 Yena Lee
Yena Lee Yunha Choi
Yunha Choi Jin-Ho Choi
Jin-Ho Choi- Department of Pediatrics, Asan Medical Center Children’s Hospital, University of Ulsan College of Medicine, Seoul, South Korea
Background: Due to remarkable progress in cancer treatment, endocrine complications are now the major medical issues facing childhood cancer survivors. Although non-central nervous system solid tumors (NCSTs) account for approximately 40% of all pediatric cancers, there have been few studies on endocrine complications associated with NCSTs. This study investigated endocrinopathies following the treatment of pediatric NCSTs.
Design and setting: Retrospective study in a single academic center.
Methods: This study analyzed 253 survivors of childhood NCSTs who were diagnosed between January of 2000 and December of 2018. The medical charts were reviewed regarding the frequency of endocrinopathies and treatment modalities. The hazard ratios were assessed by multivariable Cox regression analysis. The final height-SDS were analyzed by multivariable linear regression analysis.
Results: There were 76 patients (30%) that developed at least one endocrine complication. Forty-four patients (17.4%) experienced endocrine complications within five years of their cancer diagnosis. The most common endocrine complication was growth failure (n = 35), followed by obesity (n = 18), and primary gonadal failure (n = 16). High cumulative doses of alkylating agents increased the risk of developing at least one endocrine complication. Hematopoietic stem cell transplantation was an important risk factor for primary gonadal failure.
Conclusions: This study described the comprehensive endocrine outcomes, including growth failure, obesity, primary gonadal failure, primary hypothyroidism, dyslipidemia, and osteoporosis, following the treatment of childhood NCSTs. As endocrinopathies occurred within five years of primary tumor diagnosis, surveillance for endocrine dysfunction is required for early intervention and management.
Introduction
Pediatric non-central nervous system solid tumors (NCSTs) are a heterogeneous group of non-hematologic, extracranial cancers including neuroblastoma, soft tissue sarcoma, bone tumors, retinoblastoma, hepatoblastoma, germ cell tumors, and various other rare, solid organ tumors that predominantly occur during childhood (1). Multi-modality therapy, including surgical resection, chemotherapy, radiotherapy (RT), and autologous hematopoietic stem cell transplantation (HSCT), is required to manage children with NCSTs.
With the remarkable progress in cancer treatment, five-year childhood cancer survival rates have dramatically improved to approximately 80% in developed countries (2). With increases in survival, the late effects of cancer therapy have become a major medical issue of childhood cancer survivors. The majority of these survivors experience at least one non-endocrine or endocrine complication (3, 4). Endocrine disturbances are the most frequent complications in childhood cancer survivors, including hematologic malignancies, brain tumors, and sarcomas, with more than 50% of cancer survivors experiencing at least one hormonal disorder during their lifetime (5).
Generally, childhood survivors of acute lymphocytic leukemia achieve a normal final height with a body mass index (BMI) within the normal range (6, 7). Growth hormone deficiency (GHD) is the most common hypothalamic-pituitary disorder observed in survivors of brain tumors and those whose hypothalamic-pituitary region has been exposed to RT (8, 9). The prevalence of combined pituitary hormone deficiency has been found to be higher in patients with sellar and suprasellar tumors than in those with tumors in other locations (10).
Although NCSTs account for approximately 40% of all pediatric cancers (11), a few studies have examined their endocrine outcomes. Endocrinopathies after childhood cancer treatments, including growth failure, gonadal dysfunction, primary hypothyroidism, obesity, and disrupted bone heath, have been investigated in hematologic malignancies (12) and brain tumors (10, 13). Previous data on NCSTs were based on disease-specific cohorts. Female survivors of Wilms tumor who underwent abdominal radiation were reported to have poor fertility outcomes (14). Impaired fertility was demonstrated in Ewing sarcoma survivors as well (15). Pituitary dysfunction, after the treatment of head and neck rhabdomyosarcoma, has also been identified (16). In another study, hypothyroidism and gonadal dysfunction were the most frequent endocrine complications after the treatment of neuroblastoma (17).
To date, only one study included various NCSTs in a comprehensive analysis of endocrine outcomes (18). Therefore, this study investigated endocrine complications of pediatric patients with NCSTs and analyzed the risk factors related to endocrine complications and determinants of growth outcomes.
Subjects and Methods
Subjects
A total of 400 patients were diagnosed with NCSTs before the age of 18 years at the Department of Pediatrics in Asan Medical Center Children’s Hospital between January of 2000 and December of 2018. Inclusion criteria for this study were: 1) patients diagnosed with NCSTs between January of 2000 and December of 2018; 2) patients younger than 18 years of age at primary tumor diagnosis; 3) patients alive at the time of the study analysis, and 4) those who completed the cancer therapy more than one year previously. The treatment endpoint was defined as the completion of treatment for a primary, metastatic, or relapsed cancer. Patients who were lost to follow-up during the observational period were excluded (Figure 1). Ultimately, 253 survivors were included in this study after the exclusion of 98 patients who died during the follow-up period, a further 37 patients were lost to follow-up, and 12 patients who did not fulfill the inclusion criteria.

Figure 1 Diagram for study population.
Clinical characteristics and endocrine complications were analyzed by a retrospective medical chart review. This study was approved by the Institutional Review Board of Asan Medical Center, Seoul, Korea (IRB number 2020-1146).
Treatment Modalities and Alkylating Agent Dose (AAD) Scores of Each Patient
Treatment modalities, including operation (OP), chemotherapy, RT, autologous and allogeneic HSCT, and 131I-metaiodobenzylguanidine (131I-MIBG) therapy were reviewed. The cumulative dose and location of RT were documented. The cumulative dose (mg/m2) of alkylating agents (busulfan, cyclophosphamide, ifosfamide, melphalan, and thiotepa) was assessed by an alkylating agent dose (AAD) score. The dose distribution for each agent was divided into tertiles. A score of zero was given to patients who did not get any alkylating agent. A score of one was assigned if the patient’s dose was within the first tertile, a score of two was assigned if it was within the second tertile, and a score of three was assigned if it was within the third tertile. The scores of each agent for an individual patient were summed, the result being the summed AAD score (19, 20).
Endocrine Complications of Childhood Survivors With Non-Central Nervous System Solid Tumors (NCSTs)
Short stature was defined as the height-SDS below -2.0 SDS for the age- and gender-matched normative data from Korean references (21).Growth deceleration was defined as growth velocity below the fifth percentile for age and gender (e.g., <5 cm/year after the age of five years) or a drop in height across two or more percentiles on the growth chart before closure of the growth plate (22, 23). Insulin-like growth factor-1 (IGF-1) SDSs were calculated based on the reference values of Korean children and adolescents (24). Obesity was a BMI above 95 percentile for the age- and gender-matched reference data (21). A diagnosis of hypergonadotropic hypogonadism was based on elevated luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels with low testosterone or estradiol levels (25). Vitamin D deficiency was a level of 25-hydroxyvitamin D3 (25OHD3) less than 20 ng/mL (26). Osteoporosis was diagnosed in patients with a bone mineral density (BMD) Z-score below -2.0 on a dual-energy X-ray absorptiometry (DEXA) scan (Lunar Corp., Madison, WI, USA). BMD Z-scores were adjusted by age- and gender-matched standards for Korean children (27, 28). As the Korean reference data for children aged ≥10 years were measured by the Hologic system, the BMD data were converted with the following formula: lumbar spine BMD by Hologic system = 0.837 × lumbar spine BMD by Lunar system + 0.021; and femoral neck BMD by Hologic system = 0.913 × femoral neck BMD by Lunar system - 0.080 (29).
Survivors of NCSTs were followed-up annually at pediatric endocrine and/or hematology/oncology clinics. The following parameters were annually assessed annually: height, weight, BMI, Tanner stage, bone age, lipid profiles, serum IGF-1 and IGF binding protein 3 (IGFBP-3) levels, and thyroid function tests (thyroid-stimulating hormone [TSH], free T4). Serum LH, FSH, testosterone (male) or estradiol (female) levels were measured after pubertal age. DEXA was performed consecutively in select patients, such as adult patients with primary hypogonadism or those treated with long-term corticosteroids. The follow-up and treatment protocols were based on the previous guidelines (30–32).
