 Sharon Owino1,2
Sharon Owino1,2 Daniella D. C. Buonfiglio
Daniella D. C. Buonfiglio Gianluca Tosini
Gianluca Tosini- 1Department of Pharmacology and Toxicology Morehouse School of Medicine, Neuroscience Institute, Atlanta, GA, United States
- 2Department of Pharmacology, Emory University School of Medicine, Atlanta, GA, United States
- 3Department of Physiology and Biophysics, Institute of Biomedical Sciences-I, University of São Paulo (USP), São Paulo, Brazil
Melatonin, a hormone synthesized by both the pineal gland and retina, functions as an important modulator of a number of physiological functions. In addition to its rather well-established roles in the regulation of circadian rhythms, sleep, and reproduction, melatonin has also been identified as an important regulator of glucose metabolism. Recent genomic studies have also shown that disruption of melatonin receptors signaling may contribute to the pathogenesis of type 2 diabetes, although the exact mechanisms underlying its action remain unclear. Additionally, a large number of animal studies have highlighted a role for melatonin in the regulation of both glucose metabolism and energy balance. This review summarizes the current knowledge on the role that melatonin and its associated receptors play in the regulation of metabolism.
Introduction
Melatonin is a hormone predominantly secreted by the pineal of vertebrates. The synthesis of melatonin occurs during the night via a rather straightforward biosynthetic pathway that involves four different enzymes. In many vertebrates (e.g., fishes, amphibians, reptiles, and birds) pineal melatonin synthesis is directly controlled by the circadian clock located in the pinealocytes, whereas in the mammalian pineal gland the synthesis of melatonin is controlled by a circadian clock located in the suprachiasmatic nucleus (SCN) of the hypothalamus. Due to its lipophilic nature, melatonin is not stored within the pineal gland, but rather diffuses into the blood and cerebral spinal fluid where its levels in these fluids reflects its synthesis (1).
Melatonin acts on many different cell types within the body where it exerts its effects as an antioxidant and free radical scavenger or functions via its G-protein coupled receptors named melatonin receptor 1 (MT1) and melatonin receptor 2 (MT2) (2). MT1 and MT2 are expressed throughout the body where they regulate the entrainment of circadian rhythms, sleep, blood pressure, and reproductive functions. Since these effects of melatonin have been reviewed by other authors, in both this issue of the journal, as well as by recent reviews in other journals, we have decided to focus our review on the role that melatonin plays in the regulation metabolism. Additionally, in the last section of our review, we also summarize how modern genetic studies that have implicated melatonin receptor polymorphisms in the regulation of a number of different pathologies.
Melatonin as a Regulator of Metabolism and Body Weight
Within recent decades, a large number of animal studies using both pinealectomized rats and melatonin receptor knock out (KO) mice have begun to establish a rather unexpected role for melatonin in the regulation of glucose metabolism and energy balance. Early pinealectomy studies demonstrated that abolishing melatonin levels produces glucose intolerance and insulin resistance (3, 4). Interestingly, reintroducing exogenous melatonin into this system restored metabolic parameters to levels observed within control animals. Similarly, in mice fed a high fat diet (HFD), exogenous melatonin administration was sufficient to restore diminished insulin sensitivity and glucose tolerance (5). Consequently, another study demonstrated that daily melatonin administration was sufficient to decrease the bodyweight gain of HFD fed rats by 54% compared to HFD rats not treated with melatonin (6).
These data suggest that melatonin may be functioning, at least in part, to alleviate a number of metabolic consequences associated with diet-induced obesity (DIO). To this end, we have recently examined the effects of DIO on body weight, food intake, and related metabolic parameters within WT and MT1 KO mice fed a HFD. Our data demonstrate that DIO elicits markedly higher cumulative weight gain and hyperglycemia within MT1 KO mice. Collectively, our data highlight that signaling through MT1 may in fact regulate a number of protective metabolic responses during the course of DIO, thus raising the intriguing possibility that MT1 may offer a unique therapeutic target for counteracting metabolic consequences elicited by DIO (Figure 1).
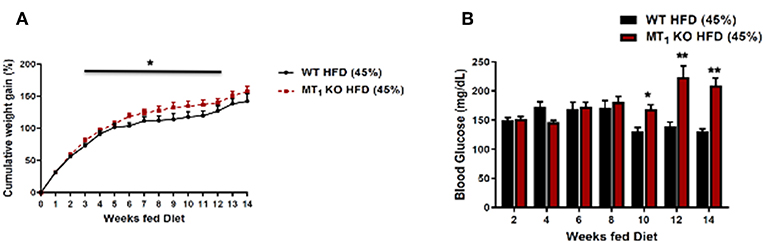
Figure 1. Removal of MT1 alters the metabolic response of C3H mice to DIO. Male mice were fed ad libitum with a HFD (D12451, 45% kcal/fat, Research Diets Inc.) from weaning at 4 weeks of age until 20 weeks of age. (A) MT1 KO mice showed a small, but significant cumulative weight gain with respect to control (B). Fasting glucose levels were significantly higher in MT1 KO mice after 10 weeks of HFD. Data are presented as mean ± SEM (n = 8–10); *p < 0.05, **p < 0.01 WT vs. MT1 KO Two-Way ANOVA (post-hoc: Holm-Sidak).
