 Xiaosen Ma
Xiaosen Ma Ming Li2†
Ming Li2† Anli Tong
Anli Tong Fen Wang
Fen Wang Yunying Cui
Yunying Cui- 1Department of Endocrinology, Key Laboratory of Endocrinology, National Health Commission of the People’s Republic of China, Peking Union Medical College Hospital, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, China
- 2Department of Clinical Laboratory, Peking Union Medical College Hospital, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, China
- 3Department of Endocrinology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 4Department of Urology, Peking Union Medical College Hospital, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, China
Pheochromocytoma/paraganglioma (PPGL) has a high genetic heterogeneity with 40% germline variants in known pathogenic genes. Data in Chinese on this aspect are scanty. To detect the genetic and clinical profile of Chinese PPGL patients, we examined the variants of 12 known germline pathogenic genes (SDHA, SDHB, SDHC, SDHD, SDHAF2, FH, VHL, RET, NF1, MAX, TMEM127, and KIF1B) by next-generation sequencing and Sanger sequencing in 314 Chinese PPGL subjects. Twenty nine percent of Chinese PPGL patients had germline variants and SDHB was the most frequently mutated (14.6%). The most frequent SDHB variants were in exon 2, exon 7, and IVS 7. Pathogenic variants were more likely to occur in metastatic PPGL patients, paragangliomas, and patients under 30, with the ratio being 50.7% (35/69), 35.9% (56/156), and 49.5% (52/105), respectively. Our cohort included 314 patients from a single setting. The genetic and clinical features of Chinese PPGL patients were unique in some aspects compared to their non-Chinese counterparts. Identification of genotype-phenotype relation can serve as an effective tool for genetic prioritization and clinical decision-making.
Introduction
Pheochromocytoma (PCC) and paraganglioma (PGL) are tumors that originate from adrenal medulla, sympathetic ganglia and parasympathetic ganglia. With the development of the next-generation sequencing (1), about 40% of PCC and PGL (PPGL) bear a relationship with the germline variants of known pathogenic genes and the germline variants are genetically highly heterogeneous (2, 3). Over the last two decades, more than 20 genes have been found to be associated with the development of hereditary PPGL. Of them, VHL, SDHB, SDHD, SDHA, and RET are the most common pathogenic genes, while several novel pathogenic genes mutated with extremely low frequency, which were only found in one or several families (4–7). At present, the variants have been profiled in different races. In 2006, a review from the European Network for the Study of Adrenal Tumors (ENS@T) summarized germline variants (VHL, SDHB, SDHD, RET, and NF1) in 166 (25.9%) of 642 PPGL patients and VHL was reportedly the most frequently mutated gene (56/642, 8.7%) (8). Recently, a research conducted in Saudi Arabia exhibited that the variant rate was 36.6% in PPGL subjects and SDHB was the most common variant (9).Collectively, these studies showed that the variant rate and underlying genetic profile, to some extent, varied with different ethnic populations. Germline profiling of Chinese PPGLs from two centers found that the variant rate was 21.7% (55/261) and VHL was the most frequently mutated gene (10). To know the profile of germline variants and genotype-phenotype correlation in Chinese PPGLs from a single center, we examined the variants in 12 known germline pathogenic genes (SDHA, SDHB, SDHC, SDHD, SDHAF2, FH, VHL, RET, NF1, MAX, TMEM127, and KIF1B) in 314 Chinese PPGLs from a single center. Our study shows that twenty nine percent of Chinese PPGLs had germline variants and SDHB was the most frequently mutated (14.6%). We also found the mutation hotspots are different with Chinese and Western populations.
Materials and Methods
Patients and Sample Collection
Our study included 314 PPGL patients from the Peking Union Medical College Hospital, Beijing, China. Most patients had undergone surgery and were histopathologically diagnosed with PPGL. For the patients with unresectable or metastatic tumor, PPGL diagnosis was established on the basis of clinical features (headache, palpitations and sweating), increased 24-h urinary catecholamine excretion or plasma metanephrines and imaging tests. 99mTc-hydrazinonicotinyl-Tyr3-0ctreotide (99mTc-HYNIC-TOC) scintigraphy, Iodine-131 metaiodobenzylguanidine scintigraphy and contrast-enhanced thoracoabdominal-pelvic computed tomography (CT) were performed for almost all PPGLs. Some patients underwent head and neck enhanced CT or head and neck enhanced MRI examination. 18F-FDG-PET/CT or 68Ga-DOATATATE-PET/CT were performed to patients suspected of metastasis or PPGLs with multiple lesions. Blood samples of the patients were collected, with informed consents obtained from the patients and approval from the medical ethics committee of the hospital.
Genomic DNA Preparation and Sanger Sequencing
Genomic DNA was extracted from the peripheral blood of PPGL patients by using blood DNA Midi Kit (Omega Bio-Tek, Norcross, Georgia, USA). For patients with metastatic PPGL, all the coding sequences and the intro-exon junctions of SDHB (NM_003000.3) were amplified and sequenced on an ABI3730 DNA analyzer (Applied Biosystems, CA, USA). For patients with head or neck PPGL, all the coding sequences and the intron-exon junctions of SDHB and SDHD (NM_003002.4) were amplified and sequenced. As for patients with clinically-diagnosed hereditary syndromes, such as multiple endocrine neoplasia Type 2 (MEN2) and Von Hippel-Lindau (VHL) disease, hot spots (10, 11, 16 exons) of RET (NM_020630.4) and all exons, including intro-exon junctions of VHL (NM_000551.3), were amplified and sequenced, respectively. The primers used for the PCR amplification were listed in Supplementary Table 1.
Next-Generation Sequencing
For all the patients except those whose variants were detected by the aforementioned Sanger sequencing, next-generation sequencing covering SDHA (NM_004168.4), SDHB (NM_003000.3), SDHC (NM_003001.5), SDHD (NM_003002.4), SDHAF2 (NM_017841.4), VHL (NM_000551.3), RET (NM_020630.4), MAX (NM_002382.5), TEMEM127 (NM_017849.4), FH (NM_000143.4), NF1 (NM_001042492.3), and KIF1B (NM_015074.3) was performed to examine the potential pathogenic germline variations.
The probes of target regions for relevant genes were designed against Agilent available online (http://www.agilent.com). Agilent SureSelectXT custom kit (Agilent Technologies, Palo Alto, CA) was used to generate sequence library according to instructions. Briefly, fragments of 180-280bp were produced by using a hydrodynamic shearing system (Covaris, Massachusetts, USA). The DNA fragments were end-repaired, A-tailed and adapter-ligated for Illumina sequencing. Then, size selection, PCR amplification and library hybridization were performed. Each captured library with an index was loaded onto the Illumina HiSeq X platform (Illumina Inc., San Diego, CA), and 150-bp paired-end reads were generated.
The fastq files were subjected to quality control to exclude unwanted sequences, including adapter-contaminated or low quality or unrecognizable nucleotides. The fastq files were aligned against the Human Reference Genome (UCSC hg19) by using the Burrows-Wheeler Aligner (BWA). Then, the SAM tools (11) were used to generate the final BAM file (12) to calculate the sequence coverage and depth by sorting the BAM files and performing repeated marking, partial realignment and base quality recalibration. At last, the Variant Call Format files obtained in the previous step were annotated by ANNOVAR (13).
Annotated variants with coverage over 10×, mutant allele frequency (MAF) < 0.01 in the 1,000 Genomes databases and in the exonic or splicing region (10 bp upstream and downstream of splicing sites) were retained (14). The synonymous variants were excluded. Non-synonymous SNVs were retained if at least two of the functional predictions by PolyPhen-2, SIFT, MutationTaster and CADD showed the SNV was damaging. “Benign” or “likely benign” variants in ClinVar were eliminated (15–18). American College of Genomic Medicine (ACMG) guidelines were used for the classification of variant pathogenicity (19). All the variants were confirmed by Sanger sequencing.
