 Jing Yang1,2,3,4
Jing Yang1,2,3,4 Zhangsuo Liu2,3,4,5*
Zhangsuo Liu2,3,4,5*- 1Department of Ophthalmology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 2Research Institute of Nephrology, Zhengzhou University, Zhengzhou, China
- 3Henan Province Research Center For Kidney Disease, Zhengzhou, China
- 4Key Laboratory of Precision Diagnosis and Treatment for Chronic Kidney Disease in Henan Province, Zhengzhou, China
- 5Department of Integrated Traditional and Western Nephrology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
Diabetic nephropathy (DN) and diabetic retinopathy (DR) are microvascular complications of diabetes. Microvascular endothelial cells are thought to be the major targets of hyperglycemic injury. In diabetic microvasculature, the intracellular hyperglycemia causes damages to the vascular endothelium, via multiple pathophysiological process consist of inflammation, endothelial cell crosstalk with podocytes/pericytes and exosomes. In addition, DN and DR diseases development are involved in several critical regulators including the cell adhesion molecules (CAMs), the vascular endothelial growth factor (VEGF) family and the Notch signal. The present review attempts to gain a deeper understanding of the pathogenesis complexities underlying the endothelial dysfunction in diabetes diabetic and retinopathy, contributing to the development of new mechanistic therapeutic strategies against diabetes-induced microvascular endothelial dysfunction.
1 Introduction
The incidence of diabetes Mellitus is increasing dramatically worldwide. Epidemiological studies have shown that the incidence of diabetes will be expected to increase by over 50% by 2045 compared with 2017 (1). Diabetic diseases will be the 7th cause of mortality by 2030 in the world (1–4). Diabetic vascular complicating diseases are the primary cause of mortality in diabetes sufferers, the most common of which are diabetic nephropathy and diabetic retinopathy. As per the epidemiology investigation of T2D in PRC’s large cities, diabetic nephropathy and diabetic retinopathy account for 39.7% and 31.5% of diabetic microangiopathy, respectively (5). Current research reveals that diabetic microangiopathy (mainly refers to diabetic nephropathy and diabetic retinopathy) cannot be fully explained by the interaction between hemodynamics and metabolic factors or molecular modification in the state of high blood glucose. Clear evidence shows that its pathogenesis is multifactorial.
The coincidence of retina and kidney pathology in diabetic patients is well recognized, and certain literatures have proposed the definition of “renal-retinal syndrome” (6), in which microvascular endothelial dysfunction is an essential common pathological mechanism in the early development phase of the two diseases: microvascular endothelial cells are the main target of hyperglycemia damage. When the blood glucose concentration increases, the microvascular endothelial cells down-regulate the glucose transport rate, leading to intracellular hyperglycemia (7, 8) and a series of critical downstream pathways such as polyol pathway flux, elevated AGEs, elevated expressing of AGEs receptors and their activation ligands, stimulation of PKC subtypes and aminohexose signal path (9–11).
2 The Critical Role of the Endothelial Barrier in the Kidney and Retina
2.1 Glomerular Endothelial Barrier
The glomerulus barrier is the most intricate biomembrane, characterized by the permission of great water filtration rate, the unrestricted passing of small and medium molecules, and complete constraint of serum ALB and large protein (12–15). The glomerular basement membrane (GBM) and glomerular capillary endothelial cells, endothelial cell surface membrane, and podocytes constitute a glomerular filtration barrier (13, 14). GBM is a special basement membrane, mainly composed of a substrate and negatively charged proteoglycans. GBM comprises a fiber net involving IV-C, laminin, nidogen/entactin and heparin sulfate protein polysaccharides (15–18).
The GBM of a glomerulus capillary is about 240-370 nm thicker than that (40-80 nm) of other vascular beds (13–15). The glomerular endothelial cells are abnormally flat, and the circum-capillary ring height is 50-150 nm (14, 15). The fenestration area of glomerular capillary endothelial cells is enormous, accounting for 20-50% of the whole endothelium surface (16–20). The type IV collagen network is the framework of GBM (19–21). The basement membrane contains many proteoglycans, mainly attached to heparan sulfate chains, which are pivotal for the selective penetration of the barrier (22–25). Recent studies have confirmed that most of the valid charge density in glomerulus barriers is situated at the endothelium or epithelium layer. The occurrence of proteinuria precedes the morphological changes of podocytes in mice, indicating that GBM is a vital component of the glomerulus barrier (26, 27). On the lumen side, the vascular wall is covered with the ESL, comprising negatively charged glycosidoproteins, GAG, and membrane-related and excreted protein polysaccharides. This layer is involved in blood coagulation, angiogenesis regulation, rheology and capillary barrier (28).
2.2 Blood-Retinal Endothelial Barrier
The blood-eye barrier plays an essential role in protecting and maintaining the best visual cell function. It provides a greatly modulated chemical milieu for the intraocular non-vascular transparent tissue and serves as a drain to discharge waste from the metabolic activities of ocular tissue (29, 30). The blood-eye barrier includes 2 primary barrier systems: the BAB and the BRB (29, 30). Resembling the BBB, BRB regulates the transport of substances in the retinal capillaries, maintaining neural homeostasis, and prevents the leakage of macromolecules and other potentially harmful substances into the retina. The integrity of BRB is closely related to the structure and function of the retina (29, 30). Evidence indicates that BRB injury causes vascular permeability transition and is highly correlated to the pathophysiological process of many blinding kidney illnesses, like DR (29–31) and retina macula degeneration (30), etc.
BRB can be classified into inner BRB (iBRB) and outer BRB (oBRB). The internal barrier mainly comprises continuous RMECs and tight junctions (TJs) (32). The outer barrier comprises adjacent retinal pigment epithelium (RPE) cells and tight junctions (30, 31, 33). In the inner and outer barriers, cells are tightly connected to restrict fluids and molecules outside the barrier, and endothelial cells along with RPE cells actively regulate the discharge of fluids and molecules. Thus, although the amino acid or aliphatic acid in plasma fluctuates significantly, their concentration in the retina remains relatively stable.
Retinal vascular endothelial cells are single-layer cells covering the vascular cavity, acting as a selective barrier between the neural retina and blood circulation and providing nutrition and oxygen to the neural retina (29–31). Retinal vascular endothelial cell connections include tight junctions, adherence junctions (AJs) and gap junctions (GJs), which together form a complex to regulate cell permeability and maintain cell polarity to mediate cell adhesion and mutual communication and maintain the local microenvironment stability of the retina (32, 33). Among them, TJs is the most critical connection structure of BRB. The sound play of BRB’s normal function mainly depends on the TJs protein between vascular endothelial cells, including a variety of proteins such as transmembrane proteins Claudins, Occludins, membrane-related proteins (such as ZO-1, ZO- 2 and ZO-3) and TJs adhesion factor, etc. (32, 33). Moreover, pericytes, astrocytes, Muller cells and microglia in the retina exert a significant impact on the normal function of BRB (29–31). Muller cells extend from the subretinal space to the vitreous and act as anatomical and functional connection scaffolds between neurons and blood vessels, which can regulate the tension of blood vessels and maintain the integrity of BRB (34). Pericytes can regulate the local bloodstream and vasopermeability of retinal capillaries and support vascular endothelial cells. The loss of Pericytes will accelerate the destruction of BRB, increase vascular permeability, and lead to angiogenic macular edema (35).
3 The Effects of Endothelium Function Disorder on the Etiopathogenesis of DN and DR
3.1 Endothelial Dysfunction in DN
DN is the most commonly seen micro vascular complicating disease of diabetic sufferers. Its incidence is about 30% in T1D patients and 40% in T2D patients. In 1983, for the first time, Mogensen reported that changes in renal microvascular in diabetic patients could cause diabetic nephropathy, characterized by glomerular endothelial cell (GEC) damage, GBM thickening, and glomerular mesangial matrix proliferation and nodular glomerulosclerosis (36–41). In recent years, many reports have revealed that approximately 12-55% of ESRDs are due to diabetes (42–44). The opening of GECs and endothelial cells around the renal tubules is fairly significant for the selective penetration of the glomerular filtration barrier and the effective passage of large amounts of urine (45–47). GECs are lined with thick and negatively-charged filar glycocalyx, forming a network with glycosaminoglycans. The glycocalyx regulates vascular permeability and fluid balance and repels blood cells from the blood vessel wall. As DN progresses, a gradual decrease of the fenestrated endothelial surface can be observed in diabetic patients. In T2D sufferers, the decrease of glomerular endothelial pores is closely related to the loss of proteinuria and GFR (45–48). In terms of diabetes, hyperglycemia and its by-products can promote the growth of pro-oxidants and induce a pro-inflammatory environment featuring GEC dysfunction, which leads to proteinuria and renal fibrosis (41–44). GEC dysfunction is a multifactorial progression, which includes increased permeability of GEC, induction of endothelial cell apoptosis, glycocalyx decomposition, and impaired crosstalk between endothelial cells and other kidney cells (podocyte for instance) (40, 43, 44). GEC damage is attributed to the incidence of microalbuminuria, and this is an early event of DN (40). Microalbuminuria is also a sign of renal and systemic endothelial dysfunction (49, 50).
3.2 Endothelial Dysfunction in Diabetic Retinopathy
Since diabetes is becoming more common worldwide, DR is the most commonly seen and most serious ocular complication in many developed countries (9, 51–57). As one of the most active tissues in terms of metabolism, the retina is extremely sensitive to fluctuations in blood glucose levels. The pathological process of diabetic retinopathy consists of 2 stages: NPDR and PDR. As an early phase, NPDR is featured by PC loss in the retina capillary, which helps generate decellularized capillaries and improves microvascular penetrability as well as exacerbates endothelial BRB damage (9, 52–54, 57). Fibrovascular preretinal membranes, intravitreous hemorrhages, and amotio retinae may result from PDR, which is an advanced stage of retinal degeneration where fragile and tortuous new blood vessels form in the retina (9, 53–56). Hyperglycemia causes REC damage and the destruction of the blood-retinal barrier, resulting in the cumulation of exocellular fluids in maculas, as well as the capillary basement membrane thickening and a resulting ECM deposition (42–44). Due to the upregulation of angiogenesis factors (like VEGF), continual damage to the retinal microvasculature leads to retinal ischemia and capillary non-perfusion (45–50). Diabetic retinopathy not only impairs vision, but also elevates the risk of other life-threatening systemic vascular complications (40, 41).
DR pathogenesis involves many factors and approaches that lead to vascular dysfunction, especially endothelial dysfunction. As a result of hyperglycemia, retinal microangiopathy, inflammation, and neurodegeneration can occur, all of which can damage the blood-retinal barrier (BRB) and decrease blood flow, leukocyte stasis, and cause endothelial damage, increased vascular permeability, and pathological angiogenesis. It manifests clinically as decellularized capillaries and retinal angioedema (9, 51). As a result of hyperglycemia, retinal endothelial cells are damaged by ischemia, oxidative stress, and the release of pro-inflammatory factors. Damage to the blood-retinal barrier is now recognized as the mechanism.
BRB injury causes solute leakage and a rise in capillary pressure, which increase the osmotic pressure of the intercellular substance and lead to macular edema. DME is the primary cause of visual loss in diabetes sufferers, especially those with NPDR of T2D. In DME, there is vessel leakage, tissue edema, and complex exudate deposition in retinas. DR’s clinical symptoms appearing before persistent high blood glucose induces the loss of autoregulation in retinal cells and the tortuous expansion of arteries and veins (58–60). In diabetic retinopathy, retinal arteriovenous dilation raises hydrostatic pressure of capillaries, causing tissue edema via the Starlin rule, resulting in capillary wall dilation (microaneurysms) and rupture (hematomas), which are all typical symptoms of the condition (60). A reduction in capillaries may increase the production of VEGF and the rest of permeability factors, causing the rupture of BRBs and the extravasation of plasmatic protein into the interstitium. In addition to the severe leakage of DME, there are also slight variations in BRB in the early stages of PCDR (Pre-clinical diabetic retinopathy) (61). PCDR occurs earlier than clinical DR that threatens vision, which takes 8-15 years to show visible symptoms. PCDR is featured by PC loss, the basal layer thickening, and vascular retraction (62, 63). The increase in retinal vasopermeability in PCDR is featured by the existence of extravascular ALB, a decrease in the intercellular junctional complex, and an increase in the number of endothelial intracellular vesicles. In addition, PCDR is featured by diffuse loss of BRB without any formation of edema, as opposed to the large leakage of DME in the late stages of NPDR, and the diffuse increase in retinal vascular permeability is considered a key process that can further lead to NPDR (64, 65). In conclusion, a better understanding of the pathogenesis of BRB loss in the early and late phases of DR is helpful in the design of more valid DME therapies and in providing potential solutions to prevent DR from developing into NPDR.
4 Classic Regulators of the Endothelial Dysfunction of Diabetic Nephropathy and Diabetic Retinopathy
4.1 Cell Adhesion Molecule (CAM)
Studies have shown that CAMs are pivotal for the pathogenesis of vascular complications. Cellular proliferation, differentiation, the forming of cellular connections and the stimulation of white blood cells in inflammation sites, are some of the functions performed by these molecules (66–69). A variety of CAMs, such as ICAM-1, VCAM-1, and PECAM-1, can be found in the cell junction and are vital for the adhesion and migration of monocytes and lymphocytes to endothelial cells (66, 67). The soluble form of ICAM-1 is a marker for EC inflammatory events and damages. PECAM-1 is pivotal for maintaining the integrity of vessels and the barrier function of endothelial cells (68). It has been demonstrated that IL-1, TNF-β, VEGF, and NF-κB can stimulate the expressing of ICAM-1 and VCAM-1 in endothelia (68, 69).
In diabetic patients, the elevated level of AGEs and ROS can activate leukocytes and increase NF-κB transcription in endothelial cells (70–73). NF-κβ also increases the expressing of ICAM-1 in high glucose (74). The increased VCAM-1 expression and ICAM-1 expression in high glucose has proven to be directly associated with the occurrence and development of DN. An increase in VCAM-1 expression has been observed in diabetic mouse models. According to cross-sectional studies, VCAM-1 circulating level and ICAM-1 circulating level in diabetes sufferers with nephropathy are higher than those in patients without renal injury (75, 76). VCAM-1 circulating levels are associated with proteinuria (76). Elevated plasma levels of ICAM-1 in T1DM patients are independently correlated with the occurrence of continuous microalbuminuria (77–84). The interaction between ICAM-1 expressed by GECs and LFA-1 is thought to be essential for migrating T cells to the kidney (78). ICAM-1 deficient db/db diabetic mice displayed a diminished ability to homing to the glomerulus as compared with ICAM-1 sufficient db/db diabetic mice (85).
Numerous evidences suggest that inflammatory events are vital for the etiopathogenesis of DR (71, 86). Furthermore, NF-κB activation promotes the inflammatory cascade in the vitreous humor and serum, thereby upregulating ET-1 and downregulating eNOS, causing REC damage, capillary stenosis, retinal ischemia, and abnormal blood flow. An activated form of NF-κB also overexpresses ICAM-1, leading to REC damage and the destruction of the blood-retinal barrier (87, 88). ICAM-1 and VCAM-1 are regulated upward in the conjunctival tissues of diabetic sufferers (71, 86, 89), and ICAM-1 is capable of acting synergistically with RAGE to realize the mediation of white blood cell recruiting in the event of acute inflammation (90). In addition to VEGF, studies have shown that there is a positive association between the sera ICAM-1 level and the degree of damage in the external limiting membrane (ELM) (91). Hence, supervising the level of serum dissolvable VCAM-1 in diabetic sufferers might contribute to evaluating the seriousness and activities of DR (92, 93).
4.2 VEGF Family
VEGF belongs to the super gene family of VEGF/PDGF, and the most widely studied member is VEGF-A (94–99) that comprises 8 exons. Utilizing variable splicing, at least 6 diverse isoforms are produced, which can be differentiated by AA lengths, including VEGF121, VEGF145, VEGF165, VEGF183, VEGF189 and VEGF206, among which the most affluent VEGF-A isoform in humans is VEGF165 (94, 99). VEGF-A is especially vital for the differentiation, proliferation and survival of endothelial cells of the vasculature, promoting endothelium-dependent vasodilation and raising vascular permeability (94, 99). VEGF-A triggers endocellular signal transduction by connecting to either VEGFR-1 or VEGFR-2, and the coreceptors NP1 and NP2. VEGFR-2 is accountable for the mediation of the majority of VEGF-A biology roles at normal status (96–99).