Previous studies have established a potential role for melatonin in the regulation of body fat by demonstrating that melatonin administration leads to a reduction in body fat content (7).
Moreover, correlations exist between aging and the reduction of pineal melatonin production, increased body weight, visceral fat, and high levels of leptin and insulin (7). In 1985, a seminal study by Bartness and Wade (8) demonstrated that in Siberian hamsters, melatonin treatment had the ability to decrease body weight, carcass lipid content, and food intake without affecting spontaneous locomotor activity. Years later, Rasmussen et al. (9) found that middle-aged rats treated with melatonin began to display decreased visceral fat, leptin, and insulin to levels that were found in young animals. In addition, in a crossover study, rats initially treated with melatonin rapidly began to increase body weight when switched to the control treatment, and rats that started as control animals rapidly decreased their body weight in response to melatonin treatment (10).
The progression of obesity is a complex process underlined by chronic inflammation (11). Interestingly, in a genetic mouse model of obesity, melatonin treatment was capable of ameliorating both inflammatory infiltration and obesity-induced adipokine alteration (12). Melatonin treatment has also been shown to reduce fat mass (and percentage of body fat) with a concomitant increase of lean mass in postmenopausal women following 1 year of treatment compared to placebo (13). Studies now suggest that melatonin treatment may stimulate lipolysis through the activation of the sympathetic nervous system, as well as stimulate intramuscular adipocyte lipolysis via activation of both extracellular signal-regulated kinase (ERK) 1/2 and protein kinase A (PKA) signaling (14, 15). Taken together, these studies begin to demonstrate a potential role for melatonin in modulating body weight, specifically through its regulation of fat mass.
To date, few studies have provided insight as to whether melatonin's regulation of body composition involves the modulation of feeding behavior. A recent study highlighted that MT1 signaling stimulates the transcription Pro-opiomelanocortin (Pomc) mRNA in the hypothalamus and pituitary and the removal of this receptor results in mice spending substantially more time feeding (16). In addition, direct melatonin infusion via intracerebroventricular cannulation has been shown to reduce food intake (17), thus providing further evidence that melatonin might play an important role in modulating the circuitry that regulates feeding behavior.
Interplay Between Melatonin and Leptin
Leptin is an adipose-derived hormone that is released in a circadian manner by adipose tissue. Plasma leptin levels peak late within the dark cycle and subsequently decrease during the light cycle (18, 19). Because of the circadian nature of melatonin's secretion—peaking at night—melatonin has been thought to be a circadian timing cue for leptin secretion. Indeed, melatonin has been shown to drive the daily rhythm of plasma leptin, and when it is absent the leptin rhythm is severely blunted in Syrian hamsters (20). Moreover, Buonfiglio et al. (17) demonstrated that the long-term absence of circulating melatonin leads to impairments in leptin signaling and leptin resistance within the hypothalamus. Obese individuals have high levels of leptin, however due to leptin resistance their regulation of food intake—and consequently body weight regulation—is impaired. Therefore, leptin resistance, which is induced either by pinealectomy or genetic removal of melatonin receptors, increases the expression of a number of orexigenic genes such as Agouti-related protein (Agrp) and Neuropeptide Y (Npy), which are modulated by leptin signaling.
These changes result in increased long-term food intake and weight gain. Interestingly, administration of exogenous melatonin prevented the negative effects induced by pinealectomy and reduced the expression of both Agrp and Orexin, thus leading to reductions in food intake, weight gain, leptin levels, and adipose fat pads (17). Consistent with these findings, Río-Lugos et al. (21) also demonstrated that melatonin treatment was able to decrease high levels of circulating leptin and adiponectin, as well as down-regulate the expression of Npy—a strong orexigenic signal—that was observed in HFD fed rats.
Finally, a recent study reported that MT1 KO mice are leptin resistant compared to controls since the administration of leptin failed to induce signal transducers and activators of transcription 3 (STAT3) phosphorylation in the arcuate nucleus (22). Consistent with these results, leptin receptor mRNA levels in the hypothalamus of MT1 KO were reduced (about 50%) with respect to mRNA levels in controls (22). Thus, the lack of MT1 signaling induces leptin resistance by down-regulation of the leptin receptor.
Melatonin and Energy Expenditure
In addition to the modulatory role of melatonin on energy intake, it seems that melatonin may also play an important role in modulating energy expenditure. In an experimental model of obesity and type 2 diabetes mellitus, using Zücker diabetic fatty rats, melatonin treatment induced browning of inguinal white adipose tissue (WAT) and increased Brown adipose tissue (BAT) weight with thermogenic properties (23, 24). Buonfiglio et al. (17) has demonstrated that pinealectomy decreased the amount of uncoupling protein 1 (UCP1) in BAT, thereby indicating lower thermogenic activity after cold exposure (25). Tan et al. (26) suggested that there is a correlation between light exposure at night and bodyweight gain in humans, and if melatonin recruits BAT in humans—as it does in other species—individuals who have their endogenous melatonin decreased by experiencing long daily photoperiods should have less functional BAT and may gain more bodyweight.
