt 2):331S−6S. doi: 10.1158/1078-0432.CCR-031212
66. Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol. (2006) 20:1996–2009. doi: 10.1210/me.2005-0525
67. Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. (2005) 146:624–32. doi: 10.1210/en.2004-1064
68. Ignatov A, Ignatov T, Roessner A, Costa SD, Kalinski T. Role of GPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer Res Treat. (2010) 123:87–96. doi: 10.1007/s10549-009-0624-6
69. Ignatov A, Ignatov T, Weissenborn C, Eggemann H, Bischoff J, Semczuk A, et al. G-protein-coupled estrogen receptor GPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res Treat. (2011) 128:457–66. doi: 10.1007/s10549-011-1584-1
70. Mo Z, Liu M, Yang F, Luo H, Li Z, Tu G, et al. GPR30 as an initiator of tamoxifen resistance in hormone-dependent breast cancer. Breast Cancer Res. (2013) 15:R114. doi: 10.1186/bcr3581
71. Ignatov T, Claus M, Nass N, Haybaeck J, Seifert B, Kalinski T, et al. G-protein-coupled estrogen receptor GPER-1 expression in hormone receptor-positive breast cancer is associated with poor benefit of tamoxifen. Breast Cancer Res Treat. (2019) 174:121–7. doi: 10.1007/s10549-018-5064-8
72. Molina L, Figueroa CD, Bhoola KD, Ehrenfeld P. GPER-1/GPR30 a novel estrogen receptor sited in the cell membrane: therapeutic coupling to breast cancer. Expert Opin Ther Targets. (2017) 21:755–66. doi: 10.1080/14728222.2017.1350264
73. De Francesco EM, Pellegrino M, Santolla MF, Lappano R, Ricchio E, Abonante S, et al. GPER mediates activation of HIF1α/VEGF signaling by estrogens. Cancer Res. (2014) 74:4053–64. doi: 10.1158/0008-5472.CAN-13-3590
74. Zekas E, Prossnitz ER. Estrogen-mediated inactivation of FOXO3a by the G protein-coupled estrogen receptor GPER. BMC Cancer. (2015) 15:702. doi: 10.1186/s12885-015-1699-6
75. Yu T, Liu M, Luo H, Wu C, Tang X, Tang S, et al. GPER mediates enhanced cell viability and motility via non-genomic signaling induced by 17β-estradiol in triple-negative breast cancer cells. J Steroid Biochem Mol Biol. (2014) 143:392–403. doi: 10.1016/j.jsbmb.2014.05.003
76. Filardo EJ, Thomas P. Minireview: G protein-coupled estrogen receptor-1, GPER-1: its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology. (2012) 153:2953–62. doi: 10.1210/en.2012-1061
77. Scaling AL, Prossnitz ER, Hathaway HJ. GPER mediates estrogen-induced signaling and proliferation in human breast epithelial cells and normal and malignant breast. Horm Cancer. (2014) 5:146–60. doi: 10.1007/s12672-014-0174-1
78. Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. (2000) 407:538–41. doi: 10.1038/35035131
79. Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. A variant of estrogen receptor-{alpha}, hER-{alpha}36: transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc Natl Acad Sci USA. (2006) 103:9063–8. doi: 10.1073/pnas.0603339103
80. Lin SL, Yan LY, Zhang XT, Yuan J, Li M, Qiao J, et al. ER-alpha36, a variant of ER-alpha, promotes tamoxifen agonist action in endometrial cancer cells via the MAPK/ERK and PI3K/Akt pathways. PLoS ONE. (2010) 5:e9013. doi: 10.1371/journal.pone.0009013
81. Johnston SR, Saccani-Jotti G, Smith IE, Salter J, Newby J, Coppen M, et al. Changes in estrogen receptor, progesterone receptor, and pS2 expression in tamoxifen-resistant human breast cancer. Cancer Res. (1995) 55:3331–8.
82. Gutierrez MC, Detre S, Johnston S, Mohsin SK, Shou J, Allred DC, et al. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J Clin Oncol. (2005) 23:2469–76. doi: 10.1200/JCO.2005.01.172
83. Kangaspeska S, Stride B, Metivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP, et al. Transient cyclical methylation of promoter DNA. Nature. (2008) 452:112–5. doi: 10.1038/nature06640
84. Ottaviano YL, Issa JP, Parl FF, Smith HS, Baylin SB, Davidson NE. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expression in human breast cancer cells. Cancer Res. (1994) 54:2552–5.
85. Yang X, Phillips DL, Ferguson AT, Nelson WG, Herman JG, Davidson NE. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. (2001) 61:7025–9.
86. Yang X, Ferguson AT, Nass SJ, Phillips DL, Butash KA, Wang SM, et al. Transcriptional activation of estrogen receptor alpha in human breast cancer cells by histone deacetylase inhibition. Cancer Res. (2000) 60:6890–4.
87. Fan J, Yin WJ, Lu JS, Wang L, Wu J, Wu FY, et al. ER alpha negative breast cancer cells restore response to endocrine therapy by combination treatment with both HDAC inhibitor and DNMT inhibitor. J Cancer Res Clin Oncol. (2008) 134:883–90. doi: 10.1007/s00432-008-0354-x
88. Badia E, Duchesne MJ, Semlali A, Fuentes M, Giamarchi C, Richard-Foy H, et al. Long-term hydroxytamoxifen treatment of an MCF-7-derived breast cancer cell line irreversibly inhibits the expression of estrogenic genes through chromatin remodeling. Cancer Res. (2000) 60:4130–8.
89. Lehmann U, Hasemeier B, Christgen M, Muller M, Romermann D, Langer F, et al. Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J Pathol. (2008) 214:17–24. doi: 10.1002/path.2251
90. Liu G, Bollig-Fischer A, Kreike B, van de Vijver MJ, Abrams J, Ethier SP, et al. Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene. (2009) 28:4491–500. doi: 10.1038/onc.2009.297
91. Wu Y, Alvarez M, Slamon DJ, Koeffler P, Vadgama JV. Caspase 8 and maspin are downregulated in breast cancer cells due to CpG site promoter methylation. BMC Cancer. (2010) 10:32. doi: 10.1186/1471-2407-10-32
92. Reid G, Metivier R, Lin CY, Denger S, Ibberson D, Ivacevic T, et al. Multiple mechanisms induce transcriptional silencing of a subset of genes, including oestrogen receptor alpha, in response to deacetylase inhibition by valproic acid and trichostatin A. Oncogene. (2005) 24:4894–907. doi: 10.1038/sj.onc.1208662
93. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
94. Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer. (2011) 104:1828–35. doi: 10.1038/bjc.2011.156
95. Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. (2013) 31:2128–35. doi: 10.1200/JCO.2012.43.7251
96. Fuqua SA, Wiltschke C, Zhang QX, Borg A, Castles CG, Friedrichs WE, et al. A hypersensitive estrogen receptor-alpha mutation in premalignant breast lesions. Cancer Res. (2000) 60:4026–9.
97. Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. (2013) 45:1439–45. doi: 10.1038/ng.2822
98. Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. (2013) 45:1446–51. doi: 10.1038/ng.2823
99. Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, et al. D538G mutation in estrogen receptor-alpha: a novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. (2013) 73:6856–64. doi: 10.1158/0008-5472.CAN-13-1197
100. Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. (2014) 20:1757–67. doi: 10.1158/1078-0432.CCR-13-2332
101. Wolf DM, Jordan VC. The estrogen receptor from a tamoxifen stimulated MCF-7 tumor variant contains a point mutation in the ligand binding domain. Breast Cancer Res Treat. (1994) 31:129–38. doi: 10.1007/BF00689683
102. Fuqua SA, Allred DC, Auchus RJ. Expression of estrogen receptor variants. J Cell Biochem Suppl. (1993) 17G:194–7. doi: 10.1002/jcb.240531135
103. Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R. ESR1 mutations—a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol. (2015) 12:573–83. doi: 10.1038/nrclinonc.2015.117
104. Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. (2013) 4:1116–30. doi: 10.1016/j.celrep.2013.08.022
105. Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. (2012) 486:353–60. doi: 10.1038/nature11143
106. Chandarlapaty S, Chen D, He W, Sung P, Samoila A, You D, et al. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol. (2016) 2:1310–5. doi: 10.1001/jamaoncol.2016.1279
107. Chung JH, Pavlick D, Hartmaier R, Schrock AB, Young L, Forcier B, et al. Hybrid capture-based genomic profiling of circulating tumor DNA from patients with estrogen receptor-positive metastatic breast cancer. Ann Oncol. (2017) 28:2866–73. doi: 10.1093/annonc/mdx490
108. Fribbens C, O'Leary B, Kilburn L, Hrebien S, Garcia-Murillas I, Beaney M, et al. Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J Clin Oncol. (2016) 34:2961–8. doi: 10.1200/JCO.2016.67.3061
109. Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Inao T, Sueta A, Fujiwara S, et al. Clinical significance of monitoring ESR1 mutations in circulating cell-free DNA in estrogen receptor positive breast cancer patients. Oncotarget. (2016) 7:32504–18. doi: 10.18632/oncotarget.8839
110. Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, et al. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell. (2018) 33:173–86.e5. doi: 10.1016/j.ccell.2018.01.004
111. Weir HM, Bradbury RH, Lawson M, Rabow AA, Buttar D, Callis RJ, et al. AZD9496: An oral estrogen receptor inhibitor that blocks the growth of ER-positive and ESR1-mutant breast tumors in preclinical models. Cancer Res. (2016) 76:3307–18. doi: 10.1158/0008-5472.CAN-15-2357
112. Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, Mazzola AM, et al. Activating. Cancer Discov. (2017) 7:277–87. doi: 10.1158/2159-8290.CD-15-1523
113. Hamilton EP, Patel MR, Armstrong AC, Baird RD, Jhaveri K, Hoch M, et al. A First-in-Human study of the new oral selective estrogen receptor degrader AZD9496 for ER. Clin Cancer Res. (2018) 24:3510–8. doi: 10.1158/1078-0432.CCR-17-3102
114. Bihani T, Patel HK, Arlt H, Tao N, Jiang H, Brown JL, et al. Elacestrant (RAD1901), a selective estrogen receptor degrader (SERD), has antitumor activity in multiple ER. Clin Cancer Res. (2017) 23:4793–804. doi: 10.1158/1078-0432.CCR-16-2561
115. Bardia A, Kabos P, Elledge R, Wang D, Shen J, Garner F, et al. Evaluation of RAD1901, a novel investigational, selective estrogen receptor degrader (SERD), for the treatment of ER-positive (ER+) advanced breast cancer. J Clin Oncol. (2017) 35(15_Suppl.):1014. doi: 10.1200/JCO.2017.35.15_suppl.1014
116. Kahraman M, Govek SP, Nagasawa JY, Lai A, Bonnefous C, Douglas K, et al. Maximizing ER-α degradation maximizes activity in a tamoxifen-resistant breast cancer model: identification of GDC-0927. ACS Med Chem Lett. (2019) 10:50–5. doi: 10.1021/acsmedchemlett.8b00414
117. Dickler MN, Villanueva R, Perez Fidalgo JA. A first-in-human phase I study to evaluate the oral selective estrogen receptor degrader (SERD), GDC-0927, in postmenopausal women with estrogen receptor positive (ER+) HER2-negative metastatic breast cancer (BC). Cancer Res. (2018) 78. doi: 10.1158/1538-7445.SABCS17-PD5-10
118. Metcalfe C, Zhou W, Guan J, Daemen A, Hafner M, Blake R, et al. Abstract GS3-05: Prospective optimization of estrogen receptor degradation yields ER ligands with variable capacities for ER transcriptional suppression. Cancer Res. (2019) 79(4 Supplement):GS3-05-GS3. doi: 10.1158/1538-7445.SABCS18-GS3-05
119. Puyang X, Furman C, Zheng GZ, Wu ZJ, Banka D, Aithal K, et al. Discovery of selective estrogen receptor covalent antagonists for the treatment of ERα. Cancer Discov. (2018) 8:1176–93. doi: 10.1158/2159-8290.CD-17-1229
