 Jelena Sikimic1
Jelena Sikimic1 Theresa Hoffmeister2
Theresa Hoffmeister2 Anne Gresch2
Anne Gresch2 Julia Kaiser1
Julia Kaiser1 Winfried Barthlen3
Winfried Barthlen3 Carmen Wolke4
Carmen Wolke4 Ilse Wieland5Uwe Lendeckel4
Ilse Wieland5Uwe Lendeckel4 Peter Krippeit-Drews1*Martina Düfer2Gisela Drews1
Peter Krippeit-Drews1*Martina Düfer2Gisela Drews1- 1Department of Pharmacology, Institute of Pharmacy, University of Tübingen, Tübingen, Germany
- 2Department of Pharmacology, Institute of Pharmaceutical and Medicinal Chemistry, University of Münster, Münster, Germany
- 3Department of Pediatric Surgery, University Medicine Greifswald, Greifswald, Germany
- 4Institute of Medical Biochemistry and Molecular Biology, University Medicine Greifswald, Greifswald, Germany
- 5Institute of Human Genetics, University Hospital Magdeburg, Magdeburg, Germany
Objective: Congenital hyperinsulinism (CHI) is a rare disease characterized by persistent hypoglycemia as a result of inappropriate insulin secretion, which can lead to irreversible neurological defects in infants. Poor efficacy and strong adverse effects of the current medications impede successful treatment. The aim of the study was to investigate new approaches to silence β-cells and thus attenuate insulin secretion.
Research Design and Methods: In the scope of our research, we tested substances more selective and more potent than the gold standard diazoxide that also interact with neuroendocrine ATP-sensitive K+ (KATP) channels. Additionally, KATP channel-independent targets as Ca2+-activated K+ channels of intermediate conductance (KCa3.1) and L-type Ca2+ channels were investigated. Experiments were performed using human islet cell clusters isolated from tissue of CHI patients (histologically classified as pathological) and islet cell clusters obtained from C57BL/6N (WT) or SUR1 knockout (SUR1-/-) mice. The cytosolic Ca2+ concentration ([Ca2+]c) was used as a parameter for the pathway regulated by electrical activity and was determined by fura-2 fluorescence. The mitochondrial membrane potential (ΔΨ) was determined by rhodamine 123 fluorescence and single channel currents were measured by the patch-clamp technique.
Results: The selective KATP channel opener NN414 (5 µM) diminished [Ca2+]c in isolated human CHI islet cell clusters and WT mouse islet cell clusters stimulated with 10 mM glucose. In islet cell clusters lacking functional KATP channels (SUR1-/-) the drug was without effect. VU0071063 (30 µM), another KATP channel opener considered to be selective, lowered [Ca2+]c in human CHI islet cell clusters. The compound was also effective in islet cell clusters from SUR1-/- mice, showing that [Ca2+]c is influenced by additional effects besides KATP channels. Contrasting to NN414, the drug depolarized ΔΨ in murine islet cell clusters pointing to severe interference with mitochondrial metabolism. An opener of KCa3.1 channels, DCEBIO (100 µM), significantly decreased [Ca2+]c in SUR1-/- and human CHI islet cell clusters. To target L-type Ca2+ channels we tested two already approved drugs, dextromethorphan (DXM) and simvastatin. DXM (100 µM) efficiently diminished [Ca2+]c in stimulated human CHI islet cell clusters as well as in stimulated SUR1-/- islet cell clusters. Similar effects on [Ca2+]c were observed in experiments with simvastatin (7.2 µM).
Conclusions: NN414 seems to provide a good alternative to the currently used KATP channel opener diazoxide. Targeting KCa3.1 channels by channel openers or L-type Ca2+ channels by DXM or simvastatin might be valuable approaches for treatment of CHI caused by mutations of KATP channels not sensitive to KATP channel openers.
Introduction
Congenital hyperinsulinism (CHI) is a rare heterogeneous genetic disorder, but the most frequent cause of severe, persistent hypoglycemia in neonates, infants and children. The main reasons for developing CHI are defects in important genes regulating pancreatic β-cell function. To date, mutations in 14 essential genes controlling insulin secretion have been reported including ABCC8 and KCNJ11. ABCC8 and KCNJ11 genes encode the KATP channel subunits SUR1 and Kir6.2, respectively, and mutations in these genes represent the most prevalent cause of CHI. Defects in these genes are responsible for the failure of β-cells to respond to normal regulatory mechanisms, leading to inappropriate and excessive insulin release despite low blood glucose concentrations resulting in frequent episodes of hypoglycemia (1, 2). There are some excellent reviews giving detailed information about molecular mechanisms underlying the pathophysiology of CHI (1–5).
Based on histopathological observations, three distinct forms of CHI are described: focal, diffuse and atypical. In focal CHI affected β-cells are localized only in small specific parts of the pancreas. Conversely, in diffuse CHI all pancreatic β-cells seem to be affected (6). If the histology of the tissue does not fit in one of the forms, it is regarded as an atypical form of CHI. It is characterized by a mosaic-like assembly of hyper-functional islets spread over the pancreas (7).
Persistent hypoglycemia is responsible for seizures and finally for severe brain damage (8). Thus, it is necessary to diagnose CHI rapidly and to start as early as possible with a suitable treatment. Treatment options include medical therapy and surgical intervention (9). First-line drug for treating CHI is the KATP channel agonist diazoxide (10). However, numerous side effects of diazoxide limit its use. Some of the most common undesired effects are Na+ and fluid retention, hypertrichosis and loss of appetite. Life threatening side effects also occur including cardiac failure, pulmonary hypertension, hyperuricemia, bone marrow suppression, and anemia (11–16). Additionally, diazoxide is only effective when KATP channels are functional (10). Alternatives to the therapy with diazoxide and novel medications include glucagon, somatostatin analogues, nifedipine, GLP1-receptor antagonists [exendin-(9–39)], and sirolimus [(17–22), reviewed in (3)]. Many of these drugs act by lowering the Ca2+ influx into β-cells (23–25). The aforementioned drugs also have numerous undesirable effects, which may be a reason for reconsidering their therapeutic usefulness: gastrointestinal symptoms, formation of gall stones, suppression of pituitary hormones, necrotizing enterocolitis, hypotension, immune suppression, thrombocytosis, impaired immune response, and many more (26–31). Recently, a new full human monoclonal antibody to the insulin receptor XMetD (also known as XOMA 358 or RZ358) has been proposed as a novel therapeutic strategy (32–35). First results in a Phase 2a clinical trial exhibited an improved glycemic control in patients with persistent hypoglycemia (36).
In patients that cannot be treated sufficiently with drugs, surgical treatment is indicated. While partial pancreatectomy is beneficial for patients with focal CHI (37, 38), in case of diffuse and drug-unresponsive CHI, near-total pancreatectomy is usually required (39, 40). Due to different post-operative complications like recurrent hypoglycemia, pancreatic exocrine insufficiency and diabetes, patients with diffuse CHI are far from being cured after surgery (41, 42). In order to reduce the development of diabetes postsurgically, a 70 to 90% resection of pancreas have been considered; however, the outcome is still unpredictable (39, 43).
Taken together, it is of great importance to explore new pharmacological options for CHI therapy in order to maintain euglycemia and reduce severe side effects from current medical and surgical treatment. Aim of this study was to find new strategies, which are able to silence β-cells by inhibiting extensive Ca2+ influx into the cell. For this purpose, new and approved drugs interacting with KATP channels and with KATP channel-independent targets have been tested on islet cell clusters obtained from biopsies of CHI patients and islet cell clusters from WT and SUR1-/- mice.
Materials and Methods
Cell and Islet Preparation
Human islets of Langerhans were obtained from different biopsies of children undergoing pancreatic surgery. Ethics approval for the study involving human participants was approved by the ethic commission of the Universitätsmedizin Greifswald (BB 050/13). Written informed consent was provided by the legal guardians of the children for the study. The islets were taken from biopsies of eight CHI patients. Genetic studies showed that seven patients had mutations in the ABCC8 gene encoding the SUR1 subunit of KATP channels. In one biopsy no mutation was found for eight genes tested (Table 1: 2). According to postsurgical evaluation of the biopsies by the Department of Pathology at the University Hospital Greifswald, the tissue was identified as pathological and assigned to the CHI type (mosaic, diffuse or focal). Islets of these pathological samples were isolated by injecting collagenase (2–4 mg/ml) into the biopsy material and by handpicking islets after digestion at 37°C. Afterward, islets were cultured in a CMRL 1066 medium with 5.5 mM glucose supplemented with 10% fetal calf serum, 100 U/ml of penicillin, 100 mg/ml of streptomycin, 10 mM HEPES, and 2 mM L-glutamine. Next day, the samples were shipped to Tübingen and/or Münster for further analysis.
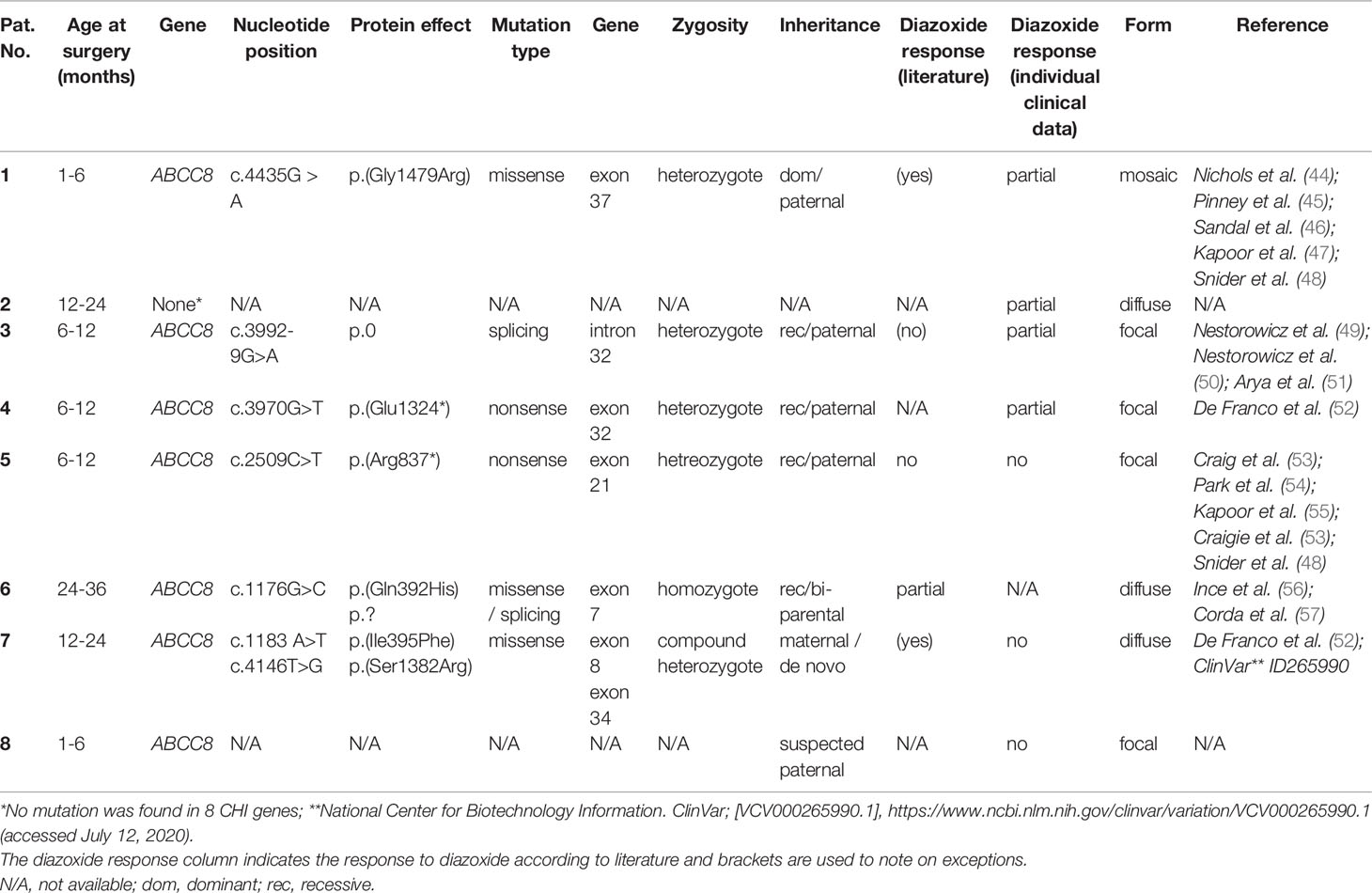
Table 1 Genetic characteristics of patients.
Mouse islets of Langerhans were isolated from adult C57BL/6N (WT) mice or SUR1 knockout (SUR1-/-) mice on a C57BL/6N background. The mice were bred in the animal facility of the Department of Pharmacology at the University of Tübingen. The principles of laboratory animal care (NIH publication no. 85-23, revised 1985) and German laws were followed. The animal study was reviewed and approved by the Regierungspräsidium Tübingen (§ 4 Abs. 3 TierSchG). Islets were isolated and cultured as previously described (58).
For experiments, human or mouse islet cell clusters of similar size were used, obtained by dispersing islets by trypsin treatment. Human and mouse islet cell clusters were kept in cell culture up to 3 days.
Solutions and Chemicals
Measurements of [Ca2+]c were performed with a bath solution, which contained (in mM): 140 NaCl, 5 KCl, 1.2 MgCl2, 2.5 CaCl2, 10 HEPES and glucose as indicated, pH 7.4 adjusted with NaOH. The same bath solution was used for the determination of the mitochondrial membrane potential (ΔΨ). The pipette solution for single channel recording contained (in mM): 130 KCl, 1.2 MgCl2, 2 CaCl2, 10 EGTA, and 10 HEPES; pH was adjusted to 7.4 with KOH. The bath solution contained (in mM): 130 KCl, 2 CaCl2, 10 EGTA, 1 Na2ATP, 1.7 MgCl2, and 20 HEPES with pH adjusted to 7.2 with KOH.
