Abstract
Copy Number Variations (CNVs) account for a large proportion of human genome and are a primary contributor to human phenotypic variation, in addition to being the molecular basis of a wide spectrum of disease. Multiallelic CNVs represent a considerable fraction of large CNVs and are strictly related to segmental duplications according to their prevalent duplicate alleles. RCCX CNV is a complex, multiallelic and tandem CNV located in the major histocompatibility complex (MHC) class III region. RCCX structure is typically defined by the copy number of a DNA segment containing a series of genes – the serine/threonine kinase 19 (STK19), the complement 4 (C4), the steroid 21-hydroxylase (CYP21), and the tenascin-X (TNX) – lie close to each other. In the Caucasian population, the most common RCCX haplotype (69%) consists of two segments containing the genes STK19-C4A-CYP21A1P-TNXA-STK19B-C4B-CYP21A2-TNXB, with a telomere-to-centromere orientation. Nonallelic homologous recombination (NAHR) plays a key role into the RCCX genetic diversity: unequal crossover facilitates large structural rearrangements and copy number changes, whereas gene conversion mediates relatively short sequence transfers. The results of these events increased the RCCX genetic diversity and are responsible of specific human diseases. This review provides an overview on RCCX complexity pointing out the molecular bases of Congenital Adrenal Hyperplasia (CAH) due to CYP21A2 deficiency, CAH-X Syndrome and disorders related to CNV of complement component C4.
Introduction
Germline Copy Number Variation (CNV) is regarded as a particular DNA fragment with variable copies compared to a reference genome and primarily includes genome duplications and deletions (1). CNVs account for a large proportion of human genome (2), greatly influence cellular phenotypes such as gene expression (3), and are accountable for a plethora of diseases, in addition to representing relevant disease risk factors (4, 5). These observations raise the possibility that CNVs could be a primary contributor to human phenotypic variation and consequently evolve under selective pressures (5). Four major mechanisms have been proposed as contributors to the generation of most CNVs, including nonallelic homologous recombination (NAHR), nonhomologous end-joining, fork stalling and template switching, and L1-mediated retrotransposition (4). Multiallelic CNVs constitute a considerable fraction of large CNVs and are strictly related to segmental duplications according to their prevalent duplicate alleles (6, 7). CNVs alleles with large, homologous, and tandem repeats are susceptible to rearrangements via NAHR mechanism (8) such as unequal crossover (9) and gene conversion (10). In this Review, we focus on the genetic complexity of the RCCX CNV discussing the molecular bases of related human diseases as Congenital Adrenal Hyperplasia (CAH).
RCCX CNV
RCCX CNV is a complex, multiallelic and tandem CNV located in the major histocompatibility complex (MHC) class III region (11, 12). It is an haplotypic structure typically defined by the copy number of a DNA segment containing a series of genes that lie close to each other: the serine/threonine kinase 19 (STK19), the complement 4 (C4), the steroid 21-hydroxylase (CYP21), and the tenascin-X (TNX) genes (13). RCCX CNV alleles commonly consist of one, two or three segments with the prevalence of approximately 17%, 69% and 14% in the Caucasian population (14). The Figure 1A shows the structure of the RCCX haplotype with two segments with the genes oriented as: STK19-C4A-CYP21A1P-TNXA-STK19B-C4B-CYP21A2-TNXB (15). STK19 gene (originally called G11 or RP), just upstream from C4A, encodes a nuclear Serine/Threonine Kinase protein recently identified as a regulator of NRAS activity (16–20). STK19B, immediately upstream from the C4B gene, consists only of 914 bases of the 3’ end of the original gene because the C4/CYP21/TNX locus duplication caused the lost of a large part of the coding DNA in this region (14, 15). C4A and C4B genes encode the two isoforms of the fourth component of serum complement (C4), an essential element for the effector arm of the humoral immune response (21). Each human C4 gene contains 41 exons, and the gene size shows a dichotomous size variation between ~22 kb and 16 kb. The longer gene is the result of the integration of the endogenous retrovirus HERV-K(C4) into intron 9 (22). Both the C4A and C4B 3’ ends lie only 2466 bp upstream the CYP21A1P and CYP21A2 transcriptional start sites, respectively. In addition, the promoter regions of CYP21 genes are located in the C4 intron 35 (23). CYP21A2 gene encodes the steroid 21-hydroxylase enzyme (cytochrome P450c21), uniquely expressed in adrenal cortex, responsible for the biosynthesis of the two principal steroid hormones, aldosterone and cortisol. Both the CYP21A2 functional gene and the CYP21A1P pseudogene consist in a total of ten exons spanning 3.4 kb. Sequence identity of 98% and approximately 96% characterizes their exons and intronic regions, respectively (24, 25).
Figure 1
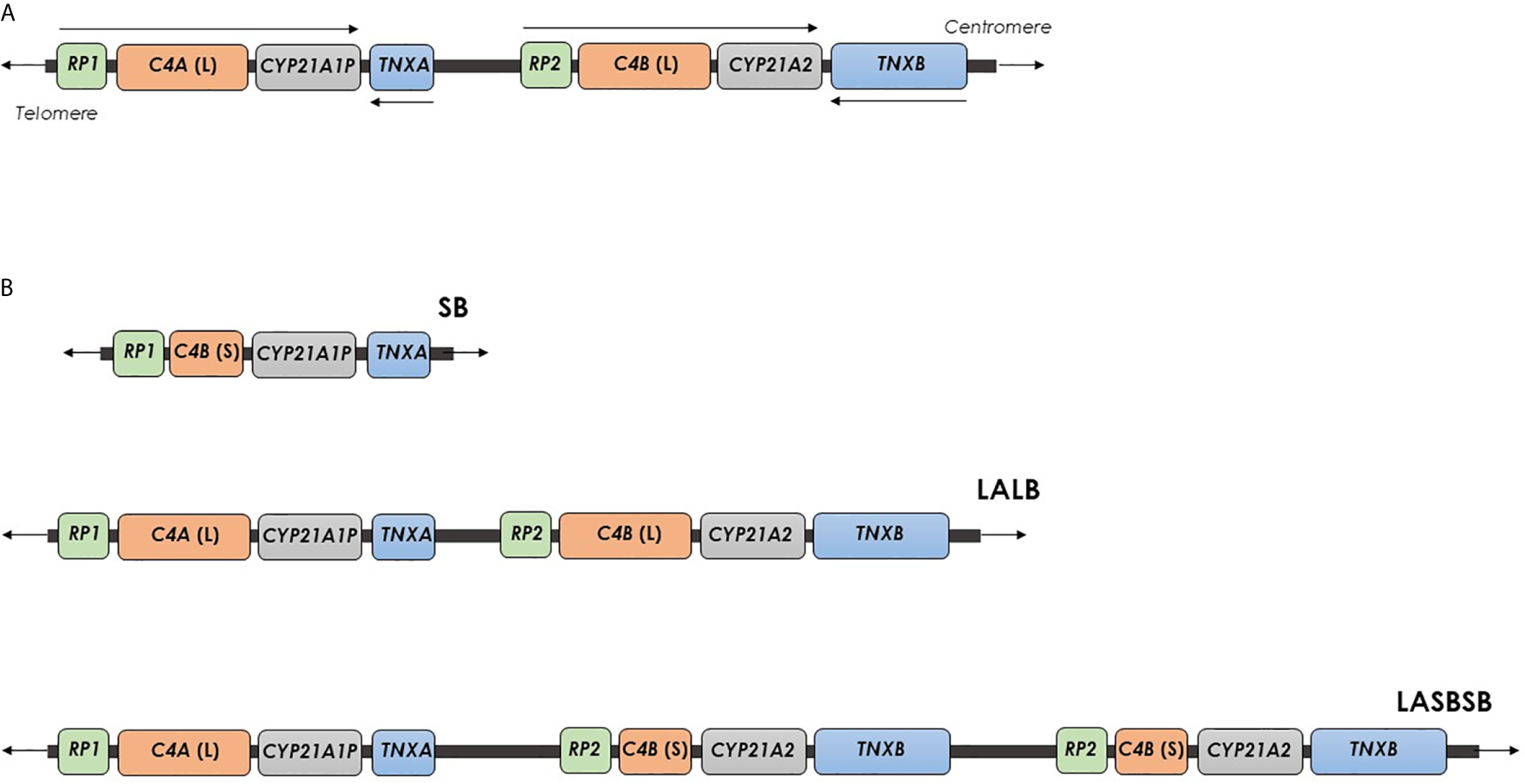
Organization of the human RCCX CNV on chromosome 6. (A)RCCX structure with two segments containing the genes STK19-C4A-CYP21A1P-TNXA-STK19B-C4B-CYP21A2-TNXB, with a telomere-to-centromere orientation. Arrows indicates the transcriptional orientation of genes. (B)RCCX structures with one, two and three segments. Each segment is indicated with two letters, the first represents the allele of the HERV-K(C4) CNV [L, Long allele (insertion) or S, Short allele (deletion)], and the second indicates the types of C4 gene (A or B). SB, A RCCX structure with one segment including a C4B gene without HERV-K(C4) insertion. LALB, A RCCX structure with two segments including two C4 genes, A and B, both with HERV-K(C4) insertion. LA SB SB: A RCCX structure with three segments including a C4A gene with HERV-K(C4) insertion and two C4B genes without HERV-K(C4) insertion.
