 Jung Soo Lim
Jung Soo Lim Samuel W. Plaska
Samuel W. Plaska Juilee Rege1
Juilee Rege1 Adina F. Turcu
Adina F. Turcu- 1Department of Molecular and Integrative Physiology, University of Michigan, Ann Arbor, MI, United States
- 2Division of Endocrinology and Metabolism, Department of Internal Medicine, Yonsei University Wonju College of Medicine, Wonju Severance Christian Hospital, Wonju, South Korea
- 3Division of Metabolism, Endocrine, and Diabetes, University of Michigan, Ann Arbor, MI, United States
Background: Somatic gene mutations that facilitate inappropriate intracellular calcium entrance have been identified in most aldosterone-producing adenomas (APAs). Studies suggest that angiotensin II and adrenocorticotropic hormone (ACTH) augment aldosterone production from APAs. Little is known, however, regarding possible variations in response to hormonal stimuli between APAs with different aldosterone-driver mutations.
Objective: To analyze the transcript expression of type 1 angiotensin II receptors (AGTR1), ACTH receptors (MC2R), and melanocortin 2 receptor accessory protein (MRAP) in APAs with known aldosterone-driver somatic mutations.
Methods: RNA was isolated from APAs with mutations in: KCNJ5 (n = 14), ATP1A1 (n = 14), CACNA1D (n = 14), and ATP2B3 (n = 5), and from normal adjacent adrenal tissue (n = 45). Transcript expression of MC2R, MRAP, AGTR1, aldosterone synthase (CYP11B2), 17α-hydroxylase/17,20-lyase (CYP17A1), and 11β-hydroxylase (CYP11B1) were quantified using quantitative RT-PCR and normalized to β-actin.
Results: Compared to adjacent normal adrenal tissue, APAs had higher transcript levels of CYP11B2 (2,216.4 [1,112.0, 2,813.5]-fold, p < 0.001), MC2R (2.88 [2.00, 4.52]-fold, p < 0.001), and AGTR1 (1.80 [1.02, 2.80]-fold, p < 0.001]), and lower transcript levels of MRAP, CYP17A1, and CYP11B1 (0.28–0.36, p < 0.001 for all). MC2R and CYP11B2 transcripts were lower in APAs with KCNJ5 vs. other mutations (p < 0.01 for both). MC2R expression correlated positively with that of AGTR1 in APAs harboring KCNJ5 and CACNA1D mutations, and with MRAP expression in APAs harboring ATPase mutations.
Conclusions: While MC2R and AGTR1 are expressed in all APAs, differences were observed based on the underlying aldosterone-driver somatic mutations. In tandem, our findings suggest that APAs with ATPase-mutations are more responsive to ACTH than KCNJ5-mutated APAs.
Introduction
Primary aldosteronism (PA) is characterized by inappropriate, renin-independent aldosterone production. PA is the most common curable form of secondary hypertension, accounting for up to 20% of resistant hypertension cases (1). Growing evidence suggests that PA increases the risk of cardiovascular and renal complications as compared to essential hypertension, independently of blood pressure control (2–4). Inappropriate mineralocorticoid receptor activation might promote the release of pro-inflammatory cytokines (5), oxidative stress (6), and, consequently, target organ damage (2, 4). Sporadic PA is broadly classified as bilateral adrenal hyperaldosteronism (BHA) or unilateral PA, which is often caused by an aldosterone-producing adenoma (APA). APAs account for 30–50% of PA cases and they can be cured by adrenalectomy, while BHA requires life-long targeted medical therapy (7). PA subtyping is typically established based on adrenal venous sampling (AVS) (7). In many centers, AVS is performed after administration of cosyntropin, a synthetic adrenocorticotropic hormone (ACTH), which enhances the confidence of successful adrenal vein catheterization and circumvents intrinsic ACTH fluctuations that might occur due to the stress of the procedure. Reports regarding the impact of ACTH on APAs, however, have been inconsistent (8–12).
Studies conducted over the past decade have identified a series of aldosterone–driver gene mutations in familial and sporadic forms of PA. Affected genes include: KCNJ5 (13), ATP1A1 (14, 15), ATP2B3 (15), CACNA1D (16), CACNA1H (17), CTNNB1 (18), and CLCN2 (19, 20). Next-generation sequencing (NGS) of aldosterone-producing areas precisely mapped using immunohistochemistry (IHC) for aldosterone synthase (CYP11B2) has revealed aldosterone-driver somatic mutations in over 90% of APAs (21–23). A shared molecular feature of the somatic mutations found in APAs is that they facilitate intracellular calcium entrance, which then stimulates aldosterone production by augmenting CYP11B2 expression (23). Nonetheless, APAs harboring different aldosterone-driver somatic mutations have distinct histopathological features (24), steroidogenic potential (25), and responses to ACTH stimulation (26).
In addition to ion channel or pump mutations, some studies suggest that the aberrant expression of receptors in APAs, such as G-protein coupled receptors (GPCRs), might contribute to their dysregulated aldosterone production (27–29). Under physiological conditions, angiotensin II, serum potassium, and, to a lesser extent, ACTH control aldosterone synthesis from the adrenal zona glomerulosa (ZG) (30, 31). Variability in type 1 angiotensin II receptor (AGTR1) and melanocortin type 2 receptor (MC2R, also known as ACTH receptor) expression, which is abundant in both APAs and normal adrenals (29), might modulate aldosterone production (30, 31). Although cellular models of aldosterone-driver mutations showed that responses to angiotensin II are increased (32, 33), data on possible variations in response to hormonal stimuli between APAs with different somatic mutations are scarce. Herein, we investigated the transcript expression of AGTR1, MC2R, and melanocortin-2-receptor accessory protein (MRAP) in APAs with known aldosterone-driver somatic mutations and in adjacent normal adrenal tissue. In addition, we assessed the relationship between aldosterone-regulators and CYP11B2 expression in APAs with different somatic mutations.
Materials and Methods
Tissue Samples
The current study included adrenals from 47 patients with APA who underwent adrenalectomy at the University of Michigan between 2004 and 2018. Patients were selected based on availability of formalin-fixed paraffin-embedded (FFPE) adrenal tumor blocks. The clinical diagnosis of PA was made according to the institutional consensus available at the time or the Endocrine Society Clinical Practice guidelines (7). All adrenal specimens were pathologically diagnosed as adrenocortical adenomas. For comparison, we used adjacent normal adrenal tissue obtained from the same patients. Because the availability of adrenal tissue adjacent to the APA was limited, cortical and medullary tissue were not dissected separately. Sections from FFPE adrenal tumor blocks were used for IHC for CYP11B2 and 17α-hydroxylase/17,20-lyase (CYP17A1) and for genetic analysis, as previously described (21). This study was approved by Institutional Review Boards at the University of Michigan (HUM00106809, HUM00024461, HUM00083056). Written informed consent was obtained from all patients who underwent adrenalectomy after February, 2011. A waiver of consent was granted for the use of archival specimens (HUM00083056).