120. Brisken C. Progesterone signalling in breast cancer: a neglected hormone coming into the limelight. Nat Rev Cancer. (2013) 13:385–96. doi: 10.1038/nrc3518
121. Owen GI, Richer JK, Tung L, Takimoto G, Horwitz KB. Progesterone regulates transcription of the p21(WAF1) cyclin- dependent kinase inhibitor gene through Sp1 and CBP/p300. J Biol Chem. (1998) 273:10696–701. doi: 10.1074/jbc.273.17.10696
122. Faivre EJ, Daniel AR, Hillard CJ, Lange CA. Progesterone receptor rapid signaling mediates serine 345 phosphorylation and tethering to specificity protein 1 transcription factors. Mol Endocrinol. (2008) 22:823–37. doi: 10.1210/me.2007-0437
123. Knutson TP, Lange CA. Tracking progesterone receptor-mediated actions in breast cancer. Pharmacol Ther. (2014) 142:114–25. doi: 10.1016/j.pharmthera.2013.11.010
124. Kastner P, Krust A, Turcotte B, Stropp U, Tora L, Gronemeyer H, et al. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J. (1990) 9:1603–14. doi: 10.1002/j.1460-2075.1990.tb08280.x
125. Petz LN, Ziegler YS, Schultz JR, Kim H, Kemper JK, Nardulli AM. Differential regulation of the human progesterone receptor gene through an estrogen response element half site and Sp1 sites. J Steroid Biochem Mol Biol. (2004) 88:113–22. doi: 10.1016/j.jsbmb.2003.11.008
126. Hilton HN, Doan TB, Graham JD, Oakes SR, Silvestri A, Santucci N, et al. Acquired convergence of hormone signaling in breast cancer: ER and PR transition from functionally distinct in normal breast to predictors of metastatic disease. Oncotarget. (2014) 5:8651–64. doi: 10.18632/oncotarget.2354
127. Branković-Magić M, Janković R, Nesković-Konstantinović Z, Nikolić-Vukosavljević D. Progesterone receptor status of breast cancer metastases. J Cancer Res Clin Oncol. (2002) 128:55–60. doi: 10.1007/s00432-001-0299-9
128. Blows FM, Driver KE, Schmidt MK, Broeks A, van Leeuwen FE, Wesseling J, et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: a collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. (2010) 7:e1000279. doi: 10.1371/journal.pmed.1000279
129. Arpino G, Weiss H, Lee AV, Schiff R, De Placido S, Osborne CK, et al. Estrogen receptor-positive, progesterone receptor-negative breast cancer: association with growth factor receptor expression and tamoxifen resistance. J Natl Cancer Inst. (2005) 97:1254–61. doi: 10.1093/jnci/dji249
130. Daniel AR, Gaviglio AL, Knutson TP, Ostrander JH, D'Assoro AB, Ravindranathan P, et al. Progesterone receptor-B enhances estrogen responsiveness of breast cancer cells via scaffolding PELP1- and estrogen receptor-containing transcription complexes. Oncogene. (2015) 34:506–15. doi: 10.1038/onc.2013.579
131. Mohammed H, Russell IA, Stark R, Rueda OM, Hickey TE, Tarulli GA, et al. Progesterone receptor modulates ERα action in breast cancer. Nature. (2015) 523:313–7. doi: 10.1038/nature14583
132. Tomiguchi M, Yamamoto Y, Yamamoto-Ibusuki M, Goto-Yamaguchi L, Fujiki Y, Fujiwara S, et al. Fibroblast growth factor receptor-1 protein expression is associated with prognosis in estrogen receptor-positive/human epidermal growth factor receptor-2-negative primary breast cancer. Cancer Sci. (2016) 107:491–8. doi: 10.1111/cas.12897
133. Templeton AJ, Diez-Gonzalez L, Ace O, Vera-Badillo F, Seruga B, Jordán J, et al. Prognostic relevance of receptor tyrosine kinase expression in breast cancer: a meta-analysis. Cancer Treat Rev. (2014) 40:1048–55. doi: 10.1016/j.ctrv.2014.08.003
134. He M, Wei MJ. Reversing multidrug resistance by tyrosine kinase inhibitors. Chin J Cancer. (2012) 31:126–33. doi: 10.5732/cjc.011.10315
135. Linggi B, Carpenter G. ErbB receptors: new insights on mechanisms and biology. Trends Cell Biol. (2006) 16:649–56. doi: 10.1016/j.tcb.2006.10.008
136. Sorlie T. Molecular portraits of breast cancer: tumour subtypes as distinct disease entities. Eur J Cancer. (2004) 40:2667–75. doi: 10.1016/j.ejca.2004.08.021
137. Migliaccio A, Di Domenico M, Castoria G, Nanayakkara M, Lombardi M, de Falco A, et al. Steroid receptor regulation of epidermal growth factor signaling through Src in breast and prostate cancer cells: steroid antagonist action. Cancer Res. (2005) 65:10585–93. doi: 10.1158/0008-5472.CAN-05-0912
138. Li G, Zhang J, Jin K, He K, Zheng Y, Xu X, et al. Estrogen receptor-alpha36 is involved in development of acquired tamoxifen resistance via regulating the growth status switch in breast cancer cells. Mol Oncol. (2013) 7:611–24. doi: 10.1016/j.molonc.2013.02.001
139. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. (2005) 307:1625–30. doi: 10.1126/science.1106943
140. Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. (2000) 14:1649–60. doi: 10.1210/mend.14.10.0532
141. Creighton CJ, Massarweh S, Huang S, Tsimelzon A, Hilsenbeck SG, Osborne CK, et al. Development of resistance to targeted therapies transforms the clinically associated molecular profile subtype of breast tumor xenografts. Cancer Res. (2008) 68:7493–501. doi: 10.1158/0008-5472.CAN-08-1404
142. Kaufman B, Mackey JR, Clemens MR, Bapsy PP, Vaid A, Wardley A, et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: results from the randomized phase III TAnDEM study. J Clin Oncol. (2009) 27:5529–37. doi: 10.1200/JCO.2008.20.6847
143. Menendez JA, Schroeder B, Peirce SK, Vellon L, Papadimitropoulou A, Espinoza I, et al. Blockade of a key region in the extracellular domain inhibits HER2 dimerization and signaling. J Natl Cancer Inst. (2015) 107:djv090. doi: 10.1093/jnci/djv090
144. Kurokawa H, Lenferink AE, Simpson JF, Pisacane PI, Sliwkowski MX, Forbes JT, et al. Inhibition of HER2/neu (erbB-2) and mitogen-activated protein kinases enhances tamoxifen action against HER2-overexpressing, tamoxifen-resistant breast cancer cells. Cancer Res. (2000) 60:5887–94.
145. Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. (2003) 95:353–61. doi: 10.1093/jnci/95.5.353
146. O'Hara J, Vareslija D, McBryan J, Bane F, Tibbitts P, Byrne C, et al. AIB1:ERalpha transcriptional activity is selectively enhanced in aromatase inhibitor-resistant breast cancer cells. Clinical Cancer Res. (2012) 18:3305–15. doi: 10.1158/1078-0432.CCR-11-3300
147. Leary AF, Drury S, Detre S, Pancholi S, Lykkesfeldt AE, Martin LA, et al. Lapatinib restores hormone sensitivity with differential effects on estrogen receptor signaling in cell models of human epidermal growth factor receptor 2-negative breast cancer with acquired endocrine resistance. Clinical Cancer Res. (2010) 16:1486–97. doi: 10.1158/1078-0432.CCR-09-1764
148. Johnston S, Pippen J Jr, Pivot X, Lichinitser M, Sadeghi S, Dieras V, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol. (2009) 27:5538–46. doi: 10.1200/JCO.2009.23.3734
149. Osborne CK, Neven P, Dirix LY, Mackey JR, Robert J, Underhill C, et al. Gefitinib or placebo in combination with tamoxifen in patients with hormone receptor-positive metastatic breast cancer: a randomized phase II study. Clin Cancer Res. (2011) 17:1147–59. doi: 10.1158/1078-0432.CCR-10-1869
150. Cristofanilli M, Valero V, Mangalik A, Royce M, Rabinowitz I, Arena FP, et al. Phase II, randomized trial to compare anastrozole combined with gefitinib or placebo in postmenopausal women with hormone receptor-positive metastatic breast cancer. Clin Cancer Res. (2010) 16:1904–14. doi: 10.1158/1078-0432.CCR-09-2282
151. Schwartzberg LS, Schwarzberg LS, Franco SX, Florance A, O'Rourke L, Maltzman J, et al. Lapatinib plus letrozole as first-line therapy for HER-2+ hormone receptor-positive metastatic breast cancer. Oncologist. (2010) 15:122–9. doi: 10.1634/theoncologist.2009-0240
152. Nayar U, Cohen O, Kapstad C, Cuoco MS, Waks AG, Wander SA, et al. Acquired HER2 mutations in ER. Nat Genet. (2019) 51:207–16. doi: 10.1038/s41588-018-0287-5
153. Croessmann S, Formisano L, Kinch LN, Gonzalez-Ericsson PI, Sudhan DR, Nagy RJ, et al. Combined blockade of activating. Clin Cancer Res. (2019) 25:277–89. doi: 10.1158/1078-0432.CCR-18-1544
154. Singh H, Walker AJ, Amiri-Kordestani L, Cheng J, Tang S, Balcazar P, et al. U.S. Food and Drug Administration approval: neratinib for the extended adjuvant treatment of early-stage HER2-positive breast cancer. Clin Cancer Res. (2018) 24:3486–91. doi: 10.1158/1078-0432.CCR-17-3628
155. Deeks ED. Neratinib: first global approval. Drugs. (2017) 77:1695–704. doi: 10.1007/s40265-017-0811-4
156. Delaloge S, Cella D, Ye Y, Buyse M, Chan A, Barrios CH, et al. Effects of neratinib on health-related quality-of-life in women with HER2-positive early-stage breast cancer: longitudinal analyses from the randomized phase III ExteNET trial. Ann Oncol. (2019). doi: 10.1093/annonc/mdz016. [Epub ahead of print]
157. Kurmasheva RT, Houghton PJ. IGF-I mediated survival pathways in normal and malignant cells. Biochim Biophys Acta. (2006) 1766:1–22. doi: 10.1016/j.bbcan.2006.05.003
158. Knowlden JM, Hutcheson IR, Barrow D, Gee JM, Nicholson RI. Insulin-like growth factor-I receptor signaling in tamoxifen-resistant breast cancer: a supporting role to the epidermal growth factor receptor. Endocrinology. (2005) 146:4609–18. doi: 10.1210/en.2005-0247
159. Wiseman LR, Johnson MD, Wakeling AE, Lykkesfeldt AE, May FE, Westley BR. Type I IGF receptor and acquired tamoxifen resistance in oestrogen-responsive human breast cancer cells. Eur J Cancer. (1993) 29A:2256–64. doi: 10.1016/0959-8049(93)90218-5
160. Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, et al. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet. (1998) 351:1393–6. doi: 10.1016/S0140-6736(97)10384-1
161. Yu H, Li BD, Smith M, Shi R, Berkel HJ, Kato I. Polymorphic CA repeats in the IGF-I gene and breast cancer. Breast Cancer Res Treat. (2001) 70:117–22. doi: 10.1023/A:1012947027213
162. Wagner K, Hemminki K, Israelsson E, Grzybowska E, Soderberg M, Pamula J, et al. Polymorphisms in the IGF-1 and IGFBP 3 promoter and the risk of breast cancer. Breast Cancer Res Treat. (2005) 92:133–40. doi: 10.1007/s10549-005-2417-x
163. Bartella V, De Marco P, Malaguarnera R, Belfiore A, Maggiolini M. New advances on the functional cross-talk between insulin-like growth factor-I and estrogen signaling in cancer. Cell Signal. (2012) 24:1515–21. doi: 10.1016/j.cellsig.2012.03.012
164. Lee AV, Weng CN, Jackson JG, Yee D. Activation of estrogen receptor-mediated gene transcription by IGF-I in human breast cancer cells. J Endocrinol. (1997) 152:39–47. doi: 10.1677/joe.0.1520039
165. Creighton CJ, Casa A, Lazard Z, Huang S, Tsimelzon A, Hilsenbeck SG, et al. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol. (2008) 26:4078–85. doi: 10.1200/JCO.2007.13.4429
166. Chong K, Subramanian A, Sharma A, Mokbel K. Measuring IGF-1, ER-alpha and EGFR expression can predict tamoxifen-resistance in ER-positive breast cancer. Anticancer Res. (2011) 31:23–32.
