 Yazeid Alhaidan1,2,3,4*
Yazeid Alhaidan1,2,3,4* Henrik Thybo Christesen2,5,6
Henrik Thybo Christesen2,5,6 Elena Lundberg7
Elena Lundberg7 Mohammed A. Al Balwi3,4,8
Mohammed A. Al Balwi3,4,8 Klaus Brusgaard1,2,9
Klaus Brusgaard1,2,9- 1Department of Clinical Genetics, Odense University Hospital, Odense, Denmark
- 2Department of Clinical Research, Faculty of Health Sciences, University of Southern Denmark, Odense, Denmark
- 3Department of Medical Genomics Research, King Abdullah International Medical Research Center, Riyadh, Saudi Arabia
- 4King Saud Bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia
- 5Hans Christian Andersen Children’s Hospital, Odense University Hospital, Odense, Denmark
- 6Odense Pancreas Center, Odense, Denmark
- 7Institute of Clinical Science, Pediatrics, Umea University, Umeå, Sweden
- 8Department of Pathology and Laboratory Medicine, King Abdulaziz Medical City, NGHA, Riyadh, Saudi Arabia
- 9Near East University, Nicosia, Cyprus
Aim: Despite the enormous efforts to understand Congenital hyperinsulinism (CHI), up to 50% of the patients are genetically unexplained. We aimed to functionally characterize a novel candidate gene in CHI.
Patient: A 4-month-old boy presented severe hyperinsulinemic hypoglycemia. A routine CHI genetic panel was negative.
Methods: A trio-based whole-exome sequencing (WES) was performed. Gene knockout in the RIN-m cell line was established by CRISPR/Cas9. Gene expression was performed using real-time PCR.
Results: Hyperinsulinemic hypoglycemia with diffuse beta-cell involvement was demonstrated in the patient, who was diazoxide-responsive. By WES, compound heterozygous variants were identified in the adenylyl cyclase 7, ADCY7 gene p.(Asp439Glu) and p.(Gly1045Arg). ADCY7 is calcium-sensitive, expressed in beta-cells and converts ATP to cAMP. The variants located in the cytoplasmic domains C1 and C2 in a highly conserved and functional amino acid region. RIN-m(-/-Adcy7) cells showed a significant increase in insulin secretion reaching 54% at low, and 49% at high glucose concentrations, compared to wild-type. In genetic expression analysis Adcy7 loss of function led to a 34.1-fold to 362.8-fold increase in mRNA levels of the insulin regulator genes Ins1 and Ins2 (p ≤ 0.0002), as well as increased glucose uptake and sensing indicated by higher mRNA levels of Scl2a2 and Gck via upregulation of Pdx1, and Foxa2 leading to the activation of the glucose stimulated-insulin secretion (GSIS) pathway.
Conclusion: This study identified a novel candidate gene, ADCY7, to cause CHI via activation of the GSIS pathway.
Introduction
Congenital hyperinsulinism (CHI) is a rare disease characterized with dysregulated insulin secretion from the pancreatic β-cells pancreatic leads to hyperinsulinemic hypoglycemia (1, 2). CHI is a heterogeneous disorder in terms of both phenotype and genotype. The rareness, severity, and complexity of CHI often lead to delayed and insufficient management with a high risk of subsequent brain damage mental retardation (3).
The estimated incidence of CHI is 1:40,000 in 50,000 newborns, but considerably higher in certain populations, e.g., 1:2,500 newborns in Saudi Arabia (4). The management of CHI is highly dependent on the major histological forms: focal or diffuse. 18F–fluoro-L-dihydroxyphenylalanine (18F–DOPA) PET/CT scan shows excellent performance in the identification of focal CHI, which is curable by limited pancreatic resections (5, 6). Milder forms of CHI response to medical treatment with diazoxide or somatostatin analogues, and an appropriate diet. Severe diffuse CHI with poor medical response, may require subtotal pancreatectomy, however imposing a high risk of subsequent diabetes (7). A medical approach is therefore encouraged, e.g. with long-acting octreotide (8).
Genetically, variants in at least nine genes have been identified as causes of CHI; ATP-Binding Cassette, subfamily C, member 8 (ABCC8) and Potassium Channel, Inwardly rectifying, subfamily J, member 11 (KCNJ11), which constitute the K+ATP channel of the beta cell, accounts for most cases (9, 10). Less frequently, activating variants in the glucokinase gene (GCK) (11), or variants in the glutamate dehydrogenase gene (GLUD1) (12), or inhibitory variants in HADH (shortchain L-3-hydroxyacyl-coenzymeA dehydrogenase) (13), the monocarboxylase transporter (SLC16A1) (14), the uncoupling protein (UCP2) (15), HNF4A (hepatocyte nuclear factor 4 alpha) (16), and HNF1 homeobox alpha (HNF1A) (17). Recently, abnormalities in three new genes have been linked to CHI hexokinase 1 (HK1), phosphoglucomutase 1 (PGM1), and phosphomannomutase 2 (PMM2) genes (18–20). In addition, several syndromes are associated with CHI such as Beckwith–Wiedemann, Sotos, Kabuki, Usher, Timothy, Costello, Trisomy 13, and Mosaic Turner syndrome (21).
Despite the enormous efforts to understand this condition, up to 50% of patients still have negative investigations in the known pathogenic genes. This poses a challenge for both the geneticist and physician, as genotype-phenotype correlations provide a basis for targeted treatment. Here, we report a patient with CHI of unknown genetic cause by routine investigations. Functional studies carried out by generating a knockout cell line identified a novel candidate gene involved in hypersecretion of insulin.
Materials and Methods
Trio-Based Whole-Exome Sequencing
DNA was extracted from whole blood using The Maxwell® RSC Blood DNA Kit from Promega. The proband was forwarded for a clinical CHI gene panel and subsequent a trio-based whole-exome sequencing (WES) approach. DNA samples obtained from our patient and his parents were subjected to exome capture using Roche NimbleGen SeqCap EZ Exome 3.5v Enrichment Kits (Roche, Hvidovre, Denmark) and sequencing was performed on the Illumina HiSeq 1500 platform. Raw reads were processed using the Burrows-Wheeler Alignment tool (BWA-MEM) v. 0.7.12, and the GATK Best Practice pipeline v. 3.3-0 was used for variant calling. A mean read depth of 63.6x was obtained.
Data Analysis
VarSeqTM (Golden Helix, Inc., Bozeman, MT) was used for downstream filtering. All variants were first filtered with a minimum of 10× coverage, non-synonymous, and presented in the exome region or splice sites, which represented 94.4% of the targeted bases. Filtered variants were then processed twice, one for each parameter as previously described (22) The first parameter, which covers the possibility of a compound heterozygous, an autosomal recessive, a multifactorial, or de novo, was set to a population frequency of ≤ 0.01 (GenomAD and ExAC). The second parameter, which covers the dominant inheritance of single nucleotide polymorphisms (SNPs) and small insertions and deletions (INDELs), was set to a frequency of ≤ 0. 000025 for CHI.