Statistical Analysis
Statistical analyses were performed using IBM SPSS Statistics for Windows version 21.0 (IBM Co., Armonk, NY, USA). Multivariable analyses were performed using Cox proportional hazards model to identify hazard ratio (HR) of endocrine complications according to the treatment modalities of NCST. Time variables, defined as duration (years) from the primary cancer diagnosis to the date of the final visit or diagnosis of the endocrine complication, were used for the Cox regression analyses. Multiple variables were evaluated by employing backward stepwise regression analysis. Kaplan-Meier curves were generated to illustrate the risk-associated cumulative incidence of endocrine dysfunction. The differences in height-SDS between initial and final value was analyzed by a paired t-test. Factors affecting final height-SDS were assessed by univariable and multivariable linear regression analysis in survivors who reached their final height (age ≥ 18 years). The Mann-Whitney test was used to compare the age at HSCT between patients with and without hypogonadism. p-value < 0.05 was considered statistically significant.
Results
Patient Characteristics and Treatment Modalities
The mean age at diagnosis of NCSTs was 4.9 ± 4.9 years (range 0–16 years). The mean age at the outcome assessment was 13.9 ± 6.7 years (range 1.5–29 years). The mean follow-up duration was 8.95 ± 4.6 years (range 1.3–22 years). Sixty-four percent of the patients were observed ≤10 years, and 36% of the study subjects continued to attend annual follow-up visits more than 10 years after their primary NCST diagnosis. Primary diagnosis included neuroblastoma (25.7%), hepatoblastoma (21.3%), osteosarcoma (16.6%), Wilms tumor (15.4%), rhabdomyosarcoma (10.7%), Ewing sarcoma (9.9%), and ectomesenchymoma (0.4%) (Table 1). Treatment modalities are summarized in Table 2. Four patients with genetic syndromes were included: Beckwith–Wiedemann syndrome (BWS, n = 2), Noonan syndrome (NS, n =1), and WAGR syndrome (n = 1).
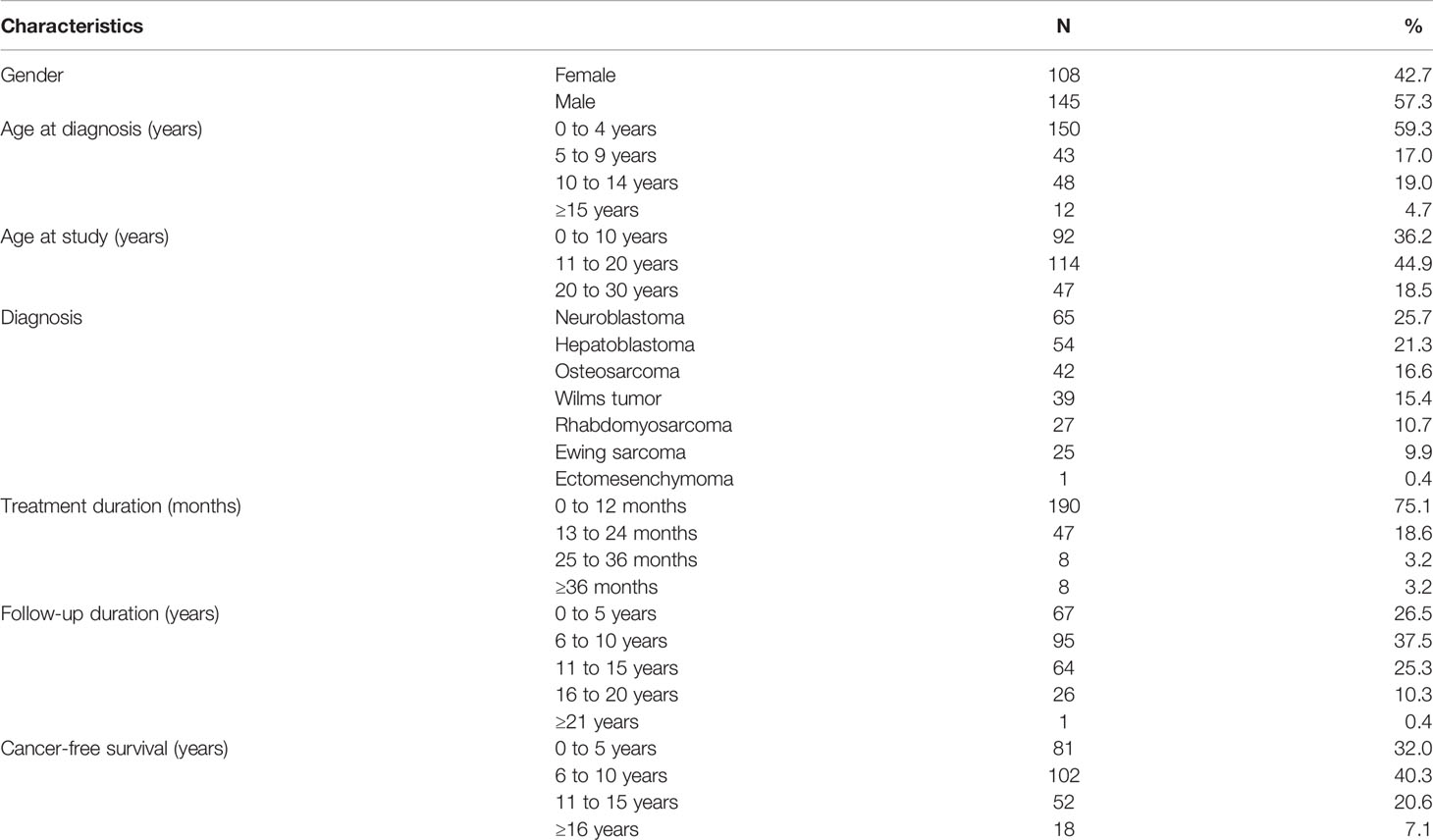
Table 1 Characteristics of childhood solid tumor survivors.
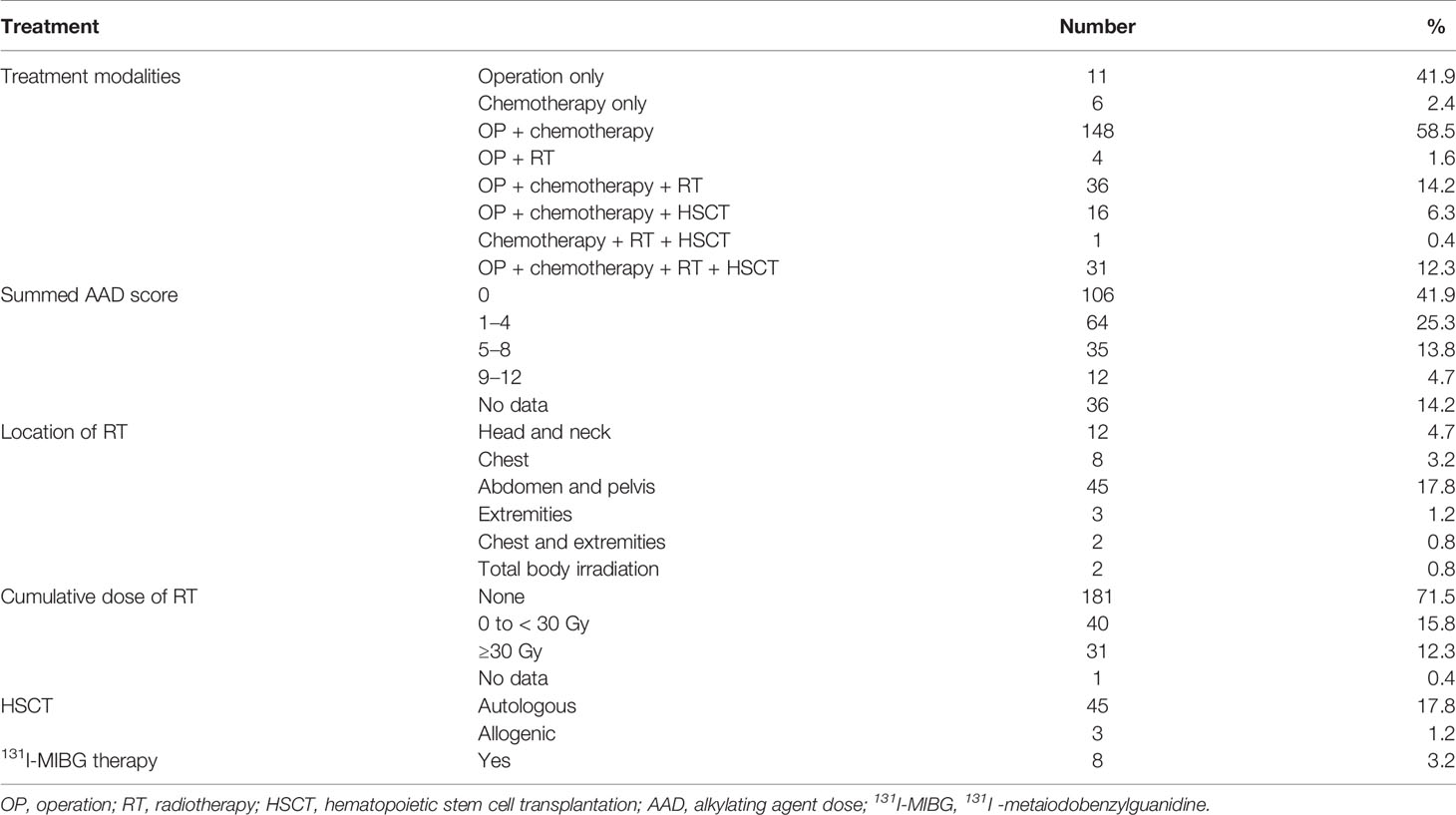
Table 2 Treatment modalities used in patients with childhood solid tumors.