DNA/RNA Isolation
Genomic DNA (gDNA) and RNA were obtained from APAs with mutations in: KCNJ5 (n = 14), ATP1A1 (n = 14), CACNA1D (n = 14), and ATP2B3 (n = 5), and from adjacent normal adrenal tissues (n = 45). Adrenocortical adenomas that displayed CYP11B2-expressing cells were considered APAs. After identification of CYP11B2-positive areas by IHC, four to nine unstained consecutive 5 µm FFPE slides were used to separately dissect corresponding CYP11B2-positive areas. Dissection of FFPE sections was performed using disposable scalpels under an Olympus SZ-40 microscope. The AllPrep DNA/RNA FFPE kit (QIAGEN, Hilden, Germany) was used to isolate gDNA and RNA, as previously described (34).
Next-Generation Sequencing
For mutation analysis, multiplexed PCR–based NGS was conducted using Ion Torrent Ampliseq sequencing (Thermo Fisher Scientific), as previously described (21, 34). The panel for library preparation included amplicons targeting the full coding regions of known aldosterone-driving genes, including the most commonly affected: KCNJ5, ATP1A1, CACNA1D, and ATP2B3. APAs with other aldosterone-driver mutations were not included in this analysis, due to their low prevalence.
Quantitative Real-Time RT-PCR (qPCR)
Total RNA was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qPCR was performed using the ABI StepOnePlus Real-Time PCR systems (Applied Biosystems). CYP11B2, CYP17A1, and CYP11B1 primer/probe mixtures were prepared as previously described (27, 35). For Human MRAP qPCR, the primer (qHsaCID0022591, Bio-Rad) was mixed with SYBR Green PCR master mix (Applied Biosystems). Primer/probe mixtures for the amplification of AGTR1 (Hs00258938_m1), MC2R (Hs00300820_s1), and β-actin (ACTB; Hs01060665_g1) were purchased from Applied Biosystems. In this study, ACTB transcript was used as a reference gene for normalization between samples. Relative quantification was determined using the comparative threshold cycle method (36). The average ΔCT value of all adjacent normal tissues was used as reference when comparing gene expression between APAs with various underlying mutations.
Statistical Analysis
Statistical analyses were conducted using SAS 9.4 (SAS Institute, Cary, NC, USA), and GraphPad Prism 8 was used to generate figures. The Kruskal-Wallis test, followed by the Dwass-Steel-Critchlow-Fligner test were employed to compare continuous variables across multiple groups. Distribution of categorical variables across groups was assessed by the Chi-square or Fisher’s exact test. Wilcoxon signed-rank test was used for paired comparison of transcript levels between APAs and the corresponding adjacent normal adrenal tissues. Correlations between gene expressions were examined with the Spearman correlation test. Two-sided p values below 0.05 were considered statistically significant.
Results
Demographic and clinical characteristics of study participants are presented in Table 1. Most patients were Caucasian, with ages between 20 and 79 years (median age 52) and 62% were men. Patients with APAs harboring KCNJ5 mutations were younger, leaner, and mostly women (Table 1).
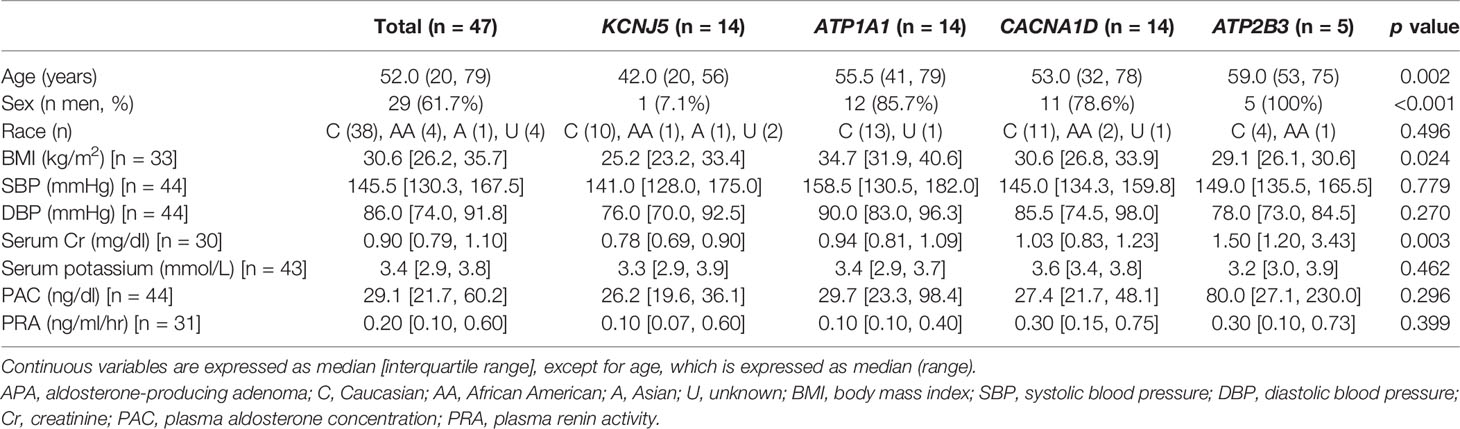
Table 1 Baseline characteristics of patients with APA participating in this study.
AGTR1, MC2R, MRAP, CYP11B2, CYP17A1, and CYP11B1 Gene Expressions in Aldosterone-Producing Adenomas
Overall, APAs displayed higher transcript levels of MC2R (2.88 [2.00, 4.52]-fold, p < 0.001), AGTR1 (1.80 [1.02, 2.80]-fold, p < 0.001), and CYP11B2 (2216.4 [1112.0, 2813.5]-fold, p < 0.001) compared to the corresponding adjacent normal adrenal tissue, and these differences remained robust in APAs with CACNA1D and ATP1A1 mutations (Table 2). AGTR1 and MC2R transcript levels were only minimally, but not significantly higher in KCNJ5-mutated APAs as compared to the paired adjacent normal adrenal tissue. Conversely, APAs had lower transcript levels of MRAP, CYP17A1, and CYP11B1 (0.28–0.36-fold, p < 0.001, Table 2) than the corresponding normal adjacent adrenal tissue and these differences were observed in all mutation subgroups.
