 Stefan Kölker*
Stefan Kölker* Florian Gleich
Florian Gleich Ulrike MützeThomas Opladen on behalf of Scientific Consortia of E-IMD, E-HOD, iNTD, U-IMD, and MetabERN
Ulrike MützeThomas Opladen on behalf of Scientific Consortia of E-IMD, E-HOD, iNTD, U-IMD, and MetabERN- Division of Child Neurology and Metabolic Medicine, Center for Child and Adolescent Medicine, Heidelberg University Hospital, Heidelberg, Germany
Rare diseases, such as inherited metabolic diseases, have been identified as a health priority within the European Union more than 20 years ago and have become an integral part of EU health programs and European Reference Networks. Having the potential to pool data, to achieve sufficient sample size, to overcome the knowledge gap on rare diseases and to foster epidemiological and clinical research, patient registries are recognized as key instruments to evidence-based medicine for individuals with rare diseases. Patient registries can be used for multiple purposes, such as (1) describing the natural history and phenotypic diversity of rare diseases, (2) improving case definition and indication to treat, (3) identifying strategies for risk stratification and early prediction of disease severity (4), evaluating the impact of preventive, diagnostic, and therapeutic strategies on individual health, health economics, and the society, and (5) informing guideline development and policy makers. In contrast to clinical trials, patient registries aim to gather real-world evidence and to achieve generalizable results based on patient cohorts with a broad phenotypic spectrum. In order to develop a consistent and sustained framework for rare disease registries, uniform core principles have been formulated and have been formalized through the European Rare Disease Registration Infrastructure. Adherence to these core principles and compliance with the European general data protection regulations ensures that data collected and stored in patient registries can be exchanged and pooled in a protected environment. To illustrate the benefits and limitations of patient registries on rare disease research this review focuses on inherited metabolic diseases.
Introduction
Any disease affecting less than 1 in 2,000 people is considered rare in the European Union (EU). Although the approximately 6,000-8,000 distinct rare diseases are highly heterogeneous regarding etiology, pathophysiology, clinical presentation, and treatability, they have in common that affected individuals are often confronted with similar problems, such as diagnostic odyssey, lack of safe and effective therapies, and scarce specialist centers, resulting in significant inequalities. On the European level, rare diseases were identified by the 2nd EU Health Program as a healthcare priority with unmet needs, highlighting patient registries as the best way to pool data in order to achieve sufficient sample sizes for epidemiological and clinical research (1).
Rare diseases such as inherited metabolic diseases have been traditionally regarded as Mendelian traits that are caused by specific pathogenic gene variations, and as experiments of nature which helped to decipher underlying mechanisms of monogenic disorders (2). Natural history studies on inherited metabolic diseases, however, often failed to identify clear-cut genotype-phenotype correlations and to predict the clinical phenotype based on the genotype or biochemical phenotype. This is partially explained by involvement of other pathways whose fluxes are changed though secondary inhibition induced by toxic metabolites or lack of substrates (3, 4). Furthermore, modifier genes (5, 6), alternative pathways (7–9), side reactions of enzymes (10), metabolic proof-reading (11), and the concomitant net effect of metabolic changes on the level of single cells, organs and the whole organism significantly contribute to the resulting phenotype. Besides these intrinsic factors, it has been increasingly recognized that metabolism can also be influenced by gene-environment and gene-nutrient interactions (12–14), and the microbiome (15, 16). As a consequence, inherited metabolic diseases are nowadays perceived as a model for complex diseases. Another important aspect that causes apparent phenotypic diversity is time, the fourth dimension setting the scene for ontogeny and ageing. Therefore, the phenotype of an individual at a certain time point of life should not be regarded as static but as a snapshot. Any phenotypic comparisons should include the individual age as an anchor point, the clinical phenotype itself being a complex moving target.
For rare diseases patient registries are thought to be key instruments to achieve a sufficient sample size for clinical research, to guide healthcare planning, and to foster the development and evaluation of diagnostic and therapeutic interventions. Patient registries significantly contribute to evidence-based personalized medicine in the field of rare diseases, since they can be used for multiple purposes, such as improvement of case definition, revision of disease classification, evaluation of indication to treat and risk stratification, as well as safety, effectiveness, feasibility, limitations and benefits of diagnostic and therapeutic strategies under real-world conditions.