Endocrine Complications Associated With NCST Treatment
Endocrine complications were found in 76 patients (Male (M):Female (F) = 30:46) (Table 3). Among them, 44 patients (17.4%) manifested endocrine complications within five years of NCST diagnosis. The risk of endocrine complications increased according to the summed AAD score (Table 4). HSCT is an important risk factor for developing at least one endocrine complication (HR = 2.44, p = 0.024). The cumulative incidence of any endocrine complication increased with a higher summed AAD score and a greater number of treatment modalities (p < 0.001, Figure 2).
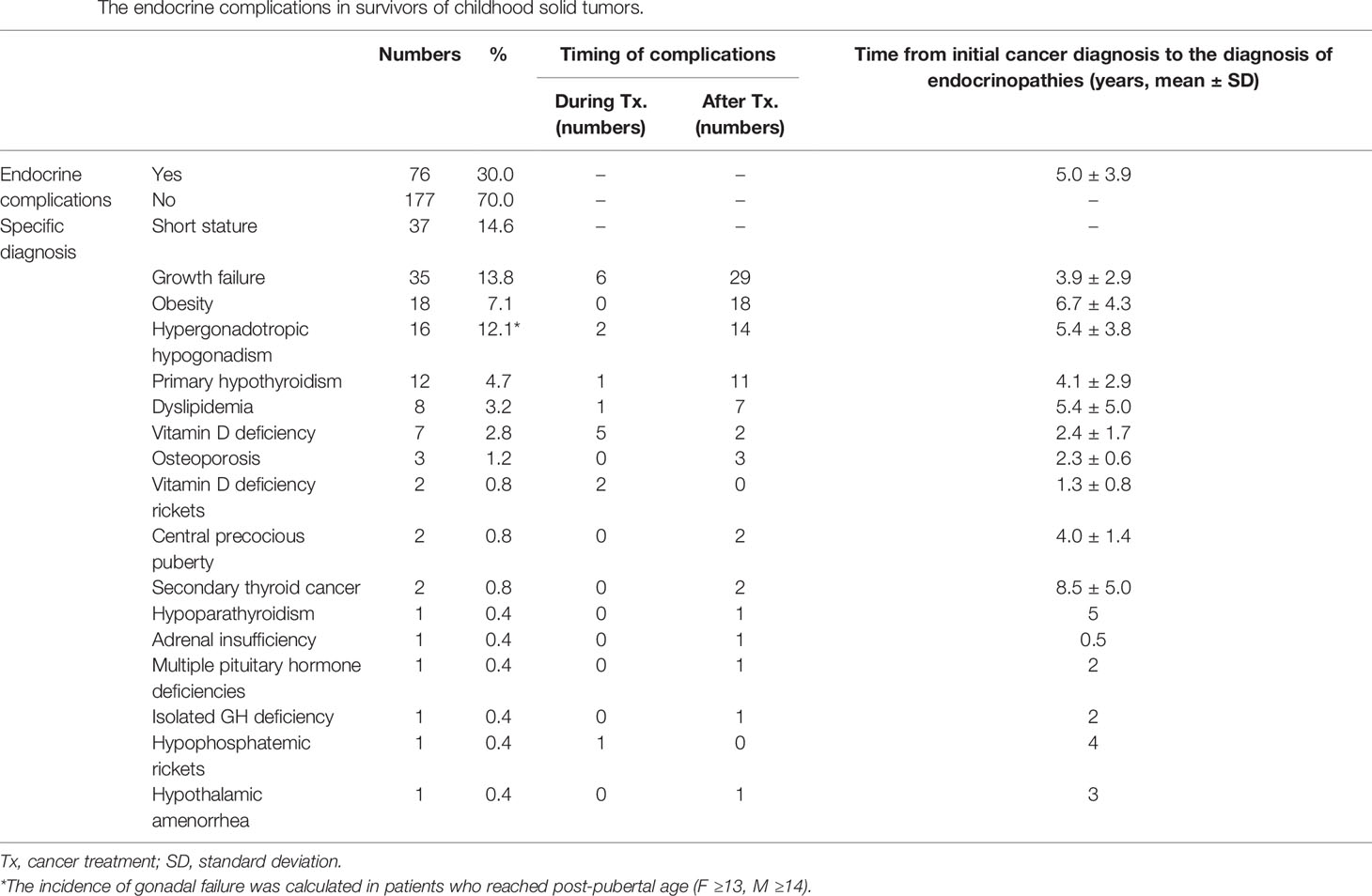
Table 3 The endocrine complications in survivors of childhood solid tumors.