NN414, diazoxide and simvastatin were obtained from Sigma-Aldrich (Schnelldorf, Germany). DCEBIO was either purchased from Tocris Bioscience (Bristol, United Kingdom) or Santa Cruz (Heidelberg, Germany), fura-2-AM from Biotrend (Köln, Germany), and dextromethorphan (DXM) from Alfa Aesar (Kandel, Germany). Rhodamine 123 (Rh123), RPMI 1640 medium, CMRL 1066 medium, Dulbecco's modified Eagle's medium, fetal calf serum (FCS), penicillin/streptomycin, glutamine, and trypsin were from Invitrogen (Karlsruhe, Germany). Collagenase used for human biopsy material was obtained from Roche Diagnostics GmbH (Mannheim, Germany). All other chemicals were obtained from Sigma-Aldrich or Carl Roth (Karlsruhe, Germany) in the purest form available.
Measurements of [Ca2+]c
Details are described in (58). In brief, islet cell clusters were loaded with 5 µM fura-2-AM for 30-35 min at 37°C. The cells were perifused with bath solution with the indicated test substances. Fluorescence was excited at 340 and 380 nm, emission was filtered (LP515) and measured by a digital camera. Cytosolic Ca2+ concentration was measured as the ratio of the fluorescence intensities (F340/F380) of the emitted light excited with 340 nm and 380 nm. A ratio, i.e., one data point, was measured every 3 s. Ca2+ in glucose-activated beta cells oscillates between a basal and a maximal concentration. Decisive for insulin secretion is the mean Ca2+ concentration. Therefore, the data points were averaged 5–8 min before the end of a maneuver, to compare [Ca2+]c under different experimental conditions.
Measurements of the Mitochondrial Membrane Potential (ΔΨ)
ΔΨ was measured by Rh123 fluorescence at 480 nm excitation wavelength as described in (59). One data point was measured every 3 s. The effects were evaluated by averaging the values of the last 60 s of each interval before solution change. At the end of each experiment FCCP (0.5 µM) was added to evaluate maximal mitochondrial depolarization. Rh123 fluorescence corresponds to the proton gradient across the inner mitochondrial membrane and thus to ATP production. A decrease in fluorescence indicates a hyperpolarization and an increase in ATP production and vice versa.
Patch-clamp recordings
Hamster cDNA encoding for the SUR1E1507K protein together with WT human cDNA for Kir6.2 was expressed in a stably transfected HEK-293 cell line (60). Cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% FCS, glutamine, 100 U/ml of penicillin, and 100 mg/ml of streptomycin. Expression was induced by addition of doxycycline (300 µM) and cells were used for characterization of channel activity from 24 to 72 h.
Patch-clamp recordings were done in the inside-out configuration. KATP currents were measured at a membrane potential of -50 mV (pipette voltage, +50 mV); inward currents are shown as downward deflections. Patch pipettes had a resistance of 6–8 MΩ. Currents were recorded with an EPC-9 patch-clamp amplifier using Patchmaster software (HEKA, Lambrecht, Germany). Analyses to estimate mean current were done offline in IgorPro 7 (Wavemetrics, Inc., Lake Oswego, OR). With diazoxide or NN414 so many channels open simultaneously that a single channel evaluation of open probability (Po) was not possible. We therefore evaluated the mean current for 20 s before the end of a maneuver.
Statistics
Each series of experiments with islet cell clusters from mice was performed with at least three independent mouse preparations. The number of preparations for recordings with human islet cell clusters varied and is indicated for every series. Box plots were generated using Graphpad Prism 8. Boxes correspond to the interquartile range, the line within the box to the median, and the cross to the mean. Whiskers correspond to the maximum and minimum values. Statistical significance of differences was assessed by Student’s t test. Multiple comparisons were made by ANOVA followed by Student-Newman-Keuls test. P values ≤ 0.05 were considered significant.
Results
Effects of Nifedipine on the Cytosolic Ca2+ Concentration in Human Islet Cell Clusters
Oscillations of the cytosolic Ca2+ concentration ([Ca2+]c) are driven by fluctuations of the membrane potential and [Ca2+]c is the trigger signal for insulin secretion. Consequently, [Ca2+]c is a very robust surrogate parameter for insulin secretion. It can be determined easily and online with few cell material, which is an enormous advantage when working with human tissue. Glucose-stimulated insulin secretion in human pancreatic β-cells is completely suppressed by pharmacologic blockage of L-type Ca2+ channels (61). This mechanism should also be functional in CHI islet cells. Thus, as control we tested the effect of the L-type Ca2+ channel blocker nifedipine on [Ca2+]c in human islet cell clusters isolated from tissue of a patient with diffuse CHI (Table 1: 7). Figure 1A presents a recording with fast oscillations of [Ca2+]c on top of a plateau in the presence of a stimulating glucose concentration of 10 mM. The addition of nifedipine at a concentration of 5 µM diminished [Ca2+]c significantly (Figure 1B).
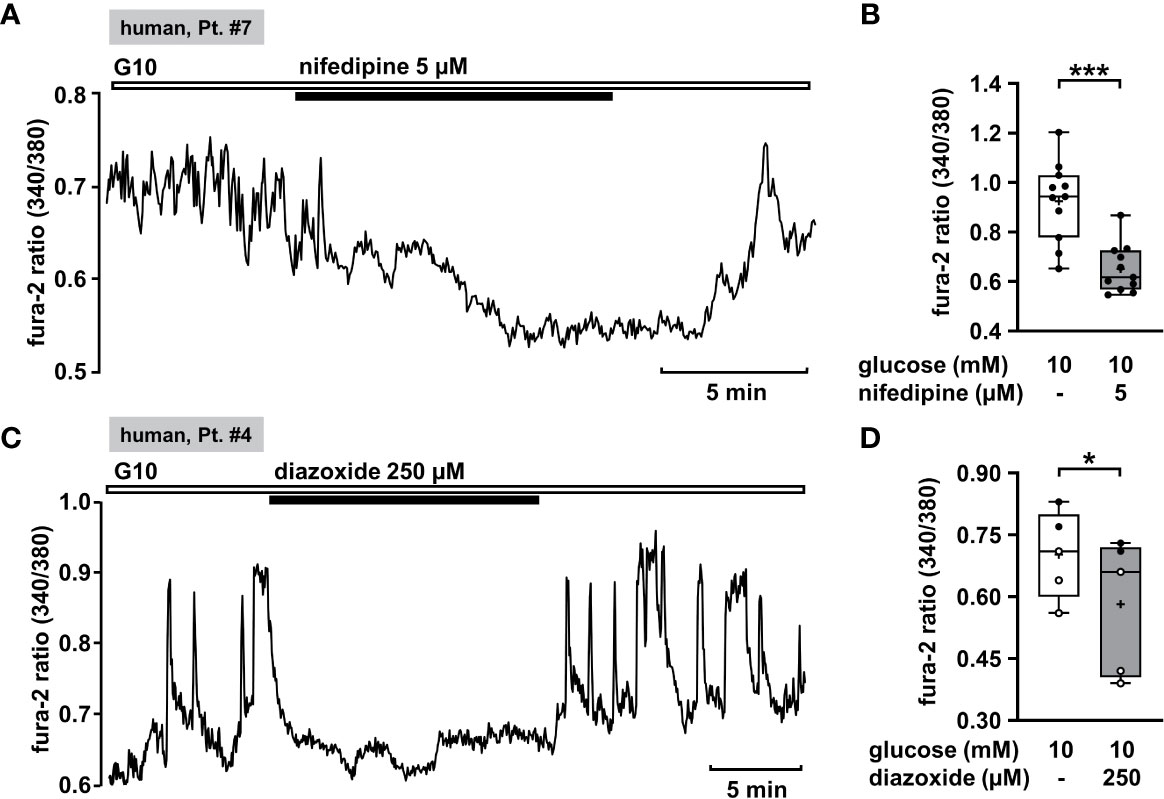
Figure 1 Nifedipine and diazoxide reduce [Ca2+]c in human CHI islet cell clusters. (A) Representative recording showing inhibition of glucose-induced oscillations of [Ca2+]c by nifedipine (5 µM) in the presence of 10 mM glucose in a human islet cell cluster isolated from pancreatic tissue of a patient with diffuse CHI (Table 1: 7, depicted as “human, Pt. #7” in the figure). (B) Summary of all experiments recorded in the presence of 10 mM glucose comprising 11 islet cell clusters isolated from pancreatic tissue of one CHI patient (Table 1: 7). (C) Representative recording showing the influence of diazoxide (250 µM) on glucose-induced oscillations of [Ca2+]c in the presence of 10 mM glucose in a human islet cell cluster isolated from pancreatic tissue of a patient with focal CHI (Table 1: 4). (D) Summary of all respective experiments from two patients, one with focal and one with mosaic form of CHI (Table 1: 1, black circles; 4, white circles) (n = 5). *p ≤ 0.05 and ***p ≤ 0.001.
KATP Channel Openers
Effects of Diazoxide on [Ca2+]c of Human Islet Cell Clusters
KATP channels (SUR1/Kir6.2) of pancreatic β-cells play a crucial role as they couple cellular metabolism to electrical activity. In electrically inactive β-cells, KATP channels are open. CHI is characterized by permanently active β-cells and thus opening of these channels is one strategy to treat it. Diazoxide is an opener of KATP channels that is already established in CHI therapy. We tested the effect of diazoxide on [Ca2+]c as a control. In human islet cell clusters isolated from two patients, one with focal and one with mosaic form of CHI (Table 1: 1 and 4), 250 µM diazoxide clearly decreased the mean fluorescence ratio (Figures 1C, D). These results show that the channels of these patients are in principle functional and can be influenced by the KATP channel opener, although mutations in the ABCC8 gene were reported to be the cause of CHI. Obviously, the complex regulation of the channels is disturbed, e.g., the sensitivity to MgATP (1).
Effects of NN414 on [Ca2+]c of Human and Mouse Islet Cell Clusters and Mitochondrial Membrane Potential of Mouse Islet Cell Clusters
The diazoxide analogue NN414 is suggested to be a selective agonist of pancreatic β-cell KATP channels, and is 100-fold more potent than diazoxide (62). Therefore, it has been proposed as useful drug for the treatment of diseases with excessive insulin secretion (62). NN414, in a concentration of 5 µM, completely abolished oscillations of [Ca2+]c and reduced [Ca2+]c to basal levels (Figure 2A). Application of 5 µM NN414 to human CHI islet cell clusters taken from three different forms (focal, diffuse, and atypical mosaic, Table 1: 1, 2, and 4) significantly lowered the mean fluorescence ratio (Figure 2B).
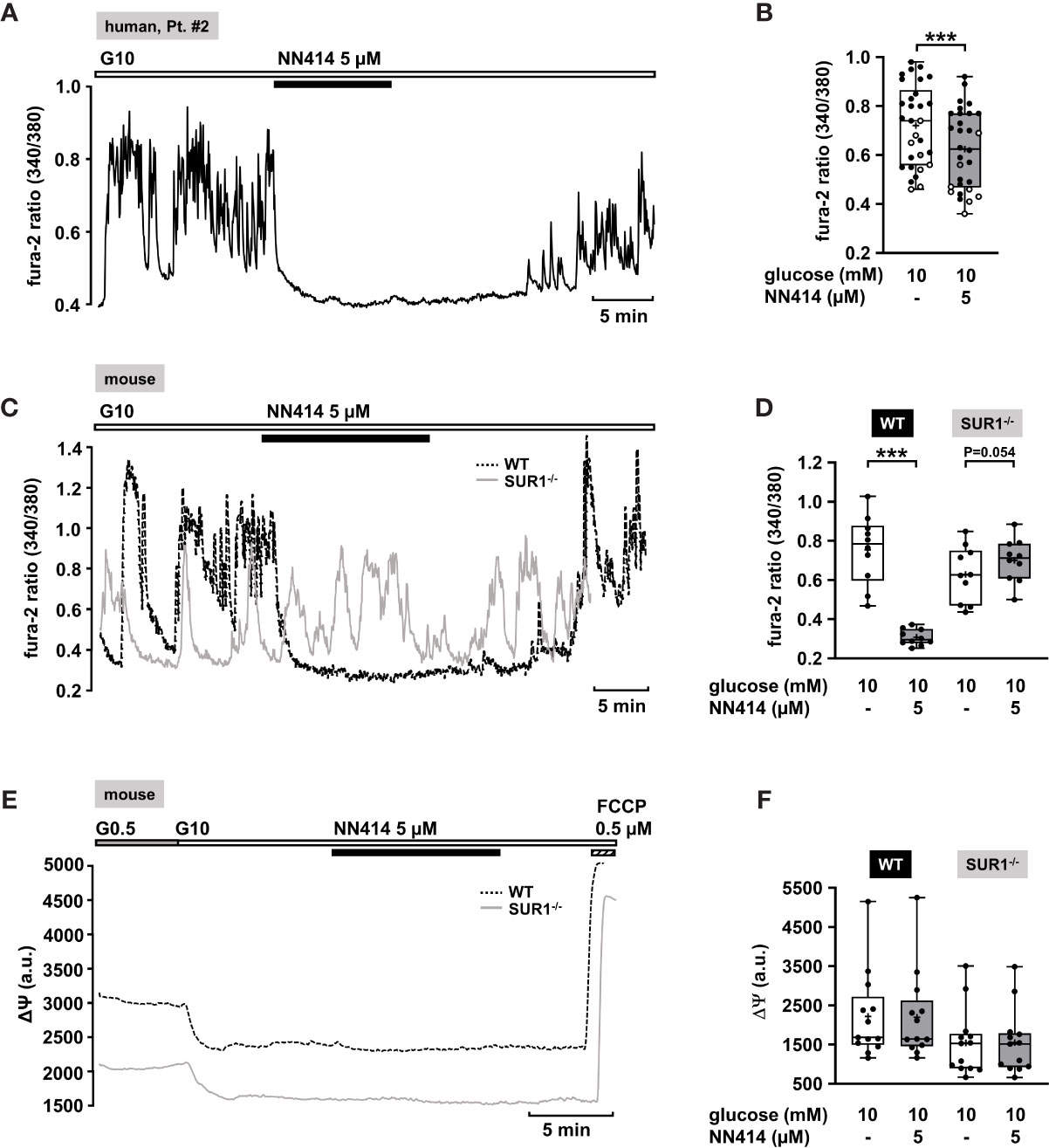
Figure 2 Effects of NN414 on [Ca2+]c and mitochondrial membrane potential ΔΨ. (A) Representative recording showing the reduction of glucose-induced oscillations of [Ca2+]c by NN414 (5 µM) in the presence of 10 mM glucose in a human islet cell cluster isolated from pancreatic tissue of a patient with diffuse CHI (Table 1: 2). (B) Summary of all respective experiments from three patients, one with diffuse, one with focal, and one with mosaic form (Table 1: 1, black circles; 2, white circles; and 4, gray circles) (n = 30). (C) Representative recordings showing the effect of NN414 (5 µM) on glucose-induced oscillations of [Ca2+]c in islet cell clusters from WT (dashed curve) and SUR1-/- (gray curve) mice. NN414 significantly reduced [Ca2+]c in islet cell clusters from WT mice, but not in islet cell clusters from SUR1-/- mice. (D) Summary of all respective experiments (n = 10 for each genotype, three different mouse preparations for each series). (E) Typical recordings showing measurement of ΔΨ in islet cell clusters obtained from WT (dashed curve) and SUR1-/- (gray curve) mice. The switch from 0.5 to 10 mM glucose hyperpolarizes ΔΨ. The addition of NN414 has no influence on ΔΨ in WT and SUR-/- islet cell clusters, respectively. (F) Summary of all experiments made under these conditions (n = 13, three different mouse preparations for each series). ***p ≤ 0.001.