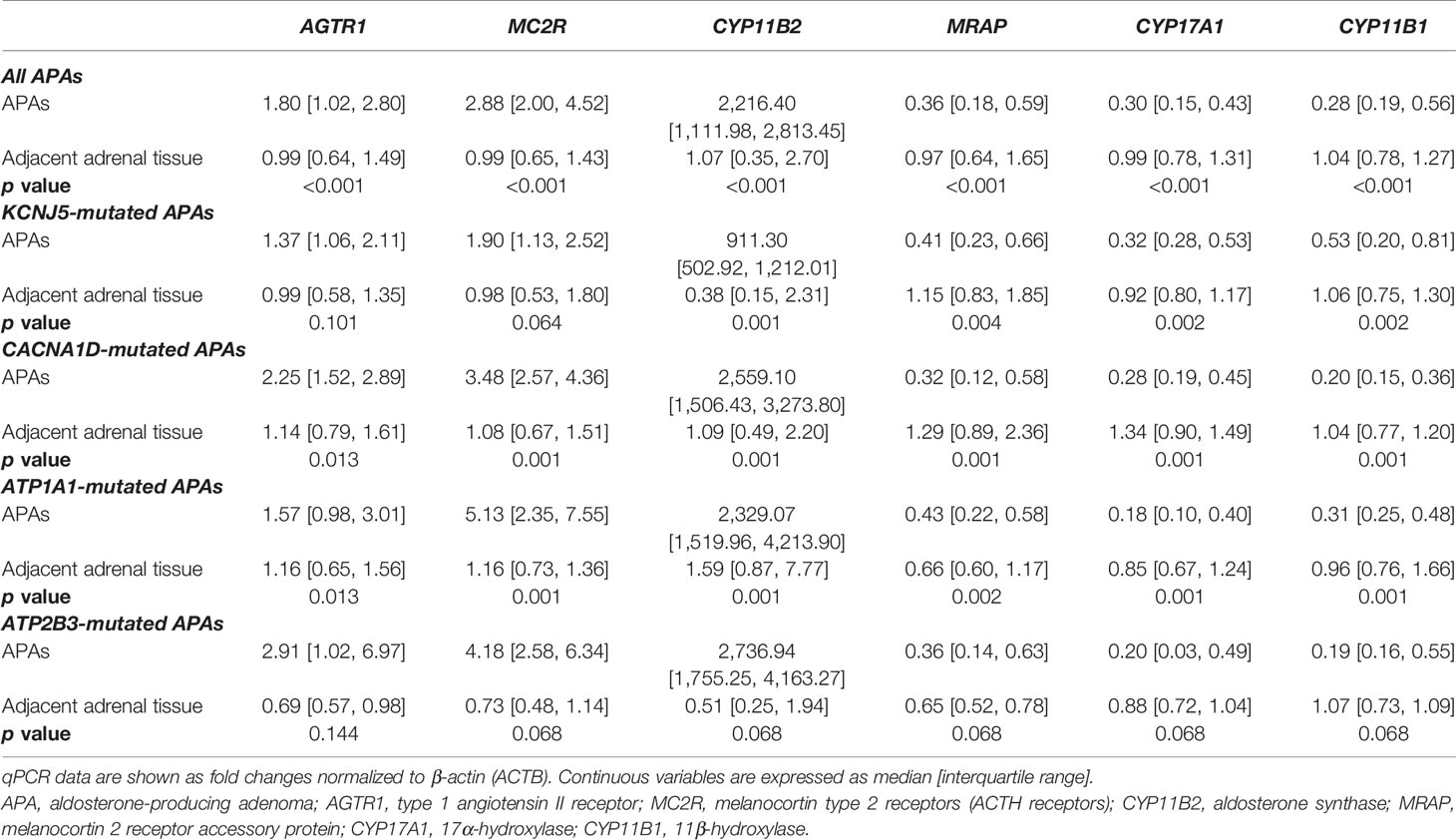
Table 2 Paired comparisons of transcript levels of AGTR1, MC2R, MRAP, and steroidogenic enzymes between APAs and adjacent normal adrenal tissue.
APAs harboring KCNJ5 mutations displayed lower MC2R and CYP11B2 mRNA expressions compared to other APAs (Figures 1B, C), while AGTR1 and MRAP transcript levels were relatively similar between mutation groups (Figures 1A, D).
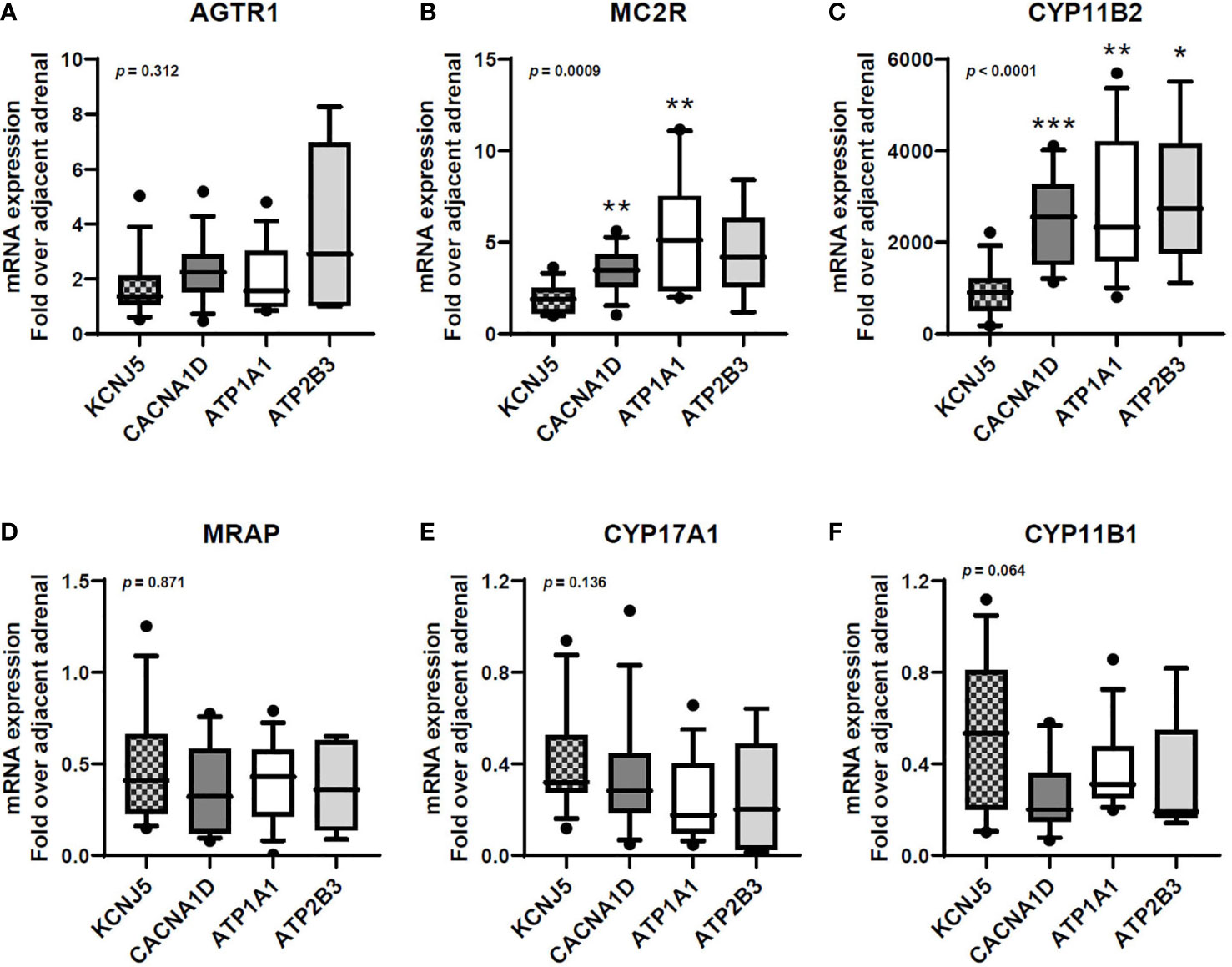
Figure 1 Transcript expression of AGTR1 (A), MC2R (B), CYP11B2 (C), MRAP (D), CYP17A1 (E), and CYP11B1 (F) in aldosterone-producing adenomas with different aldosterone-driver somatic mutations. qPCR data are shown as the fold changes normalized to β-actin (ACTB). AGTR1, type 1 angiotensin II receptor; MC2R, melanocortin type 2 receptors (ACTH receptors); CYP11B2, aldosterone synthase; MRAP, melanocortin 2 receptor accessory protein; CYP17A1, 17α-hydroxylase; CYP11B1, 11β-hydroxylase. Comparisons between groups were done using the Kruskal-Wallis test, followed by the Dwass-Steel-Critchlow-Fligner test. *p < 0.05, **p < 0.01, ***p < 0.001, compared with KCNJ5-mutated APAs. The boxes contain the 25th and 75th percentiles, the whiskers mark the 10th and 90th percentiles, and the horizontal line within the box indicates the median, and the ⚫ represent outliers.
