 Åsa Nääv1*
Åsa Nääv1* Lena Erlandsson1
Lena Erlandsson1 Christina Isaxon2
Christina Isaxon2 Eleonor Åsander Frostner3
Eleonor Åsander Frostner3 Johannes Ehinger3
Johannes Ehinger3 Moa K. Sporre4Annette M. Krais5
Moa K. Sporre4Annette M. Krais5 Bo Strandberg5Thomas Lundh5
Bo Strandberg5Thomas Lundh5 Eskil Elmér3Ebba Malmqvist4†
Eskil Elmér3Ebba Malmqvist4† Stefan R. Hansson1†
Stefan R. Hansson1†- 1Obstetrics and Gynecology, Department of Clinical Sciences Lund, Lund University, Lund, Sweden
- 2Department of Ergonomics and Aerosol Technology, Lund University, Lund, Sweden
- 3Mitochondrial Medicine, Department of Clinical Sciences Lund, Lund University, Lund, Sweden
- 4Department of Physics, Lund University, Lund, Sweden
- 5Division of Occupational and Environmental Medicine, Institution of Laboratory Medicine, Lund University, Lund, Sweden
Objective: Epidemiological studies have found air pollution to be a driver of adverse pregnancy outcomes, including gestational diabetes, low term birth weight and preeclampsia. It is unknown what biological mechanisms are involved in this process. A first trimester trophoblast cell line (HTR-8/SVneo) was exposed to various concentrations of PM2.5 (PM2.5) in order to elucidate the effect of urban particulate matter (PM) of size <2.5 μm on placental function.
Methods: PM2.5 were collected at a site representative of urban traffic and dispersed in cell media by indirect and direct sonication. The HTR-8 cells were grown under standard conditions. Cellular uptake was studied after 24 and 48 h of exposure by transmission electron microscopy (TEM). The secretion of human chorionic gonadotropin (hCG), progesterone, and Interleukin-6 (IL-6) was measured by ELISA. Changes in membrane integrity and H2O2 production were analyzed using the CellToxTM Green Cytotoxicity and ROSGloTM assays. Protease activity was evaluated by MitoToxTM assay. Mitochondrial function was assessed through high resolution respirometry in an Oroboros O2k-FluoRespirometer, and mitochondrial content was quantified by citrate synthase activity.
Results: TEM analysis depicted PM2.5 cellular uptake and localization of the PM2.5 to the mitochondria after 24 h. The cells showed aggregated cytoskeleton and generalized necrotic appearance, such as chromatin condensation, organelle swelling and signs of lost membrane integrity. The mitochondria displayed vacuolization and disruption of cristae morphology. At 48 h exposure, a significant drop in hCG secretion and a significant increase in progesterone secretion and IL-6 production occurred. At 48 h exposure, a five-fold increase in protease activity and a significant alteration of H2O2 production was observed. The HTR-8 cells exhibited evidence of increased cytotoxicity with increasing exposure time and dose of PM2.5. No significant difference in mitochondrial respiration or mitochondrial mass could be demonstrated.
Conclusion: Following exposure to air pollution, intracellular accumulation of PM may contribute to the placental dysfunction associated with pregnancy outcomes, such as preeclampsia and intrauterine growth restriction, through their direct and indirect effects on trophoblast protein secretion, hormone regulation, inflammatory response, and mitochondrial interference.
Introduction
Particulate matter (PM) is a complex mixture of suspended primary particles and agglomerates/aggregates of e.g., transition metal oxides, ammonium nitrate, sulfates, protein complexes, and organic materials including polycyclic aromatic hydrocarbons (PAHs) (1–5). PM's toxicity is multifactorial and depends on a variety of particle properties, such as composition, size, and shape as well as number, mass and surface area concentration (1–5). PM with a diameter of <2.5 μm (PM2.5) is generally associated with combustion-related emission sources (3). This fine PM, especially the size fractions <200 nm, have a high likelihood of depositing in the lungs' alveolar region and, subsequently, entering the blood circulation through the alveoli walls via translocation (6–11).
To date, a staggering 87% of the world population resides in areas that exceed the WHO air quality guidelines for PM2.5 (12). In fact, air pollution is the single largest environmental cause of disease (13). Recent studies point toward an association between air pollution exposure and a wide range of diseases such as cardiovascular events in adults (14), cerebrovascular disease (15), and adverse pregnancy outcomes such as preeclampsia (PE) (16). Air pollution has also been associated with hypertensive disorders during pregnancy (17). Furthermore, a large number of epidemiological studies have shown an association between air pollution exposure during pregnancy and the risk of developing PE, even in areas with levels below the WHO air quality guidelines (16, 18–21).
Preeclampsia is a serious pregnancy-related multisystem syndrome, commonly defined as hypertension and proteinuria after the 20th gestational week (22). It complicates roughly 3–7% of pregnancies worldwide (23, 24) and is the main cause of maternal morbidity and mortality. PE is associated with intrauterine growth restriction (IUGR), preterm birth, and perinatal deaths. Both PE and IUGR are associated with an increased long-term risk of developing cardiovascular disease and metabolic disease later in life (25, 26). The association between air pollution exposure and PE is stipulated to be driven by the oxidative stress and systemic inflammation caused or aggravated by air pollution exposure, eventually leading to the vascular endothelial injury, which is a corner stone in the etiology of PE (14, 24, 27, 28).
Despite the wealth of epidemiological evidence associating the effects of air pollution on pregnant women and the developing fetus, air pollution is still not included in the WHO Burden of Disease studies (29). The biological mechanisms and pathways by which air pollution exposure during or prior to pregnancy contributes to the development of PE remain largely unexplored. Therefore, studies deciphering the biological mechanisms contributing to the toxicity of air pollution during pregnancy are urgently needed. By investigating the influence of fine particular matter on placental trophoblast cells, we aim to identify a possible pathway by which air pollution mediates its adverse effects on the health of pregnant women and the developing fetus.
Materials and Methods
Ambient PM2.5 Preparation
Ambient PM2.5 particles were collected at a central location in Malmö, southern Sweden, over 26 days in April–May 2017. This was conducted at a height of 3, 4 m from a street crossing with an average daily traffic (2017) of 28,000 vehicles. A high-volume cascade impactor (BGI900, Mesa Labs, USA) was utilized for this process. The impactor samples air (0.9 m3/min) and collects all particles smaller than 2.5 μm on a polypropylene filter. Subsequent particle extraction followed Mesa Labs' protocol using pure methanol, and particles were then dried in a vacuum evaporator (SpeedVac HT-4X Evaporator, GeneVac, UK).
Characterization of PM2.5
During the sampling period, time-resolved mass concentration measurements of PM2.5 were conducted with a tapered element oscillating microbalance (TEOM 1400AB) with a filter dynamics measurement system, and of black carbon (soot) and organic carbon using light absorption with an aethalometer (AE33). The dried collected particles were analyzed by gas chromatography-mass spectrometry (GC-MS) for the presence of PAH (30). Further, the PM2.5 fraction was analyzed for metal compositions including aluminum (Al), arsenic (As), barium (Ba), cadmium (Cd), chromium (Cr), cobalt (Co), copper (Cu), iron (Fe), lead (Pb), manganese (Mn), nickel (Ni), thallium (Tl), vanadium (V), and zinc (Zn). For this, ~10 mg sample was dissolved in 1 ml concentrated nitric acid at 70°C for 16 h. After dilution with Milli-Q water, the metal concentrations were determined by inductively coupled plasma-mass spectrometry (ICP-MS; iCAP Q, Thermo Scientific, Bremen, GmbH) in collision cell mode with kinetic energy discrimination using helium as the collision gas. The detection limits, calculated as three times the standard deviation (SD) of the blank, were 0.05 ng (Mn, Ni, As, Cd, Tl, Pb), 0.06 ng (Ba), 0.07 ng (Cr, Cu,), 0.08 ng (V), 0.09 ng (Co), 0.15 ng (Zn), 0.70 ng (Fe), and 2.7 ng (Al). Additionally, wind trajectories were calculated by Hybrid Single Particle Lagrangian Integrated Trajectory (Hysplit, Air Resources Lab) model (31, 32) to investigate the local and the in-transported contribution to the collected PM2.5. Hysplit is run with gridded meteorological data from the Center of Environmental Predictions (NCEP) Global Data Assimilation System (GDAS). Local meteorological data from a monitoring station at Heleneholm, Malmö was also used to investigate the wind speeds and wind direction in Malmö situated around 1 km from the particle collection site.