Recent studies suggest that gut microbiota composition is also correlated with metabolic disorders and obesity (27, 28). Surprisingly, it was demonstrated that melatonin treatment following DIO in mice, was capable of modulating gut microbiota back to levels observed in lean mice, and provides beneficial effects against obesity, insulin resistance, liver steatosis, and low-grade inflammation in HFD-fed mice (29). Although there remains much that still needs to be understood regarding the role of melatonin in energy homeostasis, there is significant data in different species that now demonstrates that melatonin has an anti-obesogenic effect and that it is involved in the regulation of all three of the main steps of energy balance: energy intake, energy storage, and energy expenditure.
Melatonin Receptors Involvement in the Regulation of Glucose Metabolism
Collectively, much of the work obtained using mice lacking melatonin receptors has established a generally beneficial role for melatonin on glucose metabolism. Recent studies using these models have demonstrated that in the mouse genetic ablation of MT1 or MT2 affects glucose metabolism. MT1 KO mice display systemic insulin resistance marked by impaired skeletal muscle glucose uptake, adipose tissue glucose uptake, and significantly reduced liver insulin sensitivity (30). This effect appears to involve modulation of phosphatidylinositol-3-kinase (PI3K)-protein kinase B (AKT) activity and fits nicely with previous work demonstrating the ability of melatonin to inhibit hepatic gluconeogenesis (31), and stimulate glucose uptake within both skeletal muscle cells (32) and primary adipocytes (33). MT2 KO mice, which previously lacked a clear metabolic phenotype, were recently reported to display decreased hepatic insulin sensitivity and increased insulin secretion (34). These findings differ quite drastically from a previous report demonstrating that MT2 KO male mice are neither insulin resistant nor glucose intolerant (35) (see also Figure 2). The discrepancy between these findings may lie in the fact that the earlier metabolic characterization of MT2 KO mice utilized male mice (35) whereas the more recent study was performed utilizing female mice (35). Sex differences in insulin sensitivity have been reported for both humans and rodents (36, 37), and as such, the divergence in these findings raises the intriguing possibility that melatonin receptors may potentially be regulated differently between males and females. Additional studies need to be done to confirm that gender is in fact a variable responsible for the observed differences in insulin sensitivity within MT2 KO mice.
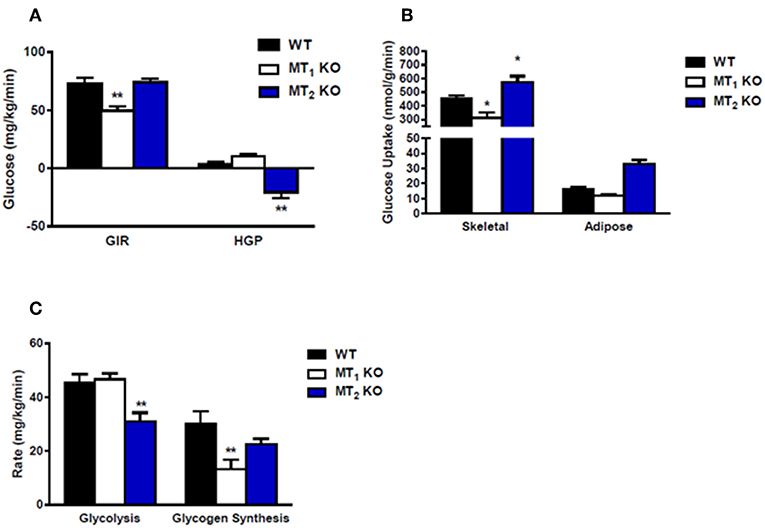
Figure 2. Loss of MT2 does not induce systemic insulin resistance in male mice. (A) Hyperinsulinemic-euglycemic clamp in awake mice. Glucose infusion rate (GIR) was not different between WT and MT2 KO mice, whereas hepatic glucose production (HGP) was significantly lower in MT2 KO mice. However, since negative HGP rates are not physiologically possible, the values observed in MT2 KO mice likely arise from a slight underestimation of glucose disposal rates. (B) Insulin-stimulated glucose uptake in skeletal muscle (gastrocnemius) and white adipose tissue (epididymal) is higher in MT2 KO with respect to WT and MT1 KO. (C) Whole body glycolysis and glycogen synthesis. Glycolysis was lower in MT2 KO mice while glycogen synthesis was not different between MT2 KO and WT. Taken together these data suggest that while male MT1KO mice show systemic insulin resistance with respect to WT, MT2 KO do not exhibit insulin resistance. Results are expressed as mean ± SEM (n = 5–7 WT, MT1 and n = 12 MT2; Two-way ANOVA *P < 0.05; **P < 0.01, post-hoc: Holm-Sidak). Hyperinsulinemic-euglycemic clamp was performed as described in Owino et al. (30).