In kidneys, VEGF-A is expressed mainly by glomerulus foot cells, and VEGF-A mRNAs can be detected as well in the distal tubule and collecting duct (100–103), while VEGFR-2 is expressed by GECs and pericapillary ECs, as well as cortex interstitial fibroblasts and renal medulla interstitial fibroblasts (104–107). Therefore, the typical VEGF signal transduction in the glomerulus can be considered as the process where podocytes secrete VEGF-A and then pass through the filter barrier contrary to urine flow and bind to VEGFR-2 expressed by glomerular endothelial cells surface (104–107). The expressing of VEGF-A by foot cells is imperative for healthy renal development. Studies have revealed that there is a paracrine VEGF/VEGFR2 regulatory loop between GEC and podocytes (105). The overall loss of VEGFR2 leads to significant kidney abnormalities and glomerular microvascular system deficiency. VEGF generated by podocytes modulates the structure and functions of adjacent endothelial cells. Experiments have shown that both the expression of VEGF and VEGFR2 in kidneys increase in the early phase of DKD (106, 107), and the inhibition of VEGF-A or VEGF receptor in diabetic animals can prevent proteinuria and alleviate glomerular damage (106, 107). Although the activation of VEGF/VEGFR in the early phase of DKD can lead to novel blood vessel formation and other glomerular damage, the loss of podocytes in the later stage induces a decrease in VEGF signal, which triggers sparse blood vessels and renal fibrosis. Recent studies have indicated that the usage VEGF antagonists can protect kidneys of most diabetic rodents (104, 107), while for cancer patients and some non-diabetic nephropathy rodents, if they receive anti-VEGF treatment, their kidneys will potentially be damaged (108).
In the etiopathogenesis of DR, VEGF is considered a vital vascular growth factor that is implicated in pathological retinal neovascularization and increases vascular permeability (61, 109–119). The majority of literature reports and clinical data demonstrate that VEGF is increased in the vitreous of PDR patients (61, 113–119). In addition to endothelial cells, other retinal cells can also produce VEGF when activated or stimulated continuously by high glucose levels, like RPEs, PCs, Müller cells, stellate cells, and glial cells (117, 118). VEGF is vital for the modulation of ocular angiogenesis and vascular permeability. In the vitreous humor and fibrovascular tissue of PDR eyes, elevated levels of VEGF can be observed (61, 119). VEGF levels in the serum and vitreous are related to blood glucose control in diabetic patients (119). There is a significant association between the increase in VEGF contents in the vitreous body and the severity of DR (61, 119). Diabetes-induced increases in the level of VEGF are considered a biomarker of DR severity. Experiments have revealed that intravitreal injection of VEGF can produce various symptoms of NPDR and PDR: nonperfused capillaries, vasodilatation, and tortuous arterioles characterized by endothelial hyperplasia and microaneurysms (61, 118, 119). The degree of damage to the external limiting membrane (ELM) is correlated positively with the level of serum VEGF, which suggests that the level of VEGF is related to the severity of DR and the degree of damage to the external limiting membrane. VEGF regulates DR-related inflammation in the early phase (118). There might be a correlation between the elevated stimulation of NF-*B in NPDR as well as PDR sufferers and the increased expression of VEGF (119). VEGF induces the expression of MCP-1, IL-8, TNF-α and ICAM-1 in retinal endothelial cells via stimulating the NF-kB pathway (61). VEGF boosts the adhesion of leukocytes to the blood vessel wall by enhancing the expressing of ICAM-1 and VCAM-1 of ECs (61, 117). Furthermore, VEGF might promote early diabetes retina white blood cell adhesion in retina arterioles by upregulating the expressing of ICAM-1 (118). It has been reported that an increase in serum VEGF levels can stimulate the formation of ROS, resulting in endothelial activation (119).
The PDGF family includes PDGF-A, PDGF-B, PDGF-C, and PDGF-D. PDGFs are secreted by endothelial cells, macrophages and epithelial cells (120–122). PDGF-C is more structurally similar to VEGF than PDGF-B, while VEGF shares approximately 25% sequence similarity to all PDGFs (123). PDGFs exert their diverse functions under physiological or pathological conditions by binding to platelet-derived growth factor receptors (PDGFRs) (124, 125). PDGF is involved in the embryonic development of many organs, including the brain, lung, retina, vasculature, and kidney, of which the most interesting is the role of PDGF in the vasculature (124–128). PDGFs play an important role in physiological angiogenesis by recruiting vascular endothelial cells, a large number of studies have shown that PDGFs and their receptors play a key role in many ocular neovascular diseases, such as proliferative diabetic retinopathy (PDR), retinopathy of prematurity (ROP), and neovascular age-related macular degeneration (NVAMD) (122, 128). Three isoforms of PDGF (AA, AB, and BB) have been shown not only to be involved in the process of neovascularization in PDR, but also to play an important role in the formation of fibrotic tissue in the retina of PDR patients (128). The concentration of PDGF-AB in the vitreous humor of patients with diabetic retinopathy is increased, and PDGF-C and PDGF-D are also abundantly expressed in the retina (128). Recent studies in ocular neovascularization models have shown that simultaneous inhibition of VEGF and PDGF, especially PDGF-B, enhances antiangiogenic effects (122–125). PDGF is also upregulated in diabetic nephropathy, and both imatinib and PDGFR-β gene knockout attenuated renal injury, especially mesangial dilation, in different diabetic nephropathy mouse models (129–132). Notably, PDGF has been extensively reported to play a beneficial repair role in diabetic foot and high glucose-induced endothelial progenitor cell injury, rather than the negative role of PDGF currently reported in DR or DN (126, 127).
4.3 Notch Signaling
The Notch signaling pathway is a highly conserved cellular signal transduction system present in most multicellular organisms. Signals are transmitted between adjacent cells through Notch receptors, which can regulate cell differentiation, proliferation and apoptosis (133–135). The Notch receptor is a single transmembrane protein encoded by the Notch gene, which is a hetero-oligomer composed of a large extracellular part, which contains a short extracellular domain through calcium-dependent non-covalent interaction with a short extracellular domain (136). Notch protein interaction between the transmembrane domain and a short intramembrane domain. Four Notch receptors (Notch 1, 2, 3, 4) have been found in mammals including humans (137). Notch ligands are also single-transmembrane proteins expressed on the cell surface, and adjacent cells transmit Notch signals through the binding of Notch receptors and ligands. There are five Notch ligands in the human body, namely Jagged 1, Jagged 2, Delta 1, Delta 3 and Delta 4 (137). After Notch ligand binds to the receptor, the activation of Notch signal is triggered, and the Notch receptor undergoes two proteolysis successively and is further transferred to the nucleus, thereby activating the transcription of target genes and exerting biological functions (138). The role of the Notch pathway as a master regulator of angiogenesis is well established, and Active Notch provides a pro-survival, anti-inflammatory and anti-atherosclerotic environment that reduces adhesion molecules such as ICAM-1 and VCAM-1 (139). Expressed to maintain endothelial integrity by participating in the formation of endothelial junction complexes (139).
In the mouse retinal vasculature, active Notch1, Jag1, Dll1 and Dll4 have been described with distinct distribution patterns (140). Cellular analysis and gene inactivation or mutation of mouse or zebrafish retinas, combined with ocular disease models, have demonstrated that Notch signaling plays an important role in embryonic and postnatal ocular angiogenesis and is involved in angiogenic ocular diseases (141). Recent findings show that NOTCH1 signaling in retinal microvascular endothelial cells induces vascular permeability in diabetes: NOTCH1 ligands JAGGED1 and DELTA LIKE-4 are secondary to hyperglycemia and activate the canonical and rapid atypical NOTCH1 pathways, ultimately disrupts adherent junctions between endothelial cells by causing diabetic endothelial dissociation (142). Additionally, modulation of Notch signaling enhanced neovascularization and reperfusion in diabetic mice by modulating EC responsiveness to VEGF. In PDR, aberrant Notch signaling may interact with VEGF signaling at multiple levels to mediate retinal microvascular endothelial cell dysfunction (143).
The vast majority of research on Notch signaling in diabetic nephropathy has focused on the effects of Notch signaling on podocytes and tubular epithelial cells (144–149). Although no studies have directly assessed the role of Notch signaling on renal arteriole function or neovascularization in diabetic nephropathy, Notch signaling does inhibit diabetic extrarenal angiogenesis by altering the sensitivity of hemangioblasts to VEGF (150, 151). Although its effect on glomerular endothelial cells in diabetic nephropathy remains to be further studied, the role of Notch signaling in microvascular endothelial cells in diabetic retinopathy and diabetic ulcers has been confirmed (152, 153).
4.4 ROS
ROS are oxygen-derived, highly reactive molecules, including free radicals such as superoxide (O 2-) or hydroxyl (•OH) and non-radical ROS such as hydrogen peroxide (H2O2) (154). ROS have physiological roles in cell signaling related to cell proliferation and survival, and are tightly regulated and balanced by cellular antioxidant responses (155). There is increasing evidence that ROS from ECs and other cell types such as vascular smooth muscle cells, myeloid cells such as neutrophils and macrophages can also stimulate angiogenesis (155). There are many sources of ROS, including NADPH oxidase (NOX), mitochondrial electron transport chain (ETC), xanthine oxidase, unconjugated endothelial nitric oxide synthase (eNOS), cytochrome P-450 oxygenase, and cyclic oxidase (156). Vascular NOX isoforms (Nox1, Nox2, Nox4, and Nox5) differ in activity and cell specificity in response to agonists, growth factors and hypoxia, and the types of ROS released upon activation (157). In ECs, ROS originate from NOX (especially Nox2 and Nox4) in the plasma membrane or the intracellular cytoplasmic compartment, as well as mitochondria, and play a key role in the angiogenic response induced by growth factors such as VEGF (158). Excessive ROS can lead to oxidative stress, which can lead to various cellular changes, which can lead to organ dysfunction and trigger diseases such as cancers, atherosclerosis and diabetic microvascular complications (159).
ROS play a direct role in diabetic nephropathy. In GECs, hyperglycemia saturates glucose metabolism and leads to activation of deleterious pathways, such as the polyol pathway, hexosamine pathway, AGE/RAGE axis, and PKC pathway, leading to overproduction of endogenous ROS (160). Oxidative stress and inflammation and their interactions are considered major pillars of CKD pathogenesis and progression. Oxidative stress promotes inflammation through the formation of pro-inflammatory oxidized lipids, AOPPs, and AGEs (advanced glycation end products), while activation of nuclear factor kappa B (NFκB) transcription factors in a pro-oxidative environment promotes the expression and recruitment of pro-inflammatory cytokines (161). Activation of leukocytes and other resident proinflammatory cells (161). Likewise, pro-inflammatory cytokines, such as tumor necrosis factor-α (TNFα), bind to their receptors on glomerular endothelial cells and trigger signaling pathways that activate the nuclear factor kappa B (NF-κB) transcription factor (162). In addition, in the context of chronic inflammation, activated leukocytes produce ROS, chlorine and nitrogen, thereby exacerbating and perpetuating oxidative stress (162). Indeed, initial ROS-mediated renal injury triggers subsequent renal and systemic inflammatory responses. Thus, in a cohort of 176 patients with CKD stages 1 to 5, serum levels of hs-CRP, interleukin-6, and malondialdehyde were significantly elevated and inversely correlated with GFR, while serum SOD and glutathione peroxidase levels were significantly reduced (161). Notably, IL-6 and hs-CRP were positively correlated with malondialdehyde, and with superoxide dismutase and glutathione peroxidase negative correlation, further supporting the relationship between inflammation and oxidative stress in CKD (161). One of the most important functions of the endothelium is the secretion of nitric oxide (NO), a relatively unstable diatomic free radical involved in a variety of biological processes, including in smooth muscle cells mediated by cyclic guanosine monophosphate (cGMP) vasodilation, inflammation and immune response (163). The relationship between NO and ROS is bidirectional, low levels of NO in the endothelium induce the expression of antioxidant genes and protect renal endothelial and mesangial cells from apoptosis and fibrosis, while on the other hand, increased ROS levels reduce Endothelial cells generate NO through inhibition and/or uncoupling of NOS enzymes (164).
Oxidative stress is the main pathogenic cause of DR, and oxidative stress can damage the integrity of cell membranes, induce apoptosis, microvascular damage and barrier damage, and ultimately lead to the development of DR (165). Polyol pathway activation represents one of the processes observed under conditions of hyperglycemia-induced oxidative stress during the pathogenesis of DR, and this pathway is also known as the sorbitol-aldose reductase pathway (165). Excessive activation of the polyol pathway leads to the accumulation of ROS, which induces oxidative stress in cells (165). Sorbitol and fructose accumulate intracellularly, leading to increased osmotic pressure, edema rupture, and impaired membrane permeability (166). The hexosamine pathway may also mediate the toxic effects of ROS in hyperglycemia. In the presence of elevated glucose levels, large amounts of ROS are produced, which may inhibit the activity of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), resulting in the influx of glycolysis products into the hexosamine pathway (167). Glucosamine production from activated hexosamine increases H2O2 production, which further leads to increased oxidation, cellular endothelial changes, increased vascular permeability, and angiogenesis. Inhibition of GAPDH also induces the activity of the AGE pathway through interaction with intracellular methylglyoxal, leading to increased retinal oxidative stress (167). Activation of the PKC pathway can lead to endothelial cell damage by increasing endothelial permeability, altering NO bioavailability, reducing prostaglandin production, inducing VEGF expression, and inducing thromboxane and endothelin-1 (ET-1) production. The hyperglycemic state induces the accumulation of ROS and the synthesis of diacylglycerol (DAG), leading to activation of the PKC pathway (31). PKC pathway activation alters NO production through eNOS expression, directly affects vascular tone and permeability, and ultimately promotes endothelial dysfunction (31). The receptor for AGEs (RAGE) also plays an important role in DR pathogenesis, as its activation mediates a wide range of biological effects, including increased ROS levels, cytokine release, and altered cellular function and death (168). AGEs and RAGEs accumulate in retinal microvessels and interact directly with intracellular proteins, leading to endothelial dysfunction (168). Additionally, during the pathogenesis of DR, ANG-II induces vasoconstriction, inflammation, oxidative stress, cellular dysfunction, angiogenesis, and fibrosis, and can activate NADPH enzyme levels, thereby increasing ROS production and directly damaging endothelial cells (169).
5 Research Hotspots of the Endothelial Dysfunction of Diabetic Nephropathy and Diabetic Retinopathy
5.1 Exosomes
Exosomes were initially found in the supernate of RBCs of in vitro cultured sheep. Exosomes, 40-100 nm (diameter) and 1.10-1.18 g/ml (density), are vesicle-like bodies that are actively secreted by cells (170–175). In 1996, Raposo found that B cells can promote T cell proliferation and inhibit tumor growth by releasing exosomes capable of expressing major histocompatibility complex (MHC) molecules (170). Exosomes are capable of carrying various protein, mRNAs, miRNAs, circRNAs, and participate in cellular interaction, motility, angiogenesis, and oncocyte proliferation (170–175). The intracellular lysosomal particles invaginate and form multivesicular bodies. When stimulated, the multi-vesicular bodies fuse with the cellular membrane and secrete out of the cell vesicles with a uniformed size at 40-100 nm in diameter. These vesicles are exosomes (171, 173–175). The formation and release of exosomes involve endosome sorting transporters and some related proteins such as CD9 and CD63 (170–172). When circulating in the blood, exosomes can act as shuttle vectors or signal transducers at their primary site and at a certain distance from the primary site (170, 172, 173). Recent studies have discovered that the exosomes in the circulation and humor of T2D angiopathy sufferers exhibit a different amount of protein and RNA content compared with that of healthy subjects, which can be taken as potential diagnostic markers and ingredients to deliver reversed high glucose damage, such as the protective role that exosomes are releasing miR-216 plays in the process of atherosclerosis through cell-to-cell communication (174–176).
Normally, exosomes can be excreted by renal cells, like podocytes, renal tubular cells, GECs, and glomerular mesangial cells (177–184). Numerous studies have indicated that changes in culture conditions may influence the composition of exosomes. For instance, under the pressure conditions of hypoxia, acidic pH, uremic toxins, high glucose, and oxidative stress, the number of exosomes increases and their composition changes (177–182). Exosomes can induce the release of cytokines and promote the aggregation of inflammatory cells. Damaged renal cells could secrete exosomes that can be transferred to other normal renal cells, altering the phenotype of normal kidney cells, and promote cell-to-cell interaction. The urinary exosome is a new biomarker for diagnosing DN (177–181). Exosomes significantly improve renal function and significantly enhance LC3 and Beclin-1 (179). As compared to healthy controls, the urine exosomes of DN patients show a new array of protein changes, including MLL3, AMBP, and VDAC1 (181). There is evidence that serum exosomes from diabetic db/db mice severely impair the endothelial function of non-diabetic db/m + mice. Proteomic analyses have revealed that arginase 1 (Arg1) is increased significantly, and it has been proven that serum exosomes deliver Arg1 to endothelial cells (185). In STZ-induced diabetic nephropathy rats, platelets with elevated CXCL7 expression can activate the mTORC1 pathway and result in glomerular endothelial dysfunction (186).
