 Carlotta Pozza1†Franz Sesti1†Carla Di Dato1
Carlotta Pozza1†Franz Sesti1†Carla Di Dato1 Emilia Sbardella1Riccardo Pofi1Francesca Schiavi2Vincenzo Bonifacio1
Emilia Sbardella1Riccardo Pofi1Francesca Schiavi2Vincenzo Bonifacio1 Andrea M. Isidori1
Andrea M. Isidori1 Antongiulio Faggiano1
Antongiulio Faggiano1 Andrea Lenzi1
Andrea Lenzi1 Elisa Giannetta1*
Elisa Giannetta1*- 1Department of Experimental Medicine, Sapienza University of Rome, Rome, Italy
- 2Familial Cancer Clinic and Oncoendocrinology, Veneto Institute of Oncology, IRCCS, Padua, Italy
Introduction: Pheochromocytomas (PCCs), paragangliomas (PGLs), ganglioneuroblastomas (GNBs), and ganglioneuromas (GNs) are neuroendocrine neoplasms (NENs) that were thought to share a common embryologic origin from neural crest cells. However, they rarely occur concurrently and recurrently. We describe the case of a 40-years-old woman with “composite PCC-GN” and multiple NENs and neuroblastic tumors.
Case presentation: The patient was first referred to our department at the age of 15 years for paroxysmal hypertension, headache, sweating, and watery diarrhea. Her personal history included the diagnosis of a pelvic GNB with lumbar–aortic lymph node metastases at 11 months. Her family history was positive for cerebral glioblastoma multiforme (father). An abdominal ultrasound showed a right adrenal mass that histologically was a “composite adrenal PCC-GN.” The symptoms disappeared after surgery. At the age of 20 years, the symptoms returned: computed tomography (CT) and 131I-metaiodobenzylguanidine (MIBG) scintigraphy showed an inter-aortocaval mass, found histologically to be an inter-aortocaval PGL. Her symptoms reappeared again at 28 years: CT and magnetic resonance imaging revealed four left adrenal gland nodules, found histologically to be multifocal PCCs with some atypia. Genetic screening for VHL, RET, NF1, Tp53, SDHD, SDHB, SDHC, SDHAF2, SDHAF3, SDHA, and TMEM127 was negative. Mutational analysis of the MAX gene revealed the presence of a novel heterozygous variant, c299G>C (p.Arg100Pro, NM_002382.5) that the bioinformatics prediction programs defined as noxious and causative of pathology.
Conclusion: This report represents the first description of a co-occurrence of multiple composite PCC-GN and neuroblastic tumors. The long timeline of the presentation of the NENs/neuroblastic tumors from infancy to adulthood requires a lifelong follow-up for this patient. Moreover, the importance of this case lies in the presence of a novel MAX gene variant deleterious, harmful, and causative of pathology, confirmed by Sanger sequencing and never been associated before with multiple composite PCC-GN. The present case underlines the importance of precision medicine and molecular diagnoses for hereditary pheochromocytomas and paragangliomas, suggesting that when they occur in early childhood, it is necessary to perform an extensive genetic investigation and a lifelong follow-up.
Introduction
Pheochromocytomas (PCCs) are chromaffin tumors arising from the adrenal medulla, while extra-adrenal chromaffin and non-chromaffin paraganglial tumors are classified as paragangliomas (PGLs) (1). PCCs, PGLs, ganglioneuroblastomas (GNBs), and ganglioneuromas (GNs) are neuroendocrine neoplasms (NENs) (1). The International Neuroblastoma Pathology Classification divides neuroblastic tumors into four categories on the basis of their morphology, clinical features, and behavior: neuroblastoma, nodular GNB, intermixed GNB, and GN (2). Neuroblastoma is a malignant tumor comprising neuroblasts with a poor Schwannian stroma, and has the lowest degree of cell maturation and differentiation. It typically undergoes spontaneous regression or differentiation into GNB and GN (3). GNB consists of ganglion cells and neuroblasts with a varying proportion of Schwannian stroma and has an intermediate malignant potential; the nodular subtype is more aggressive, has a worst prognosis and a poorer response to therapy than the intermixed subtype. GN is a benign tumor characterized by the dominance of Schwannian stroma over neuronal elements (4, 5). PCCs are rarely associated with other types of neuroblastic non-PCC tumors with the same embryologic origin, the so-called “composite pheochromocytoma” (6, 7).
From the perspective of precision medicine (8), the recommended workup for NENs includes plasma or 24-h urine fractioned metanephrine, chest/abdominal multiphasic computed tomography (CT), and magnetic resonance imaging (MRI) or fluorine-18-fluorodeoxyglucose (FDG) positron emission tomography (PET) in association with functional diagnostic tests. MIBG scintigraphy and somatostatin receptor scintigraphy as well as Gallium-68 [68Ga] SSA radiotracer PET/TC are valuable techniques especially in cases of multiple tumors and disseminated disease (9, 10). Genetic counseling and genetic testing for a number of genes involved in the pathogenesis of PCCs (NF1, RET, von Hippel-Lindau gene [VHL], SDHD, SDHC, SDHB, EGLN1/PHD2, KIF1, SDHA, IDH1, FH, HIF2, SDHAF2, and SDHAF3) are recommended when appropriate (11–15). If multiple or CT-negative tumors are suspected, a 131I-metaiodobenzylguanidine (MIBG) scintigraphy scan should be performed (16–19).
Alpha blockade (phenoxybenzamine or doxazosin) with aggressive volume repletion associated with preoperative rapid-acting intravenous alpha-adrenergic antagonists and beta-blockers are the mainstay of treatment (20). Tumor excision represents the therapy of choice of non-metastatic PCC/PGL. Whenever possible with metastatic disease, primary tumor resection should be recommended in order to alleviate cardiovascular and other symptoms from catecholamine excess or from tumor invasion. For metastatic PCCs/PGLs, there are few established molecular targeted therapies, which have or may have a positive impact. [68Ga] PET/TC gives the predictive power for the efficacy of peptide receptor radionuclide therapy (PRRT) which, togheter with 131I-MIBG, may have a benificial efficacy in unresectable disease (10, 21). Radiofrequency ablation, cryoablation, and ethanol injection may be considered in the treatment of metastatic (oligo-metastatic) PCC/PGL (22, 23). Conventional chemotherapy (Averbuch scheme and temozolomide) have been widely used (24, 25).
