 Qian Zhang
Qian Zhang Xinhua Xiao*
Xinhua Xiao*- Key Laboratory of Endocrinology, Ministry of Health, Department of Endocrinology, Peking Union Medical College, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, Beijing, China
Scope: Overnutrition in utero is a critical contributor to the susceptibility of diabetes by programming, although the exact mechanism is not clear. In this paper, we aimed to study the long-term effect of a maternal high-fat (HF) diet on offspring through epigenetic modifications.
Procedures: Five-week-old female C57BL6/J mice were fed a HF diet or control diet for 4 weeks before mating and throughout gestation and lactation. At postnatal week 3, pups continued to consume a HF or switched to a control diet for 5 weeks, resulting in four groups of offspring differing by their maternal and postweaning diets.
Results: The maternal HF diet combined with the offspring HF diet caused hyperglycemia and insulin resistance in male pups. Even after changing to the control diet, male pups exposed to the maternal HF diet still exhibited hyperglycemia and glucose intolerance. The livers of pups exposed to a maternal HF diet had a hypermethylated insulin receptor substrate 2 (Irs2) gene and a hypomethylated mitogen-activated protein kinase kinase 4 (Map2k4) gene. Correspondingly, the expression of the Irs2 gene decreased and that of Map2k4 increased in pups exposed to a maternal HF diet.
Conclusion: Maternal overnutrition programs long-term epigenetic modifications, namely, Irs2 and Map2k4 gene methylation in the offspring liver, which in turn predisposes the offspring to diabetes later in life.
Introduction
The incidence of type 2 diabetes mellitus (T2DM) is dramatically increasing. T2DM has become a major public health problem worldwide. It is clear that genetic factors play important roles in the incidence of T2DM. However, the genetic loci identified by genome-wide association studies (GWASs) can account for only a small proportion of T2DM (<10%) (1, 2). Heritable factors cannot explain the dramatic increase in T2DM, thus recent research has focused on lifestyle as a major factor for the incidence of T2DM (3).
Among lifestyle factors, prenatal and postnatal nutrition imbalances lead to epigenetic programming, which is associated with increased T2DM incidence, including both undernutrition and overnutrition (4). Maternal nutrition is important in determining susceptibility to metabolic disease (5). Epidemiological studies in humans revealed that both under- and overnutrition of the mothers during pregnancy will have far-reaching and long-term outcomes for the health of the offspring in adult life (6, 7). For example, glucose intolerance and insulin resistance were reported in offspring whose mothers were exposed to undernutrition during gestation in the well-known study of the Dutch famine, demonstrating an association (8). Similarly, maternal obesity is associated with increased susceptibility to T2DM in offspring (9, 10). Female rat pups from mothers with high-fat (HF) diet-induced obesity were more prone to obesity (11). We have reported that a maternal low-chromium diet can also program the development of diabetes in offspring (12).
Intrauterine programming has been proposed in which the maternal nutrition environment could affect the metabolism of the offspring throughout life (13). The prenatal and early postnatal period is considered a critical time for adult life. Moreover, the maternal nutrition environment and health status induce epigenetic modifications that affect the incidence of T2DM in offspring. The Developmental Origins of Health and Disease (DOHaD) concept has been proposed, and epigenetic mechanisms are considered to possibly underlie DOHaD (14, 15). Development programming in the early life period can increase the risk of T2DM in later life (14). Epigenetic modifications mainly consist of DNA methylation, histone modifications and non-coding RNAs. These epigenetic modifications can affect gene expression via environmental changes (16). DNA methylation is defined as the addition of a methyl group to a cytosine, usually in CpG islands (17). DNA methylation at CpG-rich promoters or gene regulatory regions is normally associated with the inhibition of gene expression (17).
Recently, a small number of studies have described the role of DNA methylation changes in fetal programming to metabolic disease in human and animal models. In human research, newborns with obese parents had altered methylation in multiple imprinted genes in their cord blood (18). In animal research, the results were not consistent. In one study, exposure to a maternal HF diet caused DNA hypermethylation in offspring mouse livers (19). However, Cannon et al. did not detect any effect of the maternal diet on DNA methylation in the male mouse liver (20). Another group reported that exposure of offspring to a maternal HF diet could remodel the hepatic epigenome, if they changed to a control diet during the postweaning period (21). Hence, the precise molecular mechanisms underlying the epigenetic changes induced by a maternal HF diet have not yet been thoroughly identified.
Thus, in this study, we sought to determine whether genome-wide changes in DNA methylation occur in the livers of offspring exposed to a maternal HF diet and a postweaning control diet. The liver was chosen because it is essential for maintaining metabolic homeostasis (22). We performed a genome methylation array to identify differentially methylated genes in 8-week-old postweaning control diet-fed offspring from HF diet-fed or control diet-fed dams. Differentially methylated genes were assessed in the offspring mouse genome to determine the epigenetic mechanism responsible for the maternal HF diet effects.
Materials and Methods
Animal Grouping and Treatments
All research procedures involving animals were approved by the Animal Care Committee of Peking Union Medical Hospital (Permit Number: MC-07-6004). Male and virgin female C57BL6/J mice were purchased from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China). All animals were housed under specific pathogen-free conditions. Mice were kept in a controlled environment (25 ± 1°C) under a 12-h light/dark cycle and allowed food and water ad libitum.
Five-week-old virgin females (n = 40) were divided into two groups at random. One group of mice was fed a standard AIN93G control diet (CON group, n = 20, Research Diets, Inc.; 16, 64, and 20% of calories from fat, carbohydrate, and protein, respectively), while the other group was fed a HF diet (n = 20; Research Diets, Inc.; 45, 35, and 20% of calories from fat, carbohydrate, and protein, respectively). Male mice were fed a normal diet throughout the experiment. After 4 weeks, female mice were housed overnight with males of the same age to mate at a ratio of 2:1 in each cage. The presence of a vaginal plug the following morning indicated the first day of pregnancy. During gestation and lactation, the diet scheme did not change. On postnatal day 21, one male pup was selected randomly from each dam. Male pups from control diet-fed dams were weaned onto the control diet (CON-CON, n = 10) or HF diet (CON-HF, n = 10). Meanwhile, male pups from HF diet-fed dams were weaned onto the control diet (HF-CON, n = 10) or HF diet (HF-HF, n = 10). This process created four groups of pups: CON-CON group, CON-HF group, HF-CON group, and HF-HF group. All animals were sacrificed at 8 weeks of age, and the livers were immediately collected and snap frozen in liquid nitrogen and then stored at −80°C. The animal experiment timeline is shown in Figure 1.
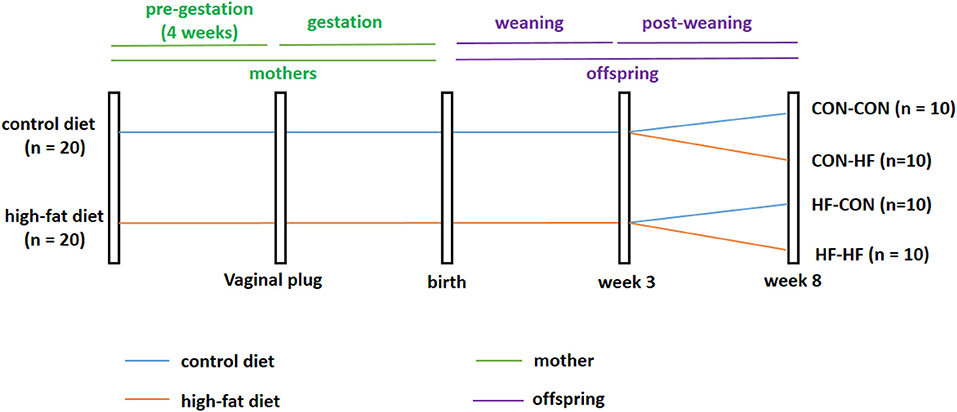
Figure 1. Timeline of animal experiment. CON-CON: control diet mother-post-weaning control diet; CON-HF, control diet mother-post-weaning high-fat diet; HF-CON, high-fat diet mother-post-weaning control diet; HF-HF, high-fat diet mother-post-weaning high-fat diet.