Studies have confirmed that intravitreal injection of exosomes derived from mesenchymal stem cells can ease DR (187–189). PPARγ is an active component of exosomes. A study demonstrated that in patients with proliferative DR, PPARγ levels was significantly increased in both aqueous humor and vitreous humor, revealing a vital effect of exosomes on DR. Various researches have tested the effects of exosomes on diabetic microangiopathy. Researchers found that exosomes in the plasma of DR patients increased the expression of cytokines and angiogenic factors, indicating that circulating exosomes might play a crucial role in carrying inflammatory factors (190, 191). A recent study has demonstrated that high glucose levels affect the anti-angiogenic exosomal miRNA levels (192). In a study on diabetic microvascular complications, exosomes that carry circRNA- cPWWP2A could non-directly regulate microvascular EC function (193). Studies have demonstrated that plasma exosomes mediate hyperglycemia-induced retinal endothelial damage through up-regulation of TLR4 signaling pathway (194). In previous researches, investigators demonstrated that circulating miR-15a contributed to the pathogenesis of diabetes and induced oxidative stress and cell damage via exosomal transport (195).
5.2 Circular RNA (circRNA)
CircRNA is a class of non-coding RNA (196–199) without a 5’end cap and a 3’end poly(A) tail, presenting a covalent bonding round architecture. circRNA can be distinguished from traditional linear ribonucleic acid. It has an enclosed helical structure and exists widely in the eukaryotic transcriptome. Most circRNAs comprise exon sequences, conservative in diverse species, and display expression overlaps in tissues and different developmental phases (197). Because of its insensitivity to nucleases, circRNA is more stable than linear ribonucleic acid, which makes it stand out in the development and application as a new clinical diagnostic marker (198, 199). CircRNA has been found to be abnormally expressed in many illnesses line malignancies, vascular illnesses, inflammatory illnesses, and neurological illnesses. CircRNA displays a diverse expression in angiogenic extreme vascular diseases, and the abnormal expression of circRNAs is related to the incidence and development of diabetic vascular complications (200, 201).
Studies have demonstrated that in HUVEC induced by HG, the level of circRNA-001175 is significantly down-regulated, and the up-regulation of circular RNA-001175 can prevent cell apoptosis induced by high glucose in HUVEC (202). In the following research, HG increased the expressing of circRNA 0054633 in HUVEC, and its down-regulation further deteriorated the endothelial cell dysfunction caused by high glucose (203). Studies have revealed that circular RNA 0054633 can protect endothelial cells from damage stimulated by high glucose through targeting the miR-218/HO-1 axis (203). Moreover, the expressing of circRNA HIPK3 was down-regulated in HG cultured HUVEC, and the overexpression of lentivirus-mediated HIPK3 inhibited HG-triggered EC death and programmed cell death via targeting miR-124 (204). In high glucose treated HUVEC, the circRNA 0068087 level is regulated upward. The knockout of circRNA 0068087 inhibits HG-triggered EC inflammation/NF-κB/NLRP3 inflammatory body signal path via the inactivation of TLR4 (205). These findings have shown that there is great potential for circRNAs to be studied as a diagnostic basis for HG-associated EC function disorder or predictive biomarkers.
Researches have recently highlighted the role of circRNA in the DN, while current evidence suggests that circRNA as a miRNA sponge plays an important role in its pathophysiology. CircRNA-15698 could increase TGF-β1 protein in DN and stimulate the synthesis of ECM-related proteins. the knockdown of circRNA-15698 could decrease the fibrillar proteins (206). In addition, a recent study has demonstrated that circLRP6 sponges miR-205 and controls mesangial cell damage in high glucose conditions (207). CircRNA has been found to affect the regulation of inflammatory events in other organs’ endothelia via consistent targets in the kidney, such as TGF-β1, which indicates that circRNA may be the key regulator in DN pathogenesis (208).
Circ_0005015 have been found to be significantly increased in both vitreous humor and plasma, and can regulate HRMECs proliferation by acting as the sponge of miR-519d-3p (209). CircHIPK3 overexpression enhances microvascular leakage and the number of acellular capillaries in diabetic retina blood vessel function disorder by regulating HRMECs (210). In diabetic conditions, c-myb can activate the axis of circHIPK3-miR-30a-3p-VEGFC, which affects the retinal microvascular endothelial function (210). In the DR model, the deficiency of cZNF609 inhibits pathological angiogenesis (211). In contrast, the circDNMT3B concentrations in FVM and human retinal microvascular EC (HRMEC) are decreased by high glucose treatment. CircDNMT3B targets MiR-20b-5p which actively regulates endothelial angiogenesis by regulating the expression of BAMBI (212). The findings of these studies further demonstrate the importance of circRNAs in the pathogenesis of diabetic retinopathy.
5.3 Interaction With Neighboring Cells
5.3.1 Crosstalk Between Renal TECs and GECs
Several studies have demonstrated that glomerulo-tubular balance and tubule-glomerular feed-back influence the development of DKD (213–218), on the foundation of which new hypoglycemic drugs could increase glucose in urine excretion, regulate BG, and thus decrease glomerulus damage in DKD patients (219). VEGF, Ang-1 and inflammation factors are abnormally secreted in DKD (220–224). Furthermore, damaged GECs produce cytokines, exosomes, and via autophagy, they induce TECs damage featuring structural changes and altered functional characteristics (225–231). Since TECs and GECs are located at a distance, their abnormal crosstalk is instrumental in the development of DKD. Microalbuminuria can be seen in early DKD because of the changes on GEC surface, including its component glycocalyx (232–234). Recent studies have shown that the paracrine communication between GECs and TECs can contribute to DKD. Recent proofs have revealed that glomerulus filter barriers and renal tubular interstitial compartments are in kinetic equilibrium, in which different types of cell damage can affect each other and result in tissue dysfunction (235). Genetic and protein expression profiles indicate that changes in diabetic glomeruli affect a number of metabolic and signaling pathways, either in individual glomerular cells or in the context of their mutual interaction (236). TECs under high glucose and proteinuria conditions induce cascade inflammation via autocrine or paracrine secretion of cytokines, miRNAs, and extracellular vesicles. Inflammatory factors cause apoptosis, necrosis, and trans-differentiation of the glomerular vascular network, causing damage to the architectures and functions of GECs. The impaired GECs induce low blood supply in the kidney tubules, thereby exacerbating the damage to TECs.
Ang-1 and Ang-2 play a vital role in mature blood vessel formation (216, 220). TECs generate Ang-1, which bind to the tyrosine kinase 2 (Tie-2) spot of GECs and reduce the endothelial function of GECs (216). VEGF is secreted by TECs and binds to VEGFR expressed by GECs, generating a VEGF/VEGFR axis, which controls the structure and function of GECs (223). The expressing of HIF-1α in TEC’s is increased, and HIF-1α enters nuclei and binds with the HIF-1β subunit to generate HIF-1, which stimulates down stream inflammation responses to induce GEC autophagy and programs cell death (236). As TECs accumulate in dense plaques, they regulate the contraction and relaxation of the glomerulus arteriole and affect GFR (237). SGLT2 expressed by proximal TECs decides the re-absorption of kidney BG (238). In DKD sufferers, SGLT2 is overexpressed due to the presence of more glucose molecules (238, 239). The SGLT-2 inhibitor can straightly decrease great GFR through the repairment of tubule-glomerular feed-back (240).
5.3.2 Crosstalk Between Pericytes and Retinal Endothelial Cells
Two major cellular components of retinal microvessels are endothelial cells (EC) and pericytes (PC). Both types of cells share the same basement membrane on the blood vessel wall. ECs and PCs are closely connected by intercellular space and communicate with other signaling factors (such as cytokines and extracellular exosomes). Interactions between these two types of cells occur in areas rich in extracellular matrix (ECM) (241–245). There are several factors affecting the mutual effect between ECs and PCs in pathological state, including changes in advanced glycation end products, increased leukocyte adhesion and inflammatory cytokines (246, 247).
It has been demonstrated that the inhibition of VE-PTP helps stabilize the ocular vascular system and prevent retinal and choroidal NV (248, 249). However, Tie2 can also be expressed by pericytes, and its signal transduction in pericytes is critical for pericyte migration, endothelial sprouting and neovascularization (248, 249). Studies have discovered that the coverage of pericytes is reduced by producing transgenic mouse strains, while inadequate pericyte coverage can lead to BRB damage and increased leakage of the developing retinal vascular system, thus impairing visual performance (250).
6 Treatment of Diabetic Nephropathy and Diabetic Retinopathy
6.1 Treatment of DN
Diabetes complications, especially DKD, can be prevented if long-term and intensive blood glucose control can be monitored from its early stage. However, intensive glycemic control in diabetic patients with pre-existing diabetic complications fails to reduce the risk of DKD progression (251–254). The studies on T2D patients and early CKD patients revealed that compared with standard treatment, all-cause death and cardio-vascular death risks for patients undergoing intensive blood glucose control were 30% and 40% higher. Long-term intensive blood glucose control can lead to hypoglycemia and cast negative impacts on curbing cardiovascular illnesses or all-cause death (251, 252). The National Kidney Foundation’s KDOQI and the KDIGO guide lines suggested that the HbA1c target value should be controlled at approximately 7.0% to avoid or postpone the development of diabetic micro-vascular complicating diseases, except for individuals suffering the risk of low blood glucose, like diabetic diseases and CKD (252).
For the treatment of high blood pressure, the JNC-8 suggests that drug intervention should be started when SBP exceeds 140 mmHg or DBP exceeds 90 mmHg with the aim to lowering these levels (255). In common patients with hypertension, including diabetes patients, initial antihypertension therapy might involve thiazide diuretics, calcium channel blockers, ACE inhibitors, or ARB. The KDIGO guidelines recommend that ACE or ARB should be used in all patients with CKD and proteinuria with a target of blood pressure lower than 130/80 mmHg. Evidence has unraveled that using ACEI or ARB to block the renin-angiotensin system can curb the development of DKD in sufferers with large albuminuria, whereas it may have the side effects of hyperkalemia and AKI (252).
Although at present there are methods for managing diabetes and hypertension using ACEI and ARB, DKD still has a large residual risk. New drugs with targeted causal links, like glomerulus hyper-filtration, inflammatory events, and fibrotic activities, have always been the main hot spot in terms of the design of novel therapies. Promising drugs include Rubista (PKC-β suppressor), Baritinib (selective JAK-1 and JAK-2 inhibitors), Pentoxifylline (antiinflammation and antifibrosis), Atrasentan (selective ETAR antagonist), and fennel one (remarkably selective non-steroidal MRA) (251). However, up till now, there is no phase 3 clinic information of those drugs yet, and no drugs have been approved for DKD. The SGLT2 suppressor, empagliflozin, can remarkably reduce mortality as well as hospitalizations for heart failure. The analyses of prespecified secondary results revealed that empagliflozin curbed the development of DKD and reduced the incidence of clinical correlation renal results in CKD phases 2-4 patients (256). In recent years, a number of randomized controlled experiments have confirmed the great role of SGLT−2 inhibitors in renal protection, suggesting that SGLT−2 inhibitors can become a new hope for improving the prognosis of DKD patients, but the mechanism of renal protection warrants further studies (256, 257). The therapeutic effect of non-steroidal mineralocorticoid receptor antagonists on DN patients is also one of the currently popular study fields (258, 259).
6.2 Treatment of Diabetic Retinopathy
Laser photocoagulation is still the primary treatment for DR that threatens vision (165, 260–266). Nevertheless, although timely and appropriate laser treatment can significantly prevent vision loss, the destruction nature of lasers is related to remarkable adverse effects. In addition, sufficient laser treatment cannot guarantee the reversal of vision loss. For that reason, investigators persistently seek novel and more valid treatments to improve vision without damaging tissues. There are two laser treatments for DR: the PRP for PDR and the macula laser photocoagulation for DME. The main function of PRP is to perform laser burning on the whole retina, preserving the central macula to facilitate the regression of retinal neovascularization and prevent its development. Although it has an undisputed effect in preventing severe vision loss, PRP is usually related to many adverse effects, like difficulty in adapting to light darkness (25%), tiny loss of vision (10%), and loss of peripheral vision (5%), color vision alteration and macular edema worsening (260, 263, 265). Besides, the results of DRS and ETDRS indicate that DR sufferers in milder phases may not benefit from laser treatment (267).
Vitreous body resection has always been the main surgical therapy for advanced retinopathy, which includes two blinding complications: chronic vitreum hemorrhage and TRD (165, 260–263). It is a double-edged sword for diabetic eyes. Although it decreases the risk of retinal blood vessel neogenesis and macula edema, it also elevates the risk of iris blood vessel neogenesis and cataract forming (260). In clinical studies of DR vitrectomy, for T1D sufferers with severe vitreous hemorrhage, those underwent vitrectomy (occurred within 1-6 months) were more likely to achieve better eyesight (261, 262) than those received vitrectomy (occurred for 1 year). However, the benefits of this operation have not been reported in T2D sufferers, which might be related to more severe macular ischemia in these patients. Vitreous body resection is also suggested as a therapy for DME which is ineffective with laser treatment, particularly when macula traction does exist (such as vitreous macular traction, detachment between the preretinal membrane and traction retinal close to the macula) (263, 265).
As a pathogenic mediator of the abnormal growth and leakage of retinal blood vessels, VEGF has always been a treatment target for DR. The concentration of VEGF in the eye is closely related to hypoxia and active neovascularization. After laser photocoagulation, its concentration decreases, and VEGF inhibitor active drugs can improve ischemia-induced animal retinal neovascularization and prevent or even realize the reversal of PDR and DME (268, 269). A random trial contrasted laser treatment + intravitreous ranibizumab injection with laser treatment + placebo injection in DME sufferers (268). In the first year, the ranibizumab group gained approximately single-line extra vision compared to laser treatment. The frequency of vision improvement in the ranibizumab group was twice that of the laser group. Notably, in contrast to the eyes mainly exposed to laser treatment alone, the eyes exposed to laser and ranibizumab treatment are less probably to show significant vision loss (268). Diabetic individuals suffer more risk of cataracts that require surgical treatment. Nevertheless, in certain sufferers, cataract surgery could aggravate macula edema and retinopathy. As a result, sometimes, it may be necessary to perform laser therapy on DR sufferers before cataract surgery or immediately posterior to cataract surgery (270). Despite the fact that antiVEGF treatment has bright clinical use prospects in DR, its longterm security in diabetic sufferers hasn’t been determined. Local adverse incidents of intravitreous antiVEGF treatment involve cataract formation, amotio retinae, intravitreous hemorrhages, infections, and possible loss of neurally formed retina cells. Because of the relatively long halflife of certain drugs and the requirement for repeated administration, if a patient are exposed to anti-VEGF drugs for a long time, the risk of systemic vascular complications in him will increase, such as stroke and non-ocular (such as stomach and kidney) bleeding (109). In addition, most anti-VEGF drugs injected into the eyes may enter the systemic circulation and endanger key blood vessels, leading to ischemia. Other systemic adverse effects involve high blood pressure, albuminuria, and damaged wound repair (271).
Inflammatory events are significant for the etiopathogenesis of DR. Similar to antiVEGF drugs, intraocular corticosteroids are broadly utilized to heal DME (165, 260, 263, 272–274). Meta-analysis results have revealed that macular edema acting intravitreal triamcinolone acetonide (long-acting corticosteroid) can moderately improve vision (272, 273). A few clinical trials about longacting steroid implants (fluocinolone or dexamethasone) have also exhibited shortterm vision improvement (274).
7 Summary
The EC barrier is essential for maintaining the normal physiological activities of kidneys and retinas and excreting abnormal protein molecules and fluids. For diabetes, high glucose impairs both the retina and glomerular microvascular endothelium. This article elucidates the pathophysiology, some pathogenic sites and biochemical/cellular pathways of the retina and kidney microvascular endothelial injury in the diabetic microenvironment (Figure 1). The key molecular pathways causing DR and DN when the damage of retina and glomerulus microvascular endothelial barrier occurs mainly include (1): Hyperglycemia causes retinal ischemia and hypoxia, oxidative stress, inflammatory cell/factor accumulation, and activation of various metabolic pathways, leading to the destruction of the endothelial barrier structure and the increase of microvascular permeability, facilitating the incidence of DR and DN.