To examine known causal genes that have been reported in the literature including related genes and pathways, a gene list was generated consisting of 6,264 genes categorized by disorders, pathways, expression, AmiGO terms, and other into 26 sublists (23). This was performed through an extensive literature review using PubMed, Ovid®, GeneCards®, and the National Center for Biotechnology Information (NCBI).Furthermore, we used gene and protein expression databases such as BioGPS and The Human Protein Atlas, protein interactions and gene network databases such as AmiGO, BioGRID, GIANT, KEGG, and Reactome, knockout mouse databases such as MGI and IMPC. However, filtering against the gene list will not replace the manual screening for all variants called; therefore, we did not consider the results of our gene list alone. Once the raw data were obtained, they were filtered and investigated individually. Variants went through serial steps ending up with a single nucleotide polymorphism variant as a potential explanation. Pathogenicity scores were determined by SIFT, PolyPhen-2, PANTHER, SNPs&GO, and nsSNPAnalyzer.
Cell Culture
RIN-m cell line was purchased from American Type Culture Collection (LGC Standards GmbH, Wesel, Germany) and maintained in RPMI-1640 medium (ATCC) supplemented with 10% fetal bovine serum (FBS: Biological Industries) and 100 U penicillin and 100 µg/ml streptomycin (P4333:SIGMA) in a humidified incubator at 5% CO2 and 37°C.
Knockout of Adcy7 by CRISPR/Cas9
CRISPR/Cas9 guide-RNA targeting exon 5 in rat Adcy7 (5′- TACCCATGGAGATGTGAGCT -3′) was manually designed using https://benchling.com and Alt-R® S.p. Cas9 Nuclease 3NLS were ordered from (Integrated DNA Technologies, BVBA, Belgium). Lipofectamine® RNAiMAX (Thermo Fischer, Naerum, Denmark) was used for transfection in Opti-MEM™ medium according to the manufacturer’s instructions. In short, knockout was performed by mixing 24 µl of 1 µM gRNA with 24 µl of 1 µM Cas9 Nuclease in 352 µl Opti-MEM® Medium for 5 minutes to assemble the RNP complex. RNP complexes were mixed with 19.2 µl Lipofectamine® RNAiMAX in 380 µl Opti-MEM® Medium for 20 minutes to form transfection complexes. This was subsequently transferred to 1.6 ml of the complete medium without an antibiotic-antimycotic solution containing 6.4x105 cells in 6 well plate. Cells were then incubated in a humidified incubator at 5% CO2 and 37°C for 72 hours prior to sequencing. Validated edited cells were then diluted and cultured at low cell concentration to isolate single cell colonies and establish a knockout Adcy7 cell line, RIN-m(-/-Adcy7).
Sanger Sequencing
Gene editing was validated using BigDye Terminator v. 3.1 cycle sequencing kit and an ABI 3730xl capillary sequencer (Thermo Fischer, Naerum, Denmark). Adcy7 primers targeting exon 5 in RIN-m cell line were ordered from Integrated DNA Technologies, BVBA, Belgium (forward 5′- ACAGGGAGGGCACATACTCT -3′, reverse 5′- AAATCCCCAGAGACACGCTC -3′).
Stimulus Insulin Secretion of β-Cells
Both RIN-m(-/-Adcy7) and RIN-mWT were cultured for 24 hours prior to stimulation. This was performed by culturing 5x105 cells in 2 ml of complete medium in 6 well plate. Krebs-Ringer-HEPES buffer (KRHB) was prepared (118 mM NaCl, 5.4 mM KCl, 2.4 mM CaCl2-2H2O, 1.2 mM MgSo4, 1.2 mM KH2Po4, 20 mM HEPES and 0.2% BSA) to be used for washing and stimulation experiments. Cells were washed twice with KRHB containing 1.1 mM glucose before starving for one hour in 1 ml KRHB containing 1.1 mM glucose. After starvation, cells were washed twice with KRHB containing 1.1 mM glucose and incubated in 2 ml KRHB containing 2mM or 10mM glucose to measure insulin secretion at low and high concentrations. Both cells and media were then collected to perform mRNA and insulin measurements. Each experiment was performed in triplicate.
Real-Time PCR
Total RNA was extracted using an RNeasy Mini Kit (Qiagen, Copenhagen). The SuperScript® III first-strand synthesis system (Thermo Fischer, Naerum, Denmark) was used for cDNA synthesis. The StepOne ™ system (Thermo Fischer, Naerum, Denmark) was used for real-time PCR using the TaqMan® PreAmp Master Mix Kit and Expression Assay ID (Adcy7; Rn01538046_m1, Actb; Rn00667869_m1, Ins1; Rn02121433_g1, Ins2; Rn01774648_g1, Prkaca; Rn01432300_g1, Prkacb; Rn01748540_g1, Rapgef4; Rn01514839_m1, Slc2a2; Rn00563565_m1, Pdx1; Rn00755591_m1, Gck; Rn00561265_m1, and Foxa2; Rn01415600_m1) (Thermo Fischer, Naerum, Denmark). All data were analyzed using β-actin (Actb) expression as an endogenous control (Supplemental Data).
Insulin Measurement by ELISA
A rat Insulin ELISA kit (cat n. ERINS) (Thermo Scientific, Naerum, Denmark) was used according to the manufacturer’s instructions. The Victor ™ X5 Multilabel Plate Reader was used to measure the absorbance (PerkinElmer, Skovlunde, Denmark).
Data Analysis
All experiments were performed in triplicate. Changes in cycle threshold (CT) and total insulin concentrations between knockout and wild-type were calculated with an unpaired t-test. Data were presented as the mean and standard error of the means (mean ± SEM).
Ethics
Oral and written consent was obtained from all participants. The study was approved by The Regional Ethical Committee of Southern Denmark (number. S-VF-20040235)
Results
Patient
A non-syndromic, Swedish Caucasian boy without Finnish inheritance was born at term, birth weight 3670 g, birth length 52 cm, with an uneventful neonatal period. He presented at age of 4 months weight 7460g (+1SDS), length 68 cm (0 SDS) with hypoglycemic convulsions and loss of consciousness. At blood glucose 1.6mM, p-insulin was 19mU/L with no ketone body detected, confirming a diagnosis of CHI. An 18F–DOPA PET/CT scan showed diffuse pancreatic involvement.
Initially, he responded to treatment with diazoxide 10 mg/kg/day, divided into 3 doses. His growth followed +1 SDS in weight and 0 SDS in height according to the Swedish growth reference. His psychomotor development was normal. Diazoxide dose was reduced to 2 mg/kg/day from 4 to 7 years of age, but increased to 4mg/kg/day at the latest follow-up age 9 years.
No variants were found by a clinical next-generation sequencing panel of nine genes related to CHI as previously described (5).
Sequencing Analysis
Trio WES analysis revealed compound heterozygous variants in the Adenylyl Cyclase 7 (ADCY7) gene. A paternal variant, NM_001114.4: c.1317C>A, p.(Asp439Glu) and a maternal variant, c.3133G>A, p.(Gly1045Arg), were found. These variants are located in the cytoplasmic domains C1 and C2, respectively, (Figure 1) in a highly conserved amino acid region (24, 25). The ConSurf server for the identification of functional regions in proteins predicted both amino acids to play a functional role and being exposed to an interaction site. The prediction software SIFT, PolyPhen-2, PANTHER, SNPs&GO, and nsSNPAnalyzer predicted both variants to be deleterious. ADCY7 is reported to be expressed in pancreatic tissue with other isoforms ADCY1, ADCY3, ADCY5, and ADCY9 (26). Adenylyl cyclases (ACs) are a family of ten different mammalian isoforms that converts ATP to cyclic AMP. ADCY7 is a membrane-bound member of this family, reported to be inhibitable by calcium.