Body Weight, Fasting Blood Glucose, Oral Glucose Tolerance Test (OGTT), and Insulin Analysis
Body weight was measured at weaning time in mothers and 8 weeks of age in pups. Fasting blood glucose (Contour TS glucometer, Bayer, Hamburg, Germany) and plasma insulin (ELISA, Millipore, Billerica, MA) levels were measured at 8 weeks of age. Insulin sensitivity was assessed using the HOMA-IR as previously described (23). After 10 h of food deprivation, the 8-week-old offspring underwent OGTT, and the blood glucose concentrations were immediately measured with a glucometer at 0, 30, 60, and 120 min post gavage (2.0 g/kg). The area under the glucose tolerance curve (AUC) of the OGTT was calculated as previously described (23).
DNA Methylation Profiling Using Array
To determine the effect of maternal HF diet on DNA methylation in offspring livers, genomic DNA was extracted from the livers of HF-CON and CON-CON pups (n = 3 in each group, selected randomly from different dams) using a DNeasy Blood & Tissue Kit (Qiagen, Fremont, CA). Samples of genomic DNA were sonicated into random fragments in a size range of ~100–500 bp. Immunoprecipitation of methylated DNA fragments (MeDIP) was performed using a mouse monoclonal anti-5-methylcytosine antibody (Diagenode). The total input and immunoprecipitated DNA were labeled with Cy3- and Cy5-labeled random 9-mers, respectively, and hybridized to an Arraystar Mouse ReqSeq Promoter Array (Arrarystar Inc., Rockville, MD), which contains 22,327 well-characterized RefSeq promoter regions [from ~-1,300 to +500 bp of the transcription start sites (TSSs)] totally covered by ~180,000 probes. Scanning was performed with an Agilent Scanner G2505C (Agilent Technologies, Waldbronn, Germany).
Methylation Enrichment and Peak-Finding
The results were obtained using a sliding-window (750 bp) peak-finding algorithm provided by NimbleScan v2.5 (Roche NimbleGen). NimbleScan detects peaks by searching for at least two probes above a minimum cutoff p-value (–log10) of 2. Peaks within 500 bp of each other are merged. The M’ value was calculated for each probe to compare differentially enriched regions between the HF-CON and CON-CON groups as follows:
M’ = Average()−Average().
The differential enrichment peaks (DEPs) called by the NimbleScan algorithm were filtered according to the following criteria:
(1) At least one of the two groups has a median (log2 MeDIP/Input) ≥0.3 and M’ > 0.
(2) At least half of the probes in a peak may have a coefficient of variability (CV) ≤ 0.8 in both groups.
To separate strong CpG islands from weak CpG islands, promoters were categorized into three levels: high CpG promoters/regions (HCPs), intermediate CpG promoters/regions (ICPs), and low CpG promoters/regions (LCPs) (24).
Pathway Analysis
DAVID Bioinformatics Resources 6.7 [http://david.abcc.ncifcrf.gov/ (25)] was used to perform Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) functional enrichment analyses for the differentially methylated genes (DMGs).
Bisulfite Sequencing PCR (BSP)
For validation of the methylation array, bisulfite conversion of genomic DNA from offspring livers in the four groups (n = 10 in each group) was conducted using a kit (Zymo Research, CA). The primers were designed using Methyl Primers Express software 1.0 (Applied Biosystems, Foster City, CA) and are shown in Table 1. The resulting PCR products were purified using a QIAquick Gel Extraction Kit (Qiagen) and cloned into the pMD18-T vector (Takara, Shiga, Japan). Individual clones were grown, and plasmids were purified using a PureLink Miniprep Kit (Invitrogen, Thermo Scientific Inc., Waltham, MA). At least 10 clones from each mouse were selected and sequenced.
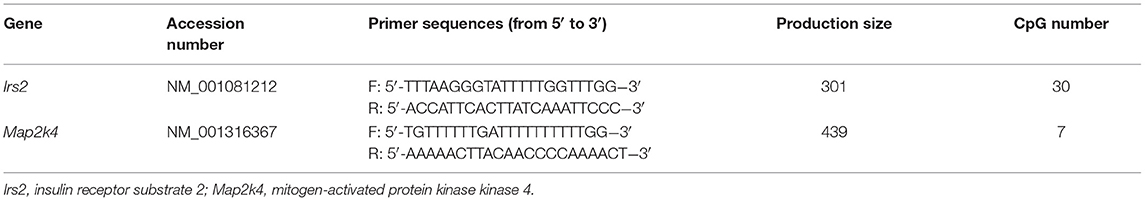
Table 1. PCR primer for bisulfite sequencing.
Quantitative Reverse-Transcription PCR
Total RNA from offspring livers in the four groups (n = 10 in each group) was isolated using a Qiagen RNeasy Mini Kit (Qiagen, Germantown, MD) according to the manufacturer's instructions. Reverse transcription was conducted with total RNA using the TaKaRa RT kit (TaKaRa, Shiga, Japan). Real-time PCR was performed on an ABI 7900 Real-Time PCR Detection System (Applied Biosystems, Foster City, CA) using the comparative Ct method (2−ΔΔCt). The relative mRNA expression levels of target genes were normalized to Gapdh. The sequences of the primers used are shown in Table 2.
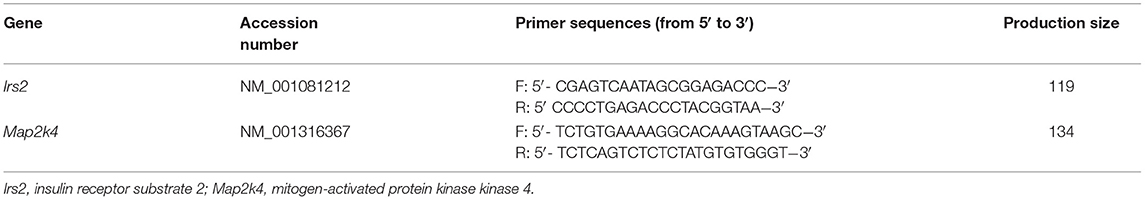
Table 2. qPCR primer.
Statistical Analysis
The results were statistically analyzed by Prism 5.0 (GraphPad Software Inc., San Diego, CA). All values are presented as the mean ± SEM. Dam body weights were compared by Student's t test. Body weight, blood glucose, plasma insulin, HOMA-IR, methylation, and mRNA expression in offspring were analyzed by two-way ANOVA (maternal diet x offspring diet). Statistical significance was defined as P < 0.05.
Results
HF Dams had Greater Body Weight
Dams in the HF group had higher body weights than those in the CON group at weaning time (23.33 ± 1.40 g vs. 19.17 ± 1.14 g, P < 0.01).
Maternal HF Diet Did Not Affect Body Weight in Male Mice
At 8 weeks of age, the mean body weight of mice in the CON-HF and HF-HF groups was significantly higher than that of CON-CON and HF-CON mice (P < 0.01, Figure 2A). There was no significant maternal HF diet effect on offspring body weight (P > 0.05, Figure 2A).