Thus far, efforts in understanding the role of melatonin on glucose metabolism have been focused heavily on pancreatic islets (38). Although valuable, this focus somewhat overshadows the effects of melatonin on other tissues (skeletal muscle, adipose tissue, liver), as well as its effects on the brain, where both melatonin receptor subtypes are highly expressed within the SCN (2). In the SCN, MT1, and MT2 function intricately together to entrain and synchronize the rhythm of the master circadian clock (39–41). It is now recognized that disruption of the circadian clock is linked to a number of metabolic disorders (42), and as such, the contribution of melatonin to clock entrainment and its potential implications on metabolism should not be overlooked in the design of future studies. The disruption of independent peripheral tissue clocks within insulin sensitive tissues has also been linked to a number of metabolic disorders (43–46). However, to date, current studies do not lend strong support to the notion that melatonin directly regulates metabolic parameters through its regulation of peripheral tissue clocks. Examination of the effect of melatonin receptor removal on clock genes within the pancreas, adipose tissue, and liver demonstrate only marginal effects on clock gene expression, amplitude and phase (47, 48).
Since the majority of the studies discussed above are performed in rodent models, another important point that must be considered in the interpretation of these results is that rodents are nocturnal and undergo a peak in melatonin synthesis during their active phase, whereas humans are diurnal and experience peak melatonin levels during their inactive “sleep phase.” This difference suggests there may exist different functional requirements for both direct and indirect effects of melatonin between rodents and humans. Interestingly, a number of studies now highlight the importance of “timing” and the “delayed effects” of melatonin in interpreting results from melatonin signaling studies. In the case of insulin signaling, this was eloquently shown in a recent study highlighting the ability of nocturnal activation of MT1 to modulate insulin sensitivity during the day (30). Moving forward in attempts to reconcile data collected from human and rodent models, it will be important to keep in mind potential differences that may exist pertaining to both the timing and functional constraints of melatonin in each species.
It is worth noting that a series of recent studies have reported that melatonin synthesis occurs in the mitochondria of neurons where MT1 receptors are also present (49). Since mitochondria are key regulators of cellular metabolism, it would be interesting to see how dysfunction of melatonin synthesis and signaling within the mitochondria may contribute to the regulation of energy metabolism and expenditure.
Role of Melatonin Receptors Variants in Human Metabolic Disorders
Over the years, genetic variants in melatonin receptors have been associated with a number of metabolic disorders such as type 2 diabetes (T2D), gestational diabetes mellitus (GDM) and obesity (50–55). Recently, there has been considerable excitement directed toward the role of melatonin in the regulation of T2D. Much of this interest has come from a series of recent genomic-wide association studies (GWAS) linking MTNR1B, the genetic locus encoding MT2, to increased fasting blood glucose levels and T2D risk (56, 57). An early study examined the association of six synonymous MTNR1B variants (G24E, L60R, V124I, R138C, R231H, and K243R) with obesity and T2D in a Danish and French population (57). None those variants were associated with T2D; however, G42E (rs8192552) was associated with decreased fasting plasma glucose. Furthermore, rs8192552 was also found to be associated with an increased prevalence of obesity, increased body mass index (BMI), and waist circumference.
In total, forty synonymous MTNR1B variants were later found to be associated with T2D. From these forty variants, four complete loss of function variants (A24P, L60R, P95L, and Y308S) – which lost the ability to bind melatonin and activate downstream Gi dependent signaling—were uncovered (58). In 2018, the same group under the lead of Karamitri reported their assessment of all forty variants on multiple pathways thought to be regulated by MTNR1B activation (58). They found that in addition to the four loss of function variants (A24P, L60R, P95L, and Y308S), four additional variants (S123R, R138C/H/L, F250V, and R316H) remarkably impaired cAMP responses (58).
A study from the DIAGRAM (DIAbetes Genetics Replication and Meta-analysis) consortium further aimed to identify a credible set of variants that overlap with FOXA2 binding sites as a causal mechanism for T2D susceptibility. Interestingly, MTNR1B variant rs10830963 was implicated as driving T2D association (59). A subsequent group did not manage to identify any association of rs10830963 and rs1387153 with T2D and fasting blood glucose (FBG) in their Indian population (60). However, when their data was subsequently stratified according to BMI, rs1387153 showed a strong association with low FBG levels in low BMI groups. Similar results were reported by Gan et al. in a Chinese population where rs10830963 didn't show an association with T2D (61). Although not associated with T2D, rs10830963 was found to be associated with GDM in a European cohort (62).
Collectively, genetic studies highlighting the association of MTNR1B to T2D risk have reconciled on the ability of two frequent variants (SNPs rs1387153 and rs10830963) to modulate insulin secretion from pancreatic beta cells (55, 57, 62–65). Interestingly, carriers of the rs10830963 risk allele express substantially higher levels of MTNR1B mRNA within their pancreatic islets when compared to non-carriers (57). This observation has fueled the general notion that increased melatonin signaling—as a consequence of increased receptor expression levels—likely inhibits pancreatic insulin secretion and increases T2D risk. These results suggest an overall negative effect of melatonin on diabetes progression; however, it is important to note that increased MTNR1B expression at the mRNA level may not correlate to increased receptor protein levels and/or receptor signaling within these islets. Moreover, these findings appear to be in conflict with a recent clinical study demonstrating that decreased nocturnal melatonin levels are in fact associated with increased, not decreased, risk for diabetes (66).