Conventional external beam radiation therapy (cEBRT) or radiotherapy/radiosurgery (gamma-knife/cyberknife) is recommended for locally unresectable disease (18, 19), in the case of bone metastases and may play a significant palliative role in oligo-metastatic disease.
In recent years, growing interest in precision medicine and molecular diagnosis concerning the expanding etiology for hereditary PCCs and PGL has led to the inclusion of SDHA, TMEM127, MAX, and SDHAF2 as susceptibility genes (26–28). Genetic testing is recommended in patients at clinically high risk who are negative for the classic gene mutations (10, 28).
We describe herein the case of a 40-years-old woman with multiple NENs, comprising a pelvic GNB, right adrenal composite PCC-GN, inter-aortocaval PGL, and multiple left adrenal PCCs, associated with a novel variant of MAX gene mutation.
Case Report
Informed consent was obtained from the patient for the collection, analysis, and publication of personal, familial, clinical, and genetic data and the conduct of genetic blood tests. This manuscript was written in accordance with CARE guidelines.
Pedigree and Family History
The patient's pedigree is shown in Figure 1. The proband is a 40-years-old Italian woman of Caucasian ancestry. Her father died from glioblastoma multiforme aged 55 years. She has no siblings. The medical history of her children was negative for any reported disease or apparent NEN-associated clinical signs. Her son (age 14 years) is negative for clinical manifestations and genetic analyses were not performed.
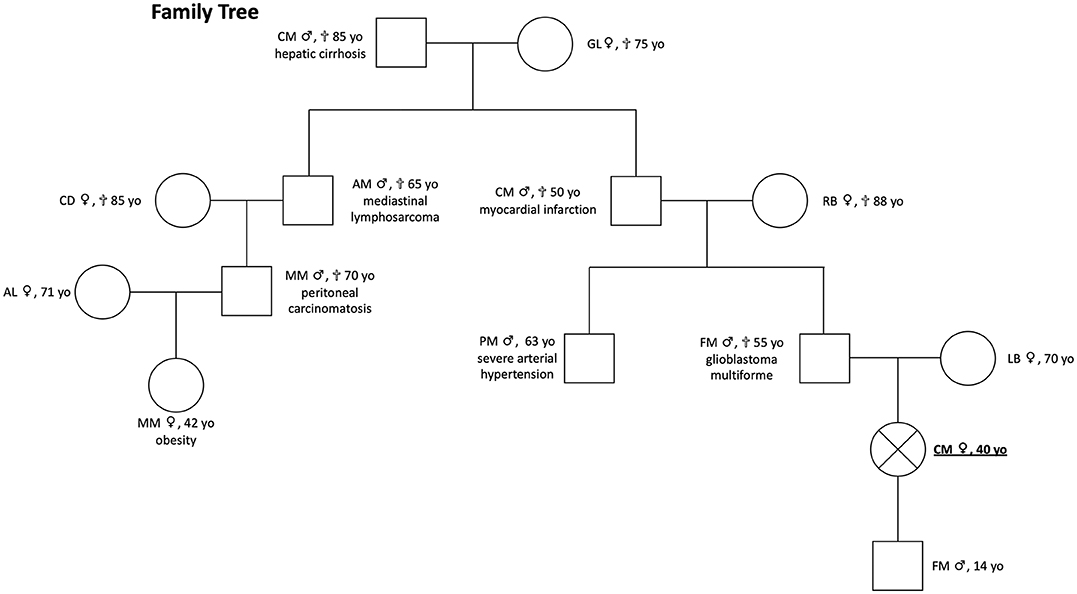
Figure 1. Patient pedigree [proband positive for MAX mutation c299G>C (p.Arg100Pro, NM_00.42382)]. She presented clinical signs of multiple PGL and composite PCCs; proband's father had a glioblastoma multiforme, and genetic testing was not performed; proband's son is negative for clinical manifestations, and genetic analyses were not performed.
Case Presentation
The timing of the multiple and composite NEN-neuroblastic tumor presentation of the present case is shown in Figure 2.
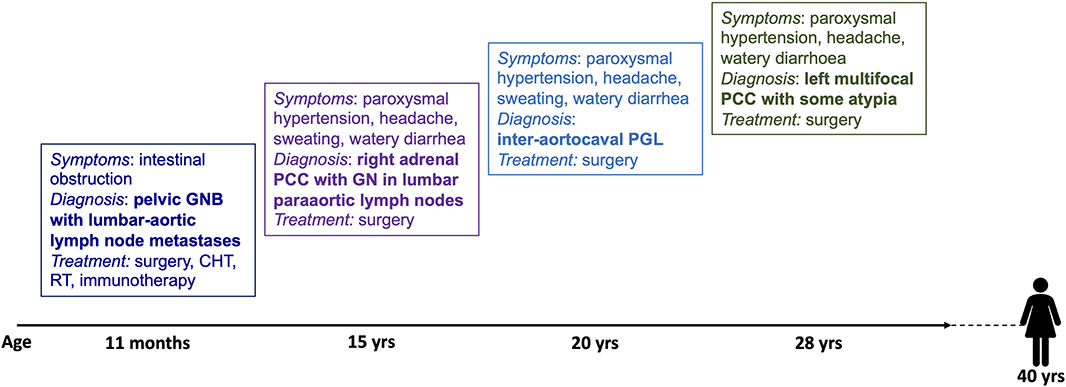
Figure 2. Timing of the presentation of multiple and composite NEN-neural tumors in this patient. GNB, ganglioneuroblastoma; CHT, chemotherapy; RT, radiotherapy; PCC, pheochromocytoma; GN, ganglioneuroma; PGL, paraganglioma.