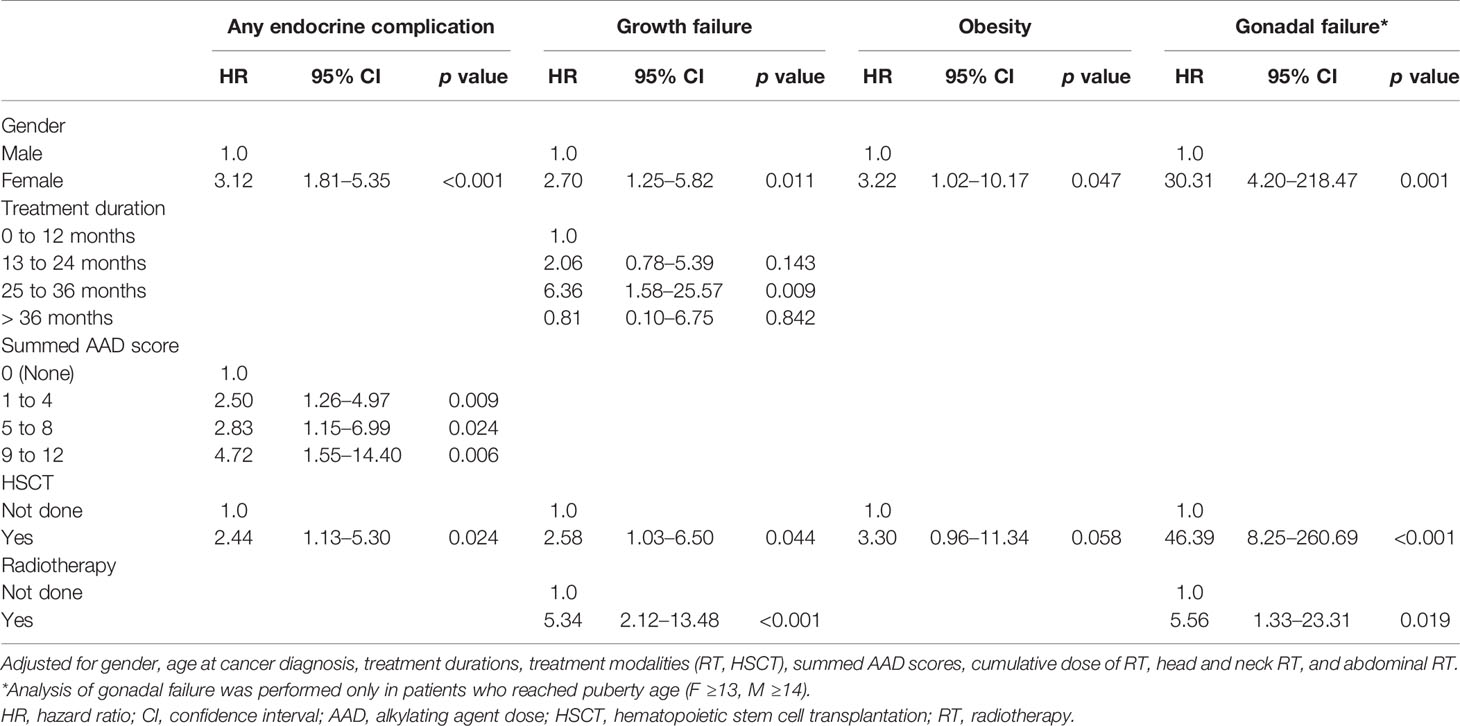
Table 4 Multivariable Cox proportional hazard analysis of endocrine complications following solid tumor treatment in childhood cancer survivors.
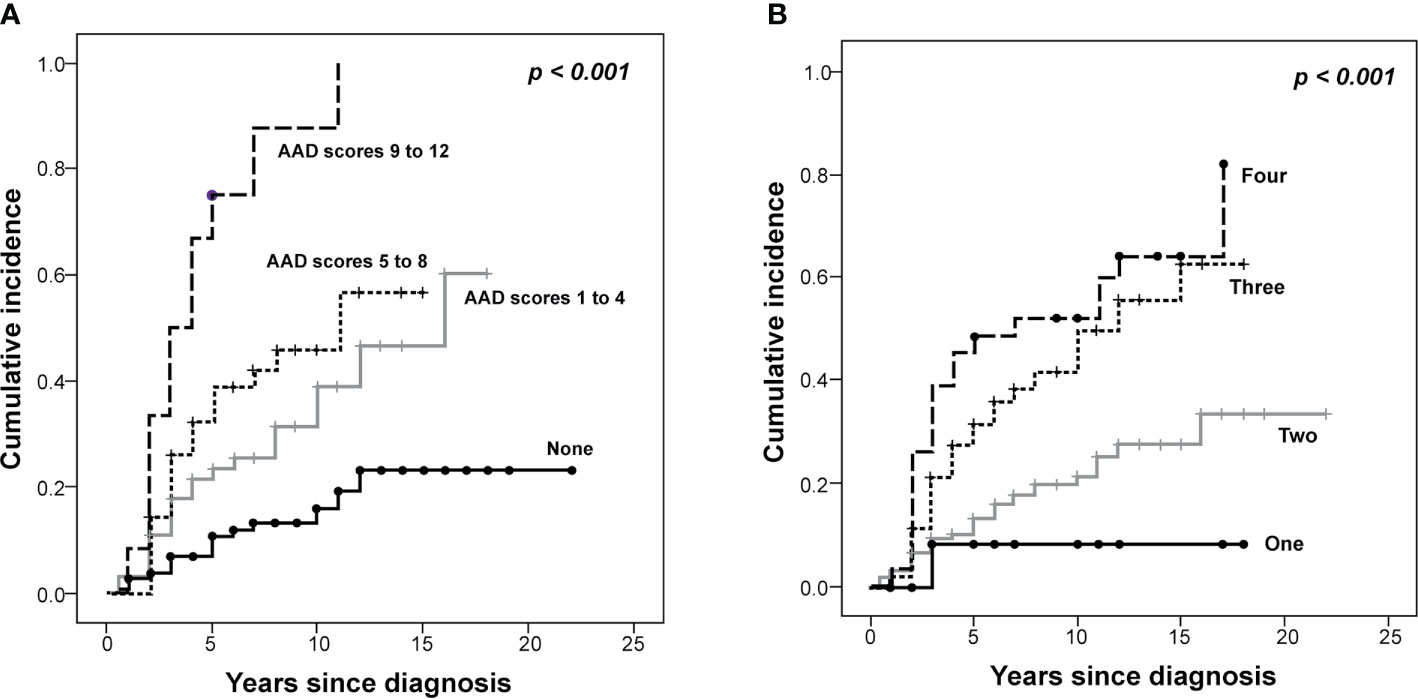
Figure 2 The Kaplan–Meier curves of the cumulative incidence of endocrine complications in survivors of childhood solid tumors. The cumulative incidence of endocrine complications is demonstrated according to the (A) alkylating agent dose (AAD) scores and the (B) number of treatment modalities (P < 0.001).
1. Short Stature and Growth Failure
Thirty-seven patients (14.6%, M:F = 15:22) had short stature at the end of their cancer treatment. Growth failure was detected in 35 patients (13.8%, M:F = 15:20). The mean duration from a diagnosis of primary cancer to the development of growth failure was 3.9 ± 2.9 years (range, 1–12 years). Among them, 26 patients were characterized by stunted growth velocity within five years, and nine patients showed decreased growth velocity 8.1 ± 2.6 years after the primary cancer diagnosis (range 5–12 years). Two female patients, who were born as a prematurity and who were small for gestational age, consistently demonstrated short stature without decreased growth velocity. Therefore, they were not included in the growth failure.
Three of the four patients with syndromic disorders (BWS [n = 1], NS [n = 1], and WAGR syndrome [n = 1]) experienced stunted growth velocity during or after NCST treatment. The height-SDS decreased from -0.32 to -2.3 in the patient with BWS, from -1.51 to -2.99 in the patient with NS, and from 0.45 to -3.15 in the patient with WAGR syndrome.
The height-SDS of patients with growth failure was decreased from -0.25 ± 1.12 to -2.47± 0.77 at the final visit (p < 0.001). The average serum IGF-1 SDS of these patients with growth failure was -1.58 ± 1.14. Among the 35 patients with growth failure, GH provocation tests were performed in four patients who underwent head and neck RT. As a result, two patients were diagnosed with GHD: one female with isolated GHD and another female with multiple pituitary hormone deficiencies (MPHD). Five survivors had a stunted pubertal growth spurt due to hypergonadotropic hypogonadism. One patient developed short stature due to the prolonged use of prednisolone to treat chronic graft-versus-host disease (GVHD).
Seven patients (7/35, 20%) were treated with recombinant human growth hormone (rhGH) therapy. Two of these were diagnosed with GHD. The other five patients with significant growth delay experienced chronic renal disease caused by cancer treatment. The rhGH therapy was started after a mean duration of three years after completion of the NCST treatment. Chronological age at initiation of rhGH therapy was 10.1 ± 2.7 years (range, 7–14 years). Height-SDS (-2.72 ± 0.78) did not significantly improve after a mean 2.4 years of rhGH treatment (-2.28 ± 0.93, p = 0.128), although serum IGF-1 SDS increased from -1.72 ± 0.56 to 0.63 ± 2.04 (p = 0.018). Adverse effects were not seen with the rhGH therapy.
Factors that increased the risk of growth failure were female gender (HR = 2.70, p = 0.011), duration of cancer treatment between 25 and 36 months (HR = 6.36, p = 0.009), HSCT (HR = 2.58, p = 0.044), and RT (HR = 5.34, p < 0.001).