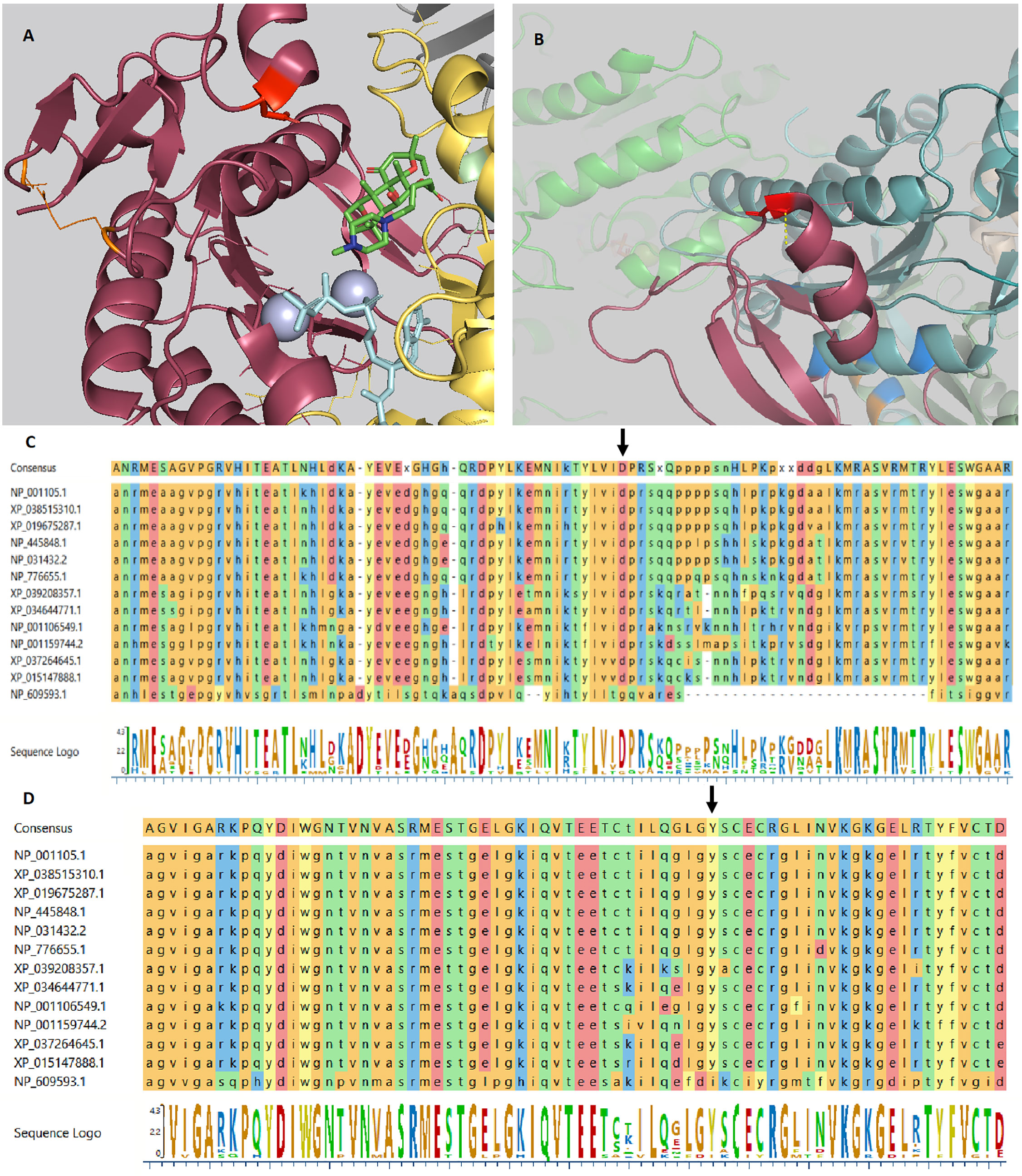
Figure 1 ADCY7 variants crystal structure. (A) The crystal structure of Gαs.VC1.IIC2 domain in complex with the substrate analog TNP-ATP based on pdb 2GVZ; ADCY5. The Asp439 residue is depicted as a red tooth stick. The adenylate cyclase inhibitor foskolin shown as green tooth sticks and the TNP-ATP substrate analog as aquamarine tooth sticks. The two manganese ions in grey. Two residues are depicted in orange are mutated in GUCY2D causing Leber congenital amaurosis 1. VC1 in salmon, IIC2 in yellow, Gαs in grey. Residues involved in VC1.IIC2 interaction shown as tooth sticks. (B) The crystal structure of the IIC2 domain based on pdb 6R4P; ADCY9. Gly1045 shown in red with a polar interaction indicated by a yellow dashed line to the backbone of residue Gly1043. Gly1043 allow a sharp turn to the loop connecting α-helix 15 and β-strand 19. Replacing Gly1045 with arginine can be predicted to prevent this turn. Residues depicted in blue are mutated in GUCY2D causing Leber congenital amaurosis 1. In the background the VC1 domain (green) can be seen. (C) Alignment of ADCY7 sequences displaying conservation from homo sapiens at the top to Drosophila melanogaster at the bottom. Asp439 indicated by an arrow. (D) Alignment of ADCY7 sequences displaying conservation from homo sapiens at the top to Drosophila melanogaster at the bottom displaying conservation of Gly1045 indicated by an arrow.
Protein Sequence Analysis
The three-dimensional protein structure of candidate genes was analyzed using PyMOL v.1.7.4 (Schrödinger, New York, USA). (PDB-ID: 2GVZ).
Cell Line Establishment
An Adcy7 knockout cell line, RIN-m(-/-Adcy7), was established and validated, (Figure 2A). A one base insertion located in exon 5 created a frameshift leading to generation of a premature termination codon located 11 codons downstream (Figure 2B). In addition, the Adcy7 mRNA level determined by real-time PCR confirmed the loss of the expression as a result of degradation via nonsense-mediated mRNA decay (Figure 2C).