A 15-years-old Caucasian girl with a history of blood and mucus-free watery diarrhea (up to four times in 24 h), headache, and sweating was referred in 1994 for paroxysmal hypertension. Her personal history included, at the age of 11 months, a GNB with lumbo-aortic lymph node metastases, which was surgically excised and then treated with chemotherapy, radiotherapy, and immunotherapy. Given the symptomatology, an endocrinological workup was performed showing normal 24-h urine vanillylmandelic acid (VMA) and total metanephrines. An abdominal ultrasound and adrenal washout CT scan demonstrated a 5.0-cm right adrenal lesion. The patient underwent abdominal surgery. The histological diagnosis was right composite adrenal PCC-GN. This diagnosis derives from the histological description of an alveolar pattern of round polygonal cells with an eosinophilic granular cytoplasm and spindle cells with a fasciculation pattern that were outlined by collagen fiber septa. The tumor cells showed round, pleomorphic nuclei with prominent nucleoli. The lymph nodes contained ganglioneuromatous tissue. After surgery, the symptomatology soon improved.
Five years later, the patient presented with a recurrence of paroxysmal hypertension. Neuroendocrine tumor marker tests were positive for 24-h urinary total metanephrines (2,168 μg/24 h; reference range <354 μg/24 h), 24-h urinary catecholamines (239 μg/24 h; reference range <100 μg/24 h), and VMA (VMA urinary spot test: positive). Serum chromogranin A was also positive (CgA 248 ng/ml; reference range <90 ng/ml), while CEA (1.20 ng/ml; reference range <5 ng/ml), NSE (4.00 ng/ml; reference range <10 ng/ml), and alfa fetoprotein (alfaFP 4.90 ng/ml; reference range <5 ng/ml) were normal. An abdominal CT scan showed a 2.2-cm inter-aortocaval right mass. MIBG scintigraphy showed a distinct hot spot in that region (Figure 3). The patient was therefore referred for surgical treatment. The histological diagnosis was inter-aortocaval PGL (see Figure 3). Histological description showed that the tumor consisted of a trabecular-nesting cell pattern with abundant basophilic granular cytoplasm. The tumor cells had oval nuclei with prominent nucleoli and were strongly positive for CgA and NSE on immunohistochemistry. A positivity to S100 antibodies was found at immunohistochemistry.
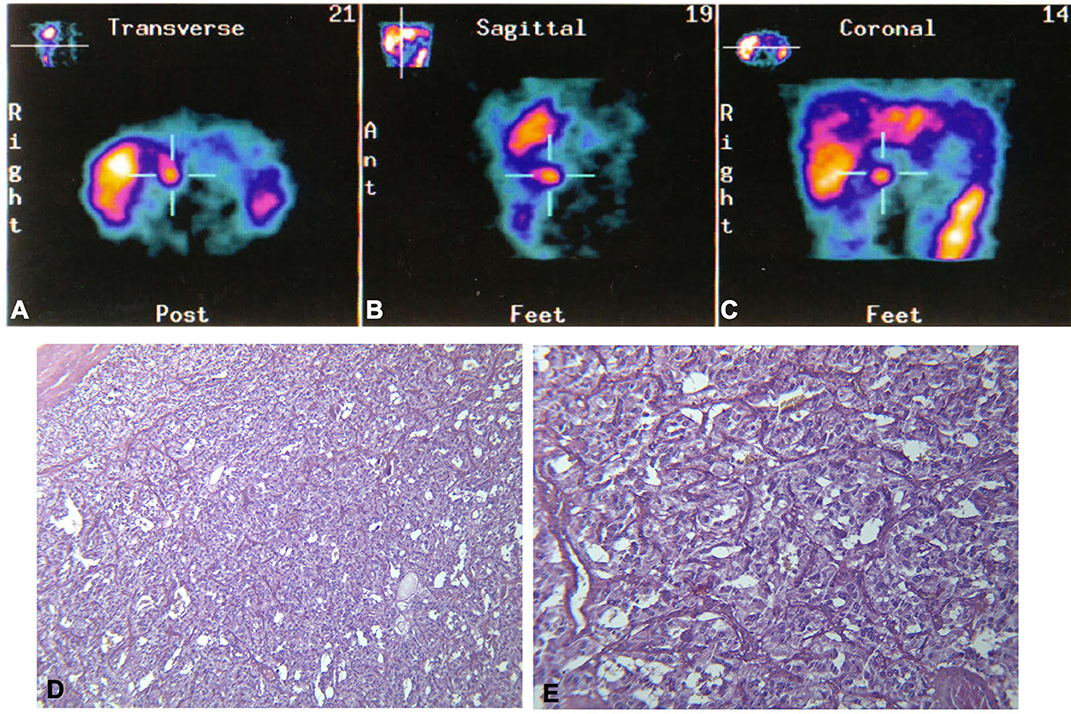
Figure 3. Top: 131I-metaiodobenzylguanidine (MIBG) scintigraphy distinctly shows a hot spot in the inter-aortocaval region, as seen in the (A) transverse, (B) sagittal, and (C) coronal scans. Bottom: Histological appearance of the paraganglioma, cords and nests of cellular elements with abundant granular basophilic cytoplasm and ovoid nuclei with prominent nucleoli. (D) EE 10×. (E) EE 20×.