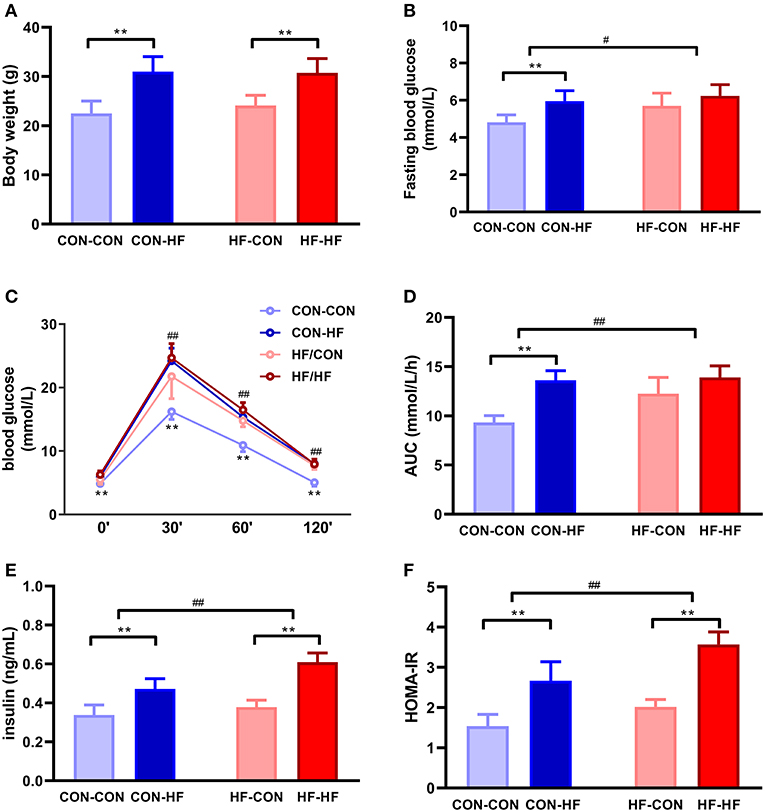
Figure 2. The effect of maternal high-fat diet on metabolic variables of male mice offspring. (A) body weight at weaning; (B) fasting blood glucose; (C) oral glucose tolerance test (OGTT); (D) area under curve (AUC) in OGTT; (E) plasma insulin; (F) HOMA-IR. **P < 0.01 offspring diet effect; #P < 0.05; ##P < 0.01 maternal diet effect. Values are mean ± SEM (n = 10). CON-CON: control diet mother-post-weaning control diet; CON-HF, control diet mother-post-weaning high-fat diet; HF-CON, high-fat diet mother-post-weaning control diet; HF-HF, high-fat diet mother-post-weaning high-fat diet.
Male Mice From HF Dams Exhibited Glucose Intolerance and Insulin Resistance
Fasting blood glucose was at similar higher levels in all HF groups (P < 0.01, Figure 2B). Offspring from HF diet-fed dams displayed higher fasting blood glucose (P < 0.05, Figure 2B). The HF diet caused significant glucose intolerance in offspring from control diet-fed dams (P < 0.01, Figure 2C). In particular, offspring from HF diet-fed dams had higher blood glucose levels at 30, 60, and 120 min and greater AUCs during OGTTs (P < 0.01, Figures 2C,D). Even when they were fed with a control diet, offspring from HF diet-fed dams had higher blood glucose levels and AUCs (P < 0.01, Figures 2C,D). The postweaning HF diet interacted with the maternal HF diet to increase fasting plasma insulin levels and HOMA-IR (P < 0.01, Figures 2E,F).
Intrauterine Environment of HF Damsaffects DNA Methylation Patterns in Male Mice
All microarray data have been deposited into the gene expression omnibus (GEO ID: GSE136814). To explore the mechanism of glucose intolerance and insulin resistance observed in the offspring from dams fed a HF diet, we performed methylation arrays on the livers of the HF-CON and CON-CON groups (n = 3). We found that a total of 1,099 differentially methylated regions (DMRs, 955 annotated genes) were identified on 20 chromosomes in the HF-CON group compared with the CON-CON group. Among these DMRs, 713 were hypermethylated and 386 were hypomethylated. Among the hypermethylated promoters, 487 (68.33%) were located in HCPs, 158 (22.1%) in ICPs, and 68 (9.5%) in LCPs. Among the hypomethylated promoters, 151 (39.1%) were located in HCPs, 105 (27.2%) in ICPs, and 130 (33.7%) in LCPs (Figure 3A). DMRs were mainly located on chromosomes 2, 4, 7, 9, and 11 (Figure 3B).
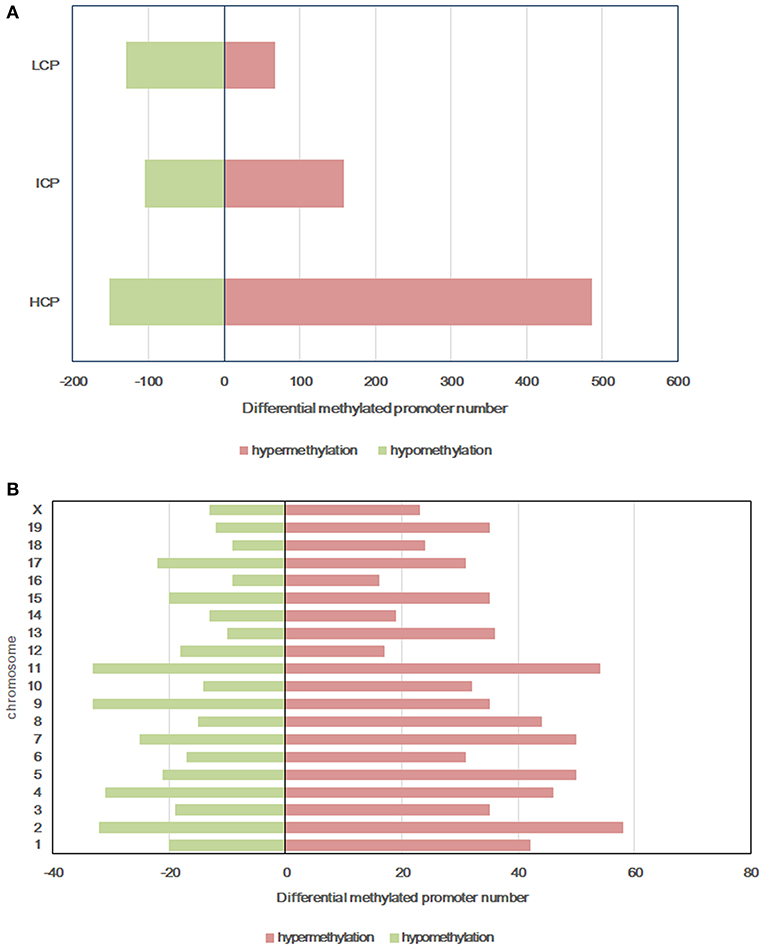
Figure 3. Differentially methylated promoters between HF-CON group and CON-CON group. (A) CpG density of differentially methylated promoters. (B) Chromosomal distribution of differentially methylated promoters. Red: differentially hypermethylated promoters; Green, differentially hypomethylated promoters. Classification of all promoters with high (HCP), intermediated (ICP), and low (LCP) CpG content.
DMR-Related Gene Analysis
To further explore the molecular mechanism by which the offspring were exposed to a HF diet in an intrauterine environment, DMR-related genes were analyzed using GO functional analysis and KEGG enrichment analysis. Significantly enriched GO terms of DMR-related genes mainly participating in molecular function are LBD domain binding, DNA binding, RNA binding, protein binding, and cadherin binding involved in cell-cell adhesion (Figure 4, Supplemental Table 1). The top 25 KEGG pathways of DMGs are enriched in the adipocytokine signaling pathway, MAPK signaling pathway, pyruvate metabolism, toxoplasmosis, hypertrophic cardiomyopathy, Rap1 signaling pathway, regulation of actin cytoskeleton, protein processing in endoplasmic reticulum, dilated cardiomyopathy, pathways in cancer, citrate cycle, TGF-beta signaling pathway, systemic lupus erythematosus, biosynthesis of antibiotics, proteoglycans in cancer, RNA transport, signaling pathway regulating pluripotency of stem cells, glycolysis/gluconeogenesis, Ras signaling pathway, FoxO signaling pathway, PI3K-Akt signaling pathway, carbon metabolism, hippo signaling pathway, and gap junction (Figure 5, Supplemental Table 2).