167. Fox EM, Miller TW, Balko JM, Kuba MG, Sánchez V, Smith RA, et al. A kinome-wide screen identifies the insulin/IGF-I receptor pathway as a mechanism of escape from hormone dependence in breast cancer. Cancer Res. (2011) 71:6773–84. doi: 10.1158/0008-5472.CAN-11-1295
168. Fagan DH, Uselman RR, Sachdev D, Yee D. Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor: implications for breast cancer treatment. Cancer Res. (2012) 72:3372–80. doi: 10.1158/0008-5472.CAN-12-0684
169. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. (2010) 10:116–29. doi: 10.1038/nrc2780
170. Brady N, Chuntova P, Bade LK, Schwertfeger KL. The FGF/FGFR axis as a therapeutic target in breast cancer. Expert Rev Endocrinol Metab. (2013) 8:391–402. doi: 10.1586/17446651.2013.811910
171. Formisano L, Stauffer KM, Young CD, Bhola NE, Guerrero-Zotano AL, Jansen VM, et al. Association of FGFR1 with ERα maintains ligand-independent ER transcription and mediates resistance to estrogen deprivation in ER. Clin Cancer Res. (2017) 23:6138–50. doi: 10.1158/1078-0432.CCR-17-1232
172. Dienstmann R, Rodon J, Prat A, Perez-Garcia J, Adamo B, Felip E, et al. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol. (2014) 25:552–63. doi: 10.1093/annonc/mdt419
173. Kwek SS, Roy R, Zhou H, Climent J, Martinez-Climent JA, Fridlyand J, et al. Co-amplified genes at 8p12 and 11q13 in breast tumors cooperate with two major pathways in oncogenesis. Oncogene. (2009) 28:1892–903. doi: 10.1038/onc.2009.34
174. Reis-Filho JS, Simpson PT, Turner NC, Lambros MB, Jones C, Mackay A, et al. FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin Cancer Res. (2006) 12:6652–62. doi: 10.1158/1078-0432.CCR-06-1164
175. Dey N, Williams C, Leyland-Jones B, De P. Mutation matters in precision medicine: a future to believe in. Cancer Treat Rev. (2017) 55:136–49. doi: 10.1016/j.ctrv.2017.03.002
176. Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. (2010) 70:2085–94. doi: 10.1158/0008-5472.CAN-09-3746
177. André F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, et al. Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res. (2013) 19:3693–702. doi: 10.1158/1078-0432.CCR-13-0190
178. Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res. (2016) 22:259–67. doi: 10.1158/1078-0432.CCR-14-3212
179. Sobhani N, Ianza A, D'Angelo A, Roviello G, Giudici F, Bortul M, et al. Current status of fibroblast growth factor receptor-targeted therapies in breast cancer. Cells. (2018) 7:E76. doi: 10.3390/cells7070076
180. Elbauomy Elsheikh S, Green AR, Lambros MB, Turner NC, Grainge MJ, Powe D, et al. FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast Cancer Res. (2007) 9:R23. doi: 10.1186/bcr1665
181. Drago JZ, Niemierko A, Spring L, Moy B, Juric D, Isakoff SJ, et al. FGFR gene amplification and response to endocrine therapy in metastatic hormone receptor positive (HR+) breast cancer. J Clin Oncol. (2017) 35(15_Suppl.):1013. doi: 10.1200/JCO.2017.35.15_suppl.1013
182. Racca FE, Zamora E, Muñoz-Couselo E, Nuciforo P, Bellet M, Peg V, et al. Prognostic and therapeutic implications of fibroblast growth factor receptors (FGFRs) 1 and 2 gene amplifications in patients (pts) with advanced breast cancer (ABC). J Clin Oncol. (2016) 34(15_Suppl.):537. doi: 10.1200/JCO.2016.34.15_suppl.537
183. Perez-Garcia J, Muñoz-Couselo E, Soberino J, Racca F, Cortes J. Targeting FGFR pathway in breast cancer. Breast. (2018) 37:126–33. doi: 10.1016/j.breast.2017.10.014
184. Sahores A, May M, Sequeira GR, Fuentes C, Jacobsen B, Lanari C, et al. Targeting FGFR with BGJ398 in breast cancer: effect on tumor growth and metastasis. Curr Cancer Drug Targets. (2018) 18:979–87. doi: 10.2174/1568009618666171214114706
185. Seckl M, Badman PD, Liu X, MacPherson IR, Zubairi IH, Baird RD, et al. RADICAL trial: A phase Ib/IIa study to assess the safety and efficacy of AZD4547 in combination with either anastrozole or letrozole in ER positive breast cancer patients progressing on these aromatase inhibitors (AIs). J Clin Oncol. (2017) 35(15_Suppl.):1059. doi: 10.1200/JCO.2017.35.15_suppl.1059
186. Musolino A, Campone M, Neven P, Denduluri N, Barrios CH, Cortes J, et al. Phase II, randomized, placebo-controlled study of dovitinib in combination with fulvestrant in postmenopausal patients with HR. Breast Cancer Res. (2017) 19:18. doi: 10.1186/s13058-017-0807-8
187. Andre F, Daly F, Azim H, Agrapart V, Fumagalli D, Gingras I, et al. Abstract OT1-03-03: FINESSE–an open, 3-cohort, phase II trial testing oral administration of lucitanib in patients with FGFR1-amplified or non-amplIfied oestrogeN rEceptor poSitive metaStatic breast cancEr. Cancer Res. (2016) 76(4 Supplement):OT1-03-OT1. doi: 10.1158/1538-7445.SABCS15-OT1-03-03
188. Formisano L, Lu Y, Jansen VM, Bauer JA, Hanker AB, Sanders ME, et al. Abstract 1008: Gain-of-function kinase library screen identifies FGFR1 amplification as a mechanism of resistance to antiestrogens and CDK4/6 inhibitors in ER+ breast cancer. Cancer Res. (2017) 77(13 Supplement):1008. doi: 10.1158/1538-7445.AM2017-1008
189. André F, Cortés J. Rationale for targeting fibroblast growth factor receptor signaling in breast cancer. Breast Cancer Res Treat. (2015) 150:1–8. doi: 10.1007/s10549-015-3301-y
190. Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. (2007) 447:1087–93. doi: 10.1038/nature05887
191. Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. (2007) 39:870–4. doi: 10.1038/ng2075
192. Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. (2013) 3:636–47. doi: 10.1158/2159-8290.CD-13-0050
193. Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. (2014) 5:4846. doi: 10.1038/ncomms5846
194. Sada K, Takano T, Yanagi S, Yamamura H. Structure and function of Syk protein-tyrosine kinase. J Biochem. (2001) 130:177–86. doi: 10.1093/oxfordjournals.jbchem.a002970
195. Fütterer K, Wong J, Grucza RA, Chan AC, Waksman G. Structural basis for Syk tyrosine kinase ubiquity in signal transduction pathways revealed by the crystal structure of its regulatory SH2 domains bound to a dually phosphorylated ITAM peptide. J Mol Biol. (1998) 281:523–37.