With respect to the C4 and CYP21, both the TNXA and TNXB genes are located in the opposite DNA strand with, consequently, an opposite transcriptional orientation. These genes partially overlap the 3’ ends of the CYP21 genes: the last exon of TNXA and TNXB lies within the 3’ untranslated region of exon 10 in CYP21A1P and CYP21A2, respectively, and contain fibronectin type III repeats (26, 27). TNXB gene, encoding the extracellular matrix protein TNX, consists of 68.2 kb of DNA and includes 44 exons (28). The TNXB gene appears to be unique in having both its 5’ and 3’ ends buried in other genes. In fact, several start sites located into or near the CREB-RP gene are responsible for the TNXB transcription initiation. The CREB-RP gene lie immediately upstream of TNXB and encoding a protein related to the CREB transcription factor (29, 30). TNXA is a duplicated section of TNXB and consists in a truncated pseudogene containing a 120 bp deletion that causes a frameshift and a premature stop codon that render the gene non-functional (31).
An haplotypic RCCX CNV structure is traditionally described by the copy number of the repeated segment of RCCX CNV (CNV allele), and, per segment, by the alleles of HERV-K(C4) CNV and the type of C4 gene (13). Usually, a RCCX segment is indicated with two letters, the first representing the alleles of the HERV-K(C4) CNV [L: long allele (insertion allele) or S: short allele (deletion allele)] and the second indicating the type of C4 gene (A or B). The multiplication of these two letters indicates the presence of two and three segments (Figure 1B) (11, 13). Very rare RCCX CNV alleles with four segments have been also reported (32, 33). In addition, in order to define the exact structure (presence or absence of HERV-K(C4) insertion and type of C4 gene) of a RCCX CNV, specific molecular approaches have been proposed (11, 34).
RCCX-Associated Diseases
The genetic diversity of the RCCX is highly attributable to NAHR: unequal crossover facilitates large structural rearrangements and copy number changes, whereas gene conversion mediates relatively short sequence transfers (9, 10). The results of these events increase the RCCX genetic diversity and are responsible of specific human diseases.
CAH Due to 21-Hydroxylase Deficiency
CAH is a group of genetic autosomal recessive disorders that affects adrenal steroidogenesis in the adrenal cortex. The vast majority of the CAH cases, approximately 95%, are related to 21-hydroxylase deficiency due to pathogenic variants accounted in CYP21A2 gene. 21-hydroxylase enzyme is responsible for the conversion of 17-hydroxyprogesterone to 11-deoxycortisol and progesterone to deoxycorticosterone (35, 36). The impairment of cortisol and aldosterone production is directly related to the clinical form of the disease that ranges from classic (CL) or severe to non-classic (NC) or mild late onset (37, 38). As above-mentioned, both the CYP21A2 gene and its CYP21A1P pseudogene are composed by a total of 10 exons, sharing a high rate of homology (25, 39). The CYP21A1P pseudogene is inactivated by multiple deleterious variants (small insertions/deletions and point pathogenic variants) responsible for the synthesis of a non-functional protein. Intergenic recombination events represent more than 95% of deleterious variants leading to 21-hydroxylase deficiency. Approximately 75% of the deleterious variants are transferred by small conversions from the pseudogene during meiosis. These conversions can involve one (microconversions) or more pseudogene variants (40–42). Differently, 5-10% of CAH alleles observed in most populations are characterized by CYP21A2 pathogenic variants that do not result in gene conversions (43–45).
The 20–25% of the cases of 21-hydroxylase deficiency is related to large misalignment due to unequal crossing over during meiosis process. This kind of event may cause gene deletion or amplification, and also broader deletions involving CYP21A2 gene and the other contiguous genes (40–42). CYP21A1P/CYP21A2 chimeric gene is the result of a recombination between CYP21A1P and CYP21A2 genes, as an unequal crossing over occurs during meiosis. Based on the C4B form of the gene, i.e. long or short, the rearrangement results into a 26 or 32 Kb deletion, encompassing the 3′ end of CYP21A1P, all of the C4B gene, and the 5′ end of the CYP21A2 gene. This event leads to a single non-functional chimeric gene containing the CYP21A1P at the 5’ end and the CYP21A2 at the 3’ end (Figure 2A). To date 9 different chimeric CYP21A1P/CYP21A2 genes have been found and characterized (46–55). In particular, two groups of chimeras, classic and attenuated, have been identified: chimeric genes where the junction site is located downstream of the c.293-13C/A>G mutation in the intron 2 (CH-1, CH-2, CH-3, CH-5, CH-6, CH-7, CH-8) are associated with the severe Salt Wasting form of CAH. In contrast, CH-4 and CH-9 chimeras, carrying the weaker CYP21A1P promoter and the sole p.(Pro30Leu) variant, are commonly related to a milder phenotype (47).