PM2.5 Dispersion
Prior to exposure experiments, 0.9 mg of dried and extracted PM2.5, as described above, was dissolved in 900 μl of cell media (CM; supplemented RPMI-1640 medium, see below) in 1.5 ml Eppendorf tubes and subjected to indirect and direct sonication. Indirect ultra sonication was performed at 4°C and at 120 W for 15 min using an Ultrasonic Cleaner water bath (Mettler Electronics), followed by direct sonication at room temperature (RT) at 50 W, 0.05 cycle, 20% amplitude for 60 s using an UP50H Ultrasonic Processor (Hielscher Ultrasound Technology). The immersion was aliquoted and diluted to desired concentration. The CM without PM, used on control cells, underwent the same protocol and maintained the same volumes. The direct sonication step followed by vortex was performed prior to each exposure.
Cell Culture
The commercially available HTR-8 (HTR-8/SVneo) cells, a human first trimester transformed trophoblast cell line (the American Type Culture Collection, ATCC Cell Lines, CRL-3271, lot number 64275781) were maintained in HyClone RPMI-1640 medium (Fisher Scientific) supplemented with 5% fetal bovine serum (FBS) (Life Technologies), 100 μg/ml streptomycin and 100 U/ml penicillin (Fisher Scientific) at 37°C in a humidified 5% CO2 incubator.
Uptake of PM2.5
HTR-8 cells at seeding density 0.3 × 106 per well in a 6-well plate were exposed to a single dose of 500 ng/ml of PM2.5 for 24–48 h. After washing with phosphate-buffered saline (PBS), the cells were trypsinized (0.25% trypsin) and pelleted by centrifugation. The cell pellet was fixed for 2 h at RT in fixative (1.5% paraformaldehyde and 1.5% glutaraldehyde in 0.1 M Sörensen buffer pH 7.2), washed once and then stored overnight at 4°C in the Sörensen buffer. The fixed samples were thereafter prepared for ultrathin sectioning and subjected to TEM as reviewed in Carlemalm (33).
Generation of Reactive Oxygen Species (ROS) and Cytotoxicity
CellTox Green® cytotoxicity assay (Promega) was employed to evaluate cytotoxicity and used according to the manufacturer's protocol. In brief, 1 × 104 cells/well were plated overnight in a 96-well plate. Cells were treated with PM2.5 (1,000, 5,000 and 10,000 ng/ml) for 48 h. The CellTox green dye was diluted 1/500 in CM and applied to the cells. After 15 min of incubation at RT, fluorescence was measured at 485/535 nm using a VICTOR3 1420 Multilabel Counter (Perkin-Elmer) plate reader. ROS was measured by ROS-Glo H2O2 Assay (Promega) according to the manufacturer's protocol. In short: 1 × 104 cells/well were plated in white, clear-bottom 96-well tissue culture plates, incubated over night to adhere and subsequently exposed to PM2.5 (5,000 and 10,000 ng/ml) for 48 h. The H2O2 substrate solution (25 μM) was added to each well and incubated for 6 h at 37°C in a CO2 incubator. With this, the H2O2 substrate reacts directly with H2O2 in the cells and generates a luciferin precursor. Thereafter, ROS-Glo Detection Solution was added and incubated for 20 min at 25°C to generate a luminescence signal. Luminescence was measured using a VICTOR3 1420 Multilabel Counter (Perkin-Elmer) plate reader.
Protein Secretion
Based on the results obtained from the CellTox Green® cytotoxicity assay, we decided to analyze the level of protein secretion from cells exposed to the following PM2.5 concentrations: 5,000 and 10,000 ng/ml. Cells were seeded at a seeding density of 0.3 × 106 per well in a 6-well plate. After 48 h of exposure, the culture supernatants were collected and used to detect the level of human chorionic gonadotropin (hCG), progesterone and IL-6. Human IL-6 was analyzed using a human IL-6-specific ELISA (Invitrogen) according to manufacturer's protocol. The hCG and progesterone analyzes were performed at the Clinical Biochemistry Laboratory at Lund Hospital, Sweden.
Mitochondrial Quantification and Function
Cells subjected to different PM exposures (50–10,000 ng/ml) were analyzed for oxygen consumption using an Oroboros O2k-FluoRespirometer (Oroboros Instruments, Innsbruck, Austria) as previously described (34). The cells were loaded into the chamber at 0.5 × 106 cells/ml in complete CM (0.89 oxygen solubility factor was used), with a total cell count of one million cells per chamber, and allowed to stabilize on routine respiration using endogenous substrates. Oligomycin (1 μg/ml) was added to inhibit the ATP-synthase, and the uncoupler protonophore carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) was then titrated to induce maximum non-coupled respiration. The experiment was terminated by adding rotenone (2 μM) and antimycin (1 μg/ml) to measure non-mitochondrial respiration, a value that was subsequently used to adjust all data for non-mitochondrial cellular oxygen consumption. Citrate Synthase (CS) activity was used as a marker of mitochondrial content as previously described (35). Samples were sonicated and loaded into a 96-well plate in assay buffer with the addition of 300 μM acetyl CoA and 100 μM 5.5-dithiobis-(2-nitrobenzoic acid). In a spectrophotometric plate reader (Bio-Rad Model 680 Microplate Reader; Bio-Rad Laboratories, Hercules, CA, USA) set to 412 nm on a kinetic program with a 1.5-min duration and 10 s intervals, the absorbance of the baseline reaction was measured. This value was deducted from the final reading for each well. Following this, 500 μM of oxaloacetate was added to each well and absorbance was measured. Kinetic plots were individually assessed for a linear change in absorbance and the CS-activity was calculated. Mitochondrial respiratory data was normalized to CS-activity.
Necrosis specific protease, Tripeptidyl peptidase II, was measured by Mitochondrial ToxGlo Assay (Promega) through fluorogenic peptide substrate bis-AAF-R110 according to manufacturer's protocol (36). In brief, cells were seeded at a density of 1 × 104 cells/well in a 96-well plate; after 2 or 48 h of PM2.5 exposure (1,000, 5,000, and 10,000 ng/ml), the cytotoxicity reagent was added to each well, mixed by orbital shaking for 1 min and then incubated at 37°C for 30 min. The fluorescent signal was subsequently measured at 485/535 nm using a VICTOR3 1420 Multilabel Counter (Perkin-Elmer) plate reader.
PAH Analysis in Cells
Cells were seeded at a seeding density of 0.3 × 106 per well in a 6-well plate and exposed to 10,000 ng/ml of PM2.5 for 48 h. After exposure, supernatants were collected and cells were harvested. Cell pellets were lysed with 500 μl of cell lysis buffer and sonicated for 30 min. Five hundred microliters of water (MilliQ) and 40 ng of internal standards were added to the lysed cells as well as to the supernatants (37). Samples were extracted twice with 2 ml of dichloromethane, and organic extracts were combined and evaporated to near dryness. To each sample, 40 ng of recover standard and 100 μl hexane were added, after which the samples were transferred to HPLC glass vials (Agilent). Analysis of PAHs was performed on a GC-MS 7890 (Agilent) as reported earlier (37).
Statistical Analysis
Data obtained through the experiments are reported as mean ± SD. Statistical significance between groups was determined by one-way analysis of variance followed by the Tukey's test using OriginPro 2017 software (OriginLab Corporation, Northampton, MA, USA). Statistical significance was defined as P < 0.05.