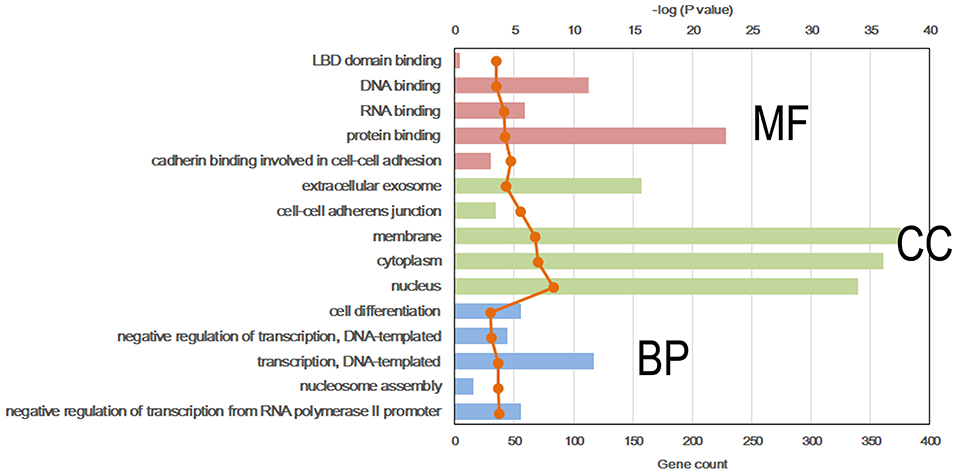
Figure 4. Top 5 significant GO term of differentially methylated genes in each classification. Red, molecular function (MF); Green, cellular component (CC); Blue, biological process (BP).
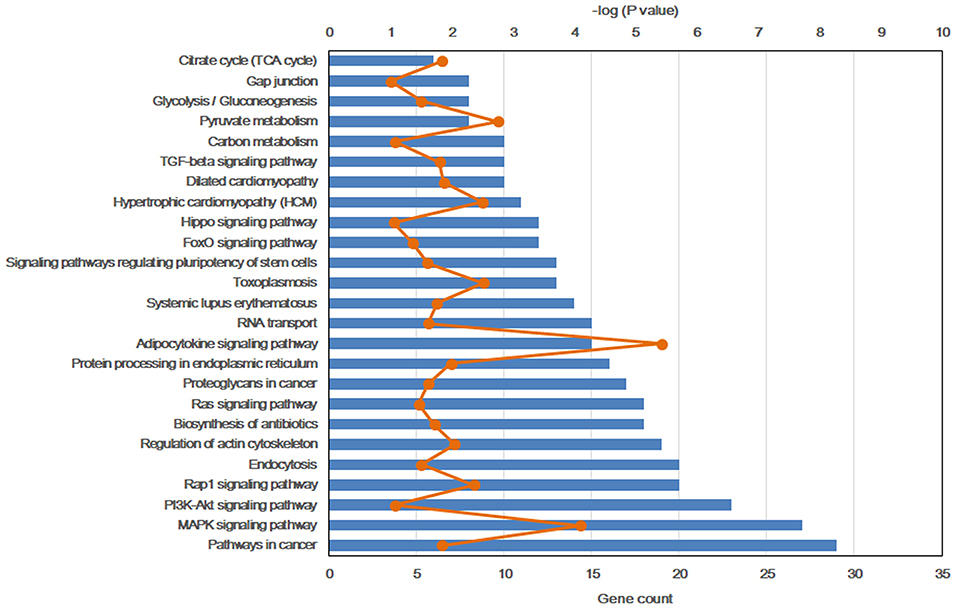
Figure 5. Top 25 significant KEGG pathways of differentially methylated genes.
Overall Differential DNA Methylation of Irs2 and Map2k4
KEGG pathway analysis revealed that insulin signaling and MAPK may be mainly enriched among the DMGs regulated by pup livers from maternal HF diet-fed mice. To verify the methylation levels of candidate genes revealed by the methylation array, we further studied Irs2 and Map2k4, which are involved in the insulin signaling pathway and the MAPK pathway. As shown in Figures 6A,B, the Irs2 gene promoter region underwent a series of hypermethylations in all HF-fed groups, and this was more pronounced in pups from HF diet dams (P < 0.01). Map2k4 gene methylation was inhibited in CON-HF mice compared with CON-CON mice (P < 0.01, Figures 6A,C). The maternal HF diet had an inhibitory effect on Map2k4 gene methylation in offspring (P < 0.01, Figures 6A,C).
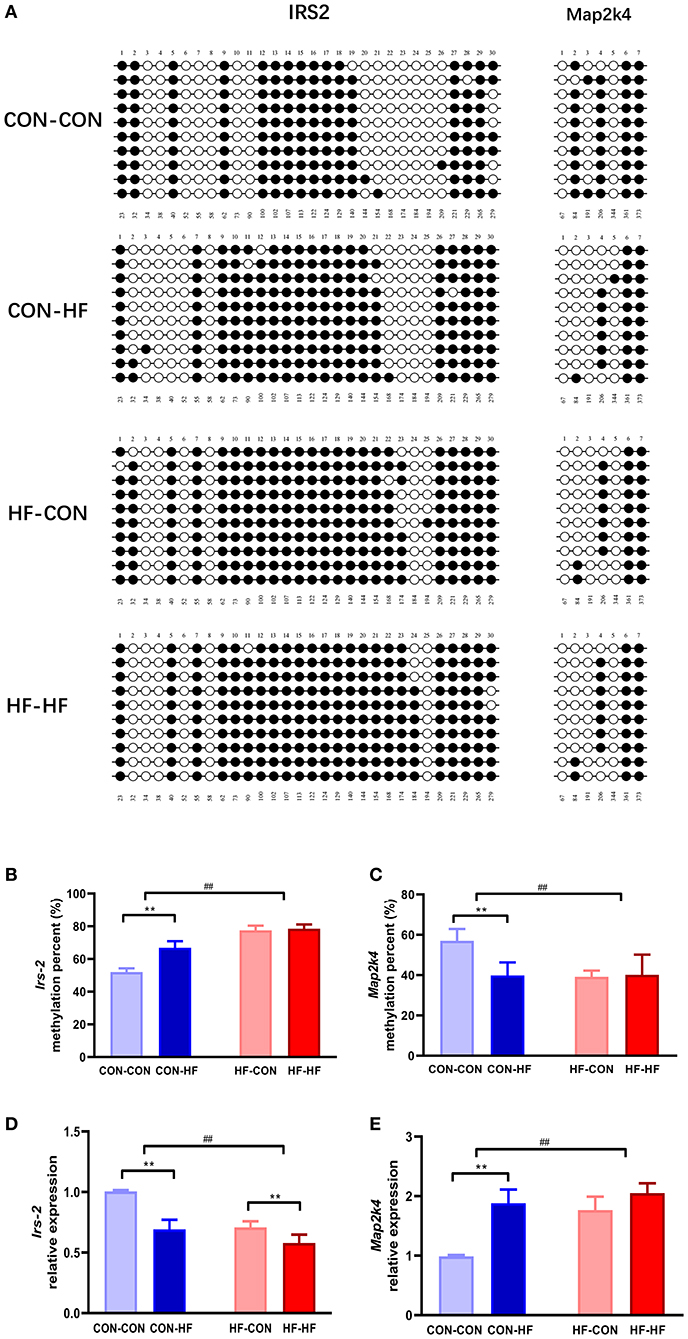
Figure 6. Validation of methylation array using bisulphite sequencing. (A) Schematic diagram of bisulphite sequencing results on 30 CpG sites on Irs2 and 7 CpG on Map2k4. Open circles indicate unmethylated CpGs, and closed circles indicate methylated CpGs. Methylation ratio of Irs2 (B) and Map2k4 (C) in different groups. Relative gene expression of Irs2 (D) and Map2k4 (E) in different groups. **P < 0.01 offspring diet effect; ##P < 0.01 maternal diet effect. Values are mean ± SEM (n = 10). CON-CON, control diet mother-postweaning control diet; CON-HF, control diet mother-postweaning high-fat diet; HF-CON, high-fat diet mother-postweaning control diet; HF-HF, high-fat diet mother-postweaning high-fat diet.