The Evolution of Rare Disease Registries
From Single Diseases to European Reference Networks (ERN) Registries
With the benefits of patient registries being widely accepted, the field of rare diseases saw a multitude of individual efforts on registry building in the past, usually realized within a national framework and focusing on single diseases or tightly defined disease groups. Successful examples on a national level include the mitoREGISTRY of the German Network for Mitochondrial Disorders (mitoNET, https://www.mitonet.org/) as well as Leukonet (https://www.leukodystrophie-info.de/), the German Network for Leukodystrophies, gathering data on mitochondrial disease since 2009 and leukodystrophies since 2003 respectively, both receiving funding from the German Federal Ministry of Education and Research (BMBF). International examples are the EUROGLYCANET, targeting congenital disorders of glycosylation (CDG) since 1999 (17) and the European Rare Kidney Disease Registry (ERKReg), observing rare kidney disease since 2018 (18) both having received funding from the European Commission, or the Urea Cycle Disorders Consortium (UCDC), having received funding from the US-American National Institutes of Health (NIH) since 2003 (19). While these are notable examples, they of course represent only a small sample of the wealth of existing registries, which currently are estimated as 793 only within the scope of Orphanet (20). Therefore the landscape of rare disease registries was highly scattered when EU policy makers recognized as early as 1999, the urgency of action on rare diseases at the European level (21), starting a process that lead to the recognition of scientific patient registries as key instruments to overcome challenges specific to rare diseases, like small sample sizes, while also envisioning the introduction of European Reference Networks as a means of organizing centers of excellence (22). This resulted in rare diseases becoming an integral part of the second EU health programme, calling for funding opportunities for the development and maintenance of rare disease registries (23). In the field of inherited metabolic diseases this development allowed the founding of the European Registry and Network for Intoxication type Metabolic Diseases (E-IMD: https://www.eimd-registry.org/; CHAFEA agreement no. 2010 12 01) in 2010, the European Network and Registry for Homocystinurias and Methylation Defects (E-HOD: https://www.ehod-registry.org/; CHAFEA agreement no. 2012 12 02) in 2012, followed by the International Working Group on Neurotransmitter-Related Disorders (iNTD: https://www.intd-registry.org/) in 2014, as expert lead international patient registries (24–26). Due to the increasing number of rare inherited metabolic diseases (currently more than 1,600 diseases according to IEMbase, URL: http://www.iembase.org/), these registries were conceptualized as multi-disease registries, logically grouping diseases by common traits of clinical manifestation or affected metabolic pathways. The registries rely on a tailored custom-made IT solution, allowing secure remote entry and central storage of pseudonymized data, while enrolling patients on the protocol of an observational study, adapted to the local legal context of participating health care providers (see Figure 1 which illustrates the modular design of electronic case report forms and study visits). The enrolment follows a top-down approach, i.e. large expert centers with great outreach in their specific countries and regions are motivated via their inherent scientific interest to join the consortia and enroll known and newly diagnosed patients on the various protocols. This process ensures an overall high quality of data through the expert-mediated enrolment and data entry process, leading to a good coverage for diseases whose management necessitates regular visits to large expert centers. On the other hand, this process could result in under-reporting where a more bottom-up approach mediated by patient organizations might be more beneficial. The associated health care providers organized themselves in scientific consortia, granting each member the right to use the entire wealth of gathered data for sufficiently elaborated projects, presented to and agreed by a member’s board, while at the same time guaranteeing strict individual ownership of data, thus still allowing single center research or evaluation of national cohorts, even including the right to opt out of common projects, if conflict mitigation mechanisms should fail.