Results
Particle Characteristics and Meteorological Data
Sources contributing to the collected PM2.5 were assessed in relation to wind direction (Figure 1). The winds during the collection period were predominantly western (48%) or eastern (50%), with only 2% calm winds/air (defined as winds <1 m/s). During these calm periods, all PM can be assumed to be locally generated. When western winds prevail, the major contributive source is Copenhagen, Denmark (Figure 2); on the other hand, air masses typically pass through southern Finland and the Baltic states before reaching Malmö, Sweden during eastern winds (Figure 3). The characteristics of the collected PM2.5 during different wind directions are displayed in Supplementary Table 1. As the table shows, the levels of PM2.5, as well as soot, are higher during calm air, indicating that the majority of all collected PM is locally generated, and is diluted to some extent when wind speeds reach more than 1 m/s. Levels of PAHs and metals in the collected particles can be found in Supplementary Table 2.
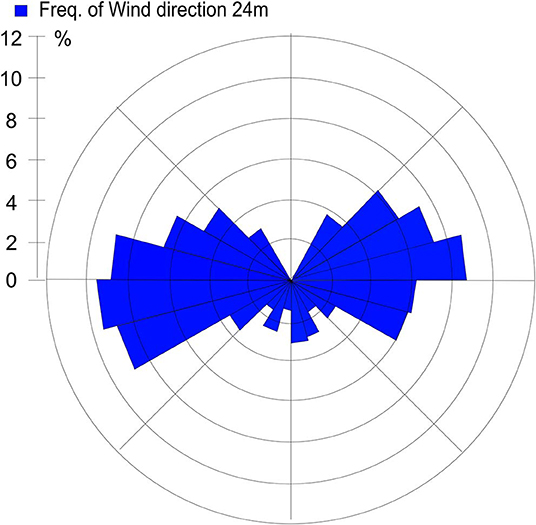
Figure 1. Wind direction frequency from Heleneholmsmasten in Malmö site during the collection period.
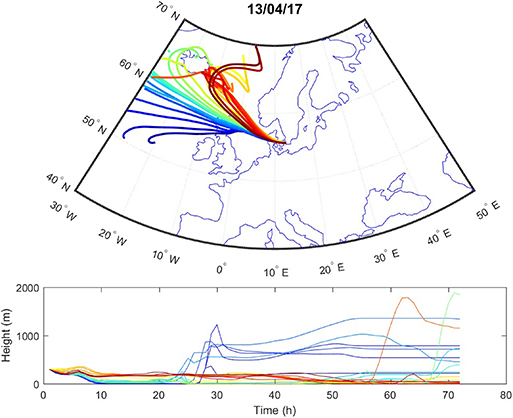
Figure 2. Typical example of air mass origins in Malmö during western winds, trajectories generated using the Hysplit software. The lines show hourly trajectories 72 h backward in time during 1 day.
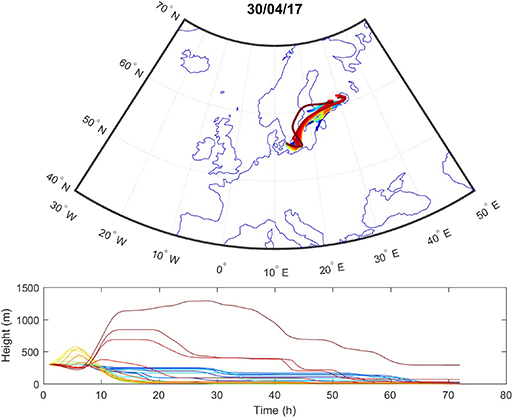
Figure 3. Typical example of air mass origins in Malmö during eastern winds, trajectories generated using the Hysplit software. The lines show hourly trajectories 72 h backward in time during 1 day.
Pregnancy-Related Hormones and Inflammatory Response
HTR-8 cells exposed to PM2.5 for 48 h showed a significant drop in secreted hCG at 5,000 ng/ml (P < 0.001) and 10,000 ng/ml (P < 0.0001; Figure 4A), as well as a dose-dependent decrease between 5,000 and 10,000 ng/ml (P < 0.05). In addition, there was a significant increase (P < 0.01) in progesterone at 5,000 and 10,000 ng/ml (Figure 4B). The PM2.5 exposure also resulted in a significant increase in secretion of IL-6 at 5,000 ng/ml (P < 0.05) and 10,000 ng/ml (P < 0.01; Figure 4C).
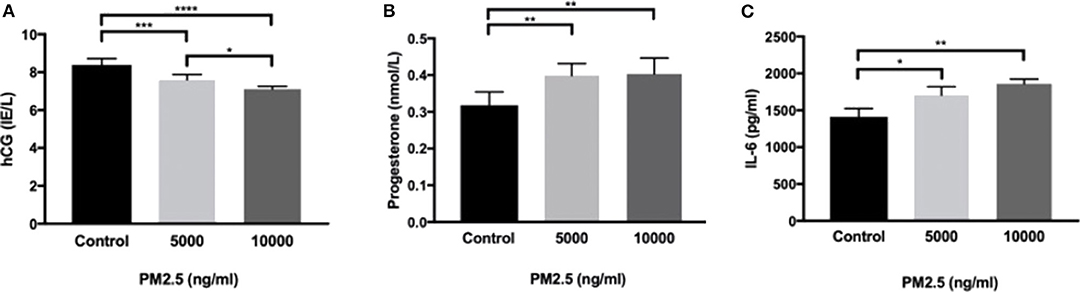
Figure 4. PM2.5 decreases hCG production and increases IL-6 and progesterone production. HTR-8 cells were exposed to PM2.5 for 48 h and subsequently measured for (A) hCG, data is presented as mean ± SD; n = 6, and *P < 0.05, ***P < 0.001, ****P < 0.0001, (B) progesterone, data is presented as mean ± SD; n = 6, and **P < 0.01, and (C) IL-6, data is presented as mean ± SD; n = 3, and *P < 0.05, **P < 0.01 secretion in the CM.
Cell Viability and H2O2 Production Affected by PM2.5 Exposure
Using the CellTox™ Green Cytotoxicity assay to detect changes in membrane integrity, PM2.5-exposed HTR-8 cells displayed significant cytotoxicity at 1,000 and 5,000 ng/ml at 48 h exposure but not at 10,000 ng/ml compared to controls. At baseline measurements (0 h), there was no difference between the groups (Figure 5A). In the HTR-8 cells, the H2O2 levels were measured after 48 h of PM2.5 exposure and showed a significant alteration of H2O2 in all PM2.5-exposed groups (Figure 5B).
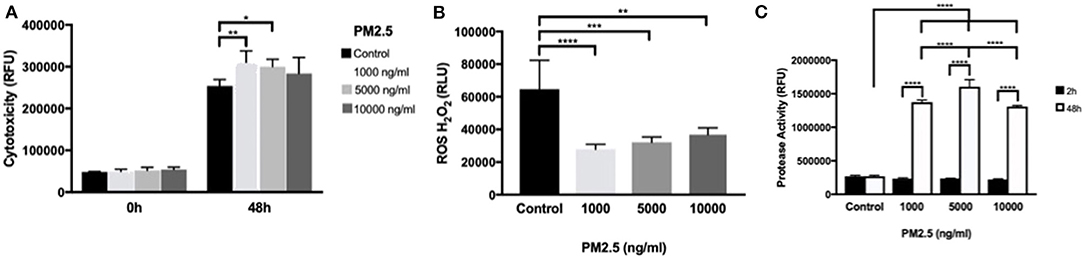
Figure 5. PM2.5 induces cytotoxicity, alters ROS production, and increases protease activity in trophoblast cells. HTR-8 cells were exposed to PM2.5 for 48 h and subsequently analyzed for (A) cytotoxicity, data is presented as mean ± SD; n = 5 in each experimental condition. *P < 0.05, **P < 0.01 (B) ROS production, data is presented as mean ± SD; n = 5 in each experimental condition. **P < 0.01, ***P < 0.001, ****P < 0.0001 (C) Protease activity, Data is presented as mean ± SD; n = 3 in each experimental condition. ***P < 0.001, ****P < 0.0001.