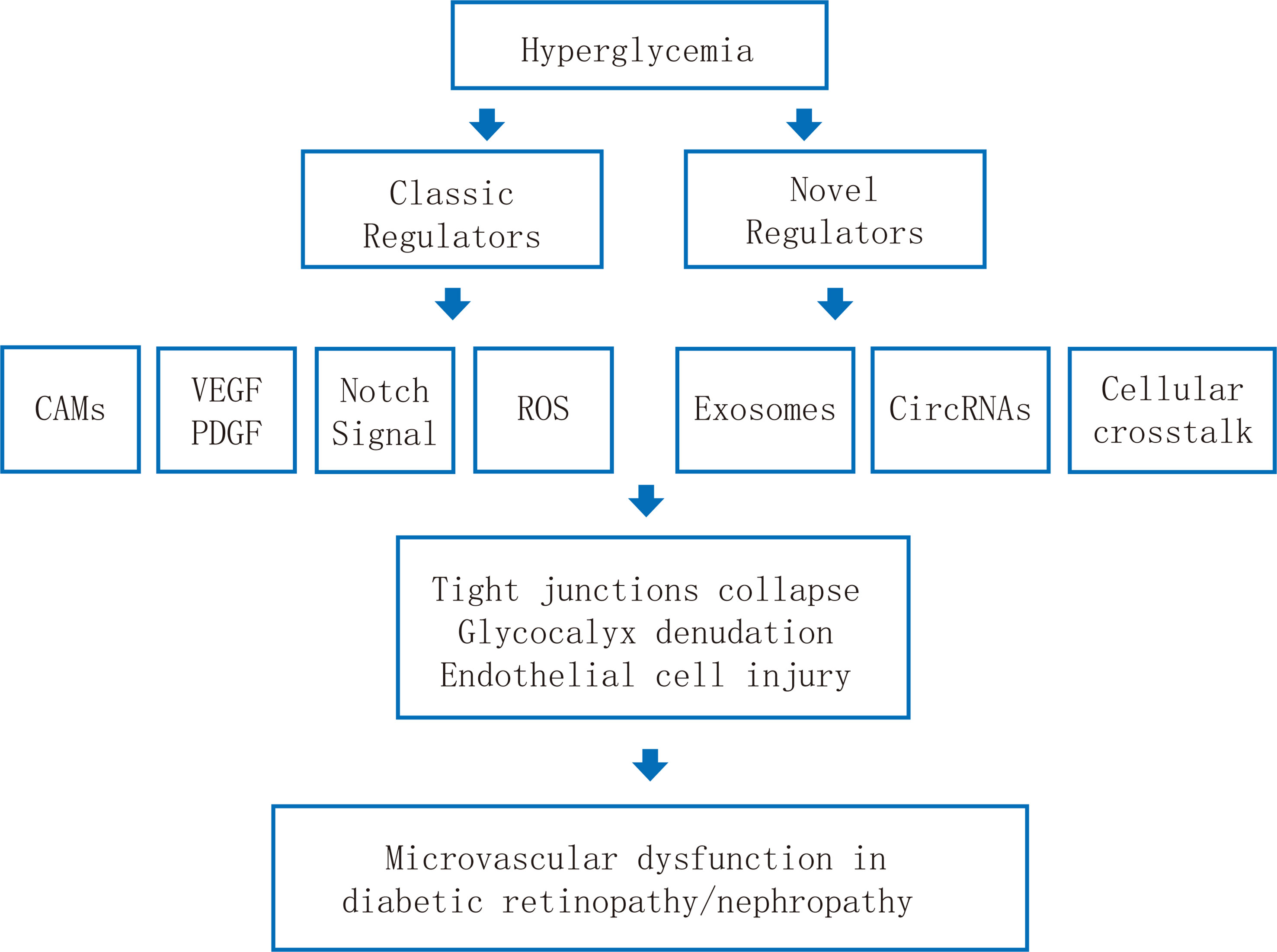
Figure 1 Schematic illustration of the pathological mechanisms of endothelial dysfunction in diabetic retinopathy and diabetic nephropathy.
(2) The retina, kidney, and circulation are rich in exosomes. High glucose leads to changes in the number and content of exosomes, thereby causing the dysfunction of target endothelial cells in the retina and kidney.
(3) circRNA is abundant in the retina, kidney and circulation. It can act as a competitive endogenous RNA to sponge target miRNA, thereby affecting mRNA expression in the ECs of the retina and kidney and promoting the pathogenesis of DR and DN.
(4) Endothelial cells in the retina and kidney can interact with neighboring cells (such as renal tubular cells, pericytes) to generate diabetic endothelial dysfunction through intercellular communication and participate in the occurrence of DR and DN. Further researches on the retina and renal endothelial barrier functions would unravel DR and DN, hence giving birth to new biomarkers, therapeutic targets, or treatments eventually.
Author Contributions
ZL conceptualized the ideas. JY performed the literature search, drafted the original manuscript, and drew the figures. All the authors approved the final version of the manuscript.
Funding
This study is supported by grants from: 1. General Program of the National Natural Science Foundation of China General Project (No.81970633); 2. the Major public welfare special projects in Henan Province (No.201300310600); 3. The National Natural Science Young Scientists Foundation of China (No.81800648) 4. Excellent Young Scientists Fund Program of the Natural Science Foundation of Henan Province (No.202300410363). 5. Henan Province Medical Science and Technology Research Program Joint Construction Project (LHGJ20200337).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Cho NH, Shaw JE, Karuranga S, Huang Y, da Rocha Fernandes JD, Ohlrogge AW, et al. IDF Diabetes Atlas: Global Estimates of Diabetes Prevalence for 2017 and Projections for 2045. Diabetes Res Clin Pract (2018) 138:271–81. doi: 10.1016/j.diabres.2018.02.023
2. Wild S, Roglic G, Green A, Sicree R, King H. Global Prevalence of Diabetes: Estimates for the Year 2000 and Projections for 2030. Diabetes Care (2004) 27(5):1047–53. doi: 10.2337/diacare.27.5.1047
3. Shaw JE, Sicree RA, Zimmet PZ. Global Estimates of the Prevalence of Diabetes for 2010 and 2030. Diabetes Res Clin Pract (2010) 87(1):4–14. doi: 10.1016/j.diabres.2009.10.007
4. Zhang P, Zhang X, Brown J, Vistisen D, Sicree R, Shaw J, et al. Global Healthcare Expenditure on Diabetes for 2010 and 2030. Diabetes Res Clin Pract (2010) 87(3):293–301. doi: 10.1016/j.diabres.2010.01.026
5. Broekhuizen LN, Lemkes BA, Mooij HL, Meuwese MC, Verberne H, Holleman F, et al. Effect of Sulodexide on Endothelial Glycocalyx and Vascular Permeability in Patients with Type 2 Diabetes Mellitus. Diabetologia (2010) 53(12):2646–55. doi: 10.1007/s00125-010-1910-x
6. Jonas JB, Wang YX, Wei WB, Xu J, You QS, Xu L. Chronic Kidney Disease and Eye Diseases: The Beijing Eye Study. Ophthalmology (2017) 124(10):1566–9. doi: 10.1016/j.ophtha.2017.04.024
7. Cheng H, Harris RC. Renal Endothelial Dysfunction in Diabetic Nephropathy. Cardiovasc Hematol Disord Drug Targets (2014) 14(1):22–33. doi: 10.2174/1871529x14666140401110841
8. Stitt AW, Curtis TM, Chen M, Medina RJ, McKay GJ, Jenkins A, et al. The Progress in Understanding and Treatment of Diabetic Retinopathy. Prog Retin Eye Res (2016) 51:156–86. doi: 10.1016/j.preteyeres.2015.08.001
9. Cheung N, Mitchell P, Wong TY. Diabetic Retinopathy. Lancet (2010) 376(9735):124–36. doi: 10.1016/S0140-6736(09)62124-3
10. Hong SB, Lee JJ, Kim SH, Suh YJ, Han JY, Kim YS, et al. The Effects of Adiponectin and Inflammatory Cytokines on Diabetic Vascular Complications in Obese and Non-Obese Patients with Type 2 Diabetes Mellitus. Diabetes Res Clin Pract (2016) 111:58–65. doi: 10.1016/j.diabres.2015.10.017
11. Rajasekar P, O'Neill CL, Eeles L, Stitt AW, Medina RJ. Epigenetic Changes in Endothelial Progenitors as a Possible Cellular Basis for Glycemic Memory in Diabetic Vascular Complications. J Diabetes Res (2015) 2015:436879. doi: 10.1155/2015/436879
12. Naylor RW, Morais MRPT, Lennon R. Complexities of the Glomerular Basement Membrane. Nat Rev Nephrol (2021) 17(2):112–7. doi: 10.1038/s41581-020-0329-y
13. Miner JH. The Glomerular Basement Membrane. Exp Cell Res (2012) 318(9):973–8. doi: 10.1016/j.yexcr.2012.02.031
14. Randles MJ, Collinson S, Starborg T, Mironov A, Krendel M, Königshausen E, et al. Three-Dimensional Electron Microscopy Reveals the Evolution of Glomerular Barrier Injury. Sci Rep (2016) 6:35068. doi: 10.1038/srep35068
15. Candiello J, Cole GJ, Halfter W. Age-Dependent Changes in the Structure, Composition and Biophysical Properties of a Human Basement Membrane. Matrix Biol (2010) 29(5):402–10. doi: 10.1016/j.matbio.2010.03.004
16. Balasubramani M, Schreiber EM, Candiello J, Balasubramani GK, Kurtz J, Halfter W. Molecular Interactions in the Retinal Basement Membrane System: A Proteomic Approach. Matrix Biol (2010) 29(6):471–83. doi: 10.1016/j.matbio.2010.04.002
17. Dane MJ, van den Berg BM, Avramut MC, Faas FG, van der Vlag J, Rops AL, et al. Glomerular Endothelial Surface Layer Acts as a Barrier Against Albumin Filtration. Am J Pathol (2013) 182(5):1532–40. doi: 10.1016/j.ajpath.2013.01.049
18. Jeansson M, Haraldsson B. Morphological and Functional Evidence for an Important Role of the Endothelial Cell Glycocalyx in the Glomerular Barrier. Am J Physiol Renal Physiol (2006) 290(1):F111–6. doi: 10.1152/ajprenal.00173.2005
19. Fidler AL, Darris CE, Chetyrkin SV, Pedchenko VK, Boudko SP, Brown KL, et al. Collagen IV and Basement Membrane at the Evolutionary Dawn of Metazoan Tissues. Elife (2017) 6:e24176. doi: 10.7554/eLife.24176
20. Cummings CF, Pedchenko V, Brown KL, Colon S, Rafi M, Jones-Paris C, et al. Extracellular Chloride Signals Collagen IV Network Assembly during Basement Membrane Formation. J Cell Biol (2016) 213(4):479–94. doi: 10.1083/jcb.201510065
21. Cosgrove D, Dufek B, Meehan DT, Delimont D, Hartnett M, Samuelson G, et al. Lysyl Oxidase Like-2 Contributes to Renal Fibrosis in Col4α3/Alport Mice. Kidney Int (2018) 94(2):303–14. doi: 10.1016/j.kint.2018.02.024
22. Fridén V, Oveland E, Tenstad O, Ebefors K, Nyström J, Nilsson UA, et al. The Glomerular Endothelial Cell Coat is Essential for Glomerular Filtration. Kidney Int (2011) 79(12):1322–30. doi: 10.1038/ki.2011.58
23. Lawrence MG, Altenburg MK, Sanford R, Willett JD, Bleasdale B, Ballou B, et al. Permeation of Macromolecules into the Renal Glomerular Basement Membrane and Capture by the Tubules. Proc Natl Acad Sci USA (2017) 114(11):2958–63. doi: 10.1073/pnas.1616457114
24. Fissell WH, Miner JH. What Is the Glomerular Ultrafiltration Barrier? J Am Soc Nephrol (2018) 29(9):2262–4. doi: 10.1681/ASN.2018050490
25. Suh JH, Miner JH. The Glomerular Basement Membrane as a Barrier to Albumin. Nat Rev Nephrol (2013) 9(8):470–7. doi: 10.1038/nrneph.2013.109
26. Suleiman H, Zhang L, Roth R, Heuser JE, Miner JH, Shaw AS, et al. Nanoscale Protein Architecture of the Kidney Glomerular Basement Membrane. Elife (2013) 2:e01149. doi: 10.7554/eLife.01149
27. Morrissey MA, Sherwood DR. An Active Role for Basement Membrane Assembly and Modification in Tissue Sculpting. J Cell Sci (2015) 128(9):1661–8. doi: 10.1242/jcs.168021
28. Unnersjö-Jess D, Scott L, Sevilla SZ, Patrakka J, Blom H, Brismar H. Confocal Super-Resolution Imaging of the Glomerular Filtration Barrier Enabled by Tissue Expansion. Kidney Int (2018) 93(4):1008–13. doi: 10.1016/j.kint.2017.09.019
29. Ockrim Z, Yorston D. Managing Diabetic Retinopathy. BMJ (2010) 341:c5400. doi: 10.1136/bmj.c5400
30. Yau JW, Rogers SL, Kawasaki R, Lamoureux EL, Kowalski JW, Bek T, et al. Global Prevalence and Major Risk Factors of Diabetic Retinopathy. Diabetes Care (2012) 35(3):556–64. doi: 10.2337/dc11-1909
31. Díaz-Coránguez M, Ramos C, Antonetti DA. The Inner Blood-Retinal Barrier: Cellular Basis and Development. Vision Res (2017) 139:123–37. doi: 10.1016/j.visres.2017.05.009
32. Tervonen A, Vainio I, Nymark S, Hyttinen J. Prediction of Passive Drug Permeability Across the Blood-Retinal Barrier. Pharm Res (2014) 31(9):2297–311. doi: 10.1007/s11095-014-1325-3
33. Cunha-Vaz J. The Blood-Retinal Barrier in the Management of Retinal Disease: EURETINA Award Lecture. Ophthalmologica (2017) 237(1):1–10. doi: 10.1159/000455809
34. Santiago AR, Boia R, Aires ID, Ambrósio AF, Fernandes R. Sweet Stress: Coping With Vascular Dysfunction in Diabetic Retinopathy. Front Physiol (2018) 9:820. doi: 10.3389/fphys.2018.00820
35. Hudson N, Campbell M. Inner Blood-Retinal Barrier Regulation in Retinopathies. Adv Exp Med Biol (2019) 1185:329–33. doi: 10.1007/978-3-030-27378-1_54
36. Runkle EA, Antonetti DA. The Blood-Retinal Barrier: Structure and Functional Significance. Methods Mol Biol (2011) 686:133–48. doi: 10.1007/978-1-60761-938-3_5
37. Le YZ. VEGF Production and Signaling in Müller Glia are Critical to Modulating Vascular Function and Neuronal Integrity in Diabetic Retinopathy and Hypoxic Retinal Vascular Diseases. Vision Res (2017) 139:108–14. doi: 10.1016/j.visres.2017.05.005
38. Santos GSP, Prazeres PHDM, Mintz A, Birbrair A. Role of Pericytes in the Retina. Eye (2018) 32:483–6. doi: 10.1038/eye.2017.220
39. Braunwald E. Diabetes, Heart Failure, and Renal Dysfunction: The Vicious Circles. Prog Cardiovasc Dis (2019) 62:298–302. doi: 10.1016/j.pcad.2019.07.003
40. Mogensen CE, Christensen CK, Vittinghus E. The Stages in Diabetic Renal Disease. With Emphasis on the Stage of Incipient Diabetic Nephropathy. Diabetes (1983) 32:64–78. doi: 10.2337/diab.32.2.s64
41. Andrésdóttir G, Jensen ML, Carstensen B, Parving HH, Rossing K, Hansen TW, et al. Improved Survival and Renal Prognosis of Patients with Type 2 Diabetes and Nephropathy with Improved Control of Risk Factors. Diabetes Care (2014) 37:1660–7. doi: 10.2337/dc13-2036
42. Papadopoulou-Marketou N, Chrousos GP, Kanaka-Gantenbein C. Diabetic Nephropathy in type 1 Diabetes: A Review of Early Natural History, Pathogenesis, and Diagnosis. Diabetes Metab Res Rev (2017) 33:27457509. doi: 10.1002/dmrr.2841
43. Alicic RZ, Rooney MT, Tuttle KR. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin J Am Soc Nephrol (2017) 12:2032–45. doi: 10.2215/CJN.11491116
44. Stehouwer CD. Endothelial Dysfunction in Diabetic Nephropathy: State of the Art and Potential Significance for Non-diabetic Renal Disease. Nephrol Dial Transplant (2004) 19:778–81. doi: 10.1093/ndt/gfh015
275. Cooper ME. Pathogenesis, Prevention, and Treatment of Diabetic Nephropathy. Lancet (1998) 352:213–9. doi: 10.1016/S0140-6736(98)01346-4
45. Takiyama Y, Haneda M. Hypoxia in Diabetic Kidneys. BioMed Res Int (2014) 2014:837421. doi: 10.1155/2014/837421
46. Gilbert RE. The Endothelium in Diabetic Nephropathy. Curr Atheroscler Rep (2014) 16:410. doi: 10.1007/s11883-014-0410-8
47. Reidy K, Kang HM, Hostetter T, Susztak K. Molecular Mechanisms of Diabetic Kidney Disease. J Clin Invest (2014) 124:2333–40. doi: 10.1172/JCI72271
48. Weil EJ, Lemley KV, Mason CC, Yee B, Jones LI, Blouch K, et al. Podocyte Detachment and Reduced Glomerular Capillary Endothelial Fenestration Promote Kidney Disease in Type 2 Diabetic Nephropathy. Kidney Int (2012) 82:1010–7. doi: 10.1038/ki.2012.234
49. Swärd P, Rippe B. Acute and Sustained Actions of Hyperglycaemia on Endothelial and Glomerular Barrier Permeability. Acta Physiol (2012) 204:294–307. doi: 10.1111/j.1748-1716.2011.02343.x
50. Daroux M, Prévost G, Maillard-Lefebvre H, Gaxatte C, D'Agati VD, Schmidt AM, et al. Advanced Glycation end-Products: Implications for Diabetic and Non-Diabetic Nephropathies. Diabetes Metab (2010) 36:1–10. doi: 10.1016/j.diabet.2009.06.005
51. Paeng J, Park J, Um JE, Nam BY, Kang HY, Kim S, et al. The Locally Activated Renin-Angiotensin System Is Involved in Albumin Permeability in Glomerular Endothelial Cells Under High Glucose Conditions. Nephrol Dial Transplant (2017) 32:61–72. doi: 10.1093/ndt/gfw089
52. Forsblom C, Harjutsalo V, Thorn LM, Wadén J, Tolonen N, Saraheimo M, et al. Competing-Risk Analysis of ESRD and Death among Patients with type 1 Diabetes and Macroalbuminuria. J Am Soc Nephrol (2011) 22:537–44. doi: 10.1681/ASN.2010020194
53. Abougalambou SS, Abougalambou AS. Prevalence and Risk Factors of Microalbuminuria in Type 2 Diabetes Mellitus Outpatients at University Sains Malaysia Hospital. Diabetes Metab Syndr (2013) 7:64–7. doi: 10.1016/j.dsx.2013.02.034
54. Shah AR, Gardner TW. Diabetic Retinopathy: Research to Clinical Practice. Clin Diabetes Endocrinol (2017) 3:9. doi: 10.1186/s40842-017-0047-y
55. Cheung N, Mitchell P, Wong TY. Diabetic Retinopathy. Lancet (2010) 376:124–36. doi: 10.1016/S0140-6736(09)62124-3
56. Klein R, Lee KE, Gangnon RE, Klein BE. The 25-year Incidence of Visual Impairment in type 1 Diabetes Mellitus the Wisconsin Epidemiologic Study of Diabetic Retinopathy. Ophthalmology (2010) 117:63–70. doi: 10.1016/j.ophtha.2009.06.051
57. Moss SE, Klein R, Klein BE. The 14-year Incidence of Visual Loss in a Diabetic Population. Ophthalmology (1998) 105:998–1003. doi: 10.1016/S0161-6420(98)96025-0