Correlations Between Aldosterone Regulators and Steroidogenic Enzymes in Aldosterone-Producing Adenomas
Overall, APA CYP11B2 expression correlated positively with MC2R (r = 0.77, p < 0.0001) and AGTR1 (r = 0.52, p = 0.0002, Figure 2), and inversely with CYP17A1 and CYP11B1 (r = −0.3, p < 0.05 for both). The strongest correlations between CYP11B2 and both MC2R and AGTR1 were observed in ATP1A1-mutated APAs (r = 0.77, p = 0.001 and r = 0.61, p = 0.021, respectively).
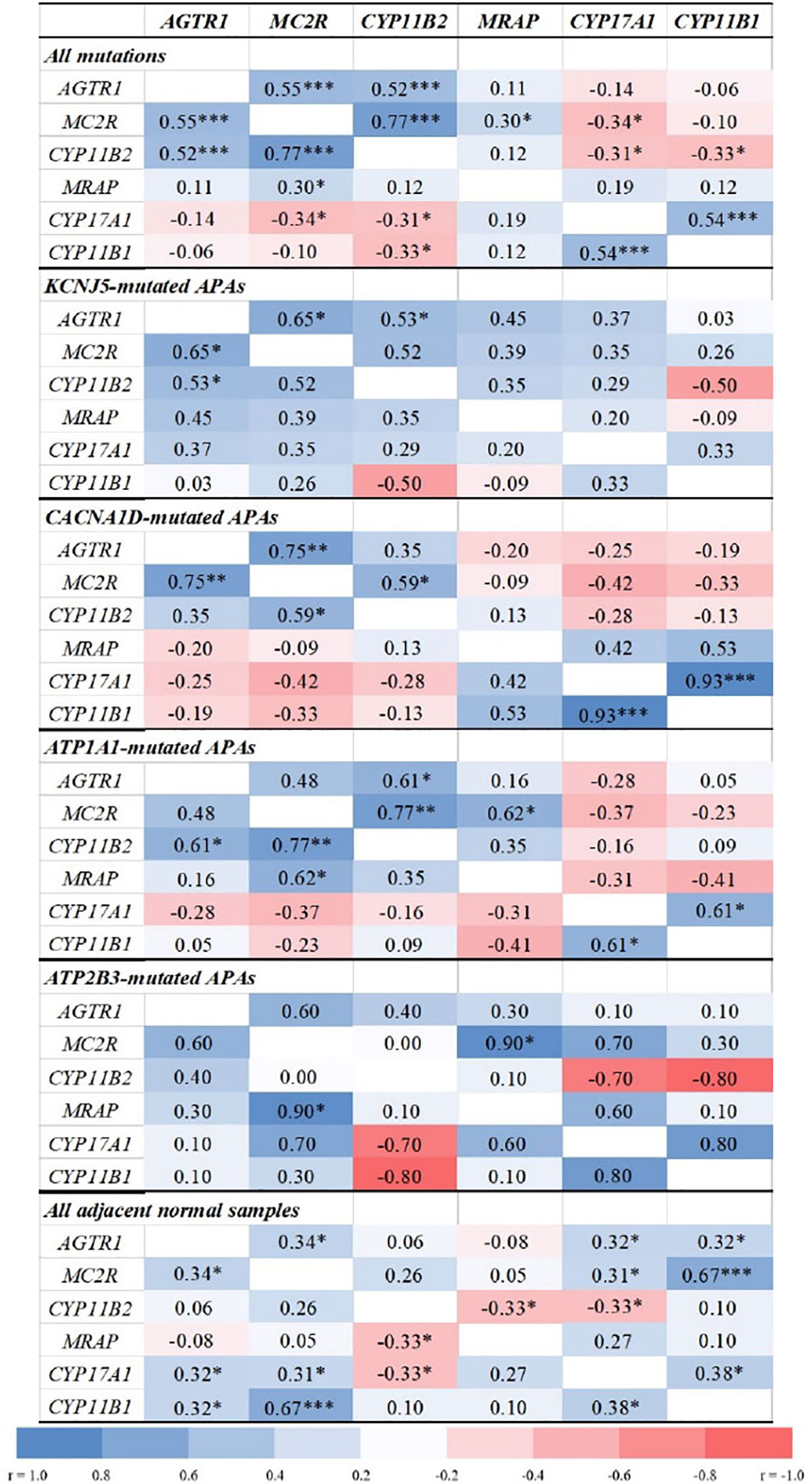
Figure 2 Correlations between transcript levels of AGTR1, MC2R, MRAP, and steroidogenic enzymes in aldosterone-producing adenomas and adjacent normal adrenal tissue. AGTR1, type 1 angiotensin II receptor; MC2R, melanocortin type 2 receptors (ACTH receptors); CYP11B2, aldosterone synthase; MRAP, melanocortin 2 receptor accessory protein; CYP17A1, 17α-hydroxylase; CYP11B1, 11β-hydroxylase. Correlation analyses were done using the Spearman correlation test. *p < 0.05, **p < 0.01, ***p < 0.001.
APAs with CACNA1D and KCNJ5 mutations displayed tight positive correlations between MC2R and AGTR1 transcripts (r = 0.75, p = 0.002 and r = 0.65, p = 0.012, respectively), while no significant correlations were found in APAs with ATPase mutations. Conversely, MC2R and MRAP expressions correlated positively only in ATP1A1- and ATP2B3-mutated APAs (r = 0.62, p = 0.018 and r = 0.90, p = 0.037, respectively).
Discussion
In this study, we delineate differential gene expression of the primary aldosterone regulatory receptors in APAs with different underlying mutations. We found that APAs displayed higher mRNA expression of both MC2R and AGTR1 than adjacent normal adrenal tissue. In addition, we show that the expression patterns of MC2R and AGTR1, and their associations with CYP11B2 transcripts differ between APAs with various underlying aldosterone-driver somatic mutations.