To further investigate cell viability, dead cell protease activity was measured at 2 and 48 h of PM2.5 exposure. Protease activity significantly increased (P < 0.0001) with cell culture duration, and there was a significant difference in protease activity in all concentrations of PM2.5 compared to controls in additions to significant differences between PM2.5 doses, with peak measurements at 5,000 ng/ml (Figure 5C).
PM2.5 Uptake: Organelle Localization and Morphological Changes
Transmission electron microscopy of control HTR-8 cells revealed dark and homogenous cytoplasm and mitochondria with intact inner and outer membranes and well-ordered organelle morphology (Figure 6A, Supplementary Figure 3). In comparison, PM2.5-exposed cells displayed several structural changes including mitochondrial vacuolization, aggregated cytoskeleton, chromatin condensation, dilated ER structures, and autophagosomes. The cytoplasm of exposed cells was also lighter in color compared to control cells and contained numerous vacuoles. Finally, the uptake of PM2.5 particles was visualized within the inner mitochondrial membranes of exposed cells (Figures 6B,C, Supplementary Figures 2, 3). Cells were analyzed after 24 and 48 h following exposure, but no differences were noted related to exposure duration, with all of the above-mentioned morphological changes seen in both groups.
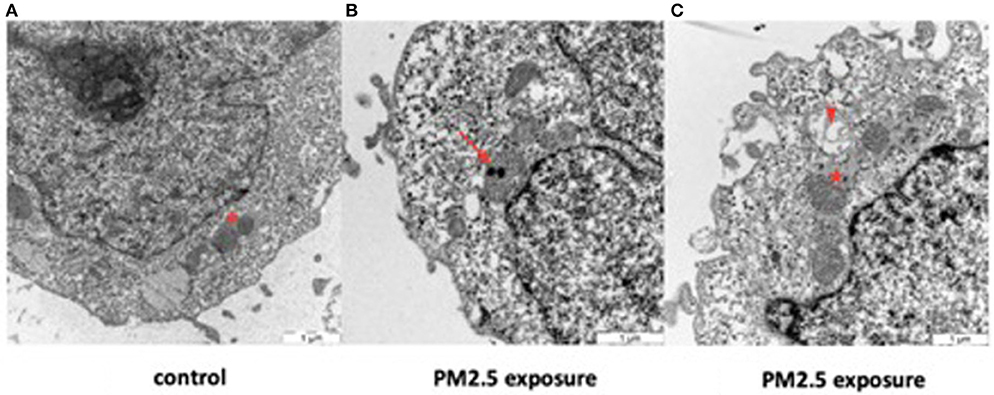
Figure 6. Effect of PM2.5 on mitochondrial morphology in HTR-8 cells. Cells were exposed to a single dose of 500 ng/ml of PM2.5 for 24 or 48 h and subsequently observed through TEM. No differences were seen between the 24 and 48 h exposure groups. Unexposed cells (A) showed mitochondria with normal organelle morphology and intact inner and outer membranes (asterisk). In contrast, exposed cells (B,C) had internalized PM2.5 particles within the inner membrane of the mitochondria (arrow) and displayed mitochondrial vacuolization (arrowhead), dilated ER structures with large pools of ER lumen (star), and various degrees of mitochondrial membrane disruption. Scale bar = 1 μm.
Mitochondrial Function
Neither routine (endogenous) respiration, oligomycin-induced LEAK respiration, maximum non-coupled respiration induced by the protonophore FCCP, nor the control ratio (oligomycin-induced LEAK respiration/maximum non-coupled respiration, Supplementary Figure 1) differed significantly between control cells and cells exposed to various concentrations of PM2.5 for 48 h. Data was normalized to cell number and specific mitochondrial content (citrate synthase activity), respectively, and analyzed without significant findings or trends considered to be consistent or relevant.
PAH Analysis
Exposed cells and supernatants were analyzed for a set of 16 PAHs previously detected in the collected PM2.5 (Supplementary Table 2). The PAHs could only be detected in cell lysates and not in the supernatants. While PAHs levels were close to the detection limit in cell lysates of untreated cells, PAH concentrations ranged from 7 to 35 ng per sample in cells treated with 10,000 ng PM2.5 (Table 1). Specifically, levels were highest for benzo(b)fluoranthene (35 ng per 10,000 ng PM2.5), pyrene (30 ng per 10,000 ng PM2.5) and fluoranthene (27 ng per 10,000 ng PM2.5), followed by phenanthrene, benzo(k)fluoranthene, benzo(a)pyrene, dibenzo(a,h)anthracene, benzo(g,h,i)perylene (13–15 ng per 10,000 ng PM2.5), and anthracene (7 ng per 10,000 ng PM2.5; Table 1). The high concentrations of benzo(b)fluoranthene, pyrene, and fluoranthene at the cellular level are in concordance with the results from the initial PAH analysis of the collected PM2.5 particles (Supplementary Table 2).
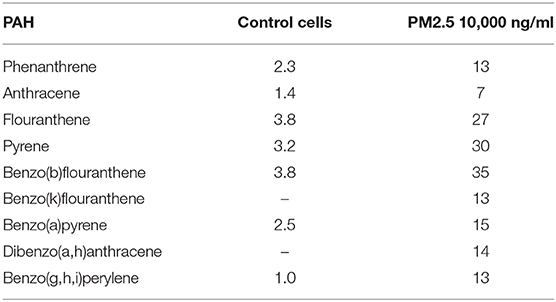
Table 1. PAH in PM2.5 exposed cells.
Discussion
Ambient air pollution in the form of particles is the single largest environmental health threat of our time (13). Studies on the effects of air pollution on human health pertaining to biological pathways and biological effects have mainly focused on the respiratory organs; therefore, more studies on adverse health effects during pregnancy are highly warranted (38). In fact, a growing body of evidence suggests an association between air pollution and birth- and pregnancy outcomes (17, 39). Even though all components of air pollution are harmful to human health, particulate matter in particular can consist of, and carry, a broad range of toxic substances that, depending on particle size and particle solubility, can penetrate the respiratory tract and gain access to the blood stream (40, 41). These such PM compositions are particularly dangerous for pregnancies.
Pregnancy encompasses a period in which susceptibility to exposure-related alterations is heightened, initiating adverse processes that can have life-long implications for both mother and child. Throughout pregnancy, the placenta serves as a gatekeeper between mother and fetus. Indeed, the fetus is indirectly in contact with the same environmental stressors as the mother through the maternal blood circulation. This makes the placenta a useful, albeit temporary, organ for studies on accumulated pregnancy exposures, such as ambient air pollution.
Indeed, a recent study reports evidence of inhaled PM being translocated from the lungs to the placenta (42). Although the placenta's actual exposure to PM2.5 is unknown, we attempted to study doses relevant to human exposure. Levels up to 5,000 ng/ml, for example, could correspond to 25 μg/m3, which is the WHO target for short term exposure (24 h), and 10,000 ng/ml might correspond to levels in a more polluted city with PM2.5 concentrations of 50 μg/m3. Previous studies have estimated the physiological range to be similar to the doses used in this study (43).
PM exposure in general has been linked to inhibition of phagocytosis, stimulation of inflammatory response, and increases in levels of oxidative stress (44). Even specific toxic compounds connected to PM, such as PAHs, are known to be carcinogenic and genotoxic and have also been shown to disrupt endocrine functions (45, 46). In addition, several studies have demonstrated that PAH exposure during pregnancy is associated with adverse pregnancy outcomes, such as small for gestational age (SGA) or IUGR (47). In this study, the molecular effects of PM2.5 on first trimester trophoblast cells were evaluated by studying mechanisms related to cytotoxicity, cellular disruption, mitochondrial function, inflammatory response, and hormone production. We have previously conducted a study of the effects of PM2.5 on trophoblast cells showing a reduction in cellular growth, endoplasmic reticulum stress, and altered protein expression (43). In this study, we confirm our previous results as well as broaden the scope to include additional organelle systems.