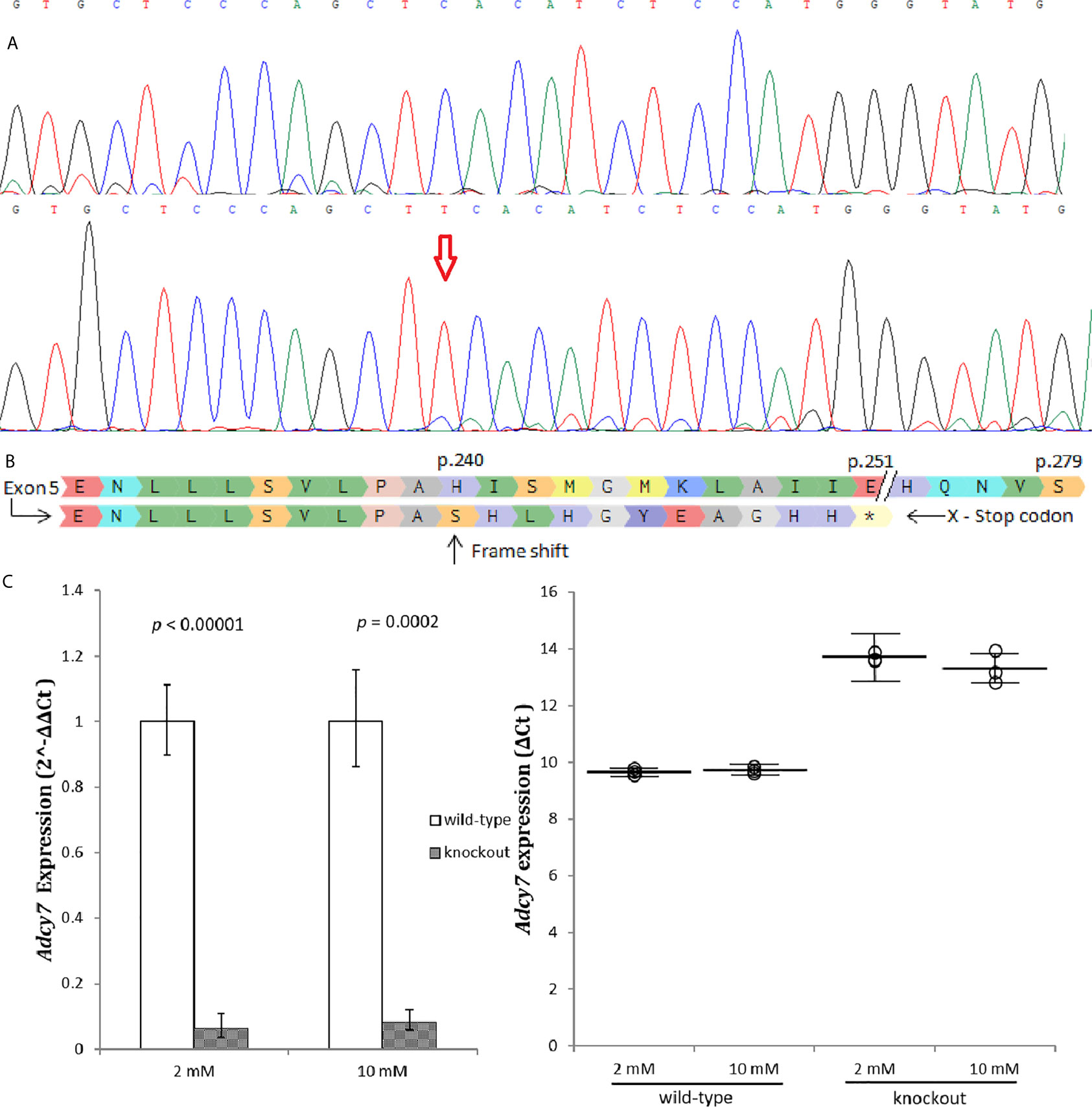
Figure 2 Adcy7 knockout establishment. (A) Sanger sequencing wild-type RIN-m(Adcy7/Adcy7) (Top) compared to the mutant cell line (bottom) confirmed an insertion creating a frameshift. (B) Amino acid sequencing with a reference (top) shows indel creating a frameshift causing a premature termination codon located in 11 codons downstream. (C) Adcy7 mRNA levels established by quantitative RT-PCR indicating a very low expression level. The CT values were normalized to β-actin to yield ΔCT and 2^-ΔΔCT values presented in dots and bar chart, respectively. Each dot represents an individual sample, whereas the horizontal lines represent the mean and standard error of the mean. ΔCT values were then calculated and presented as 2^-ΔΔCT values (fold change) in a bar chart. Unpaired t-test with delta CT and SE ± values were used for statistic test.
Adcy7 Knockout Shows an Excessive Insulin Expression and Secretion
To investigate whether Adcy7 influences the insulin secretion genes Ins1 and Ins2, cells were incubated in low 2 mM and high 10 mM glucose concentration and mRNA was measured. The levels of both Ins1 and Ins2 mRNA displayed a statistically significant increase in absence of Adcy7 gene expression. The regulator of insulin gene expression, Ins1 was significantly increased by 362.8 fold (p<0.00001) at low glucose media 2 mM, and by 320.9 fold (p<0.00001) at high glucose media 10 mM (Figure 3A). The secretion regulator Ins2 was also dramatically increased by 43.6 fold (p=0.00001) at low glucose concentration and by 34.1 fold (p=0.00002) at high glucose concentration for RIN-m(-/-Adcy7) cells compared to the wild-type (Figure 3B).
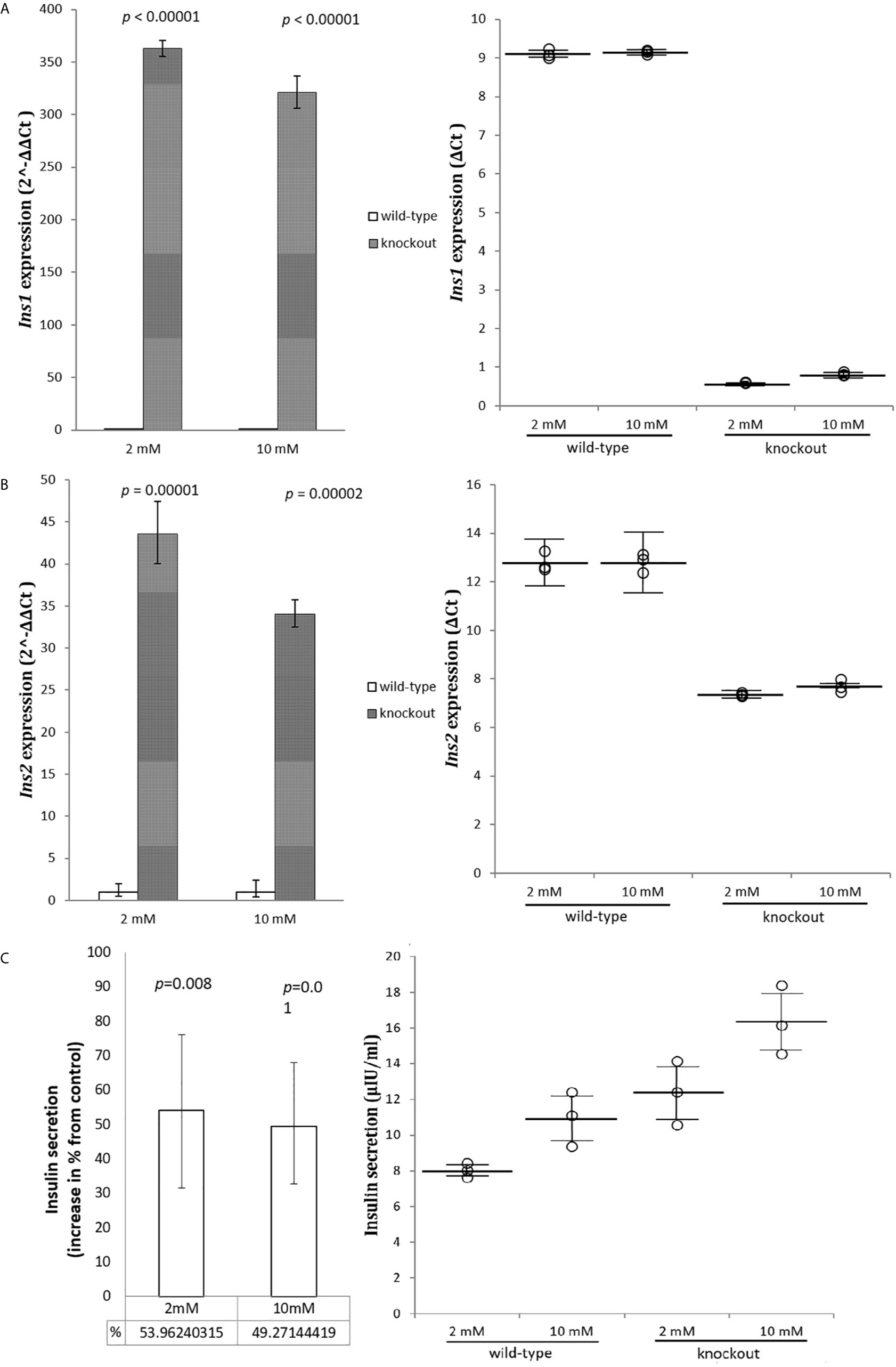
Figure 3 Insulin genes and secretion. RIN-m(-/-Adcy7) mRNA level established by quantitative RT-PCR. The CT values were normalized to β-actin to yield ΔCT and 2^-ΔΔCT values presented in dots and bar chart, respectively. Each dot represents an individual sample, whereas the horizontal lines represent the mean and standard error of the mean. ΔCT values were then calculated and presented as 2^-ΔΔCT values (fold change) in a bar chart. (A) presents the mRNA level of Ins1 when incubated in low glucose level 2mM and high glucose level 10mM, while (B) shows the expression level of Ins2 under the same condition. (C) illustrates insulin secretion when incubated for one hour with 2mM glucose and 10mM glucose compared to the control. Unpaired t-test with mean of ELISA or delta CT and SE ± values were used for statistic test.