Figure 2
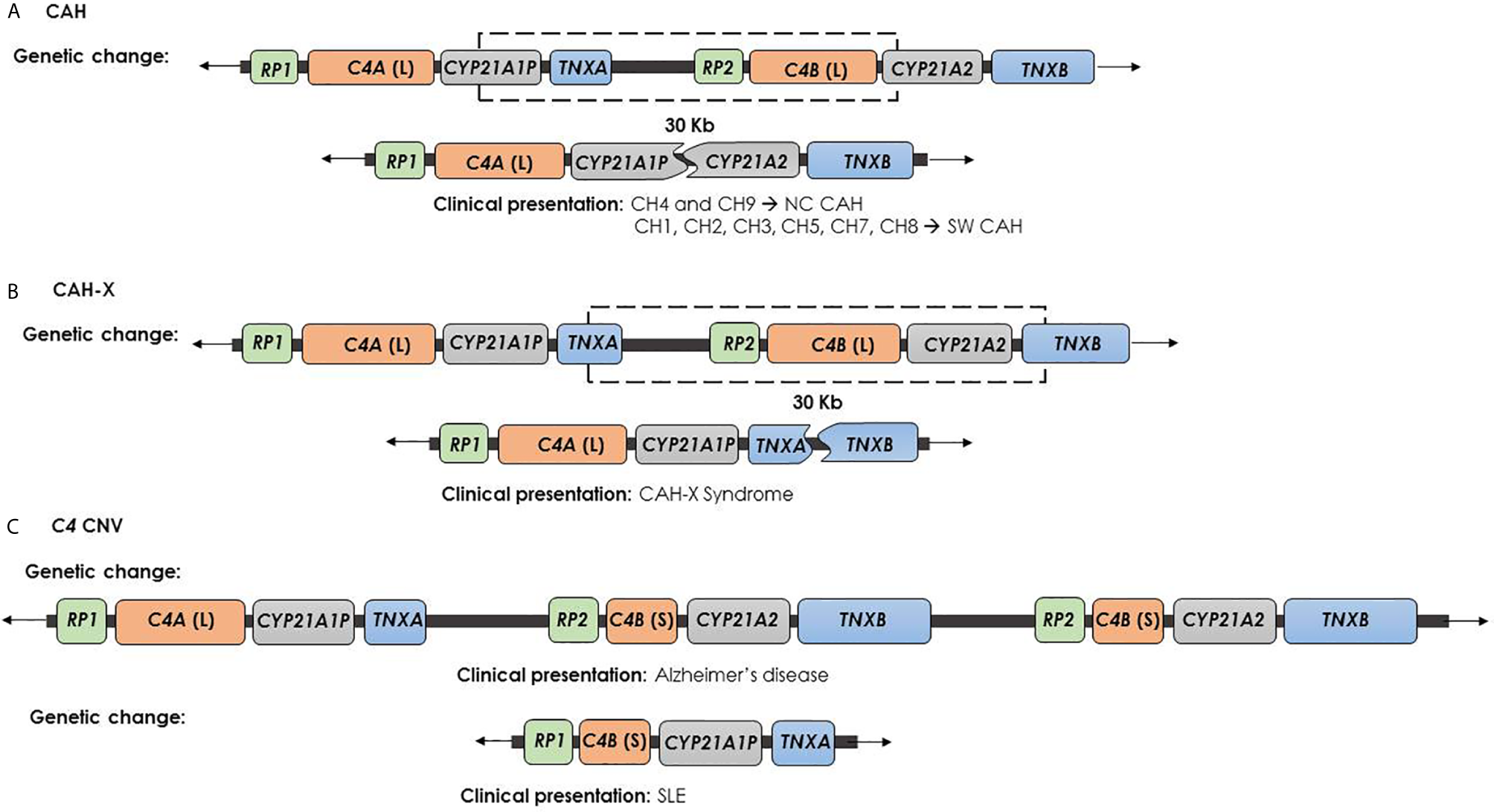
Genetic changes and clinical presentation. (A) CAH chimera (CYP21A1P/CYP21A2) is caused by recombination between CYP21A1P and CYP21A2 and causes the impairment of CYP21A2 and the deletion of C4B, but leaves safe the TNXB gene. CH-1, CH-2, CH-3, CH-5, CH-6, CH-7 and CH-8 chimeras are involved in the severe SW form of CAH. In contrast, CH-4 and CH-9 chimeras are generally related to a milder phenotype. (B) CAH-X chimera (TNXA/TNXB) is caused by recombination between TNXA and TNXB and produces the complete deletion of CYP21A2 gene, the impairment of TNXB gene and the deletion of C4B. This contiguous deletion is termed CAH-X and causes CAH-X syndrome. (C) Psychiatric and autoimmune diseases due to C4 CNV: the high C4B copy number has been described as Alzheimer’s disease risk factor, while the C4A deficiency was related to SLE.
Unequal crossover is also the cause of copy number changes of RCCX segment. The most well-known case is an haplotypic RCCX CNV structure containing three distinct segments with two CYP21A2 gene copies and one CYP21A1P pseudogene copy (56–62). Generally, the CYP21A2 gene located downstream the TNXA gene shows a wild-type nucleotide sequence, or carries one or more deleterious variants. Conversely, the presence of the CYP21A2 p.(Gln319Ter) mutation characterized the gene copy located next to TNXB gene (13, 57–64). To date, 8 different haplotypes with two active CYP21A2 genes on a chromosome 6 have been detected (63). The absence of a clear correlation between genotype and phenotype observed in many individuals is solved by the existence of these rare haplotypes, underlying the need of the RCCX CNV assessment in the molecular diagnosis of 21-hydroxylase deficiency (56, 65, 66).
Finally, the complete deletion of CYP21A2 gene can occur as the result of an unequal crossing over between TNXA and TNXB genes. This event produces a chromosome with two copies of CYP21A2 gene and a chromosome where the arrangement of the RCCX segment shows the C4-CYP21A1P-TNXA/TNXB sequence, lacking CYP21A2 gene copy. This condition is associated to the CAH-X Syndrome (67).
CAH-X Syndrome
Ehlers-Danlos syndromes (EDS) are a clinically and genetically heterogeneous group of heritable connective tissue disorders characterized by joint hypermobility (JH), skin hyperextensibility, and tissue fragility. EDS is typically caused by autosomal dominant mutations in collagen-encoding genes or in genes encoding collagen-modifying enzymes (68). Tenascin-X deficiency causes a clinically distinct form of EDS due to homozygous or compound heterozygous pathogenic variants in the TNXB gene. Pathogenic variants account in the coding region of the EGF-like repeats or the fibronectin type III domain of the tenascin protein. The clinical phenotype resembles the classical EDS type with a pattern of autosomal recessive inheritance (69, 70). Heterozygosity for severe TNXB mutations causes TNXB haploinsufficiency and it is related to hypermobility type EDS (hEDS), characterized by JH, recurring joint dislocations, joint pain and structural cardiac valve abnormality (71). The CAH-X term was first used for the description of a specific subgroup of CAH affected subjects showing an EDS phenotype caused by CYP21A2 monoallelic deletion extending into the TNXB gene (72). The result of this 30 Kb deletion, caused by a recombination event between TNXA and TNXB genes, is a chimeric TNXA/TNXB gene (Figure 2B) (73). To date, three TNXA/TNXB chimeras that differ in the junction site and result in a contiguous CYP21A2 and TNXB gene deletion (CH-1 to CH-3) have been reported (72, 74, 75). CAH-X CH-1 is characterized by a TNXA pseudogene derived 120-bp deletion in exon 35 that causes the non-functionality of the gene and also results in decreased TNX expression in both dermal and serum, claiming an haploinsufficiency mechanism (69, 72). CAH-X CH-2 is characterized by the variant c.12174C>G (p.Cys4058Trp) (exon 40) derived from TNXA pseudogene. This substitution deletes a cysteine residue and leads to the loss of a critical disulfide bond in the tertiary structure of the TNX C-terminal fibrinogen-like domain (74). The third chimera, termed CAH-X CH-3, has TNXB exons 41-44 substituted by TNXA and it is characterized by a cluster of 3 closely linked variants also derived from TNXA pseudogene: the c.12218G>A (p.Arg4073His) in exon 41 and the c.12514G>A (p.Asp4172Asn) and the c.12524G>A (p.Ser4175Asn) in exon 43 (75). Computational studies showed that the p.(Arg4073His) variant interferes with TNX fibrinogen-like domain stability. In particular, the arginine 4073 is predicted to form a cation-pi interaction with the p.Phe4080 residue, which is lost in the p.(Arg4073His) change, penalizing the folding energy with a loss of 35 kcal/mol. The remaining variants in the cluster did not significantly affect the folding energies in the models (75). Differently to CAH-X CH-1 chimera, CH-2 and CH-3 not reduce the TNX expression but produce altered proteins and are associated with a dominant-negative effect.