The final height of 12 of the 90 patients (13.3%) was below -2.0 SDS. To assess the factors that affected final height, univariable and multivariable linear regression analyses were performed in the 90 patients (M:F = 60:30) who reached final height (Table 1 in Supplementary Material). As a result, as treatment duration increased by a month, the final height-SDS decreased by -0.063 SDS (B = -0.063, p = 0.029). In addition, the final height decreased by -0.163 SDS when the summed AAD score increased by a point (B = -0.163, p = 0.028). The initial height-SDS was found to positively correlate with the final height-SDS (B = 0.996, p < 0.001).
2. Obesity and Dyslipidemia
Obesity was observed in 18 patients (7.1%, M:F = 8:10). They were not obese at their initial cancer diagnosis, but developed obesity during or after NSCT treatment. The mean duration from initial cancer diagnosis to the development of primary hypothyroidism was 6.7 ± 4.3 years (range, 2-16 years). Multivariable Cox regression analysis revealed that female gender increased the risk of obesity (HR = 3.22, p = 0.047). In addition, HSCT increased the risk of obesity; however, this finding did not have statistical significance (HR = 3.30, p = 0.058).
Eight patients (3.2%, M:F = 4:4) were diagnosed with dyslipidemia. On average, it took 5.4 ± 5.0 years for dyslipidemia to present after the initial diagnosis of cancer (range, 2-17 years). Six of the patients were obese when they developed dyslipidemia. Five of them exhibited high low-density lipoprotein (LDL)-cholesterol levels (169.2 ± 10.1 mg/dL), and six patients showed hypertriglyceridemia (405.2 ± 155.0 mg/dL). Three exhibited mixed dyslipidemia with hypertriglyceridemia and hypercholesterolemia.
3. Hypergonadotropic Hypogonadism and Precocious Puberty
At the last follow-up, 132 patients (52%) were over 13 years of age for females and 14 years of age for males. Of these, 16 patients (12.1%, M:F = 2:12) developed hypergonadotropic hypogonadism. The mean duration from initial cancer diagnosis to the development of gonadal dysfunction was 5.4 ± 3.8 years (range, 2–12 years). Female patients manifested primary ovarian failure 4.9 ± 3.7 years after initial cancer diagnosis, while male patients were diagnosed with gonadal dysfunction after 9.0 ± 2.8 years.
The mean LH and FSH levels of these patients at diagnosis of hypogonadism were 20.5 ± 18.7 mIU/mL (range, 5.5–72.4 mIU/mL) and 54.6 ± 37.1 mIU/mL (range, 5.9–132 mIU/mL), respectively. The mean levels of estradiol in females and testosterone in males were 5.4 ± 2.3 pg/mL (range, 4.0–11.0 pg/mL) and 4.6 ± 3.4 ng/mL, respectively. There were 14 patients (one male and 13 females) under sex hormone replacement therapy.
One female with facial rhabdomyosarcoma who underwent head and neck RT showed MPHD, including GHD, central hypothyroidism, adrenal insufficiency, and hypogonadotropic hypogonadism. A 15-year-old female with osteosarcoma manifested hypothalamic amenorrhea. Two patients with hepatoblastoma developed central precocious puberty (CPP) at seven years of age, which was three and five years after they completed cancer treatments.
Factors that increased the risk of gonadal failure in patients of post-pubertal age were female gender (HR = 30.31, p = 0.001), HSCT (HR = 46.39, p < 0.001), and RT (HR = 5.56, p = 0.019) using multivariable Cox regression analysis. The patients who developed hypergonadotropic hypogonadism underwent HSCT at 10.6 ± 3.7 years on average while those without hypogonadism underwent HSCT at 5.8 ± 4.8 years of age (p = 0.002).
4. Primary Hypothyroidism
Primary hypothyroidism was observed in 12 survivors (4.7%, M:F = 5:7). The mean duration from initial cancer diagnosis to the development of primary hypothyroidism was 4.1 ± 2.9 years (range, 2–12 years). All these patients were treated with RT and HSCT and were on levothyroxine therapy. Eleven of the 12 patients (91.7%) with hypothyroidism were survivors of neuroblastoma; the remaining patient had Ewing sarcoma. Among the eight patients who underwent 131I-MIBG therapy, six (6/8, 75%) developed overt hypothyroidism and have been treated with levothyroxine.
5. Vitamin D Deficiency and Osteoporosis
Vitamin D deficiency was detected in seven patients (2.8%, M:F = 2:5). The mean duration from initial cancer diagnosis to the development of primary hypothyroidism was 2.4 ± 1.7 years (range, 0.5–5 years). The mean 25OHD3 level was 8.31 ± 3.7 ng/mL. Two patients showed radiologically confirmed rickets. One patient demonstrated renal tubulopathy caused by chemotherapeutic agents, leading to hypophosphatemic rickets.
Osteoporosis was found in three of the 23 survivors who underwent a DEXA scan (13%, M:F = 1:2). On average, it took 2.3 ± 0.6 years for osteoporosis to develop after the initial NCST diagnosis (range, 2–3 years). The mean BMD Z-scores of the femur neck and L-spine were -3.1 ± 0.3 and -2.1 ± 1.2, respectively. One female patient with high-risk neuroblastoma had multiple hormonal deficiencies, including primary hypothyroidism, vitamin D deficiency, and hypergonadotropic hypogonadism. A male patient with osteoporosis had been treated with prednisolone for chronic GVHD following allogeneic HSCT. Another female with Ewing sarcoma had primary ovarian failure after autologous HSCT.
Discussion
This study comprehensively analyzed endocrine complications in 253 childhood survivors of NCSTs. In this study, 30% of survivors developed at least one endocrine complication during the follow-up period, similar to a previous study on neuroblastoma (28%) and rhabdomyosarcoma of the head and neck (35%) (16). Shalitin et al. (18) described how 31.7% of survivors with NCSTs manifested at least one endocrine abnormality during a nine-year observation period, which is a comparable prevalence with our cohort.
In the present study, impaired growth was the most common endocrine complication after NCST treatment. Knijnenburg et al. (33) reported that 8.9% of 573 cancer survivors had a height ≤ -2 SDS at adulthood, which was slightly lower than the value obtained in our study. A treatment duration of between 25 and 36 months, HSCT, and exposure to RT were identified as the main risk factors for growth failure in the current study. Longer treatment duration implies tumor recurrence or metastasis that requires a more intensive treatment regimen, which, in turn, leads to impaired growth rate. The decrease in the risk of impaired growth in patients whose duration of treatment was ≥ 36 months was presumed to be due to a higher rate of mortality in these patients.
Growth failure in childhood NCST survivors was not associated with the hypothalamic-pituitary axis because only a few patients underwent head and neck RT, as in our cohort with two GHD patients. Multivariable linear regression analysis demonstrated that the summed AAD score negatively correlated with final height. Thus, it can be presumed that stunted growth velocity is caused by the direct skeletal toxicity of chemotherapeutic agents on the growth plate (17, 34). Doxorubicin inhibits chondrocyte destruction in vitro (35), which subsequently inhibits bone formation (34). Actinomycin D and purine antimetabolites reduced the proliferation of epiphyseal chondrocytes in vitro (36). Trans-retinoic acid induced premature epiphyseal closure in patients with high-risk neuroblastoma (37). Cyclophosphamide causes apoptosis in the proliferative zones of rat growth plates (38). In addition, a higher AAD score suggests an advanced primary cancer, which can impair general health, subsequently leading to growth failure. Decreased serum IGF-1 levels due to malnutrition during high-dose polychemotherapy and dexamethasone can also affect linear growth (34, 39). Another explanation for a decreased final height is the early onset of puberty or diminished pubertal growth spurt due to hypogonadism (40, 41).