SDHB Immunohistochemistry
Immunohistochemical procedures of SDHB (ZM-0162, Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing, China) were performed on formalin-fixed, paraffin-embedded (FFPE) tissue sections of PPGL according to the manufacturer’s recommendations (1:50 dilution). Positive SDHB staining was defined as strong granular staining in cytoplasm; negative SDHB as cytoplasm showing no staining with positive staining for the internal control.
Multiplex Ligation Dependent Probe Amplification
Screening for large deletions of SDHx was carried out using the P226‐D1 Multiplex Ligation dependent Probe Amplification (MLPA) kit, following the manufacturer’s protocol (MRC‐Holland, Amsterdam, Netherlands). This P226‐D1 probemix contains probes for all exons of the SDHB, SDHC, SDHD, SDHAF2, and SDHAF1 genes. In addition, 13 reference probes are included in this probemix, detecting several different autosomal chromosomal locations.
Large deletions of VHL was detected using MLPA kit (P016-C2 VHL obtained from MRC‐Holland, Netherlands), following the protocols described by the manufacturer. This P016-C2 probemix contains 9 probes for VHL, 6 probes for genes located close to VHL, 2 probes on 3p which are further telomeric or centromeric from VHL, and 12 reference probes detecting sequences on other chromosomes.
Final products were separated using ABI 3730xl capillary electrophoresis (Life technologies, CA USA) and the electropherogram was evaluated using Coffalyser.Net.
Statistical Analysis
Continuous variables with non-normal distribution were expressed as median (25%, 75%). Two independent samples were compared by using Mann-Whitney test. Categorical variables were presented as frequency counts and percentages. Association between two categorical variables was determined by Chi-square test. Kaplan-Meier survival analysis was employed as appropriate. A P < 0.05 was considered to be statistically significant. SPSS version 23.0 for Windows was used for all statistical analysis. All unverified VUS were not included in the genotype-phenotypic analysis.
Results
Patient Characteristics
Our study included 314 PPGL patients (165 females and 149 males), with a median age of 38 at diagnosis, ranging from 5 to 68. The clinical and genetic features of the patients are shown in Figure 1 and Tables 1 and 2. In all, 314 patients, 69 (22.0%) had metastatic diseases. In addition, 156 (49.7%) patients had PGL in head and neck, thoracic, retroperitoneal, and pelvic locations; and 158 (50.3%) had PCC which contained 20 bilateral PCC; and 17 patients had tumors in both adrenal and extra-adrenal glands.
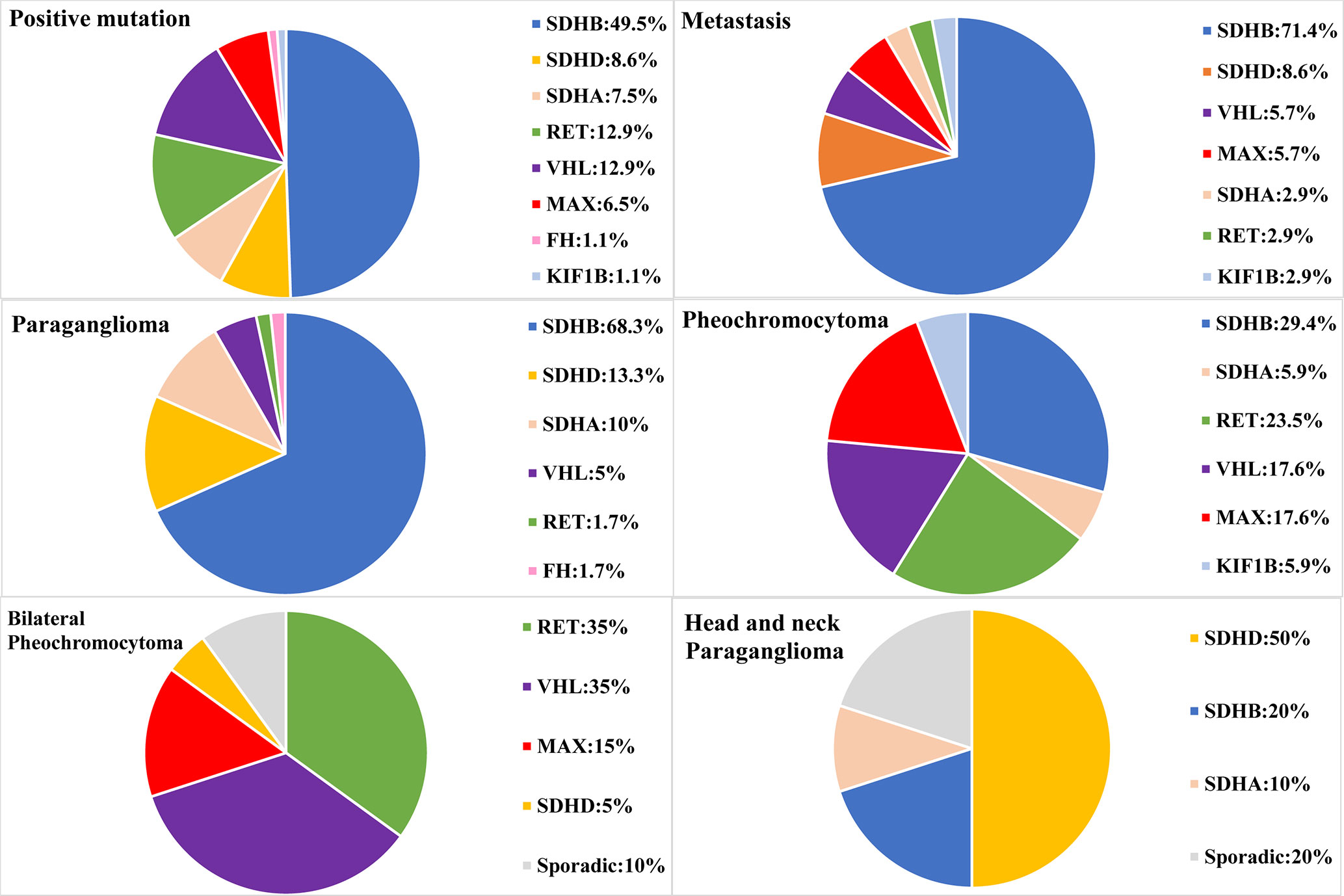
Figure 1 Genetic characteristics of PPGL patients with different phenotypes. Circular statistical graphic was used to illustrate the proportion of mutated gene in different clinical characteristic of PPGL.