Although MTNR1A variants have not been directly associated with T2D, a few studies have reported an association of MTNR1A variants with polycystic ovary syndrome (PCOS). Patients affected by PCOS often develop insulin resistance and T2D. Finally, it is worth mentioning that a reduction in MTNR1A mRNAs has been observed in the liver of T2D patients that were unable to control glucose levels (30). Thus, experimental evidence also supports a possible role for MT1 in T2D (30, 53, 67).
Conclusions and Future Perspective
The experimental evidence accumulated thus far indicates that melatonin plays an important role in the regulation of glucose and metabolism are quite solid with a substantial amount of work conducted in human studies. Nonetheless, there continues to be a need for more conclusive studies to fully elucidate the exact role which melatonin and its associated receptors contribute to the regulation of different metabolic processes within the body.
Author Contributions
SO, DB, CT, and GT wrote the paper. SO drew the figures.
Funding
This work was supported by grants from São Paulo Research Foundation (FAPESP) 2017/01882-8 to DB; National Institutes of Health (NIH) grant T-32 HL103104 to GT; and by 5U54NS083932, S21MD000101, G12-RR03034, U54RR026137 to Morehouse School of Medicine.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Tricoire H, Locatelli A, Chemineau P, Malpaux B. Melatonin enters the cerebrospinal fluid through the pineal recess. Endocrinology. (2002) 143:84–90. doi: 10.1210/endo.143.1.8585
2. Jockers R, Delagrange P, Dubocovich ML, Markus RP, Renault N, Tosini G, et al. Update on melatonin receptors: IUPHAR Review 20. Br J Pharmacol. (2016) 173:2702–25. doi: 10.1111/bph.13536
3. Lima FB, Machado UF, Bartol I, Seraphim PM, Sumida DH, Moraes SM, et al. Pinealectomy causes glucose intolerance and decreases adipose cell responsiveness to insulin in rats. Am J Physiol. (1998) 275(6 Pt 1):E934–41. doi: 10.1152/ajpendo.1998.275.6.E934
4. Nogueira TC, Lellis-Santos C, Jesus DS, Taneda M, Rodrigues SC, Amaral FG, et al. Absence of melatonin induces night-time hepatic insulin resistance and increased gluconeogenesis due to stimulation of nocturnal unfolded protein response. Endocrinology. (2011) 152:1253–63. doi: 10.1210/en.2010-1088
5. Nogueira TC, Lellis-Santos C, Jesus DS, Taneda M, Rodrigues SC, Amaral FG. Melatonin improves glucose homeostasis and endothelial vascular function in high-fat diet-fed insulin-resistant mice. Endocrinology. (2009) 150:5311–7. doi: 10.1210/en.2009-0425
6. Prunet-Marcassus B, Desbazeille M, Bros A, Louche K, Delagrange P, Renard P, et al. Melatonin reduces body weight gain in Sprague Dawley rats with diet-induced obesity. Endocrinology. (2003) 144:5347–52. doi: 10.1210/en.2003-0693
7. Prunet-Marcassus B, Desbazeille M, Bros A, Louche K, Delagrange P, Renard P, et al. Melatonin, energy metabolism, and obesity: a review. J Pineal Res. (2014) 56:371–81. doi: 10.1111/jpi.12137
8. Prunet-Marcassus B, Desbazeille M, Bros A, Louche K, Delagrange P, Renard P, et al. Body weight, food intake and energy regulation in exercising and melatonin-treated Siberian hamsters. Physiol Behav. (1985) 35:805–8. doi: 10.1016/0031-9384(85)90415-9
9. Rasmussen DD, Boldt BM, Wilkinson CW, Yellon SM, Matsumoto AM. Daily melatonin administration at middle age suppresses male rat visceral fat, plasma leptin, and plasma insulin to youthful levels. Endocrinology. (1999) 140:1009–2. doi: 10.1210/endo.140.2.6674
10. Wolden-Hanson T, Mitton DR, McCants RL, Yellon SM, Wilkinson CW, Matsumoto AM, et al. Daily melatonin administration to middle-aged male rats suppresses body weight, intraabdominal adiposity, and plasma leptin and insulin independent of food intake and total body fat. Endocrinology. (2000) 141:487–97. doi: 10.1210/en.141.2.487
11. Balland E, Cowley MA. New insights in leptin resistance mechanisms in mice. Front Neuroendocrinol. (2015) 39:59–65. doi: 10.1016/j.yfrne.2015.09.004
12. Favero G, Stacchiotti A, Castrezzati S, Bonomini F, Albanese M, Rezzani R, et al. Melatonin reduces obesity and restores adipokine patterns and metabolism in obese (ob/ob) mice. Nutr Res. (2015) 35:891–900. doi: 10.1016/j.nutres.2015.07.001
13. Amstrup AK, Sikjaer T, Pedersen SB, Heickendorff L, Mosekilde L, Rejnmark L. Reduced fat mass and increased lean mass in response to 1 year of melatonin treatment in postmenopausal women: a randomized placebo-controlled trial. Clin Endocrinol. (2016) 84:342–7. doi: 10.1111/cen.12942