To test whether NN414 specifically interferes with KATP channels, we studied the effects of this compound on [Ca2+]c with islet cell clusters of WT mice and mice lacking functional KATP channels (SUR1-/- mice) (63). As expected, 5 µM NN414 abolished [Ca2+]c oscillations in islet cell clusters of WT mice (Figure 2C, black trace) and reduced the mean fluorescence ratio (Figure 2D, left part). By contrast, NN414 hardly affected [Ca2+]c oscillations and did not decrease the mean fluorescence ratio in islet cell clusters obtained from SUR1-/- mice (Figures 2C, gray trace, 2D, right part).
Some KATP channel openers affect mitochondrial function in addition to their direct influence on KATP channels (64). To address this point, comparative experiments with islet cell clusters of the two mouse genotypes were performed evaluating a possible effect of NN414 on the mitochondrial membrane potential. Figure 2E shows typical recordings of ΔΨ for a WT and a SUR1-/- islet cell cluster. The switch from 0.5 to 10 mM glucose is accompanied by a strong decrease in Rh123 fluorescence reflecting hyperpolarization of ΔΨ and thus ATP production upon the increase of glucose concentration (65, 66). This maneuver was performed in each cell cluster to test for glucose responsiveness. At the end of each experiment, the uncoupler FCCP was applied to evaluate maximal mitochondrial depolarization. Neither in WT nor in islet cell clusters from SUR1-/- mice NN414 (5 µM) exerted any effect on ΔΨ (Figure 2F).
Diazoxide and NN414 Open KATP Channels Carrying a CHI Mutation
Mutations in the KATP channel subunits are the most common cause of CHI. However, they do not necessarily lead to diazoxide unresponsiveness. Response to diazoxide is even observed in patients in whom non-response would be predicted (67). Moreover, focal CHI is clinically heterogeneous and responsiveness or resistance to diazoxide was reported for patients with the same mutation in KATP channels (68). It is unclear whether these clinical effects are due to interference of diazoxide with the mutated channels or off-target effects. As an example illustrating the efficacy of KATP channel openers on mutant KATP channels, we used SUR1E1507K/WT Kir 6.2 channels since the SURE1507K mutation leads to CHI (69), but patients with this Glu to Lys mutation respond well to diazoxide (45). SUR1E1507K/WT Kir 6.2 channels were expressed in HEK-293 cells, and diazoxide was tested in comparison to NN414. Figure 3A shows that when inside/out patches from cells expressing SUR1E1507K/WT Kir 6.2 channels were pulled into nucleotide-free medium, numerous channels were activated as nucleotides inhibiting channel activity by antagonism on their WT pores were washed away (see start of the experiment before ATP application). Addition of 1 mM ATP rapidly inhibited channel activity as expected. Application of diazoxide (340 µM) or NN414 (5 µM) in the presence of ATP led to opening of SUR1E1507K/WT Kir 6.2 channels. (Figures 3B, C), showing that KATP channel agonists can directly affect mutated channels.
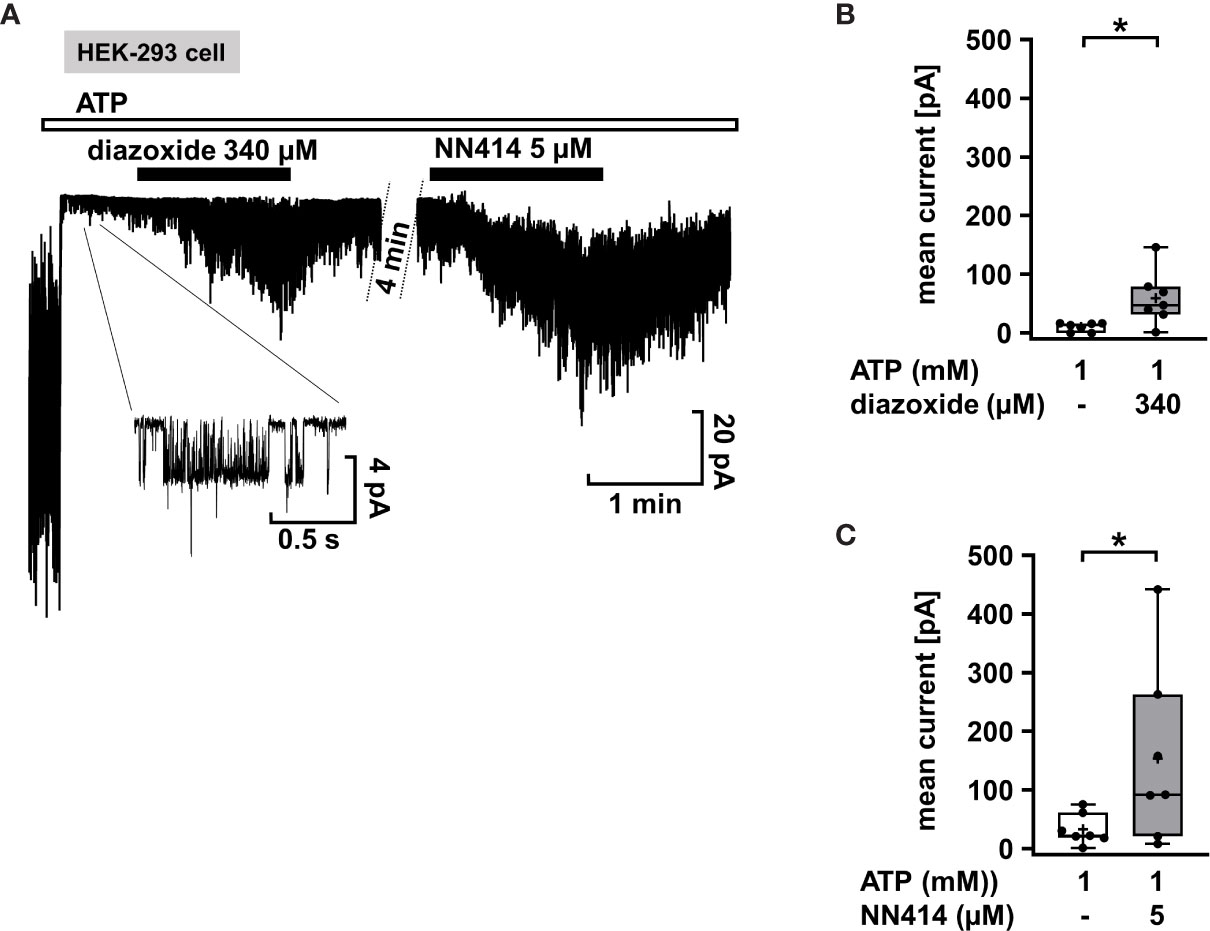
Figure 3 Diazoxide and NN414 open mutated KATP channels. (A) Representative trace showing the activation of SUR1E1507K/WT Kir 6.2 channels expressed in HEK-293 cells by the channel agonists diazoxide and NN414. This mutation is associated with CHI. At the beginning of the experiment the patch was pulled in nucleotide-free medium, which activates numerous channels as inhibitory nucleotides leave the pore. Addition of ATP rapidly inhibits almost all channel activity. Concurrent application of diazoxide (340 µM) or NN414 (5 µM) enhances channel activity. (B, C) Summary of all experiments with diazoxide (n = 7) and NN414 (n = 7), respectively. *p ≤ 0.05. The inset shows single channel openings at extended scales. The channel has an amplitude of about 4 pA, giving, at 50 mV driving force, a conductance of 80 pS, which is typical for KATP channels under these conditions. Four min under control conditions of the continuous recording were taken out for the clarity of the figure.
VU0071063 Silences Islet Cell Clusters in a KATP Channel-Dependent and -Independent Manner
Recently, Raphemot et al. discovered a novel xanthine derivative, VU0071063 that directly and selectively activates KATP channels (70). They found that VU0071063 is more potent and activates KATP channels with a faster kinetic than diazoxide. These findings encouraged us to test its effect on changes in [Ca2+]c on human islet cell clusters from CHI patients. Administration of VU0071063 (30 µM) to islet cells clusters from pancreatic tissue with a focal or diffuse lesion (Table 1: 2 and 4) induced a prompt reduction of [Ca2+]c in all four measurements. Figure 4A shows a typical example. The mean fluorescence ratio clearly changed (Figure 4B). Due to the limited pathological material, which explains the low number of experiments, we did not perform a statistical test with this data.
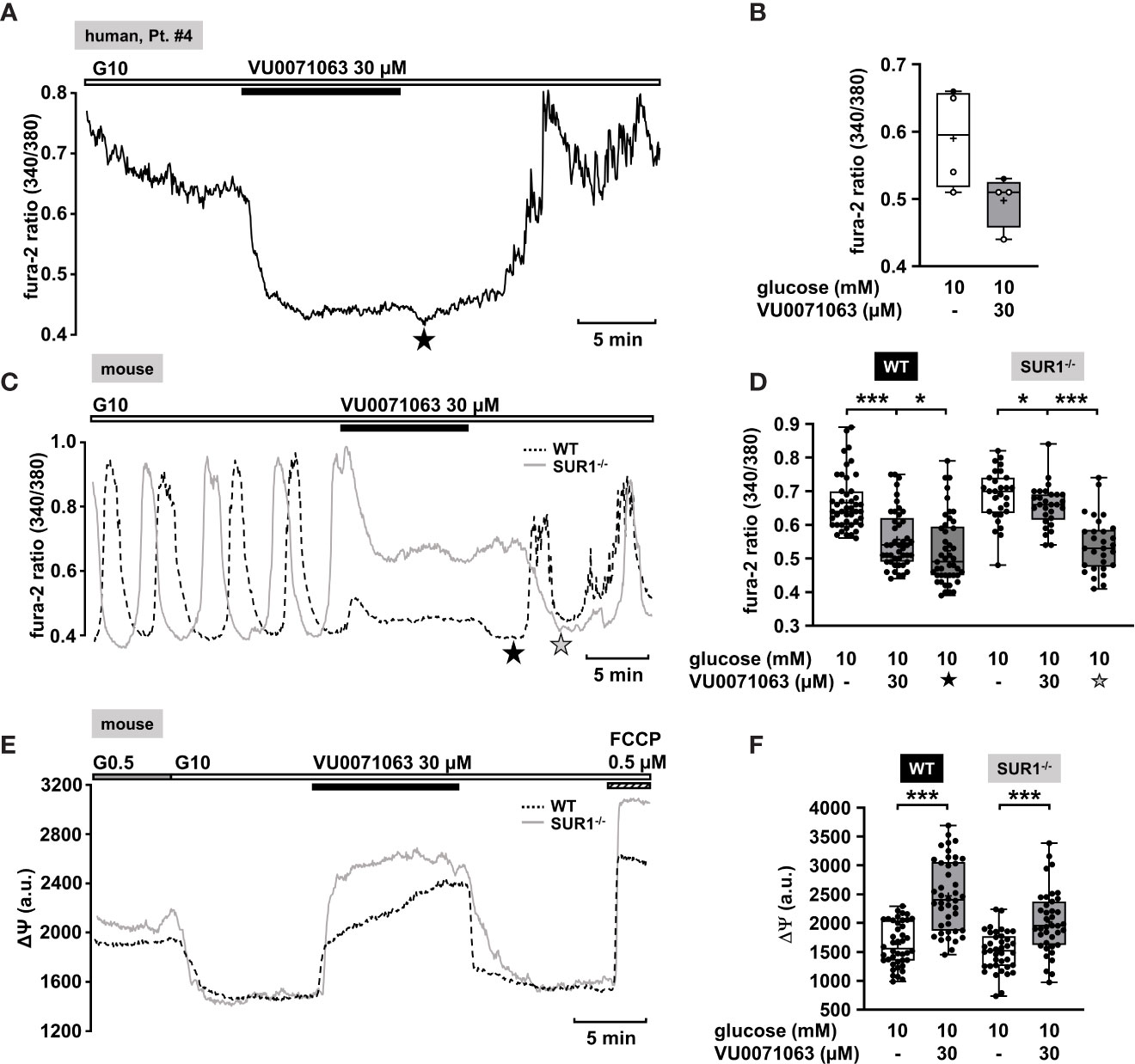
Figure 4 KATP channel-dependent and -independent effects of VU0071063. (A) Representative recording showing the reduction of [Ca2+]c by VU0071063 (30 µM) in the presence of 10 mM glucose in a human islet cell cluster isolated from pancreatic tissue of a CHI patients with a focal lesion (Table 1: 4). The star depicts the nadir after wash-out of VU0071063. (B) Summary of four experiments obtained from two patients, one with focal and one with diffuse form of CHI. VU0071063 rapidly reduced [Ca2+]c in all 4 experiments, but due to the low number of experiments, the effect is not significant. (Table 1: 2, black circles; 4, white circles). (C) Representative recordings showing the effect of VU0071063 (30 µM) on oscillations of [Ca2+]c induced by 10 mM glucose in islet cell clusters from WT (dashed curve) and SUR1-/- (gray curve) mice. VU0071063 significantly reduced [Ca2+]c in islet cell clusters from SUR1-/- mice, revealing KATP channel-independent effects of the compound. Note the drop in [Ca2+]c after removal of VU0071063 (black star: WT, gray star: SUR1-/-). (D) Summary of all respective experiments; n = 45 and 29 for WT and SUR-/- islet cell clusters. (E) Representative recordings showing the effect of VU0071063 (30 µM) on the mitochondrial membrane potential (ΔΨ) in islet cell clusters obtained from WT (dashed curve) and SUR1-/- (gray curve) mice. (F) Summary of all respective experiments; n = 42 and 39 for WT and SUR-/- islet cell clusters. Cell cluster were isolated from three WT and three SUR1-/- mice. *p ≤ 0.05 and ***p ≤ 0.001.