196. Geahlen RL. Syk and pTyr'd: signaling through the B cell antigen receptor. Biochim Biophys Acta. (2009) 1793:1115–27. doi: 10.1016/j.bbamcr.2009.03.004
197. Coopman PJ, Mueller SC. The Syk tyrosine kinase: a new negative regulator in tumor growth and progression. Cancer Lett. (2006) 241:159–73. doi: 10.1016/j.canlet.2005.11.004
198. Elkak A, Al Sarakbi W, Mokbel K. SYK expression in human breast cancer. J Carcinog. (2005) 4:7. doi: 10.1186/1477-3163-4-7
199. Kawakami Y, Kitaura J, Yao L, McHenry RW, Newton AC, Kang S, et al. A Ras activation pathway dependent on Syk phosphorylation of protein kinase C. Proc Natl Acad Sci USA. (2003) 100:9470–5. doi: 10.1073/pnas.1633695100
200. Coopman PJ, Do MT, Barth M, Bowden ET, Hayes AJ, Basyuk E, et al. The Syk tyrosine kinase suppresses malignant growth of human breast cancer cells. Nature. (2000) 406:742–7. doi: 10.1038/35021086
201. Toyama T, Iwase H, Yamashita H, Hara Y, Omoto Y, Sugiura H, et al. Reduced expression of the Syk gene is correlated with poor prognosis in human breast cancer. Cancer Lett. (2003) 189:97–102. doi: 10.1016/S0304-3835(02)00463-9
202. Moroni M, Soldatenkov V, Zhang L, Zhang Y, Stoica G, Gehan E, et al. Progressive loss of Syk and abnormal proliferation in breast cancer cells. Cancer Res. (2004) 64:7346–54. doi: 10.1158/0008-5472.CAN-03-3520
203. Blancato J, Graves A, Rashidi B, Moroni M, Tchobe L, Ozdemirli M, et al. SYK allelic loss and the role of Syk-regulated genes in breast cancer survival. PLoS ONE. (2014) 9:e87610. doi: 10.1371/journal.pone.0087610
204. Fueyo J, Alonso MM, Parker Kerrigan BC, Gomez-Manzano C. Linking inflammation and cancer: the unexpected SYK world. Neuro Oncol. (2018) 20:582–3. doi: 10.1093/neuonc/noy036
205. Mullard A. FDA approves first-in-class SYK inhibitor. Nat Rev Drug Discov. (2018) 17:385. doi: 10.1038/nrd.2018.96
206. Coffey G, Rani A, Betz A, Pak Y, Haberstock-Debic H, Pandey A, et al. PRT062607 achieves complete inhibition of the spleen tyrosine kinase at tolerated exposures following oral dosing in healthy volunteers. J Clin Pharmacol. (2017) 57:194–210. doi: 10.1002/jcph.794
207. Coffey GP, Feng J, Betz A, Pandey A, Birrell M, Leeds JM, et al. Cerdulatinib pharmacodynamics and relationships to tumor response following oral dosing in patients with relapsed/refractory B-cell malignancies. Clin Cancer Res. (2019) 25:1174–1184. doi: 10.1158/1078-0432.CCR-18-1047
208. Braselmann S, Taylor V, Zhao H, Wang S, Sylvain C, Baluom M, et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J Pharmacol Exp Ther. (2006) 319:998–1008. doi: 10.1124/jpet.106.109058
209. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. (2009) 9:631–43. doi: 10.1038/nrc2713
210. Butt AJ, McNeil CM, Musgrove EA, Sutherland RL. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: the potential roles of c-Myc, cyclin D1 and cyclin E. Endocr Relat Cancer. (2005) 12 Suppl. 1:S47–59. doi: 10.1677/erc.1.00993
211. Span PN, Tjan-Heijnen VC, Manders P, Beex LV, Sweep CG. Cyclin-E is a strong predictor of endocrine therapy failure in human breast cancer. Oncogene. (2003) 22:4898–904. doi: 10.1038/sj.onc.1206818
212. Mukherjee S, Conrad SE. c-Myc suppresses p21WAF1/CIP1 expression during estrogen signaling and antiestrogen resistance in human breast cancer cells. J Biol Chem. (2005) 280:17617–25. doi: 10.1074/jbc.M502278200
213. Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. (2008) 8:253–67. doi: 10.1038/nrc2347
214. Pérez-Tenorio G, Berglund F, Esguerra Merca A, Nordenskjöld B, Rutqvist LE, Skoog L, et al. Cytoplasmic p21WAF1/CIP1 correlates with Akt activation and poor response to tamoxifen in breast cancer. Int J Oncol. (2006) 28:1031–42. doi: 10.3892/ijo.28.5.1031
215. Miller TW, Balko JM, Fox EM, Ghazoui Z, Dunbier A, Anderson H, et al. ERα-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov. (2011) 1:338-51. doi: 10.1158/2159-8290.CD-11-0101
216. Caldon CE, Daly RJ, Sutherland RL, Musgrove EA. Cell cycle control in breast cancer cells. J Cell Biochem. (2006) 97:261-74. doi: 10.1002/jcb.20690
217. Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. (2004) 18:2699-711. doi: 10.1101/gad.1256504
218. Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol. (1995) 15:2612-24. doi: 10.1128/MCB.15.5.2612
219. Ferguson RL, Maller JL. Centrosomal localization of cyclin E-Cdk2 is required for initiation of DNA synthesis. Curr Biol. (2010) 20:856-60. doi: 10.1016/j.cub.2010.03.028
220. Polychronis A, Sinnett HD, Hadjiminas D, Singhal H, Mansi JL, Shivapatham D, et al. Preoperative gefitinib versus gefitinib and anastrozole in postmenopausal patients with oestrogen-receptor positive and epidermal-growth-factor-receptor-positive primary breast cancer: a double-blind placebo-controlled phase II randomised trial. Lancet Oncol. (2005) 6:383–91. doi: 10.1016/S1470-2045(05)70176-5
221. Lukas J, Bartkova J, Bartek J. Convergence of mitogenic signalling cascades from diverse classes of receptors at the cyclin D-cyclin-dependent kinase-pRb-controlled G1 checkpoint. Mol Cell Biol. (1996) 16:6917–25. doi: 10.1128/MCB.16.12.6917
222. Thangavel C, Dean JL, Ertel A, Knudsen KE, Aldaz CM, Witkiewicz AK, et al. Therapeutically activating RB: reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr Relat Cancer. (2011) 18:333–45. doi: 10.1530/ERC-10-0262
223. Guerrero-Zotano AL, Stricker TP, Formisano L, Hutchinson KE, Stover DG, Lee KM, et al. ER. Clin Cancer Res. (2018) 24:2517–29. doi: 10.1158/1078-0432.CCR-17-2904
224. Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. (2004) 3:1427–38.
225. Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. (2009) 11:R77. doi: 10.1186/bcr2419
226. Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. (2016) 375:1925–36. doi: 10.1056/NEJMoa1607303
227. Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. (2017) 35:3638–46. doi: 10.1200/JCO.2017.75.6155
228. Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. (2016) 375:1738–48. doi: 10.1056/NEJMoa1609709
229. Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. (2016) 17:425–39. doi: 10.1016/S1470-2045(15)00613-0
230. Rugo HS, Rumble RB, Macrae E, Barton DL, Connolly HK, Dickler MN, et al. Endocrine therapy for hormone receptor-positive metastatic breast cancer: American Society of Clinical Oncology guideline. J Clin Oncol. (2016) 34:3069–103. doi: 10.1200/JCO.2016.67.1487
231. Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. Results of a randomized phase 2 study of PD 0332991, a cyclin-dependent kinase (CDK) 4/6 inhibitor, in combination with letrozole vs letrozole alone for first-line treatment of ER+/HER2– advanced breast cancer (BC). Cancer Res. (2012) 72:24 Suppl.:Abstract nr S1–6. doi: 10.1158/0008-5472.SABCS12-S1-6
232. Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. (2015) 16:25–35. doi: 10.1016/S1470-2045(14)71159-3
233. Rugo HS, Turner NC, Finn RS, Joy AA, Verma S, Harbeck N, et al. Palbociclib plus endocrine therapy in older women with HR+/HER2- advanced breast cancer: a pooled analysis of randomised PALOMA clinical studies. Eur J Cancer. (2018) 101:123–33. doi: 10.1016/j.ejca.2018.05.017
234. Turner NC, Finn RS, Martin M, Im SA, DeMichele A, Ettl J, et al. Clinical considerations of the role of palbociclib in the management of advanced breast cancer patients with and without visceral metastases. Ann Oncol. (2018) 29:669–80. doi: 10.1093/annonc/mdx797
235. Curigliano G, Gómez Pardo P, Meric-Bernstam F, Conte P, Lolkema MP, Beck JT, et al. Ribociclib plus letrozole in early breast cancer: a presurgical, window-of-opportunity study. Breast. (2016) 28:191–8. doi: 10.1016/j.breast.2016.06.008
236. Rinnerthaler G, Gampenrieder SP, Greil R. SABCS 2017: lifestyle factors, hormone receptor-positive advanced disease, liquid biopsies, and prognosis. Memo. (2018) 11:208–12. doi: 10.1007/s12254-018-0433-x
237. Rossi L, McCartney A, Risi E, Malorni L, Biganzoli L, Di Leo A. Managing advanced HR-positive, HER2-negative breast cancer with CDK4/6 inhibitors in post-menopausal patients: is there a best sequence? Ther Adv Med Oncol. (2018) 10:1758835918815591. doi: 10.1177/1758835918815591
238. Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. (2016) 6:740–53. doi: 10.1158/2159-8290.CD-16-0095
239. Sammons SL, Topping DL, Blackwell KL. HR+, HER2- advanced breast cancer and CDK4/6 inhibitors: mode of action, clinical activity, and safety profiles. Curr Cancer Drug Targets. (2017) 17:637–49. doi: 10.2174/1568009617666170330120452
240. Nur Husna SM, Tan HT, Mohamud R, Dyhl-Polk A, Wong KK. Inhibitors targeting CDK4/6, PARP and PI3K in breast cancer: a review. Ther Adv Med Oncol. (2018) 10:1758835918808509. doi: 10.1177/1758835918808509
241. Dickler MN, Tolaney SM, Rugo HS, Cortés J, Diéras V, Patt D, et al. MONARCH 1, a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, in patients with refractory HR. Clin Cancer Res. (2017) 23:5218–24. doi: 10.1158/1078-0432.CCR-17-0754
242. Sledge GW, Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2- advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. (2017) 35:2875–84. doi: 10.1200/JCO.2017.73.7585
243. Johnston S, Martin M, Di Leo A, Im SA, Awada A, Forrester T, et al. MONARCH 3 final PFS: a randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer. (2019) 5:5. doi: 10.1038/s41523-018-0097-z
244. de Groot AF, Kuijpers CJ, Kroep JR. CDK4/6 inhibition in early and metastatic breast cancer: a review. Cancer Treat Rev. (2017) 60:130–8. doi: 10.1016/j.ctrv.2017.09.003
245. Pernas S, Tolaney SM, Winer EP, Goel S. CDK4/6 inhibition in breast cancer: current practice and future directions. Ther Adv Med Oncol. (2018) 10:1758835918786451. doi: 10.1177/1758835918786451
246. Pan H, Gray R, Braybrooke J, Davies C, Taylor C, McGale P, et al. 20-Year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. N Engl J Med. (2017) 377:1836–46. doi: 10.1056/NEJMoa1701830
247. Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell. (2016) 29:255–69. doi: 10.1016/j.ccell.2016.02.006
248. Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. (2014) 26:136–49. doi: 10.1016/j.ccr.2014.05.020
249. Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. (2016) 76:2301–13. doi: 10.1158/0008-5472.CAN-15-0728
250. Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. (2006) 24:1770–83. doi: 10.1200/JCO.2005.03.7689
251. Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. (2017) 548:471–5. doi: 10.1038/nature23465
252. Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. (2018) 8:216–33. doi: 10.1158/2159-8290.CD-17-0915
253. Teo ZL, Versaci S, Dushyanthen S, Caramia F, Savas P, Mintoff CP, et al. Combined CDK4/6 and PI3Kα inhibition is synergistic and immunogenic in triple-negative breast cancer. Cancer Res. (2017) 77:6340–52. doi: 10.1158/0008-5472.CAN-17-2210
254. Li Z, Razavi P, Li Q, Toy W, Liu B, Ping C, et al. Loss of the FAT1 tumor suppressor promotes resistance to CDK4/6 inhibitors via the hippo pathway. Cancer Cell. (2018) 34:893–905.e8. doi: 10.1016/j.ccell.2018.11.006
255. Veeraraghavan J, Tan Y, Cao XX, Kim JA, Wang X, Chamness GC, et al. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat Commun. (2014) 5:4577. doi: 10.1038/ncomms5577
256. Hartmaier RJ, Trabucco SE, Priedigkeit N, Chung JH, Parachoniak CA, Vanden Borre P, et al. Recurrent hyperactive ESR1 fusion proteins in endocrine therapy-resistant breast cancer. Ann Oncol. (2018) 29:872–80. doi: 10.1093/annonc/mdy025
257. Lei JT, Shao J, Zhang J, Iglesia M, Chan DW, Cao J, et al. Functional annotation of ESR1 gene fusions in estrogen receptor-positive breast cancer. Cell Rep. (2018) 24:1434–44.e7. doi: 10.1016/j.celrep.2018.07.009
258. Wardell SE, Ellis MJ, Alley HM, Eisele K, VanArsdale T, Dann SG, et al. Efficacy of SERD/SERM hybrid-CDK4/6 inhibitor combinations in models of endocrine therapy-resistant breast cancer. Clin Cancer Res. (2015) 21:5121–30. doi: 10.1158/1078-0432.CCR-15-0360
259. O'Leary B, Cutts RJ, Liu Y, Hrebien S, Huang X, Fenwick K, et al. The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA-3 trial. Cancer Discov. (2018) 8:1390–403. doi: 10.1158/2159-8290.CD-18-0264
260. Burris HA III. Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol. (2013) 71:829–42. doi: 10.1007/s00280-012-2043-3
261. Gonzalez-Angulo AM, Blumenschein GR Jr. Defining biomarkers to predict sensitivity to PI3K/Akt/mTOR pathway inhibitors in breast cancer. Cancer Treat Rev. (2013) 39:313–20. doi: 10.1016/j.ctrv.2012.11.002
262. Hernandez-Aya LF, Gonzalez-Angulo AM. Targeting the phosphatidylinositol 3-kinase signaling pathway in breast cancer. Oncologist. (2011) 16:404–14. doi: 10.1634/theoncologist.2010-0402
263. Loi S, Michiels S, Lambrechts D, Fumagalli D, Claes B, Kellokumpu-Lehtinen PL, et al. Somatic mutation profiling and associations with prognosis and trastuzumab benefit in early breast cancer. J Natl Cancer Inst. (2013) 105:960–7. doi: 10.1093/jnci/djt121
264. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. (2004) 304:554. doi: 10.1126/science.1096502
265. Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. (2005) 5:921–9. doi: 10.1038/nrc1753
266. Fox EM, Arteaga CL, Miller TW. Abrogating endocrine resistance by targeting ERalpha and PI3K in breast cancer. Front Oncol. (2012) 2:145. doi: 10.3389/fonc.2012.00145
267. Fu X, Osborne CK, Schiff R. Biology and therapeutic potential of PI3K signaling in ER+/HER2-negative breast cancer. Breast. (2013) 22 Suppl. 2:S12–8. doi: 10.1016/j.breast.2013.08.001
268. Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol. (2009) 27:2278–87. doi: 10.1200/JCO.2008.20.0766
269. Breuleux M, Klopfenstein M, Stephan C, Doughty CA, Barys L, Maira SM, et al. Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Mol Cancer Ther. (2009) 8:742–53. doi: 10.1158/1535-7163.MCT-08-0668
270. Baselga J, Semiglazov V, van Dam P, Manikhas A, Bellet M, Mayordomo J, et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol. (2009) 27:2630–7. doi: 10.1200/JCO.2008.18.8391
271. Baselga J, Campone M, Piccart M, Burris HA III, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. (2012) 366:520–9. doi: 10.1056/NEJMoa1109653
272. Ribas R, Pancholi S, Rani A, Schuster E, Guest SK, Nikitorowicz-Buniak J, et al. Targeting tumour re-wiring by triple blockade of mTORC1, epidermal growth factor, and oestrogen receptor signalling pathways in endocrine-resistant breast cancer. Breast Cancer Res. (2018) 20:44. doi: 10.1186/s13058-018-0983-1
273. Bachelot T, Bourgier C, Cropet C, Ray-Coquard I, Ferrero JM, Freyer G, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol. (2012) 30:2718–24. doi: 10.1200/JCO.2011.39.0708
274. Baselga J, Im SA, Iwata H, Cortés J, De Laurentiis M, Jiang Z, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. (2017) 18:904–16.
275. Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. (2015) 15:7–24. doi: 10.1038/nrc3860
276. Andre F, Campone M, Ciruelos EM, Iwata H, Loibl S, Rugo HS, et al. SOLAR-1: A phase III study of alpelisib + fulvestrant in men and postmenopausal women with HR+/HER2– advanced breast cancer (BC) progressing on or after prior aromatase inhibitor therapy. J Clin Oncol. (2016) 34(15_Suppl.):TPS618-TPS. doi: 10.1200/JCO.2016.34.15_suppl.TPS618
277. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. (2005) 434:913–7. doi: 10.1038/nature03443
278. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. (2005) 434:917–21. doi: 10.1038/nature03445
279. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. (2010) 10:293–301. doi: 10.1038/nrc2812
280. Tutt A, Tovey H, Cheang MCU, Kernaghan S, Kilburn L, Gazinska P, et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT Trial. Nat Med. (2018) 24:628–37. doi: 10.1038/s41591-018-0009-7
281. De Felice F, Tombolini V, Marampon F, Musella A, Marchetti C. Defective DNA repair mechanisms in prostate cancer: impact of olaparib. Drug Des Devel Ther. (2017) 11:547–52. doi: 10.2147/DDDT.S110264
282. Kristeleit R, Shapiro GI, Burris HA, Oza AM, LoRusso P, Patel MR, et al. A phase I–II study of the oral PARP inhibitor rucaparib in patients with germline. Clin Cancer Res. (2017) 23:4095–106. doi: 10.1158/1078-0432.CCR-16-2796
283. Brown JS, Kaye SB, Yap TA. PARP inhibitors: the race is on. Br J Cancer. (2016) 114:713–5. doi: 10.1038/bjc.2016.67
284. Brown JS, O'Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. (2017) 7:20–37. doi: 10.1158/2159-8290.CD-16-0860
285. Robson ME, Tung N, Conte P, Im SA, Senkus E, Xu B, et al. OlympiAD final overall survival and tolerability results: olaparib versus chemotherapy treatment of physician's choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann Oncol. (2019). doi: 10.1093/annonc/mdz012. [Epub ahead of print]
286. Ettl J, Quek RGW, Lee KH, Rugo HS, Hurvitz S, Gonçalves A, et al. Quality of life with talazoparib versus physician's choice of chemotherapy in patients with advanced breast cancer and germline BRCA1/2 mutation: patient-reported outcomes from the EMBRACA phase III trial. Ann Oncol. (2018) 29:1939–47. doi: 10.1093/annonc/mdy257
287. Rubin SM. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem Sci. (2013) 38:12–9. doi: 10.1016/j.tibs.2012.10.007
288. Wiśnik E, Płoszaj T, Robaszkiewicz A. Downregulation of PARP1 transcription by promoter-associated E2F4-RBL2-HDAC1-BRM complex contributes to repression of pluripotency stem cell factors in human monocytes. Sci Rep. (2017) 7:9483. doi: 10.1038/s41598-017-10307-z
289. Tempka D, Tokarz P, Chmielewska K, Kluska M, Pietrzak J, Rygielska Z, et al. Downregulation of PARP1 transcription by CDK4/6 inhibitors sensitizes human lung cancer cells to anticancer drug-induced death by impairing OGG1-dependent base excision repair. Redox Biol. (2018) 15:316–26. doi: 10.1016/j.redox.2017.12.017
290. Emberley ED, Bennett M, Chen J, Gross M, Huang T, Makkouk A, et al. Abstract 3509: the glutaminase inhibitor CB-839 synergizes with CDK4/6 and PARP inhibitors in pre-clinical tumor models. Cancer Res. (2018) 78(13 Supplement):3509. doi: 10.1158/1538-7445.AM2018-3509
291. Chen D, Washbrook E, Sarwar N, Bates GJ, Pace PE, Thirunuvakkarasu V, et al. Phosphorylation of human estrogen receptor alpha at serine 118 by two distinct signal transduction pathways revealed by phosphorylation-specific antisera. Oncogene. (2002) 21:4921–31. doi: 10.1038/sj.onc.1205420
292. Driggers PH, Segars JH. Estrogen action and cytoplasmic signaling pathways. Part II: the role of growth factors and phosphorylation in estrogen signaling. Trends Endocrinol Metab. (2002) 13:422–7. doi: 10.1016/S1043-2760(02)00634-3
293. Chung YL, Sheu ML, Yang SC, Lin CH, Yen SH. Resistance to tamoxifen-induced apoptosis is associated with direct interaction between Her2/neu and cell membrane estrogen receptor in breast cancer. Int J Cancer. (2002) 97:306–12. doi: 10.1002/ijc.1614
294. Osborne CK, Schiff R. Growth factor receptor cross-talk with estrogen receptor as a mechanism for tamoxifen resistance in breast cancer. Breast. (2003) 12:362–7. doi: 10.1016/S0960-9776(03)00137-1
295. Britton DJ, Hutcheson IR, Knowlden JM, Barrow D, Giles M, McClelland RA, et al. Bidirectional cross talk between ERalpha and EGFR signalling pathways regulates tamoxifen-resistant growth. Breast Cancer Res Treat. (2006) 96:131–46. doi: 10.1007/s10549-005-9070-2
296. McGlynn LM, Kirkegaard T, Edwards J, Tovey S, Cameron D, Twelves C, et al. Ras/Raf-1/MAPK pathway mediates response to tamoxifen but not chemotherapy in breast cancer patients. Clin Cancer Res. (2009) 15:1487–95. doi: 10.1158/1078-0432.CCR-07-4967
297. Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. (2004) 96:926–35. doi: 10.1093/jnci/djh166
298. Bayliss J, Hilger A, Vishnu P, Diehl K, El-Ashry D. Reversal of the estrogen receptor negative phenotype in breast cancer and restoration of antiestrogen response. Clin Cancer Res. (2007) 13:7029–36. doi: 10.1158/1078-0432.CCR-07-0587
299. Iorns E, Turner NC, Elliott R, Syed N, Garrone O, Gasco M, et al. Identification of CDK10 as an important determinant of resistance to endocrine therapy for breast cancer. Cancer Cell. (2008) 13:91–104. doi: 10.1016/j.ccr.2008.01.001
300. Wallace MD, Pfefferle AD, Shen L, McNairn AJ, Cerami EG, Fallon BL, et al. Comparative oncogenomics implicates the neurofibromin 1 gene (NF1) as a breast cancer driver. Genetics. (2012) 192:385–96. doi: 10.1534/genetics.112.142802
301. Sharif S, Moran A, Huson SM, Iddenden R, Shenton A, Howard E, et al. Women with neurofibromatosis 1 are at a moderately increased risk of developing breast cancer and should be considered for early screening. J Med Genet. (2007) 44:481–4. doi: 10.1136/jmg.2007.049346
302. Wang X, Levin AM, Smolinski SE, Vigneau FD, Levin NK, Tainsky MA. Breast cancer and other neoplasms in women with neurofibromatosis type 1: a retrospective review of cases in the Detroit metropolitan area. Am J Med Genet A. (2012) 158A:3061–4. doi: 10.1002/ajmg.a.35560
303. Teschendorff AE, Journée M, Absil PA, Sepulchre R, Caldas C. Elucidating the altered transcriptional programs in breast cancer using independent component analysis. PLoS Comput Biol. (2007) 3:e161. doi: 10.1371/journal.pcbi.0030161
304. Ogata H, Sato H, Takatsuka J, De Luca LM. Human breast cancer MDA-MB-231 cells fail to express the neurofibromin protein, lack its type I mRNA isoform and show accumulation of P-MAPK and activated Ras. Cancer Lett. (2001) 172:159–64. doi: 10.1016/S0304-3835(01)00648-6
305. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. (2012) 486:400–4. doi: 10.1038/nature11017
306. Mendes-Pereira AM, Sims D, Dexter T, Fenwick K, Assiotis I, Kozarewa I, et al. Genome-wide functional screen identifies a compendium of genes affecting sensitivity to tamoxifen. Proc Natl Acad Sci USA. (2012) 109:2730–5. doi: 10.1073/pnas.1018872108
307. Sokol ES, Feng YX, Jin DX, Basudan A, Lee AV, Atkinson JM, et al. Loss of function of NF1 is a mechanism of acquired resistance to endocrine therapy in lobular breast cancer. Ann Oncol. (2019) 30:115–23. doi: 10.1093/annonc/mdy497
308. Verbeek BS, Vroom TM, Adriaansen-Slot SS, Ottenhoff-Kalff AE, Geertzema JG, Hennipman A, et al. c-Src protein expression is increased in human breast cancer. An immunohistochemical and biochemical analysis. J Pathol. (1996) 180:383–8. doi: 10.1002/(SICI)1096-9896(199612)180:4<383::AID-PATH686>3.0.CO;2-N
309. Browne AL, Charmsaz S, Varešlija D, Fagan A, Cosgrove N, Cocchiglia S, et al. Network analysis of SRC-1 reveals a novel transcription factor hub which regulates endocrine resistant breast cancer. Oncogene. (2018) 37:2008–21. doi: 10.1038/s41388-017-0042-x
310. Stashi E, York B, O'Malley BW. Steroid receptor coactivators: servants and masters for control of systems metabolism. Trends Endocrinol Metab. (2014) 25:337–47. doi: 10.1016/j.tem.2014.05.004
311. McIlroy M, McCartan D, Early S, O Gaora P, Pennington S, Hill AD, et al. Interaction of developmental transcription factor HOXC11 with steroid receptor coactivator SRC-1 mediates resistance to endocrine therapy in breast cancer [corrected]. Cancer Res. (2010) 70:1585–94. doi: 10.1158/0008-5472.CAN-09-3713
312. Fleming FJ, Myers E, Kelly G, Crotty TB, McDermott EW, O'Higgins NJ, et al. Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; a predictive role for SRC-1. J Clin Pathol. (2004) 57:1069–74. doi: 10.1136/jcp.2004.016733