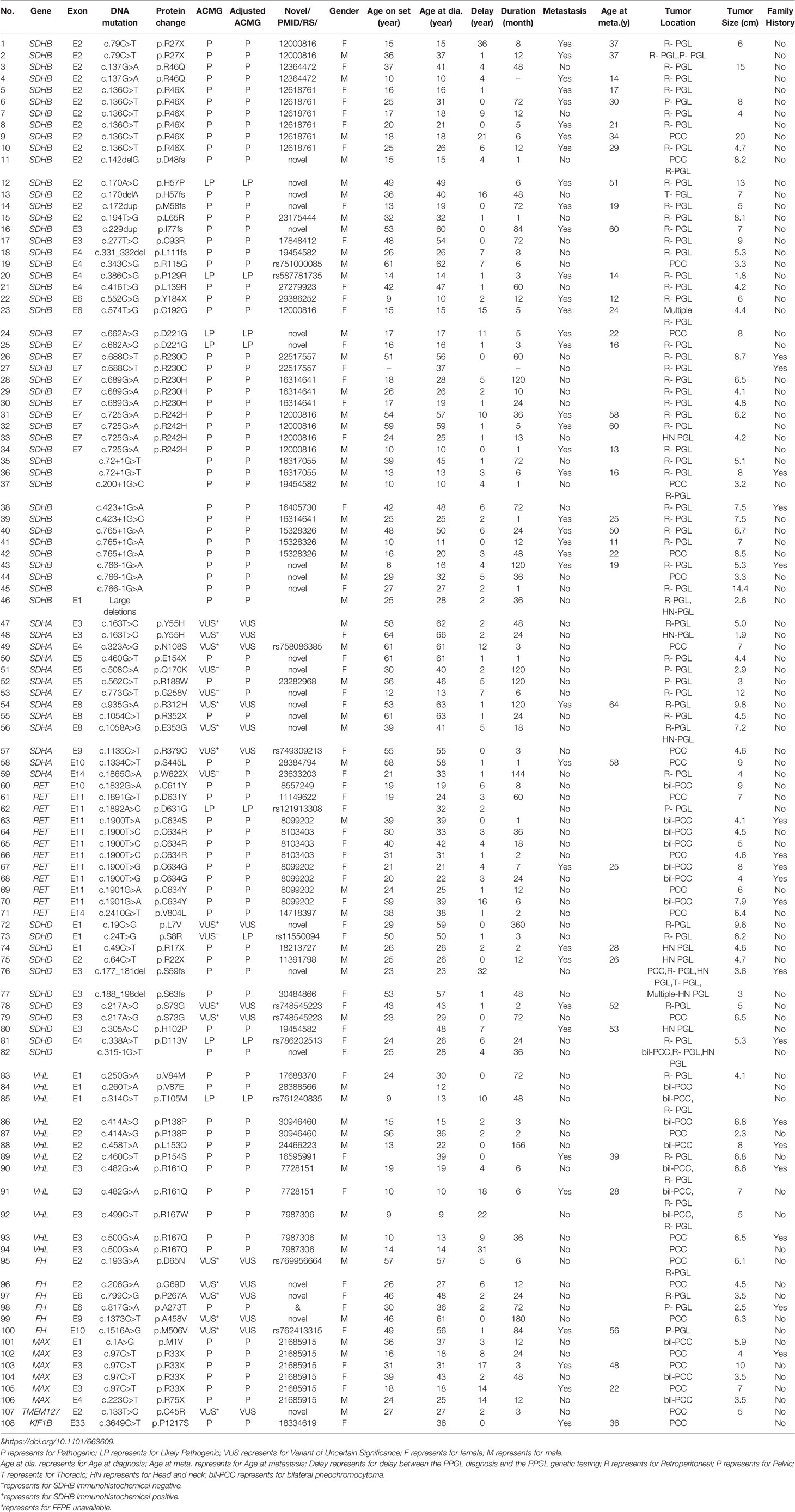
Table 1 Clinical and genetic characteristics of variant-positive patients.
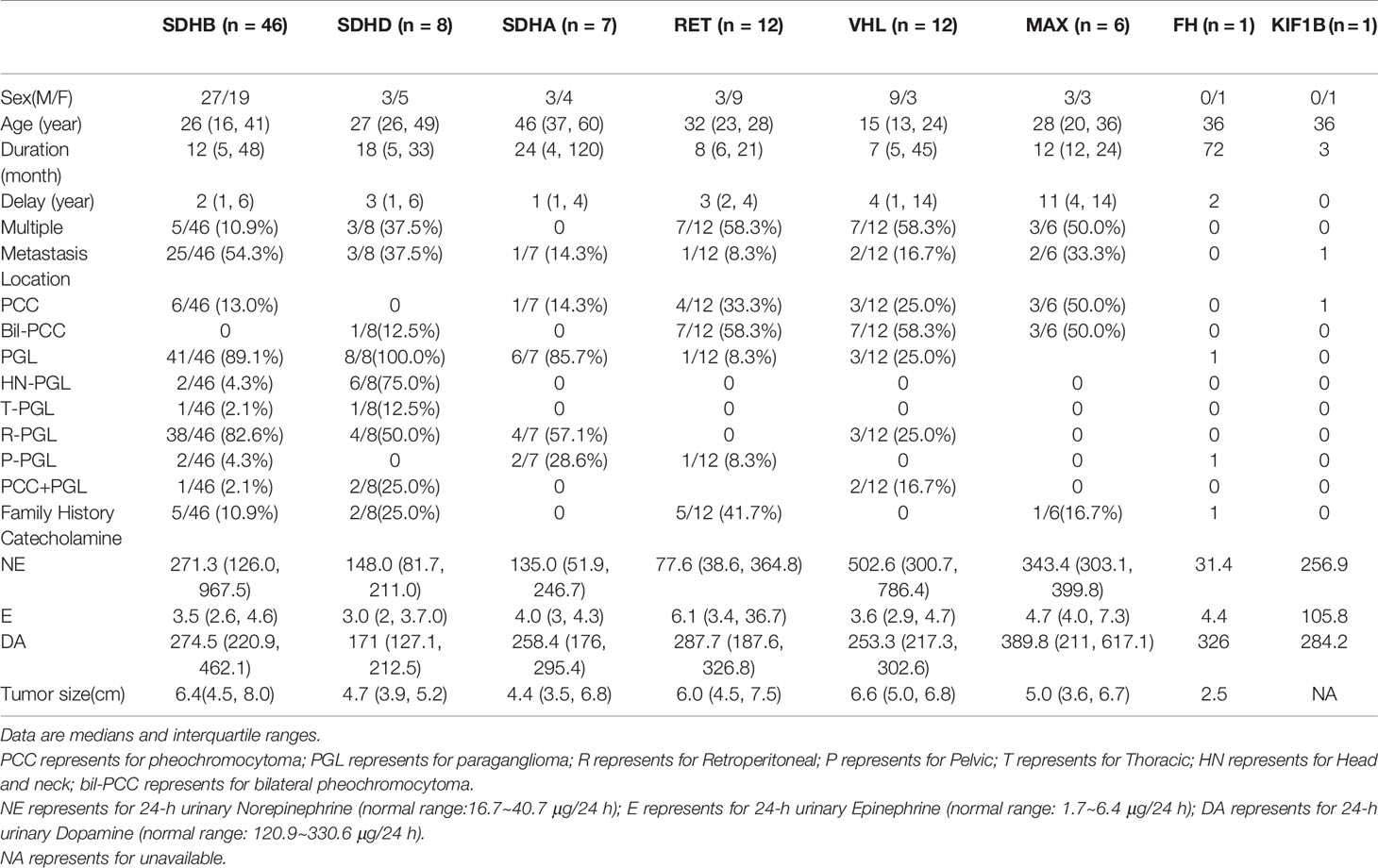
Table 2 Clinical characteristics of different pathogenic genes in PPGL patients.
The Profile of Gene Variants
We found 107 variants in 314 cases (34.1%) (Table 1). Among them, 76/107 variants (65.4%) were previously reported as pathogenic or likely pathogenic, 8/107 variants were uncertainly significant, and 23/107 variants (21.5%) were novel. According to the guidelines of ACMG, 13/23 (56.5%) novel variations were classified as pathogenic or likely pathogenic, 10 variants remained of uncertain significance. SDHB immunohistochemistry was performed on available FFEP tissue sections with VUS of SDHx. Negative immunostaining of SDHB was presented in 3 VUS of SDHA and one VUS of SDHD (Figure 2). Positive immunostaining of SDHB was presented in 2 VUS of SDHA and 2 VUS of SDHD, which suggests variants may not be pathogenic (Supplementary Figure S1).