In relation to inflammatory response, Interleukin 6 (IL-6) a pro-inflammatory cytokine and, an anti-inflammatory myokine was examined. It has effects on both cells from the immune system and other cell types, such as hepatocytes (48). Interleukin-6 is mainly secreted by monocytes but is also produced by trophoblast cells in vivo (49) as well as in vitro by the HTR-8 trophoblast cell line (50). Interleukin 6 binds to the IL-6 receptor, expressed either on the surface of cells or in a soluble form released by several cell types. This pro-inflammatory cytokine, together with IL-8 and TNF-a, have been suggested as potential inflammatory markers for PE (51), increased plasma levels has been associated with adverse pregnancy outcomes such as PE (52, 53). Our results demonstrate that PM2.5 exposure elevates the production of IL-6 in the HTR-8 cell line. Previous studies have shown that IL-6 stimulation of trophoblast cells induces an increase in hCG production (54). The results from our study diverge from these findings: cells treated with PM2.5 responded with an increased production of IL-6 and a decreased production of hCG. This might be due to possible PM2.5 interference with the steroidogenesis in the trophoblast cells or other upstream mechanisms. The increased production of IL-6 indicate that the trophoblast cells were in a heightened inflammatory state; moreover, they lacked the necessary increase in hCG, which is pivotal to maintaining viable and healthy placental development. In fact, pregnancy-associated diseases such as PE, IUGR and preterm births, all have defect placentation in combination with aggravated inflammatory responses as common denominators (55, 56). One could, therefore, speculate that the imbalance between the increased inflammation caused by IL-6 and the absence of hCG increase could be part of the explanation as to why air pollution has been linked to these pregnancy complications (57) as well as PE and IUGR (17, 58). During the initial phase of pregnancy progesterone is produced by corpus luteum, but by the end of the first trimester the progesterone production is in full taken over by the trophoblast cells. In this study we used a first trimester cell line, and it is somewhat surprising that we observe an increase in progesterone. The progesterone and hCG production in trophoblasts are interconnected by a range of mediators (59). The fact that hCG and progesterone jointly drive the trophoblasts to form a syncytium in the very early stages in pregnancy, and that disturbances have been observed in PE and IUGR, our increase in progesterone it is an interesting finding (60, 61). The levels of progesterone were significantly elevated, although close to the detection limit of the assay.
Experimental studies have established an association between exposure to PM2.5 containing high levels of metals or PAH and increased ROS production in the exposed cells (62). Correspondingly, our previous and current results illustrate that trophoblast cells absorb PAHs and, thereafter, exhibit an altered production of ROS (43). Considering that ROS is a major driver of the progression of PE, through systemic inflammation and vascular endothelial damage, these results are unexpected and allude to the complexity of PM2.5's impact on biological systems.
The mitochondria are central to upholding the oxidation-reduction balance, also called redox balance in the cell, being both a target for, and producer of ROS (63). Enhanced levels of ROS tilt the cellular redox balance, which in turn trigger inflammation and eventually apoptosis (64, 65). Exposure to PM2.5 has been shown to target mitochondria specifically and induce mitochondrial ROS production in human lung cells (66). The smaller particles, ultrafine particles (UFPs), have been shown to localize to mitochondria of endothelial cells, where they induce major structural damage (67). Combustion-related particles have also been shown to cause mitochondrial structural damage in lung cells (68, 69). Studies on placentas exposed to higher levels of air pollution show placental mitochondrial DNA methylation (70) and decreased placental mitochondrial DNA content (71). Such mitochondrial dysfunction has been proposed to be one of the mechanisms contributing to the dysfunctional placenta in PE (72).
Our results confirm that PM2.5 specifically targets the mitochondria in the trophoblast cells, visualized by the localization of the particles to this organelle. The structural damage of the mitochondria observed by TEM is a direct indicator of mitochondrial dysfunction. Potential explanations for this dysfunction could include PM2.5's physical presence localized to the mitochondria, its interference with redox balance and molecular processes in the mitochondria and/or its chemical PAH content. Indeed, the chemical content of the particles may add to the cellular toxicity observed. Our findings showed that the same PAHs were found in the PM2.5 particles were also discovered in the, suggesting that PAH may disorb, or leak, from the particles in a biological matrix, thus being able to cause cell damage. Despite the limited sample size of the PAH analysis on cell lysates, these results clearly indicate that there is an uptake of PAH into the cells.
No dose-dependent effect of exposure to PM2.5 on mitochondrial respiration was found in this study. We did, however, notice a somewhat surprising decline in H2O2 production in the exposed cells. One could speculate that this might be due to the increased cytotoxicity present in the PM2.5 exposed cells, which would effectively leave fewer viable cells able to produce H2O2. Cells exposed to a single dose of PM2.5 showed a significant decrease in cell count after 7 days (Supplementary Figure 4). Cytotoxicity at the highest doses is not statistically significant compared to the control, therefore it would be difficult to draw any conclusions. One could speculate if reduced proliferation as a result of exposure to high doses of PM2.5, could explain this finding. Similar speculations of reduced proliferation can be made regarding the protease activity being lower in the higher fraction of PM2.5. In the PM2.5 exposed groups there was a trend of increasing ROS-production with increasing dosage. However, due to the small sample size, statistical significance was not upheld. Further studies are needed to determine the effects of PM2.5 on ROS-production. In addition to the local effects of the PM2.5 in the mitochondria, pro-inflammatory factors such as IL-6 have been suggested to induce mitochondrial damage (73). The results from this study not only confirms that the particles were internalized by the cells, as observed by TEM imaging (Figure 6), but also showed that PAHs could be released from the particles inside the cells, thus having a possible role in inducing the cellular toxicity observed.
The HTR-8 trophoblast cell line does not form a syncytia, a potential drawback of this study, however the HTR-8 trophoblast cell line is a first trimester cell line and hence displaying age typical characteristics for the period of gestation when the pathological changes to the placenta associated with PE is thought to occur. Future avenues to explore could entail syncytia forming trophoblast, placental organoid cultures and/or the dual-placenta-perfusion model.
To understand the correlation between PM2.5 exposure and PE, as well as other adverse pregnancy outcomes, it is essential to gain further knowledge of the mechanisms through which PM2.5 interferes with placental function during pregnancy. Air pollution has already been linked to placental transcriptome changes (74), placental DNA methylation (75) and a reduced placenta weight (76). Further, women exposed to high levels of air pollution from cooking with solid fuels have shown chronic placental hypoxia (77) and fetal thrombotic vasculopathy (78). Adding to this body of knowledge, our data suggest that exposure to PM2.5 in vitro has the potential to induce cellular toxicity as well as hormone dysregulation, oxidative damage, inflammatory response, and interference with mitochondria in first trimester placental cells (HTR-8 cells). Future studies will have to elucidate if theses findings are translatable to in vivo conditions.
Public Health Impact
The effects of air pollution on pregnancy and birth outcomes are not negligible. One study estimated that 11% of term low birth weight cases would be avoided if the PM2.5 concentration was reduced by 5 μg/m3 (58). Another suggests that 11% of all PE cases were attributable to exhaust emissions in Malmö, Sweden (79). Indeed, the PM2.5 used in our present study was collected in real-life conditions in Malmö, Sweden in order to mimic the actual exposure pregnant women face in an urban setting. Our findings on the subsequent effects on human trophoblasts cells are suggestive of underlying placental mechanisms potentially contributing to the adverse effects air pollution exposure exerts during pregnancy.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
ÅN, LE, EM, CI, BS, AK, TL, and SH conceived and designed the experiments. ÅN, LE, AK, EÅ, CI, and MS performed the experiments. ÅN, LE, EM, CI, MS, EE, JE, BS, TL, AK, and SH analyzed the data. ÅN, LE, EM, and SH wrote the paper. ÅN, LE, CI, MS, EÅ, JE, AK, BS, TL, EE, EM, and SH revised the manuscript. ÅN, LE, CI, MS, EÅ, JE, AK, BS, TL, EE, EM, and SH approved the final version of the manuscript.