313. Hiscox S, Morgan L, Green TP, Barrow D, Gee J, Nicholson RI. Elevated Src activity promotes cellular invasion and motility in tamoxifen resistant breast cancer cells. Breast Cancer Res Treat. (2006) 97:263–74. doi: 10.1007/s10549-005-9120-9
314. Mayer EL, Baurain JF, Sparano J, Strauss L, Campone M, Fumoleau P, et al. A phase 2 trial of dasatinib in patients with advanced HER2-positive and/or hormone receptor-positive breast cancer. Clin Cancer Res. (2011) 17:6897–904. doi: 10.1158/1078-0432.CCR-11-0070
315. Finn RS. Targeting Src in breast cancer. Ann Oncol. (2008) 19:1379–86. doi: 10.1093/annonc/mdn291
316. Morgan L, Gee J, Pumford S, Farrow L, Finlay P, Robertson J, et al. Elevated Src kinase activity attenuates Tamoxifen response in vitro and is associated with poor prognosis clinically. Cancer Biol Ther. (2009) 8:1550–8. doi: 10.4161/cbt.8.16.8954
317. Frame MC. Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta. (2002) 1602:114–30. doi: 10.1016/S0304-419X(02)00040-9
318. Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. (2009) 6:587–95. doi: 10.1038/nrclinonc.2009.129
319. Le Romancer M, Treilleux I, Leconte N, Robin-Lespinasse Y, Sentis S, Bouchekioua-Bouzaghou K, et al. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol Cell. (2008) 31:212–21. doi: 10.1016/j.molcel.2008.05.025
320. Manavathi B, Acconcia F, Rayala SK, Kumar R. An inherent role of microtubule network in the action of nuclear receptor. Proc Natl Acad Sci USA. (2006) 103:15981–6. doi: 10.1073/pnas.0607445103
321. Vallabhaneni S, Nair BC, Cortez V, Challa R, Chakravarty D, Tekmal RR, et al. Significance of ER-Src axis in hormonal therapy resistance. Breast Cancer Res Treat. (2011) 130:377–85. doi: 10.1007/s10549-010-1312-2
322. Song RX, Zhang Z, Santen RJ. Estrogen rapid action via protein complex formation involving ERalpha and Src. Trends Endocrinol Metab. (2005) 16:347–53. doi: 10.1016/j.tem.2005.06.010
323. Cheskis BJ, Greger J, Cooch N, McNally C, McLarney S, Lam HS, et al. MNAR plays an important role in ERa activation of Src/MAPK and PI3K/Akt signaling pathways. Steroids. (2008) 73:901–5. doi: 10.1016/j.steroids.2007.12.028
324. Tian M, Schiemann WP. TGF-β stimulation of EMT programs elicits non-genomic ER-α activity and anti-estrogen resistance in breast cancer cells. J Cancer Metastasis Treat. (2017) 3:150–60. doi: 10.20517/2394-4722.2017.38
325. Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer. (2002) 2:101–12. doi: 10.1038/nrc721
326. Castoria G, Giovannelli P, Lombardi M, De Rosa C, Giraldi T, de Falco A, et al. Tyrosine phosphorylation of estradiol receptor by Src regulates its hormone-dependent nuclear export and cell cycle progression in breast cancer cells. Oncogene. (2012) 31:4868–77. doi: 10.1038/onc.2011.642
327. Fan P, Wang J, Santen RJ, Yue W. Long-term treatment with tamoxifen facilitates translocation of estrogen receptor alpha out of the nucleus and enhances its interaction with EGFR in MCF-7 breast cancer cells. Cancer Res. (2007) 67:1352–60. doi: 10.1158/0008-5472.CAN-06-1020
328. Furth PA. STAT signaling in different breast cancer sub-types. Mol Cell Endocrinol. (2014) 382:612–5. doi: 10.1016/j.mce.2013.03.023
329. Rani A, Murphy JJ. STAT5 in cancer and immunity. J Interferon Cytokine Res. (2016) 36:226–37. doi: 10.1089/jir.2015.0054
330. Wingelhofer B, Neubauer HA, Valent P, Han X, Constantinescu SN, Gunning PT, et al. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia. (2018) 32:1713–26. doi: 10.1038/s41375-018-0117-x
331. Silva CM, Shupnik MA. Integration of steroid and growth factor pathways in breast cancer: focus on signal transducers and activators of transcription and their potential role in resistance. Mol Endocrinol. (2007) 21:1499–512. doi: 10.1210/me.2007-0109
332. Litterst CM, Kliem S, Marilley D, Pfitzner E. NCoA-1/SRC-1 is an essential coactivator of STAT5 that binds to the FDL motif in the alpha-helical region of the STAT5 transactivation domain. J Biol Chem. (2003) 278:45340–51. doi: 10.1074/jbc.M303644200
333. Litterst CM, Pfitzner E. Transcriptional activation by STAT6 requires the direct interaction with NCoA-1. J Biol Chem. (2001) 276:45713–21. doi: 10.1074/jbc.M108132200
334. Clevenger CV. Roles and regulation of stat family transcription factors in human breast cancer. Am J Pathol. (2004) 165:1449–60. doi: 10.1016/S0002-9440(10)63403-7
335. Yeh YT, Ou-Yang F, Chen IF, Yang SF, Wang YY, Chuang HY, et al. STAT3 ser727 phosphorylation and its association with negative estrogen receptor status in breast infiltrating ductal carcinoma. Int J Cancer. (2006) 118:2943–7. doi: 10.1002/ijc.21771
336. Yamashita H, Nishio M, Ando Y, Zhang Z, Hamaguchi M, Mita K, et al. Stat5 expression predicts response to endocrine therapy and improves survival in estrogen receptor-positive breast cancer. Endocr Relat Cancer. (2006) 13:885–93. doi: 10.1677/erc.1.01095
337. Huang R, Faratian D, Sims AH, Wilson D, Thomas JS, Harrison DJ, et al. Increased STAT1 signaling in endocrine-resistant breast cancer. PLoS ONE. (2014) 9:e94226. doi: 10.1371/journal.pone.0094226
338. Zhou Y, Eppenberger-Castori S, Eppenberger U, Benz CC. The NFkappaB pathway and endocrine-resistant breast cancer. Endocr Relat Cancer. (2005) 12 Suppl. 1:S37–46. doi: 10.1677/erc.1.00977
339. Murohashi M, Hinohara K, Kuroda M, Isagawa T, Tsuji S, Kobayashi S, et al. Gene set enrichment analysis provides insight into novel signalling pathways in breast cancer stem cells. Br J Cancer. (2010) 102:206–12. doi: 10.1038/sj.bjc.6605468
340. Helbig G, Christopherson KW, Bhat-Nakshatri P, Kumar S, Kishimoto H, Miller KD, et al. NF-kappaB promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J Biol Chem. (2003) 278:21631–8. doi: 10.1074/jbc.M300609200
341. Cogswell PC, Guttridge DC, Funkhouser WK, Baldwin AS Jr. Selective activation of NF-kappa B subunits in human breast cancer: potential roles for NF-kappa B2/p52 and for Bcl-3. Oncogene. (2000) 19:1123–31. doi: 10.1038/sj.onc.1203412
342. Yde CW, Emdal KB, Guerra B, Lykkesfeldt AE. NFκB signaling is important for growth of antiestrogen resistant breast cancer cells. Breast Cancer Res Treat. (2012) 135:67–78. doi: 10.1007/s10549-012-2053-1
343. Nehra R, Riggins RB, Shajahan AN, Zwart A, Crawford AC, Clarke R. BCL2 and CASP8 regulation by NF-kappaB differentially affect mitochondrial function and cell fate in antiestrogen-sensitive and -resistant breast cancer cells. FASEB J. (2010) 24:2040–55. doi: 10.1096/fj.09-138305
344. Franco HL, Nagari A, Kraus WL. TNFα signaling exposes latent estrogen receptor binding sites to alter the breast cancer cell transcriptome. Mol Cell. (2015) 58:21–34. doi: 10.1016/j.molcel.2015.02.001
345. Pratt MA, Bishop TE, White D, Yasvinski G, Menard M, Niu MY, et al. Estrogen withdrawal-induced NF-kappaB activity and bcl-3 expression in breast cancer cells: roles in growth and hormone independence. Mol Cell Biol. (2003) 23:6887–900. doi: 10.1128/MCB.23.19.6887-6900.2003
346. Gionet N, Jansson D, Mader S, Pratt MA. NF-kappaB and estrogen receptor alpha interactions: differential function in estrogen receptor-negative and -positive hormone-independent breast cancer cells. J Cell Biochem. (2009) 107:448–59. doi: 10.1002/jcb.22141
347. Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. (2009) 139:693–706. doi: 10.1016/j.cell.2009.10.014
348. Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. (2010) 120:485–97. doi: 10.1172/JCI39397
349. Yamaguchi N, Nakayama Y. Down-regulation of Forkhead box protein A1 (FOXA1) leads to cancer stem cell-like properties in tamoxifen-resistant breast cancer cells through induction of interleukin-6. J Biol Chem. (2017) 292:8136–48. doi: 10.1074/jbc.M116.763276
350. Lu L, Yuan X, Zhang Q, Zhang H, Shen B. LMTK3 knockdown retards cell growth and invasion and promotes apoptosis in thyroid cancer. Mol Med Rep. (2017) 15:2015–22. doi: 10.3892/mmr.2017.6262
351. Xu H, Sun H, Zhang H, Liu J, Fan F, Li Y, et al. An ShRNA based genetic screen identified Sesn2 as a potential tumor suppressor in lung cancer via suppression of Akt-mTOR-p70S6K signaling. PLoS ONE. (2015) 10:e0124033. doi: 10.1371/journal.pone.0124033
352. Giamas G, Filipović A, Jacob J, Messier W, Zhang H, Yang D, et al. Kinome screening for regulators of the estrogen receptor identifies LMTK3 as a new therapeutic target in breast cancer. Nat Med. (2011) 17:715–9. doi: 10.1038/nm.2351