Under physiological conditions, angiotensin II induces Gi-mediated cell membrane depolarization and increases intracellular calcium signaling, thereby stimulating acute steroid production as a result of increased steroidogenic acute regulatory protein (StAR) protein expression (31). Furthermore, this elevation in intracellular calcium activates a cascade of signaling events that lead to increased CYP11B2 transcription and aldosterone secretion from ZG cells (30, 37). Although PA is theoretically renin-independent, aldosterone excess may also result from aberrant receptor expression within APAs and/or hypersensitivity to physiological stimuli. A variety of autocrine and paracrine regulatory factors (38) can activate ectopic or aberrant receptors, which may govern aldosterone secretion independently from the suppressed renin-angiotensin system (29, 39). Indeed, mRNA expressions of AGTRI and MC2R were previously reported to be higher in APA tissues compared to healthy adult adrenals (27, 29, 40). The effects of posture, angiotensin II infusion, and angiotensin converting enzyme inhibitors have been shown to differ in APA when compared to BHA, although results have been variable (7, 41–43). In our study, AGTR1 transcript levels tended to be higher in APAs as compared to adjacent normal adrenal tissue. Tunny and colleagues found that angiotensin II-unresponsive APAs were more common in women, while those responsive to angiotensin II were more prevalent in men (41). Indeed, we herein found that KCNJ5-mutated APAs, which are most prevalent in women of all races (44–46), expressed AGTR1 transcript levels comparable to those found in the corresponding normal adrenal tissue.
In contrast with angiotensin II and potassium, ACTH stimulates aldosterone secretion acutely but transiently (31, 47). Aldosterone production follows a circadian rhythm that parallels that of ACTH both in normal individuals, as well as in patients with PA (48, 49). In patients with aldosterone-secreting tumors, plasma aldosterone concentration starts to fall around mid-morning, as ACTH levels decrease, in spite of upright posture (39). The relative impact of ACTH on aldosterone production from APA vs. BHA and normal ZG cells remains incompletely understood. Small studies suggest that APAs might be more sensitive to ACTH stimulation and suppression than BHA and normal adrenals (49). Asian studies (50–52) indicated that the response of aldosterone to cosyntropin stimulation, with or without a priori overnight suppression with 1mg dexamethasone, is higher in patients with APA than in those with BHA. Nevertheless, AVS data have shown that aldosterone lateralization might be apparent only prior to or exclusively after cosyntropin stimulation (8, 9, 53). Washout of a baseline aldosterone gradient between the two adrenal glands following cosyntropin stimulation indicates a relatively higher response from either normal ZG cells or from asymmetrical BHA. Conversely, amplification of a baseline aldosterone lateralization points towards a highly ACTH-sensitive APA.
The impact of ACTH on aldosterone secretion is dependent on the expression of MC2R in CYP11B2-positive cells (31). As ACTH is the primary regulator of cortisol synthesis, MC2R is abundantly expressed in the zona fasciculata (ZF) cells (54). Previous studies have shown that APAs have higher MC2R transcript levels than normal adrenal tissue, non-functional adrenal adenomas, or carcinomas (27, 29, 40, 55–57), although the levels reported have been somewhat variable. Our study is the first to quantify the expression of MC2R and AGTR1 transcript levels in APAs confirmed by CYP11B2 IHC. Non-functional cortical adenomas can be present in patients with PA, and these tumors display lower MC2R expression than APAs or normal cortical tissue (40, 55); this might explain previously reported variability of MC2R expression in presumed APAs that were not functionally confirmed by examining CYP11B2 expression. Another cause of variability relates to the APA genotype. While all APAs had higher transcript levels of MC2R compared to adjacent normal adrenal tissue, KCNJ5-mutated APAs displayed lower MC2R transcripts than other APAs. Considering that BHA are often caused by multiple APCCs that harbor CACNA1D mutations (58), it is not surprising that East Asians studies that assessed the aldosterone response to ACTH stimulation or suppression in patients with APA vs. BHA found considerable overlap. As confirmed by several cohorts, KCNJ5 mutations account for the vast majority of APAs in East Asian populations (45, 59). In line with these findings, we have previously reported that aldosterone lateralization during AVS often dampens following cosyntropin stimulation in patients with APAs harboring KCNJ5 mutations, while the opposite happens in patients with ATPase mutations (26).
ACTH binds to its MC2R, and induces the activation of adenylate cyclase and the generation of intracellular cAMP (54, 60). Subsequently, the increased cAMP activates protein kinase A, which augments CREB phosphorylation and CYP11B2 transcription (30, 31). MRAP, a small transmembrane protein, is an essential factor in regulating trafficking and functional expression of the MC2R in the adrenal gland (61, 62). Both MC2R and MRAP are known to be highly expressed in the undifferentiated zone as well as the ZF cells (63). Furthermore, the acute steroidogenic responses to ACTH stimulation depend on adequate amounts of MC2R and MRAP on the plasma membrane surface (61). In this study, MC2R transcripts correlated positively with MRAP expression only in ATPase-mutated APAs. These findings further support the high responsivity of ATPase-mutated APAs to cosyntropin observed during AVS (26), in contrast with KCNJ5 or CACNA1D-mutated APAs. Conversely, MC2R transcript levels correlated positively with those of AGTR1 in APAs harboring KCNJ5 or CACNA1D mutations, but not in those with ATPase mutations. Together these results highlight molecular differences between APAs, which go beyond those illustrated by recent histopathological studies (23, 24). Additional downstream molecular mechanisms might be impacted differently by various aldosterone-driver mutations and deserve further investigation. For example, in vitro studies suggest that angiotensin II upregulates NR4A1, NR4A2, and NR4A3 gene expression (64, 65), and that NR4A2 and NR4A3 are upregulated in cell models overexpressing KCNJ5 mutations (66, 67). Other transcriptome and methylome variations have been shown between APA with and without KCNJ5 mutations (68). In addition, differences in the expression of inhibitory regulators, such as dopamine receptors (69, 70) across APAs with various aldosterone-driver mutations deserve further investigation.
In summary, we found that ACTH and angiotensin II receptors are expressed in functionally confirmed APAs harboring the four most common aldosterone-driver somatic mutations. Additionally, we show that these key aldosterone regulatory receptors display several differences in expression across APAs with distinct underlying mutations. Specifically, KCNJ5-mutated APAs express lower mRNA transcript levels of both MC2R and CYP11B2 as compared to other APAs, and they display no association between MC2R and MRAP expression, possibly explaining their relatively modest response to cosyntropin stimulation observed during AVS. Conversely, ATP1A1-mutated APAs showed robust positive correlation of MC2R with both MRAP and CYP11B2 expression, supporting their ACTH-sensitivity. The relatively small number of tissue samples and individual variability from APAs with distinct somatic mutation are limitation of our study. Another important limitation is the lack of protein translation assessment, and thus conclusions regarding protein function remain limited. Such studies will be critical once highly selective human MC2R antibodies become available. Nevertheless, this initial study provides insight into the possible actions of ACTH and angiotensin II in APA with various aldosterone-driver mutations.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics Statement
This research was reviewed and approved by the Institutional Review Boards at the University of Michigan (HUM00106809, HUM00024461, HUM00083056). Written informed consent was obtained from all patients who underwent adrenalectomy prior to February, 2011. A waiver of consent was granted for the use of archival specimens (HUM00083056).