14. Ryu V, Zarebidaki E, Albers HE, Xue B, Bartness TJ. Short photoperiod reverses obesity in Siberian hamsters via sympathetically induced lipolysis and Browning in adipose tissue. Physiol Behav. (2018) 190:11–20. doi: 10.1016/j.physbeh.2017.07.011
15. Liu K, Yu W, Wei W, Zhang X, Tian Y, Sherif M, et al. Melatonin reduces intramuscular fat deposition by promoting lipolysis and increasing mitochondrial function. J Lip Res. (2018) 60:767–82 doi: 10.1194/jlr.M087619
16. Fischer C, Mueller T, Pfeffer M, Wicht H, von Gall C, Korf HW. Melatonin receptor 1 deficiency affects feeding dynamics and pro-opiomelanocortin expression in the arcuate nucleus and pituitary of mice. Neuroendocrinology. (2017) 105:35–43. doi: 10.1159/000448333
17. Buonfiglio D, Parthimos R, Dantas R, Cerqueira Silva R, Gomes G, Andrade-Silva J, et al. Melatonin absence leads to long-term leptin resistance and overweight in rats. Front. Endocrinol. (2018) 9:122. doi: 10.3389/fendo.2018.00122
18. Saladin R, De Vos P, Guerre-Millo M, Leturque A, Girard J, et al. Transient increase in obese gene expression after food intake or insulin administration. Nature. (1995) 377:527–9. doi: 10.1038/377527a0
19. Ahima RS, Prabakaran D, Flier JS. Postnatal leptin surge and regulation of circadian rhythm of leptin by feeding. Implications for energy homeostasis and neuroendocrine function. J Clin Invest. (1998) 101:1020–7. doi: 10.1172/JCI1176
20. Chakir I, Dumont S, Pévet P, Ouarour A, Challet E, Vuillez P. Pineal melatonin is a circadian time-giver for leptin rhythm in Syrian hamsters. Front Neurosci. (2015) 9:190. doi: 10.3389/fnins.2015.00190
21. Ríos-Lugo MJ, Jiménez-Ortega V, Cano-Barquilla P, Mateos PF, Spinedi EJ, Cardinali DP, et al. Melatonin counteracts changes in hypothalamic gene expression of signals regulating feeding behavior in high-fat fed rats. Hormone Mol Biol Clin Invest. (2015) 21:175–83. doi: 10.1515/hmbci-2014-0041
22. Buonfiglio D, Tchio C, Furigo I, Donato J, Baba K, Cipolla-Neto J, et al. Removing melatonin receptor type 1 signaling leads to selective leptin resistance in the arcuate nucleus. J Pineal Res. (2019):e12580. doi: 10.1111/jpi.12580
23. Jiménez-Aranda A, Fernández-Vázquez G, Campos D, Tassi M, Velasco-Perez L, Tan DX, et al. Melatonin induces browning of inguinal white adipose tissue in Zucker diabetic fatty rats. J Pineal Res. (2013) 55:416–23. doi: 10.1111/jpi.12089
24. Fernández Vázquez G, Reiter RJ, Agil A. Melatonin increases brown adipose tissue mass and function in Zucker diabetic fatty rats: implications for obesity control. J Pineal Res. (2018) 64:e12472. doi: 10.1111/jpi.12472
25. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. (2004) 84:277–359. doi: 10.1152/physrev.00015.2003
26. Tan DX, Manchester LC, Fuentes-Broto L, Paredes SD, Reiter RJ. Significance and application of melatonin in the regulation of brown adipose tissue metabolism: relation to human obesity. Obes Rev. (2011) 12:167–88. doi: 10.1111/j.1467-789X.2010.00756.x
27. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. (2006) 444:1027–31. doi: 10.1038/nature05414
28. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. (2006) 444:1022–3. doi: 10.1038/4441022a
29. Xu P, Wang J, Hong F, Wang S, Jin X, Xue T, et al. Melatonin prevents obesity through modulation of gut microbiota in mice. J Pineal Res. (2017) 62:e12399. doi: 10.1111/jpi.12399
30. Owino S, Sánchez-Bretaño A, Tchio C, Cecon E, Karamitri A, Dam J, et al. Nocturnal activation of melatonin receptor type 1 signaling modulates diurnal insulin sensitivity via regulation of PI3K activity. J Pineal Res. (2018) 64. doi: 10.1111/jpi.12462
31. Faria JA, Kinote A, Ignacio-Souza LM, de Araújo TM, Razolli DS, Doneda DL, et al. Melatonin acts through MT1/MT2 receptors to activate hypothalamic Akt and suppress hepatic gluconeogenesis in rats. Am J Physiol Endocrinol Metab. (2013) 305:E230–42. doi: 10.1152/ajpendo.00094.2013
32. Ha E, Yim SV, Chung JH, Yoon KS, Kang I, Cho YH, et al. Melatonin stimulates glucose transport via insulin receptor substrate-1/phosphatidylinositol 3-kinase pathway in C2C12 murine skeletal muscle cells. J Pineal Res. (2006) 41:67–72. doi: 10.1111/j.1600-079X.2006.00334.x
33. Lima FB, Matsushita DH, Hell NS, Dolnikoff MS, Okamoto MM, Cipolla Neto J. The regulation of insulin action in isolated adipocytes. Role of the periodicity of food intake, time of day and melatonin. Braz J Med Biol Res. (1994) 27:995–1000.