Gene Expression of Irs2 and Map2k4
qPCR was applied to confirm the abnormal expression of Irs2 and Map2k4 in the livers from offspring exposed to a HF diet in utero or in adulthood. As illustrated in Figure 6D, the relative expression of Irs2 mRNA was decreased markedly by both a maternal HF diet and postweaning HF diet (P < 0.01). Map2k4 mRNA expression increased significantly in HF diet-fed mice (P < 0.01, Figure 6E). Furthermore, those exposed to maternal HF diet displayed increased Map2k4 mRNA levels (P < 0.01, Figure 6E).
Discussion
Our results showed that the postweaning HF diet increased body weight in mice. However, the maternal HF diet did not change pup body weight at 8 weeks of age. While some studies reported that HF diet-fed offspring exposed to a maternal HF diet had higher body weight in the adult period (26), other studies did not report any difference (27). This discrepancy may be because of different dietary components, fat sources or the strain and sex of the mice used. Our results revealed that both the maternal HF diet and postweaning HF diet led to glucose intolerance in male mice. Similar observations have been made in a previous study (26, 28–30). A systematic review of animal models found that male offspring exposed to a maternal HF diet independent of maternal obesity, birth weight or postweaning macronutrient intake had glucose intolerance (31). Additionally, a HF diet during fetal life, particularly if combined with the same insult during the suckling period, can induce the type 2 diabetes phenotype (32–34). Maternal obesity interacted with the postweaning HF diet to induce higher levels of glucose intolerance in offspring rodents (35, 36). This programming effect may have sex differences and lead to liver transcriptome changes (37). We also found that fasting insulin levels increased significantly both in mice exposed to the maternal HF diet and postweaning HF diet. In several studies on rats, offspring exposed in utero to a HF diet had increased fasting insulin levels (34, 38). In our study, both maternal HF diet and postweaning HF diet caused insulin resistance in pups. The discordance between an increased HOMA index and lack of an increase in plasma insulin levels may be because of the high fasting blood glucose levels in pups exposed to a maternal HF diet. Previous studies have reported that offspring exposed to a maternal HF diet in utero and/or postnatally developed manifestations of insulin resistance (29, 39).
A HF diet may affect the epigenetic status through several pathways. On the one hand, a HF diet can provoke inflammation and hormone secretion and alter DNA methylation (40). On the other hand, a HF diet can also act directly on epigenetic modification and methylation pathways. A short-term HF diet in healthy men induces widespread DNA methylation changes in the skeletal muscle. These changes were only partially reversed after 6–8 weeks (41). Roux-en-Y gastric bypass (RYGB)-induced weight loss is associated with the restoration of DNA methylation changes caused by obesity in skeletal muscle (42) and adipose tissue (43, 44). These DNA methylation changes involve pathways that control lipid metabolism and mitochondrial function in skeletal muscle (42), adipogenesis (43), and insulin-mediated glucose (44) uptake in adipose tissue.
A maternal HF diet may also induce DNA methylation changes in several metabolic genes (45) and their expression (37) in male offspring livers. In rats, the hepatic cell cycle inhibitor Cdkn1a was hypomethylated in offspring from HF diet-fed mothers (46). Increased methylation of the leptin promoter and decreased methylation of Ppar-α was also observed in the liver of female offspring from HF diet-fed dams (47). The maternal diet also increased Ppar-γ and liver X receptor α (LXRα) DNA methylation levels in male mouse livers (48). The present study used genomic DNA methylation technologies to examine DNA methylation profiles in mice exposed to a HF diet. First, to address whether maternal HF diet exposure induced methylation changes, we compared HF-CON and CON-CON mice. One thousand 99 DMRs were identified, and gene-associated DMRs clustered mainly in 25 pathways. We then compared DNA methylation in the identified regions among all four groups of mice to uncover the impact of HF diet intake regardless of timing. From these metabolic pathways, we concluded that the HF diet decreases Map2k4 DNA methylation and increases Irs2 DNA methylation, as all of the HF-CON, CON-HF, and HF-HF groups generally had lower Map2k4 DNA methylation and higher Irs2 DNA methylation than the CON-CON group.
Previous studies revealed that exposure to both undernutrition and overnutrition in utero induces gene-specific DNA methylation modification (49–52) in animal models. Several clinical studies revealed that genome-wide DNA methylation significantly changed in offspring born to overweight, obese or malnourished mothers (53–57). These DNA methylation modifications in offspring cord blood involve cardiovascular, inflammatory and apoptosis pathways (55, 57). Moreover, exposure to a maternal HF diet in utero might affect glucose and lipid metabolism of female offspring through epigenetic modifications to adiponectin and leptin genes even for multiple continuous generations (58). Exposure to normal diet in utero in the subsequent generations after HF diet exposure for three generations did not completely reverse the changes (59). However, maternal short-term transition from a HF diet to a normal diet before and during pregnancy and lactation without weight loss is not beneficial and even aggravated offspring obesity (60).
These observed methylation modifications affected by a maternal HF diet may result from several mechanisms during early life. In the perinatal period, de novo methyltransferases, such as Dnm3a and Dnm3b, block de novo methylation, which is important in normal development and disease (61). However, during the postweaning period, DNA methyltransferase DNMT1 carried out de novo and non-CG methylation (62). A HF diet in utero may affect these methyltransferases. Folate- and methyl-deficient diets in utero affect methyltransferase expression, including DNMT1 and DNMT3 (63–66). The HF diet affected the expression of DNMTs and their binding to the leptin promoter (67).
The molecular pathway analysis based on the KEGG database revealed that the differentially methylated Mapk gene was enriched. In our research, the Map2k4 gene was hypomethylated in pups exposed to a maternal HF diet. Map2k4 gene expression increased in the HF-CON, CON-HF, and HF-HF groups compared with expression in the CON-CON group. MAPKs consist of p38 MAPK, JNK, and ERK (68, 69). Activated MAPKs can inhibit expression of multiple targets, including insulin receptor substrate (IRS) proteins (70, 71) and resulting in the inhibition of insulin activity (72, 73).
Our results demonstrated that the Irs2 methylation level increased in offspring exposed to a HF diet in utero. Moreover, Irs2 gene expression was reduced in the livers of mice exposed to a HF diet as adults and in utero. IRS proteins are key components in the insulin signaling pathway (74, 75). Once stimulated by insulin, IRS is phosphorylated and then triggers intracellular signaling through the recruitment of proteins with the Src homology-2 domain, including PI3K, Grb-2, Nck, fyn, and Shp-2, among others (74, 76–78). Murine experiments reveal that targeted depletion of IRS-1 or IRS-2 leads to insulin resistance (79–82). Clinical and animal experiments prove that T2D patients and insulin-resistant rodents have defects in the phosphorylation of IRS proteins in vivo and in vitro (83, 84). This evidence proves that IRS defects are the molecular basis for insulin resistance. Hence, our results demonstrated that the maternal HF diet activated hepatic Irs2 methylation and reduced Irs2 gene expression. These findings suggest that the potent insulin resistance induced by a maternal HF diet may occur via activation of the key insulin signaling pathway molecule IRS, methylation, and reduced Irs2 expression.
Conclusions
In summary, we characterize the epigenetic alteration profile in the livers of postweaning control diet-fed offspring exposed to a maternal HF diet throughout gestation and lactation. In particular, decreases in Map2k4 DNA methylation and increases in Irs2 DNA methylation may play a central role in the livers of pups exposed to a maternal HF diet (Figure 7). More evidence needs to focus on the association of the epigenetic genome-wide status of the liver with the diabetes status in offspring. Furthermore, detailed studies exploring the function of candidate genes in the regulation of the liver are needed and could contribute to the prevention of the morbidity of metabolic-related diseases resulting from maternal HF diets.