Eight years later, the patient presented with a recurrence of paroxysmal hypertension. The endocrinological workup was positive for 24-h urinary total metanephrines (1,989 μg/24 h), serum noradrenaline (2,100 pg/ml; reference range 300–900 pg/ml), and serum adrenaline (156 pg/ml; reference range 0–100). Serum CgA (260 ng/ml) and alfaFP (5.31 ng/ml) were also positive. CEA (3.00 ng/ml) and NSE (7.70 ng/ml) were normal. Adrenal washout CT scans showed four non-adenomatous left adrenal masses between 10 and 18 mm (Figure 4). Abdominal MRI confirmed these lesions with significantly high signal T2-hyperintensity, suggesting a neuroendocrine (NE) origin. Surgical excision was performed. The diagnosis was multifocal left PCCs with some atypia. The macroscopic observation showed four adrenal nodules measured 18, 20, 25, and 27 mm that were enclosed in a thin capsule with a reddish-tan surface and having multiple hemorrhagic areas. Histologically, spindle cells were found in 30% of the neoplasm, with prominent nucleoli. The nodules were strongly positive for CgA and synaptophysin on immunohistochemistry. Large polygonal cells with eosinophilic granular cytoplasm and strong atypia were found in 40% of the nodule tissue. The Ki-67 proliferation index was >5%. There were no sustentacular cells. The symptoms disappeared and plasma and urinary catecholamines and 24-h urinary metanephrines returned to normal levels.
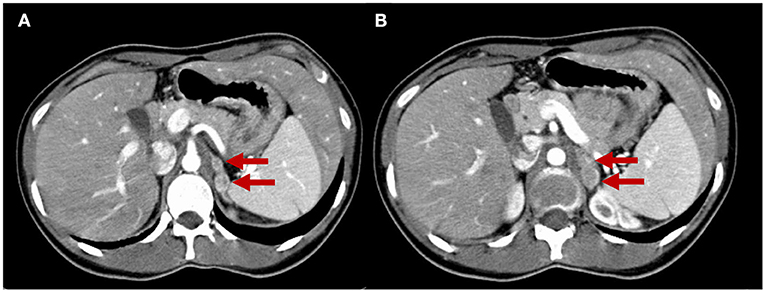
Figure 4. Adrenal CT scan. The two slices (A,B) show four nodules in the left adrenal gland measuring from 10 to 18 mm that are unattenuated after contrast administration (red arrows), compatible with left multifocal adrenal pheochromocytomas.
The patient is currently in good health. She is in follow-up with no evidence of metastases and recurrence. The trend of NE markers over the course of the clinical history is shown in Figure S1. A review of the pathology slides of all the resected tumors confirmed the four diagnoses, which can be summarized as metastatic GNB, bilateral metastatic PCCs, and inter-aortocaval PGL.
Methods
Genetic Analyses
Next-Generation Sequencing was performed for VHL, RET, NF1, Tp53, SDHD, SDHB, SDHC, SDHAF2, SDHAF3, SDHA, TMEM127, and MAX genes in the Molecular Diagnostic Laboratory for Hereditary Tumors, Veneto Institute of Oncology, Padova and Genetic Ames Group Laboratory, Naples, Italy. Mutation of the MAX gene was confirmed by Integrative Genomics Viewer (IGV), a high-performance visualization tool. Consequently, the MAX gene was also analyzed for intragenic mutations by Sanger sequencing.
Results
No mutations were found in VHL, RET, NF1, Tp53, SDHD, SDHB, SDHC, SDHAF2, SDHAF3, SDHA, or TMEM127. In the MAX gene, a novel heterozygous variant, c299G>C (p.Arg100Pro, NM_002382.5), was found. This variant of germline mutation (from peripheral blood) was analyzed with the bioinformatics prediction programs “Sift,” “PolyPhen,” and “Mutation Taster” at Genetic Ames Group Laboratory, Naples, Italy. See Table 1. The programs have defined that this variant is deleterious, harmful, and causative of pathology. For diagnostic completeness, this variant has been confirmed by Sanger sequencing. The variant is not currently reported in the ClinVAr and dbSNP databases and in GnomAD database. See Figure S2.
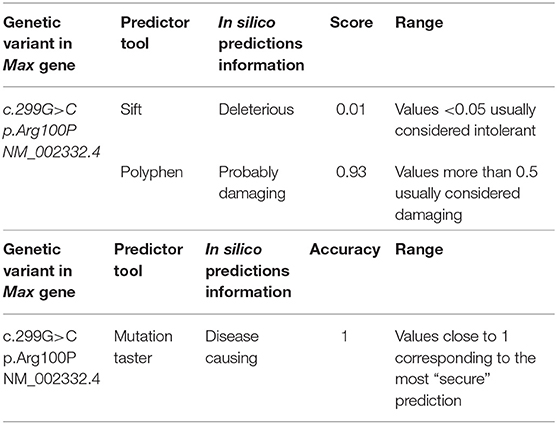
Table 1. In silico prediction table.
Discussion
The lesson from this case report highlights several important messages.
First, the metachronic occurrence of PCCs/PGL and neuroblastic tumors. Although previously PCCs, PGLs, and neuroblastic tumors were thought to share the same embryonic origin from neural crest cells, several recent studies showed that the progenitor cells have partly overlapping origin and that chromaffin cells of adrenal medulla arise from peripheral glial stem cells or Schwann cell precursors (29). The simultaneous presence of PCCs and PGLs is rarely found and it is reported in RET-positive patients with MEN2B and also in SDH mutated patients (30).
To our knowledge, 34 composite PCC-GN cases are reported in literature (6, 31–45). Most of these did not involve any genetic factors, while four cases involved an association with MEN2 (31, 42), VHL (40), or NF1 (43) syndrome.
In this view, the second message of scientific interest is represented by the association of the new described MAX gene heterozygous variant, c299G>C (p.Arg100Pro, NM_002382.5), with the tumors.
An association between a MAX gene mutation and composite PCC with a ganglioneuromatous component has never been described in the literature, while just one composite case has been described involving PGL, in an adolescent who had been successfully treated for a stage IV-S neuroblastoma 15 years earlier (46).
MAX is a crucial component of the MYC-MAX-MXD1 transcription factor network, which regulates cell proliferation and differentiation and apoptosis (47). The formation of MAX-MXD1 heterodimers counteracts the dimerization of MYC with MAX, which would act as a transcriptional activator (47). MAX is thus a tumor suppressor gene and its mutation favors the development of hereditary PCCs and PGLs (28, 46–48).