Author Contributions
JSL, WER, and AFT conceived and designed the study. JSL and SP performed the experiment. JSL and AFT analyzed the data. JSL, JR, WR, and ADT interpreted the data. JL and AFT drafted and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
AFT was supported by grants 1K08DK109116 from the NIDDK and DDCF_2019087 from the Doris Duke Charitable Foundation. WER was supported by grant R01DK106618 from the NIDDK.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SM declared a past co-authorship with one of the authors WR to the handling editor.
Acknowledgments
We thank Ms. Sarah Brand and former University of Michigan Adrenal Research Team members for assistance with regulatory processes and patient consent; Dr. Tom Giordano and Ms. Michelle Vinco for assistance with case identification and slide preparation; Ms. Amy R. Blinder for technical assistance; and all study participants.
References
1. Douma S, Petidis K, Doumas M, Papaefthimiou P, Triantafyllou A, Kartali N, et al. Prevalence of primary hyperaldosteronism in resistant hypertension: a retrospective observational study. Lancet (2008) 371:1921–6. doi: 10.1016/S0140-6736(08)60834-X
2. Monticone S, D’Ascenzo F, Moretti C, Williams TA, Veglio F, Gaita F, et al. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: a systematic review and meta-analysis. Lancet Diabetes Endocrinol (2018) 6:41–50. doi: 10.1016/S2213-8587(17)30319-4
3. Savard S, Amar L, Plouin PF, Steichen O. Cardiovascular complications associated with primary aldosteronism: a controlled cross-sectional study. Hypertension (2013) 62:331–6. doi: 10.1161/HYPERTENSIONAHA.113.01060
4. Mulatero P, Monticone S, Bertello C, Viola A, Tizzani D, Iannaccone A, et al. Long-term cardio- and cerebrovascular events in patients with primary aldosteronism. J Clin Endocrinol Metab (2013) 98:4826–33. doi: 10.1210/jc.2013-2805
5. Lim JS, Park S, Park SI, Oh YT, Choi E, Kim JY, et al. Cardiac Dysfunction in Association with Increased Inflammatory Markers in Primary Aldosteronism. Endocrinol Metab (Seoul) (2016) 31:567–76. doi: 10.3803/EnM.2016.31.4.567
6. Zia AA, Kamalov G, Newman KP, McGee JE, Bhattacharya SK, Ahokas RA, et al. From aldosteronism to oxidative stress: the role of excessive intracellular calcium accumulation. Hypertens Res (2010) 33:1091–101. doi: 10.1038/hr.2010.159
7. Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, et al. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2016) 101:1889–916. doi: 10.1210/jc.2015-4061
8. El Ghorayeb N, Mazzuco TL, Bourdeau I, Mailhot JP, Zhu PS, Therasse E, et al. Basal and Post-ACTH Aldosterone and Its Ratios Are Useful During Adrenal Vein Sampling in Primary Aldosteronism. J Clin Endocrinol Metab (2016) 101:1826–35. doi: 10.1210/jc.2015-3915
9. Rossi GP, Pitter G, Bernante P, Motta R, Feltrin G, Miotto D. Adrenal vein sampling for primary aldosteronism: the assessment of selectivity and lateralization of aldosterone excess baseline and after adrenocorticotropic hormone (ACTH) stimulation. J Hypertens (2008) 26:989–97. doi: 10.1097/HJH.0b013e3282f9e66a
10. Seccia TM, Miotto D, De Toni R, Pitter G, Mantero F, Pessina AC, et al. Adrenocorticotropic hormone stimulation during adrenal vein sampling for identifying surgically curable subtypes of primary aldosteronism: comparison of 3 different protocols. Hypertension (2009) 53:761–6. doi: 10.1161/HYPERTENSIONAHA.108.128553
11. Young WF, Stanson AW, Thompson GB, Grant CS, Farley DR, van Heerden JA. Role for adrenal venous sampling in primary aldosteronism. Surgery (2004) 136:1227–35. doi: 10.1016/j.surg.2004.06.051
12. Wolley MJ, Ahmed AH, Gordon RD, Stowasser M. Does ACTH improve the diagnostic performance of adrenal vein sampling for subtyping primary aldosteronism? Clin Endocrinol (Oxf) (2016) 85:703–9. doi: 10.1111/cen.13110
13. Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science (2011) 331:768–72. doi: 10.1126/science.1198785
14. Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet (2013) 45:1055–60. doi: 10.1038/ng.2716
15. Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet (2013) 45:440–4, 4e1-2. doi: 10.1038/ng.2550
16. Scholl UI, Goh G, Stolting G, de Oliveira RC, Choi M, Overton JD, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet (2013) 45:1050–4. doi: 10.1038/ng.2695
17. Scholl UI, Stolting G, Nelson-Williams C, Vichot AA, Choi M, Loring E, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife (2015) 4:e06315. doi: 10.7554/eLife.06315
18. Wu VC, Wang SM, Chueh SJ, Yang SY, Huang KH, Lin YH, et al. The prevalence of CTNNB1 mutations in primary aldosteronism and consequences for clinical outcomes. Sci Rep (2017) 7:39121. doi: 10.1038/srep39121
19. Scholl UI, Stolting G, Schewe J, Thiel A, Tan H, Nelson-Williams C, et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet (2018) 50:349–54. doi: 10.1038/s41588-018-0048-5
20. Fernandes-Rosa FL, Daniil G, Orozco IJ, Goppner C, El Zein R, Jain V, et al. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat Genet (2018) 50:355–61. doi: 10.1038/s41588-018-0053-8
21. Nanba K, Omata K, Else T, Beck PCC, Nanba AT, Turcu AF, et al. Targeted Molecular Characterization of Aldosterone-Producing Adenomas in White Americans. J Clin Endocrinol Metab (2018) 103:3869–76. doi: 10.1210/jc.2018-01004
22. Nanba K, Omata K, Gomez-Sanchez CE, Stratakis CA, Demidowich AP, Suzuki M, et al. Genetic Characteristics of Aldosterone-Producing Adenomas in Blacks. Hypertension (2019) 73:885–92. doi: 10.1161/HYPERTENSIONAHA.118.12070
23. De Sousa K, Boulkroun S, Baron S, Nanba K, Wack M, Rainey WE, et al. Genetic, Cellular, and Molecular Heterogeneity in Adrenals With Aldosterone-Producing Adenoma. Hypertension (2020) 75:1034–44. doi: 10.1161/HYPERTENSIONAHA.119.14177