In the human islet cell clusters, a drop of [Ca2+]c was noticed directly after withdrawal of VU0071063 (Figure 4A, asterisk). Presumably, this drop is due to ATP-dependent sequestration of Ca2+ into the ER (71). This suggests that VU0071063 affects additional targets besides KATP channels. To evaluate this assumption, [Ca2+]c of islet cell clusters from WT mice and SUR1-/- mice was measured. VU0071063 (30 µM) suppressed Ca2+ oscillations and lowered [Ca2+]c in both genotypes (Figures 4C–F). Note that the effect was weaker in the cells of the knock-out mice. Like in human islet cell clusters, the drug further reduced [Ca2+]c after its removal in both WT and SUR1-/- mouse islet cell clusters (Figure 4C, asterisks). This points to alterations in mitochondrial metabolism, which can cause changes in KATP channel activity independent of any direct interaction with the channel proteins (72). As the mitochondrial membrane potential is for the most part directly linked to ATP production (65), we evaluated effects of VU0071063 on ΔΨ. Similar to the experiments described above, a rise in the glucose concentration caused a decrease of the fluorescence signal (Figure 4E). In islet cell clusters of WT mice and of SUR1-/- mice 30 µM VU0071063 strongly and reversibly depolarized mitochondrial membrane potential (Figures 4E, F).
KATP Channel-Independent Drugs
In the following part we present drugs and potential strategies, which could be effective in CHI patients non-responsive to KATP channel openers.
KCa3.1 Channel Openers as a Potential Approach
In addition to KATP and voltage-gated K+ channels, pancreatic β-cells express K+ channels regulated by the cytosolic Ca2+ concentration (KCa) (72). Depending on their single channel conductance, there are three groups whose existence has been detected in pancreatic β-cells (73–76). It has been demonstrated that the KCa channels of intermediate conductance (KCa3.1, SK4) play an important role in the K+ current (Kslow) that contributes to β-cell hyperpolarization at the end of a burst phase with electrical activity (66, 74, 77, 78). Previous results from Düfer et al. (74) demonstrated that activation of KCa3.1 channels hyperpolarized the membrane potential of pancreatic β-cells from WT mice. Since about 50% of Kslow is KATP current (79), the sulfonylurea-insensitive KCa component could be even more significant in β-cells lacking functional KATP channels, which resembles the situation in CHI channelopathies.
To verify this assumption, we evaluated the effect of the KCa3.1 opener DCEBIO on islet cell clusters isolated from SUR1-/- mice. DCEBIO (100 µM) effectively abolished the glucose-induced oscillations of [Ca2+]c (Figure 5A) and reduced the mean fluorescence ratio (Figure 5B). Next, we tested the effect of DCEBIO on human islet cell clusters from tissue of pancreatectomies. DCEBIO was tested on human islet cell clusters isolated from pancreatic tissues with mosaic and diffuse forms of CHI (Table 1: 1 and 2). The compound suppressed the oscillations of [Ca2+]c (Figure 5C) and significantly decreased the mean fluorescence ratio (Figure 5D).
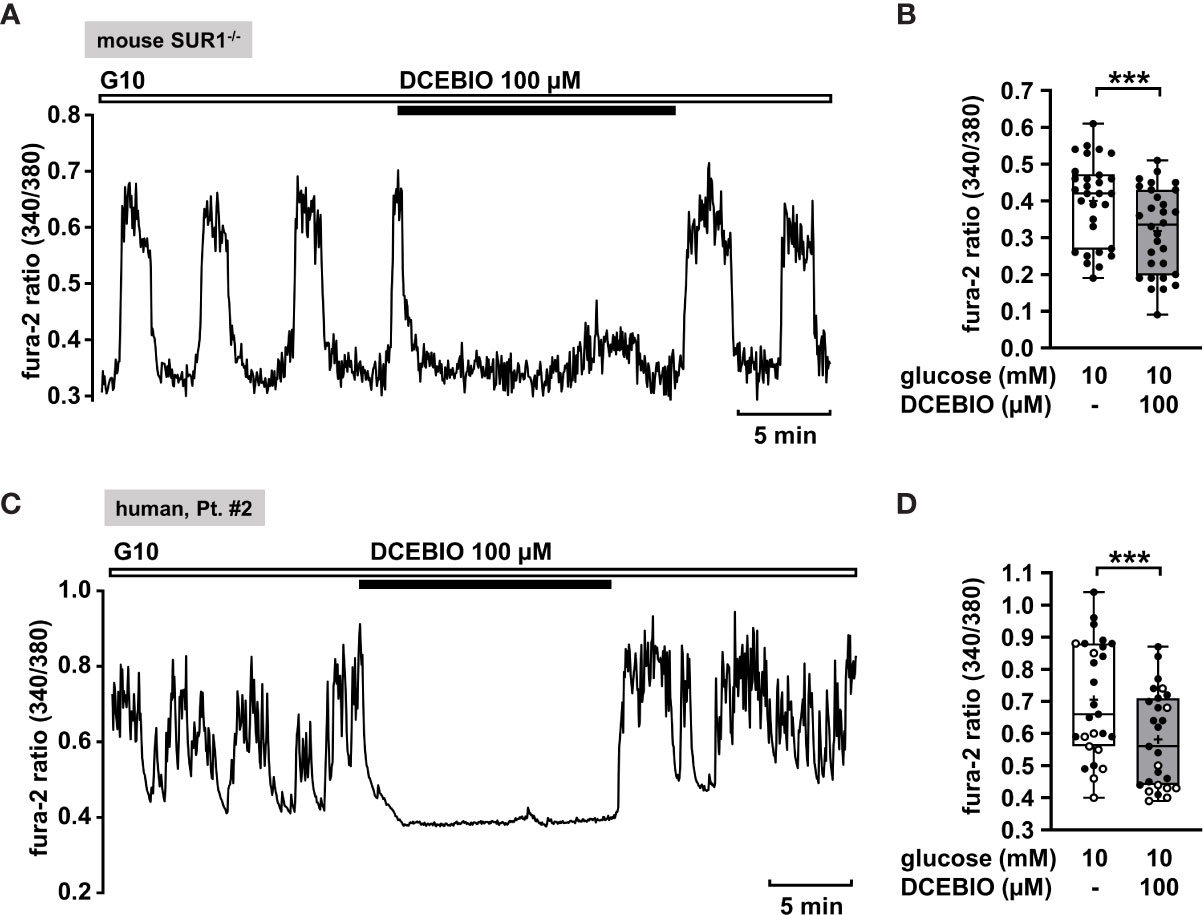
Figure 5 The KCa3.1 channel opener DCEBIO reduces [Ca2+]c in islet cell clusters isolated from SUR1-/- mice and in human islet cell clusters. (A) Representative recording showing rapid inhibition of glucose-induced oscillations of [Ca2+]c by DCEBIO (100 µM) in the presence of 10 mM glucose in islet cell clusters from SUR1-/- mice. (B) Summary of all respective experiments; n = 30. Islet cell clusters were obtained from three different SUR1-/- mice preparations. ***p ≤ 0.001. (C) Representative recording showing the reduction of glucose-induced oscillations of [Ca2+]c by DCEBIO (100 µM) in the presence of 10 mM glucose in a human islet cell cluster isolated from pancreatic tissue affected by diffuse CHI (Table 1: 2). (D) Summary of all respective experiments from biopsies of two CHI patients, one with diffuse, one with mosaic form (Table 1: 1, black circles; 2, white circles) (n = 27). ***p ≤ 0.001.
Effect of Dextromethorphan on [Ca2+]c of Human Islet Cell Clusters
Dextromethorphan (DXM) is a known antagonist of NMDA receptors. Active NMDA receptors can activate other ion channels, like Ca2+-activated K+ channels or KATP channels and thus potentiate K+ outflow (80). A block of NMDA receptors leads to prolonged depolarization and increases insulin secretion (81). Lesser-known is its ability to directly inhibit L-type Ca2+ channels. Carpenter et al. found that DXM moderately inhibits L-type Ca2+ channels, thereby lowering [Ca2+]c. This effect was observed with permanently depolarized cells under stimulating glucose concentrations (82). Since permanent depolarization is a characteristic of CHI β-cells, DXM may offer a possibility to rescue, i.e., silence the overstimulated cells.
The measurement in Figure 6A shows a recording of [Ca2+]c of a permanently depolarized islet cell cluster from a SUR1-/- mouse in the presence of 10 mM glucose and application of 100 µM DXM. The drug significantly lowered the mean fluorescence ratio (Figure 6B). The mean fluorescence, application of DXM rapidly reduced [Ca2+]c in an islet cell cluster from a patient with diffuse CHI (Figure 6C). The mean fluorescence ratio measured in islet cell clusters obtained from two patients with focal and two patients with diffuse CHI (Table 1: 3, 5, 6 and 7) was lowered (Figure 6D).
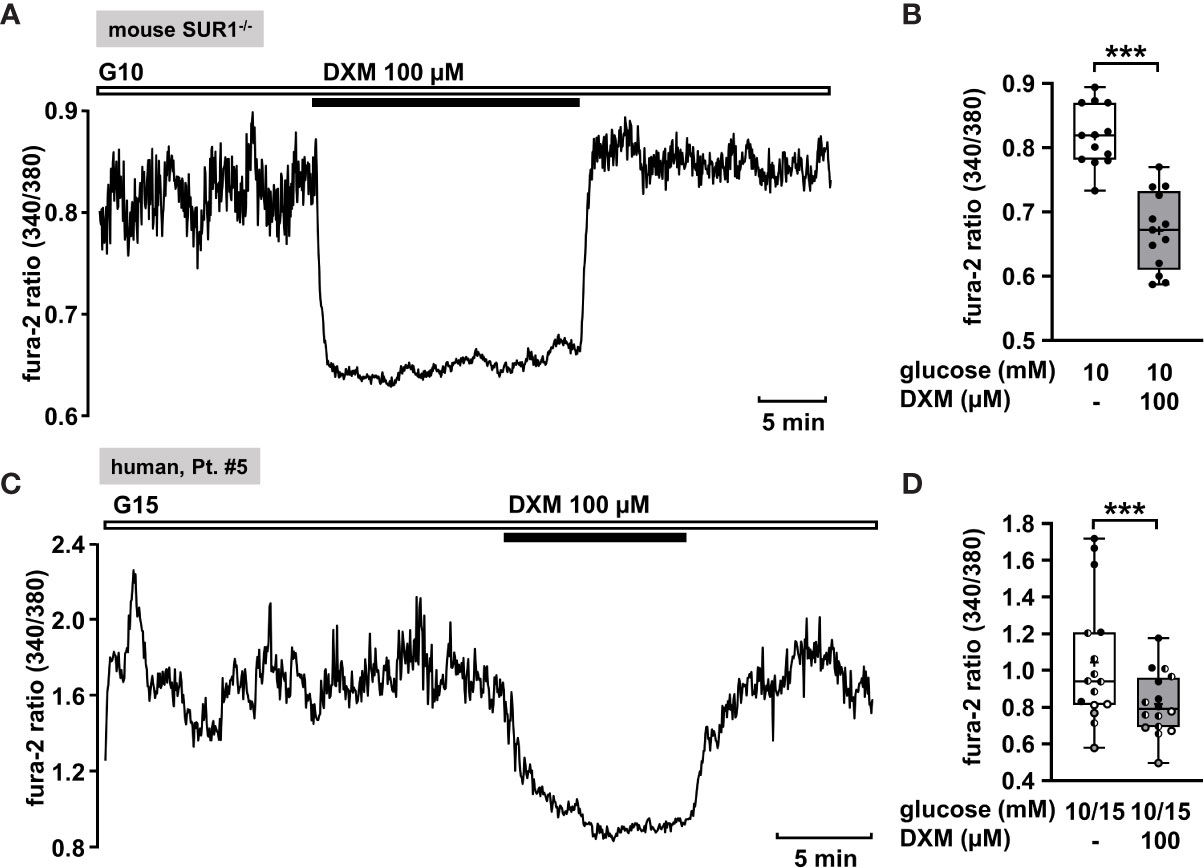
Figure 6 DXM lowers [Ca2+]c in islet cell clusters lacking functional KATP channels. (A) Representative recording showing a rapid decrease of [Ca2+]c by DXM (100 µM) in the presence of 10 mM glucose in an islet cell cluster from a SUR1-/- mouse. (B) Summary of all respective experiments (n = 13) with different cell cluster from three SUR1-/- mice. ***p ≤ 0.001. (C) Representative recording showing reduction of [Ca2+]c by DXM (100 µM) in the presence of 15 mM glucose in a human islet cell cluster isolated from pancreatic tissue of a patient with focal CHI (Table 1: 5). (D) Summary of all respective experiments obtained from biopsies of two patients with focal and two patients with diffuse CHI (Table 1: 3, gray circles; 5, black circles; 6, white circles; and 7, hatched circles) (n = 16). ***p ≤ 0.001.