34. Tuomi T, Nagorny CLF, Singh P, Bennet H, Yu Q, Alenkvist I, et al. Increased melatonin signaling is a risk factor for type 2 diabetes. Cell Metab. (2016) 23:1067–77. doi: 10.1016/j.cmet.2016.04.009
35. Contreras-Alcantara S, Baba K, Tosini G. Removal of melatonin receptor type 1 induces insulin resistance in the mouse. Obesity. (2010) 18:1861–3. doi: 10.1038/oby.2010.24
36. Macotela Y, Boucher J, Tran TT, Kahn CR. Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes. (2009) 58:803–812. doi: 10.2337/db08-1054
37. Geer EB, Shen W. Gender differences in insulin resistance, body composition, and energy balance. Gend Med. (2009) 6 Suppl 1:60–75. doi: 10.1016/j.genm.2009.02.002
38. Karamitri A, Jockers R. Melatonin in type 2 diabetes mellitus and obesity. Nat Rev Endocrinol. (2019) 15:105–25. doi: 10.1038/s41574-018-0130-1
39. Shibata S, Cassone VM, Moore RY. Effects of melatonin on neuronal activity in the rat suprachiasmatic nucleus in vitro. Neurosci Lett. (1989) 97:140–144. doi: 10.1016/0304-3940(89)90153-5
40. Stehle J, Vanecek J, Vollrath L. Effects of melatonin on spontaneous electrical activity of neurons in rat suprachiasmatic nuclei: an in vitro iontophoretic study. J Neural Transm. (1989) 78:173–7. doi: 10.1007/BF01252503
41. Benloucif S, Dubocovich ML. Melatonin and light induce phase shifts of circadian activity rhythms in the C3H/HeN mouse. J Biol Rhythms. (1996) 11:113–25. doi: 10.1177/074873049601100204
42. Stenvers DJ, Scheer FAJL, Schrauwen P, la Fleur SE, Kalsbeek A. Circadian clocks and insulin resistance. Nat Rev Endocrinol. (2018) 15:75–89 doi: 10.1038/s41574-018-0122-1
43. Dyar KA, Ciciliot S, Wright LE, Biensø RS, Tagliazucchi GM, Patel V, et al. Muscle insulin sensitivity and glucose metabolism are controlled by the intrinsic muscle clock. Mol Metab. (2014) 3:29–41. doi: 10.1016/j.molmet.2013.10.005
44. Lamia KA, Storch KF, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci USA. (2008) 105:15172–77. doi: 10.1073/pnas.0806717105
45. Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. (2010) 466:627–31. doi: 10.1038/nature09253
46. Paschos GK, Ibrahim S, Song WL, Kunieda T, Grant G, Reyes TM, et al. Obesity in mice with adipocyte-specific deletion of clock component Arntl. Nat Med. (2012) 18:1768–77. doi: 10.1038/nm.2979
47. Mühlbauer E, Gross E, Labucay K, Wolgast S, Peschke E. Loss of melatonin signalling and its impact on circadian rhythms in mouse organs regulating blood glucose. Eur J Pharmacol. (2009) 606:61–71. doi: 10.1016/j.ejphar.2009.01.029
48. Owino S, Contreras-Alcantara S, Baba K, Tosini G. Melatonin signaling controls the daily rhythm in blood glucose levels independent of peripheral clocks. PLoS ONE. (2016) 11:e0148214. doi: 10.1371/journal.pone.0148214
49. Suofu Y, Li W, Jean-Alphonse FG, Jia J, Khattar NK, Li J, et al. Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release. Proc Natl Acad Sci USA. (2017) 114:E7997–E8006. doi: 10.1073/pnas.1705768114
50. Wu L, Cui L, Tam WH, Ma RC, Wang CC. Genetic variants associated with gestational diabetes mellitus: a meta-analysis and subgroup analysis. Sci Rep. (2016) 6:30539. doi: 10.1038/srep30539
51. Tarnowski M, Malinowski D, Safranow K, Dziedziejko V, Pawlik A. MTNR1A and MTNR1B gene polymorphisms in women with gestational diabetes. Gynecol Endocrinol. (2017) 33:395–8. doi: 10.1080/09513590.2016.1276556
52. Song X, Sun X, Ma G, Sun Y, Shi Y, Du Y, et al. Family association study between melatonin receptor gene polymorphisms and polycystic ovary syndrome in Han Chinese. Eur J Obstet Gynecol Reprod Biol. (2015) 195:108–12. doi: 10.1016/j.ejogrb.2015.09.043
53. Li C, Qiao B, Zhan Y, Peng W, Chen ZJ, Sun L, et al. Association between genetic variations in MTNR1A and MTNR1B genes and gestational diabetes mellitus in Han Chinese women. Gynecol Obstet Invest. (2013) 76:221–27. doi: 10.1159/000355521