Funding
This article was funded by Swedish Research Council FORTE 2016-00572 and part of ReproUnion collaborative study, co-financed by the European Union, Interreg V ÖKS. and ALF, Region Skåne, Vetenskapsrådet and Wallenberg foundation.
Conflict of Interest
SH holds patents for the diagnosis and treatment of preeclampsia. SH is co-founder of the companies Preelumina Diagnostics AB and A1M pharma AB.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge Malmö City Environmental Department.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2020.00075/full#supplementary-material
Abbreviations
PE, preeclampsia; PM2.5, particulate matter of size <2.5 μm; PAHs, polycyclic aromatic hydrocarbons; PM, particulate matter; IL-6, Interleukin 6; ROS, Reactive Oxygen Species; TEM, transmission electron microscopy; RT, room temperature; hCG, human chorionic gonadotropin; SD, standard deviation; PBS, phosphate-buffered saline; CS, citrate synthase; IUGR, intra uterine growth restriction; NCEP, Centre of Environmental Predictions; GDAS, global data assimilation system.
References
1. Dominici F, McDermott A, Zeger SL, Samet JM. National maps of the effects of particulate matter on mortality: exploring geographical variation. Environ Health Perspect. (2003) 111:39–44. doi: 10.1289/ehp.5181
2. Lippmann M. Toxicological and epidemiological studies on effects of airborne fibers: coherence and public [corrected] health implications. Crit Rev Toxicol. (2014) 44:643–95. doi: 10.3109/10408444.2014.928266
3. Monn C, Becker S. Cytotoxicity and induction of proinflammatory cytokines from human monocytes exposed to fine (PM2.5) and coarse particles (PM10-2.5) in outdoor and indoor air. Toxicol Appl Pharmacol. (1999) 155:245–52. doi: 10.1006/taap.1998.8591
4. Schneider J, Hock N, Weimer S, Borrmann S, Kirchner U, Vogt R, et al. Nucleation particles in diesel exhaust: composition inferred from in situ mass spectrometric analysis. Environ Sci Technol. (2005) 39:6153–61. doi: 10.1021/es049427m
5. Utell MJ, Frampton MW, Zareba W, Devlin RB, Cascio WE. Cardiovascular effects associated with air pollution: potential mechanisms and methods of testing. Inhal Toxicol. (2002) 14:1231–47. doi: 10.1080/08958370290084881
6. Balasubramanian SK, Poh KW, Ong CN, Kreyling WG, Ong WY, Yu LE. The effect of primary particle size on biodistribution of inhaled gold nano-agglomerates. Biomaterials. (2013) 34:5439–52. doi: 10.1016/j.biomaterials.2013.03.080
7. Geiser M, Kreyling WG. Deposition and biokinetics of inhaled nanoparticles. Part Fibre Toxicol. (2010) 7:2. doi: 10.1186/1743-8977-7-2
8. Kreyling WG. Dosimetry of nanomaterials after different routes of exposure. Toxicol Lett. (2013) 221:S6. doi: 10.1016/j.toxlet.2013.06.022
9. Nemmar A, Vanbilloen H, Hoylaerts MF, Hoet H, Verbruggen A, Nemery B. Passage of intratracheally instilled ultrafine particles from the lung into the systemic circulation in hamster. Am J Respir Crit Care Med. (2001) 164:1665–8. doi: 10.1164/ajrccm.164.9.2101036
10. Oberdörster G, Sharp Z, Atudorei V, Elder A, Gelein R, Lunts A, et al. Extrapulmonary translocation of ultrafine carbon particles following whole-body inhalation exposure of rats. J Toxicol Environ Health A. (2002) 65:1531–43. doi: 10.1080/00984100290071658
11. Semmler-Behnke M, Lipka J, Wenk A, Hirn S, Schäffler M, Tian F, et al. Size dependent translocation and fetal accumulation of gold nanoparticles from maternal blood in the rat. Part Fibre Toxicol. (2014) 11:33. doi: 10.1186/s12989-014-0033-9
12. Brauer M, Freedman G, Frostad J, van Donkelaar A, Martin RV, Dentener F, et al. Ambient air pollution exposure estimation for the global burden of disease 2013. Environ Sci Technol. (2016) 50:79–88. doi: 10.1021/acs.est.5b03709
13. Forouzanfar MH, Afshin A, Alexander LT, Ross Anderson H, Bhutta ZA, Biryukov S, et al. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. (2016) 388:1659–724. doi: 10.1016/S0140-6736(16)31679-8
14. Shah ASV, Langrish JP, Nair H, McAllister DA, Hunter AL, Donaldson K, et al. Global association of air pollution and heart failure: a systematic review and meta-analysis. Lancet. (2013) 382:1039–48. doi: 10.1016/S0140-6736(13)60898-3
15. Stafoggia M, Cesaroni G, Peters A, Andersen ZJ, Badaloni C, Beelen R, et al. Long-term exposure to ambient air pollution and incidence of cerebrovascular events: results from 11 European cohorts within the ESCAPE project. Environ Health Perspect. (2014) 122:919–25. doi: 10.1289/ehp.1307301
16. Pedersen M, Halldorsson TI, Olsen SF, Hjortebjerg D, Ketzel M, Grandström C, et al. Impact of road traffic pollution on pre-eclampsia and pregnancy-induced hypertensive disorders. Epidemiology. (2017) 28:99–106. doi: 10.1097/EDE.0000000000000555
17. Pedersen M, Stayner L, Slama R, Sørensen M, Figueras F, Nieuwenhuijsen MJ, et al. Ambient air pollution and pregnancy-induced hypertensive disorders: a systematic review and meta-analysis. Hypertension. (2014) 64:494–500. doi: 10.1161/HYPERTENSIONAHA.114.03545
18. Dadvand P, Figueras F, Basagana X, Beelen R, Martinez D, Cirach M, et al. Ambient air pollution and preeclampsia: a spatiotemporal analysis. Environ Health Perspect. (2013) 121, 1365–71. doi: 10.1289/ehp.1206430
19. Nahidi F, Gholami R, Rashidi Y, Alavi Majd H. Relationship between air pollution and pre-eclampsia in pregnant women: a case-control study. East Mediterr Health J. (2014) 19:60–6.