All the TNXA/TNXB chimeras cause EDS in monoallelic or biallelic form regardless of CAH status, although patients with CAH usually show more severe EDS manifestations with respect to carriers without CAH (69, 72, 74–76). Approximately 10% of patients with CAH due to 21-hydroxylase deficiency are affected by CAH-X (74). Recently, Marino et al. reported that the overall prevalence of CAH-X in a large cohort of Argentine CAH patients was 14%, which was similar to that previously found in a large cohort from the National Institutes of Health and in the Chinese population (15% and 14% respectively) (77–79). In addition, Lao et al. reported a particularly high prevalence (29.2%) of CAH-X in 21-hydroxylase deficient patients carrying the 30 kb deletion (78).
Regarding clinical manifestations, CAH-X affected subjects show generalized JH, subluxation and chronic arthralgia, while cardiac abnormalities have been observed in about 25% (80). More severe clinical manifestations were found in patients with a biallelic than in those with a monoallelic form (8, 10). In addition, compared to haploinsufficiency, a dominant-negative effect causes a more severe phenotype displayed by greater skin and joint involvement (74). The diagnosis of EDS due to CAH-X relies mainly on clinical evaluations including physical examination for JH, skin characteristics and imaging. A serum tenascin-X test, based on enzyme-linked immunosorbent assay, has been developed to identify complete deficiency, but it is not accurate in identifying heterozygous forms (69, 81). Molecular diagnosis represents a valid support to the clinical evaluation of CAH-X and, in this context, Sanger sequencing results to be the most reliable an informative method for all TNXB variations, even if it is laborious and expensive (82).
Complement Component C4 CNV
Complement component C4 is a central protein in the classical and lectin pathways within the complement system (83). The two isotypes of C4, which differ by only four amino acids, demonstrate differential chemical reactivities: C4A displays higher affinity for amino group-containing antigens or immune complexes, and C4B for hydroxyl group-containing antigens (84, 85). In the general population, the most common RCCX haplotype consists of two segments with two C4 in tandem genes coding for C4A and C4B. So, approximately 60% of healthy individuals have two C4A and two C4B genes (14, 86, 87). However, deletions and duplications of C4 genes are well documented and the human C4 locus has been identified as a functional CNV hotspot within the RCCX region. C4 isotypes involvement is described in several pathological conditions (88). For instance, an high C4A gene dosage represents a relevant schizophrenia risk factor, while both C4A or C4B high copy number is related to Alzheimer’s disease (89, 90) (Figure 2C). The presence of one C4A or C4B gene is called heterozygous C4A or C4B deficiency, while the presence of no functional C4A or C4B genes causes complete C4A or C4B deficiency and is called homozygous C4 deficiency (14). Homozygous deficiencies of complement C4A or C4B are detected in 1-10% of populations. Homozygous deficiency of C4A has been reported to associate with increased frequency of autoimmune diseases, whereas homozygous C4B deficiency has been associated with increased susceptibility of bacterial and enveloped viral infections (91, 92). Many studies support the association between homozygous C4A deficiency and systemic lupus erythematosus (SLE) (93–97) (Figure 2C).
C4 structural variations frequently arise in CAH affected subjects with relevant clinical implications, particularly in relation to psychiatric morbidity and autoimmunity (98, 99). Moreover, Lao et al. reported in a cohort of 145 CAH subjects with 21-hydroxylase deficiency, the correlation between C4A copy number and the externalization of psychiatric comorbidity (98). Interestingly, authors specified that C4B copy number was the determinant of C4 serum levels in CAH patients because C4B copy number varied in CAH patients carrying the 30-Kb deletion and in NC patients carrying the p.(Val282Leu) variant. In fact, as a consequence of 30 Kb deletion, both C4B and CYP21A2 genes are frequently lost concurrently, producing a CYP21A1P/CYP21A2 or CYP21A1P-TNXA/TNXB chimera (Figures 2A, B). Conversely, the known association of the NC p.(Val282Leu) variant with high total C4 copy number was found to be due to a duplication of C4B gene, not C4A (98, 100).
Recently, Falhammar et al. reported an increased prevalence of autoimmune disorders in a large cohort of Swedish patients with 21-hydroxylase deficiency (99). However, some limitations of the study were point out. In particular, the relatively young age of the patients and the possible protective effects of glucocorticoid treatment may have led to underestimates in the lifetime risks for autoimmune disorders (99).
The complex genetics of human histocompatibility complex provides evidences that RCCX genotype being related to C4 could represent a further risk factor for additional illnesses in CAH affected subjects with 21-hydroxylase deficiency. However, the role of the C4 gene dosage related to CYP21A2 genotype in CAH patients needs to further investigations.
Discussion
RCCX CNV represents a complex, multiallelic and tandem CNV in the MHC class III region. Genetic recombination events typically affect this genomic region due to the peculiar co-presence of genes and pseudogenes with high sequence homology, causing frequent misalignment during meiosis. The challenging related to the molecular diagnosis of 21-hydroxylase deficiency, owed to the complexity of the RCCX CNV structure, are well documented. For this reason, it is essential to refer to effective guidelines for the standardization of molecular genetic testing of CAH due to CYP21A2 defects (101). In addition, as recently suggested, including CAH-X chimeras determination in 21-hydroxylase deficiency molecular testing would be particularly beneficial for individuals carrying an allele with the “30Kb deletion”. In fact, a very early CAH-X diagnosis could be offered to young children before hypermobility evaluation is applicable, and to enable early screening for cardiac defects (102). However, a reflection is currently in progress on the need to carry out further studies in order to broader the knowledge and the expertise on CAH-X before including respective methods in routine diagnostic procedures (103, 104).