58. Fenwick EK, Pesudovs K, Rees G, Dirani M, Kawasaki R, Wong TY, et al. The Impact of Diabetic Retinopathy: Understanding the Patient's Perspective. Br J Ophthalmol (2011) 95:774–82. doi: 10.1136/bjo.2010.191312
59. Wong TY, Klein R, Islam FM, Cotch MF, Folsom AR, Klein BE, et al. Diabetic Retinopathy in a Multi-Ethnic Cohort in the United States. Am J Ophthalmol (2006) 141:446–55. doi: 10.1016/j.ajo.2005.08.063
60. Cheloni R, Gandolfi SA, Signorelli C, Odone A. Global Prevalence of Diabetic Retinopathy: Protocol for a Systematic Review and Meta-Analysis. BMJ Open (2019) 9(3):e022188. doi: 10.1136/bmjopen-2018-022188
61. Klein R, Klein BE, Moss SE, Davis MD, DeMets DL. The Wisconsin Epidemiologic Study of Diabetic Retinopathy. II. Prevalence and Risk of Diabetic Retinopathy when Age at Diagnosis Is Less than 30 years. Arch Ophthalmol (1984) 102(4):520–6. doi: 10.1001/archopht.1984.01040030398010
62. Cheloni R, Gandolfi SA, Signorelli C, Odone A. Global Prevalence of Diabetic Retinopathy: Protocol for a Systematic Review and Meta-Analysis. BMJ Open (2019) 9(3):e022188. doi: 10.1136/bmjopen-2018-022188
63. Nanayakkara N, Curtis AJ, Heritier S, Gadowski AM, Pavkov ME, Kenealy T, et al. Impact of Age at type 2 Diabetes Mellitus Diagnosis on Mortality and Vascular Complications: Systematic Review and Meta-Analyses. Diabetologia (2021) 64(2):275–87. doi: 10.1007/s00125-020-05319-w
64. Fangueiro JF, Silva AM, Garcia ML, Souto EB. Current Nanotechnology Approaches for the Treatment and Management of Diabetic Retinopathy. Eur J Pharm Biopharm (2015) 95(Pt B):307–22. doi: 10.1016/j.ejpb.2014.12.023
65. Kur J, Newman EA, Chan-Ling T. Cellular and Physiological Mechanisms Underlying Blood Flow Regulation in the Retina and Choroid in Health and Disease. Prog Retin Eye Res (2012) 31(5):377–406. doi: 10.1016/j.preteyeres.2012.04.004
66. Yang Y, Liu Y, Li Y, Chen Z, Xiong Y, Zhou T, et al. MicroRNA-15b Targets VEGF and Inhibits Angiogenesis in Proliferative Diabetic Retinopathy. J Clin Endocrinol Metab (2020) 105(11):3404–15. doi: 10.1210/clinem/dgaa538
67. Bain SC, Klufas MA, Ho A, Matthews DR. Worsening of Diabetic Retinopathy with Rapid Improvement in Systemic Glucose Control: A Review. Diabetes Obes Metab (2019) 21(3):454–66. doi: 10.1111/dom.13538
68. Taurone S, Ralli M, Nebbioso M, Greco A, Artico M, Attanasio G, et al. The Role of Inflammation in Diabetic Retinopathy: A Review. Eur Rev Med Pharmacol Sci (2020) 24(20):10319–29. doi: 10.26355/eurrev_202010_23379
69. Kowluru RA, Koppolu P, Chakrabarti S, Chen S. Diabetes-Induced Activation of Nuclear Transcriptional Factor in the Retina, and Its Inhibition by Antioxidants. Free Radic Res (2003) 37(11):1169–80. doi: 10.1080/10715760310001604189
70. Yuuki T, Kanda T, Kimura Y, Kotajima N, Tamura J, Kobayashi I, et al. Inflammatory Cytokines in Vitreous Fluid and Serum of Patients with Diabetic Vitreoretinopathy. J Diabetes Complications (2001) 15(5):257–9. doi: 10.1016/s1056-8727(01)00155-6
71. Whitehead M, Wickremasinghe S, Osborne A, Van Wijngaarden P, Martin KR. Diabetic Retinopathy: A Complex Pathophysiology Requiring Novel Therapeutic Strategies. Expert Opin Biol Ther (2018) 18(12):1257–70. doi: 10.1080/14712598.2018.1545836
72. Yeh PT, Huang HW, Yang CM, Yang WS, Yang CH. Astaxanthin Inhibits Expression of Retinal Oxidative Stress and Inflammatory Mediators in Streptozotocin-Induced Diabetic Rats. PloS One (2016) 11(1):e0146438. doi: 10.1371/journal.pone.0146438
73. Chawla A, Chawla R, Jaggi S. Microvasular and Macrovascular Complications in Diabetes Mellitus: Distinct or Continuum? Indian J Endocrinol Metab (2016) 20(4):546–51. doi: 10.4103/2230-8210.183480
74. Barrett EJ, Liu Z, Khamaisi M, King GL, Klein R, Klein BEK, et al. Diabetic Microvascular Disease: An Endocrine Society Scientific Statement. J Clin Endocrinol Metab (2017) 102(12):4343–410. doi: 10.1210/jc.2017-01922
75. Kibel A, Selthofer-Relatic K, Drenjancevic I, Bacun T, Bosnjak I, Kibel D, et al. Coronary Microvascular Dysfunction in Diabetes Mellitus. J Int Med Res (2017) 45(6):1901–29. doi: 10.1177/0300060516675504
76. Rosenson RS, Chen Q, Najera SD, Lee ML, Cho DJ. Ticagrelor and the Prevention of Microvascular Complications in Diabetes Patients with Lower Extremity Arterial Disease; Rationale and Design of the Hema-Kinesis Trial. Cardiovasc Drugs Ther (2018) 32(5):443–51. doi: 10.1007/s10557-018-6815-9
77. Giacco F, Brownlee M. Oxidative Stress and Diabetic Complications. Circ Res (2010) 107(9):1058–70. doi: 10.1161/CIRCRESAHA.110.223545
78. Nellaiappan K, Preeti K, Khatri DK, Singh SB. Diabetic Complications: An Update on Pathobiology and Therapeutic Strategies. Curr Diabetes Rev (2022) 18(1):e030821192146. doi: 10.2174/1573399817666210309104203
79. Adki KM, Kulkarni YA. Potential Biomarkers in Diabetic Retinopathy. Curr Diabetes Rev (2020) 16(9):971–83. doi: 10.2174/1573399816666200217092022
80. Kowluru RA, Kowluru A, Mishra M, Kumar B. Oxidative Stress and Epigenetic Modifications in the Pathogenesis of Diabetic Retinopathy. Prog Retin Eye Res (2015) 48:40–61. doi: 10.1016/j.preteyeres.2015.05.001
81. Yao Y, Du J, Li R, Zhao L, Luo N, Zhai JY, et al. Association between ICAM-1 Level and Diabetic Retinopathy: A Review and Meta-Analysis. Postgrad Med J (2019) 95(1121):162–8. doi: 10.1136/postgradmedj-2018-136102
82. Hu J, Dziumbla S, Lin J, Bibli SI, Zukunft S, de Mos J, et al. Inhibition of Soluble Epoxide Hydrolase Prevents Diabetic Retinopathy. Nature (2017) 552(7684):248–52. doi: 10.1038/nature25013
83. Zhang Y, Qu Y, Niu T, Wang H, Liu K. O-GlcNAc Modification of Sp1 Mediates Hyperglycaemia-induced ICAM-1 up-Regulation in Endothelial Cells. Biochem Biophys Res Commun (2017) 484(1):79–84. doi: 10.1016/j.bbrc.2017.01.068
84. Lv Z, Li Y, Wu Y, Qu Y. Association of ICAM-1 and HMGA1 Gene Variants with Retinopathy in Type 2 Diabetes Mellitus Among Chinese Individuals. Curr Eye Res (2016) 41(8):1118–22. doi: 10.3109/02713683.2015.1094093
85. Njeim R, Azar WS, Fares AH, Azar ST, Kfoury Kassouf H, Eid AA. NETosis Contributes to the Pathogenesis of Diabetes and Its Complications. J Mol Endocrinol (2020) 65(4):R65–76. doi: 10.1530/JME-20-0128
86. Borgohain MP, Lahkar M, Ahmed S, Chowdhury L, Kumar S, Pant R, et al. Small Molecule Inhibiting Nuclear Factor-kB Ameliorates Oxidative Stress and Suppresses Renal Inflammation in Early Stage of Alloxan-Induced Diabetic Nephropathy in Rat. Basic Clin Pharmacol Toxicol (2017) 120(5):442–9. doi: 10.1111/bcpt.12718
87. Shihab PK, Al-Roub A, Al-Ghanim M, Al-Mass A, Behbehani K, Ahmad R. TLR2 and AP-1/NF-kappaB Are Involved in the Regulation of MMP-9 Elicited by Heat Killed Listeria Monocytogenes in Human Monocytic THP-1 cells. J Inflammation (2015) 12:32. doi: 10.1186/s12950-015-0077-0
88. Zhang P, Yu C, Yu J, Li Z, Lan HY, Zhou Q. Arid2-IR Promotes NF-κB-Mediated Renal Inflammation by Targeting NLRC5 Transcription. Cell Mol Life Sci (2021) 78(5):2387–404. doi: 10.1007/s00018-020-03659-9
89. Patel S, Santani D. Role of NF-kappa B in the Pathogenesis of Diabetes and its Associated Complications. Pharmacol Rep (2009) 61(4):595–603. doi: 10.1016/s1734-1140(09)70111-2
90. Wan RJ, Li YH. MicroRNA−146a/NAPDH oxidase4 Decreases Reactive Oxygen Species Generation and Inflammation in a Diabetic Nephropathy Model. Mol Med Rep (2018) 17(3):4759–66. doi: 10.3892/mmr.2018.8407
91. Tang LQ, Ni WJ, Cai M, Ding HH, Liu S, Zhang ST. Renoprotective Effects of Berberine and Its Potential Effect on the Expression of β-Arrestins and Intercellular Adhesion Molecule-1 and Vascular Cell Adhesion Molecule-1 in Streptozocin-Diabetic Nephropathy Rats. J Diabetes (2016) 8(5):693–700. doi: 10.1111/1753-0407.12349
92. Mooyaart AL, Valk EJ, van Es LA, Bruijn JA, de Heer E, Freedman BI, et al. Genetic Associations in Diabetic Nephropathy: A Meta-Analysis. Diabetologia (2011) 54(3):544–53. doi: 10.1007/s00125-010-1996-1
93. Nazir N, Siddiqui K, Al-Qasim S, Al-Naqeb D. Meta-Analysis of Diabetic Nephropathy Associated Genetic Variants in Inflammation and Angiogenesis Involved in Different Biochemical Pathways. BMC Med Genet (2014) 15:103. doi: 10.1186/s12881-014-0103-8
94. Murakami H, Tamasawa N, Matsui J, Yamato K, JingZhi G, Suda T. Plasma Levels of Soluble Vascular Adhesion Molecule-1 and Cholesterol Oxidation Product in Type 2 Diabetic Patients with Nephropathy. J Atheroscler Thromb (2001) 8(1):21–4. doi: 10.5551/jat1994.8.21
95. Jenkins AJ, Zhang SX, Rowley KG, Karschimkus CS, Nelson CL, Chung JS, et al. Increased Serum Pigment Epithelium-Derived Factor Is Associated with Microvascular Complications, Vascular Stiffness and Inflammation in Type 1 Diabetes. Diabetes Med (2007) 24(12):1345–51. doi: 10.1111/j.1464-5491.2007.02281.x
96. Okada S, Shikata K, Matsuda M, Ogawa D, Usui H, Kido Y, et al. Intercellular Adhesion Molecule-1-Deficient Mice Are Resistant Against Renal Injury After Induction of Diabetes. Diabetes (2003) 52(10):2586–93. doi: 10.2337/diabetes.52.10.2586
97. Harjunpää H, Llort Asens M, Guenther C, Fagerholm SC. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front Immunol (2019) 10:1078. doi: 10.3389/fimmu.2019.01078
98. Karimi Z, Kahe F, Jamil A, Marszalek J, Ghanbari A, Afarideh M, et al. Intercellular Adhesion Molecule-1 in Diabetic Patients With and Without Microalbuminuria. Diabetes Metab Syndr (2018) 12(3):365–8. doi: 10.1016/j.dsx.2017.12.028
99. Zhang X, Seman NA, Falhammar H, Brismar K, Gu HF. Genetic and Biological Effects of ICAM-1 E469K Polymorphism in Diabetic Kidney Disease. J Diabetes Res (2020) 2020:8305460. doi: 10.1155/2020/8305460
100. Ciobanu DM, Mircea PA, Bala C, Rusu A, Vesa Ş, Roman G. Intercellular Adhesion Molecule-1 (ICAM-1) Associates With 24-hour Ambulatory Blood Pressure Variability in Type 2 Diabetes and Controls. Cytokine (2019) 116:134–8. doi: 10.1016/j.cyto.2019.01.006
101. Fathollahi A, Massoud A, Amirzargar AA, Aghili B, Nasli Esfahani E, Rezaei N. sICAM-1, sVCAM-1 and sE-Selectin Levels in Type 1 Diabetes. Fetal Pediatr Pathol (2018) 37(1):69–73. doi: 10.1080/15513815.2017.1405467
102. Luo SF, Chin CY, Ho LJ, Tseng WY, Kuo CF, Lai JH. Monosodium Urate Crystals Induced ICAM-1 Expression and Cell-Cell Adhesion in Renal Mesangial Cells: Implications for the Pathogenesis of Gouty Nephropathy. J Microbiol Immunol Infect (2020) 53(1):23–32. doi: 10.1016/j.jmii.2017.12.004
103. Borgohain MP, Lahkar M, Ahmed S, Chowdhury L, Kumar S, Pant R, et al. Small Molecule Inhibiting Nuclear Factor-kB Ameliorates Oxidative Stress and Suppresses Renal Inflammation in Early Stage of Alloxan-Induced Diabetic Nephropathy in Rat. Basic Clin Pharmacol Toxicol (2017) 120(5):442–9. doi: 10.1111/bcpt.12718
104. Garibotto G, Carta A, Picciotto D, Viazzi F, Verzola D. Toll-like Receptor-4 Signaling Mediates Inflammation and Tissue Injury in Diabetic Nephropathy. J Nephrol (2017) 30(6):719–27. doi: 10.1007/s40620-017-0432-8
105. Spencer BG, Estevez JJ, Liu E, Craig JE, Finnie JW. Pericytes, Inflammation, and Diabetic Retinopathy. Inflammopharmacology (2020) 28(3):697–709. doi: 10.1007/s10787-019-00647-9
106. Joy SS, Siddiqui K. Molecular and Pathophysiological Mechanisms of Diabetic Retinopathy in Relation to Adhesion Molecules. Curr Diabetes Rev (2019) 15(5):363–71. doi: 10.2174/1573399814666181017103844
107. Xia JP, Liu SQ, Wang S. Intravitreal Conbercept Improves Outcome of Proliferative Diabetic Retinopathy Through Inhibiting Inflammation and Oxidative Stress. Life Sci (2021) 265:118795. doi: 10.1016/j.lfs.2020.118795
108. Turan M, Turan G. Immunoreactivity of ICAM-1, MMP-2, and Nesfatin-1 in Lens Epithelial Cells of Patients with Diabetes Mellitus With or Without Diabetic Retinopathy. Eur J Ophthalmol (2022) 32(1):255–62. doi: 10.1177/1120672120966559
109. Bharadwaj AS, Stempel AJ, Olivas A, Franzese SE, Ashander LM, Ma Y, et al. Molecular Signals Involved in Human B Cell Migration into the Retina: In Vitro Investigation of ICAM-1, VCAM-1, and CXCL13. Ocul Immunol Inflammation (2017) 25(6):811–9. doi: 10.1080/09273948.2016.1180401
110. Siveen KS, Prabhu K, Krishnankutty R, Kuttikrishnan S, Tsakou M, Alali FQ, et al. Vascular Endothelial Growth Factor (VEGF) Signaling in Tumour Vascularization: Potential and Challenges. Curr Vasc Pharmacol (2017) 15(4):339–51. doi: 10.2174/1570161115666170105124038
111. Byrne AM, Bouchier-Hayes DJ, Harmey JH. Angiogenic and Cell Survival Functions of Vascular Endothelial Growth Factor (VEGF). J Cell Mol Med (2005) 9(4):777–94. doi: 10.1111/j.1582-4934.2005.tb00379.x
112. Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, et al. Ephrin-B2 Controls VEGF-induced Angiogenesis and Lymphangiogenesis. Nature (2010) 465(7297):483–6. doi: 10.1038/nature09002
113. Karaman S, Leppänen VM, Alitalo K. Vascular Endothelial Growth Factor Signaling in Development and Disease. Development (2018) 145(14):dev151019. doi: 10.1242/dev.151019
114. Williams D, Argáez C. Acute, Sustained, Intraocular Pressure Increases Following Anti-Vascular Endothelial Growth Factor Treatment for Retinal Conditions: A Review of Clinical Evidence and Guidelines. Ottawa (ON: Canadian Agency for Drugs and Technologies in Health (2019).