Germline mutations affecting the MYC-associated protein X (MAX) gene are considered a major genetic predisposition factor for the development of hereditary PCC and/or PGL (49). The only tumor suppressor genes known to show a “parent-of-origin” phenotype are the recently described genes SDHAF2 and MAX, located on chromosome 11q12.2 and 14q23, respectively (50).
MAX mutations cause hereditary and sporadic PCC/PGL. Genotype–phenotype associations suggested that MAX mutations were associated with bilateral PCC and with an apparent paternal transmission of the disease (47).
Little is known about genetic alterations in sporadic tumors. Mutations in PCC/PGL susceptibility genes are detrimental for neuronal precursor cells. This could explain the apparent rarity of somatic mutations in these genes in apparently sporadic PCC/PGL (51).
Third, the genetic pattern is confirmed by the intermediate biochemical phenotype that in NENs' patients seems to be associated with a germline MAX gene mutation (52). Given that the patient showed a noradrenalin secretion as biochemical phenotype, “cluster 2” of PCC/PGL would appear to be delineated (10). Indeed, before MAX gene identification, our patient had undergone various genetic tests over the years (RET for associations with MEN2; VHL for associations with Von Hippel Lindau Syndrome; NF1 for associations with Neurofibromatosis type 1; and Tp53 for associations with Li Fraumeni Syndrome), all proved negative. Furthermore, SDH gene mutation analyses performed to explore PGL context (53) were also negative.
Literature analysis showed that the case of a young woman with adrenergic phenotype and bilateral PCC with PGL associated to a germline mutation in MAX gene (c.70_73delAAAC/p.Lys24fs*40) was reported by Shibata et al. (54). Recently, a case of a 49-years-old woman with bilateral PCC and adrenergic phenotype plus pituitary prolactinoma was also associated to a pathogenic MAX mutation c.296-1G>T (NM_002382), which has not been previously described (55).
Moreover, a 25-years-old patient with multiple PCCs associated with adrenal medullary hyperplasia and with a non-sense germline MAX mutation was described (56).
Other data from literature reported a complex MAX rearrangement with the loss of the wild-type MAX and FUT8 in a family with malignant PCCs, renal oncocytoma, and erythrocytosis (57).
Given the role of MAX as a tumor suppressor gene, the novel heterozygous variant identified and the relative youth of the patient, her follow-up will require considerable attention, given the risk of other endocrine and non-endocrine neoplasms and because loss of MAX function is correlated with greater aggressiveness and metastatic potential (47) than the other mutated pathways involved in these types of tumor. The significance of the MAX variant (c.299G>C; p.Arg100Pro) is currently unknown (49).
In addition to the multiplicity of tumor lesions, the earliness of onset also represents a peculiar characteristic (46, 58).
The mean onset age among the known described patients was 33 years (range 13–58 years), while in our case, the first presentation was in infancy. This is a crucial point in relation to MAX gene penetrance. Indeed, nowadays no reliable penetrance estimations are available for MAX mutation carriers.
Our patient had no family history for PCC/PGL. Given that her father died of glioblastoma multiforme, the present study may suggest an evidence of paternal mode of transmission in MAX mutation carriers. If this mode of inheritance was confirmed, disease is only passed on to children by their father, resulting in the phenomenon of generation-skipping. In this view, it would be even more important to analyze whether the mutation was transmitted to the patient's son, who would be able to transmit the disease to the offspring as well as the MAX genetic mutation (28).
The limitations of this case include the fact that the assessments performed on the patient over the last 25 years are obviously not all in line with the current guidelines. The 24-h urinary VMA measurement is no longer recommended for the diagnosis of PCC (9). For this reason, no clinical significance can be attributed to the normal concentration of 24-h urinary VMA at the time of the second and the third tumor. In fact, a false-negative rate of 41% has been described in PEHO (progressive encephalopathy with edema, arrhythmia and optic atrophy) and neuroblastoma patients (59). In addition, MIBG scintigraphy is less efficacious than FDG- or F-DOPA-PET for the diagnosis of PGLs; however, the latter techniques were not available at the time of the patient's earlier workups.
In conclusion, we describe, for the first time, a novel heterozygous MAX gene variant: c299G>C (p.Arg100Pro, NM_00.42382), associated with the occurrence of multiple and composite PCC/neuroblastic tumors, occurred yet from infancy in a young women. The molecular mechanism of the MAX gene and the impact of MAX mutations require thorough investigation, to enable the prognosis of affected patients to be promptly established and targeted treatments to be developed.
Ethics Statement
The patient gave her written consent to sample collection, genetic testing, and the use of genetic test data for the purposes of research. The sample collection and preparation protocols were approved by the Department of Experimental Medicine Sapienza University of Rome, Italy. Institutional review board review and approval was not required for this case report. Consent for publication has been obtained from the patient, including permission for the details/images to be available on the Internet and viewable by the general public.
Author Contributions
EG, CP, and FSe conceived and designed the study. FSe, CD, ES, and RP collected the clinical samples and analyzed and interpreted the patient's data. FSc carried out the genetic analyses that first demonstrated the presence of the MAX gene mutation in this patient in 2011. CP, FSe, AF, and EG co-wrote the manuscript. VB, AI, and AL contributed to the revision of the manuscript. All authors have read and approved the final manuscript.
Funding
The genetic analyses were supported by the ministerial research project PRIN 2017Z3N3YC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MP declared a past co-authorship with one of the authors FS to the handling editor.
Acknowledgments
We wish to thank Dr. Giovanni Savarese, Dr. Raffaella Ruggiero, and Dr. Luisa Circelli of the Genetic Ames Group, Napoli, Italy, for identifying the novel heterozygous MAX gene variant. We also wish to thank the NETTARE Unit—NeuroEndocrine Tumor TAsk foRcE of Sapienza University of Rome, Italy, led by AL, AI, and EG, for integrating the patient's multidisciplinary clinical, diagnostic, and therapeutic management and follow-up. We would like to thank Marie-Hélène Hayles MITI for revision of the English text.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2020.00234/full#supplementary-material
Figure S1. Trend of NE markers in relation to neoplastic occurrence over the course of the clinical history. CgA, Chromogranin A (<90 ng/mL); U catecholamines, urinary catecholamines (<100 mcg/24 h); U metanephrines, urinary metanephrines (<354 mcg/24 h); VMA, vanillylmandelic acid (urinary VMA spot test: positivity was expressed graphically as a value of 100). The endocrinological work-up in 2008 included serum assessment of noradrenaline and adrenaline, which were not expressed in the panel, while CgA, VMA and U metanephrines were reported.