Figure 2 SDHB immunohistochemistry. (A) PPGL with SDHA mutation (c.C508A). (B) PPGL with SDHA mutation (c.G773T). (C) PPGL with SDHA mutation (c.G1865A). (D) PPGL with SDHD mutation (c.T24G). (E) PPGL with an exon 1 large deletion of SDHB. (F) PPGL with RET mutation. (G) Normal adrenal medulla. Note: Absence of SDHB immunostaining in the tumour cells (A–E), with positive staining in the normal cells of the intratumoral fibrovascular network. Strong granular staining in PPGL with RET mutation and normal adrenal medulla (F, G).
In 207 cases without variants in 12 common pathogenic genes. SDHB immunohistochemistry was performed in 96/207 FFPE tissue sections, only one sample presented negative immunostaining of SDHB. MLPA analysis of the SDHx was then used in the immunohistochemistry negative sample and another 43 samples which contain multiple, metastatic PPGL. MLPA analysis of the VHL was used in early onset (under 20 years old) (n = 19), bilateral PCC (n = 2) or PCC and PGL (n = 6) in succession. We did not detect any deletions and duplications of VHL. Deletions of SDHB gene were detected in the negative immunostaining sample, affecting exon 1.
Altogether, 93/314(29.6%) of PPGLs had germline mutations in 12 known pathogenic genes.SDHB was the most frequently mutated gene (46/314, 14.6%), followed by RET (12/314, 3.8%), VHL (12/314, 3.8%), SDHD (8/314, 2.5%), SDHA (7/314, 2.2%), MAX (6/314, 1.9%), FH (1/314, 0.3%), and KIF1B (1/314, 0.3%).
SDHB Variants
SDHB was the most commonly mutated gene in our series (46/93 variants, 49.5%) with a median age at diagnosis of 26. Compared to sporadic PPGL, PPGL with SDHB variants were more likely to develop distant metastases or PGL (Table 3). 25/46(54.3%) of cases developed metastasis with the median duration of 12 (5, 51) months. Eighty-five percent (39/46) of the cases were PGL, 5/46 of the cases were PCC and 2/46 of the cases had both PCC and PGL (Table 2). In our cohort, SDHB variants were mainly located in exon 2 and exon 7, which accounted for 26/46 (56.5%) of all SDHB variants. Among them, codon 46 in exon 2 and codons 230 and 242 in exon 7 were hot variant sites, and were found in 8, 5, and 4 patients, respectively. Besides, variants in IVS7 (c.765+1G>A and c.766−1G>A) were also common in our study and the rate was 6/46 (13.0%).Four unrelated individuals had a family history of PPGL. Two PPGL complicated with pancreatic neuroendocrine tumor and renal clear cell carcinoma, respectively.
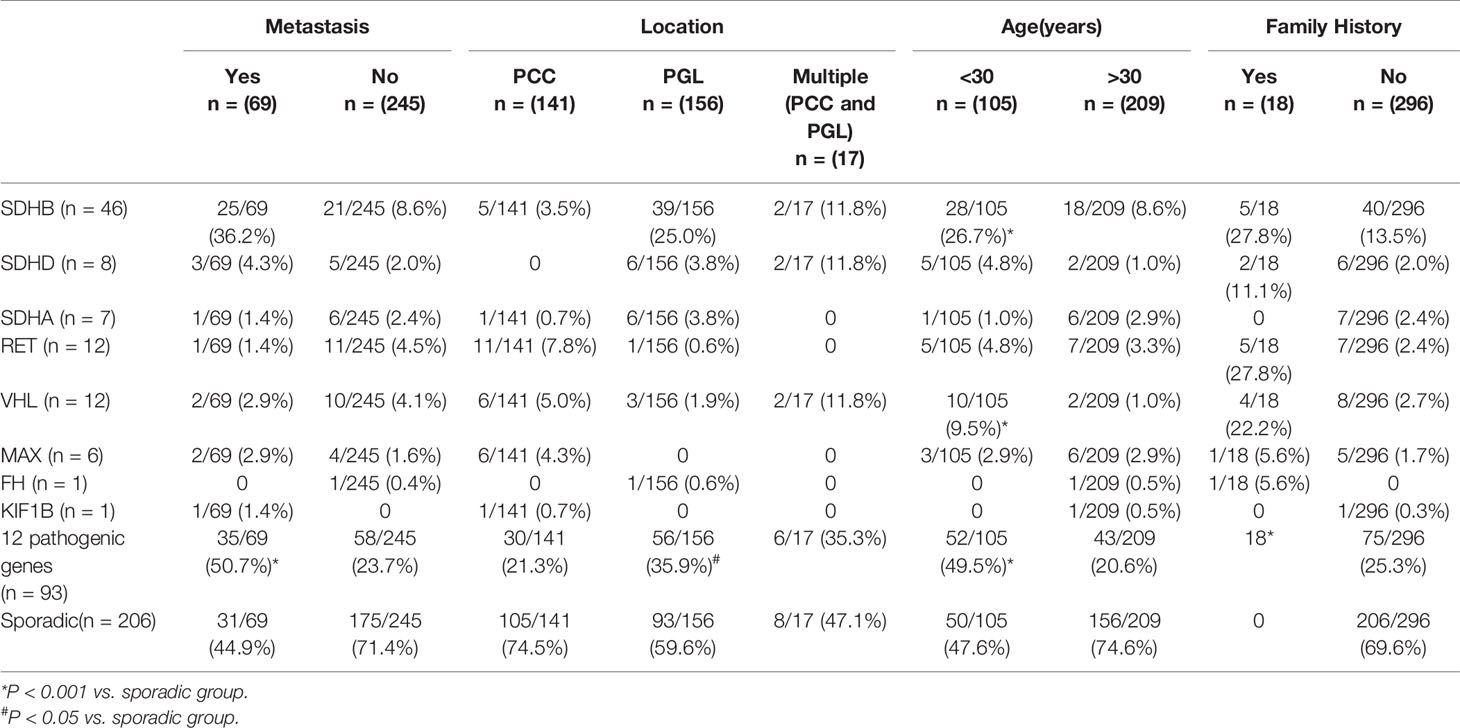
Table 3 Genotype-phenotype correlation in patients with PPGL.
The risk of metastasis in SDHB-mutated and sporadic PPGL patients was shown in Figure 3. There was a statistically significant difference in the risk of development into distant metastasis between patients with SDHB variants and sporadic PPGLs (P <.001). For PPGLs with SDHB variants, the risk of metastasis by the age of 60 was close to 60%, while in patients with sporadic PPGL, the risk was roughly 20% (P <.001) (Figure 3).