Finally, novel and larger studies are required in order to elucidate the role of C4 dosage in several disorders, especially in CAH patients with 21-hydroxylase deficiency.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
LF and EP researched and wrote a first draft of the review. PC and CC revised the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Zarrei M MacDonald JR Merico D Scherer SW . A Copy Number Variation Map of the Human Genome. Nat Rev Genet (2015) 16:172–83. doi: 10.1038/nrg3871
2
Handsaker RE Van Doren V Berman JR Genovese G Kashin S Boettger LM et al . Large Multiallelic Copy Number Variations in Humans. Nat Genet (2015) 47:296–303. doi: 10.1038/ng.3200
3
Stranger BE Forrest MS Dunning M Ingle CE Beazley C Thorne N et al . Relative Impact of Nucleotide and Copy Number Variation on Gene Expression Phenotypes. Science (2007) 9:848–53. doi: 10.1126/science.1136678
4
Hu L Yao X Huang H Guo Z Cheng X Xu Y et al . Clinical Significance of Germline Copy Number Variation in Susceptibility of Human Diseases. J Genet Genomics (2018) 45:3–12. doi: 10.1016/j.jgg.2018.01.001
5
Saitou M Gokcumen O . An Evolutionary Perspective on the Impact of Genomic Copy Number Variation on Human Health. J Mol Evol (2020) 88:104–19. doi: 10.1007/s00239-019-09911-6
6
Conrad DF Pinto D Redon R Feuk L Gokcumen O Zhang Y et al . Origins and Functional Impact of Copy Number Variation in the Human Genome. Nature (2010) 464:704–12. doi: 10.1038/nature08516
7
Redon R Ishikawa S Fitch KR Feuk L Perry GH Andrews TD et al . Global Variation in Copy Number in the Human Genome. Nature (2006) 444:444–54. doi: 10.1038/nature05329
8
Dittwald P Gambin T Szafranski P Li J Amato S Divon MY et al . NAHR-Mediated Copy-Number Variants in a Clinical Population: Mechanistic Insights Into Both Genomic Disorders and Mendelizing Traits. Genome Res (2013) 23:1395–409. doi: 10.1101/gr.152454.112
9
Lupski JR Stankiewicz P . Genomic Disorders: Molecular Mechanisms for Rearrangements and Conveyed Phenotypes. PloS Genet (2005) 1:e49. doi: 10.1371/journal.pgen.0010049
10
Chen JM Férec C Cooper DN . Gene Conversion in Human Genetic Disease. Genes (Basel) (2010) 1:550–63. doi: 10.3390/genes1030550
11
Bánlaki Z Szabó JA Szilágyi Á Patócs A Prohászka Z Füst G et al . Intraspecific Evolution of Human RCCX Copy Number Variation Traced by Haplotypes of the CYP21A2 Gene. Genome Biol Evol (2013) 5:98–112. doi: 10.1093/gbe/evs121
12
Horton R Wilming L Rand V Lovering RC Bruford EA Khodiyar VK et al . Gene Map of the Extended Human MHC. Nat Rev Genet (2004) 5:889–99. doi: 10.1038/nrg1489
13
Doleschall M Luczay A Koncz K Hadzsiev K Erhardt É Szilágyi Á et al . A Unique Haplotype of RCCX Copy Number Variation: From the Clinics of Congenital Adrenal Hyperplasia to Evolutionary Genetics. Eur J Hum Genet (2017) 25:702–10. doi: 10.1038/ejhg.2017.38
14
Blanchong CA Zhou B Rupert KL Chung EK Jones KN Sotos JF et al . Deficiencies of Human Complement Component C4A and C4B and Heterozygosity in Length Variants of RP-C4-CYP21-TNX (RCCX) Modules in Caucasians. The Load of RCCX Genetic Diversity on Major Histocompatibility Complex-Associated Disease. J Exp Med (2000) 191:2183–96. doi: 10.1084/jem.191.12.2183
15
Bánlaki Z Doleschall M Rajczy K Fust G Szilágyi A . Fine-Tuned Characterization of RCCX Copy Number Variants and Their Relationship With Extended MHC Haplotypes. Genes Immun (2012) 13:530–5. doi: 10.1038/gene.2012.29
16
Sargent CA Anderson MJ Hsieh SL Kendall E Gomez-Escobar N Campbell RD . Characterisation of the Novel Gene G11 Lying Adjacent to the Complement C4A Gene in the Human Major Histocompatibility Complex. Hum Mol Genet (1994) 3:481–88. doi: 10.1093/hmg/3.3.481
17
Shen L Wu LC Sanlioglu S Chen R Mendoza AR Dangel AW et al . Structure and Genetics of the Partially Duplicated Gene RP Located Immediately Upstream of the Complement C4A and the C4B Genes in the HLA Class III Region. Molecular Cloning, Exon-Intron Structure, Composite Retroposon, and Breakpoint of Gene Duplication. J Biol Chem (1994) 269:8466–76. doi: 10.1016/S0021-9258(17)37217-4
18
Gomez-Escobar N Chou CF Lin WW Hsieh SL Campbell RD . The G11 Gene Located in the Major Histocompatibility Complex Encodes a Novel Nuclear Serine/Threonine Protein Kinase. J Biol Chem (1998) 273:30954–60. doi: 10.1074/jbc.273.47.30954
19
Yin C Zhu B Zhang T Liu T Chen S Liu Y et al . Pharmacological Targeting of STK19 Inhibits Oncogenic NRAS-Driven Melanomagenesis. Cell (2019) 176:1113–27. doi: 10.1016/j.cell.2019.01.002
20
Gimple RC Wang X . RAS: Striking at the Core of the Oncogenic Circuitry. Front Oncol (2019) 9:965. doi: 10.3389/fonc.2019.00965
21
Carroll MC . Complement and Humoral Immunity. Vaccine (2008) 26:I28–33. doi: 10.1016/j.vaccine.2008.11.022
22
Yu CY Belt KT Giles CM Campbell RD Porter RR . Structural Basis of the Polymorphism of Human Complement Components C4A and C4B: Gene Size, Reactivity and Antigenicity. EMBO J (1986) 5:2873–81. doi: 10.1002/j.1460-2075.1986.tb04582.x
23
Wijesuriya SD Zhang G Dardis A Miller WL . Transcriptional Regulatory Elements of the Human Gene for Cytochrome P450c21 (Steroid 21-Hydroxylase) Lie Within Intron 35 of the Linked C4B Gene. J Biol Chem (1999) 274:38097–106. doi: 10.1074/jbc.274.53.38097
24
White PC Grossberger D Onufer BJ Chaplin DD New MI Dupont B et al . Two Genes Encoding Steroid 21-Hydroxylase are Located Near the Genes Encoding the Fourth Component of Complement in Man. Proc Natl Acad Sci U.S.A. (1985) 82:1089–93. doi: 10.1073/pnas.82.4.1089