Recent studies did not show a significant increase in the development of second neoplasms of the central nervous system in childhood cancer survivors who were treated with rhGH (42–45). Similarly, long-term follow-up data did not demonstrate a significant change in the risk of tumor recurrence or an increased risks of mortality in survivors treated with rhGH (46, 47). Traditionally, the recommendation has been that clinicians should start rhGH therapy for survivors of malignant tumors at least one year after the childhood cancer survivor is disease free (31). In our cohort, the mean duration for the commencement of rhGH therapy was three years after completion of NCST treatment. Of seven patients who were treated with rhGH, two patients were diagnosed with GHD, and the remaining five had chronic kidney disease (CKD). The efficacy of rhGH therapy in patients with CKD has previously been demonstrated (48). The efficacy of rhGH therapy could not be assessed in our cohort owing to the relatively short periods of rhGH treatment (a mean of 2.4 years) and the small number of patients.
The incidence of obesity in the current cohort (7.1%) was substantially higher than that in a previous study on NCST survivors (1.4%) (18). However, a much higher incidence (i.e., obesity of 31.7%) was reported in research on childhood cancer survivors (49). The prevalence of obesity in childhood survivors with acute lymphoblastic leukemia was shown to be substantially high (29%–69%) in one meta-analysis (50). The pathogenesis of dyslipidemia and obesity is mediated by factors related to anticancer treatment (chemotherapy, corticosteroids, and RT) and additional factors (i.e., genetic, dietary, or individual) (51). A high risk of obesity was associated with female gender and the initiation of cancer treatment at a young age (52). Thus, it is recommended that lipid profiles should be monitored in childhood cancer survivors (53).
Among patients who reached puberty, the prevalence of hypergonadotropic hypogonadism (12.1%) was higher than previous data on hematologic malignancies, lymphoma, and several extra-cranial solid tumors (6.3%) (54). As in previous research (54), the preservation of gonadal function was associated with a younger age at HSCT. In our study, we found significant difference in HSCT age between the two groups with or without hypogonadism, suggesting that gonadal function may be preserved if chemotherapy and/or HSCT are administered at a young age. The HR for primary gonadal dysfunction in female survivors was considerably high in our study. This difference in prevalence might be associated with underdiagnosed cases of male gonadal dysfunction and the peculiar amenorrhea symptoms in female patients. In males, Leydig cells were relatively resistant to chemotherapy, and the symptoms of Leydig cell failure tend to be subclinical in the majority of cases (30, 55). RT is a well-established cause of ovarian failure. TBI, rather than abdominal RT, was a risk factor for primary ovarian failure (56). In our cohort, exposure to RT and HSCT increased the risk of gonadal failure. RT has a direct harmful effect on oocytes, as even less than 4 Gy has been responsible for the death of 50% of human oocytes (57). Primary gonadal failure following HSCT is caused by conditioning regimens, including TBI and/or high dose chemotherapy (58). As gonadal insufficiency has a close association with HSCT, but not with a higher AAD score in our study, specific conditioning agents, such as busulfan or melphalan, may impair gonadal function rather than the total dose of alkylating agents. A previous research has demonstrated that a busulfan-based conditioning regimen has a higher risk of primary ovarian failure (59).
Primary hypothyroidism occurred in 9.9% of survivors in a previous study of NCSTs (18), which is higher than our study (4.7%). Previously reported risk factors of hypothyroidism include neck irradiation (60), HSCT, alkylating agents (18), and 131I-MIBG therapy (17, 61). Given that all patients with hypothyroidism had a history of RT during cancer treatment, hypothyroidism and radiotherapy were closely associated in our cohort.
Childhood cancer survivors face significant bone health disruptions (62). Besides the direct harmful effect of chemotherapeutic agents on bones (34), concomitant GHD, hypogonadism, and hyperthyroidism influence BMD deficits (22). Some chemotherapeutic agents, such as cyclophosphamide and cisplatin, cause renal damage and the dysregulation of calcium and vitamin D metabolism (63). As with the finding of the current study, in other research, the prevalence of vitamin D deficiency was high owing to poor sun exposure and an insufficient intake of vitamin D and calcium (64). The 25OHD3 levels of childhood cancer survivors must be monitored to prevent osteoporosis and fractures beyond the tumor site.
Although the risk of endocrine complications is significantly increased in cancer survivors, they are likely to be lost to follow-up if they do not have current health problems. Therefore, education on the late effects of cancer treatment should be emphasized when managing this at-risk population (65).
There were several limitations to this study. As this study was performed retrospectively, data on the cumulative dosages of RT or chemotherapy were missing. BMD and serum 25OHD3 levels were not screened in all subjects. Twenty seven percent of patients were followed-up less than 5 years, which might be too short duration to confirm the long-term consequences of cancer treatment. In addition, the primary tumors of this cohort were heterogeneous, which may hamper the unified analysis of endocrine outcomes. Finally, we did not consider staging and NCST relapses in the analyses.
In conclusion, this study analyzed the endocrine outcomes of 253 childhood NCST survivors. HSCT and high AAD scores increased the risk of developing at least one endocrine complication. Since high-dose myeloablative chemotherapy was mainly used as the pre-conditioning regimen prior to HSCT, alkylating agents rather than TBI was associated with endocrine complications. Growth failure was the most common endocrine complication. Factors affecting the final height were the initial height, treatment duration and cumulative dose of alkylating agents. HSCT was highly related to gonadal dysfunction. Lifelong surveillance for endocrine events is highly recommended in patients with NCSTs who were treated with high-dose polychemotherapy.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author Contributions
YL and J-HC designed the research and wrote the manuscript. YL analyzed the data. JS, YC, HK, K-NK, HI, and H-WY collected the data. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (2017R1D1A1B03029638).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.610730/full#supplementary-material
References
1. Scotting PJ, Walker DA, Perilongo G. Childhood solid tumours: a developmental disorder. Nat Rev Cancer (2005) 5:481–8. doi: 10.1038/nrc1633
2. Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin (2014) 64:83–103. doi: 10.3322/caac.21219
3. Geenen MM, Cardous-Ubbink MC, Kremer LC, van den Bos C, van der Pal HJ, Heinen RC, et al. Medical assessment of adverse health outcomes in long-term survivors of childhood cancer. JAMA (2007) 297:2705–15. doi: 10.1001/jama.297.24.2705