Modified cells showed a significant increase in insulin secretion reaching 54% at low, and 49% at high glucose concentration, respectively, compared to the wild-type (p=0.008, p=0.01), (Figure 3C).
Insulin Secretion Pathway
As Adcy7 converts ATP to cyclic AMP and pyrophosphate, we examined whether the loss of the Adcy7 function influences insulin secretion via the cAMP pathway.
Protein Kinase cAMP-Activated Catalytic Subunits alpha (Prkaca) and beta (Prkacb) and Rap Guanine Nucleotide Exchange Factor 4 (Rapgef4) mRNA, which are highly expressed in pancreatic tissue, were measured as per their role on cAMP activation (27). mRNA levels for the PRKACs subunits indicated no change in the expression level (Figure 4A) while Rapgef4 decreased slightly, but significant (Figure 4B). These results indicated no activation of the cAMP pathway in RIN-m(-/-Adcy7) cells as the cause to increase insulin secretion.
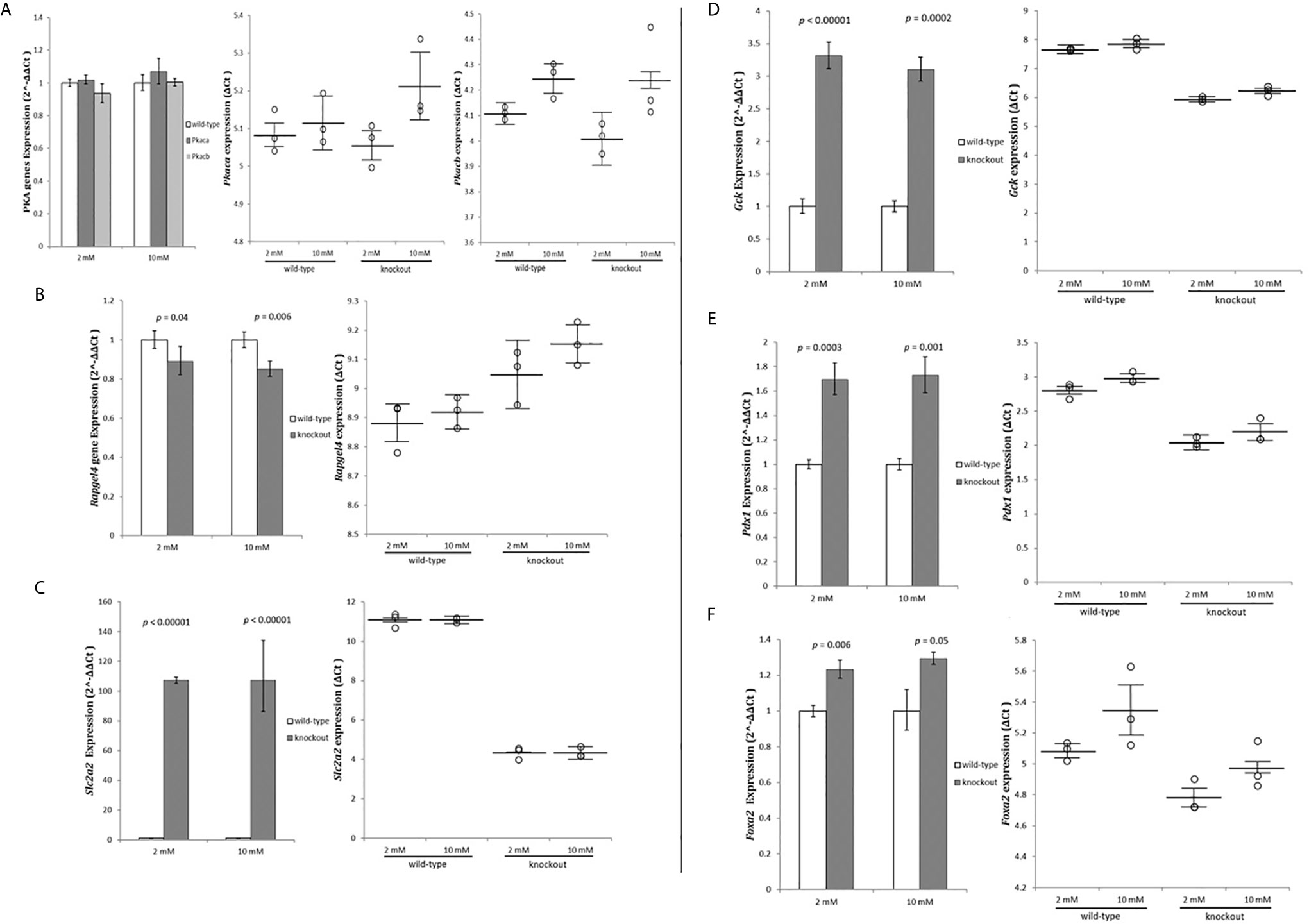
Figure 4 Exome data analysis strategy. RIN-m(-/-Adcy7) mRNA level under 2mM glucose and 10mM glucose concentration established by quantitative RT-PCR. The CT values were normalized to β-actin to yield ΔCT and 2^-ΔΔCT values presented in dots and bar chart, respectively. Each dot represents an individual sample, whereas the horizontal lines represent the mean and standard error of the mean. ΔCT values were then calculated and presented as 2^-ΔΔCT values (fold change) in a bar chart. (A) PRKACs alpha (Prkaca) and beta (Prkacb) expressions, (B) Rapgef4, (C) Slc2a2, (D) Gck, (E) Pdx1, (F) Foxa2. Unpaired t-test with delta CT and SE ± values were used for statistic test.
Next, we investigated whether the glucose stimulated-insulin secretion (GSIS) pathway was activated. The glucose uptake and the sensing genes Solute Carrier Family 2 Member 2 (SCL2A2) and Glucokinase (GCK) mRNA levels were measured. Glucose transporter member 2 (Glut2) protein, was extremely upregulated (107.3 fold - p<0.00001) and (107.5 fold - p<0.00001) for both low and high glucose concentrations, respectively (Figure 4C). Furthermore, glucokinase protein, which phosphorylates glucose in the first step of GSIS in β-cells, was also significantly upregulated (3.32 fold - p<0.00001) during low glucose conditions and (3.1 fold - p=0.0002) during high glucose level (Figure 4D).