Statins as a Potential Strategy to Silence Human Islet Cell Clusters
Lipid-lowering statins are inhibitors of the enzyme HMG-CoA-reductase, which plays a significant role in cholesterol synthesis by converting HMG-CoA to mevalonate. For these drugs it has been reported that they increase the risk of type 2 diabetes (83). Different studies have been conducted in order to enlighten the mechanism how the statins impair insulin secretion. In the study using β-cells isolated from rats, Yada et al. (84) showed that simvastatin in a concentration of 3 µg/ml acutely blocked L-type Ca2+ channels, thus lowering insulin secretion. Furthermore, Yaluri et al. demonstrated that simvastatin diminished glucose-stimulated insulin secretion and [Ca2+]c in MIN6 β-cells via multiple mechanisms (85).
Hence, we considered simvastatin as a potential therapeutic strategy to treat CHI. In order to confirm that simvastatin shows its effect when functional KATP channels are lacking, we measured [Ca2+]c in islet cell clusters from SUR1-/- mice (Figure 7A). Simvastatin in a concentration of 7.2 µM [according to the concentration of 3 µg/ml that was used in the study of Yada et al. (84)] rapidly decreased the glucose-stimulated Ca2+ oscillations and diminished the mean fluorescence ratio (Figure 7B). Further, we tested simvastatin on human islet cell clusters. Figure 7C shows a representative measurement of [Ca2+]c in an islet cell cluster isolated from pancreatic tissue of a patient with focal CHI. The mean fluorescence ratio markedly declined (Figure 7D). The biopsy material was obtained from two patients with focal and two patients with diffuse CHI (Table 1: 5 – 8).
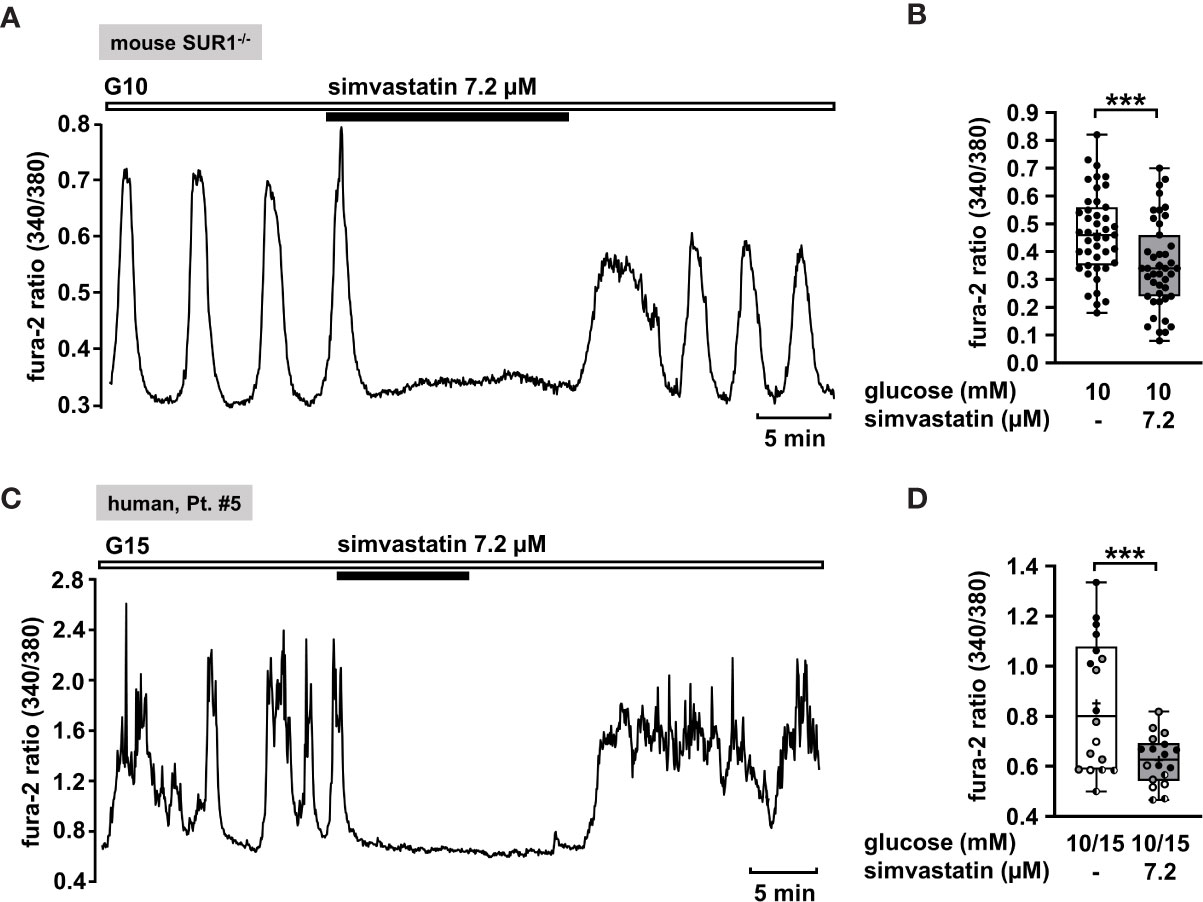
Figure 7 Simvastatin as a potential strategy to silence islet cell clusters affected by CHI. (A) Representative recording showing rapid inhibition of glucose-induced oscillations of [Ca2+]c by simvastatin (7.2 µM) in the presence of 10 mM glucose in an islet cell cluster from SUR1-/- mice. (B) Summary of all respective experiments (n = 43) with islet cell clusters obtained from three SUR1-/- mice. ***p ≤ 0.001. (C) Representative recording showing the reduction of glucose-induced oscillations of [Ca2+]c by simvastatin (7.2 µM) in the presence of 15 mM glucose in a human islet cell cluster isolated from pancreatic tissue of a patient affected by focal CHI (Table 1: 5). (D) Summary of all respective experiments from biopsies of two patients with focal and two patients with diffuse CHI (Table 1: 5, black circles; 6, white circles; 7, gray circles; and 8, hatched circles) (n = 18). ***p ≤ 0.001.
Discussion
Possible KATP Channel-Dependent Strategies to Treat CHI
In the present study islet cell clusters isolated from biopsies of CHI patients were used to search for new strategies to treat the disease. Pancreatic islet cell clusters isolated from either focal, diffuse or atypical pancreatic tissue were used as material. We are aware that these are distinct diseases. Due to the limited material we did not separate our results according to the different CHI forms. Noteworthy, the aim of the study was not to suggest novel drugs for CHI treatment but to optimize existing approaches, to search for novel targets and concepts for future drug development. Islet cell clusters were sensitive to the L-type Ca2+ channel blocker nifedipine. This maneuver resulted in a decrease of [Ca2+]c as expected from numerous observations with murine β-cells and insulin-secreting tumor cell lines as well as the restricted number of studies with human β-cells. This shows that the biopsy material is suitable to receive reliable and reproducible results. This is also confirmed by the results observed with diazoxide used as gold standard in CHI treatment. Noteworthy, nifedipine has been used for the treatment of diazoxide-unresponsive CHI (19, 29), but due to reported hypotension in patients with mutations in the ABCC8 gene, it is not commonly recommended for the treatment of CHI (29, 86).
Diazoxide is usually effective in all forms of CHI including severe cases caused by mutations in the genes encoding KATP channels (ABCC8 and KCNJ11), e.g., in (2, 67, 87, 88). Our sample cohort was derived from patients harboring ABCC8 missense mutations (patients 1, 6, and 7), a mutation affecting splicing (patient 3) and two nonsense mutations (patients 4,5). The missense and splicing mutations studied possibly allow production of SUR1 proteins albeit at reduced function or level. For nonsense mutation c.3970G>T p.(Glu1324*) detected in exon 32 of patient 4, nonsense-mediated RNA decay (NMD) has been predicted in silico, however, clinically the patient was reported to be diazoxide-responsive at dosage 8mg/kg/d. This may suggest escape of NMD with this particular mutation and generation of a truncated SUR1 protein lacking the last encoded six exons but retaining residual channel function. For one patient (patient 2) no mutation was found in the KATP channel genes (ABCC8 and KCNJ11) or in other CHI genes tested. This is not unusual in clinical routine. As this patient displayed a partial response to diazoxide (see Table 1) it was included in the study despite the unknown genetic background. Serious adverse drug effects are a consequence of the non-selectivity of diazoxide for pancreatic KATP (SUR1/Kir6.2) channels (89). Besides pancreatic KATP channels, the drug activates those of smooth muscles (SUR2B/Kir6.2 and SUR2B/Kir6.1) and exerts weak stimulatory effects on KATP channels of the cardiac muscle (SUR2A/Kir6.2) (90). One of the most common adverse effects of diazoxide, hirsutism, could be explained by activating both SUR1/Kir6.2 and SUR2B/Kir6.2 channels in hair follicles (91, 92).
NN414
As one strategy to improve CHI therapy we tested KATP channel openers more specific for β-cells. In comparison to diazoxide, NN414 is reported to be a selective SUR1 agonist, 100-fold more potent than diazoxide, suggesting that the drug is effective at much lower concentrations (62). Early, prediabetic stages of type 2 diabetes mellitus (T2DM) are normally characterized by compensatory hypersecretion of insulin. KATP channel openers have been suggested as beneficial medication to counteract excessive hormone release in prediabetic patients as insulin hypersecretion may cause or contribute to the development of glucose intolerance and β-cell degeneration in T2DM (93). NN414 has been used in numerous in vitro and in vivo studies to achieve β-cell rest, thereby preserving β-cell function and preventing apoptosis (94, 95). An animal in vivo study revealed a significant potential of NN414 in the treatment of disorders resulting from excessive insulin release (96). Alemzadeh et al. showed in a 6-week study that NN414 reduced hyperinsulinemia and improved glucose responsiveness in Zucker obese rats in a dose-dependent manner. NN414 entered human clinical trials for the treatment of T2DM. In healthy subjects, it inhibited insulin release, was well tolerated, and did not induce clinically relevant changes in safety parameters besides side effects on the gastrointestinal tract (97). NN414 was advanced in phase 2 of clinical trials where it showed a tendency to improve β-cell secretory function in diabetic patients (98, 99). The clinical trial was stopped because of elevated liver enzymes in treated patients (99, 100). The SUR1 selectivity, the low doses, and the reproducible Ca2+-lowering effect observed in our study in islet cell clusters from biopsy material (Figures 2A, B) suggest to consider NN414 as a potential alternative to diazoxide for the treatment of CHI with at least partially functioning KATP channels. Of note, this paper is not intended to characterize different types of CHI with respect to their diazoxide responsiveness or to recommend general treatment of all CHI types with NN414. Liver enzymes have to be monitored during therapy with NN414, but moderate elevation of their plasma concentration is no criterion to exclude the drug, although it would be desirable to develop NN414 analogues without this side effect. Notably, increased concentrations of circulating liver enzymes is one of the most reported side effects for octreotide that is used off-label as second-line therapeutic in the long-term management of CHI and for sirolimus that is proposed for patients resistant to diazoxide and octreotide (28, 101, 102), reviewed in (3).
VU0071063
Recently, a novel KATP channel activator, VU0071063 was discovered (70). VU0071063 is reported to be more selective for SUR1/Kir6.2 channels than for SUR2A/Kir6.2 and SUR2A/Kir6.1 channels. It has been demonstrated that it opens SUR1/Kir6.2 channels with a higher potency than diazoxide (70). VU0071063 was shown to activate KATP channels expressed in HEK-293 cells and to reduce glucose-stimulated Ca2+ influx in murine β-cells (70). Our data demonstrate at a first glance a beneficial characteristic of VU0071063 in human islet cell clusters isolated from CHI patients (Figures 4A, B), supporting the idea of a direct activation of KATP channels in pancreatic islets. By contrast, the observation that removal of VU0071063 from the solution initiated a transient drop in [Ca2+]c suggests that the drug does not selectively interfere with KATP channels, but also with ATP production (71). This assumption is supported by the following observations: 1) VU0071063 strongly and reversibly depolarized ΔΨ in both, WT and SUR1-/- islet cell clusters, which points to inhibition of ATP formation. 2) The removal of VU0071063 was followed by a transient drop in [Ca2+]c in WT and SUR1-/- islet cell clusters, which is presumably due to ATP-dependent SERCA activation. VU0071063 rapidly and significantly decreased [Ca2+]c in SUR1-/- islet cell clusters, too. This seems paradoxical as the ATP depletion leads to Ca2+ release out of the Ca2+ stores; however, the decreased [Ca2+]c during application of the drug might be secondary to Ca2+-dependent inactivation of L-type Ca2+ channels. Our data suggest that the Ca2+-lowering effect of VU0071063 is caused by a dual mechanism: 1) direct opening of KATP channels and indirect opening of KATP channels by ATP depletion; 2) interference with SERCA function and Ca2+ release, thereby mediating unpredictable interactions with other ion channels. In conclusion, since VU0071063 raises the expectation of detrimental effects on mitochondria, thereby impairing all ATP-dependent processes, this compound seems not to be suitable for use in humans without structural modifications avoiding this side effect. Noteworthy, NN414 did not affect ΔΨ (compare Figures 2E, F to 4E, F) and did hardly change [Ca2+]c in SUR1-/- islet cell clusters (compare Figures 2C, Dto 4C, D). These differences clearly show that the effects of NN414 are, in contrast to those of VU0071063, caused by a specific interference with KATP channels.