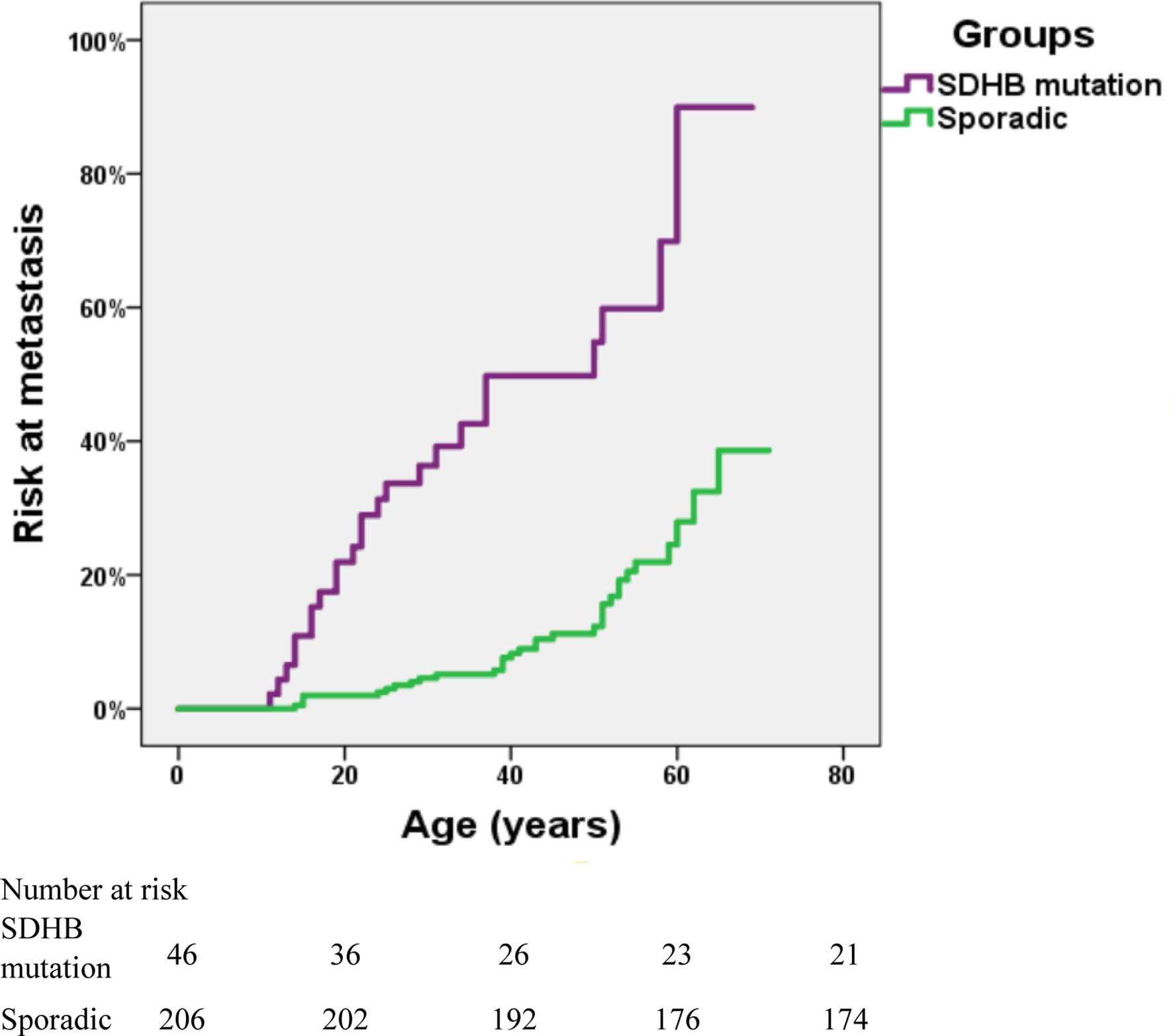
Figure 3 Risk of metastasis in SDHB-mutated and sporadic PPGL patients. Kaplan-Meier analysis includes 46 SDHB (succinate dehydrogenase subunit B) mutated PPGLs and 206 apparently sporadic PPGLs.
SDHA Variants
There were 13 cases with SDHA variants. Seven patients with pathogenic SDHA variants were diagnosed at a median age of 46 (range: from 13 to 63 years). Most of the patients had PGL (6/7, 85.7%), and of them, 4 had retroperitoneal PGL and 2 had pelvic PGL. One cases developed metastasis (Table 2). Exon 5 tended to have recurrent variants. There was no family history or syndrome in PPGLs with SDHA variants.
SDHD Variants
There were 11 cases with SDHD variants while 8 variants were verified as pathogenic and 3 remained VUS. Cases carrying pathogenic SDHD mutation were diagnosed at a median age of 27 (range: from 23 to 57 years), and 99mTc-HYNIC-TOC scintigraphy was used for the detection of head and neck PGL or multiple PGL in SDHD mutation carriers. All cases had PGL. PGL was in head and neck in 4 and in retroperitoneal cavity in 2. Thirty seven percent (3/8) of cases had multiple PPGL. The mutation type of 3 cases with multiple PPGL were framshifts or splicing. Three cases with single nucleotide variants, mostly (2/3, 66.7%) nonsence mutation, tend to develop into single head and neck PGL and metastasis. Two patients had both PCC and PGL.The variant scattered across the 4 exons of SDHD. Two cases had a family history of PPGL, and one case had pituitary growth hormone adenoma in addition to PPGL.
RET Variants
RET variants were found in 12 patients. Ninety one percent (11/12) of patients had PCC, among which, 7 had bilateral PCC. Variants in codon 631 and codon 634 in exon 11 of RET, were found in 10/12 (83.3%) cases. Forty one percent (5/12) had PPGL family history. In addition to PPGL, 6 patients also had clinical manifestations of medullary thyroid carcinoma. One patient had both PCC and primary hyperparathyroidism.
VHL Variants
VHL variants were detected in 12 patients. Six cases had PCC, including 3 bilateral PCC. Two patients had retroperitoneal PGL, and 4 patients had both bilateral PCC and retroperitoneal PGL (Table 1). Two metastatic PPGL which had no proof of renal cell carcinoma were also found in VHL mutated patients. Variants in codon 161 and 167, the most frequently reported mutant sites, were detected in 5/12(41.7%) patients. Four patients had family history of PPGL.And two cases had other clinical manifestations of VHL disease, such as bilateral retinoblastoma, cerebellar hemangioblastoma, renal clear cell carcinoma, and pancreatic neuroendocrine tumor in addition to PPGL.
Compared to sporadic PPGLs, pathogenic germline variants, especially SDHB and VHL, were more likely to be detected in patients under 30 (P <.001).
Other Gene Variants
Six cases had MAX variants, and all of them were PCC and half of the PCC were bilateral. Five variants were nonsense and one disrupted the MAX protein by affecting the initial methionine (c.1A>G). The most frequent variant was c.97C>T, p.Arg33Ter (4/6, 66.7%), and 2 of the 4 patients with such variant had metastatic PPGL. One of the patients with MAX variants had family history. In addition to PCC, two patients had pituitary growth hormone/prolactin adenoma and thyroid papillary carcinoma, respectively. One case with a family history of PPGL carried a FH pathogenic variant. A patient with KIF1B variant had metastatic PCC.
Comparison Between Apparently Sporadic PPGLs and Their Counterparts
In our cohort, further analysis was conducted in 206 patients with apparently sporadic PPGL (i.e., patients without variants in 12 known pathogenic genes) and 25 cases with family history or syndromic presentation (Table 4). Compared to apparently sporadic PPGLs, patients with family history or syndromic presentation had an earlier age of onset (P <.000), a higher level of norepinephrine (P <.038) and were more likely to develop multiple PPGL (P <.000). Besides, PCC was more common in apparently sporadic PPGLs (P = 0.014).