4. Oeffinger KC, Mertens AC, Sklar CA, Kawashima T, Hudson MM, Meadows AT, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med (2006) 355:1572–82. doi: 10.1056/NEJMsa060185
5. Brignardello E, Felicetti F, Castiglione A, Chiabotto P, Corrias A, Fagioli F, et al. Endocrine health conditions in adult survivors of childhood cancer: the need for specialized adult-focused follow-up clinics. Eur J Endocrinol (2013) 168:465–72. doi: 10.1530/EJE-12-1043
6. Bruzzi P, Predieri B, Corrias A, Marsciani A, Street ME, Rossidivita A, et al. Final height and body mass index in adult survivors of childhood acute lymphoblastic leukemia treated without cranial radiotherapy: a retrospective longitudinal multicenter Italian study. BMC Pediatr (2014) 14:236. doi: 10.1186/1471-2431-14-236
7. Bruzzi P, Bigi E, Predieri B, Bonvicini F, Cenciarelli V, Felici F, et al. Long-term effects on growth, development, and metabolism of ALL treatment in childhood. Expert Rev Endocrinol Metab (2019) 14:49–61. doi: 10.1080/17446651.2019.1561271
8. Chemaitilly W, Li Z, Huang S, Ness KK, Clark KL, Green DM, et al. Anterior hypopituitarism in adult survivors of childhood cancers treated with cranial radiotherapy: a report from the St Jude Lifetime Cohort study. J Clin Oncol (2015) 33:492–500. doi: 10.1200/jco.2014.56.7933
9. Laughton SJ, Merchant TE, Sklar CA, Kun LE, Fouladi M, Broniscer A, et al. Endocrine outcomes for children with embryonal brain tumors after risk-adapted craniospinal and conformal primary-site irradiation and high-dose chemotherapy with stem-cell rescue on the SJMB-96 trial. J Clin Oncol (2008) 26:1112–8. doi: 10.1200/jco.2008.13.5293
10. Seo GH, Choi JH, Kim YM, Koh KN, Im HJ, Ra YS, et al. Long-term endocrine effects and trends in body mass index changes in patients with childhood-onset brain tumors. J Neurooncol (2018) 138:55–62. doi: 10.1007/s11060-018-2765-0
11. Allen-Rhoades W, Whittle SB, Rainusso N. Pediatric solid tumors of infancy: an overview. Pediatr Rev (2018) 39:57–67. doi: 10.1542/pir.2017-0057
12. Howard SC, Pui CH. Endocrine complications in pediatric patients with acute lymphoblastic leukemia. Blood Rev (2002) 16:225–43. doi: 10.1016/s0268-960x(02)00042-5
13. Clement SC, Schouten-van Meeteren AY, Boot AM, Claahsen-van der Grinten HL, Granzen B, Sen Han K, et al. Prevalence and risk factors of early endocrine disorders in childhood brain tumor survivors: a nationwide, multicenter study. J Clin Oncol (2016) 34:4362–70. doi: 10.1200/JCO.2016.67.5025
14. Wright KD, Green DM, Daw NC. Late effects of treatment for wilms tumor. Pediatr Hematol Oncol (2009) 26:407–13. doi: 10.3109/08880010903019344
15. Ginsberg JP, Goodman P, Leisenring W, Ness KK, Meyers PA, Wolden SL, et al. Long-term survivors of childhood Ewing sarcoma: report from the childhood cancer survivor study. J Natl Cancer Inst (2010) 102:1272–83. doi: 10.1093/jnci/djq278
16. Clement SC, Schoot RA, Slater O, Chisholm JC, Abela C, Balm AJM, et al. Endocrine disorders among long-term survivors of childhood head and neck rhabdomyosarcoma. Eur J Cancer (2016) 54:1–10. doi: 10.1016/j.ejca.2015.10.064
17. Geurten C, Geurten M, Hoyoux C, Lebrethon MC. Endocrine consequences of neuroblastoma treatment in children: 20 years’ experience of a single center. J Pediatr Endocrinol Metab (2019) 32:347–54. doi: 10.1515/jpem-2018-0273
18. Shalitin S, Laur E, Lebenthal Y, Ash S, Yaniv I, Phillip M. Endocrine complications and components of the metabolic syndrome in survivors of childhood malignant non-brain solid tumors. Horm Res Paediatr (2014) 81:32–42. doi: 10.1159/000355577
19. Tucker MA, Meadows AT, Boice JD Jr., Stovall M, Oberlin O, Stone BJ, et al. Leukemia after therapy with alkylating agents for childhood cancer. J Natl Cancer Inst (1987) 78:459–64. doi: 10.1093/jnci/78.3.459
20. Green DM, Nolan VG, Goodman PJ, Whitton JA, Srivastava D, Leisenring WM, et al. The cyclophosphamide equivalent dose as an approach for quantifying alkylating agent exposure: a report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer (2014) 61:53–67. doi: 10.1002/pbc.24679
21. Kim JH, Yun S, Hwang SS, Shim JO, Chae HW, Lee YJ, et al. The 2017 Korean national growth charts for children and adolescents: development, improvement, and prospects. Korean J Pediatr (2018) 61:135–49. doi: 10.3345/kjp.2018.61.5.135
22. Nandagopal R, Laverdiere C, Mulrooney D, Hudson MM, Meacham L. Endocrine late effects of childhood cancer therapy: a report from the Children’s Oncology Group. Horm Res (2008) 69:65–74. doi: 10.1159/000111809
23. Haymond M, Kappelgaard AM, Czernichow P, Biller BM, Takano K, Kiess W. Early recognition of growth abnormalities permitting early intervention. Acta Paediatr (2013) 102:787–96. doi: 10.1111/apa.12266
24. Hyun SE, Lee BC, Suh BK, Chung SC, Ko CW, Kim HS, et al. Reference values for serum levels of insulin-like growth factor-I and insulin-like growth factor binding protein-3 in Korean children and adolescents. Clin Biochem (2012) 45:16–21. doi: 10.1016/j.clinbiochem.2011.10.003
25. Viswanathan V, Eugster EA. Etiology and treatment of hypogonadism in adolescents. Pediatr Clin North Am (2011) 58:1181–2000. doi: 10.1016/j.pcl.2011.07.009
26. Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab (2011) 96:1911–30. doi: 10.1210/jc.2011-0385
27. Lim JS, Hwang JS, Lee JA, Kim DH, Park KD, Cheon GJ, et al. Bone mineral density according to age, bone age, and pubertal stages in Korean children and adolescents. J Clin Densitom (2010) 13:68–76. doi: 10.1016/j.jocd.2009.09.006
28. Kang MJ, Hong HS, Chung SJ, Lee YA, Shin CH, Yang SW. Body composition and bone density reference data for Korean children, adolescents, and young adults according to age and sex: results of the 2009-2010 Korean National Health and Nutrition Examination Survey (KNHANES). J Bone Miner Metab (2016) 34:429–39. doi: 10.1007/s00774-015-0686-y
29. Fan B, Lu Y, Genant H, Fuerst T, Shepherd J. Does standardized BMD still remove differences between Hologic and GE-Lunar state-of-the-art DXA systems? Osteoporos Int (2010) 21:1227–36. doi: 10.1007/s00198-009-1062-3
30. Chemaitilly W, Cohen LE. Diagnosis of endocrine disease: endocrine late-effects of childhood cancer and its treatments. Eur J Endocrinol (2017) 176:R183–203. doi: 10.1530/EJE-17-0054
31. Sklar CA, Antal Z, Chemaitilly W, Cohen LE, Follin C, Meacham LR, et al. Hypothalamic-Pituitary and Growth Disorders in Survivors of Childhood Cancer: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2018) 103:2761–84. doi: 10.1210/jc.2018-01175
32. Chemaitilly W, Sklar CA. Childhood Cancer Treatments and Associated Endocrine Late Effects: A Concise Guide for the Pediatric Endocrinologist. Horm Res Paediatr (2019) 91:74–82. doi: 10.1159/000493943
33. Knijnenburg SL, Raemaekers S, van den Berg H, van Dijk IW, Lieverst JA, van der Pal HJ, et al. Final height in survivors of childhood cancer compared with Height Standard Deviation Scores at diagnosis. Ann Oncol (2013) 24:1119–26. doi: 10.1093/annonc/mds580
34. van Leeuwen BL, Kamps WA, Jansen HW, Hoekstra HJ. The effect of chemotherapy on the growing skeleton. Cancer Treat Rev (2000) 26:363–76. doi: 10.1053/ctrv.2000.0180
35. Rainsford KD. Doxorubicin is a potent inhibitor of interleukin 1 induced cartilage proteoglycan resorption in-vitro. J Pharm Pharmacol (1989) 41:60–3. doi: 10.1111/j.2042-7158.1989.tb06332.x
36. Robson H, Anderson E, Eden OB, Isaksson O, Shalet S. Chemotherapeutic agents used in the treatment of childhood malignancies have direct effects on growth plate chondrocyte proliferation. J Endocrinol (1998) 157:225–35. doi: 10.1677/joe.0.1570225