25
White PC New MI Dupont B . Structure of Human Steroid 21-Hydroxylase Genes. Proc Natl Acad Sci U.S.A. (1986) 83:5111–5. doi: 10.1073/pnas.83.14.5111
26
Morel Y Bristow J Gitelman SE Miller WL . Transcript Encoded on the Opposite Strand of the Human Steroid 21-Hydroxylase/Complement Component C4 Gene Locus. Proc Natl Acad Sci USA (1989) 86:6582–6. doi: 10.1073/pnas.86.17.6582
27
Matsumoto K Arai M Ishihara N Ando A Inoko H Ikemura T . Cluster of Fibronectin Type III Repeats Found in the Human Major Histocompatibility Complex Class III Region Shows the Highest Homology With the Repeats in an Extracellular Matrix Protein, Tenascin. Genomics (1992) 12:485–91. doi: 10.1016/0888-7543(92)90438-x
28
Bristow J Tee MK Gitelman SE Mellon SH Miller WL . Tenascin-X: A Novel Extracellular Matrix Protein Encoded by the Human XB Gene Overlapping P450c21B. J Cell Biol (1993) 122:265–78. doi: 10.1083/jcb.122.1.265
29
Speek M Barry F Miller WL . Alternate Promoters and Alternate Splicing of Human Tenascin-X, a Gene With 5’ and 3’ Ends Buried in Other Genes. Hum Mol Genet (1996) 5:1749–58. doi: 10.1093/hmg/5.11.1749
30
Min J Shukla H Kozono H Bronson SK Weissman SM Chaplin DD . A Novel Creb Family Gene Telomeric of HLA-DRA in the HLA Complex. Genomics (1995) 30:149–56. doi: 10.1006/geno.1995.9891
31
Gitelman SE Bristow J and Miller WL . Mechanism and Consequences of the Duplication of the Human C4/P450c21/gene X Locus. Mol Cell Biol (1992) 12:2124–34. doi: 10.1128/mcb.12.7.3313-b
32
Koppens PF Hoogenboezem T Degenhart HJ . Duplication of the CYP21A2 Gene Complicates Mutation Analysis of Steroid 21-Hydroxylase Deficiency: Characteristics of Three Unusual Haplotypes. Hum Genet (2002) 111:405–10. doi: 10.1007/s00439-002-0810-7
33
Chung EK Yang Y Rennebohm RM Lokki ML Higgins GC Jones KN et al . Genetic Sophistication of Human Complement Components C4A and C4B and RP-C4-CYP21-TNX (RCCX) Modules in the Major Histocompatibility Complex. Am J Hum Genet (2002) 71:823–37. doi: 10.1086/342777
34
Sekar A Bialas AR de Rivera H Davis A Hammond TR Kamitaki N et al . Schizophrenia Risk From Complex Variation of Complement Component 4. Nature (2016) 530:177–83. doi: 10.1038/nature16549
35
Parsa AA New MI . Steroid 21-Hydroxylase Deficiency in Congenital Adrenal Hyperplasia. J Steroid Biochem Mol Biol (2017) 165:2–11. doi: 10.1016/j.jsbmb.2016.06.015
36
El-Maouche D Arlt W Merke DP . Congenital Adrenal Hyperplasia. Lancet (2017) 17:31431–9. doi: 10.1016/S0140-6736(17)31431-9
37
Speiser PW . Nonclassic Adrenal Hyperplasia. Rev Endocr Metab Disord (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
38
Bachelot A Chakthoura Z Rouxel A Dulon J Touraine P . Classical Forms of Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency in Adults. Horm Res (2008) 69:203–11. doi: 10.1159/000113020
39
Higashi Y Yoshioka H Yamane M Gotoh O Fujii-Kuriyama Y . Complete Nucleotide Sequence of Two Steroid 21-Hydroxylase Genes Tandemly Arranged in Human Chromosome: A Pseudogene and a Genuine Gene. Proc Natl Acad Sci USA (1986) 83:2841–5. doi: 10.1073/pnas.83.9.2841
40
Higashi Y Tanae A Inoue H Fujii-Kuriyama Y . Evidence for Frequent Gene Conversion in the Steroid 21-Hydroxylase P-450(C21) Gene: Implications for Steroid 21-Hydroxylase Deficiency. Am J Hum Genet (1988) 42:17–25.
41
Tusié-Luna MT White PC . Gene Conversions and Unequal Crossovers Between CYP21 (Steroid 21-Hydroxylase Gene) and CYP21P Involve Different Mechanisms. Proc Natl Acad Sci USA (1995) 92:10796–800. doi: 10.1073/pnas.92.23.10796
42
Pignatelli D Carvalho BL Palmeiro A Barros A Guerreiro SG Macut D . The Complexities in Genotyping of Congenital Adrenal Hyperplasia: 21-Hydroxylase Deficiency. Front Endocrinol (Lausanne) (2019) 10:432. doi: 10.3389/fendo.2019.00432
43
Concolino P Costella A . Congenital Adrenal Hyperplasia (CAH) Due to 21-Hydroxylase Deficiency: A Comprehensive Focus on 233 Pathogenic Variants of CYP21A2 Gene. Mol Diagn Ther (2018) 22:261–80. doi: 10.1007/s40291-018-0319-y
44
Concolino P Paragliola RM . Molecular Analysis of 21-Hydroxylase Deficiency Reveals Two Novel Severe Genotypes in Affected Newborns. Mol Diagn Ther (2021) 25:327–37. doi: 10.1007/s40291-021-00520-y
45
Simonetti L Bruque CD Fernández CS Benavides-Mori B Delea M Kolomenski JE et al . CYP21A2 Mutation Update: Comprehensive Analysis of Databases and Published Genetic Variants. Hum Mutat (2018) 39:5–22. doi: 10.1002/humu.23351
46
Lee HH . The Chimeric CYP21P/CYP21 Gene and 21-Hydroxylase Deficiency. J Hum Genet (2004) 49:65–72. doi: 10.1007/s10038-003-0115-2
47
Chen W Xu Z Sullivan A Finkielstain GP Van Ryzin C Merke DP et al . Junction Site Analysis of Chimeric CYP21A1P/CYP21A2 Genes in 21-Hydroxylase Deficiency. Clin Chem (2012) 58:421–30. doi: 10.1373/clinchem.2011.174037
48
Concolino P Mello E Minucci A Giardina E Zuppi C Toscano V et al . A New CYP21A1P/CYP21A2 Chimeric Gene Identified in an Italian Woman Suffering From Classical Congenital Adrenal Hyperplasia Form. BMC Med Genet (2009) 10:72. doi: 10.1186/1471-2350-10-72
49
Vrzalová Z Hrubá Z Hrabincová ES Vrábelová S Votava F Koloušková S et al . Chimeric CYP21A1P/CYP21A2 Genes Identified in Czech Patients With Congenital Adrenal Hyperplasia. Eur J Med Genet (2011) 54:112–7. doi: 10.1016/j.ejmg.2010.10.005
50
Chu X Braun-Heimer L Rittner C Schneider PM . Identification of the Recombination Site Within the Steroid 21-Hydroxylase Gene (CYP21) of the HLA-B47, DR7 Haplotype. Exp Clin Immunogenet (1992) 9:80–5.