To investigate the causes of the extreme upregulation of insulin and GSIS genes, Pancreatic and duodenal homeobox 1 (Pdx1), and Forkhead Box Protein A2 (Foxa2) genes expression were measured. Pdx1 was significantly up-regulated by 1.7 fold (p=0.0003) in 2 mM glucose and 1.73 fold (p=0.001) in 10 mM compared to the wild-type (Figure 4E). On the other hand, Foxa2, which binds to Pdx1 to regulate genes responsible for maintaining the mature β-cell function, were slightly but significantly increased by 1.23 fold (p=0.006) and 1.29 fold (p=0.05) when incubated with 2 mM and 10 mM glucose concentration, respectively, compared to the control (Figure 4F).
Discussion
In this article, we identified ADCY7 as a novel candidate genetic cause of CHI. ADCY7 encodes a membrane-bound adenylate cyclase that converts ATP to cyclic AMP and pyrophosphate. We showed that Adcy7 loss of function leads to excessive insulin secretion and activated the glucose stimulated-insulin secretion pathway.
Mammalian adenylyl cyclases (ACs) consist of nine membrane-bound proteins AC isoforms, AC1–AC9, coded by ADCY1-ADCY9 (28). ACs are constructed by two clusters of six transmembrane domains each followed by a cytoplasmic domain C1 and C2 (29). Our patient’s ADCY7 variants were predicted to disrupt the function of the highly conserved domain C1 and C2 justifying further loss-of-function studies. The Asp439 variant is shown to be reside in the gate to the activating domain in VC1 thus likely influencing regulation of substrate uptake (Figure 1A). Gly1045 allow a sharp turn of the loop connecting α-helix 15 and β-strand 19 necessary for IIC2 confirmation (Figure 1B).
Several studies have been linking AC members with insulin secretion and sensitivity. For instance, A gain of function of Adcy3 in mutagenized mice resistant to diet-induced obesity caused a reduction in both body weight, fat mass, insulin, and glucose levels compared to wild-type when subjected to high fat diet (30). On the contrary, whole-body Adcy5 knockout mice exhibited lower weight, lower fasting glucose, improved glucose tolerance, and increased insulin sensitivity (31). In pancreatic islet study by Hodson and his colleagues, ADCY5 silencing led to impaired glucose-induced cAMP increases and lower ATP concentrations at high glucose levels, in keeping with reduced mRNA expression of ADCY5 in patients with the ADCY5 SNP rs11708067, which increases the risk of type 2 diabetes (32). At least in part, differences in tissue and cell type-specific localization of the individual AC may explain the diversity of regulatory features by different AC members in cAMP pathway signaling (24, 33). Currently, no study reported the association of ADCY7 with insulin secretion and sensitivity. In the present study, ADCY7 loss of function was associated with increased insulin secretion, but not with evidence of cAMP alterations at glucose concentrations up to 10 mM. This was in line with findings of Hodson et al., where cAMP changes after ADCY5 silencing only occurred at higher glucose levels (32). More studies are needed to understand the differences between the different ACs in glucose metabolism. Compared to the wild-type, our RIN-m(-/-Adcy7) demonstrated an increase in the acute insulin activity leading to a 54% higher secretion during hypoglycemia in vitro. These results mimic the pathophysiology of CHI.
Genetic analysis using RT-PCR gene expression indicated that Adcy7 loss of expression upregulates the insulin regulator genes, Ins1 and Ins2, as well as glucose uptake and sensing by regulating Scl2a2 and Gck via upregulation of Pdx1.
PDX1 is a transcriptional factor that regulates the insulin gene. Transfection of a small interfering RNA specific for Pdx1 in pancreatic islets insulinoma cell line shows that Pdx1 directly regulates the insulin transcription promotor (34). Dox-induced Pdx1 expression in embryonic stem cells increased Ins1 and Ins2 mRNA compared to the non-induced cells and corresponding upregulated by 140% compared to the insulinoma cell line βTC6 (35).
Insulin secretion from the pancreatic β cell is a tightly regulated process by insulin-tropic factors stimulating intracellular cAMP, or by glucose sensing and uptake. We examined whether intracellular cAMP activation is responsible for increasing insulin secretion in RIN-m(-/-Adcy7) cells, similar to the role of ADCY3 haploinsufficiency in a previous report (36). The mRNA level for PRKACs subunits did not show significant changes, while Rapgef4 decreased only slightly. These results indicated no robust evidence of a cAMP activation pathway in RIN-m(-/-Adcy7) cells up to 10 mM glucose concentration. Subsequently, the expression analysis of key components of glucose uptake and sensing, Scl2a2, and Gck, revealed an extreme increase in mRNA levels leading to the activation of the glucose stimulated-insulin secretion (GSIS) pathway. This due to overexpression of PDX1 gene (37), seen in RIN-m(-/-Adcy7). In Pdx1 overexpressed mouse βPdx1; Ins2Akita, insulin secretion, Gck mRNA, and Slc2a2 localization was significantly increaseed compared to the non-modified Ins2Akita mouse (38). Foxa2 on the other hand, which binds to Pdx1 to regulate genes responsible for maintaining mature β-cell function (39), was also significantly increased. However, the exact mechanism by which ADCY7 leads to excessive insulin secretion through GSIS is still not explained by only increasing SLC2A2 and GCK expression during hypoglycemia conditions. In addition, the similarity of insulin secretion percentage between two glucose concentrations compared to wild-type as well as the expression changes seen in this study may also suggest loss of glucose sensitivity rather than an increased glucose phosphorylation. Further analyses with e.g. KCl, sulphonylurea and diazoxide, Gck activity, downstream analyses of both GSIS and cAMP pathways using a full gene expression profile as well as a protein study are needed to fully explain the ADCY7 mechanism.
RIN-m rat pancreas/islet cell line is a well-characterized cell line that secretes insulin with some concerns raised regarding GSIS and glucose sensitivity. Due to the limitation of a stable and well-characterized human pancreatic cell line, several published studies used RIN-m to investigate target genes concerning insulin secretion and GSIS (40–42). Santina Bruzzone et al. presented the effects of Abscisic acid (ABA) on insulin secretion using RIN-m and human pancreatic islets, concluding that they both show the same effect mediating cAMP on different glucose level (43). In addition, pre-treatment and starvation have shown different insulin reactions toward glucose in the RIN-m cell line (42). In this study, RIN-m wild-type and RIN-m(-/-Adcy7) cultured in 10 mM demonstrated 36.3%, and 32.3% higher in insulin secretion compared to 2 mM, respectively. Nonetheless, the ADCY7 role in glucose induced insulin secretion needs to be further tested in other cell types, e.g. human pancreatic islets, EndoC-βH1 and rat pancreatic cells, BRIN-BD11 and INS-1.
In conclusion, our study identified a novel candidate gene ADCY7 to cause CHI. The loss of Adcy7 function in the RIN-m cell line resulted in higher insulin secretion. The complexity of the relationship between ADCY7 and insulin secretion was at least in part explained by the changes in glucose sensing and glucose uptake in β-cells, which regulates insulin secretion via the glucose stimulated-insulin secretion pathway. Our finding will potentially open new doors for future work on the adenylyl cyclase complex with an insulin-signaling pathway to obtain better understanding of signaling pathway; secondly to develop a new therapeutic treatment triggering this pathway for patients with diabetes or congenital hyperinsulinism.