In summary, our data with KATP channel agonists demonstrate that these drugs can be effective in different forms of CHI caused by mutations in KATP channels. As shown by patch-clamp experiments (Figure 3), diazoxide and NN414 act as direct channel openers in mutated KATP channels with a dominant mutation comparable to WT channels. With respect to specificity, dosage, and expected side effects, NN414 seems superior to diazoxide. VU0071063 is unsuitable because of its multiple and yet not completely understood mode of action and therefore potential adverse side effects.
Possible KATP Channel-Independent Strategies to Treat CHI
Current Second- And Third-Line Therapy Regimen Targeting KATP Channel Independent Pathways
There are mutations in ABCC8 or KCNJ11 genes known to disrupt the expression of KATP channels at the cell surface (4, 103, 104). In this case openers, e.g., diazoxide, are ineffective in the treatment of CHI (51, 105). For these patients it is indispensable to find drugs targeting mechanisms, which can induce β-cell rest and inhibit insulin release independent of KATP channels. Currently available alternatives to diazoxide therapy are somatostatin analogues (octreotide, octreotide-LAR, and lanreotide), sirolimus and exendin-(9–39) (3). Octreotide, a short-acting synthetic somatostatin analogue, inhibits insulin secretion by binding to and activating somatostatin receptors 2 and 5 (SSTR2 and SSTR5) (106). Activation of SSTRs shows multifactorial modulation of β-cells, which involves inhibition of the adenylate cyclase/cAMP pathway, activation of G protein-activated inwardly rectifying K+ (GIRK) channels, decrease in Ca2+ influx via P/Q-type Ca2+ channels and inhibition of exocytosis (24, 107, 108). Infants respond well to initial doses of octreotide, but tachyphylaxis after a few doses makes it not suitable for the long-term treatment. Long-acting somatostatin analogues (octreotide-LAR and lanreotide) have similar effects as octreotide but have the advantage that they are given once every 4 weeks, which improves therapy compliance and quality of life (21, 109, 110). However, due to a similar mechanism of action as octreotide, long-acting somatostatin analogues show similar side effects (111). Sirolimus, a mammalian target of rapamycin (mTOR) inhibitor, is an immunosuppressive and anti-proliferative agent that has been used in patients with diffuse CHI, unresponsive to diazoxide and octreotide therapy (22, 112). It suppresses insulin release by different mechanisms, which have not been fully elucidated (30). It has been proposed that downregulation of mTOR pathway decreases insulin production in pancreatic β-cells and β-cell growth and may restore ketogenesis (112, 113). Furthermore, upregulation of liver gluconeogenesis by sirolimus contributes to insulin resistance (114). However, severe and life-threatening side effects reported for the above-mentioned drugs restrict their use.
Opening of KCa3.1 Channels
Beside KATP channels, Ca2+-activated K+ channels of intermediate conductance (KCa3.1, SK4) contribute to Kslow, the hyperpolarizing current that terminates bursts of action potentials in β-cells (74, 78). Accordingly, KCa3.1 channels may become predominant regulators of membrane potential and insulin secretion when functional KATP channels are absent, which makes these channels ideal as drug targets in CHI. Our data show that an opener of KCa3.1 channels, DCEBIO, was able to strongly reduce [Ca2+]c in SUR1-/- islet cell clusters. Furthermore, DCEBIO was highly effective in silencing human islet cell clusters obtained from pancreatic tissue of CHI patients (Figure 5). These experiments provide valuable support for the idea of targeting KCa3.1 channels in the treatment of CHI. To follow this strategy would of course require the search for new KCa3.1 channel openers with high selectivity for β-cells since unspecific KCa3.1 channel openers are expected to exert severe side effects in numerous organs (e.g., lung, cells of the hematopoietic system, and salivary glands) (115–117).
DXM as L-type Ca2+ Channel Antagonist
The block of NMDA receptors by DXM is expected to increase insulin secretion (81). However, DXM has a higher affinity for L-type Ca2+ channels than to its known target, the NMDA receptor (118). As mentioned before, DXM moderately inhibits L-type Ca2+ channels, thereby lowering [Ca2+]c in permanently depolarized β-cells under stimulating glucose concentrations (82, 119). This is exactly what we observe in our experiments with depolarized islet cell clusters isolated from pancreatic tissue of CHI patients and with depolarized islet cell clusters from SUR1-/- mice (Figure 6). Thus, DXM could be an alternative strategy for the treatment of CHI especially in the diffuse form of CHI. In patients with focal lesions, where only a distinct portion of β-cells, i.e., the focal ones, is dysregulated, the drug could cause an undesired increase in [Ca2+]c and insulin secretion in healthy islets. The benefits of this drug are that it is already available as a pharmaceutical and that inhibition of L-type Ca2+ channels by e.g., nifedipine is already a proved treatment of CHI (19, 29). Considering side effects of nifedipine, like hypotension, the moderate effect of DXM on L-type Ca2+ channels could be of advantage (82).
Simvastatin
It was shown that simvastatin lowers insulin secretion by blocking L-type Ca2+ channels (84, 85). The effect of simvastatin on [Ca2+]c was comparable with that of nifedipine in the insulin-secreting cell line MIN-6 (85). This interaction might contribute to the increased risk to develop diabetes mellitus under a cholesterol-lowering therapy with statins (83). With respect to CHI patients, this side effect could constitute a suitable therapeutic approach. Our results obtained from experiments with islet cell clusters isolated from SUR1-/- mice and human islet cell clusters isolated from patients with CHI indeed point toward a possible beneficial effect of simvastatin in the treatment of CHI (Figure 7). Noteworthy, statins are widely used and well tolerated in the long-term therapy. In contrast to nifedipine, which affects the cardiovascular system, statins are safe with respect to blood pressure or heart rate (120). Additionally, it is proposed that statins induce hepatic gluconeogenesis in human liver cells by activation of the pregnane X receptor (PXR) (121, 122), which could also counteract hypoglycemic conditions in CHI patients.
Conclusions
There is a clear need to develop novel approaches to prevent hypoglycemia in CHI patients and to establish better therapies with less side effects for the different forms of CHI. In this study, we had access to biopsy material of CHI patients and give suggestions, which drugs or targets should be studied in future. Promising results were obtained for NN414, DCEBIO, DXM and simvastatin.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors upon request.
Ethics Statement
Ethics approval for the study involving human participants was approved by the ethic commission of the Universitätsmedizin Greifswald (BB 050/13). Written informed consent was provided by the legal guardians of the children for the study. The animal study was reviewed and approved by the Ethics Committee of Regierungspraesidium Tuebingen.
Author Contributions
JS researched the data and wrote and edited the manuscript. TH, AG, and JK researched the data, contributed to the discussion, and edited the manuscript. WB, CW, and UL provided human β-cells and contributed to discussion. IW supported the manuscript as an expert in CHI genetics and contributed to the discussion. MD and PK-D contributed to discussion and study design and edited the manuscript. GD designed the study, wrote and edited the manuscript, and contributed to discussion. GD is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the DFG (DR225/11-1 to GD and INST 211/647-1 FUGG to MD).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Dr. Frank Dombrowski and colleagues (Institute of Pathology, University of Greifswald, Greifswald, Germany) for providing the biopsy material. We are grateful to Prof. Dr. Bryan, Seattle, for providing mutated KATP channels expressed in HEK-293 cells. We acknowledge support by Open Access Publishing Fund of University of Tübingen.
References
1. Dunne MJ, Cosgrove KE, Shepherd RM, Aynsley-Green A, Lindley KJ. Hyperinsulinism in infancy: from basic science to clinical disease. Physiol Rev (2004) 84(1):239–75. doi: 10.1152/physrev.00022.2003
2. Banerjee I, Salomon-Estebanez M, Shah P, Nicholson J, Cosgrove KE, Dunne MJ. Therapies and outcomes of congenital hyperinsulinism-induced hypoglycaemia. Diabetes Med (2019) 36(1):9–21. doi: 10.1111/dme.13823
3. Demirbilek H, Rahman SA, Buyukyilmaz GG, Hussain K. Diagnosis and treatment of hyperinsulinaemic hypoglycaemia and its implications for paediatric endocrinology. Int J Pediatr Endocrinol (2017) 2017:9. doi: 10.1186/s13633-017-0048-8
4. Vajravelu ME, De León DD. Genetic characteristics of patients with congenital hyperinsulinism. Curr Opin Pediatr (2018) 30(4):568–75. doi: 10.1097/MOP.0000000000000645
5. Galcheva S, Demirbilek H, Al-Khawaga S, Hussain K. The Genetic and Molecular Mechanisms of Congenital Hyperinsulinism. Front Endocrinol (2019) 10:111. doi: 10.3389/fendo.2019.00111
6. Rahier J, Guiot Y, Sempoux C. Morphologic analysis of focal and diffuse forms of congenital hyperinsulinism. Semin Pediatr Surg (2011) 20(1):3–12. doi: 10.1053/j.sempedsurg.2010.10.010
7. Sempoux C, Capito C, Bellanne-Chantelot C, Verkarre V, de Lonlay P, Aigrain Y, et al. Morphological mosaicism of the pancreatic islets: a novel anatomopathological form of persistent hyperinsulinemic hypoglycemia of infancy. J Clin Endocrinol Metab (2011) 96(12):3785–93. doi: 10.1210/jc.2010-3032
8. Kapoor RR, Flanagan SE, James C, Shield J, Ellard S, Hussain K. Hyperinsulinaemic hypoglycaemia. Arch Dis Child (2009) 94(6):450–7. doi: 10.1136/adc.2008.148171
9. Yorifuji T, Horikawa R, Hasegawa T, Adachi M, Soneda S, Minagawa M, et al. Clinical practice guidelines for congenital hyperinsulinism. Clin Pediatr Endocrinol (2017) 26(3):127–52. doi: 10.1297/cpe.26.127
10. Aynsley-Green A, Hussain K, Hall J, Saudubray JM, Nihoul-Fekete C, De Lonlay-Debeney P, et al. Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal (2000) 82(2):F98–F107. doi: 10.1136/fn.82.2.F98
11. Shah P, Rahman SA, Demirbilek H, Guemes M, Hussain K. Hyperinsulinaemic hypoglycaemia in children and adults. Lancet Diabetes Endocrinol (2017) 5(9):729–42. doi: 10.1016/S2213-8587(16)30323-0
12. Welters A, Lerch C, Kummer S, Marquard J, Salgin B, Mayatepek E, et al. Long-term medical treatment in congenital hyperinsulinism: a descriptive analysis in a large cohort of patients from different clinical centers. Orphanet J Rare Dis (2015) 10:150. doi: 10.1186/s13023-015-0367-x
13. Nebesio TD, Hoover WC, Caldwell RL, Nitu ME, Eugster EA. Development of pulmonary hypertension in an infant treated with diazoxide. J Pediatr Endocrinol Metab (2007) 20(8):939–44. doi: 10.1515/JPEM.2007.20.8.939
14. Yildizdas D, Erdem S, Kucukosmanoglu O, Yilmaz M, Yuksel B. Pulmonary hypertension, heart failure and neutropenia due to diazoxide therapy. Adv Ther (2008) 25(5):515–9. doi: 10.1007/s12325-008-0049-3
15. Demirel F, Unal S, Cetin II, Esen I, Arasli A. Pulmonary hypertension and reopening of the ductus arteriosus in an infant treated with diazoxide. J Pediatr Endocrinol Metab (2011) 24(7-8):603–5. doi: 10.1515/jpem.2011.238
16. Timlin MR, Black AB, Delaney HM, Matos RI, Percival CS. Development of Pulmonary Hypertension During Treatment with Diazoxide: A Case Series and Literature Review. Pediatr Cardiol (2017) 38(6):1247–50. doi: 10.1007/s00246-017-1652-3
17. Mohnike K, Blankenstein O, Pfuetzner A, Potzsch S, Schober E, Steiner S, et al. Long-term non-surgical therapy of severe persistent congenital hyperinsulinism with glucagon. Horm Res (2008) 70(1):59–64. doi: 10.1159/000129680
18. Neylon OM, Moran MM, Pellicano A, Nightingale M, O’Connell MA. Successful subcutaneous glucagon use for persistent hypoglycaemia in congenital hyperinsulinism. J Pediatr Endocrinol Metab (2013) 26(11-12):1157–61. doi: 10.1515/jpem-2013-0115
19. Eichmann D, Hufnagel M, Quick P, Santer R. Treatment of hyperinsulinaemic hypoglycaemia with nifedipine. Eur J Pediatr (1999) 158(3):204–6. doi: 10.1007/s004310051049
20. Modan-Moses D, Koren I, Mazor-Aronovitch K, Pinhas-Hamiel O, Landau H. Treatment of congenital hyperinsulinism with lanreotide acetate (Somatuline Autogel). J Clin Endocrinol Metab (2011) 96(8):2312–7. doi: 10.1210/jc.2011-0605
21. Le Quan Sang KH, Arnoux JB, Mamoune A, Saint-Martin C, Bellanne-Chantelot C, Valayannopoulos V, et al. Successful treatment of congenital hyperinsulinism with long-acting release octreotide. Eur J Endocrinol (2012) 166(2):333–9. doi: 10.1530/EJE-11-0874
22. Senniappan S, Brown RE, Hussain K. Sirolimus in severe hyperinsulinemic hypoglycemia. N Engl J Med (2014) 370(25):2448–9. doi: 10.1056/NEJMc1404716
23. Baş F, Darendeliler F, Demirkol D, Bundak R, Saka N, Günöz H. Successful therapy with calcium channel blocker (nifedipine) in persistent neonatal hyperinsulinemic hypoglycemia of infancy. J Pediatr Endocrinol Metab (1999) 12(6):873–8. doi: 10.1515/JPEM.1999.12.6.873
24. Kailey B, van de Bunt M, Cheley S, Johnson PR, MacDonald PE, Gloyn AL, et al. SSTR2 is the functionally dominant somatostatin receptor in human pancreatic beta- and alpha-cells. Am J Physiol Endocrinol Metab (2012) 303(9):E1107–16. doi: 10.1152/ajpendo.00207.2012
25. Gromada J, Brock B, Schmitz O, Rorsman P. Glucagon-like peptide-1: regulation of insulin secretion and therapeutic potential. Basic Clin Pharmacol Toxicol (2004) 95(6):252–62. doi: 10.1111/j.1742-7843.2004.t01-1-pto950502.x
26. Hawkes CP, Adzick NS, Palladino AA, De Leon DD. Late Presentation of Fulminant Necrotizing Enterocolitis in a Child with Hyperinsulinism on Octreotide Therapy. Horm Res Paediatr (2016) 86(2):131–6. doi: 10.1159/000443959
27. McMahon AW, Wharton GT, Thornton P, De Leon DD. Octreotide use and safety in infants with hyperinsulinism. Pharmacoepidemiol Drug Saf (2017) 26(1):26–31. doi: 10.1002/pds.4144
28. Demirbilek H, Shah P, Arya VB, Hinchey L, Flanagan SE, Ellard S, et al. Long-term follow-up of children with congenital hyperinsulinism on octreotide therapy. J Clin Endocrinol Metab (2014) 99(10):3660–7. doi: 10.1210/jc.2014-1866
29. Durmaz E, Flanagan SE, Parlak M, Ellard S, Akcurin S, Bircan I. A combination of nifedipine and octreotide treatment in an hyperinsulinemic hypoglycemic infant. J Clin Res Pediatr Endocrinol (2014) 6(2):119–21. doi: 10.4274/jcrpe.1230
30. Szymanowski M, Estebanez MS, Padidela R, Han B, Mosinska K, Stevens A, et al. mTOR Inhibitors for the Treatment of Severe Congenital Hyperinsulinism: Perspectives on Limited Therapeutic Success. J Clin Endocrinol Metab (2016) 101(12):4719–29. doi: 10.1210/jc.2016-2711
31. Banerjee I, De Leon D, Dunne MJ. Extreme caution on the use of sirolimus for the congenital hyperinsulinism in infancy patient. Orphanet J Rare Dis (2017) 12(1):70. doi: 10.1186/s13023-017-0621-5
32. Corbin JA, Bhaskar V, Goldfine ID, Bedinger DH, Lau A, Michelson K, et al. Improved glucose metabolism in vitro and in vivo by an allosteric monoclonal antibody that increases insulin receptor binding affinity. PloS One (2014) 9(2):e88684. doi: 10.1371/journal.pone.0088684
33. Corbin JA, Bhaskar V, Goldfine ID, Issafras H, Bedinger DH, Lau A, et al. Inhibition of insulin receptor function by a human, allosteric monoclonal antibody: a potential new approach for the treatment of hyperinsulinemic hypoglycemia. MAbs (2014) 6(1):262–72. doi: 10.4161/mabs.26871
34. Issafras H, Bedinger DH, Corbin JA, Goldfine ID, Bhaskar V, White ML, et al. Selective allosteric antibodies to the insulin receptor for the treatment of hyperglycemic and hypoglycemic disorders. J Diabetes Sci Technol (2014) 8(4):865–73. doi: 10.1177/1932296814529886
35. Patel P, Charles L, Corbin J, Goldfine ID, Johnson K, Rubin P, et al. A unique allosteric insulin receptor monoclonal antibody that prevents hypoglycemia in the SUR-1(-/-) mouse model of KATP hyperinsulinism. MAbs (2018) 10(5):796–802. doi: 10.1080/19420862.2018.1457599
36. Johnson KW, Neale AC, Gordon A, De León-Crutchlow DDC, Hussain K, Mohnike KL, et al. Activity of Xoma 358, an Inhibitor of Insulin Action Following Short-Term Administration to Congenital Hyperinsulinism Patients. Endocrine Rev June (2017) 38(3).