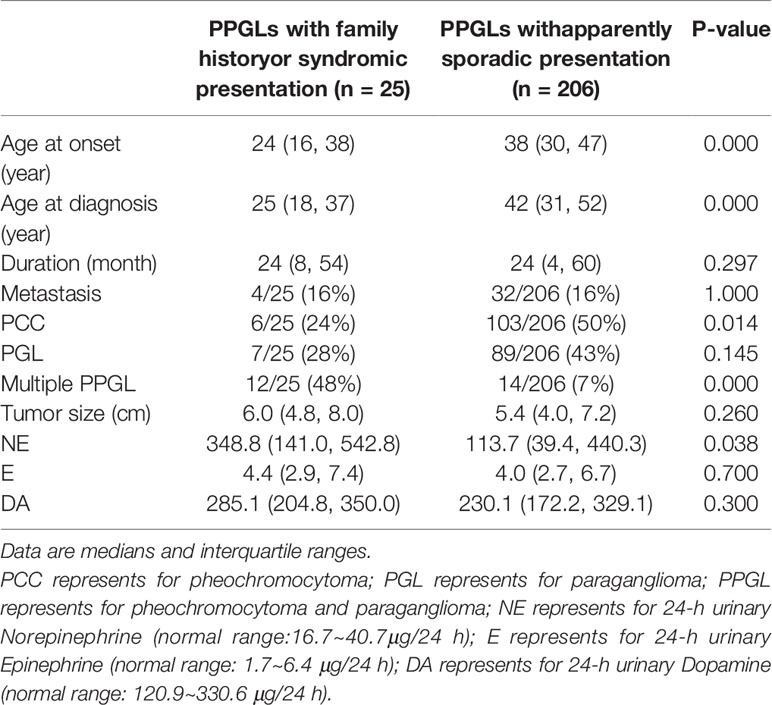
Table 4 Comparison of the clinical characteristics between PPGLs with family history or syndromic presentation and apparently sporadic PPGLs.
Variants in PCC and PGL
There were 141 PCC in 314 PPGLs. The 141 PCC patients included 15 bilateral PCC and 21 metastatic PCC patients. Thirty PCC patients (30/141, 21.3%) had germline variants (Figure 1 and Table 3). The top two most frequently mutated genes were RET (11/141, 7.8%) and VHL (6/141, 4.3%), followed by MAX (6/141, 4.3%), SDHB (5/141, 3.5%), SDHA (1/141, 0.7%), and KIF1B (1/141, 0.7%).
Twenty bilateral PCC were included in our cohort (Figure 1), 5 were complicated with PGL. 7/20 (35%) had RET variants, 7/20 (35%) had VHL variants, 3/20 (15%) had MAX variants, 1/20 (5%) had SDHD variant, and 2/20 (10%) had no known germline variants (Table 2).
One hundred and fifty six PGL patients were included in our research, involving 129 retroperitoneal PGL, 18 pelvic PGL, 9 head and neck PGL, and 4 thoracic PGL (Table 2). Thirty six percent (56/156) of the PGLs had known pathogenic gene variants (Table 3). In 129 retroperitoneal PGL (Table 2), 34.1% of them had germline mutations. The most frequently mutated gene was SDHB (36/129, 27.9%), followed by SDHA (4/129, 3.1%), VHL (2/129, 1.6%), SDHD (1/129, 0.8%), and RET (1/129, 0.8%). In 18 pelvic PGL patients, 33.3% of them had germline variants, including SDHB (2/18, 11.1%), SDHA (2/18, 11.1%), RET (1/18, 5.6%), and FH (1/18, 5.6%). In 9 patients with head and neck PGL (Figure 1), 66.7% of the patients had germline variants, including SDHD (4/9, 44.4%) and SDHB (2/9, 22.2%). Thoracic PGL were related to SDHB (1/4, 25%) and sporadic PPGL (3/4, 75%). Five patients had both bilateral PCC and PGL. All of them had pathogenic variants, of which 4 carried VHL mutation and 1 carried SDHD mutation.
Compared to sporadic PPGLs, PPGLs with variants in known pathogenic genes were more likely to develop PGL (P <.001).
Genetic Alterations in Metastatic PPGLs
Pathogenic germline variants were more likely to occur in metastatic PPGL than in their non-metastatic counterparts [35/69 (50.7%) vs. 58/245 (23.7%), P <.001]. Moreover, distant metastasis occurred more often in PGL than in PCC [45/156 (28.8%) in PGL vs. 21/141 (14.9%) in PCC, P = 0.004]. In metastatic PPGL, SDHB (25/69, 36.2%) was the most commonly mutated gene (Figure 1 and Table 3). SDHD (3/69, 4.3%), MAX (2/69, 2.9%), and VHL (2/69, 2.9%) had relatively higher frequency of variants. SDHA, RET, and KIF1B were also found to have variants, with a rate of 1.4% (1/69), while we did not found mutations in FH in metastatic PPGLs. Of note, 31/69 (44.9%) metastatic PPGL did not possess 12 known pathogenic genes (Figure 1 and Table 3).
Catecholamine Phenotype in PPGLs
Twenty four–hour urinary catecholamine excretion charactered elevated NE or elevated NE and DA in SDHB mutated PPGLs. Most patients carrying mutations in SDHD, SDHA, or MAX had elevated NE.
Discussion
In this cohort of Chinese PPGL patients, we profiled the genetic variants and demonstrated genotype-phenotype correlation in Chinese PPGL patients. We found that germline variant rate in PPGL patients was up to 29.6%, and SDHB was the most frequently mutated gene. The genetic and clinical features of Chinese PPGL patients were unique in some aspects compared to their non-Chinese counterparts.
The overall variant rates of pathogenic genes in PPGLs in our cohort were similar to those reported previously, standing somewhere between 11% to over 40% (7, 20, 21). A study conducted in Italy examined 10 genes in 501 PPGL patients and found that germline variant rate was 32.1%, and VHL was the most frequently mutated gene (22). A recent study from Saudi Arabia reveals that the variant rate was 36.6%, and SDHB was the most common variant (9). Jiang J et al. recently reported 719 PPGLs from two centers in China, of the 719 PPGLs, 266 cases were included in the analysis of germline mutations. Almost twenty one percent of PPGL had pathogenic germline mutations and VHL was mostly mutated in PPGL at a rate of 23/261(8.8%) in germline level (10). Compared to the research of Jiang J et al., our single center had a higher mutation rate of 29.6% and SDHB was the most commonly mutated gene in our center. The difference may come from that patients were referred to our hospital from all over the country mostly for their relative complicated or hard-treated diseases. Moreover, the proportion of metastatic PPGL and extra-adrenal PGL in a cohort might also affect the profile of genetic variants.
It is noteworthy that the variant hotspots of SDHB varied with different cohorts. According to TCGA data from 173 PPGL patients (23), the detection rate of SDHB germline variant was 9.8% (17/173). Among the 17 SDHB mutated PPGLs, p.Arg46Ter variant, which was the most frequently mutated one in our SDHB mutated PPGLs (6/45, 13.3%), occurred in only one patient (1/17, 5.9%). And the most frequent SDHB variant in TCGA data was p.Ile127Ser [29.4% (5/17)], which was not detected in our cohort. In a research conducted in Saudi Arabia, the most common SDHB variant was p.Arg90Ter, occurring in 57% of all the SDHB mutated cases, while in our series, no case had such variant (9). A national study from the Netherlands revealed that IVS4 (c.423+1G>A) was the most common variant site and was detected in 16/83 (19%) SDHB mutated patients, while the variant was found in 2/45 (4.4%) SDHB mutated patient in our cohort (24). In addition, in our study, novel variants c.765-1G>A and p.Asp221Gly in SDHB were recurrent. Exon 2, exon 7, and the splicing of IVS7 (c.765+1G>A and c.766-1G>A) were the most commonly mutated domains in our cohorts, and the finding was different from the results of other large sample researches (24–27). All the aforementioned studies, including ours, strongly suggest that the hotspots of SDHB variant vary in different races or countries, and it may be feasible to preferentially detect some specific sites of SDHB gene for metastatic PPGL patients according to the variant hotspots to achieve better cost-effectiveness and efficiency. The clinical features of patients with SDHB variants in our series mimicked those in other studies, and SDHB variant is associated with development of metastasis and extra-adrenal tumors.
Our results revealed that patients under 30 had a high germline variant rate (52.6%), suggesting that it is necessary to screen the genes in young patients. On the other hand, with SDHA and SDHD variants, corresponding tumors tend to affect elderly patients (23). It has been reported that the SDHD variant carrier had a penetrance rate of 86% at the age of 50 (22, 24, 25). In our study, only 2/8 (25%) SDHD-mutated patients had family history of PPGL. Family history may be underestimated because it was obtained by enquiring rather than systematic examination and gene detection in family members.