Data Availability Statement
The original contributions presented in the study are included in the Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The study was approved by The Regional Ethical Committee of Southern Denmark (number. S-VF-20040235). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
KB, HC, and MB were involved in planning and supervising the project. HC and EL collected and provided the clinical data. YA and KB designed the experiments. YA performed the experiments and analyzed the data. YA drafted the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This project was funded by Region of Southern Denmark and The University of Southern Denmark.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank our laboratory staff; Jette Møller for running exome sequencing and Martin J. Larsen for performing the sequencing alignment.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.657873/full#supplementary-material
References
1. Kapoor RR, Flanagan SE, James C, Shield J, Ellard S, Hussain K. Hyperinsulinaemic Hypoglycaemia. Arch Dis Child (2009) 94:450–7. doi: 10.1136/adc.2008.148171
2. Rahman SA, Nessa A, Hussain K. Molecular Mechanisms of Congenital Hyperinsulinism. J Mol Endocrinol (2015) 54:R119–29. doi: 10.1530/JME-15-0016
3. Helleskov A, Melikyan M, Globa E, Shcherderkina I, Poertner F, Larsen AM, et al. Both Low Blood Glucose and Insufficient Treatment Confer Risk of Neurodevelopmental Impairment in Congenital Hyperinsulinism: A Multinational Cohort Study. Front Endocrinol (Lausanne) (2017) 8:156. doi: 10.3389/fendo.2017.00156
4. Arnoux JB, Verkarre V, Saint-Martin C, Montravers F, Brassier A, Valayannopoulos V, et al. Congenital Hyperinsulinism: Current Trends in Diagnosis and Therapy. Orphanet J Rare Dis (2011) 6:63. doi: 10.1186/1750-1172-6-63
5. Christiansen CD, Petersen H, Nielsen AL, Detlefsen S, Brusgaard K, Rasmussen L, et al. 18f-Dopa PET/CT and 68Ga-DOTANOC PET/CT Scans as Diagnostic Tools in Focal Congenital Hyperinsulinism: A Blinded Evaluation. Eur J Nucl Med Mol Imaging (2018) 45:250–61. doi: 10.1007/s00259-017-3867-1
6. Pierro A, Nah SA. Surgical Management of Congenital Hyperinsulinism of Infancy. Semin Pediatr Surg (2011) 20:50–3. doi: 10.1053/j.sempedsurg.2010.10.009
7. Beltrand J, Caquard M, Arnoux JB, Laborde K, Velho G, Verkarre V, et al. Glucose Metabolism in 105 Children and Adolescents After Pancreatectomy for Congenital Hyperinsulinism. Diabetes Care (2012) 35:198–203. doi: 10.2337/dc11-1296
8. van der Steen I, van Albada ME, Mohnike K, Christesen HT, Empting S, Salomon-Estebanez M, et al. A Multicenter Experience With Long-Acting Somatostatin Analogues in Patients With Congenital Hyperinsulinism. Horm Res Paediatr (2018) 89:82–9. doi: 10.1159/000485184
9. Thomas P, Ye Y, Lightner E. Mutation of the Pancreatic Islet Inward Rectifier Kir6.2 Also Leads to Familial Persistent Hyperinsulinemic Hypoglycemia of Infancy. Hum. Mol Genet (1996) 5:1809–12. doi: 10.1093/hmg/5.11.1809
10. Thomas PM, Cote GJ, Wohllk N, Haddad B, Mathew PM, Rabl W, et al. Mutations in the Sulfonylurea Receptor Gene in Familial Persistent Hyperinsulinemic Hypoglycemia of Infancy. Science (1995) 268:426–9. doi: 10.1126/science.7716548
11. Christesen HB, Tribble ND, Molven A, Siddiqui J, Sandal T, Brusgaard K, et al. Activating Glucokinase (GCK) Mutations as a Cause of Medically Responsive Congenital Hyperinsulinism: Prevalence in Children and Characterisation of a Novel GCK Mutation. Eur J Endocrinol (2008) 159:27–34. doi: 10.1530/EJE-08-0203
12. Stanley CA, Lieu YK, Hsu BY, Burlina AB, Greenberg CR, Hopwood NJ, et al. Hyperinsulinism and Hyperammonemia in Infants With Regulatory Mutations of the Glutamate Dehydrogenase Gene. N Engl J Med (1998) 338:1352–7. doi: 10.1056/NEJM199805073381904
13. Clayton PT, Eaton S, Aynsley-Green A, Edginton M, Hussain K, Krywawych S, et al. Hyperinsulinism in Short-Chain L-3-hydroxyacyl-CoA Dehydrogenase Deficiency Reveals the Importance of Beta-Oxidation in Insulin Secretion. J Clin Invest (2001) 108:457–65. doi: 10.1172/JCI200111294
14. Otonkoski T, Jiao H, Kaminen-Ahola N, Tapia-Paez I, Ullah MS, et al. Physical Exercise-Induced Hypoglycemia Caused by Failed Silencing of Monocarboxylate Transporter 1 in Pancreatic Beta Cells. Am J Hum Genet (2007) 81:467–74. doi: 10.1086/520960
15. Gonzalez-Barroso MM, Giurgea I, Bouillaud F, Anedda A, Bellanne-Chantelot C, Hubert L, et al. Mutations in UCP2 in Congenital Hyperinsulinism Reveal a Role for Regulation of Insulin Secretion. PLoS One (2008) 3:e3850. doi: 10.1371/journal.pone.0003850
16. Pearson ER, Boj SF, Steele AM, Barrett T, Stals K, Shield JP, et al. Macrosomia and Hyperinsulinaemic Hypoglycaemia in Patients With Heterozygous Mutations in the HNF4A Gene. PLoS Med (2007) 4:e118. doi: 10.1371/journal.pmed.0040118
17. Stanescu DE, Hughes N, Kaplan B, Stanley CA, De Leon DD. Novel Presentations of Congenital Hyperinsulinism Due to Mutations in the MODY Genes: HNF1A and HNF4A. J Clin Endocrinol Metab (2012) 97:E2026–30. doi: 10.1210/jc.2012-1356
18. Pinney SE, Ganapathy K, Bradfield J, Stokes D, Sasson A, Mackiewicz K, et al. Dominant Form of Congenital Hyperinsulinism Maps to HK1 Region on 10q. Horm Res Paediatr (2013) 80:18–27. doi: 10.1159/000351943
19. Tegtmeyer LC, Rust S, van Scherpenzeel M, Ng BG, Losfeld ME, Timal S, et al. Multiple Phenotypes in Phosphoglucomutase 1 Deficiency. N Engl J Med (2014) 370:533–42. doi: 10.1056/NEJMc1403446
20. Cabezas OR, Flanagan SE, Stanescu H, Garcia-Martinez E, Caswell R, Lango-Allen H, et al. Polycystic Kidney Disease With Hyperinsulinemic Hypoglycemia Caused by a Promoter Mutation in Phosphomannomutase 2. J Am Soc Nephrol (2017) 28:2529–39. doi: 10.1681/ASN.2016121312