115. Cai W, Salvador-Reyes LA, Zhang W, Chen QY, Matthew S, Ratnayake R, et al. Apratyramide, a Marine-Derived Peptidic Stimulator of VEGF-A and Other Growth Factors with Potential Application in Wound Healing. ACS Chem Biol (2018) 13(1):91–9. doi: 10.1021/acschembio.7b00827
116. Khanna P, Soh HJ, Chen CH, Saxena R, Amin S, Naughton M, et al. ACE2 Abrogates Tumor Resistance to VEGFR Inhibitors Suggesting Angiotensin-(1-7) as a Therapy for Clear Cell Renal Cell Carcinoma. Sci Transl Med (2021) 13(577):eabc0170. doi: 10.1126/scitranslmed.abc0170
117. Tung CW, Hsu YC, Shih YH, Chang PJ, Lin CL. Glomerular Mesangial Cell and Podocyte Injuries in Diabetic Nephropathy. Nephrology (2018) 23(Suppl 4):32–7. doi: 10.1111/nep.13451
118. Zhang A, Fang H, Chen J, He L, Chen Y. Role of VEGF-A and LRG1 in Abnormal Angiogenesis Associated With Diabetic Nephropathy. Front Physiol (2020) 11:1064. doi: 10.3389/fphys.2020.01064
119. Nakagawa T, Sato W, Kosugi T, Johnson RJ. Uncoupling of VEGF with Endothelial NO as a Potential Mechanism for Abnormal Angiogenesis in the Diabetic Nephropathy. J Diabetes Res (2013) 2013:184539. doi: 10.1155/2013/184539
120. Singh K, Sandler S, Espes D. The Increased Circulating Plasma Levels of Vascular Endothelial Growth Factor in Patients with Type 1 Diabetes Do Not Correlate to Metabolic Control. J Diabetes Res (2017) 2017:6192896. doi: 10.1155/2017/6192896
121. Majumder S, Advani A. VEGF and the Diabetic Kidney: More than too Much of a Good Thing. J Diabetes Complications (2017) 31(1):273–9. doi: 10.1016/j.jdiacomp.2016.10.020
122. Li X, Wu TT, Chen J, Qiu W. Elevated Expression Levels of Serum Insulin-Like Growth Factor-1, Tumor Necrosis Factor-α and Vascular Endothelial Growth Factor 165 might Exacerbate Type 2 Diabetic Nephropathy. J Diabetes Investig (2017) 8(1):108–14. doi: 10.1111/jdi.12542
123. Mahdy RA, Nada WM, Hadhoud KM, El-Tarhony SA. The Role of Vascular Endothelial Growth Factor in the Progression of Diabetic Vascular Complications. Eye (2010) 24(10):1576–84. doi: 10.1038/eye.2010.86
124. Frezzetti D, Gallo M, Maiello MR, D'Alessio A, Esposito C, Chicchinelli N, et al. VEGF as a Potential Target in Lung Cancer. Expert Opin Ther Targets (2017) 21(10):959–66. doi: 10.1080/14728222.2017.1371137
125. Wang W, Lo ACY. Diabetic Retinopathy: Pathophysiology and Treatments. Int J Mol Sci (2018) 19(6):1816. doi: 10.3390/ijms19061816
126. Zhang D, Lv FL, Wang GH. Effects of HIF-1α on Diabetic Retinopathy Angiogenesis and VEGF Expression. Eur Rev Med Pharmacol Sci (2018) 22(16):5071–6. doi: 10.26355/eurrev_201808_15699
127. Jenkins AJ, Joglekar MV, Hardikar AA, Keech AC, O'Neal DN, Januszewski AS. Biomarkers in Diabetic Retinopathy. Rev Diabetes Stud (2015) 12(1-2):159–95. doi: 10.1900/RDS.2015.12.159
128. Leley SP, Ciulla TA, Bhatwadekar AD. Diabetic Retinopathy in the Aging Population: A Perspective of Pathogenesis and Treatment. Clin Interv Aging (2021) 16:1367–78. doi: 10.2147/CIA.S297494
129. Itatani Y, Kawada K, Yamamoto T, Sakai Y. Resistance to Anti-Angiogenic Therapy in Cancer-Alterations to Anti-VEGF Pathway. Int J Mol Sci (2018) 19(4):1232. doi: 10.3390/ijms19041232
130. Youngblood H, Robinson R, Sharma A, Sharma S. Proteomic Biomarkers of Retinal Inflammation in Diabetic Retinopathy. Int J Mol Sci (2019) 20(19):4755. doi: 10.3390/ijms20194755
131. Mansour SE, Browning DJ, Wong K, Flynn HW Jr, Bhavsar AR. The Evolving Treatment of Diabetic Retinopathy. Clin Ophthalmol (2020) 14:653–78. doi: 10.2147/OPTH.S236637
132. Huang H, He J, Johnson D, Wei Y, Liu Y, Wang S, et al. Deletion of Placental Growth Factor Prevents Diabetic Retinopathy and Is Associated with Akt Activation and HIF1α-VEGF Pathway Inhibition. Diabetes (2015) 64(1):200–12. doi: 10.2337/db14-0016
133. Yan HT, Su GF. Expression and Significance of HIF-1 α and VEGF in Rats with Diabetic Retinopathy. Asian Pac J Trop Med (2014) 7(3):237–40. doi: 10.1016/S1995-7645(14)60028-6
134. Li JK, Wei F, Jin XH, Dai YM, Cui HS, Li YM. Changes in Vitreous VEGF, bFGF and Fibrosis in Proliferative Diabetic Retinopathy after Intravitreal Bevacizumab. Int J Ophthalmol (2015) 8(6):1202–6. doi: 10.3980/j.issn.2222-3959.2015.06.22
135. Bahrami B, Hong T, Gilles MC, Chang A. Anti-VEGF Therapy for Diabetic Eye Diseases. Asia Pac J Ophthalmol (2017) 6(6):535–45. doi: 10.22608/APO.2017350
136. Yang Y, Liu Y, Li Y, Chen Z, Xiong Y, Zhou T, et al. MicroRNA-15b Targets VEGF and Inhibits Angiogenesis in Proliferative Diabetic Retinopathy. J Clin Endocrinol Metab (2020) 105(11):3404–15. doi: 10.1210/clinem/dgaa538
137. Papadopoulos N, Lennartsson J. The PDGF/PDGFR Pathway as a Drug Target. Mol Aspects Med (2018) 62:75–88. doi: 10.1016/j.mam.2017.11.007
138. Kumar A, Li X. PDGF-C and PDGF-D in Ocular Diseases. Mol Aspects Med (2018) 62:33–43. doi: 10.1016/j.mam.2017.10.002
139. Folestad E, Kunath A, Wågsäter D. PDGF-C and PDGF-D Signaling in Vascular Diseases and Animal Models. Mol Aspects Med (2018) 62:1–11. doi: 10.1016/j.mam.2018.01.005
141. Lefevere E, Van Hove I, Sergeys J, Steel DHW, Schlingemann R, Moons L, et al. PDGF as an Important Initiator for Neurite Outgrowth Associated with Fibrovascular Membranes in Proliferative Diabetic Retinopathy. Curr Eye Res (2022) 47(2):277–86. doi: 10.1080/02713683.2021.1966479
142. Certelli A, Valente P, Uccelli A, Grosso A, Di Maggio N, D'Amico R, et al. Robust Angiogenesis and Arteriogenesis in the Skin of Diabetic Mice by Transient Delivery of Engineered VEGF and PDGF-BB Proteins in Fibrin Hydrogels. Front Bioeng Biotechnol (2021) 9:688467. doi: 10.3389/fbioe.2021.688467
143. Li Y, Zhi K, Han S, Li X, Li M, Lian W, et al. TUG1 Enhances High Glucose-Impaired Endothelial Progenitor Cell Function Via miR-29c-3p/PDGF-BB/Wnt Signaling. Stem Cell Res Ther (2020) 11(1):441. doi: 10.1186/s13287-020-01958-3
144. Togliatto G, Dentelli P, Rosso A, Lombardo G, Gili M, Gallo S, et al. PDGF-BB Carried by Endothelial Cell-Derived Extracellular Vesicles Reduces Vascular Smooth Muscle Cell Apoptosis in Diabetes. Diabetes (2018) 67(4):704–16. doi: 10.2337/db17-0371
145. Muhiddin HS, Kamaruddin MI, Ichsan AM, Budu. Vitreous and Serum Concentrations of Vascular Endothelial Growth Factor and Platelet-Derived Growth Factor in Proliferative Diabetic Retinopathy. Clin Ophthalmol (2020) 14:1547–52. doi: 10.2147/OPTH.S248812
146. Uehara G, Suzuki D, Toyoda M, Umezono T, Sakai H. Glomerular Expression of Platelet-Derived Growth Factor (PDGF)-A, -B Chain and PDGF Receptor-Alpha, -Beta in Human Diabetic Nephropathy. Clin Exp Nephrol (2004) 8(1):36–42. doi: 10.1007/s10157-003-0265-8
147. Wang QY, Guan QH, Chen FQ. The Changes of Platelet-Derived Growth Factor-BB (PDGF-BB) in T2DM and Its Clinical Significance for Early Diagnosis of Diabetic Nephropathy. Diabetes Res Clin Pract (2009) 85(2):166–70. doi: 10.1016/j.diabres.2009.05.008
148. Lassila M, Jandeleit-Dahm K, Seah KK, Smith CM, Calkin AC, Allen TJ, et al. Imatinib Attenuates Diabetic Nephropathy in Apolipoprotein E-knockout Mice. J Am Soc Nephrol (2005) 16(2):363–73. doi: 10.1681/ASN.2004050392
149. Boor P, Ostendorf T, Floege J. PDGF and the Progression of Renal Disease. Nephrol Dial Transplant (2014) 29(Suppl 1):i45–54. doi: 10.1093/ndt/gft273
150. Akil A, Gutiérrez-García AK, Guenter R, Rose JB, Beck AW, Chen H, et al. Notch Signaling in Vascular Endothelial Cells, Angiogenesis, and Tumor Progression: An Update and Prospective. Front Cell Dev Biol (2021) 9:642352. doi: 10.3389/fcell.2021.642352
151. Fortini F, Vieceli Dalla Sega F, Marracino L, Severi P, Rapezzi C, Rizzo P, et al. Well-Known and Novel Players in Endothelial Dysfunction: Updates on a Notch(ed) Landscape. Biomedicines (2021) 9(8):997. doi: 10.3390/biomedicines9080997
152. Fernández-Chacón M, García-González I, Mühleder S, Benedito R. Role of Notch in Endothelial Biology. Angiogenesis (2021) 24(2):237–50. doi: 10.1007/s10456-021-09793-7
153. Mack JJ, Iruela-Arispe ML. NOTCH Regulation of the Endothelial Cell Phenotype. Curr Opin Hematol (2018) 25(3):212–8. doi: 10.1097/MOH.0000000000000425
154. Gomez AH, Joshi S, Yang Y, Tune JD, Zhao MT, Yang H. Bioengineering Systems for Modulating Notch Signaling in Cardiovascular Development, Disease, and Regeneration. J Cardiovasc Dev Dis (2021) 8(10):125. doi: 10.3390/jcdd8100125
155. Tian DY, Jin XR, Zeng X, Wang Y. Notch Signaling in Endothelial Cells: Is It the Therapeutic Target for Vascular Neointimal Hyperplasia? Int J Mol Sci (2017) 18(8):1615. doi: 10.3390/ijms18081615
156. Lobov I, Mikhailova N. The Role of Dll4/Notch Signaling in Normal and Pathological Ocular Angiogenesis: Dll4 Controls Blood Vessel Sprouting and Vessel Remodeling in Normal and Pathological Conditions. J Ophthalmol (2018) 2018:3565292. doi: 10.1155/2018/3565292
157. Fan J, Shen W, Lee SR, Mathai AE, Zhang R, Xu G, et al. Targeting the Notch and TGF-β Signaling Pathways to Prevent Retinal Fibrosis in Vitro and in Vivo. Theranostics (2020) 10(18):7956–73. doi: 10.7150/thno.45192
158. Rangasamy S, Monickaraj F, Legendre C, Cabrera AP, Llaci L, Bilagody C, et al. Transcriptomics Analysis of Pericytes from Retinas of Diabetic Animals Reveals Novel Genes and Molecular Pathways Relevant to Blood-Retinal Barrier Alterations in Diabetic Retinopathy. Exp Eye Res (2020) 195:108043. doi: 10.1016/j.exer.2020.108043
159. Zhu DD, Wang YZ, Zou C, She XP, Zheng Z. The Role of Uric Acid in the Pathogenesis of Diabetic Retinopathy Based on Notch Pathway. Biochem Biophys Res Commun (2018) 503(2):921–9. doi: 10.1016/j.bbrc.2018.06.097
160. Miloudi K, Oubaha M, Ménard C, Dejda A, Guber V, Cagnone G, et al. NOTCH1 Signaling Induces Pathological Vascular Permeability in Diabetic Retinopathy. Proc Natl Acad Sci USA (2019) 116(10):4538–47. doi: 10.1073/pnas.1814711116
161. Zoja C, Xinaris C, Macconi D. Diabetic Nephropathy: Novel Molecular Mechanisms and Therapeutic Targets. Front Pharmacol (2020) 11:586892. doi: 10.3389/fphar.2020.586892
162. Sun J, Zhao F, Zhang W, Lv J, Lv J, Yin A. BMSCs and miR-124a Ameliorated Diabetic Nephropathy Via Inhibiting Notch Signalling Pathway. J Cell Mol Med (2018) 22(10):4840–55. doi: 10.1111/jcmm.13747
163. Liu M, Liang K, Zhen J, Zhou M, Wang X, Wang Z, et al. Sirt6 Deficiency Exacerbates Podocyte Injury and Proteinuria through Targeting Notch Signaling. Nat Commun (2017) 8(1):413. doi: 10.1038/s41467-017-00498-4
164. Nishad R, Mukhi D, Tahaseen SV, Mungamuri SK, Pasupulati AK. Growth Hormone Induces Notch1 Signaling in Podocytes and Contributes to Proteinuria in Diabetic Nephropathy. J Biol Chem (2019) 294(44):16109–22. doi: 10.1074/jbc.RA119.008966
165. He Y, Zhang M, Wu Y, Jiang H, Fu H, Cai Y, et al. Aberrant Activation of Notch-1 Signaling Inhibits Podocyte Restoration After Islet Transplantation in a Rat Model of Diabetic Nephropathy. Cell Death Dis (2018) 9(10):950. doi: 10.1038/s41419-018-0985-z
166. Matoba K, Kawanami D, Nagai Y, Takeda Y, Akamine T, Ishizawa S, et al. Rho-Kinase Blockade Attenuates Podocyte Apoptosis by Inhibiting the Notch Signaling Pathway in Diabetic Nephropathy. Int J Mol Sci (2017) 18(8):1795. doi: 10.3390/ijms18081795
167. Liu M, Ning X, Li R, Yang Z, Yang X, Sun S, et al. Signalling Pathways Involved in Hypoxia-Induced Renal Fibrosis. J Cell Mol Med (2017) 21(7):1248–59. doi: 10.1111/jcmm.13060