In our cohort, the hotspot variants of RET and VHL genes were similar to those reported previously, and were at codons 631 and 634 in exon 11 of RET gene and at codons 161 and 167 in exon 3 of VHL gene (26, 27). Of note, a synonymous variant recently reported in VHL was found in two patients in our study (28). RET and VHL gene variants mainly cause PCC, and they are principally responsible for development of bilateral PCC, accounting for 70% in all bilateral PCC in our study. Previous studies also found PGL in RET- and VHL-mutated patients (26, 27). In our series, only one patient (1/12, 8.3%) with RET variant developed PGL. But in VHL-mutated patients, 50% (6/12 cases) had retroperitoneal PGL. The rate was higher than that of previous research (10%–20%) (26). Our study suggested that although VHL and RET gene variants mainly lead to adrenal PCC, they may also result in extra-adrenal PGL. This is especially true of VHL variant, which needs to be screened carefully in clinical practice.
Up to now, only 12 PPGL patients with FH gene variants have been reported (29–31). In our cohort, we identified six FH variants in 314 individuals with PPGL. Only one of the FH variants was identified to be pathogenic variants against ACMG. The first published report showed that metastatic PPGLs accounted for 60% (3/5 cases) in FH-mutated PPGLs. Nonetheless, no evidence of metastasis of the PPGL with FH mutation in our cohort has been found till now.
MAX has been recently described at a total of ~40 PPGLs (9, 32–34). In our study, 6 patients had MAX variant, with the ratio being 1.9% in all PPGL patients. All MAX-mutated patients developed PCC in our cohort, which was coincident with previous studies (35). The frequent variant c.97C>T was also common in European and American populations (8/23 vs. 4/6 in our study) (35). Interestingly, of all the 4 PPGL cases carrying the same variant site c.97C>T, two were metastatic cases, while previous studies (26, 35) showed that patients carrying c.97C>T did not develop metastasis. This might be a unique genotype-phenotype correlation in Chinese.
KIF1B gene variant had been reported previously in a familial PCC patient. Both the proband and his grandfather had bilateral PCC without distant metastasis. Besides, the proband was also diagnosed with a neuroblastoma and a large well-differentiated leiomyosarcoma successively (36). Our research found a case of KIF1B variant who had a unilateral PCC with a metastatic lesion. Of note, the variant site had been previously reported in a patient with neuroblastoma (37). Our case suggested that the germline variants of the KIF1B gene could also pose risk of metastatic PCC.
Metastatic PPGL was reported to make up 2% to 13% (vs. 21/141, 14.9% in our study) in PCC and 2.4% to 50% (vs. 45/156, 28.8% in our study) in PGL (38). In our cohort, 50.7% (35/69) of metastatic PPGLs had hereditary germline variants, and were mostly caused by SDHB variant, followed by SDHD, VHL, MAX, SDHA, RET, and KIF1B. The results were in line with previous reports (26). The median age at diagnose in metastatic PPGLs was 7 years older (32 years vs. 39 years in our study) than the previously reported age (38, 39). PPGLs with SDHB variants need more attention since they are highly predisposed to distant metastasis.
Our study had several limitations. The research focused on the most common 12 pathogenic genes of PPGL and registered a variant detection rate of 29.6%. Our study did not include rare pathogenic genes such as DLST, SLC25A11 and DNMT3A, etc., which might lower the detection rate of pathogenic mutations. Furthermore, next-generation sequencing used in this study could not detect large deletions of some pathogenic genes, such as SDHx and VHL. Nevertheless, we added SDHB immunohistochemistry technique and MLPA detection to all samples with multiple PPGL or metastasis or early onset that are most likely to have large deletions. The proportion of large deletions in SDHx was 1.6% (1/61), lower than previously reported.
Conclusions
In conclusion, our study profiled the genetic variants and identified the corresponding clinical characteristics of PPGL in Chinese subjects. SDHB mutation hotspots are different between Chinese and other races. The genotype-phenotype relationship in Chinese is helpful in the prioritization of genetic tests and clinical decision-making.
Data Availability Statement
The raw sequence data reported in the manuscript have been deposited in the Genome Sequence Archive in National Genomics Data Center, Beijing Institute of Genomics (China National Center for Bioinformation), Chinese Academy of Sciences, under accession number HRA000415 that are accessible at https://bigd.big.ac.cn/gsa-human/s/sKCKPT0A.
Ethics Statement
The studies involving human participants were reviewed and approved by Medical Ethics Committee at Peking Union Medical College Hospital and written informed consent to participate in the study was provided by participants or their legal guardians/next of kin. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
XM conducted the experiments and drafted the manuscript. ML conducted the experiments and analyzed the data. FW, YC, SC, XZ, and YZ collected the specimens. YL and AT reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by CAMS Innovation Fund for Medical Sciences (CIFMS), grant number 2017-I2M-1-001.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are indebted to Dr. Zhengpei Zeng in Department of Endocrinology and Dr. Hanzhong Li in Department of Urology for providing expert advice in treating the patients.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2020.574662/full#supplementary-material. The Supplementary Material (Table_1.xlsx) for this article can be found online at: https://doi.org/10.6084/m9.figshare.12034230.v1
References
1. Ben Aim L, Pigny P, Castro-Vega LJ, Buffet A, Amar L, Bertherat J, et al. Targeted next-generation sequencing detects rare genetic events in pheochromocytoma and paraganglioma. J Med Genet (2019) 56(8):513–20. doi: 10.1136/jmedgenet-2018-105714
2. Toledo RA, Burnichon N, Cascon A, Benn DE, Bayley JP, Welander J, et al. Consensus Statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol (2017) 13(4):233–47. doi: 10.1038/nrendo.2016.185
3. Toledo RA, Dahia PL. Next-generation sequencing for the diagnosis of hereditary pheochromocytoma and paraganglioma syndromes. Curr Opin Endocrinol Diabetes Obes (2015) 22(3):169–79. doi: 10.1097/med.0000000000000150
4. Remacha L, Curras-Freixes M, Torres-Ruiz R, Schiavi F, Torres-Perez R, Calsina B, et al. Gain-of-function mutations in DNMT3A in patients with paraganglioma. Genet Med (2018) 20(12):1644–51. doi: 10.1038/s41436-018-0003-y
5. Remacha L, Pirman D, Mahoney CE, Coloma J, Calsina B, Curras-Freixes M, et al. Recurrent Germline DLST Mutations in Individuals with Multiple Pheochromocytomas and Paragangliomas. Am J Hum Genet (2019) 104(5):1008–10. doi: 10.1016/j.ajhg.2019.04.010
6. Buffet A, Morin A, Castro-Vega LJ, Habarou F, Lussey-Lepoutre C, Letouze E, et al. Germline Mutations in the Mitochondrial 2-Oxoglutarate/Malate Carrier SLC25A11 Gene Confer a Predisposition to Metastatic Paragangliomas. Cancer Res (2018) 78(8):1914–22. doi: 10.1158/0008-5472.Can-17-2463
7. Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer (2014) 14(2):108–19. doi: 10.1038/nrc3648
8. Gimenez-Roqueplo AP, Lehnert H, Mannelli M, Neumann H, Opocher G, Maher ER, et al. Phaeochromocytoma, new genes and screening strategies. Clin Endocrinol (Oxf) (2006) 65(6):699–705. doi: 10.1111/j.1365-2265.2006.02714.x
9. Albattal S, Alswailem M, Moria Y, Al-Hindi H, Dasouki M, Abouelhoda M, et al. Mutational profile and genotype/phenotype correlation of non-familial pheochromocytoma and paraganglioma. Oncotarget (2019) 10(57):5919–31. doi: 10.18632/oncotarget.27194
10. Jiang J, Zhang J, Pang Y, Bechmann N, Li M, Monteagudo M, et al. Sino-European Differences in the Genetic Landscape and Clinical Presentation of Pheochromocytoma and Paraganglioma. J Clin Endocrinol Metab (2020) 105(10):3295–307. doi: 10.1210/clinem/dgaa502
11. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics (2009) 25(16):2078–9. doi: 10.1093/bioinformatics/btp352
12. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (2009) 25(14):1754–60. doi: 10.1093/bioinformatics/btp324
13. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res (2010) 38(16):e164. doi: 10.1093/nar/gkq603
14. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res (2001) 29(1):308–11. doi: 10.1093/nar/29.1.308
15. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet (2013) 76(7):20. doi: 10.1002/0471142905.hg0720s76
16. Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res (2003) 31(13):3812–4. doi: 10.1093/nar/gkg509
17. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods (2010) 7(8):575–6. doi: 10.1038/nmeth0810-575
18. Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet (2014) 46(3):310–5. doi: 10.1038/ng.2892
19. Bahcall OG. Genetic testing. ACMG guides on the interpretation of sequence variants. Nat Rev Genet (2015) 16(5):256–7. doi: 10.1038/nrg3940
20. Brito JP, Asi N, Bancos I, Gionfriddo MR, Zeballos-Palacios CL, Leppin AL, et al. Testing for germline mutations in sporadic pheochromocytoma/paraganglioma: a systematic review. Clin Endocrinol (Oxf) (2015) 82(3):338–45. doi: 10.1111/cen.12530
21. Crona J, Lamarca A, Ghosal S, Welin S, Skogseid B, Pacak K. Genotype-phenotype correlations in pheochromocytoma and paraganglioma: a systematic review and individual patient meta-analysis. Endocr Relat Cancer (2019) 26(5):539–50. doi: 10.1530/erc-19-0024
22. Mannelli M, Castellano M, Schiavi F, Filetti S, Giacche M, Mori L, et al. Clinically guided genetic screening in a large cohort of italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab (2009) 94(5):1541–7. doi: 10.1210/jc.2008-2419
23. Cascon A, Remacha L, Calsina B, Robledo M. Pheochromocytomas and Paragangliomas: Bypassing Cellular Respiration. Cancers (Basel) (2019) 11(5):683. doi: 10.3390/cancers11050683
24. Ricketts CJ, Forman JR, Rattenberry E, Bradshaw N, Lalloo F, Izatt L, et al. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat (2010) 31(1):41–51. doi: 10.1002/humu.21136
25. Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. Jama (2004) 292(8):943–51. doi: 10.1001/jama.292.8.943
26. Neumann HPH, Young WF Jr., Eng C. Pheochromocytoma and Paraganglioma. N Engl J Med (2019) 381(6):552–65. doi: 10.1056/NEJMra1806651
27. Castinetti F, Qi XP, Walz MK, Maia AL, Sanso G, Peczkowska M, et al. Outcomes of adrenal-sparing surgery or total adrenalectomy in phaeochromocytoma associated with multiple endocrine neoplasia type 2: an international retrospective population-based study. Lancet Oncol (2014) 15(6):648–55. doi: 10.1016/s1470-2045(14)70154-8
28. Flores SK, Cheng Z, Jasper AM, Natori K, Okamoto T, Tanabe A, et al. A synonymous VHL variant in exon 2 confers susceptibility to familial pheochromocytoma and von Hippel-Lindau disease. J Clin Endocrinol Metab (2019) 104(9):3826–34. doi: 10.1210/jc.2019-00235
29. Richter S, Gieldon L, Pang Y, Peitzsch M, Huynh T, Leton R, et al. Metabolome-guided genomics to identify pathogenic variants in isocitrate dehydrogenase, fumarate hydratase, and succinate dehydrogenase genes in pheochromocytoma and paraganglioma. Genet Med (2019) 21(3):705–17. doi: 10.1038/s41436-018-0106-5
30. Clark GR, Sciacovelli M, Gaude E, Walsh DM, Kirby G, Simpson MA, et al. Germline FH mutations presenting with pheochromocytoma. J Clin Endocrinol Metab (2014) 99(10):E2046–50. doi: 10.1210/jc.2014-1659
31. Muller M, Ferlicot S, Guillaud-Bataille M, Le Teuff G, Genestie C, Deveaux S, et al. Reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma syndrome in French FH mutation carriers. Clin Genet (2017) 92(6):606–15. doi: 10.1111/cge.13014
32. Taieb D, Jha A, Guerin C, Pang Y, Adams KT, Chen CC, et al. 18F-FDOPA PET/CT Imaging of MAX-Related Pheochromocytoma. J Clin Endocrinol Metab (2018) 103(4):1574–82. doi: 10.1210/jc.2017-02324
33. Daly AF, Castermans E, Oudijk L, Guitelman MA, Beckers P, Potorac I, et al. Pheochromocytomas and pituitary adenomas in three patients with MAX exon deletions. Endocr Relat Cancer (2018) 25(5):L37–l42. doi: 10.1530/erc-18-0065
34. Chang X, Li Z, Ma X, Cui Y, Chen S, Tong A. A Novel Phenotype of Germline Pathogenic Variants in MAX: Concurrence of Pheochromocytoma and Ganglioneuroma in a Chinese Family and Literature Review. Front Endocrinol (Lausanne) (2020) 11:558. doi: 10.3389/fendo.2020.00558
35. Burnichon N, Cascon A, Schiavi F, Morales NP, Comino-Mendez I, Abermil N, et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res (2012) 18(10):2828–37. doi: 10.1158/1078-0432.Ccr-12-0160
36. Yeh IT, Lenci RE, Qin Y, Buddavarapu K, Ligon AH, Leteurtre E, et al. A germline mutation of the KIF1B beta gene on 1p36 in a family with neural and nonneural tumors. Hum Genet (2008) 124(3):279–85. doi: 10.1007/s00439-008-0553-1
37. Schlisio S, Kenchappa RS, Vredeveld LC, George RE, Stewart R, Greulich H, et al. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev (2008) 22(7):884–93. doi: 10.1101/gad.1648608
38. Hamidi O, Young WF Jr., Iniguez-Ariza NM, Kittah NE, Gruber L, Bancos C, et al. Malignant Pheochromocytoma and Paraganglioma: 272 Patients Over 55 Years. J Clin Endocrinol Metab (2017) 102(9):3296–305. doi: 10.1210/jc.2017-00992
Keywords: pheochromocytoma, paraganglioma, Chinese, genetics, genotype-phenotype relation
Citation: Ma X, Li M, Tong A, Wang F, Cui Y, Zhang X, Zhang Y, Chen S and Li Y (2020) Genetic and Clinical Profiles of Pheochromocytoma and Paraganglioma: A Single Center Study. Front. Endocrinol. 11:574662. doi: 10.3389/fendo.2020.574662
Received: 20 June 2020; Accepted: 03 November 2020;
Published: 11 December 2020.
Edited by:
Lee E. Eiden, National Institutes of Health (NIH), United StatesReviewed by:
Stephanie Fliedner, University Medical Center Schleswig-Holstein, GermanyAnne-Paule Gimenez-Roqueplo, Assistance Publique Hopitaux De Paris, France
Copyright © 2020 Ma, Li, Tong, Wang, Cui, Zhang, Zhang, Chen and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anli Tong, dG9uZ2FubGlAaG90bWFpbC5jb20=
†These authors have contributed equally to this work and share first authorship