24. Ono Y, Yamazaki Y, Omata K, Else T, Tomlins SA, Rhayem Y, et al. Histological Characterization of Aldosterone-producing Adrenocortical Adenomas with Different Somatic Mutations. J Clin Endocrinol Metab (2020) 105(3):e282-9. doi: 10.1210/clinem/dgz235
25. Williams TA, Peitzsch M, Dietz AS, Dekkers T, Bidlingmaier M, Riester A, et al. Genotype-Specific Steroid Profiles Associated With Aldosterone-Producing Adenomas. Hypertension (2016) 67:139–45. doi: 10.1161/HYPERTENSIONAHA.115.06186
26. Wannachalee T, Zhao L, Nanba K, Nanba AT, Shields JJ, Rainey WE, et al. Three Discrete Patterns of Primary Aldosteronism Lateralization in Response to Cosyntropin During Adrenal Vein Sampling. J Clin Endocrinol Metab (2019) 104:5867–76. doi: 10.1210/jc.2019-01182
27. Ye P, Mariniello B, Mantero F, Shibata H, Rainey WE. G-protein-coupled receptors in aldosterone-producing adenomas: a potential cause of hyperaldosteronism. J Endocrinol (2007) 195:39–48. doi: 10.1677/JOE-07-0037
28. Perraudin V, Delarue C, Lefebvre H, Do Rego JL, Vaudry H, Kuhn JM. Evidence for a role of vasopressin in the control of aldosterone secretion in primary aldosteronism: in vitro and in vivo studies. J Clin Endocrinol Metab (2006) 91:1566–72. doi: 10.1210/jc.2005-1453
29. Zwermann O, Suttmann Y, Bidlingmaier M, Beuschlein F, Reincke M. Screening for membrane hormone receptor expression in primary aldosteronism. Eur J Endocrinol (2009) 160:443–51. doi: 10.1530/EJE-08-0711
30. Hattangady NG, Olala LO, Bollag WB, Rainey WE. Acute and chronic regulation of aldosterone production. Mol Cell Endocrinol (2012) 350:151–62. doi: 10.1016/j.mce.2011.07.034
31. El Ghorayeb N, Bourdeau I, Lacroix A. Role of ACTH and Other Hormones in the Regulation of Aldosterone Production in Primary Aldosteronism. Front Endocrinol (Lausanne) (2016) 7:72. doi: 10.3389/fendo.2016.00072
32. Oki K, Plonczynski MW, Lam ML, Gomez-Sanchez EP, Gomez-Sanchez CE. The potassium channel, Kir3.4 participates in angiotensin II-stimulated aldosterone production by a human adrenocortical cell line. Endocrinology (2012) 153:4328–35. doi: 10.1210/en.2012-1241
33. Reimer EN, Walenda G, Seidel E, Scholl UI. CACNA1H(M1549V) Mutant Calcium Channel Causes Autonomous Aldosterone Production in HAC15 Cells and Is Inhibited by Mibefradil. Endocrinology (2016) 157:3016–22. doi: 10.1210/en.2016-1170
34. Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, et al. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc Natl Acad Sci U.S.A. (2015) 112:E4591–9. doi: 10.1073/pnas.1505529112
35. Bassett MH, Mayhew B, Rehman K, White PC, Mantero F, Arnaldi G, et al. Expression profiles for steroidogenic enzymes in adrenocortical disease. J Clin Endocrinol Metab (2005) 90:5446–55. doi: 10.1210/jc.2005-0836
36. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (2001) 25:402–8. doi: 10.1006/meth.2001.1262
37. Gambaryan S, Butt E, Tas P, Smolenski A, Allolio B, Walter U. Regulation of aldosterone production from zona glomerulosa cells by ANG II and cAMP: evidence for PKA-independent activation of CaMK by cAMP. Am J Physiol Endocrinol Metab (2006) 290:E423–33. doi: 10.1152/ajpendo.00128.2005
38. Lefebvre H, Prevost G, Louiset E. Autocrine/paracrine regulatory mechanisms in adrenocortical neoplasms responsible for primary adrenal hypercorticism. Eur J Endocrinol (2013) 169:R115–38. doi: 10.1530/EJE-13-0308
39. St-Jean M, Ghorayeb NE, Bourdeau I, Lacroix A. Aberrant G-protein coupled hormone receptor in adrenal diseases. Best Pract Res Clin Endocrinol Metab (2018) 32:165–87. doi: 10.1016/j.beem.2018.01.003
40. Schubert B, Fassnacht M, Beuschlein F, Zenkert S, Allolio B, Reincke M. Angiotensin II type 1 receptor and ACTH receptor expression in human adrenocortical neoplasms. Clin Endocrinol (Oxf) (2001) 54:627–32. doi: 10.1046/j.1365-2265.2001.01253.x
41. Tunny TJ, Klemm SA, Stowasser M, Gordon RD. Angiotensin-responsive aldosterone-producing adenomas: postoperative disappearance of aldosterone response to angiotensin. Clin Exp Pharmacol Physiol (1993) 20:306–9. doi: 10.1111/j.1440-1681.1993.tb01690.x
42. Mantero F, Fallo F, Opocher G, Armanini D, Boscaro M, Scaroni C. Effect of angiotensin II and converting enzyme inhibitor (captopril) on blood pressure, plasma renin activity and aldosterone in primary aldosteronism. Clin Sci (Lond) (1981) 61 Suppl 7:289s–93s. doi: 10.1042/cs061289s
43. Stowasser M, Klemm SA, Tunny TJ, Gordon RD. Plasma aldosterone response to ACTH in subtypes of primary aldosteronism. Clin Exp Pharmacol Physiol (1995) 22:460–2. doi: 10.1111/j.1440-1681.1995.tb02044.x
44. Williams TA, Monticone S, Mulatero P. KCNJ5 mutations are the most frequent genetic alteration in primary aldosteronism. Hypertension (2015) 65:507–9. doi: 10.1161/HYPERTENSIONAHA.114.04636
45. Hong AR, Kim JH, Song YS, Lee KE, Seo SH, Seong MW, et al. Genetics of Aldosterone-Producing Adenoma in Korean Patients. PloS One (2016) 11:e0147590. doi: 10.1371/journal.pone.0147590
46. Fernandes-Rosa FL, Williams TA, Riester A, Steichen O, Beuschlein F, Boulkroun S, et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension (2014) 64:354–61. doi: 10.1161/HYPERTENSIONAHA.114.03419
47. Funder JW. The Potential of ACTH in the Genesis of Primary Aldosteronism. Front Endocrinol (Lausanne) (2016) 7:40. doi: 10.3389/fendo.2016.00040
48. Schambelan M, Brust NL, Chang BC, Slater KL, Biglieri EG. Circadian rhythm and effect of posture on plasma aldosterone concentration in primary aldosteronism. J Clin Endocrinol Metab (1976) 43:115–31. doi: 10.1210/jcem-43-1-115