353. Stebbing J, Filipovic A, Lit LC, Blighe K, Grothey A, Xu Y, et al. LMTK3 is implicated in endocrine resistance via multiple signaling pathways. Oncogene. (2013) 32:3371–80. doi: 10.1038/onc.2012.343
354. Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. (2016) 352:175–80. doi: 10.1126/science.aaf4405
355. D'Ignazio L, Batie M, Rocha S. Hypoxia and Inflammation in cancer, focus on HIF and NF-κB. Biomedicines. (2017) 5:21. doi: 10.3390/biomedicines5020021
356. Yang J, AlTahan A, Jones DT, Buffa FM, Bridges E, Interiano RB, et al. Estrogen receptor-α directly regulates the hypoxia-inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc Natl Acad Sci USA. (2015) 112:15172–7. doi: 10.1073/pnas.1422015112
357. Kawazu M, Saso K, Tong KI, McQuire T, Goto K, Son DO, et al. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS ONE. (2011) 6:e17830. doi: 10.1371/journal.pone.0017830
358. Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci USA. (2012) 109:E3367–76. doi: 10.1073/pnas.1217394109
359. Masoud GN, Li W. HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B. (2015) 5:378–89. doi: 10.1016/j.apsb.2015.05.007
360. Jia X, Hong Q, Lei L, Li D, Li J, Mo M, et al. Basal and therapy-driven hypoxia-inducible factor-1α confers resistance to endocrine therapy in estrogen receptor-positive breast cancer. Oncotarget. (2015) 6:8648–62. doi: 10.18632/oncotarget.3257
361. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. (2003) 3:721–32. doi: 10.1038/nrc1187
362. Generali D, Buffa FM, Berruti A, Brizzi MP, Campo L, Bonardi S, et al. Phosphorylated ERalpha, HIF-1alpha, and MAPK signaling as predictors of primary endocrine treatment response and resistance in patients with breast cancer. J Clin Oncol. (2009) 27:227–34. doi: 10.1200/JCO.2007.13.7083
363. de Beça FF, Caetano P, Gerhard R, Alvarenga CA, Gomes M, Paredes J, et al. Cancer stem cells markers CD44, CD24 and ALDH1 in breast cancer special histological types. J Clin Pathol. (2013) 66:187–91. doi: 10.1136/jclinpath-2012-201169
364. Fu X, Jeselsohn R, Pereira R, Hollingsworth EF, Creighton CJ, Li F, et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc Natl Acad Sci USA. (2016) 113:E6600–E9. doi: 10.1073/pnas.1612835113
365. Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. (2005) 122:33–43. doi: 10.1016/j.cell.2005.05.008
366. Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science. (2017) 355:1324–30. doi: 10.1126/science.aah6893
367. Jeselsohn R, Cornwell M, Pun M, Buchwalter G, Nguyen M, Bango C, et al. Embryonic transcription factor SOX9 drives breast cancer endocrine resistance. Proc Natl Acad Sci USA. (2017) 114:E4482–E91. doi: 10.1073/pnas.1620993114
368. Hong D, Fritz AJ, Finstad KH, Fitzgerald MP, Weinheimer A, Viens AL, et al. Suppression of breast cancer stem cells and tumor growth by the RUNX1 transcription factor. Mol Cancer Res. (2018) 16:1952–64. doi: 10.1158/1541-7786.MCR-18-0135
369. Browne G, Taipaleenmäki H, Bishop NM, Madasu SC, Shaw LM, van Wijnen AJ, et al. Runx1 is associated with breast cancer progression in MMTV-PyMT transgenic mice and its depletion in vitro inhibits migration and invasion. J Cell Physiol. (2015) 230:2522–32. doi: 10.1002/jcp.24989
370. Xue Y, Lai L, Lian W, Tu X, Zhou J, Dong P, et al. SOX9/FXYD3/Src axis is critical for ER+ breast cancer stem cell function. Mol Cancer Res. (2018) 17:238–49. doi: 10.1158/1541-7786.MCR-18-0610
371. Yun J, Pannuti A, Espinoza I, Zhu H, Hicks C, Zhu X, et al. Crosstalk between PKCalpha and Notch-4 in endocrine-resistant breast cancer cells. Oncogenesis. (2013) 2:e60. doi: 10.1038/oncsis.2013.26
372. Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Role of notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. (2004) 6:R605–15. doi: 10.1186/bcr920
373. Stylianou S, Clarke RB, Brennan K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. (2006) 66:1517–25. doi: 10.1158/0008-5472.CAN-05-3054
374. D'Angelo RC, Ouzounova M, Davis A, Choi D, Tchuenkam SM, Kim G, et al. Notch reporter activity in breast cancer cell lines identifies a subset of cells with stem cell activity. Mol Cancer Ther. (2015) 14:779–87. doi: 10.1158/1535-7163.MCT-14-0228
375. Simmons MJ, Serra R, Hermance N, Kelliher MA. NOTCH1 inhibition in vivo results in mammary tumor regression and reduced mammary tumorsphere-forming activity in vitro. Breast Cancer Res. (2012) 14:R126. doi: 10.1186/bcr3321
376. Magnani L, Stoeck A, Zhang X, Lanczky A, Mirabella AC, Wang TL, et al. Genome-wide reprogramming of the chromatin landscape underlies endocrine therapy resistance in breast cancer. Proc Natl Acad Sci USA. (2013) 110:E1490–9. doi: 10.1073/pnas.1219992110
377. Rizzo P, Miao H, D'Souza G, Osipo C, Song LL, Yun J, et al. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. (2008) 68:5226–35. doi: 10.1158/0008-5472.CAN-07-5744
378. Lombardo Y, Faronato M, Filipovic A, Vircillo V, Magnani L, Coombes RC. Nicastrin and Notch4 drive endocrine therapy resistance and epithelial-mesenchymal transition in MCF7 breast cancer cells. Breast Cancer Res. (2014) 16:R62. doi: 10.1186/bcr3675
379. Corominas-Faja B, Cufí S, Oliveras-Ferraros C, Cuyàs E, López-Bonet E, Lupu R, et al. Nuclear reprogramming of luminal-like breast cancer cells generates Sox2-overexpressing cancer stem-like cellular states harboring transcriptional activation of the mTOR pathway. Cell Cycle. (2013) 12:3109–24. doi: 10.4161/cc.26173
380. Leis O, Eguiara A, Lopez-Arribillaga E, Alberdi MJ, Hernandez-Garcia S, Elorriaga K, et al. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. (2012) 31:1354–65. doi: 10.1038/onc.2011.338
381. Rani A, Greenlaw R, Smith RA, Galustian C. HES1 in immunity and cancer. Cytokine Growth Factor Rev. (2016) 30:113–7. doi: 10.1016/j.cytogfr.2016.03.010
382. Jang GB, Kim JY, Cho SD, Park KS, Jung JY, Lee HY, et al. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci Rep. (2015) 5:12465. doi: 10.1038/srep12465
383. Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. (2011) 12:247–54. doi: 10.1038/ni.1995
384. Yu H, Jove R. The STATs of cancer–new molecular targets come of age. Nat Rev Cancer. (2004) 4:97–105. doi: 10.1038/nrc1275
385. Bui QT, Im JH, Jeong SB, Kim YM, Lim SC, Kim B, et al. Essential role of Notch4/STAT3 signaling in epithelial-mesenchymal transition of tamoxifen-resistant human breast cancer. Cancer Lett. (2017) 390:115–25. doi: 10.1016/j.canlet.2017.01.014
386. Ng JM, Curran T. The Hedgehog's tale: developing strategies for targeting cancer. Nat Rev Cancer. (2011) 11:493–501. doi: 10.1038/nrc3079
387. Clark AS, West K, Streicher S, Dennis PA. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol Cancer Ther. (2002) 1:707–17.
388. Ramaswamy B, Lu Y, Teng KY, Nuovo G, Li X, Shapiro CL, et al. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res. (2012) 72:5048–59. doi: 10.1158/0008-5472.CAN-12-1248
389. Lu Y, Li J, Cheng J, Lubahn DB. Genes targeted by the Hedgehog-signaling pathway can be regulated by estrogen related receptor beta. BMC Mol Biol. (2015) 16:19. doi: 10.1186/s12867-015-0047-3
390. Vesuna F, Bergman Y, Raman V. Genomic pathways modulated by Twist in breast cancer. BMC Cancer. (2017) 17:52. doi: 10.1186/s12885-016-3033-3
391. Vesuna F, van Diest P, Chen JH, Raman V. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem Biophys Res Commun. (2008) 367:235–41. doi: 10.1016/j.bbrc.2007.11.151
392. Dhasarathy A, Kajita M, Wade PA. The transcription factor snail mediates epithelial to mesenchymal transitions by repression of estrogen receptor-alpha. Mol Endocrinol. (2007) 21:2907–18. doi: 10.1210/me.2007-0293
393. Dhasarathy A, Phadke D, Mav D, Shah RR, Wade PA. The transcription factors Snail and Slug activate the transforming growth factor-beta signaling pathway in breast cancer. PLoS ONE. (2011) 6:e26514. doi: 10.1371/journal.pone.0026514
394. Ohshiro K, Schwartz AM, Levine PH, Kumar R. Alternate estrogen receptors promote invasion of inflammatory breast cancer cells via non-genomic signaling. PLoS ONE. (2012) 7:e30725. doi: 10.1371/journal.pone.0030725
395. Vegvari A, Shavkunov AS, Fehniger TE, Grabau D, Nimeus E, Marko-Varga G. Localization of tamoxifen in human breast cancer tumors by MALDI mass spectrometry imaging. Clin Transl Med. (2016) 5:10. doi: 10.1186/s40169-016-0090-9
396. Gerber M, Astre C, Ségala C, Saintot M, Scali J, Simony-Lafontaine J, et al. Tumor progression and oxidant-antioxidant status. Cancer Lett. (1997) 114:211–4.
397. Poulsen HE, Prieme H, Loft S. Role of oxidative DNA damage in cancer initiation and promotion. Eur J Cancer. Prev. (1998) 7:9–16.