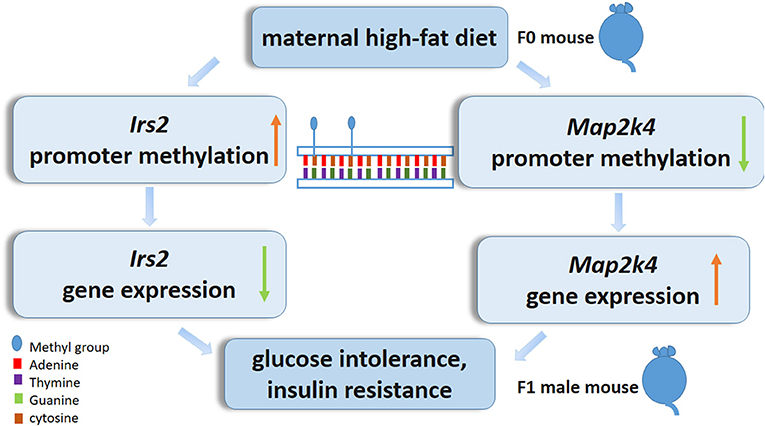
Figure 7. Epigenetic mechanism of high-fat diet in utero and adult on offspring. In utero expose to high-fat diet modify Irs2 and Map2k4 gene methylation and gene expression in offspring, led glucose intolerance, and insulin resistance.
Data Availability Statement
All microarray data have been deposited into the gene expression omnibus (GEO ID: GSE136814) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE136814).
Ethics Statement
All research procedures involving animals were approved by the Animal Care Committee of Peking Union Medical Hospital (Permit Number: MC-07-6004).
Author Contributions
XX conceived and designed the experiments. QZ, JZ, TW, and XW performed the experiments. MY, ML, and FP analyzed the data. XX contributed reagents, materials, and analysis tools. QZ wrote the paper.
Funding
This work was supported by the grants from National Key R&D Program of China (2017YFC1309603), National Key Research and Development Program of China (2016YFA0101002), Medical Epigenetics Research Center, Chinese Academy of Medical Sciences (2017PT31036 and 2018PT31021), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (Nos. 2017PT32020 and 2018PT32001), National Natural Science Foundation of China (Nos. 81170736, 81570715, 81870579, and 81870545), National Natural Science Foundation for Young Scholars of China (No. 81300649), China Scholarship Council foundation (201308110443), PUMC Youth Fund (33320140022), and Fundamental Research Funds for the Central Universities, and Scientific Activities Foundation for Selected Returned Overseas Professionals of Human Resources and Social Security Ministry.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are very grateful to Beijing Compass Biotechnology Company for excellent technical assistance with the microarray experiments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2019.00871/full#supplementary-material
References
1. Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. (2015) 518:197–206. doi: 10.1038/nature14177
2. Raciti GA, Longo M, Parrillo L, Ciccarelli M, Mirra P, Ungaro P, et al. Understanding type 2 diabetes: from genetics to epigenetics. Acta Diabetol. (2015) 52:821–7. doi: 10.1007/s00592-015-0741-0
3. Cheng Z, Zheng L, Almeida FA. Epigenetic reprogramming in metabolic disorders: nutritional factors and beyond. J Nutr Biochem. (2018) 54:1–10. doi: 10.1016/j.jnutbio.2017.10.004
4. van Dijk SJ, Tellam RL, Morrison JL, Muhlhausler BS, Molloy PL. Recent developments on the role of epigenetics in obesity and metabolic disease. Clin Epigenetics. (2015) 7:66. doi: 10.1186/s13148-015-0101-5
5. Barker DJ. Maternal nutrition, fetal nutrition, and disease in later life. Nutrition. (1997) 13:807–13. doi: 10.1016/S0899-9007(97)00193-7
6. Barker DJ. The origins of the developmental origins theory. J Intern Med. (2007) 261:412–7. doi: 10.1111/j.1365-2796.2007.01809.x
7. Oken E, Taveras EM, Kleinman KP, Rich-Edwards JW, Gillman MW. Gestational weight gain and child adiposity at age 3 years. Am J Obstet Gynecol. (2007) 196:322.e321–328. doi: 10.1016/j.ajog.2006.11.027
8. Roseboom T, de Rooij S, Painter R. The Dutch famine and its long-term consequences for adult health. Early Hum Dev. (2006) 82:485–91. doi: 10.1016/j.earlhumdev.2006.07.001
9. Juonala M, Jaaskelainen P, Sabin MA, Viikari JS, Kahonen M, Lehtimaki T, et al. Higher maternal body mass index is associated with an increased risk for later type 2 diabetes in offspring. J Pediatr. (2013) 162:918–23.e911. doi: 10.1016/j.jpeds.2012.10.062
10. Paliy O, Piyathilake CJ, Kozyrskyj A, Celep G, Marotta F, Rastmanesh R. Excess body weight during pregnancy and offspring obesity: potential mechanisms. Nutrition. (2014) 30:245–51. doi: 10.1016/j.nut.2013.05.011
11. Bahari H, Caruso V, Morris MJ. Late-onset exercise in female rat offspring ameliorates the detrimental metabolic impact of maternal obesity. Endocrinology. (2013) 154:3610–21. doi: 10.1210/en.2013-1059
12. Zhang Q, Sun X, Xiao X, Zheng J, Li M, Yu M, et al. Dietary chromium restriction of pregnant mice changes the methylation status of hepatic genes involved with insulin signaling in adult male offspring. PLoS ONE. (2017) 12:e0169889. doi: 10.1371/journal.pone.0169889
13. Lucas A. Role of nutritional programming in determining adult morbidity. Arch Dis Child. (1994) 71:288–90. doi: 10.1136/adc.71.4.288
14. Barker DJ. The fetal and infant origins of adult disease. Bmj. (1990) 301:1111. doi: 10.1136/bmj.301.6761.1111
15. Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. (2008) 359:61–73. doi: 10.1056/NEJMra0708473
16. Marx V. Epigenetics: reading the second genomic code. Nature. (2012) 491:143–7. doi: 10.1038/491143a
17. Jang HS, Shin WJ, Lee JE, Do JT. CpG and Non-CpG methylation in epigenetic gene regulation and brain function. Genes. (2017) 8:148. doi: 10.3390/genes8060148
18. Soubry A, Murphy SK, Wang F, Huang Z, Vidal AC, Fuemmeler BF, et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int J Obes. (2015) 39:650–7. doi: 10.1038/ijo.2013.193
19. Seki Y, Suzuki M, Guo X, Glenn AS, Vuguin PM, Fiallo A, et al. In utero exposure to a high-fat diet programs hepatic hypermethylation and gene dysregulation and development of metabolic syndrome in male mice. Endocrinology. (2017) 158:2860–72. doi: 10.1210/en.2017-00334
20. Cannon MV, Buchner DA, Hester J, Miller H, Sehayek E, Nadeau JH, et al. Maternal nutrition induces pervasive gene expression changes but no detectable DNA methylation differences in the liver of adult offspring. PLoS ONE. (2014) 9:e90335. doi: 10.1371/journal.pone.0090335
21. Moody L, Chen H, Pan YX. Postnatal diet remodels hepatic DNA methylation in metabolic pathways established by a maternal high-fat diet. Epigenomics. (2017) 9:1387–402. doi: 10.2217/epi-2017-0066
22. Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol. (2012) 56:952–64. doi: 10.1016/j.jhep.2011.08.025
23. Zhang Q, Sun X, Xiao X, Zheng J, Li M, Yu M, et al. Maternal chromium restriction induces insulin resistance in adult mice offspring through miRNA. Int J Mol Med. (2018) 41:1547–59. doi: 10.3892/ijmm.2017.3328
24. Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. (2007) 39:457–66. doi: 10.1038/ng1990
25. Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. (2003) 4:P3. doi: 10.1186/gb-2003-4-9-r60
26. Srinivasan M, Katewa SD, Palaniyappan A, Pandya JD, Patel MS. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am J Physiol Endocrinol Metab. (2006) 291:E792–9. doi: 10.1152/ajpendo.00078.2006
27. Dias-Rocha CP, Almeida MM, Santana EM, Costa JCB, Franco JG, Pazos-Moura CC, et al. Maternal high-fat diet induces sex-specific endocannabinoid system changes in newborn rats and programs adiposity, energy expenditure and food preference in adulthood. J Nutr Biochem. (2018) 51:56–68. doi: 10.1016/j.jnutbio.2017.09.019