37. Barthlen W, Varol E, Empting S, Wieland I, Zenker M, Mohnike W, et al. Surgery in Focal Congenital Hyperinsulinism (CHI) - The “Hyperinsulinism Germany International” Experience in 30 Children. Pediatr Endocrinol Rev (2016) 14(2):129–37. doi: 10.17458/PER.2016.BVE.Surgeryinfocal
38. Adzick NS, Thornton PS, Stanley CA, Kaye RD, Ruchelli E. A multidisciplinary approach to the focal form of congenital hyperinsulinism leads to successful treatment by partial pancreatectomy. J Pediatr Surg (2004) 39(3):270–5. doi: 10.1016/j.jpedsurg.2003.11.019
39. Barthlen W, Mohnike W, Mohnike K. Techniques in pediatric surgery: congenital hyperinsulinism. Horm Res Paediatr (2010) 74(6):438–43. doi: 10.1159/000321902
40. Adzick NS, De León DD, States LJ, Lord K, Bhatti TR, Becker SA, et al. Surgical treatment of congenital hyperinsulinism: Results from 500 pancreatectomies in neonates and children. J Pediatr Surg (2019) 54(1):27–32. doi: 10.1016/j.jpedsurg.2018.10.030
41. Lovvorn HN,3, Nance ML, Ferry RJ Jr., Stolte L, Baker L, O’Neill JA Jr., et al. Congenital hyperinsulinism and the surgeon: lessons learned over 35 years. J Pediatr Surg (1999) 34(5):786–92; discussion 92-3. doi: 10.1016/S0022-3468(99)90374-3
42. Meissner T, Wendel U, Burgard P, Schaetzle S, Mayatepek E. Long-term follow-up of 114 patients with congenital hyperinsulinism. Eur J Endocrinol (2003) 149(1):43–51. doi: 10.1530/eje.0.1490043
43. Barthlen W. Surgery in congenital hyperinsulinism-tips and tricks not only for surgeons. A practical guide. Semin Pediatr Surg (2011) 20(1):56–9. doi: 10.1053/j.sempedsurg.2010.10.002
44. Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JPt, Gonzalez G, et al. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science (1996) 272(5269):1785–7. doi: 10.1126/science.272.5269.1785
45. Pinney SE, MacMullen C, Becker S, Lin YW, Hanna C, Thornton P, et al. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J Clin Invest (2008) 118(8):2877–86. doi: 10.1172/JCI35414
46. Sandal T, Laborie LB, Brusgaard K, Eide SA, Christesen HB, Sovik O, et al. The spectrum of ABCC8 mutations in Norwegian patients with congenital hyperinsulinism of infancy. Clin Genet (2009) 75(5):440–8. doi: 10.1111/j.1399-0004.2009.01152.x
47. Kapoor RR, Flanagan SE, James CT, McKiernan J, Thomas AM, Harmer SC, et al. Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant ABCC8/KCNJ11 mutations. Diabetologia (2011) 54(10):2575–83. doi: 10.1007/s00125-011-2207-4
48. Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, et al. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab (2013) 98(2):E355–63. doi: 10.1210/jc.2012-2169
49. Nestorowicz A, Wilson BA, Schoor KP, Inoue H, Glaser B, Landau H, et al. Mutations in the sulonylurea receptor gene are associated with familial hyperinsulinism in Ashkenazi Jews. Hum Mol Genet (1996) 5(11):1813–22. doi: 10.1093/hmg/5.11.1813
50. Nestorowicz A, Glaser B, Wilson BA, Shyng SL, Nichols CG, Stanley CA, et al. Genetic heterogeneity in familial hyperinsulinism. Hum Mol Genet (1998) 7(7):1119–28. doi: 10.1093/hmg/7.7.1119
51. Arya VB, Guemes M, Nessa A, Alam S, Shah P, Gilbert C, et al. Clinical and histological heterogeneity of congenital hyperinsulinism due to paternally inherited heterozygous ABCC8/KCNJ11 mutations. Eur J Endocrinol (2014) 171(6):685–95. doi: 10.1530/EJE-14-0353
52. De Franco E, Saint-Martin C, Brusgaard K, Knight Johnson AE, Aguilar-Bryan L, et al. Update of variants identified in the pancreatic β-cell KATP channel genes KCNJ11 and ABCC8 in individuals with congenital hyperinsulinism and diabetes. Hum Mutat (2020) 41(5):884–905. doi: 10.1002/humu.23995
53. Craigie RJ, Salomon-Estebanez M, Yau D, Han B, Mal W, Newbould M, et al. Clinical Diversity in Focal Congenital Hyperinsulinism in Infancy Correlates With Histological Heterogeneity of Islet Cell Lesions. Front Endocrinol (Lausanne) (2018) 9:619:619. doi: 10.3389/fendo.2018.00619
54. Park SE, Flanagan SE, Hussain K, Ellard S, Shin CH, Yang SW. Characterization of ABCC8 and KCNJ11 gene mutations and phenotypes in Korean patients with congenital hyperinsulinism. Eur J Endocrinol (2011) 164(6):919–26. doi: 10.1530/eje-11-0160
55. Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K. Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur J Endocrinol (2013) 168:557–64. doi: 10.1530/EJE-12-0673
56. Ince DA, Sahin NM, Ecevit A, Kurt A, Kinik ST, Flanagan SE, et al. Congenital hyperinsulinism in a newborn with a novel homozygous mutation (p.Q392H) in the ABCC8 gene. J Pediatr Endocrinol Metab (2014) 27(11–12):1253–5. doi: 10.1515/jpem-2014-0072
57. Corda H, Kummer S, Welters A, Teig N, Klee D, Mayatepek E, et al. Treatment with long-acting lanreotide autogel in early infancy in patients with severe noenatal hyperinuslinism. Orphanet J Rare Dis (2017) 12:108. doi: 10.1186/s13023-017-0653-x
58. Gier B, Krippeit-Drews P, Sheiko T, Aguilar-Bryan L, Bryan J, Düfer M, et al. Suppression of KATP channel activity protects murine pancreatic beta cells against oxidative stress. J Clin Invest (2009) 119(11):3246–56. doi: 10.1172/JCI38817
59. Edalat A, Schulte-Mecklenbeck P, Bauer C, Undank S, Krippeit-Drews P, Drews G, et al. Mitochondrial succinate dehydrogenase is involved in stimulus-secretion coupling and endogenous ROS formation in murine beta cells. Diabetologia (2015) 58(7):1532–41. doi: 10.1007/s00125-015-3577-9
60. Sikimic J, McMillen TS, Bleile C, Dastvan F, Quast U, Krippeit-Drews P, et al. ATP binding without hydrolysis switches sulfonylurea receptor 1 (SUR1) to outward-facing conformations that activate KATP channels. J Biol Chem (2019) 294(10):3707–19. doi: 10.1074/jbc.RA118.005236
61. Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, et al. Voltage-gated ion channels in human pancreatic beta-cells: electrophysiological characterization and role in insulin secretion. Diabetes (2008) 57(6):1618–28. doi: 10.2337/db07-0991
62. Dabrowski M, Larsen T, Ashcroft FM, Bondo Hansen J, Wahl P. Potent and selective activation of the pancreatic beta-cell type K(ATP) channel by two novel diazoxide analogues. Diabetologia. (2003) 46(10):1375–82. doi: 10.1007/s00125-003-1198-1
63. Seghers V, Nakazaki M, DeMayo F, Aguilar-Bryan L, Bryan J. Sur1 knockout mice. A model for KATP channel-independent regulation of insulin secretion. J Biol Chem (2000) 275(13):9270–7. doi: 10.1074/jbc.275.13.9270
64. Kullin M, Li Z, Bondo Hansen J, Welsh N, Karlsson FA, Sandler S. Protection of rat pancreatic islets by potassium channel openers against alloxan, sodium nitroprusside and interleukin-1beta mediated suppression–possible involvement of the mitochondrial membrane potential. Diabetologia. (2003) 46(1):80–8. doi: 10.1007/s00125-002-0997-0
65. Drews G, Bauer C, Edalat A, Düfer M, Krippeit-Drews P. Evidence against a Ca(2+)-induced potentiation of dehydrogenase activity in pancreatic beta-cells. Pflugers Arch (2015) 467(11):2389–97. doi: 10.1007/s00424-015-1707-3
66. Krippeit-Drews P, Düfer M, Drews G. Parallel oscillations of intracellular calcium activity and mitochondrial membrane potential in mouse pancreatic B-cells. Biochem Biophys Res Commun (2000) 267(1):179–83. doi: 10.1006/bbrc.1999.1921
67. Kiff S, Babb C, Guemes M, Dastamani A, Gilbert C, Flanagan SE, et al. Partial diazoxide responsiveness in a neonate with hyperinsulinism due to homozygous ABCC8 mutation. Endocrinol Diabetes Metab Case Rep (2019) 2019. doi: 10.1530/EDM-18-0120
68. Ismail D, Kapoor RR, Smith VV, Ashworth M, Blankenstein O, Pierro A, et al. The heterogeneity of focal forms of congenital hyperinsulinism. J Clin Endocrinol Metab (2012) 97(1):E94–9. doi: 10.1210/jc.2011-1628
69. Huopio H, Reimann F, Ashfield R, Komulainen J, Lenko HL, Rahier J, et al. Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. J Clin Invest (2000) 106(7):897–906. doi: 10.1172/JCI9804
70. Raphemot R, Swale DR, Dadi PK, Jacobson DA, Cooper P, Wojtovich AP, et al. Direct activation of beta-cell KATP channels with a novel xanthine derivative. Mol Pharmacol (2014) 85(6):858–65. doi: 10.1124/mol.114.091884
71. Maczewsky J, Sikimic J, Bauer C, Krippeit-Drews P, Wolke C, Lendeckel U, et al. The LXR Ligand T0901317 Acutely Inhibits Insulin Secretion by Affecting Mitochondrial Metabolism. Endocrinology (2017) 158(7):2145–54. doi: 10.1210/en.2016-1941
72. Drews G, Krippeit-Drews P, Düfer M. Electrophysiology of islet cells. Adv Exp Med Biol (2010) 654:115–63. doi: 10.1007/978-90-481-3271-3_7
73. Tamarina NA, Wang Y, Mariotto L, Kuznetsov A, Bond C, Adelman J, et al. Small-conductance calcium-activated K+ channels are expressed in pancreatic islets and regulate glucose responses. Diabetes (2003) 52(8):2000–6. doi: 10.2337/diabetes.52.8.2000
74. Düfer M, Gier B, Wolpers D, Krippeit-Drews P, Ruth P, Drews G. Enhanced glucose tolerance by SK4 channel inhibition in pancreatic beta-cells. Diabetes. (2009) 58(8):1835–43. doi: 10.2337/db08-1324
75. Jacobson DA, Mendez F, Thompson M, Torres J, Cochet O, Philipson LH. Calcium-activated and voltage-gated potassium channels of the pancreatic islet impart distinct and complementary roles during secretagogue induced electrical responses. J Physiol (2010) 588(Pt 18):3525–37. doi: 10.1113/jphysiol.2010.190207
76. Düfer M, Neye Y, Hörth K, Krippeit-Drews P, Hennige A, Widmer H, et al. BK channels affect glucose homeostasis and cell viability of murine pancreatic beta cells. Diabetologia (2011) 54(2):423–32. doi: 10.1007/s00125-010-1936-0
77. Goforth PB, Bertram R, Khan FA, Zhang M, Sherman A, Satin LS. Calcium-activated K+Channels of Mouse β-cells are Controlled by Both Store and Cytoplasmic Ca2+. J Gen Physiol (2002) 120(3):307–22. doi: 10.1085/jgp.20028581
78. Göpel SO, Kanno T, Barg S, Eliasson L, Galvanovskis J, Renstrom E, et al. Activation of Ca(2+)-dependent K(+) channels contributes to rhythmic firing of action potentials in mouse pancreatic beta cells. J Gen Physiol (1999) 114(6):759–70. doi: 10.1085/jgp.114.6.759