51
Helmberg A Tabarelli M Fuchs MA Keller E Dobler G Schnegg I et al . Identification of Molecular Defects Causing Congenital Adrenal Hyperplasia by Cloning and Differential Hybridization of Polymerase Chain Reaction-Amplified 21-Hydroxylase (CYP21) Genes. DNA Cell Biol (1992) 11:359–68. doi: 10.1089/dna.1992.11.359
52
Lee HH Lee YJ Chan P Lin CY . Use of PCR-Based Amplification Analysis as a Substitute for the Southern Blot Method for CYP21 Deletion Detection in Congenital Adrenal Hyperplasia. Clin Chem (2004) 50:1074–6. doi: 10.1373/clinchem.2003.028597
53
Lee HH Chang SF Lee YJ Raskin S Lin SJ Chao MC et al . Deletion of the C4-CYP21 Repeat Module Leading to the Formation of a Chimeric CYP21P/CYP21 Gene in a 9.3-Kb Fragment as a Cause of Steroid 21-Hydroxylase Deficiency. Clin Chem (2003) 49:319–22. doi: 10.1373/49.2.319
54
White PC New MI Dupont B . HLA-Linked Congenital Adrenal Hyperplasia Results From a Defective Gene Encoding a Cytochrome P-450 Specific for Steroid 21-Hydroxylation. Proc Natl Acad Sci USA (1984) 81:7505–9. doi: 10.1073/pnas.81.23.7505
55
L’Allemand D Tardy V Grüters A Schnabel D Krude H Morel Y . How a Patient Homozygous for a 30-Kb Deletion of the C4-CYP 21 Genomic Region can Have a Nonclassic Form of 21-Hydroxylase Deficiency. J Clin Endocrinol Metab (2000) 85:4562–7. doi: 10.1210/jcem.85.12.7018
56
Ezquieta B Beneyto M Mun˜oz-Pacheco R Barrio R Oyarzabal M Lechuga JL et al . Gene Duplications in 21-Hydroxylase Deficiency: The Importance of Accurate Molecular Diagnosis in Carrier Detection and Prenatal Diagnosis. Prenat Diagn (2006) 26:1172–8. doi: 10.1002/pd.1584
57
Kharrat M Riahi A Maazoul F M’rad R Chaabouni H . Detection of a Frequent Duplicated CYP21A2 Gene Carrying a Q318X Mutation in a General Population With Quantitative PCR Methods. Diagn Mol Pathol (2011) 20:123–7. doi: 10.1097/PDM.0b013e3181f24807
58
Parajes S Quinteiro C Domı´nguez F Loidi L . High Frequency of Copy Number Variations and Sequence Variants at CYP21A2 Locus: Implication for the Genetic Diagnosis of 21-Hydroxylase Deficiency. PloS One (2008) 3:e2138. doi: 10.1371/journal.pone.0002138
59
Wedell A Stengler B Luthman H . Characterization of Mutations on the Rare Duplicated C4/CYP21 Haplotype in Steroid 21-Hydroxylase Deficiency. Hum Genet (1994) 94:50–4. doi: 10.1007/BF02272841
60
Kleinle S Lang R Fischer GF Vierhapper H Waldhauser F Fo¨dinger M et al . Duplications of the Functional CYP21A2 Gene Are Primarily Restricted to Q318X Alleles: Evidence for a Founder Effect. J Clin Endocrinol Metab (2009) 94:3954–8. doi: 10.1210/jc.2009-0487
61
Koppens PF Hoogenboezem T Degenhart HJ . CYP21 and CYP21P Variability in Steroid 21-Hydroxylase Deficiency Patients and in the General Population in the Netherlands. Eur J Hum Genet (2000) 8:827–36. doi: 10.1038/sj.ejhg.5200543
62
Concolino P Mello E Minucci A Giardina B Capoluongo E . Genes, Pseudogenes and Like Genes: The Case of 21-Hydroxylase in Italian Population. Clin Chim Acta (2013) 424:85–9. doi: 10.1016/j.cca.2013.05.019
63
Concolino P . A Rare CYP21A2 Haplotype Clarifies the Phenotype-Genotype Discrepancy in an Italian Patient With Non Classical Congenital Adrenal Hyperplasia (NC-CAH). Mol Biol Rep (2020) 47:3049–52. doi: 10.1007/s11033-020-05379-6
64
Tsai LP Cheng CF Chuang SH Lee HH . Analysis of the CYP21A1P Pseudogene: Indication of Mutational Diversity and CYP21A2-Like and Duplicated CYP21A2 Genes. Anal Biochem (2011) 413:133–41. doi: 10.1016/j.ab.2011.02.016
65
Lekarev O Tafuri K Lane AH Zhu G Nakamoto JM BullerBurckle AM et al . Erroneous Prenatal Diagnosis of Congenital Adrenal Hyperplasia Owing to a Duplication of the CYP21A2 Gene. J Perinatol (2013) 33:76–8. doi: 10.1038/jp.2012.5
66
Sani I Rossodivita AN Mariani M Costella A Molinario R Concolino P et al . CYP21A2 Genetics: When Genotype Does Not Fit Phenotype. Clin Biochem (2016) 49:524–5. doi: 10.1016/j.clinbiochem.2015.07.022
67
Lee HH Lee YJ Lin CY . PCR-Based Detection of the CYP21 Deletion and TNXA/TNXB Hybrid in the RCCX Module. Genomics (2004) 83:944–50. doi: 10.1016/j.ygeno.2003.11.006
68
Malfait F Francomano C Byers P Belmont J Berglund B Black J et al . The 2017 International Classification of the Ehlers-Danlos Syndromes. Am J Med Genet C Semin Med Genet (2017) 175:8–26. doi: 10.1002/ajmg.c.31552
69
Schalkwijk J Zweers MC Steijlen PM Dean WB Taylor G van Vlijmen IM et al . A Recessive Form of the Ehlers-Danlos Syndrome Caused by Tenascin-X Deficiency. N Engl J Med (2001) 345:1167–75. doi: 10.1056/NEJMoa002939
70
Lindor NM Bristow J . Tenascin-X Deficiency in Autosomal Recessive Ehlers-Danlos Syndrome. Am J Med Genet A (2005) 135:75–80. doi: 10.1002/ajmg.a.30671
71
Lao Q Mallappa A Rueda Faucz F Joyal E Veeraraghavan P Chen W et al . A TNXB Splice Donor Site Variant as a Cause of Hypermobility Type Ehlers-Danlos syndrome in Patients With Congenital Adrenal Hyperplasia. Mol Genet Genomic Med (2021) 9:e1556. doi: 10.1002/mgg3.1556
72
Merke DP Chen W Morissette R Xu Z Van Ryzin C Sachdev V et al . Tenascin-X Haploinsufficiency Associated With Ehlers-Danlos Syndrome in Patients With Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab (2013) 98:E379–87. doi: 10.1210/jc.2012-3148
73
Lee HH . Chimeric CYP21P/CYP21 and TNXA/TNXB Genes in the RCCX Module. Mol Genet Metab (2005) 84:4–8. doi: 10.1016/j.ymgme.2004.09.009
74
Morissette R Chen W Perritt AF Dreiling JL Arai AE Sachdev V et al . Broadening the Spectrum of Ehlers Danlos Syndrome in Patients With Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab (2015) 100:E1143–52. doi: 10.1210/jc.2015-2232
75
Chen W Perritt AF Morissette R Dreiling JL Bohn MF Mallappa A et al . Ehlers-Danlos Syndrome Caused by Biallelic TNXB Variants in Patients With Congenital Adrenal Hyperplasia. Hum Mutat (2016) 37:893–7. doi: 10.1002/humu.23028
76
Burch GH Gong Y Liu W Dettman RW Curry CJ Smith L et al . Tenascin-X Deficiency Is Associated With Ehlers-Danlos Syndrome. Nat Genet (1997) 17:104–8. doi: 10.1038/ng0997-104
77
Marino R Garrido NP Ramirez P Notaristéfano G Moresco A Touzon MS et al . Ehlers-Danlos Syndrome: Molecular and Clinical Characterization of TNXA/TNXB Chimeras in Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab (2021) 22:dgab033. doi: 10.1210/clinem/dgab033
78
Lao Q Brookner B Merke DP . High-Throughput Screening for CYP21A1P-TNXA/TNXB Chimeric Genes Responsible for Ehlers-Danlos Syndrome in Patients With Congenital Adrenal Hyperplasia. J Mol Diagn (2019) 21:924–31. doi: 10.1016/j.jmoldx.2019.06.001
79
Gao Y Lu L Yu B Mao J Wang X Nie M et al . The Prevalence of the Chimeric TNXA/TNXB Gene and Clinical Symptoms of Ehlers-Danlos Syndrome With 21-Hydroxylase Deficiency. J Clin Endocrinol Metab (2020) 105:dgaa199. doi: 10.1210/clinem/dgaa199
80
Miller WL Merke DP . Tenascin-X, Congenital Adrenal Hyperplasia, and the CAH-X Syndrome. Horm Res Paediatr (2018) 89:352–61. doi: 10.1159/000481911