49. Sonoyama T, Sone M, Tamura N, Honda K, Taura D, Kojima K, et al. Role of endogenous ACTH on circadian aldosterone rhythm in patients with primary aldosteronism. Endocr Connect (2014) 3:173–9. doi: 10.1530/EC-14-0086
50. Sonoyama T, Sone M, Miyashita K, Tamura N, Yamahara K, Park K, et al. Significance of adrenocorticotropin stimulation test in the diagnosis of an aldosterone-producing adenoma. J Clin Endocrinol Metab (2011) 96:2771–8. doi: 10.1210/jc.2011-0573
51. Inoue K, Omura M, Sugisawa C, Tsurutani Y, Saito J, Nishikawa T. Clinical Utility of the Adrenocorticotropin Stimulation Test with/without Dexamethasone Suppression for Definitive and Subtype Diagnosis of Primary Aldosteronism. Int J Mol Sci (2017) 18(5):948. doi: 10.3390/ijms18050948
52. Jiang Y, Zhang C, Wang W, Su T, Zhou W, Jiang L, et al. Diagnostic value of ACTH stimulation test in determining the subtypes of primary aldosteronism. J Clin Endocrinol Metab (2015) 100:1837–44. doi: 10.1210/jc.2014-3551
53. Nanba AT, Nanba K, Byrd JB, Shields JJ, Giordano TJ, Miller BS, et al. Discordance between imaging and immunohistochemistry in unilateral primary aldosteronism. Clin Endocrinol (2017) 87(6):665–72. doi: 10.1111/cen.13442
54. Chan LF, Metherell LA, Clark AJ. Effects of melanocortins on adrenal gland physiology. Eur J Pharmacol (2011) 660:171–80. doi: 10.1016/j.ejphar.2010.11.041
55. Reincke M, Beuschlein F, Latronico AC, Arlt W, Chrousos GP, Allolio B. Expression of adrenocorticotrophic hormone receptor mRNA in human adrenocortical neoplasms: correlation with P450scc expression. Clin Endocrinol (1997) 46:619–26. doi: 10.1046/j.1365-2265.1997.1991009.x
56. Arnaldi G, Mancini V, Costantini C, Giovagnetti M, Petrelli M, Masini A, et al. ACTH receptor mRNA in human adrenocortical tumors: overexpression in aldosteronomas. Endocr Res (1998) 24:845–9. doi: 10.3109/07435809809032695
57. Murakami M, Yoshimoto T, Nakabayashi K, Tsuchiya K, Minami I, Bouchi R, et al. Integration of transcriptome and methylome analysis of aldosterone-producing adenomas. Eur J Endocrinol/European Fed Endocr Societies (2015) 173:185–95. doi: 10.1530/EJE-15-0148
58. Omata K, Satoh F, Morimoto R, Ito S, Yamazaki Y, Nakamura Y, et al. Cellular and Genetic Causes of Idiopathic Hyperaldosteronism. Hypertension (2018) 72:874–80. doi: 10.1161/HYPERTENSIONAHA.118.11086
59. Taguchi R, Yamada M, Nakajima Y, Satoh T, Hashimoto K, Shibusawa N, et al. Expression and mutations of KCNJ5 mRNA in Japanese patients with aldosterone-producing adenomas. J Clin Endocrinol Metab (2012) 97:1311–9. doi: 10.1210/jc.2011-2885
60. de Joussineau C, Sahut-Barnola I, Levy I, Saloustros E, Val P, Stratakis CA, et al. The cAMP pathway and the control of adrenocortical development and growth. Mol Cell Endocrinol (2012) 351:28–36. doi: 10.1016/j.mce.2011.10.006
61. Novoselova TV, Jackson D, Campbell DC, Clark AJ, Chan LF. Melanocortin receptor accessory proteins in adrenal gland physiology and beyond. J Endocrinol (2013) 217:R1–11. doi: 10.1530/JOE-12-0501
62. Cooray SN, Almiro Do Vale I, Leung KY, Webb TR, Chapple JP, Egertova M, et al. The melanocortin 2 receptor accessory protein exists as a homodimer and is essential for the function of the melanocortin 2 receptor in the mouse y1 cell line. Endocrinology (2008) 149:1935–41. doi: 10.1210/en.2007-1463
63. Gorrigan RJ, Guasti L, King P, Clark AJ, Chan LF. Localisation of the melanocortin-2-receptor and its accessory proteins in the developing and adult adrenal gland. J Mol Endocrinol (2011) 46:227–32. doi: 10.1530/JME-11-0011
64. Romero DG, Plonczynski M, Vergara GR, Gomez-Sanchez EP, Gomez-Sanchez CE. Angiotensin II early regulated genes in H295R human adrenocortical cells. Physiol Genomics (2004) 19:106–16. doi: 10.1152/physiolgenomics.00097.2004
65. Bassett MH, Suzuki T, Sasano H, White PC, Rainey WE. The orphan nuclear receptors NURR1 and NGFIB regulate adrenal aldosterone production. Mol Endocrinol (2004) 18:279–90. doi: 10.1210/me.2003-0005
66. Monticone S, Hattangady NG, Nishimoto K, Mantero F, Rubin B, Cicala MV, et al. Effect of KCNJ5 mutations on gene expression in aldosterone-producing adenomas and adrenocortical cells. J Clin Endocrinol Metab (2012) 97:E1567–72. doi: 10.1210/jc.2011-3132
67. Hattangady NG, Karashima S, Yuan L, Ponce-Balbuena D, Jalife J, Gomez-Sanchez CE, et al. Mutated KCNJ5 activates the acute and chronic regulatory steps in aldosterone production. J Mol Endocrinol (2016) 57:1–11. doi: 10.1530/JME-15-0324
68. Murakami M, Yoshimoto T, Nakabayashi K, Nakano Y, Fukaishi T, Tsuchiya K, et al. Molecular characteristics of the KCNJ5 mutated aldosterone-producing adenomas. Endocr Relat Cancer (2017) 24:531–41. doi: 10.1530/ERC-17-0117
69. Chang HW, Huang CY, Yang SY, Wu VC, Chu TS, Chen YM, et al. Role of D2 dopamine receptor in adrenal cortical cell proliferation and aldosterone-producing adenoma tumorigenesis. J Mol Endocrinol (2014) 52:87–96. doi: 10.1530/JME-13-0044
Keywords: primary aldosteronism, aldosterone, angiotensin, adrenocorticotropic hormone (ACTH), adrenal, adrenal cortex
Citation: Lim JS, Plaska SW, Rege J, Rainey WE and Turcu AF (2021) Aldosterone-Regulating Receptors and Aldosterone-Driver Somatic Mutations. Front. Endocrinol. 12:644382. doi: 10.3389/fendo.2021.644382
Received: 21 December 2020; Accepted: 08 February 2021;
Published: 16 March 2021.
Edited by:
Vin-Cent Wu, National Taiwan University, TaiwanReviewed by:
Masanori Murakami, Tokyo Medical and Dental University, JapanSilvia Monticone, University of Turin, Italy
Copyright © 2021 Lim, Plaska, Rege, Rainey and Turcu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adina F. Turcu, YXR1cmN1QHVtaWNoLmVkdQ==