20. Malmqvist E, Jensen EL, Westerberg K, Stroh E, Rittner R, Gustafsson S, et al. Gestational diabetes and preeclampsia in association with air pollution at levels below current air quality guidelines. Environ Health Perspect. (2013) 121:488–93. doi: 10.1289/ehp.1205736
21. Ritz B, Wilhelm M, Hoggatt KJ, Ghosh JKC. Ambient air pollution and preterm birth in the environment and pregnancy outcomes study at the University of California, Los Angeles. Am J Epidemiol. (2007) 166:1045–52. doi: 10.1093/aje/kwm181
22. Brown MA, Magee LA, Kenny LC, Karumanchi SA, McCarthy FP, et al. The hypertensive disorders of pregnancy: ISSHP classification, diagnosis and management recommendations for international practice. Pregnancy Hypertens. (2018) 13:291–310. doi: 10.1016/j.preghy.2018.05.004
23. Ananth CV, Keyes KM, Wapner RJ. Pre-eclampsia rates in the United States, 1980-2010: age-period-cohort analysis. BMJ. (2013) 347. doi: 10.1136/bmj.f6564
24. Roberts CL, Ford JB, Algert CS, Antonsen S, Chalmers J, Cnattingius S, et al. Population-based trends in pregnancy hypertension and pre-eclampsia: an international comparative study. BMJ Open. (2011) 1:101. doi: 10.1136/bmjopen-2011-000101
25. Behrens I, Basit S, Lykke JA, Ranthe MF, Wohlfahrt J, Bundgaard H, et al. Association between hypertensive disorders of pregnancy and later risk of cardiomyopathy. JAMA. (2016) 315:1026–33. doi: 10.1001/jama.2016.1869
26. Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. (2010) 376:631–44. doi: 10.1016/S0140-6736(10)60279-6
27. Montiel-Dávalos A, de Jesús Ibarra-Sánchez M, Ventura-Gallegos J, Alfaro-Moreno E, López-Marure R. Oxidative stress and apoptosis are induced in human endothelial cells exposed to urban particulate matter. Toxicol Vitro. (2010) 24:135–41. doi: 10.1016/j.tiv.2009.08.004
28. Hansson SR, Nääv Å, Erlandsson L. Oxidative stress in preeclampsia and the role of free fetal hemoglobin. Front Physiol. (2015) 5:516. doi: 10.3389/fphys.2014.00516
29. Gakidou E, Afshin A, Alemu Abajobir A, Hassen Abate K, Abbafati C, Abbas KM, et al. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. (2017) 390:1345–422. doi: 10.1016/S0140-6736(17)32366-8
30. Kliucininkas L, Martuzevicius D, Krugly E, Prasauskas T, Kauneliene V, Molnar P, et al. Indoor and outdoor concentrations of fine particles, particle-bound PAHs and volatile organic compounds in Kaunas, Lithuania. J Environ Monit. (2011) 13:182–91. doi: 10.1039/C0EM00260G
31. Stein AF, Draxler RR, Rolph GD, Stunder BJB, Cohen MD. Noaa's hysplit atmospheric transport and dispersion modeling system. B Am Meteorol Soc. (2015) 2059–77. doi: 10.1175/BAMS-D-14-00110.1
32. Suess HE. Radiocarbon concentration in modern wood. Science. (1955) 122:415–7. doi: 10.1126/science.122.3166.415-a
33. Carlemalm E. Lowicryl resins in microbiology. J Struct Biol. (1990) 104:189–91. doi: 10.1016/1047-8477(90)90075-N
34. Sjövall F, Morota S, Persson J, Hansson MJ, Elmér E. Patients with sepsis exhibit increased mitochondrial respiratory capacity in peripheral blood immune cells. Crit Care. (2013) 17:R152. doi: 10.1186/cc12831
35. Ehinger JK, Morota S, Hansson MJ, Paul G, Elmér E. Mitochondrial dysfunction in blood cells from amyotrophic lateral sclerosis patients. J Neurol. (2015) 262:1493–503. doi: 10.1007/s00415-015-7737-0
36. Niles AL, Moravec RA, Hesselberth E, Scurria MA, Daily WJ, Riss TL. A homogeneous assay to measure live and dead cells in the same sample by detecting different protease biomarkers. Anal Biochem. (2007) 366:197–206. doi: 10.1016/j.ab.2007.04.007
37. Strandberg B, Julander A, Sjöström M, Lewné M, Koca Akdeva H, Bigert C. Evaluation of polyurethane foam passive air sampler (PUF) as a tool for occupational PAH measurements. Chemosphere. (2018) 190:35–42. doi: 10.1016/j.chemosphere.2017.09.106
38. REVIHAAP. Review of Evidence on Health Aspects of Air Pollution – REVIHAAP Project. Technical report. World Health Organization; Regional Office for Europe (2013). Available online at: www.euro.who.int (accessed April 15, 2019).
39. Stieb DM, Chen L, Eshoul M, Judek S. Ambient air pollution, birth weight and preterm birth: a systematic review and meta-analysis. Environ Res. (2012) 117:100–11. doi: 10.1016/j.envres.2012.05.007
40. Brown JS, Zeman KL, Bennett WD. Ultrafine particle deposition and clearance in the healthy and obstructed lung. Am J Respir Crit Care Med. (2002) 166:1240–7. doi: 10.1164/rccm.200205-399OC
41. Franck U, Odeh S, Wiedensohler A, Wehner B, Herbarth O. The effect of particle size on cardiovascular disorders–the smaller the worse. Sci Total Environ. (2011) 409:4217–21. doi: 10.1016/j.scitotenv.2011.05.049
42. Liu N, Miyashita L, McPhail G, Thangaratinam S, Grigg J. Do Inhaled Carbonaceous Particles Translocate From the Lung to the Placenta? Abstract no: PA360, Occupational and Environmental Lung Diseases: Asthma and the Airways. Paris: European Respiratory Society International Congress (2018). doi: 10.1183/13993003.congress-2018.PA360
43. Familari M, Nääv Å, Erlandsson L, de Longh RU, Isaxon C, Strandberg B, et al. Exposure of trophoblast cells to fine particulate matter air pollution leads to growth inhibition, inflammation and ER stress. PLoS ONE. (2019) 14:e0218799. doi: 10.1371/journal.pone.0218799
44. Terzano C, Di Stefano F, Conti V, Graziani E, Petroianni E. Air pollution ultrafine particles: toxicity beyond the lung. Eur Rev Med Pharmacol Sci. (2010) 14:809–21.
45. Boström CE, Gerde P, Hanberg A, Jernström A, Johansson C, Kyrklund T, et al. Cancer risk assessment, indicators, and guidelines for polycyclic aromatic hydrocarbons in the ambient air. Environ Health Perspect. (2002) 110(Suppl 3):451–88. doi: 10.1289/ehp.110-1241197
46. Kazeto Y, Place AR, Trant JM. Effects of endocrine disrupting chemicals on the expression of CYP19 genes in Zebrafish (Danio rerio) juveniles. Aquat Toxicol. (2004) 69:25–34. doi: 10.1016/j.aquatox.2004.04.008
47. Stieb DM, Chen L, Beckerman BS, Jerrett M, Crouse DL, Rasugu Omariba DW, et al. Associations of Pregnancy Outcomes and PM2.5 in a National Canadian Study. Environ Health Perspect. (2016) 124:243–9. doi: 10.1289/ehp.1408995
48. Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci. (2012) 8:1237–47. doi: 10.7150/ijbs.4989
49. Park H-R, Loch-Caruso R. Protective effect of nuclear factor E2-related factor 2 on inflammatory cytokine response to brominated diphenyl ether-47 in the HTR-8/SVneo human first trimester extravillous trophoblast cell line. Toxicol Appl Pharmacol. (2014) 281:67–77. doi: 10.1016/j.taap.2014.09.015
50. Svinarich DM, Bitonti OM, Romero R, Gonik B. Induction and posttranslational expression of cytokines in a first-trimester trophoblast cell line by lipopolysaccharide. Am J Obstet Gynecol. (1996) 175(4 Pt 1):970–3. doi: 10.1016/S0002-9378(96)80034-2
51. Black KD, Horowitz JA. Inflammatory markers and preeclampsia: a systematic review. Nurs Res. (2018) 67:242–51. doi: 10.1097/NNR.0000000000000285
52. Ouyang Y-Q, Li S-J, Zhang Q, Cai H-B, Chen H. Interactions between inflammatory and oxidative stress in preeclampsia. Hypertens Pregnancy. (2009) 28:56–62. doi: 10.1080/10641950802233064