168. Mukherjee M, Fogarty E, Janga M, Surendran K. Notch Signaling in Kidney Development, Maintenance, and Disease. Biomolecules (2019) 9(11):692. doi: 10.3390/biom9110692
169. Shao H, Li Y, Pastar I, Xiao M, Prokupets R, Liu S, et al. Notch1 Signaling Determines the Plasticity and Function of Fibroblasts in Diabetic Wounds. Life Sci Alliance (2020) 3(12):e202000769. doi: 10.26508/lsa.202000769
170. Ebrahim N, Dessouky AA, Mostafa O, Hassouna A, Yousef MM, Seleem Y, et al. Adipose Mesenchymal Stem Cells Combined with Platelet-Rich Plasma Accelerate Diabetic Wound Healing by Modulating the Notch Pathway. Stem Cell Res Ther (2021) 12(1):392. doi: 10.1186/s13287-021-02454-y
171. Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA. Cellular Death, Reactive Oxygen Species (ROS) and Diabetic Complications. Cell Death Dis (2018) 9(2):119. doi: 10.1038/s41419-017-0135-z
172. Fukai T, Ushio-Fukai M. Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells (2020) 9(8):1849. doi: 10.3390/cells9081849
173. Ochoa CD, Wu RF, Terada LS. ROS Signaling and ER Stress in Cardiovascular Disease. Mol Aspects Med (2018) 63:18–29. doi: 10.1016/j.mam.2018.03.002
174. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ Res (2018) 122(6):877–902. doi: 10.1161/CIRCRESAHA.117.311401
175. Grandl G, Wolfrum C. Hemostasis, Endothelial Stress, Inflammation, and the Metabolic Syndrome. Semin Immunopathol (2018) 40(2):215–24. doi: 10.1007/s00281-017-0666-5
176. Zhang Y, Murugesan P, Huang K, Cai H. NADPH Oxidases and Oxidase Crosstalk in Cardiovascular Diseases: Novel Therapeutic Targets. Nat Rev Cardiol (2020) 17(3):170–94. doi: 10.1038/s41569-019-0260-8
177. Lassén E, Daehn IS. Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction. Int J Mol Sci (2020) 21(24):9456. doi: 10.3390/ijms21249456
178. Duni A, Liakopoulos V, Roumeliotis S, Peschos D, Dounousi E. Oxidative Stress in the Pathogenesis and Evolution of Chronic Kidney Disease: Untangling Ariadne's Thread. Int J Mol Sci (2019) 20(15):3711. doi: 10.3390/ijms20153711
179. Lee SR, An EJ, Kim J, Bae YS. Function of NADPH Oxidases in Diabetic Nephropathy and Development of Nox Inhibitors. Biomol Ther (2020) 28(1):25–33. doi: 10.4062/biomolther.2019.188
180. Sifuentes-Franco S, Padilla-Tejeda DE, Carrillo-Ibarra S, Miranda-Díaz AG. Oxidative Stress, Apoptosis, and Mitochondrial Function in Diabetic Nephropathy. Int J Endocrinol (2018) 2018:1875870. doi: 10.1155/2018/1875870
181. Schiffer TA, Friederich-Persson M. Mitochondrial Reactive Oxygen Species and Kidney Hypoxia in the Development of Diabetic Nephropathy. Front Physiol (2017) 8:211. doi: 10.3389/fphys.2017.00211
182. Kang Q, Yang C. Oxidative Stress and Diabetic Retinopathy: Molecular Mechanisms, Pathogenetic Role and Therapeutic Implications. Redox Biol (2020) 37:101799. doi: 10.1016/j.redox.2020.101799
183. Wu MY, Yiang GT, Lai TT, Li CJ. The Oxidative Stress and Mitochondrial Dysfunction during the Pathogenesis of Diabetic Retinopathy. Oxid Med Cell Longev (2018) 2018:3420187. doi: 10.1155/2018/3420187
184. Kowluru RA. Diabetic Retinopathy and NADPH Oxidase-2: A Sweet Slippery Road. Antioxidants (2021) 10(5):783. doi: 10.3390/antiox10050783
185. Santiago AR, Boia R, Aires ID, Ambrósio AF, Fernandes R. Sweet Stress: Coping With Vascular Dysfunction in Diabetic Retinopathy. Front Physiol (2018) 9:820. doi: 10.3389/fphys.2018.00820
186. Robles-Rivera RR, Castellanos-González JA, Olvera-Montaño C, Flores-Martin RA, López-Contreras AK, Arevalo-Simental DE, et al. Adjuvant Therapies in Diabetic Retinopathy as an Early Approach to Delay Its Progression: The Importance of Oxidative Stress and Inflammation. Oxid Med Cell Longev (2020) 2020:3096470. doi: 10.1155/2020/3096470
187. Ruan Y, Jiang S, Musayeva A, Gericke A. Oxidative Stress and Vascular Dysfunction in the Retina: Therapeutic Strategies. Antioxidants (2020) 9(8):761. doi: 10.3390/antiox9080761
188. Pegtel DM, Gould SJ. Exosomes. Annu Rev Biochem (2019) 88:487–514. doi: 10.1146/annurev-biochem-013118-111902
189. Théry C, Zitvogel L, Amigorena S. Exosomes: Composition, Biogenesis and Function. Nat Rev Immunol (2002) 2(8):569–79. doi: 10.1038/nri855
190. Jeppesen DK, Fenix AM, Franklin JL, Higginbotham JN, Zhang Q, Zimmerman LJ, et al. Reassessment of Exosome Composition. Cell (2019) 177(2):428–45.e18. doi: 10.1016/j.cell.2019.02.029
191. He C, Zheng S, Luo Y, Wang B. Exosome Theranostics: Biology and Translational Medicine. Theranostics (2018) 8(1):237–55. doi: 10.7150/thno.21945
192. Raposo G, Stoorvogel W. Extracellular Vesicles: Exosomes, Microvesicles, and Friends. J Cell Biol (2013) 200(4):373–83. doi: 10.1083/jcb.201211138
193. Mashouri L, Yousefi H, Aref AR, Ahadi AM, Molaei F, Alahari SK. Exosomes: Composition, Biogenesis, and Mechanisms in Cancer Metastasis and Drug Resistance. Mol Cancer (2019) 18(1):75. doi: 10.1186/s12943-019-0991-5
194. Castaño C, Novials A, Párrizas M. Exosomes and Diabetes. Diabetes Metab Res Rev (2019) 35(3):e3107. doi: 10.1002/dmrr.3107
195. Wang K, Yang C, Shi J, Gao T. Ox-LDL-induced lncRNA MALAT1 Promotes Autophagy in Human Umbilical Vein Endothelial Cells by Sponging miR-216a-5p and Regulating Beclin-1 Expression. Eur J Pharmacol (2019) 858:172338. doi: 10.1016/j.ejphar.2019.04.019
196. Lu Y, Liu D, Feng Q, Liu Z. Diabetic Nephropathy: Perspective on Extracellular Vesicles. Front Immunol (2020) 11:943. doi: 10.3389/fimmu.2020.00943
197. Chen J, Zhang Q, Liu D, Liu Z. Exosomes: Advances, Development and Potential Therapeutic Strategies in Diabetic Nephropathy. Metabolism (2021) 122:154834. doi: 10.1016/j.metabol.2021.154834
198. Thongboonkerd V. Roles for Exosome in Various Kidney Diseases and Disorders. Front Pharmacol (2020) 10:1655. doi: 10.3389/fphar.2019.01655
199. Sun H, Yao W, Tang Y, Zhuang W, Wu D, Huang S, et al. Urinary Exosomes as a Novel Biomarker for Evaluation of α-lipoic Acid's Protective Effect in Early Diabetic Nephropathy. J Clin Lab Anal (2017) 31(6):e22129. doi: 10.1002/jcla.22129
200. Sinha N, Kumar V, Puri V, Nada R, Rastogi A, Jha V, et al. Urinary Exosomes: Potential Biomarkers for Diabetic Nephropathy. Nephrology (2020) 25(12):881–7. doi: 10.1111/nep.13720
201. Ebrahim N E, Ahmed IA, Hussien NI, Dessouky AA, Farid AS, Elshazly AM, et al. Mesenchymal Stem Cell-Derived Exosomes Ameliorated Diabetic Nephropathy by Autophagy Induction through the mTOR Signaling Pathway. Cells (2018) 7(12):226. doi: 10.3390/cells7120226
202. Khan NU, Lin J, Liu X, Li H, Lu W, Zhong Z, et al. Insights into Predicting Diabetic Nephropathy Using Urinary Biomarkers. Biochim Biophys Acta Proteins Proteom (2020) 1868(10):140475. doi: 10.1016/j.bbapap.2020.140475
203. Vitorino R, Ferreira R, Guedes S, Amado F, Thongboonkerd V. What Can Urinary Exosomes Tell Us? Cell Mol Life Sci (2021) 78(7):3265–83. doi: 10.1007/s00018-020-03739-w
204. Zhang H, Liu J, Qu D, Wang L, Wong CM, Lau CW, et al. Serum Exosomes Mediate Delivery of Arginase 1 as a Novel Mechanism for Endothelial Dysfunction in Diabetes. Proc Natl Acad Sci USA (2018) 115(29):E6927–36. doi: 10.1073/pnas.1721521115
205. Zhang Y, Ma KL, Gong YX, Wang GH, Hu ZB, Liu L, et al. Platelet Microparticles Mediate Glomerular Endothelial Injury in Early Diabetic Nephropathy. J Am Soc Nephrol (2018) 29(11):2671–95. doi: 10.1681/ASN.2018040368
206. Mathew B, Ravindran S, Liu X, Torres L, Chennakesavalu M, Huang CC, et al. Mesenchymal Stem Cell-Derived Extracellular vesicles and Retinal Ischemia-Reperfusion. Biomaterials (2019) 197:146–60. doi: 10.1016/j.biomaterials.2019.01.016
207. Mathew B, Torres LA, Gamboa Acha L, Tran S, Liu A, Patel R, et al. Uptake and Distribution of Administered Bone Marrow Mesenchymal Stem Cell Extracellular Vesicles in Retina. Cells (2021) 10(4):730. doi: 10.3390/cells10040730
208. Moisseiev E, Anderson JD, Oltjen S, Goswami M, Zawadzki RJ, Nolta JA, et al. Protective Effect of Intravitreal Administration of Exosomes Derived from Mesenchymal Stem Cells on Retinal Ischemia. Curr Eye Res (2017) 42(10):1358–67. doi: 10.1080/02713683.2017.1319491
209. Katome T, Namekata K, Mitamura Y, Semba K, Egawa M, Naito T, et al. Expression of Intraocular Peroxisome Proliferator-Activated Receptor Gamma in Patients with Proliferative Diabetic Retinopathy. J Diabetes Complications (2015) 29(2):275–81. doi: 10.1016/j.jdiacomp.2014.10.010
210. Olejarz W, Kubiak-Tomaszewska G, Chrzanowska A, Lorenc T. Exosomes in Angiogenesis and Anti-Angiogenic Therapy in Cancers. Int J Mol Sci (2020) 21(16):5840. doi: 10.3390/ijms21165840
211. Chen Z, Wang H, Xia Y, Yan F, Lu Y. Therapeutic Potential of Mesenchymal Cell-Derived miRNA-150-5p-Expressing Exosomes in Rheumatoid Arthritis Mediated by the Modulation of MMP14 and VEGF. J Immunol (2018) 201(8):2472–82. doi: 10.4049/jimmunol.1800304
212. Maisto R, Trotta MC, Petrillo F, Izzo S, Cuomo G, Alfano R, et al. Resolvin D1 Modulates the Intracellular VEGF-Related miRNAs of Retinal Photoreceptors Challenged With High Glucose. Front Pharmacol (2020) 11:235. doi: 10.3389/fphar.2020.00235
213. Liu C, Ge HM, Liu BH, Dong R, Shan K, Chen X, et al. Targeting Pericyte-Endothelial Cell Crosstalk by Circular RNA-cPWWP2A Inhibition Aggravates Diabetes-Induced Microvascular Dysfunction. Proc Natl Acad Sci USA (2019) 116(15):7455–64. doi: 10.1073/pnas.1814874116
214. Zhang W, Dong X, Wang T, Kong Y. Exosomes Derived from Platelet-Rich Plasma Mediate Hyperglycemia-Induced Retinal Endothelial Injury Via Targeting the TLR4 Signaling Pathway. Exp Eye Res (2019) 189:107813. doi: 10.1016/j.exer.2019.107813
215. Kamalden TA, Macgregor-Das AM, Kannan SM, Dunkerly-Eyring B, Khaliddin N, Xu Z, et al. Exosomal MicroRNA-15a Transfer from the Pancreas Augments Diabetic Complications by Inducing Oxidative Stress. Antioxid Redox Signal (2017) 27(13):913–30. doi: 10.1089/ars.2016.6844
216. Chen L, Wang C, Sun H, Wang J, Liang Y, Wang Y, et al. The Bioinformatics Toolbox for circRNA Discovery and Analysis. Brief Bioinform (2021) 22(2):1706–28. doi: 10.1093/bib/bbaa001
217. Xu X, Zhang J, Tian Y, Gao Y, Dong X, Chen W, et al. CircRNA Inhibits DNA Damage Repair by Interacting with Host Gene. Mol Cancer (2020) 19(1):128. doi: 10.1186/s12943-020-01246-x
218. Meng S, Zhou H, Feng Z, Xu Z, Tang Y, Li P, et al. CircRNA: Functions and Properties of a Novel Potential Biomarker for Cancer. Mol Cancer (2017) 16(1):94. doi: 10.1186/s12943-017-0663-2
219. Cao M, Zhang L, Wang JH, Zeng H, Peng Y, Zou J, et al. Identifying circRNA-Associated-ceRNA Networks in Retinal Neovascularization in Mice. Int J Med Sci (2019) 16(10):1356–65. doi: 10.7150/ijms.35149
220. Zhang W, Xu W, Feng Y, Zhou X. Non-coding RNA Involvement in the Pathogenesis of Diabetic Cardiomyopathy. J Cell Mol Med (2019) 23(9):5859–67. doi: 10.1111/jcmm.14510
221. Yan Q, He X, Kuang G, Ou C. CircRNA cPWWP2A: An Emerging Player in Diabetes Mellitus. J Cell Commun Signal (2020) 14(3):351–3. doi: 10.1007/s12079-020-00570-7
222. Pei X, Ye S, Jin G, Yu Y. Overexpression of circRNA-001175 Promotes Proliferation and Angiogenesis and Inhibits Apoptosis of the Human Umbilical Vein Endothelial Cells (HUVECs) induced by High Glucose. Int J Clin Exp Pathol (2018) 11(1):359–66.