Figure S2. Sequencing of the MAX gene. Sequences of MAX mutation in genomic DNA from a venous blood sample. (A) shows Sanger image of MAX mutation analysis; (B) shows the MAX mutation analyzed with the Integrative Genomics Viewer (IGV).
Abbreviations
alfaFP, alfa fetoprotein; CgA, chromogranin A; CT, computed tomography; FDG, fluorine-18-fluorodeoxyglucose; GN, ganglioneuroma; GNB, ganglioneuroblastoma; MIBG, 131I-metaiodobenzylguanidine; MRI, magnetic resonance imaging; NEN, neuroendocrine neoplasm; PET, positron emission tomography; PCC, pheochromocytoma, PGL, paragangliomas. VMA, 24-h urine vanillylmandelic acid.
References
1. Lloyd RV, Osamura RY, Klöppel G, Rosai J. WHO Classification of Tumours of Endocrine Organs. 4th ed. Lyon (2017).
2. Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B, et al. The international neuroblastoma pathology classification (the shimada system). Cancer. (1999) 86:364–72. doi: 10.1002/(SICI)1097-0142(19990715)86:2<364::AID-CNCR21>3.0.CO;2-7
3. Brodeur GM, Bagatell R. Mechanisms of neuroblastoma regression. Nat Rev Clinical Oncol. (2014) 11:704–13. doi: 10.1038/nrclinonc.2014.168
4. He WG, Yan Y, Tang W, Cai R, Ren G. Clinical and biological features of neuroblastic tumors: a comparison of neuroblastoma and ganglioneuroblastoma. Oncotarget. (2017) 8:37730–9. doi: 10.18632/oncotarget.17146
5. Lam AK. Update on adrenal tumours in 2017 World Health Organization (WHO) of endocrine tumours. Endocr Pathol. (2017) 28:213–27. doi: 10.1007/s12022-017-9484-5
6. Khan AN, Solomon SS, Childress RD. Composite pheochromocytoma-ganglioneuroma: a rare experiment of nature. Endocr. Prac. (2010) 16:291–9. doi: 10.4158/EP09205.RA
7. Comstock JM, Willmore-Payne C, Holden JA, Coffin CM. Composite pheochromocytoma: a clinicopathologic and molecular comparison with ordinary pheochromocytoma and neuroblastoma. Am J Clin Pathol. (2009) 132:69–73. doi: 10.1309/AJCPN76VTIGWPOAG
8. Gaudenzi G, Dicitore A, Carra S, Saronni D, Pozza C, Giannetta E, et al. Management of endocrine disease: precision medicine in neuroendocrine neoplasms: an update on current management and future perspectives. Eur J Endocrinol. (2019) 181:R1–10. doi: 10.1530/EJE-19-0021
9. Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metabol. (2014) 99:1915–42. doi: 10.1210/jc.2014-1498
10. Nolting S, Ullrich M, Pietzsch J, Ziegler CG, Eisenhofer G, Grossman A, et al. Current management of pheochromocytoma/paraganglioma: a guide for the practicing clinician in the era of precision medicine. Cancers. (2019) 11:1505. doi: 10.3390/cancers11101505
11. Sbardella E, Cranston T, Isidori AM, Shine B, Pal A, Jafar-Mohammadi B, et al. Routine genetic screening with a multi-gene panel in patients with pheochromocytomas. Endocrine. (2018) 59:175–82. doi: 10.1007/s12020-017-1310-9
12. Dwight T, Na U, Kim E, Zhu Y, Richardson AL, Robinson BG, et al. Analysis of SDHAF3 in familial and sporadic pheochromocytoma and paraganglioma. BMC Cancer. (2017) 17:497. doi: 10.1186/s12885-017-3486-z
13. Bader HL, Hsu T. Inactivation of the tumor suppressor gene von Hippel-Lindau (VHL) in granulocytes contributes to development of liver hemangiomas in a mouse model. BMC Cancer. (2016) 16:797. doi: 10.1186/s12885-016-2802-3
14. Neumann HP, Young WF Jr, Krauss T, Bayley JP, Schiavi F, Opocher G, et al. 65 YEARS OF THE DOUBLE HELIX: genetics informs precision practice in the diagnosis and management of pheochromocytoma. Endocr Relat Cancer. (2018) 25:T201–19. doi: 10.1530/ERC-18-0085
15. Alrezk R, Suarez A, Tena I, Pacak K. Update of pheochromocytoma syndromes: genetics, biochemical evaluation, and imaging. Front Endocrinol. (2018) 9:515. doi: 10.3389/fendo.2018.00515
16. Pavel M, de Herder WW. ENETS consensus guidelines for the standard of care in neuroendocrine tumors. Neuroendocrinology. (2017) 105:193–5. doi: 10.1159/000457957
17. Sundin A, Arnold R, Baudin E, Cwikla JB, Eriksson B, Fanti S, et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: radiological, nuclear medicine & hybrid imaging. Neuroendocrinology. (2017) 105:212–44. doi: 10.1159/000471879
18. Kulke MH, Shah MH, Benson AB 3rd, Bergsland E, Berlin JD, Blaszkowsky LS, et al. Neuroendocrine tumors, version 1.2015. JNCCN. (2015) 13:78–108. doi: 10.6004/jnccn.2015.0011
19. Shah MH, Goldner WS, Halfdanarson TR, Bergsland E, Berlin JD, Halperin D, et al. NCCN guidelines insights: neuroendocrine and adrenal tumors, version 2.2018. JNCCN. (2018) 16:693–702. doi: 10.6004/jnccn.2018.0056