28. Dunn GA, Bale TL. Maternal high-fat diet promotes body length increases and insulin insensitivity in second-generation mice. Endocrinology. (2009) 150:4999–5009. doi: 10.1210/en.2009-0500
29. Ashino NG, Saito KN, Souza FD, Nakutz FS, Roman EA, Velloso LA, et al. Maternal high-fat feeding through pregnancy and lactation predisposes mouse offspring to molecular insulin resistance and fatty liver. J Nutr Biochem. (2012) 23:341–8. doi: 10.1016/j.jnutbio.2010.12.011
30. Dong M, Zheng Q, Ford SP, Nathanielsz PW, Ren J. Maternal obesity, lipotoxicity and cardiovascular diseases in offspring. J Mol Cell Cardiol. (2013) 55:111–6. doi: 10.1016/j.yjmcc.2012.08.023
31. Ainge H, Thompson C, Ozanne SE, Rooney KB. A systematic review on animal models of maternal high fat feeding and offspring glycaemic control. Int J Obes. (2011) 35:325–35. doi: 10.1038/ijo.2010.149
32. Gniuli D, Calcagno A, Caristo ME, Mancuso A, Macchi V, Mingrone G, et al. Effects of high-fat diet exposure during fetal life on type 2 diabetes development in the progeny. J Lipid Res. (2008) 49:1936–45. doi: 10.1194/jlr.M800033-JLR200
33. Parente LB, Aguila MB, Mandarim-de-Lacerda CA. Deleterious effects of high-fat diet on perinatal and postweaning periods in adult rat offspring. Clin Nutr. (2008) 27:623–34. doi: 10.1016/j.clnu.2008.05.005
34. Cerf ME, Louw J. High fat programming induces glucose intolerance in weanling Wistar rats. Horm Metab Res. (2010) 42:307–10. doi: 10.1055/s-0030-1248303
35. Chen H, Simar D, Morris MJ. Hypothalamic neuroendocrine circuitry is programmed by maternal obesity: interaction with postnatal nutritional environment. PLoS ONE. (2009) 4:e6259. doi: 10.1371/journal.pone.0006259
36. Uddin GM, Youngson NA, Doyle BM, Sinclair DA, Morris MJ. Nicotinamide mononucleotide. (NMN) supplementation ameliorates the impact of maternal obesity in mice: comparison with exercise. Sci Rep. (2017) 7:15063. doi: 10.1038/s41598-017-14866-z
37. Lomas-Soria C, Reyes-Castro LA, Rodriguez-Gonzalez GL, Ibanez CA, Bautista CJ, Cox LA, et al. Maternal obesity has sex-dependent effects on insulin, glucose and lipid metabolism and the liver transcriptome in young adult rat offspring. J Physiol. (2018) 596:4611–28. doi: 10.1113/JP276372
38. Mitra A, Alvers KM, Crump EM, Rowland NE. Effect of high-fat diet during gestation, lactation, or postweaning on physiological and behavioral indexes in borderline hypertensive rats. Am J Physiol Regul Integr Comp Physiol. (2009) 296:R20–28. doi: 10.1152/ajpregu.90553.2008
39. Volpato AM, Schultz A, Magalhaes-da-Costa E, Correia ML, Aguila MB, Mandarim-de-Lacerda CA. Maternal high-fat diet programs for metabolic disturbances in offspring despite leptin sensitivity. Neuroendocrinology. (2012) 96:272–84. doi: 10.1159/000336377
40. Burdge GC, Hanson MA, Slater-Jefferies JL, Lillycrop KA. Epigenetic regulation of transcription: a mechanism for inducing variations in phenotype. (fetal programming) by differences in nutrition during early life? Br J Nutr. (2007) 97:1036–46. doi: 10.1017/S0007114507682920
41. Jacobsen SC, Brons C, Bork-Jensen J, Ribel-Madsen R, Yang B, Lara E, et al. Effects of short-term high-fat overfeeding on genome-wide DNA methylation in the skeletal muscle of healthy young men. Diabetologia. (2012) 55:3341–9. doi: 10.1007/s00125-012-2717-8
42. Barres R, Kirchner H, Rasmussen M, Yan J, Kantor FR, Krook A, et al. Weight loss after gastric bypass surgery in human obesity remodels promoter methylation. Cell Rep. (2013) 3:1020–7. doi: 10.1016/j.celrep.2013.03.018
43. Dahlman I, Sinha I, Gao H, Brodin D, Thorell A, Ryden M, et al. The fat cell epigenetic signature in post-obese women is characterized by global hypomethylation and differential DNA methylation of adipogenesis genes. Int J Obes. (2015) 39:910–9. doi: 10.1038/ijo.2015.31
44. Multhaup ML, Seldin MM, Jaffe AE, Lei X, Kirchner H, Mondal P, et al. Mouse-human experimental epigenetic analysis unmasks dietary targets and genetic liability for diabetic phenotypes. Cell Metab. (2015) 21:138–49. doi: 10.1016/j.cmet.2014.12.014
45. Keleher MR, Zaidi R, Shah S, Oakley ME, Pavlatos C, El Idrissi S, et al. Maternal high-fat diet associated with altered gene expression, DNA methylation, and obesity risk in mouse offspring. PLoS ONE. (2018) 13:e0192606. doi: 10.1371/journal.pone.0192606
46. Dudley KJ, Sloboda DM, Connor KL, Beltrand J, Vickers MH. Offspring of mothers fed a high fat diet display hepatic cell cycle inhibition and associated changes in gene expression and DNA methylation. PLoS ONE. (2011) 6:e21662. doi: 10.1371/journal.pone.0021662
47. Ge ZJ, Luo SM, Lin F, Liang QX, Huang L, Wei YC, et al. DNA methylation in oocytes and liver of female mice and their offspring: effects of high-fat-diet-induced obesity. Environ Health Perspect. (2014) 122:159–64. doi: 10.1289/ehp.1307047
48. Yu HL, Dong S, Gao LF, Li L, Xi YD, Ma WW, et al. Global DNA methylation was changed by a maternal high-lipid, high-energy diet during gestation and lactation in male adult mice liver. Br J Nutr. (2015) 113:1032–9. doi: 10.1017/S0007114515000252
49. Plagemann A, Roepke K, Harder T, Brunn M, Harder A, Wittrock-Staar M, et al. Epigenetic malprogramming of the insulin receptor promoter due to developmental overfeeding. J Perinat Med. (2010) 38:393–400. doi: 10.1515/jpm.2010.051
50. Vucetic Z, Kimmel J, Totoki K, Hollenbeck E, Reyes TM. Maternal high-fat diet alters methylation and gene expression of dopamine and opioid-related genes. Endocrinology. (2010) 151:4756–64. doi: 10.1210/en.2010-0505
51. Begum G, Stevens A, Smith EB, Connor K, Challis JR, Bloomfield F, et al. Epigenetic changes in fetal hypothalamic energy regulating pathways are associated with maternal undernutrition and twinning. FASEB J. (2012) 26:1694–703. doi: 10.1096/fj.11-198762
52. Mahmood S, Smiraglia DJ, Srinivasan M, Patel MS. Epigenetic changes in hypothalamic appetite regulatory genes may underlie the developmental programming for obesity in rat neonates subjected to a high-carbohydrate dietary modification. J Dev Orig Health Dis. (2013) 4:479–90. doi: 10.1017/S2040174413000238
53. Guenard F, Deshaies Y, Cianflone K, Kral JG, Marceau P, Vohl MC. Differential methylation in glucoregulatory genes of offspring born before vs. after maternal gastrointestinal bypass surgery. Proc Natl Acad Sci USA. (2013) 110:11439–44. doi: 10.1073/pnas.1216959110
54. Liu X, Chen Q, Tsai HJ, Wang G, Hong X, Zhou Y, et al. Maternal preconception body mass index and offspring cord blood DNA methylation: exploration of early life origins of disease. Environ Mol Mutagen. (2014) 55:223–30. doi: 10.1002/em.21827