223. Pan L, Lian W, Zhang X, Han S, Cao C, Li X, et al. Human Circular RNA−0054633 Regulates High Glucose−Induced Vascular Endothelial Cell Dysfunction through the microRNA−218/Roundabout 1 and microRNA−218/heme Oxygenase−1 Axes. Int J Mol Med (2018) 42(1):597–06. doi: 10.3892/ijmm.2018.3625
224. Cao Y, Yuan G, Zhang Y, Lu R. High Glucose-Induced circHIPK3 Downregulation Mediates Endothelial Cell Injury. Biochem Biophys Res Commun (2018) 507(1-4):362–8. doi: 10.1016/j.bbrc.2018.11.041
225. Cheng J, Liu Q, Hu N, Zheng F, Zhang X, Ni Y, et al. Downregulation of hsa_circ_0068087 Ameliorates TLR4/NF-κB/NLRP3 Inflammasome-Mediated Inflammation and Endothelial Cell Dysfunction in High Glucose Conditioned by Sponging miR-197. Gene (2019) 709:1–7. doi: 10.1016/j.gene.2019.05.012
226. Hu W, Han Q, Zhao L, Wang L. Circular RNA circRNA_15698 Aggravates the Extracellular Matrix of Diabetic Nephropathy Mesangial Cells Via miR-185/TGF-β1. J Cell Physiol (2019) 234(2):1469–76. doi: 10.1002/jcp.26959
227. Chen B, Li Y, Liu Y, Xu Z. circLRP6 Regulates High Glucose-Induced Proliferation, Oxidative Stress, ECM Accumulation, and Inflammation in Mesangial Cells. J Cell Physiol (2019) 234(11):21249–59. doi: 10.1002/jcp.28730
228. Jin J, Sun H, Shi C, Yang H, Wu Y, Li W, et al. Circular RNA in Renal Diseases. J Cell Mol Med (2020) 24(12):6523–33. doi: 10.1111/jcmm.15295
229. Zhang SJ, Chen X, Li CP, Li XM, Liu C, Liu BH, et al. Identification and Characterization of Circular RNAs as a New Class of Putative Biomarkers in Diabetes Retinopathy. Invest Ophthalmol Vis Sci (2017) 58(14):6500–9. doi: 10.1167/iovs.17-22698
230. Shan K, Liu C, Liu BH, Chen X, Dong R, Liu X, et al. Circular Noncoding RNA HIPK3 Mediates Retinal Vascular Dysfunction in Diabetes Mellitus. Circulation (2017) 136(17):1629–42. doi: 10.1161/CIRCULATIONAHA.117.029004
231. Liu C, Yao MD, Li CP, Shan K, Yang H, Wang JJ, et al. Silencing Of Circular RNA-ZNF609 Ameliorates Vascular Endothelial Dysfunction. Theranostics (2017) 7(11):2863–77. doi: 10.7150/thno.19353
232. Zhu K, Hu X, Chen H, Li F, Yin N, Liu AL, et al. Downregulation of circRNA DMNT3B Contributes to Diabetic Retinal Vascular Dysfunction through Targeting miR-20b-5p and BAMBI. EBioMedicine (2019) 49:341–53. doi: 10.1016/j.ebiom.2019.10.004
233. Schiessl IM. The Role of Tubule-Interstitial Crosstalk in Renal Injury and Recovery. Semin Nephrol (2020) 40(2):216–31. doi: 10.1016/j.semnephrol.2020.01.012
234. Andrade-Oliveira V, Foresto-Neto O, Watanabe IKM, Zatz R, Câmara NOS. Inflammation in Renal Diseases: New and Old Players. Front Pharmacol (2019) 10:1192. doi: 10.3389/fphar.2019.01192
235. Eelen G, de Zeeuw P, Treps L, Harjes U, Wong BW, Carmeliet P. Endothelial Cell Metabolism. Physiol Rev (2018) 98(1):3–58. doi: 10.1152/physrev.00001.2017
236. Pierce RW, Shabanova V, Canarie M, Pinto M, da Silva YS, Bhandari V, et al. Angiopoietin Level Trajectories in Toddlers With Severe Sepsis and Septic Shock and Their Effect on Capillary Endothelium. Shock (2019) 51(3):298–305. doi: 10.1097/SHK.0000000000001172
237. Rudman-Melnick V, Adam M, Potter A, Chokshi SM, Ma Q, Drake KA, et al. Single-Cell Profiling of AKI in a Murine Model Reveals Novel Transcriptional Signatures, Profibrotic Phenotype, and Epithelial-to-Stromal Crosstalk. J Am Soc Nephrol (2020) 31(12):2793–814. doi: 10.1681/ASN.2020010052
238. Dimke H, Sparks MA, Thomson BR, Frische S, Coffman TM, Quaggin SE. Tubulovascular Cross-Talk by Vascular Endothelial Growth Factor a Maintains Peritubular Microvasculature in Kidney. J Am Soc Nephrol (2015) 26(5):1027–38. doi: 10.1681/ASN.2014010060
239. Matoba K, Takeda Y, Nagai Y, Kawanami D, Utsunomiya K, Nishimura R. Unraveling the Role of Inflammation in the Pathogenesis of Diabetic Kidney Disease. Int J Mol Sci (2019) 20(14):3393. doi: 10.3390/ijms20143393
240. Messerli FH, Bangalore S, Bavishi C, Rimoldi SF. Angiotensin-Converting Enzyme Inhibitors in Hypertension: To Use or Not to Use? J Am Coll Cardiol (2018) 71(13):1474–82. doi: 10.1016/j.jacc.2018.01.058
241. Qian J, Zhong J, Yan M, Cheng P, Shi H, Hao C, et al. Circulating α-Klotho is Related to Plasma Aldosterone and Its Follow-Up Change Predicts CKD Progression. Kidney Blood Press Res (2018) 43(3):836–46. doi: 10.1159/000490138
242. de Seigneux S, Courbebaisse M, Rutkowski JM, Wilhelm-Bals A, Metzger M, Khodo SN, et al. Proteinuria Increases Plasma Phosphate by Altering Its Tubular Handling. J Am Soc Nephrol (2015) 26(7):1608–18. doi: 10.1681/ASN.2014010104
243. Byron A, Randles MJ, Humphries JD, Mironov A, Hamidi H, Harris S, et al. Glomerular Cell Cross-Talk Influences Composition and Assembly of Extracellular Matrix. J Am Soc Nephrol (2014) 25(5):953–66. doi: 10.1681/ASN.2013070795
244. Lappas M. Runt-Related Transcription Factor 1 (RUNX1) Deficiency Attenuates Inflammation-Induced Pro-Inflammatory and Pro-Labour Mediators in Myometrium. Mol Cell Endocrinol (2018) 473:61–71. doi: 10.1016/j.mce.2018.01.003
245. Wang Y, Sun M, Wang Z, Li X, Zhu Y, Li Y. Omentin-1 Ameliorates the Attachment of the Leukocyte THP-1 Cells to HUVECs by Targeting the Transcriptional Factor KLF2. Biochem Biophys Res Commun (2018) 498(1):152–6. doi: 10.1016/j.bbrc.2018.02.012
246. Zhong F, Chen H, Wei C, Zhang W, Li Z, Jain MK, et al. Reduced Krüppel-like Factor 2 Expression may Aggravate the Endothelial Injury of Diabetic Nephropathy. Kidney Int (2015) 87(2):382–95. doi: 10.1038/ki.2014.286
247. Horne SJ, Vasquez JM, Guo Y, Ly V, Piret SE, Leonardo AR, et al. Podocyte-Specific Loss of Krüppel-Like Factor 6 Increases Mitochondrial Injury in Diabetic Kidney Disease. Diabetes (2018) 67(11):2420–33. doi: 10.2337/db17-0958
248. Wen Y, Lu X, Ren J, Privratsky JR, Yang B, Rudemiller NP, et al. KLF4 in Macrophages Attenuates TNFα-Mediated Kidney Injury and Fibrosis. J Am Soc Nephrol (2019) 30(10):1925–38. doi: 10.1681/ASN.2019020111
249. Kuosmanen SM, Kansanen E, Kaikkonen MU, Sihvola V, Pulkkinen K, Jyrkkänen HK, et al. NRF2 Regulates Endothelial Glycolysis and Proliferation with miR-93 and Mediates the Effects of Oxidized Phospholipids on Endothelial Activation. Nucleic Acids Res (2018) 46(3):1124–38. doi: 10.1093/nar/gkx1155
250. Hu HJ, Lin XL, Liu MH, Fan XJ, Zou WW. Curcumin Mediates Reversion of HGF-induced Epithelial-Mesenchymal Transition Via Inhibition of c-Met Expression in DU145 Cells. Oncol Lett (2016) 11(2):1499–505. doi: 10.3892/ol.2015.4063
251. Ising C, Koehler S, Brähler S, Merkwirth C, Höhne M, Baris OR, et al. Inhibition of Insulin/IGF-1 Receptor Signaling Protects from Mitochondria-Mediated Kidney Failure. EMBO Mol Med (2015) 7(3):275–87. doi: 10.15252/emmm.201404916
252. Lässer C, Jang SC, Lötvall J. Subpopulations of Extracellular Vesicles and their Therapeutic Potential. Mol Aspects Med (2018) 60:1–14. doi: 10.1016/j.mam.2018.02.002
253. Lv LL, Feng Y, Tang TT, Liu BC. New Insight into the Role of Extracellular Vesicles in Kidney Disease. J Cell Mol Med (2019) 23(2):731–9. doi: 10.1111/jcmm.14101
254. Lv LL, Feng Y, Wu M, Wang B, Li ZL, Zhong X, et al. Exosomal miRNA-19b-3p of Tubular Epithelial Cells Promotes M1 Macrophage Activation in Kidney Injury. Cell Death Differ (2020) 27(1):210–26. doi: 10.1038/s41418-019-0349-y
255. Wang T, Weinbaum S, Weinstein AM. Regulation of Glomerulotubular Balance: Flow-Activated Proximal Tubule Function. Pflugers Arch (2017) 469(5-6):643–54. doi: 10.1007/s00424-017-1960-8
256. Chen Y, Jiang S, Zou J, Zhong Y, Ding X. Silencing HIF-1α Aggravates Growth Inhibition and Necrosis of Proximal Renal Tubular Epithelial Cell under Hypoxia. Ren Fail (2016) 38(10):1726–34. doi: 10.1080/0886022X.2016.1229994
257. Liu WJ, Shen TT, Chen RH, Wu HL, Wang YJ, Deng JK, et al. Autophagy-Lysosome Pathway in Renal Tubular Epithelial Cells Is Disrupted by Advanced Glycation End Products in Diabetic Nephropathy. J Biol Chem (2015) 290(33):20499–510. doi: 10.1074/jbc.M115.666354
258. DeFronzo RA, Norton L, Abdul-Ghani M. Renal, Metabolic and Cardiovascular Considerations of SGLT2 Inhibition. Nat Rev Nephrol (2017) 13(1):11–26. doi: 10.1038/nrneph.2016.170
259. Nespoux J, Vallon V. SGLT2 Inhibition and Kidney Protection. Clin Sci (2018) 132(12):1329–39. doi: 10.1042/CS20171298
260. Tentolouris A, Vlachakis P, Tzeravini E, Eleftheriadou I, Tentolouris N. SGLT2 Inhibitors: A Review of Their Antidiabetic and Cardioprotective Effects. Int J Environ Res Public Health (2019) 16(16):2965. doi: 10.3390/ijerph16162965
261. Armulik A, Abramsson A, Betsholtz C. Endothelial/Pericyte Interactions. Circ Res (2005) 97(6):512–23. doi: 10.1161/01.RES.0000182903.16652.d7
262. Trost A, Bruckner D, Rivera FJ, Reitsamer HA. Pericytes in the Retina. Adv Exp Med Biol (2019) 1122:1–26. doi: 10.1007/978-3-030-11093-2_1
263. Caporali A, Martello A, Miscianinov V, Maselli D, Vono R, Spinetti G. Contribution of Pericyte Paracrine Regulation of the Endothelium to Angiogenesis. Pharmacol Ther (2017) 171:56–64. doi: 10.1016/j.pharmthera.2016.10.001
264. Monickaraj F, McGuire P, Das A. Cathepsin D Plays a Role in Endothelial-Pericyte Interactions During Alteration of the Blood-Retinal Barrier in Diabetic Retinopathy. FASEB J (2018) 32(5):2539–48. doi: 10.1096/fj.201700781RR
265. Huang H. Pericyte-Endothelial Interactions in the Retinal Microvasculature. Int J Mol Sci (2020) 21(19):7413. doi: 10.3390/ijms21197413
266. Selvam S, Kumar T, Fruttiger M. Retinal Vasculature Development in Health and Disease. Prog Retin Eye Res (2018) 63:1–19. doi: 10.1016/j.preteyeres.2017.11.001
267. Sweeney M, Foldes G. It Takes Two: Endothelial-Perivascular Cell Cross-Talk in Vascular Development and Disease. Front Cardiovasc Med (2018) 5:154. doi: 10.3389/fcvm.2018.00154
268. Ferro Desideri L, Traverso CE, Nicolò M. The Emerging Role of the Angiopoietin-Tie Pathway as Therapeutic Target for Treating Retinal Diseases. Expert Opin Ther Targets (2022) 26(2):145–54. doi: 10.1080/14728222.2022.203612
269. Campochiaro PA, Sophie R, Tolentino M, Miller DM, Browning D, Boyer DS, et al. Treatment of Diabetic Macular Edema with an Inhibitor of Vascular Endothelial-Protein Tyrosine Phosphatase that Activates Tie2. Ophthalmology (2015) 122(3):545–54. doi: 10.1016/j.ophtha.2014.09.023
270. Hussain RM, Neiweem AE, Kansara V, Harris A, Ciulla TA. Tie-2/Angiopoietin Pathway Modulation as a Therapeutic Strategy for Retinal Disease. Expert Opin Investig Drugs (2019) 28(10):861–9. doi: 10.1080/13543784.2019.1667333
271. Shaw I, Rider S, Mullins J, Hughes J, Péault B. Pericytes in the Renal Vasculature: Roles in Health and Disease. Nat Rev Nephrol (2018) 14(8):521–34. doi: 10.1038/s41581-018-0032-4
272. Doshi SM, Friedman AN. Diagnosis and Management of Type 2 Diabetic Kidney Disease. Clin J Am Soc Nephrol (2017) 12(8):1366–73. doi: 10.2215/CJN.11111016
273. Umanath K, Lewis JB. Update on Diabetic Nephropathy: Core Curriculum 2018. Am J Kidney Dis (2018) 71(6):884–95. doi: 10.1053/j.ajkd.2017.10.026
Keywords: diabetic retinopathy (DR), diabetic nephropathy (DN), diabetic endothelial dysfunction, CircRNAs, exosomes, cellular crosstalk
Citation: Yang J and Liu Z (2022) Mechanistic Pathogenesis of Endothelial Dysfunction in Diabetic Nephropathy and Retinopathy. Front. Endocrinol. 13:816400. doi: 10.3389/fendo.2022.816400
Received: 16 November 2021; Accepted: 28 March 2022;
Published: 25 May 2022.
Edited by:
Ana Stancic, University of Belgrade, SerbiaReviewed by:
Jelena Kotur-Stevuljević, University of Belgrade, SerbiaHu Huang, University of Missouri, United States
Copyright © 2022 Yang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhangsuo Liu, emhhbmdzdW9saXVAenp1LmVkdS5jbg==