79. Kanno T, Rorsman P, Göpel SO. Glucose-dependent regulation of rhythmic action potential firing in pancreatic β-cells by kATP-channel modulation. J Physiol (2002) 545(2):501–7. doi: 10.1113/jphysiol.2002.031344
80. Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci (2005) 8(5):642–9. doi: 10.1038/nn1449
81. Marquard J, Otter S, Welters A, Stirban A, Fischer A, Eglinger J, et al. Characterization of pancreatic NMDA receptors as possible drug targets for diabetes treatment. Nat Med (2015) 21(4):363–72. doi: 10.1038/nm.3822
82. Carpenter CL, Marks SS, Watson DL, Greenberg DA. Dextromethorphan and dextrorphan as calcium channel antagonists. Brain Res (1988) 439(1-2):372–5. doi: 10.1016/0006-8993(88)91497-7
83. Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, de Craen AJ, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet (2010) 375(9716):735–42. doi: 10.1016/S0140-6736(10)60991-9
84. Yada T, Nakata M, Shiraishi T, Kakei M. Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L-type Ca2+ channels in rat islet beta-cells. Br J Pharmacol (1999) 126(5):1205–13. doi: 10.1038/sj.bjp.0702397
85. Yaluri N, Modi S, Lopez Rodriguez M, Stancakova A, Kuusisto J, Kokkola T, et al. Simvastatin Impairs Insulin Secretion by Multiple Mechanisms in MIN6 Cells. PloS One (2015) 10(11):e0142902. doi: 10.1371/journal.pone.0142902
86. Güemes M, Shah P, Silvera S, Morgan K, Gilbert C, Hinchey L, et al. Assessment of Nifedipine Therapy in Hyperinsulinemic Hypoglycemia due to Mutations in the ABCC8 Gene. J Clin Endocrinol Metab (2017) 102(3):822–30. doi: 10.1210/jc.2016-2916
87. Maiorana A, Barbetti F, Boiani A, Rufini V, Pizzoferro M, Francalanci P, et al. Focal congenital hyperinsulinism managed by medical treatment: a diagnostic algorithm based on molecular genetic screening. Clin Endocrinol (2014) 81(5):679–88. doi: 10.1111/cen.12400
88. Dekel B, Lubin D, Modan-Moses D, Quint J, Glaser B, Meyerovitch J. Compound heterozygosity for the common sulfonylurea receptor mutations can cause mild diazoxide-sensitive hyperinsulinism. Clin Pediatr (Phila) (2002) 41(3):183–6. doi: 10.1177/000992280204100310
89. Coetzee WA. Multiplicity of effectors of the cardioprotective agent, diazoxide. Pharmacol Ther (2013) 140(2):167–75. doi: 10.1016/j.pharmthera.2013.06.007
90. D’Hahan N, Moreau C, Prost AL, Jacquet H, Alekseev AE, Terzic A, et al. Pharmacological plasticity of cardiac ATP-sensitive potassium channels toward diazoxide revealed by ADP. Proc Natl Acad Sci U.S.A. (1999) 96(21):12162–7. doi: 10.1073/pnas.96.21.12162
91. Davies GC, Thornton MJ, Jenner TJ, Chen YJ, Hansen JB, Carr RD, et al. Novel and established potassium channel openers stimulate hair growth in vitro: implications for their modes of action in hair follicles. J Invest Dermatol (2005) 124(4):686–94. doi: 10.1111/j.0022-202X.2005.23643.x
92. Burton JL, Schutt WH, Caldwell IW. Hypertrichosis due to diazoxide. Br J Dermatol (1975) 93(6):707–11. doi: 10.1111/j.1365-2133.1975.tb05123.x
93. Hansen JB, Arkhammar PO, Bodvarsdottir TB, Wahl P. Inhibition of insulin secretion as a new drug target in the treatment of metabolic disorders. Curr Med Chem (2004) 11(12):1595–615. doi: 10.2174/0929867043365026
94. Ritzel RA, Hansen JB, Veldhuis JD, Butler PC. Induction of beta-cell rest by a Kir6.2/SUR1-selective K(ATP)-channel opener preserves beta-cell insulin stores and insulin secretion in human islets cultured at high (11 mM) glucose. J Clin Endocrinol Metab (2004) 89(2):795–805. doi: 10.1210/jc.2003-031120
95. Nielsen FE, Bodvarsdottir TB, Worsaae A, MacKay P, Stidsen CE, Boonen HC, et al. 6-Chloro-3-alkylamino-4H-thieno[3,2-e]-1,2,4-thiadiazine 1,1-dioxide derivatives potently and selectively activate ATP sensitive potassium channels of pancreatic beta-cells. J Med Chem (2002) 45(19):4171–87. doi: 10.1021/jm0208121
96. Alemzadeh R, Fledelius C, Bodvarsdottir T, Sturis J. Attenuation of hyperinsulinemia by NN414, a SUR1/Kir6.2 selective K+-adenosine triphosphate channel opener, improves glucose tolerance and lipid profile in obese Zucker rats. Metabolism. (2004) 53(4):441–7. doi: 10.1016/j.metabol.2003.10.027
97. Zdravkovic M, Kruse M, Rost KL, Moss J, Kecskes A, Dyrberg T. The effects of NN414, a SUR1/Kir6.2 selective potassium channel opener, in healthy male subjects. J Clin Pharmacol (2005) 45(7):763–72. doi: 10.1177/0091270005276947
99. Sarabu R, Tilley J. Recent advances in therapeutic approaches to type 2 diabetes. In: Doherty AM, editor. Annual Reports in Medicinal Chemistry, vol. 40. Elsevier (2005). p. 167–81. doi: 10.1016/50065-7743(04)39004-4
100. Zdravkovic M, Kruse M, Rost KL, Moss J, Kecskes A. The effects of NN414, a SUR1/Kir6.2 selective potassium channel opener in subjects with type 2 diabetes. Exp Clin Endocrinol Diabetes (2007) 115(6):405–6. doi: 10.1055/s-2007-973062
101. Koren I, Riskin A, Barthlen W, Gillis D. Hepatitis in an infant treated with octreotide for congenital hyperinsulinism. J Pediatr Endocrinol Metab (2013) 26(1-2):183–5. doi: 10.1515/jpem-2012-0372
102. Ben-Ari J, Greenberg M, Nemet D, Edelstein E, Eliakim A. Octreotide-induced hepatitis in a child with persistent hyperinsulinemia hypoglycemia of infancy. J Pediatr Endocrinol Metab (2013) 26(1-2):179–82. doi: 10.1515/jpem-2012-0349
103. Glaser B, Thornton P, Otonkoski T, Junien C. Genetics of neonatal hyperinsulinism. Arch Dis Child Fetal Neonatal Ed (2000) 82(2):F79–86. doi: 10.1136/fn.82.2.F79
104. Saint-Martin C, Arnoux JB, de Lonlay P, Bellanne-Chantelot C. KATP channel mutations in congenital hyperinsulinism. Semin Pediatr Surg (2011) 20(1):18–22. doi: 10.1053/j.sempedsurg.2010.10.012
105. Macmullen CM, Zhou Q, Snider KE, Tewson PH, Becker SA, Aziz AR, et al. Diazoxide-unresponsive congenital hyperinsulinism in children with dominant mutations of the beta-cell sulfonylurea receptor SUR1. Diabetes (2011) 60(6):1797–804. doi: 10.2337/db10-1631
107. Doyle ME, Egan JM. Pharmacological agents that directly modulate insulin secretion. Pharmacol Rev (2003) 55(1):105–31. doi: 10.1124/pr.55.1.7
108. Braun M. The somatostatin receptor in human pancreatic beta-cells. Vitam Horm (2014) 95:165–93. doi: 10.1016/B978-0-12-800174-5.00007-7
109. Shah P, Rahman SA, McElroy S, Gilbert C, Morgan K, Hinchey L, et al. Use of Long-Acting Somatostatin Analogue (Lanreotide) in an Adolescent with Diazoxide-Responsive Congenital Hyperinsulinism and Its Psychological Impact. Horm Res Paediatr (2015) 84(5):355–60. doi: 10.1159/000439131
110. Dastamani A, Guemes M, Pitfield C, Morgan K, Rajab M, Rottenburger C, et al. The Use of a Long-Acting Somatostatin Analogue (Lanreotide) in Three Children with Focal Forms of Congenital Hyperinsulinaemic Hypoglycaemia. Horm Res Paediatr (2019) 91(1):56–61. doi: 10.1159/000491101
111. van der Steen I, van Albada ME, Mohnike K, Christesen HT, Empting S, Salomon-Estebanez M, et al. A Multicenter Experience with Long-Acting Somatostatin Analogues in Patients with Congenital Hyperinsulinism. Horm Res Paediatr (2018) 89(2):82–9. doi: 10.1159/000485184
112. Güemes M, Dastamani A, Ashworth M, Morgan K, Ellard S, Flanagan SE, et al. Sirolimus: Efficacy and Complications in Children With Hyperinsulinemic Hypoglycemia: A 5-Year Follow-Up Study. J Endocr Soc (2019) 3(4):699–713. doi: 10.1530/ey.16.2.4
113. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. (2006) 124(3):471–84. doi: 10.1016/j.cell.2006.01.016
114. Houde VP, Brule S, Festuccia WT, Blanchard PG, Bellmann K, Deshaies Y, et al. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes (2010) 59(6):1338–48. doi: 10.2337/db09-1324
115. Hoffman JF, Joiner W, Nehrke K, Potapova O, Foye K, Wickrema A. The hSK4 (KCNN4) isoform is the Ca2+-activated K+ channel (Gardos channel) in human red blood cells. Proc Natl Acad Sci U S A (2003) 100(12):7366–71. doi: 10.1073/pnas.1232342100
116. Begenisich T, Nakamoto T, Ovitt CE, Nehrke K, Brugnara C, Alper SL, et al. Physiological roles of the intermediate conductance, Ca2+-activated potassium channel Kcnn4. J Biol Chem (2004) 279(46):47681–7. doi: 10.1074/jbc.M409627200
117. Jensen BS, Strobaek D, Christophersen P, Jorgensen TD, Hansen C, Silahtaroglu A, et al. Characterization of the cloned human intermediate-conductance Ca2+-activated K+ channel. Am J Physiol (1998) 275(3):C848–56. doi: 10.1152/ajpcell.1998.275.3.C848
118. Jaffe DB, Marks SS, Greenberg DA. Antagonist drug selectivity for radioligand binding sites on voltage-gated and N-methyl-D-aspartate receptor-gated Ca2+ channels. Neurosci Lett (1989) 105(1-2):227–32. doi: 10.1016/0304-3940(89)90042-6
119. Gresch A, Düfer M. Dextromethorphan and dextrorphan influence insulin secretion by interacting with KATP and L-type Ca(2+) channels in pancreatic beta-cells. J Pharmacol Exp Ther (2020) 375(1):10–20. doi: 10.1124/jpet.120.265835
120. Thompson PD, Panza G, Zaleski A, Taylor B. Statin-Associated Side Effects. J Am Coll Cardiol (2016) 67(20):2395–410. doi: 10.1016/j.jacc.2016.02.071
121. Gotoh S, Negishi M. Statin-activated nuclear receptor PXR promotes SGK2 dephosphorylation by scaffolding PP2C to induce hepatic gluconeogenesis. Sci Rep (2015) 5:14076. doi: 10.1038/srep14076
Keywords: congenital hyperinsulinism, KATP channels, diazoxide, NN414, L-type Ca2+ channels, KCa3.1 channels
Citation: Sikimic J, Hoffmeister T, Gresch A, Kaiser J, Barthlen W, Wolke C, Wieland I, Lendeckel U, Krippeit-Drews P, Düfer M and Drews G (2020) Possible New Strategies for the Treatment of Congenital Hyperinsulinism. Front. Endocrinol. 11:545638. doi: 10.3389/fendo.2020.545638
Received: 25 March 2020; Accepted: 02 October 2020;
Published: 27 October 2020.
Edited by:
Jeff M. P. Holly, University of Bristol, United KingdomReviewed by:
Charles Alfred Stanley, Children’s Hospital of Philadelphia, United StatesHana Zemkova, Institute of Physiology (ASCR), Czechia
Copyright © 2020 Sikimic, Hoffmeister, Gresch, Kaiser, Barthlen, Wolke, Wieland, Lendeckel, Krippeit-Drews, Düfer and Drews. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Krippeit-Drews, cGV0ZXIua3JpcHBlaXQtZHJld3NAdW5pLXR1ZWJpbmdlbi5kZQ==