53. Vrachnis N, Vitoratos N, Iliodromiti Z, Sifakis S, Deligeoroglou E, Creatsas G. Intrauterine inflammation and preterm delivery. Ann N Y Acad Sci. (2010) 1205:118–22. doi: 10.1111/j.1749-6632.2010.05684.x
54. Nishino E, Matsuzaki N, Masuhiro K, Kameda T, Taniguchi T, Takagi T. Trophoblast-derived interleukin-6 (IL-6) regulates human chorionic gonadotropin release through IL-6 receptor on human trophoblasts. J Clin Endocrinol Metab. (1990) 71:436–41. doi: 10.1210/jcem-71-2-436
55. Fisher SJ. Why is placentation abnormal in preeclampsia? Am J Obstet Gynecol. (2015) 213(4 Suppl):115–22. doi: 10.1016/j.ajog.2015.08.042
56. Roberts JM, Escudero C. The placenta in preeclampsia. Pregnancy Hypertens. (2012) 2:72–83. doi: 10.1016/j.preghy.2012.01.001
57. Grippo A, Zhang J, Chu L, Guo Y, Qiao L, Zhang J, et al. Air pollution exposure during pregnancy and spontaneous abortion and stillbirth. Rev Environ Health. (2018) 33:247–64. doi: 10.1515/reveh-2017-0033
58. Pedersen M, Giorgis-Allemand L, Bernard C, Aguilera I, Nybo Andersen A-M, Ballester F, et al. Ambient air pollution and low birthweight: a European cohort study (ESCAPE). Lancet Respir Med. (2013) 1:695–704. doi: 10.1016/S2213-2600(13)70192-9
59. Gauster M, Moser G, Orendi K, Huppertz B. Factors involved in regulating trophoblast fusion: potential role in the development of preeclampsia. Placenta. (2009) 30:S49–54. doi: 10.1016/j.placenta.2008.10.011
60. Sullivan MHF. Endocrine cell lines from the placenta. Mol Cell Endocrinol. (2004) 228:103–19. doi: 10.1016/j.mce.2003.03.001
61. Tuckey RC. Progesterone synthesis by the human placenta. Placenta. (2005) 26:273–81. doi: 10.1016/j.placenta.2004.06.012
62. Manzo S, Rocco A, Carotenuto R, De Luca Picione F, Miglietta ML, Rametta G, et al. Investigation of ZnO nanoparticles' ecotoxicological effects towards different soil organisms. Environ Sci Pollut Res Int. (2011) 18:756–63. doi: 10.1007/s11356-010-0421-0
63. Blajszczak C, Bonini MG. Mitochondria targeting by environmental stressors: Implications for redox cellular signaling. Toxicology. (2017) 391:84–9. doi: 10.1016/j.tox.2017.07.013
64. Upadhyay D, Panduri V, Ghio A, Kamp DW. Particulate matter induces alveolar epithelial cell DNA damage and apoptosis: role of free radicals and the mitochondria. Am J Respir Cell Mol Biol. (2003) 29:180–7. doi: 10.1165/rcmb.2002-0269OC
65. Gosset P, Garcon G, Casset A, Fleurisse L, Hannothiaux MH, Creusy C, et al. Benzo(a)pyrene-coated onto Fe2O3 particles-induced apoptotic events in the lungs of Spraque-Dawley rats. Toxicol Lett. (2003) 143:223–32. doi: 10.1016/S0378-4274(03)00153-X
66. Rodriquez-Cotto RI, Ortiz-Martínez MG, Jiménez-Vélez BD. Organic extracts from African dust storms stimulate oxidative stress and induce inflammatory responses in human lung cells through Nrf2 but nog NF-kB. Environ Toxicol Pharmacol. (2015) 39:845–56. doi: 10.1016/j.etap.2015.02.015
67. Li N, Sioutas C, Cho A, Schmitz D, Misra C, Sempf J, et al. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ Health Perspect. (2003) 111:455–60. doi: 10.1289/ehp.6000
68. Bhargava A, Tamrakar S, Aglawa A, Lad H, Srivastava RK, Mishra DK, et al. Ultrafine particulate matter impairs mitochondrial redox homeostasis and activates phosphatidylinositol 3-kinase mediated DNA damage responses in lymphocytes. Environ Pollut. (2018) 234:406–19. doi: 10.1016/j.envpol.2017.11.093
69. Bove H, Devoght J, Rasking L, Peters M, Slenders E, Roeffaers M, et al. Combustion-derived particles inhibit in vitro human lung fibroblast-mediated matrix remodeling. J Nanobiotechnol. (2018) 16:82. doi: 10.1186/s12951-018-0410-x
70. Janssen BG, Byun HM, Gyselaers W, Lefebvre W, Baccarelli AA, Nawrot TS. Placental mitochondrial methylation and exposure to airborne particulate matter in the early life environment: An ENVIRONAGE birth cohort study. Epigenetics. (2015) 10:536–44. doi: 10.1080/15592294.2015.1048412
71. Janssen BG, Munters E, Pieters N, Smeets K, Cox B, Cuypers A, et al. Placental mitochondrial DNA content and particulate air pollution during in utero life. Environ Health Perspect. (2012) 120:1346–52. doi: 10.1289/ehp.1104458
72. Teran E, Hernández I, Tana L, Teran S, Galaviz-Hernandez C, Sosa-Macías M. Mitochondria and Coenzyme Q10 in the pathogenesis of preeclampsia. Front Physiol. (2018) 9:1561. doi: 10.3389/fphys.2018.01561
73. Zell R, Geck P, Werdan K, Boekstegers. TNF-alpha and IL-1 alpha inhibit both pyruvate dehydrogenase activity and mitochondrial function in cardiomyocytes: evidence for primary impairment of mitochondrial function. Mol Cell Biochem. (1997) 177:61–7. doi: 10.1023/A:1006896832582
74. Saenen ND, Plusquin M, Bijnens E, Janssen BG, Gyselaers W, Cox B, et al. In utero fine particle air pollution and placental expression of genes in the brain-derived neurotrophic factor signaling pathway: an ENVIRONAGE birth cohort study. Environ Health Perspect. (2015) 123:834–40. doi: 10.1289/ehp.1408549
75. Janssen BG, Godderis L, Pieters N, Poels K, Kicinski M, Cuypers A, et al. Placental DNA hypomethylation in association with particulate air pollution in early life. Part Fibre Toxicol. (2013) 10:22. doi: 10.1186/1743-8977-10-22
76. Ghasemi-Tehrani H, Fallah S, Mozafarian N, Miranzadeh S, Sadeghi S, Azidhak A. Effect of exposure to air pollution on placental weight in Isfahan-Iran. J Family Reprod Health. (2017) 11:90–6.
77. Dutta A, Khramtsova G, Brito K, Alexander D, Mueller A, Chinthala S, et al. Household air pollution andchronic hypoxia in the placenta of pregnant Nigerian women: a randomized controlled ethanol Cookstove intervention. Sci Total Environ. (2018) 619–620:212–20. doi: 10.1016/j.scitotenv.2017.11.091
78. Wylie BJ, Matechi E, Kishashu Y, Fawzi W, Premji Z, Coull BA, et al. Placental pathology associated with household air pollution in a cohort of pregnant women from Dar es Salaam, Tanzania. Environ Health Perspect. (2017) 125:134–40. doi: 10.1289/EHP256
Keywords: placenta, trophoblast cells, air pollution, PM2.5, preeclampsia, inflammation, polycyclic aromatic hydrocarbons, mitochondria
Citation: Nääv Å, Erlandsson L, Isaxon C, Åsander Frostner E, Ehinger J, Sporre MK, Krais AM, Strandberg B, Lundh T, Elmér E, Malmqvist E and Hansson SR (2020) Urban PM2.5 Induces Cellular Toxicity, Hormone Dysregulation, Oxidative Damage, Inflammation, and Mitochondrial Interference in the HRT8 Trophoblast Cell Line. Front. Endocrinol. 11:75. doi: 10.3389/fendo.2020.00075
Received: 26 July 2019; Accepted: 04 February 2020;
Published: 12 March 2020.
Edited by:
GianLuca Colussi, University of Udine, ItalyReviewed by:
Ana Claudia Zenclussen, University Hospital Magdeburg, GermanyMartin Gauster, Medical University Graz, Austria
Ekkehard Schleussner, University Hospital Jena, Germany
Copyright © 2020 Nääv, Erlandsson, Isaxon, Åsander Frostner, Ehinger, Sporre, Krais, Strandberg, Lundh, Elmér, Malmqvist and Hansson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Åsa Nääv, YXNhLm5hYXZAbWVkLmx1LnNl
†These authors have contributed equally to this work