81
Zweers MC Bristow J Steijlen PM Dean WB Hamel BC Otero M et al . Haploinsufficiency of TNXB Is Associated With Hypermobility Type of Ehlers-Danlos Syndrome. Am J Hum Genet (2003) 73:214–7. doi: 10.1086/376564
82
Lao Q Merke DP . Letter to the Editor From Lao and Merke: “Ehlers-Danlos Syndrome: Molecular and Clinical Characterization of TNXA/TNXB Chimeras in Congenital Adrenal Hyperplasia”. J Clin Endocrinol Metab (2021) 106:e2835–e2836. doi: 10.1210/clinem/dgab280
83
Mortensen S Kidmose RT Petersen SV Szilágyi Á Prohászka Z Andersen GR . Structural Basis for the Function of Complement Component C4 Within the Classical and Lectin Pathways of Complement. J Immunol (2015) 194:5488–96. doi: 10.4049/jimmunol.1500087
84
Blanchong CA Chung EK Rupert KL Yang Y Yang Z Zhou B et al . Genetic, Structural and Functional Diversities of Human Complement Components C4A and C4B and Their Mouse Homologues, Slp and C4. Int Immunopharmacol (2001) 1:365–92. doi: 10.1016/s1567-5769(01)00019-4
85
Law SK Dodds AW Porter RR . A Comparison of the Properties of Two Classes, C4A and C4B, of the Human Complement Component C4. EMBO J (1984) 3:1819–23. doi: 10.1002/j.1460-2075.1984.tb02052.x
86
Chung EK Yang Y Rupert KL Jones KN Rennebohm RM Blanchong CA et al . Determining the One, Two, Three, or Four Long and Short Loci of Human Complement C4 in a Major Histocompatibility Complex Haplotype Encoding C4A or C4B Proteins. Am J Hum Genet (2002) 71:810–22. doi: 10.1086/342778
87
Chung EK Wu YL Yang Y Zhou B Yu CY . Human Complement Components C4A and C4B Genetic Diversities: Complex Genotypes and Phenotypes. Curr Protoc Immunol (2005). doi: 10.1002/0471142735.im1308s68
88
Ricklin D Hajishengallis G Yang K Lambris JD . Complement: A Key System for Immune Surveillance and Homeostasis. Nat Immunol (2010) 11:785–97. doi: 10.1038/ni.1923
89
Yilmaz M Yalcin E Presumey J Aw E Ma M Whelan CW et al . Overexpression of Schizophrenia Susceptibility Factor Human Complement C4A Promotes Excessive Synaptic Loss and Behavioral Changes in Mice. Nat Neurosci (2021) 24:214–24. doi: 10.1038/s41593-020-00763-8
90
Zorzetto M Datturi F Divizia L Pistono C Campo I De Silvestri A et al . Complement C4A and C4B Gene Copy Number Study in Alzheimer’s Disease Patients. Curr Alzheimer Res (2017) 14:303–8. doi: 10.2174/1567205013666161013091934
91
Li N Zhang J Liao D Yang L Wang Y Hou S . Association Between C4, C4A, and C4B Copy Number Variations and Susceptibility to Autoimmune Diseases: A Meta-Analysis. Sci Rep (2017) 7:42628. doi: 10.1038/srep42628
92
Samano ES Ribeiro Lde M Gorescu RG Rocha KC Grumach AS . Involvement of C4 Allotypes in the Pathogenesis of Human Diseases. Rev do Hosp das Clin (2004) 59:138–44. doi: 10.1590/s0041-87812004000300009
93
Wu YL Yang Y Chung EK Zhou B Kitzmiller KJ Savelli SL et al . Phenotypes, Genotypes and Disease Susceptibility Associated With Gene Copy Number Variations: Complement C4 CNVs in European American Healthy Subjects and Those With Systemic Lupus Erythematosus. Cytogenet Genome Res (2008) 123:131–41. doi: 10.1159/000184700
94
Yang Y Chung EK Wu YL Savelli SL Nagaraja HN Zhou B et al . Gene Copy-Number Variation and Associated Polymorphisms of Complement Component C4 in Human Systemic Lupus Erythematosus (SLE): Low Copy Number Is a Risk Factor for and High Copy Number is a Protective Factor Against SLE Susceptibility in European Americans. Am J Hum Genet (2007) 80:1037–54. doi: 10.1086/518257
95
Jüptner M Flachsbart F Caliebe A Lieb W Schreiber S Zeuner R et al . Low Copy Numbers of Complement C4 and Homozygous Deficiency of C4A May Predispose to Severe Disease and Earlier Disease Onset in Patients With Systemic Lupus Erythematosus. Lupus (2018) 27:600–9. doi: 10.1177/0961203317735187
96
Yang Y Chung EK Zhou B Lhotta K Hebert LA Birmingham DJ et al . The Intricate Role of Complement Component C4 in Human Systemic Lupus Erythematosus. Curr Dir Autoimmun (2004) 7:98–132. doi: 10.1159/000075689
97
Boteva L Morris DL Cortés-Hernández J Martin J Vyse TJ Fernando MM . Genetically Determined Partial Complement C4 Deficiency States Are Not Independent Risk Factors for SLE in UK and Spanish Populations. Am J Hum Genet (2012) 90:445–56. doi: 10.1016/j.ajhg.2012.01.012
98
Lao Q Jardin MD Jayakrishnan R Ernst M Merke DP . Complement Component 4 Variations may Influence Psychopathology Risk in Patients With Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. Hum Genet (2018) 137:955–60. doi: 10.1007/s00439-018-1959-z
99
Falhammar H Frisén L Hirschberg AL Nordenskjöld A Almqvist C Nordenström A . Increased Risk of Autoimmune Disorders in 21-Hydroxylase Deficiency: A Swedish Population-Based National Cohort Study. J Endocr Soc (2019) 3:1039–52. doi: 10.1210/js.2019-00122
100
Chen W Xu Z Nishitani M Van Ryzin C McDonnell NB Merke DP . Complement Component 4 Copy Number Variation and CYP21A2 Genotype Associations in Patients With Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. Hum Genet (2012) 131:1889–94. doi: 10.1007/s00439-012-1217-8
101
Baumgartner-Parzer S Witsch-Baumgartner M Hoeppner W . EMQN Best Practice Guidelines for Molecular Genetic Testing and Reporting of 21-Hydroxylase Deficiency. Eur J Hum Genet (2020) 28:1341–67. doi: 10.1038/s41431-020-0653-5
102
Lao Q Merke DP . Molecular Genetic Testing of Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency Should Include CAH-X Chimeras. Eur J Hum Genet (2021) 29:1047–8. doi: 10.1038/s41431-021-00870-5
103
Baumgartner-Parzer S Witsch-Baumgartner M Hoeppner W . Reply to Lao Q and Merke Dp. Eur J Hum Genet (2021) 29:1045–6. doi: 10.1038/s41431-021-00869-y
104
Szilagyi A Fust G . Diseases Associated With the Low Copy Number of the C4B Gene Encoding C4, the Fourth Component of Complement. Cytogenet Genome Res (2008) 123:118–30. doi: 10.1159/000184699
Summary
Keywords
RCCX, haplotypes, Congenital Adrenal Hyperplasia (CAH), CAH-X, Copy Number Variation (CNV), Complement Component C4
Citation
Carrozza C, Foca L, De Paolis E and Concolino P (2021) Genes and Pseudogenes: Complexity of the RCCX Locus and Disease. Front. Endocrinol. 12:709758. doi: 10.3389/fendo.2021.709758
Received
14 May 2021
Accepted
19 July 2021
Published
30 July 2021
Volume
12 - 2021
Edited by
Liliana Dain, Centro Nacional de Genética Médica, Argentina
Reviewed by
Mirjana Kocova, Saints Cyril and Methodius University of Skopje, North Macedonia; Shin Matsubara, Suntory Foundation for Life Sciences, Japan
Updates
Copyright
© 2021 Carrozza, Foca, De Paolis and Concolino.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Concolino, paola.concolino@policlinicogemelli.it
This article was submitted to Cellular Endocrinology, a section of the journal Frontiers in Endocrinology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.