37. Hobbie WL, Mostoufi SM, Carlson CA, Gruccio D, Ginsberg JP. Prevalence of advanced bone age in a cohort of patients who received cis-retinoic acid for high-risk neuroblastoma. Pediatr Blood Cancer (2011) 56:474–6. doi: 10.1002/pbc.22839
38. Xian CJ, Cool JC, van Gangelen J, Foster BK, Howarth GS. Effects of etoposide and cyclophosphamide acute chemotherapy on growth plate and metaphyseal bone in rats. Cancer Biol Ther (2007) 6:170–7. doi: 10.4161/cbt.6.2.3576
39. Bath LF, Crofton PM, Evans AE, Ranke MB, Elmlinger MW, Kelnar CJ, et al. Bone turnover and growth during and after chemotherapy in children with solid tumors. Pediatr Res (2004) 55:224–30. doi: 10.1203/01.PDR.0000100903.83472.09
40. Groot-Loonen JJ, van Setten P, Otten BJ, vant Hof MA, Lippens RJ, Stoelinga GB. Shortened and diminished pubertal growth in boys and girls treated for acute lymphoblastic leukaemia. Acta Paediatr (1996) 85:1091–5. doi: 10.1111/j.1651-2227.1996.tb14223.x
41. Moell C, Garwicz S, Westgren U, Wiebe T. Disturbed pubertal growth in girls treated for acute lymphoblastic leukemia. Pediatr Hematol Oncol (1987) 4:1–5. doi: 10.3109/08880018709141243
42. Patterson BC, Chen Y, Sklar CA, Neglia J, Yasui Y, Mertens A, et al. Growth hormone exposure as a risk factor for the development of subsequent neoplasms of the central nervous system: a report from the childhood cancer survivor study. J Clin Endocrinol Metab (2014) 99:2030–7. doi: 10.1210/jc.2013-4159
43. Mackenzie S, Craven T, Gattamaneni HR, Swindell R, Shalet SM, Brabant G. Long-term safety of growth hormone replacement after CNS irradiation. J Clin Endocrinol Metab (2011) 96:2756–61. doi: 10.1210/jc.2011-0112
44. Sklar CA, Mertens AC, Mitby P, Occhiogrosso G, Qin J, Heller G, et al. Risk of disease recurrence and second neoplasms in survivors of childhood cancer treated with growth hormone: a report from the Childhood Cancer Survivor Study. J Clin Endocrinol Metab (2002) 87:3136–41. doi: 10.1210/jcem.87.7.8606
45. Ergun-Longmire B, Mertens AC, Mitby P, Qin J, Heller G, Shi W, et al. Growth hormone treatment and risk of second neoplasms in the childhood cancer survivor. J Clin Endocrinol Metab (2006) 91:3494–8. doi: 10.1210/jc.2006-0656
46. Tamhane S, Sfeir JG, Kittah NEN, Jasim S, Chemaitilly W, Cohen LE, et al. GH Therapy in Childhood Cancer Survivors: A Systematic Review and Meta-Analysis. J Clin Endocrinol Metab (2018) 103:2794–801. doi: 10.1210/jc.2018-01205
47. Swerdlow AJ, Reddingius RE, Higgins CD, Spoudeas HA, Phipps K, Qiao Z, et al. Growth hormone treatment of children with brain tumors and risk of tumor recurrence. J Clin Endocrinol Metab (2000) 85:4444–9. doi: 10.1210/jcem.85.12.7044
48. Haffner D, Schaefer F, Nissel R, Wühl E, Tönshoff B, Mehls O. Effect of growth hormone treatment on the adult height of children with chronic renal failure. N Engl J Med (2000) 343:923–30. doi: 10.1056/NEJM200009283431304
49. Nathan PC, Jovcevska V, Ness KK, Mammone D’Agostino N, Staneland P, Urbach SL, et al. The prevalence of overweight and obesity in pediatric survivors of cancer. J Pediatr (2006) 149:518–25. doi: 10.1016/j.jpeds.2006.06.039
50. Zhang FF, Kelly MJ, Saltzman E, Must A, Roberts SB, Parsons SK. Obesity in pediatric ALL survivors: a meta-analysis. Pediatrics (2014) 133:e704–15. doi: 10.1542/peds.2013-3332
51. Rosen GP, Nguyen HT, Shaibi GQ. Metabolic syndrome in pediatric cancer survivors: a mechanistic review. Pediatr Blood Cancer (2013) 60:1922–8. doi: 10.1002/pbc.24703
52. Haddy RI, Haddy TB. Lifetime follow-up care after childhood cancer. J Am Board Family Med (2010) 23:647–54. doi: 10.3122/jabfm.2010.05.100031
53. Barg E, Połubok J, Hetman M, Gonera A, Jasielska O, Sęga-Pondel D, et al. Metabolic Disturbances in Children Treated for Solid Tumors. Nutrients (2019) 11:3062. doi: 10.3390/nu11123062
54. Chemaitilly W, Mertens AC, Mitby P, Whitton J, Stovall M, Yasui Y, et al. Acute ovarian failure in the childhood cancer survivor study. J Clin Endocrinol Metab (2006) 91:1723–8. doi: 10.1210/jc.2006-0020
55. Orio F, Muscogiuri G, Palomba S, Serio B, Sessa M, Giudice V, et al. Endocrinopathies after allogeneic and autologous transplantation of hematopoietic stem cells. ScientificWorldJournal (2014) 2014:282147. doi: 10.1155/2014/282147
56. Cohen LE, Gordon JH, Popovsky EY, Gunawardene S, Duffey-Lind E, Lehmann LE, et al. Late effects in children treated with intensive multimodal therapy for high-risk neuroblastoma: high incidence of endocrine and growth problems. Bone Marrow Transpl (2014) 49:502–8. doi: 10.1038/bmt.2013.218
57. Wallace WH, Shalet SM, Crowne EC, Morris-Jones PH, Gattamaneni HR. Ovarian failure following abdominal irradiation in childhood: natural history and prognosis. Clin Oncol (R Coll Radiol) (1989) 1:75–9. doi: 10.1016/s0936-6555(89)80039-1
58. Chow EJ, Anderson L, Baker KS, Bhatia S, Guilcher GM, Huang JT, et al. Late effects surveillance recommendations among survivors of childhood hematopoietic cell transplantation: a Children’s Oncology Group Report. Biol Blood Marrow Transpl (2016) 22:782–95. doi: 10.1016/j.bbmt.2016.01.023
59. Cho WK, Lee JW, Chung NG, Jung MH, Cho B, Suh BK, et al. Primary ovarian dysfunction after hematopoietic stem cell transplantation during childhood: busulfan-based conditioning is a major concern. J Pediatr Endocrinol Metab (2011) 24:1031–5. doi: 10.1515/jpem.2011.339
60. Rose SR, Horne VE, Howell J, Lawson SA, Rutter MM, Trotman GE, et al. Late endocrine effects of childhood cancer. Nat Rev Endocrinol (2016) 12:319–36. doi: 10.1038/nrendo.2016.45
61. Picco P, Garaventa A, Claudiani F, Gattorno M, De Bernardi B, Borrone C. Primary hypothyroidism as a consequence of 131-I-metaiodobenzylguanidine treatment for children with neuroblastoma. Cancer (1995) 76:1662–4. doi: 10.1002/1097-0142(19951101)76:9<1662::aid-cncr2820760924>3.0.co;2-v
62. Kang MJ, Lim JS. Bone mineral density deficits in childhood cancer survivors: pathophysiology, prevalence, screening, and management. Korean J Pediatr (2013) 56:60–7. doi: 10.3345/kjp.2013.56.2.60
63. van Dorp W, van Beek RD, Laven JS, Pieters R, de Muinck Keizer-Schrama SM, van den Heuvel-Eibrink MM. Long-term endocrine side effects of childhood Hodgkin’s lymphoma treatment: a review. Hum Reprod Update (2012) 18:12–28. doi: 10.1093/humupd/dmr038
64. Fouda A, Kandil S, Boujettif K, Fayea N. Hypovitamininosis D in Childhood Cancer Survivors: Importance of Vitamin D Supplementation and Measurement Over Different Points of Time. J Pediatr Hematol Oncol (2018) 40:e83–90. doi: 10.1097/mph.0000000000001060
Keywords: cancer survivors, child, endocrine system disease, risk factors, alkylating agents, hematopoietic stem cell transplantation, solid tumors
Citation: Lee Y, Shin J, Choi Y, Kim H, Koh K-N, Im HJ, Yoo H-W and Choi J-H (2021) Endocrine Complications in Children and Adolescents With Non-Central Nervous System Solid Tumors. Front. Endocrinol. 12:610730. doi: 10.3389/fendo.2021.610730
Received: 27 September 2020; Accepted: 03 March 2021;
Published: 17 March 2021.
Edited by:
Valentino Cherubini, Azienda Ospedaliero Universitaria Ospedali Riuniti, ItalyReviewed by:
Barbara Predieri, University of Modena and Reggio Emilia, ItalyYardena Tenenbaum Rakover, Ha’Emek Medical Center, Israel
Copyright © 2021 Lee, Shin, Choi, Kim, Koh, Im, Yoo and Choi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin-Ho Choi, amhjQGFtYy5zZW91bC5rcg==