20. Canu L, Parenti G, De Filpo G, Mannelli M. Pheochromocytomas and paragangliomas as causes of endocrine hypertension. Front Endocrinol. (2019) 10:333. doi: 10.3389/fendo.2019.00333
21. Mak IYF, Hayes AR, Khoo B, Grossman A. Peptide receptor radionuclide therapy as a novel treatment for metastatic and invasive phaeochromocytoma and paraganglioma. Neuroendocrinology. (2019) 109:287–98. doi: 10.1159/000499497
22. Gravel G, Leboulleux S, Tselikas L, Fassio F, Berraf M, Berdelou A, et al. Prevention of serious skeletal-related events by interventional radiology techniques in patients with malignant paraganglioma and pheochromocytoma. Endocrine. (2018) 59:547–54. doi: 10.1007/s12020-017-1515-y
23. Kohlenberg J, Welch B, Hamidi O, Callstrom M, Morris J, Sprung J, et al. Efficacy and safety of ablative therapy in the treatment of patients with metastatic pheochromocytoma and paraganglioma. Cancers. (2019) 11:195. doi: 10.3390/cancers11020195
24. Nolting S, Grossman A, Pacak K. Metastatic Phaeochromocytoma: Spinning Towards More Promising Treatment Options. Exp Clin Endocrinol Diabet. (2019) 127:117–28. doi: 10.1055/a-0715-1888
25. Tong A, Li M, Cui Y, Ma X, Wang H, Li Y. Temozolomide is a potential therapeutic tool for patients with metastatic pheochromocytoma/paraganglioma-case report and review of the literature. Front Endocrinol. (2020) 11:61. doi: 10.3389/fendo.2020.00061
26. Jha A, de Luna K, Balili CA, Millo C, Paraiso CA, Ling A, et al. Clinical, diagnostic, and treatment characteristics of SDHA-Related metastatic pheochromocytoma and paraganglioma. Front Oncol. (2019) 9:53. doi: 10.3389/fonc.2019.00053
27. Burnichon N, Lepoutre-Lussey C, Laffaire J, Gadessaud N, Molinie V, Hernigou A, et al. A novel TMEM127 mutation in a patient with familial bilateral pheochromocytoma. Eur J Edocrinol. (2011) 164:141–5. doi: 10.1530/EJE-10-0758
28. Bausch B, Schiavi F, Ni Y, Welander J, Patocs A, Ngeow J, et al. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for Gene-Informed Prevention. JAMA Oncol. (2017) 3:1204–12. doi: 10.1001/jamaoncol.2017.0223
29. Koopman K, Gaal J, de Krijger RR. Pheochromocytomas and paragangliomas: new developments with regard to classification, genetics, and cell of origin. Cancers. (2019) 11:e1070. doi: 10.3390/cancers11081070
30. Hoekstra AS, Devilee P, Bayley JP. Models of parent-of-origin tumorigenesis in hereditary paraganglioma. Semin Cell Dev Biol. (2015) 43:117–24. doi: 10.1016/j.semcdb.2015.05.011
31. Efared B, Atsame-Ebang G, Tahirou S, Mazaz K, Hammas N, El Fatemi H, et al. Bilateral pheochromocytoma with ganglioneuroma component associated with multiple neuroendocrine neoplasia type 2A: a case report. J Med Case Rep. (2017) 11:208. doi: 10.1186/s13256-017-1364-6
32. Zhang BY, Zhao M, Li B, Zhang JM. Diverse proportion in composite pheochromocytoma-ganglioneuroma may induce varied clinical symptom: comparison of two cases. Int J Clin Exp Pathol. (2015) 8:15369–74.
33. Shida Y, Igawa T, Abe K, Hakariya T, Takehara K, Onita T, et al. Composite pheochromocytoma of the adrenal gland: a case series. BMC Res Notes. (2015) 8:257. doi: 10.1186/s13104-015-1233-6
34. Shawa H, Elsayes KM, Javadi S, Sircar K, Jimenez C, Habra MA. Clinical and radiologic features of pheochromocytoma/ganglioneuroma composite tumors: a case series with comparative analysis. Endocr Pract. (2014) 20:864–9. doi: 10.4158/EP14010.OR
35. Rao RN, Singla N, Yadav K. Composite pheochromocytoma-ganglioneuroma of the adrenal gland: a case report with immunohistochemical study. Urology Ann. (2013) 5:115–8. doi: 10.4103/0974-7796.110011
36. Hu J, Wu J, Cai L, Jiang L, Lang Z, Qu G, et al. Retroperitoneal composite pheochromocytoma-ganglioneuroma : a case report and review of literature. Diagnostic Pathol. (2013) 8:63. doi: 10.1186/1746-1596-8-63
37. Majumder S, Grabska J, Trikudanathan G, Kowalczyk P, Stoica-Mustafa E, Dasanu CA. Functional 'composite' pheochromocytoma-ganglioneuroma presenting as a pancreatic mass. Pancreatol. (2012) 12:211–4. doi: 10.1016/j.pan.2012.02.001
38. Lau SK, Chu PG, Weiss LM. Mixed cortical adenoma and composite pheochromocytoma-ganglioneuroma: an unusual corticomedullary tumor of the adrenal gland. Ann Diagnostic Pathol. (2011) 15:185–9. doi: 10.1016/j.anndiagpath.2010.02.005
39. Tohme CA, Mattar WE, Ghorra CS. Extra-adrenal composite pheochromocytoma-ganglioneuroma. Saudi Med J. (2006) 27:1594–7.