54. Bonnefond A, Clément N, Fawcett K, Yengo L, Vaillant E, Guillaume JL, et al.Bonnefond A, Clement N, Fawcett K, et al. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat Genet. (2012) 44:297–301. doi: 10.1038/ng.1053
55. Andersson EA, Holst B, Sparsø T, Grarup N, Banasik K, Holmkvist J, et al. MTNR1B G24E variant associates With BMI and fasting plasma glucose in the general population in studies of 22,142 Europeans. Diabetes. (2010) 59:1539–48. doi: 10.2337/db09-1757
56. Bouatia-Naji N, Bonnefond A, Cavalcanti-Proença C, Sparsø T, Holmkvist J, Marchand M, et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet. (2009) 41:89–94. doi: 10.1038/ng.277
57. Lyssenko V, Nagorny CL, Erdos MR, Wierup N, Jonsson A, Spégel P, et al. Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat Genet. (2009) 41:82–8. doi: 10.1038/ng.288
58. Karamitri A, Plouffe B, Bonnefond A, Chen M, Gallion J, Guillaume JL, et al. Type 2 diabetes-associated variants of the MT2 melatonin receptor affect distinct modes of signaling. Sci Signal. (2018) 11:eaan6622. doi: 10.1126/scisignal.aan6622
59. Gaulton KJ, Ferreira T, Lee Y, Raimondo A, Mägi R, Reschen ME, et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat Genet. (2015) 47:1415–25. doi: 10.1038/ng.3437
60. Been LF, Hatfield JL, Shankar A, Aston CE, Ralhan S, Wander GS, et al. A low frequency variant within the GWAS locus of MTNR1B affects fasting glucose concentrations: genetic risk is modulated by obesity. Nutr Metab Cardiovasc Dis. (2012) 22:944–51. doi: 10.1016/j.numecd.2011.01.006
61. Gan W, Walters RG, Holmes MV, Bragg F, Millwood IY, Banasik K, et al. Evaluation of type 2 diabetes genetic risk variants in Chinese adults: findings from 93,000 individuals from the China Kadoorie Biobank. Diabetologia. (2016) 59:1446–57. doi: 10.1007/s00125-016-3920-9
62. Rosta K, Al-Aissa Z, Hadarits O, Harreiter J, Nádasdi Á, Kelemen F, et al. Association study with 77 SNPs confirms the robust role for the rs10830963/G of MTNR1B variant and identifies two novel associations in gestational diabetes mellitus development. PLoS ONE. (2017) 12:e0169781. doi: 10.1371/journal.pone.0169781
63. Prokopenko I, Langenberg C, Florez JC, Saxena R, Soranzo N, Thorleifsson G, et al. Variants in MTNR1B influence fasting glucose levels. Nat Genet. (2009) 41:77–81. doi: 10.1038/ng.290
64. Sparsø T, Bonnefond A, Andersson E, Bouatia-Naji N, Holmkvist J, Wegner L, et al. G-allele of intronic rs10830963 in MTNR1B confers increased risk of impaired fasting glycemia and type 2 diabetes through an impaired glucose-stimulated insulin release: studies involving 19,605 Europeans. Diabetes. (2009) 58:1450–6. doi: 10.2337/db08-1660
65. Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. (2010) 42:579–89. doi: 10.1038/ng.609
66. McMullan CJ, Schernhammer ES, Rimm EB, Hu FB, Forman JP. Melatonin secretion and the incidence of type 2 diabetes. JAMA. (2013) 309:1388–96. doi: 10.1001/jama.2013.2710
Keywords: melatonin, MT1, MT2, diabetes, leptin
Citation: Owino S, Buonfiglio DDC, Tchio C and Tosini G (2019) Melatonin Signaling a Key Regulator of Glucose Homeostasis and Energy Metabolism. Front. Endocrinol. 10:488. doi: 10.3389/fendo.2019.00488
Received: 18 March 2019; Accepted: 04 July 2019;
Published: 17 July 2019.
Edited by:
James M. Olcese, Florida State University, United StatesReviewed by:
Krystyna Skwarlo-Sonta, University of Warsaw, PolandDun-Xian Tan, The University of Texas Health Science Center at San Antonio, United States
Copyright © 2019 Owino, Buonfiglio, Tchio and Tosini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gianluca Tosini, Z3Rvc2luaUBtc20uZWR1