398. Guo C, Li X, Ye M, Xu F, Yu J, Xie C, et al. Discriminating patients with early-stage breast cancer from benign lesions by detection of oxidative DNA damage biomarker in urine. Oncotarget. (2017) 8:53100–9. doi: 10.18632/oncotarget.17831
399. Sova H, Jukkola-Vuorinen A, Puistola U, Kauppila S, Karihtala P. 8-Hydroxydeoxyguanosine: a new potential independent prognostic factor in breast cancer. Br J Cancer. (2010) 102:1018–23. doi: 10.1038/sj.bjc.6605565
400. Yeh CC, Hou MF, Wu SH, Tsai SM, Lin SK, Hou LA, et al. A study of glutathione status in the blood and tissues of patients with breast cancer. Cell Biochem Funct. (2006) 24:555–9. doi: 10.1002/cbf.1275
401. Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev. (2013) 2013:972913. doi: 10.1155/2013/972913
402. Fan P, Griffith OL, Agboke FA, Anur P, Zou X, McDaniel RE, et al. c-Src modulates estrogen-induced stress and apoptosis in estrogen-deprived breast cancer cells. Cancer Res. (2013) 73:4510–20. doi: 10.1158/0008-5472.CAN-12-4152
403. Zou X, Santa-Maria CA, O'Brien J, Gius D, Zhu Y. Manganese superoxide dismutase acetylation and dysregulation, due to loss of SIRT3 activity, promote a luminal B-like breast carcinogenic-permissive phenotype. Antioxid Redox Signal. (2016) 25:326–36. doi: 10.1089/ars.2016.6641
404. Bekele RT, Venkatraman G, Liu RZ, Tang X, Mi S, Benesch MG, et al. Oxidative stress contributes to the tamoxifen-induced killing of breast cancer cells: implications for tamoxifen therapy and resistance. Sci Rep. (2016) 6:21164. doi: 10.1038/srep21164
405. Jordan VC, Collins MM, Rowsby L, Prestwich G. A monohydroxylated metabolite of tamoxifen with potent antioestrogenic activity. J Endocrinol. (1977) 75:305–16. doi: 10.1677/joe.0.0750305
406. Stearns V, Johnson MD, Rae JM, Morocho A, Novielli A, Bhargava P, et al. Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J Natl Cancer Inst. (2003) 95:1758–64. doi: 10.1093/jnci/djg108
407. Nowell S, Sweeney C, Winters M, Stone A, Lang NP, Hutchins LF, et al. Association between sulfotransferase 1A1 genotype and survival of breast cancer patients receiving tamoxifen therapy. J Natl Cancer Inst. (2002) 94:1635–40. doi: 10.1093/jnci/94.21.1635
408. Jin Y, Desta Z, Stearns V, Ward B, Ho H, Lee KH, et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J Natl Cancer Inst. (2005) 97:30–9. doi: 10.1093/jnci/dji005
409. Sacco K, Grech G. Actionable pharmacogenetic markers for prediction and prognosis in breast cancer. EPMA J. (2015) 6:15. doi: 10.1186/s13167-015-0037-z
410. Wegman P, Vainikka L, Stål O, Nordenskjöld B, Skoog L, Rutqvist LE, et al. Genotype of metabolic enzymes and the benefit of tamoxifen in postmenopausal breast cancer patients. Breast Cancer Res. (2005) 7:R284–90. doi: 10.1186/bcr993
411. Lim HS, Ju Lee H, Seok Lee K, Sook Lee E, Jang IJ, Ro J. Clinical implications of CYP2D6 genotypes predictive of tamoxifen pharmacokinetics in metastatic breast cancer. J Clin Oncol. (2007) 25:3837–45. doi: 10.1200/JCO.2007.11.4850
412. Lum DW, Perel P, Hingorani AD, Holmes MV. CYP2D6 genotype and tamoxifen response for breast cancer: a systematic review and meta-analysis. PLoS ONE. (2013) 8:e76648. doi: 10.1371/journal.pone.0076648
413. Thuy Phuong NT, Kim JW, Kim JA, Jeon JS, Lee JY, Xu WJ, et al. Role of the CYP3A4-mediated 11,12-epoxyeicosatrienoic acid pathway in the development of tamoxifen-resistant breast cancer. Oncotarget. (2017) 8:71054–69. doi: 10.18632/oncotarget.20329
414. Goetz MP, Suman VJ, Reid JM, Northfelt DW, Mahr MA, Ralya AT, et al. First-in-human phase I study of the tamoxifen metabolite Z-endoxifen in women with endocrine-refractory metastatic breast cancer. J Clin Oncol. (2017) 35:3391–400. doi: 10.1200/JCO.2017.73.3246
415. Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. (1996) 56: 4625–9.
416. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). (2014) 6:1670–90. doi: 10.3390/cancers6031670
417. Baumgarten SC, Frasor J. Minireview: Inflammation: an instigator of more aggressive estrogen receptor (ER) positive breast cancers. Mol Endocrinol. (2012) 26:360–71. doi: 10.1210/me.2011-1302
418. Sas L, Lardon F, Vermeulen PB, Hauspy J, Van Dam P, Pauwels P, et al. The interaction between ER and NFκB in resistance to endocrine therapy. Breast Cancer Res. (2012) 14:212. doi: 10.1186/bcr3196
419. deGraffenried LA, Chandrasekar B, Friedrichs WE, Donzis E, Silva J, Hidalgo M, et al. NF-kappa B inhibition markedly enhances sensitivity of resistant breast cancer tumor cells to tamoxifen. Ann Oncol. (2004) 15:885–90. doi: 10.1093/annonc/mdh232
420. Zhu P, Baek SH, Bourk EM, Ohgi KA, Garcia-Bassets I, Sanjo H, et al. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell. (2006) 124:615–29. doi: 10.1016/j.cell.2005.12.032
421. Stender JD, Nwachukwu JC, Kastrati I, Kim Y, Strid T, Yakir M, et al. Structural and molecular mechanisms of cytokine-mediated endocrine resistance in human breast cancer cells. Mol Cell. (2017) 65:1122–35.e5. doi: 10.1016/j.molcel.2017.02.008
422. Rugo HS, Delord JP, Im SA, Ott PA, Piha-Paul SA, Bedard PL, et al. Safety and antitumor activity of pembrolizumab in patients with estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer. Clin Cancer Res. (2018) 24:2804–11. doi: 10.1158/1078-0432.CCR-17-3452
423. Plaks V, Boldajipour B, Linnemann JR, Nguyen NH, Kersten K, Wolf Y, et al. Adaptive immune regulation of mammary postnatal organogenesis. Dev Cell. (2015) 34:493–504. doi: 10.1016/j.devcel.2015.07.015
424. Yau C, Esserman L, Moore DH, Waldman F, Sninsky J, Benz CC. A multigene predictor of metastatic outcome in early stage hormone receptor-negative and triple-negative breast cancer. Breast Cancer Res. (2010) 12:R85. doi: 10.1186/bcr2753
425. Desmedt C, Haibe-Kains B, Wirapati P, Buyse M, Larsimont D, Bontempi G, et al. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res. (2008) 14:5158–65. doi: 10.1158/1078-0432.CCR-07-4756
426. Dunbier AK, Ghazoui Z, Anderson H, Salter J, Nerurkar A, Osin P, et al. Molecular profiling of aromatase inhibitor-treated postmenopausal breast tumors identifies immune-related correlates of resistance. Clin Cancer Res. (2013) 19:2775–86. doi: 10.1158/1078-0432.CCR-12-1000
427. Law AMK, Lim E, Ormandy CJ, Gallego-Ortega D. The innate and adaptive infiltrating immune systems as targets for breast cancer immunotherapy. Endocr Relat Cancer. (2017) 24:X1. doi: 10.1530/ERC-16-0404e
428. Domchek SM, Recio A, Mick R, Clark CE, Carpenter EL, Fox KR, et al. Telomerase-specific T-cell immunity in breast cancer: effect of vaccination on tumor immunosurveillance. Cancer Res. (2007) 67:10546–55. doi: 10.1158/0008-5472.CAN-07-2765
429. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. (2012) 12:253–68. doi: 10.1038/nri3175
430. Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. (2005) 65:7065–70. doi: 10.1158/0008-5472.CAN-05-1783
431. Cittelly DM, Das PM, Spoelstra NS, Edgerton SM, Richer JK, Thor AD, et al. Downregulation of miR-342 is associated with tamoxifen resistant breast tumors. Mol Cancer. (2010) 9:317. doi: 10.1186/1476-4598-9-317
432. Miller TE, Ghoshal K, Ramaswamy B, Roy S, Datta J, Shapiro CL, et al. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J Biol Chem. (2008) 283:29897–903. doi: 10.1074/jbc.M804612200
433. Bergamaschi A, Katzenellenbogen BS. Tamoxifen downregulation of miR-451 increases 14-3-3zeta and promotes breast cancer cell survival and endocrine resistance. Oncogene. (2012) 31:39–47. doi: 10.1038/onc.2011.223
434. Wei Y, Lai X, Yu S, Chen S, Ma Y, Zhang Y, et al. Exosomal miR-221/222 enhances tamoxifen resistance in recipient ER-positive breast cancer cells. Breast Cancer Res Treat. (2014) 147:423–31. doi: 10.1007/s10549-014-3037-0
435. Sachdeva M, Wu H, Ru P, Hwang L, Trieu V, Mo YY. MicroRNA-101-mediated Akt activation and estrogen-independent growth. Oncogene. (2011) 30:822–31. doi: 10.1038/onc.2010.463
436. Ward A, Shukla K, Balwierz A, Soons Z, König R, Sahin O, et al. MicroRNA-519a is a novel oncomir conferring tamoxifen resistance by targeting a network of tumour-suppressor genes in ER+ breast cancer. J Pathol. (2014) 233:368–79. doi: 10.1002/path.4363
437. Liu ZR, Song Y, Wan LH, Zhang YY, Zhou LM. Over-expression of miR-451a can enhance the sensitivity of breast cancer cells to tamoxifen by regulating 14-3-3ζ, estrogen receptor α, and autophagy. Life Sci. (2016) 149:104–13. doi: 10.1016/j.lfs.2016.02.059
438. Lü M, Ding K, Zhang G, Yin M, Yao G, Tian H, et al. MicroRNA-320a sensitizes tamoxifen-resistant breast cancer cells to tamoxifen by targeting ARPP-19 and ERRγ. Sci Rep. (2015) 5:8735. doi: 10.1038/srep08735
439. Cui J, Yang Y, Li H, Leng Y, Qian K, Huang Q, et al. MiR-873 regulates ERα transcriptional activity and tamoxifen resistance via targeting CDK3 in breast cancer cells. Oncogene. (2015) 34:4018. doi: 10.1038/onc.2014.430
440. Ward A, Balwierz A, Zhang JD, Küblbeck M, Pawitan Y, Hielscher T, et al. Re-expression of microRNA-375 reverses both tamoxifen resistance and accompanying EMT-like properties in breast cancer. Oncogene. (2013) 32:1173–82. doi: 10.1038/onc.2012.128
441. Roscigno G, Puoti I, Giordano I, Donnarumma E, Russo V, Affinito A, et al. MiR-24 induces chemotherapy resistance and hypoxic advantage in breast cancer. Oncotarget. (2017) 8:19507–21. doi: 10.18632/oncotarget.14470
442. Yáñez-Mó M, Siljander PR, Andreu Z, Zavec AB, Borràs FE, Buzas EI, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. (2015) 4:27066. doi: 10.3402/jev.v4.27066
443. Balaj L, Lessard R, Dai L, Cho YJ, Pomeroy SL, Breakefield XO, et al. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat Commun. (2011) 2:180. doi: 10.1038/ncomms1180
Keywords: endocrine resistance, breast cancer, signaling, estrogen (E2), estrogen receptor
Citation: Rani A, Stebbing J, Giamas G and Murphy J (2019) Endocrine Resistance in Hormone Receptor Positive Breast Cancer–From Mechanism to Therapy. Front. Endocrinol. 10:245. doi: 10.3389/fendo.2019.00245
Received: 26 October 2018; Accepted: 28 March 2019;
Published: 24 May 2019.
Edited by:
Anindita Chakrabarty, Shiv Nadar University, IndiaReviewed by:
Eva Surmacz, Temple University, United StatesAriella Hanker, University of Texas Southwestern Medical Center, United States
Copyright © 2019 Rani, Stebbing, Giamas and Murphy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aradhana Rani, YS5yYW5pQHdlc3RtaW5zdGVyLmFjLnVr