55. Morales E, Groom A, Lawlor DA, Relton CL. DNA methylation signatures in cord blood associated with maternal gestational weight gain: results from the ALSPAC cohort. BMC Res Notes. (2014) 7:278. doi: 10.1186/1756-0500-7-278
56. Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. (2014) 5:5592. doi: 10.1038/ncomms6592
57. Sharp GC, Lawlor DA, Richmond RC, Fraser A, Simpkin A, Suderman M, et al. Maternal pre-pregnancy BMI and gestational weight gain, offspring DNA methylation and later offspring adiposity: findings from the Avon Longitudinal Study of Parents and Children. Int J Epidemiol. (2015) 44:1288–304. doi: 10.1093/ije/dyv042
58. Block T, El-Osta A. Epigenetic programming, early life nutrition and the risk of metabolic disease. Atherosclerosis. (2017) 266:31–40. doi: 10.1016/j.atherosclerosis.2017.09.003
59. Masuyama H, Mitsui T, Nobumoto E, Hiramatsu Y. The effects of high-fat diet exposure in utero on the obesogenic and diabetogenic traits through epigenetic changes in adiponectin and leptin gene expression for multiple generations in female mice. Endocrinology. (2015) 156:2482–91. doi: 10.1210/en.2014-2020
60. Fu Q, Olson P, Rasmussen D, Keith B, Williamson M, Zhang KK, et al. A short-term transition from a high-fat diet to a normal-fat diet before pregnancy exacerbates female mouse offspring obesity. Int J Obes.. (2016) 40:564–72. doi: 10.1038/ijo.2015.236
61. Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. (1999) 99:247–57. doi: 10.1016/S0092-8674(00)81656-6
62. Pradhan S, Bacolla A, Wells RD, Roberts RJ. Recombinant human DNA. (cytosine-5) methyltransferase. I Expression, purification, and comparison of de novo and maintenance methylation. J Biol Chem. (1999) 274:33002–10. doi: 10.1074/jbc.274.46.33002
63. Ghoshal K, Li X, Datta J, Bai S, Pogribny I, Pogribny M, et al. A folate- and methyl-deficient diet alters the expression of DNA methyltransferases and methyl CpG binding proteins involved in epigenetic gene silencing in livers of F344 rats. J Nutr. (2006) 136:1522–7. doi: 10.1093/jn/136.6.1522
64. Kovacheva VP, Mellott TJ, Davison JM, Wagner N, Lopez-Coviella I, Schnitzler AC, et al. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J Biol Chem. (2007) 282:31777–88. doi: 10.1074/jbc.M705539200
65. Pogribny IP, Tryndyak VP, Bagnyukova TV, Melnyk S, Montgomery B, Ross SA, et al. Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. J Hepatol. (2009) 51:176–86. doi: 10.1016/j.jhep.2009.03.021
66. Ding YB, He JL, Liu XQ, Chen XM, Long CL, Wang YX. Expression of DNA methyltransferases in the mouse uterus during early pregnancy and susceptibility to dietary folate deficiency. Reproduction. (2012) 144:91–100. doi: 10.1530/REP-12-0006
67. Xia L, Wang C, Lu Y, Fan C, Ding X, Fu H, et al. Time-specific changes in DNA methyltransferases associated with the leptin promoter during the development of obesity. Nutr Hosp. (2014) 30:1248–55.
68. Guastamacchia E, Resta F, Triggiani V, Liso A, Licchelli B, Ghiyasaldin S, et al. Evidence for a putative relationship between type 2 diabetes and neoplasia with particular reference to breast cancer: role of hormones, growth factors and specific receptors. Curr Drug Targets Immune Endocr Metabol Disord. (2004) 4:59–66. doi: 10.2174/1568008043339965
69. Giovannucci E, Michaud D. The role of obesity and related metabolic disturbances in cancers of the colon, prostate, and pancreas. Gastroenterology. (2007) 132:2208–25. doi: 10.1053/j.gastro.2007.03.050
70. Paz K, Hemi R, LeRoith D, Karasik A, Elhanany E, Kanety H, et al. A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J Biol Chem. (1997) 272:29911–8. doi: 10.1074/jbc.272.47.29911
71. Birnbaum MJ. Turning down insulin signaling. J Clin Invest. (2001) 108:655–9. doi: 10.1172/JCI200113714
72. Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. (2002) 23:599–622. doi: 10.1210/er.2001-0039
73. Evans JL, Maddux BA, Goldfine ID. The molecular basis for oxidative stress-induced insulin resistance. Antioxid Redox Signal. (2005) 7:1040–52. doi: 10.1089/ars.2005.7.1040
74. White MF. The insulin signalling system and the IRS proteins. Diabetologia. (1997) 40 (Suppl. 2):S2–17. doi: 10.1007/s001250051387
75. White MF. The IRS-signaling system: a network of docking proteins that mediate insulin and cytokine action. Recent Prog Horm Res. (1998) 53:119–38. doi: 10.1007/978-3-642-80481-6_8
76. Pawson T, Gish GD. SH2 and SH3 domains: from structure to function. Cell. (1992) 71:359–62. doi: 10.1016/0092-8674(92)90504-6
77. Cherniack AD, Klarlund JK, Conway BR, Czech MP. Disassembly of Son-of-sevenless proteins from Grb2 during p21ras desensitization by insulin. J Biol Chem. (1995) 270:1485–8. doi: 10.1074/jbc.270.4.1485
78. Hadari YR, Kouhara H, Lax I, Schlessinger J. Binding of Shp2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation. Mol Cell Biol. (1998) 18:3966–73. doi: 10.1128/MCB.18.7.3966
79. Patti ME, Sun XJ, Bruening JC, Araki E, Lipes MA, White MF, et al. 4PS/insulin receptor substrate (IRS)-2 is the alternative substrate of the insulin receptor in IRS-1-deficient mice. J Biol Chem. (1995) 270:24670–3. doi: 10.1074/jbc.270.42.24670
80. Tobe K, Tamemoto H, Yamauchi T, Aizawa S, Yazaki Y, Kadowaki T. Identification of a 190-kDa protein as a novel substrate for the insulin receptor kinase functionally similar to insulin receptor substrate-1. J Biol Chem. (1995) 270:5698–701. doi: 10.1074/jbc.270.11.5698
81. Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. (1998) 391:900–4. doi: 10.1038/36116
82. Backhed F. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: the normal gut microbiota in health and disease. Clin Exp Immunol. (2010) 160:80–4. doi: 10.1111/j.1365-2249.2010.04123.x
83. Gao J, Katagiri H, Ishigaki Y, Yamada T, Ogihara T, Imai J, et al. Involvement of apolipoprotein E in excess fat accumulation and insulin resistance. Diabetes. (2007) 56:24–33. doi: 10.2337/db06-0144
Keywords: DNA methylation, insulin receptor substrate, MAPK, maternal high fat diet, epigenetics
Citation: Zhang Q, Xiao X, Zheng J, Li M, Yu M, Ping F, Wang T and Wang X (2019) A Maternal High-Fat Diet Induces DNA Methylation Changes That Contribute to Glucose Intolerance in Offspring. Front. Endocrinol. 10:871. doi: 10.3389/fendo.2019.00871
Received: 29 September 2019; Accepted: 28 November 2019;
Published: 13 December 2019.
Edited by:
Undurti Narasimha Das, UND Life Sciences LLC, United StatesReviewed by:
Margaret Morris, University of New South Wales, AustraliaVenu Lagishetty, University of California, Los Angeles, United States
Copyright © 2019 Zhang, Xiao, Zheng, Li, Yu, Ping, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinhua Xiao, eGlhb3hoMjAxNEB2aXAuMTYzLmNvbQ==