21. Hussain K. Diagnosis and Management of Hyperinsulinaemic Hypoglycaemia of Infancy. Horm Res (2008) 69:2–13. doi: 10.1159/000111789
22. Alhaidan Y, Christesen HT, Højlund K, Al Balwi MA, Brusgaard K. A Novel Gene in Early Childhood Diabetes: EDEM2 Silencing Decreases SLC2A2 and PXD1 Expression, Leading to Impaired Insulin Secretion. Mol Genet Genomics (2020) 295:1253–62. doi: 10.1007/s00438-020-01695-5
23. Alhaidan Y, Larsen MJ, Schou AJ, Stenlid MH, Balwi HT, Christesen Y, et al. Exome Sequencing Revealed DNA Variants in NCOR1, IGF2BP1, SGLT2 and NEK11 as Potential Novel Causes of Ketotic Hypoglycemia in Children. Sci Rep (2020) 10(1):2114. doi: 10.1038/s41598-020-58845-3
24. Iyengar R. Molecular and Functional Diversity of Mammalian Gs-stimulated Adenylyl Cyclases. FASEB J (1993) 7:768–75. doi: 10.1096/fasebj.7.9.8330684
25. Yan SZ, Beeler JA, Chen Y, Shelton RK, Tang WJ. The Regulation of Type 7 Adenylyl Cyclase by its C1b Region and Escherichia Coli Peptidylprolyl Isomerase, Slyd. J Biol Chem (2001) 276:8500–6. doi: 10.1074/jbc.M010361200
26. Ludwig MG, Seuwen K. Characterization of the Human Adenylyl Cyclase Gene Family: cDNA, Gene Structure, and Tissue Distribution of the Nine Isoforms. J Recept Signal Transduction Res (2002) 22:79–110. doi: 10.1081/RRS-120014589
27. Fujimoto K, Shibasaki T, Yokoi N, Kashima Y, Matsumoto M, Sasaki T, et al. Piccolo, a Ca2+ Sensor in Pancreatic Beta-Cells. Involvement of Camp-GEFII.Rim2. Piccolo Complex in cAMP-dependent Exocytosis. J Biol Chem (2002) 277:50497–502. doi: 10.1074/jbc.M210146200
28. Sunahara RK, Dessauer CW, Gilman AG. Complexity and Diversity of Mammalian Adenylyl Cyclases. Annu Rev Pharmacol Toxicol (1996) 36:461–80. doi: 10.1146/annurev.pa.36.040196.002333
29. Tang W-J, Hurley JH. Catalytic Mechanism and Regulation of Mammalian Adenylyl Cyclases. Mol Pharmacol (1998) 54:231–40. doi: 10.1124/mol.54.2.231
30. Pitman JL, Wheeler MC, Lloyd DJ, Walker JR, Glynne RJ, Gekakis N. A Gain-of-Function Mutation in Adenylate Cyclase 3 Protects Mice From Diet-Induced Obesity. PLoS One (2014) 9:e110226. doi: 10.1371/journal.pone.0110226
31. Ho D, Zhao X, Yan L, Yuan C, Zong H, Vatner DE, et al. Adenylyl Cyclase Type 5 Deficiency Protects Against Diet-Induced Obesity and Insulin Resistance. Diabetes (2015) 64:2636–45. doi: 10.2337/db14-0494
32. Hodson DJ, Mitchell RK, Marselli L, Pullen TJ, Gimeno Brias S, Semplici F, et al. ADCY5 Couples Glucose to Insulin Secretion in Human Islets. Diabetes (2014) 63:3009–21. doi: 10.2337/db13-1607
33. Hanoune J, Defer N. Regulation and Role of Adenylyl Cyclase Isoforms. Annu Rev Pharmacol Toxicol (2001) 41:145–74. doi: 10.1146/annurev.pharmtox.41.1.145
34. Iype T, Francis J, Garmey JC, Schisler JC, Nesher R, Weir GC, et al. Mechanism of Insulin Gene Regulation by the Pancreatic Transcription Factor Pdx-1: Application of pre-mRNA Analysis and Chromatin Immunoprecipitation to Assess Formation of Functional Transcriptional Complexes. J Biol Chem (2005) 280:16798–807. doi: 10.1074/jbc.M414381200
35. Kubo A, Stull R, Takeuchi M, Bonham K, Gouon-Evans V, Sho M, et al. Pdx1 and Ngn3 Overexpression Enhances Pancreatic Differentiation of Mouse Es Cell-Derived Endoderm Population. PLoS One (2011) 6:e24058. doi: 10.1371/journal.pone.0024058
36. Tong T, Shen Y, Lee H-W, Yu R, Park T. Adenylyl Cyclase 3 Haploinsufficiency Confers Susceptibility to Diet-Induced Obesity and Insulin Resistance in Mice. Sci Rep (2016) 6:34179. doi: 10.1038/srep34179
37. Watada H, Kajimoto Y, Umayahara Y, Matsuoka T, Kaneto H, Fujitani Y, et al. The Human Glucokinase Gene Beta-Cell-Type Promoter: An Essential Role of Insulin Promoter Factor 1/PDX-1 in its Activation in HIT-T15 Cells. Diabetes (1996) 45:1478–88. doi: 10.2337/diabetes.45.11.1478
38. Yamamoto Y, Miyatsuka T, Sasaki S, Miyashita K, Kubo F, Shimo N, et al. Preserving Expression of Pdx1 Improves β-Cell Failure in Diabetic Mice. Biochem Biophys Res Commun (2017) 483:418–24. doi: 10.1016/j.bbrc.2016.12.128
39. Wu KL, Gannon M, Peshavaria M, Offield MF, Henderson E, Ray M, et al. Hepatocyte Nuclear Factor 3beta is Involved in Pancreatic Beta-Cell-Specific Transcription of the Pdx-1 Gene. Mol Cell Biol (1997) 17:6002–13. doi: 10.1128/MCB.17.10.6002
40. Amoreaux WJL, Cuttitta C, Santora A, Blaize JF, Tachjadi J, Idrissi E. Taurine Regulates Insulin Release From Pancreatic Beta Cell Lines. J Biomed Sci (2010) 17(Suppl 1):S11. doi: 10.1186/1423-0127-17-S1-S11
41. Yan H, Li Y, Yang Y, Zhang Z, Zhang G, Sun Y, et al. Protective Effects of Andrographolide Derivative AL-1 on High Glucose-Induced Oxidative Stress in RIN-m Cells. Curr Pharm Des (2016) 22:499–505. doi: 10.2174/1381612821666150921110716
42. L’Amoreaux WJ, Cuttitta C, Santora A, Blaize JF, Tachjadi J, El Idrissi A. Taurine Regulates Insulin Release From Pancreatic Beta Cell Lines. J Biomed Sci (2010) 17 Suppl 1:S11. doi: 10.1186/1423-0127-17-S1-S11
Keywords: genetics, metabolomics, congenital hyperinsulinism, hyperinsulinemic hypoglycemia, pediatrics, adenylyl cyclase
Citation: Alhaidan Y, Christesen HT, Lundberg E, Balwi MAA and Brusgaard K (2021) CRISPR/Cas9 ADCY7 Knockout Stimulates the Insulin Secretion Pathway Leading to Excessive Insulin Secretion. Front. Endocrinol. 12:657873. doi: 10.3389/fendo.2021.657873
Received: 26 January 2021; Accepted: 18 May 2021;
Published: 11 June 2021.
Edited by:
Åke Sjöholm, Gävle Hospital, SwedenReviewed by:
Amanda Ackermann, Children’s Hospital of Philadelphia, United StatesDiana Stanescu, Children’s Hospital of Philadelphia, United States
Copyright © 2021 Alhaidan, Christesen, Lundberg, Balwi and Brusgaard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yazeid Alhaidan, eWF6ZWlkQG1haWwubmV0LnNh