40. Bernini GP, Moretti A, Mannelli M, Ercolino T, Bardini M, Caramella D, et al. Unique association of non-functioning pheochromocytoma, ganglioneuroma, adrenal cortical adenoma, hepatic and vertebral hemangiomas in a patient with a new intronic variant in the VHL gene. J Endocrinol Invest. (2005) 28:1032–7. doi: 10.1007/BF03345345
41. Lam KY, Lo CY. Composite pheochromocytoma-ganglioneuroma of the adrenal gland: an uncommon entity with distinctive clinicopathologic features. Endocr Pathol. (1999) 10:343–52. doi: 10.1007/BF02739777
42. Brady S, Lechan RM, Schwaitzberg SD, Dayal Y, Ziar J, Tischler AS. Composite pheochromocytoma/ganglioneuroma of the adrenal gland associated with multiple endocrine neoplasia 2A: case report with immunohistochemical analysis. Am J Surg Pathol. (1997) 21:102–8. doi: 10.1097/00000478-199701000-00011
43. Chetty R, Duhig JD. Bilateral pheochromocytoma-ganglioneuroma of the adrenal in type 1 neurofibromatosis. Am J Surg Pathol. (1993) 17:837–41. doi: 10.1097/00000478-199308000-00009
44. Kragel PJ, Johnston CA. Pheochromocytoma-ganglioneuroma of the adrenal. Arch Pathol Lab Med. (1985) 109:470–2.
45. Mendelsohn G, Eggleston JC, Olson JL, Said SI, Baylin SB. Vasoactive intestinal peptide and its relationship to ganglion cell differentiation in neuroblastic tumors. Lab Invest J Tech Methods Pathol. (1979) 41:144–9.
46. Lopez-Andreu JA, Castel V, Verdeguer A, Muro MD, Esquembre C, Ferris J. Neuroblastoma IV-S followed by extra-adrenal pheochromocytoma 15 years later. Med Pediatr Oncol. (1995) 24:388–91. doi: 10.1002/mpo.2950240609
47. Comino-Mendez I, Gracia-Aznarez FJ, Schiavi F, Landa I, Leandro-Garcia LJ, Leton R, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. (2011) 43:663–7. doi: 10.1038/ng.861
48. Burnichon N, Cascon A, Schiavi F, Morales NP, Comino-Mendez I, Abermil N, et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res. (2012) 18:2828–37. doi: 10.1158/1078-0432.CCR-12-0160
49. Comino-Mendez I, Leandro-Garcia LJ, Montoya G, Inglada-Perez L, de Cubas AA, Curras-Freixes M, et al. Functional and in silico assessment of MAX variants of unknown significance. J Mol Med. (2015) 93:1247–55. doi: 10.1007/s00109-015-1306-y
50. Peczkowska M, Kowalska A, Sygut J, Waligorski D, Malinoc A, Janaszek-Sitkowska H, et al. Testing new susceptibility genes in the cohort of apparently sporadic phaeochromocytoma/paraganglioma patients with clinical characteristics of hereditary syndromes. Clin Endocrinol. (2013) 79:817–23. doi: 10.1111/cen.12218
51. Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell. (2005) 8:155–67. doi: 10.1016/j.ccr.2005.06.015
52. Qin N, de Cubas AA, Garcia-Martin R, Richter S, Peitzsch M, Menschikowski M, et al. Opposing effects of HIF1alpha and HIF2alpha on chromaffin cell phenotypic features and tumor cell proliferation: insights from myc-associated factor x. Int J Cancer. (2014) 135:2054–64. doi: 10.1002/ijc.28868
53. Nozieres C, Walter T, Joly MO, Giraud S, Scoazec JY, Borson-Chazot F, et al. A SDHB malignant paraganglioma with dramatic response to temozolomide-capecitabine. Eur J Endocrinol. (2012) 166:1107–11. doi: 10.1530/EJE-11-1098
54. Shibata M, Inaishi T, Miyajima N, Adachi Y, Takano Y, Nakanishi K, et al. Synchronous bilateral pheochromocytomas and paraganglioma with novel germline mutation in MAX: a case report. Surg Case Rep. (2017) 3:131. doi: 10.1186/s40792-017-0408-x
55. Roszko KL, Blouch E, Blake M, Powers JF, Tischler AS, Hodin R, et al. Case Report of a Prolactinoma in a Patient With a Novel MAX Mutation and Bilateral Pheochromocytomas. J Endocr Soc. (2017) 1:1401–7. doi: 10.1210/js.2017-00135
56. Romanet P, Guerin C, Pedini P, Essamet W, Castinetti F, Sebag F, et al. Pathological and genetic characterization of bilateral adrenomedullary hyperplasia in a patient with germline max mutation. Endocr Pathol. (2017) 28:302–7. doi: 10.1007/s12022-016-9460-5
57. Korpershoek E, Koffy D, Eussen BH, Oudijk L, Papathomas TG, van Nederveen FH, et al. Complex max rearrangement in a family with malignant pheochromocytoma, renal oncocytoma, and erythrocytosis. J Clin Endocrinol Metab. (2016) 101:453–60. doi: 10.1210/jc.2015-2592
58. McHugh K. Renal and adrenal tumours in children. Cancer Imag. (2007) 7:41–51. doi: 10.1102/1470-7330.2007.0007
Keywords: composite NEN, pheochromocytoma, ganglioneuroblastoma, MAX gene, paraganglioma
Citation: Pozza C, Sesti F, Di Dato C, Sbardella E, Pofi R, Schiavi F, Bonifacio V, Isidori AM, Faggiano A, Lenzi A and Giannetta E (2020) A Novel MAX Gene Mutation Variant in a Patient With Multiple and “Composite” Neuroendocrine–Neuroblastic Tumors. Front. Endocrinol. 11:234. doi: 10.3389/fendo.2020.00234
Received: 20 November 2019; Accepted: 31 March 2020;
Published: 19 May 2020.
Edited by:
Antonino Belfiore, University of Catania, ItalyReviewed by:
Massimo Mannelli, University of Florence, ItalyMariola Pęczkowska, Cardinal Stefan Wyszynski Institute of Cardiology, Poland
Copyright © 2020 Pozza, Sesti, Di Dato, Sbardella, Pofi, Schiavi, Bonifacio, Isidori, Faggiano, Lenzi and Giannetta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisa Giannetta, ZWxpc2EuZ2lhbm5ldHRhQHVuaXJvbWExLml0
†These authors have contributed equally to this work