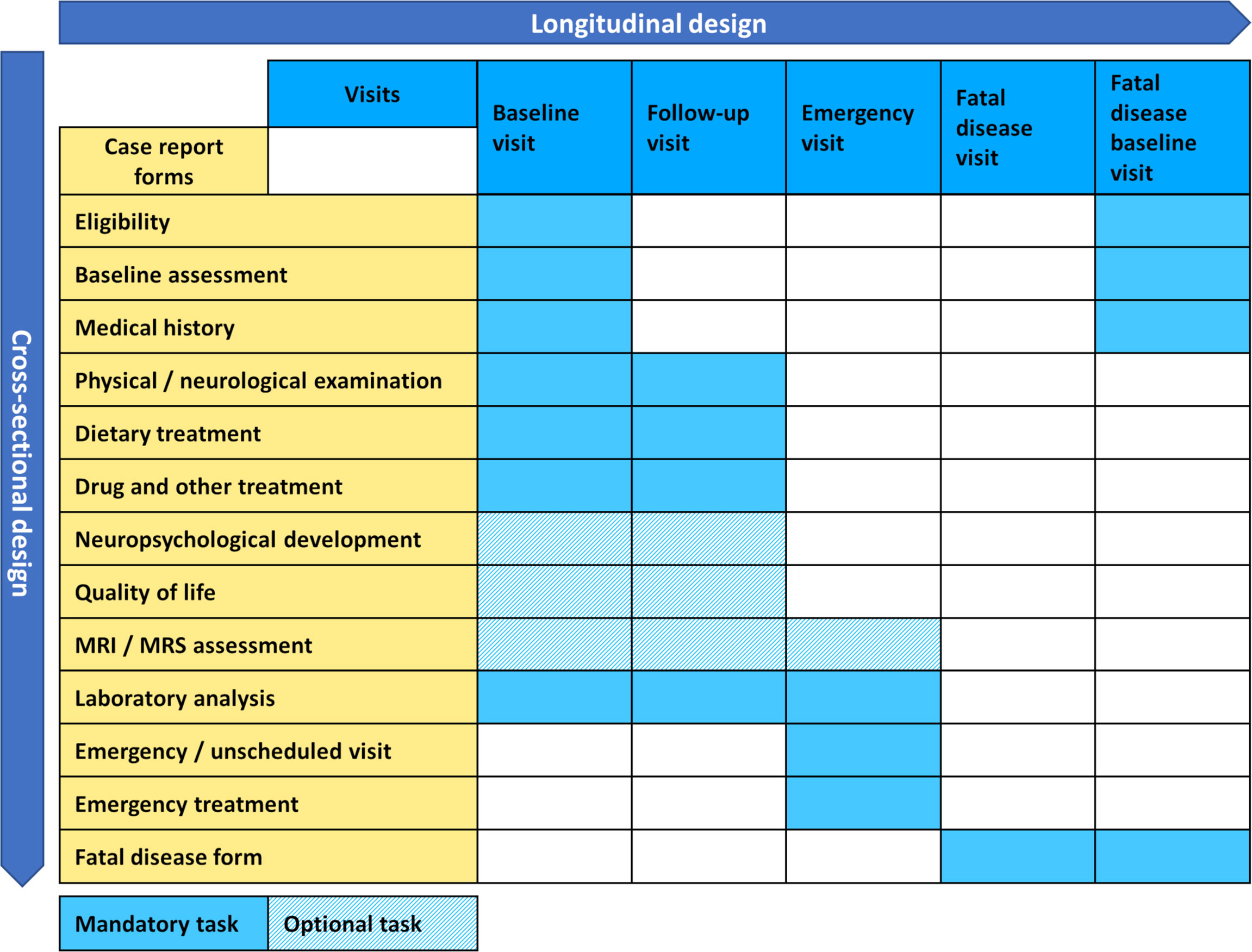
Figure 1 General design of the E-IMD, E-HOD, iNTD and U-IMD registries.
The data model employed by each registry is the result of the collaborative effort of experts as well as patient organizations in the field, being perfectly suitable for capturing clinical phenotypes, but nevertheless meeting more recently established standards of semantic interoperability only to a restricted extent.
European Rare Disease Registration Infrastructure (ERDRI) and Semantic Interoperability
Concomitantly to promoting individual registry building efforts the European Commission also furthered the development of the underlying framework for actions in the field of rare diseases, by establishing the advisory body of the European Union Committee of Experts on Rare Diseases (EUCERD) among others (27). The resulting EUCERD core recommendations on rare disease patient registration and data collection emphasize the need for establishing minimum common data sets, which should be uniform not only among disease specific registries but also across the entire spectrum of rare diseases, stress the critical importance of semantic interoperability between registries, which can be achieved by using standardized and controlled languages like OMIM code for case definition or SNOMED-CT for describing clinical phenotypes and endorse the introduction of a common European patient identifier (28). These principles should later materialize in the form of the European Rare Disease Registration Infrastructure (ERDRI), an integrated set of tools and recommendations presented on the European Platform for Rare Diseases. The central instrument of ERDRI is the Common Data Elements (CDE), a set of defined variables that should form the core of all European rare disease registries along with concrete recommendations on the usage of controlled dictionaries like Orpha or Alpha code for coding diseases, the nomenclature of the Human Genome Variation Society (HGVS) for describing the genetic phenotype and the Human Phenotype Ontology (HPO) for the clinical phenotype. The CDEs are embedded with further tools comprising an European metadata repository (ERDRI.mdr), an European reference database for rare disease registries (ERDRI.dor) and the introduction of an unique European patient identifier (European Platform on Rare Diseases Registration (29). With European Reference Networks having become a reality, by the time the ERDRI tools were released, the Unified Registry for Inherited Metabolic Disorders (U-IMD; CHAFEA agreement no. 777259) could be implemented as the first European and international registry encompassing all inherited metabolic diseases. U-IMD implements the ERDRI and serves as the official registry of the European Reference Network for Hereditary Metabolic Disorders (MetabERN) (see Figure 2 for an illustration the combined global outreach of the European IMD registries and Table 1 for the combined current enrolment numbers). Apart from introducing a new registry, the U-IMD action also aims at providing the expertise for implementing a higher level of semantic interoperability to the existing IMD registries. In this way U-IMD works as a complement and enhancement to the existing structures, avoiding duplication much as possible for a generic approach (30). The hurdles represented by lack of semantic interoperability were experienced by E-IMD in its collaborative effort to establish the largest continuously followed cohort of urea cycle disorders together with the American Urea Cycle Disorders Consortium (UCDC). Although surmountable the datasets required extensive work-up by clinicians to correctly classify clinical abnormalities since semantic interoperability of both registries was incomplete (31). Introduction of HPO terms and other controlled and structured dictionaries could have significantly shortened the mapping process (32). With these lessons in mind the adaption process facilitated by U-IMD includes the introduction of the EDRI tools to the E-IMD, E-HOD and iNTD registries, with special emphasis enhancing semantic interoperability between the registry and to other data sources with a comparable standard. U-IMD itself ensured interoperability to other ERN registries already during its design, going as far as mirroring the data model of the European Rare Kidney Disease Reference Network (ERKNet) since both ERNs share metabolic nephropathies as target diseases (30).
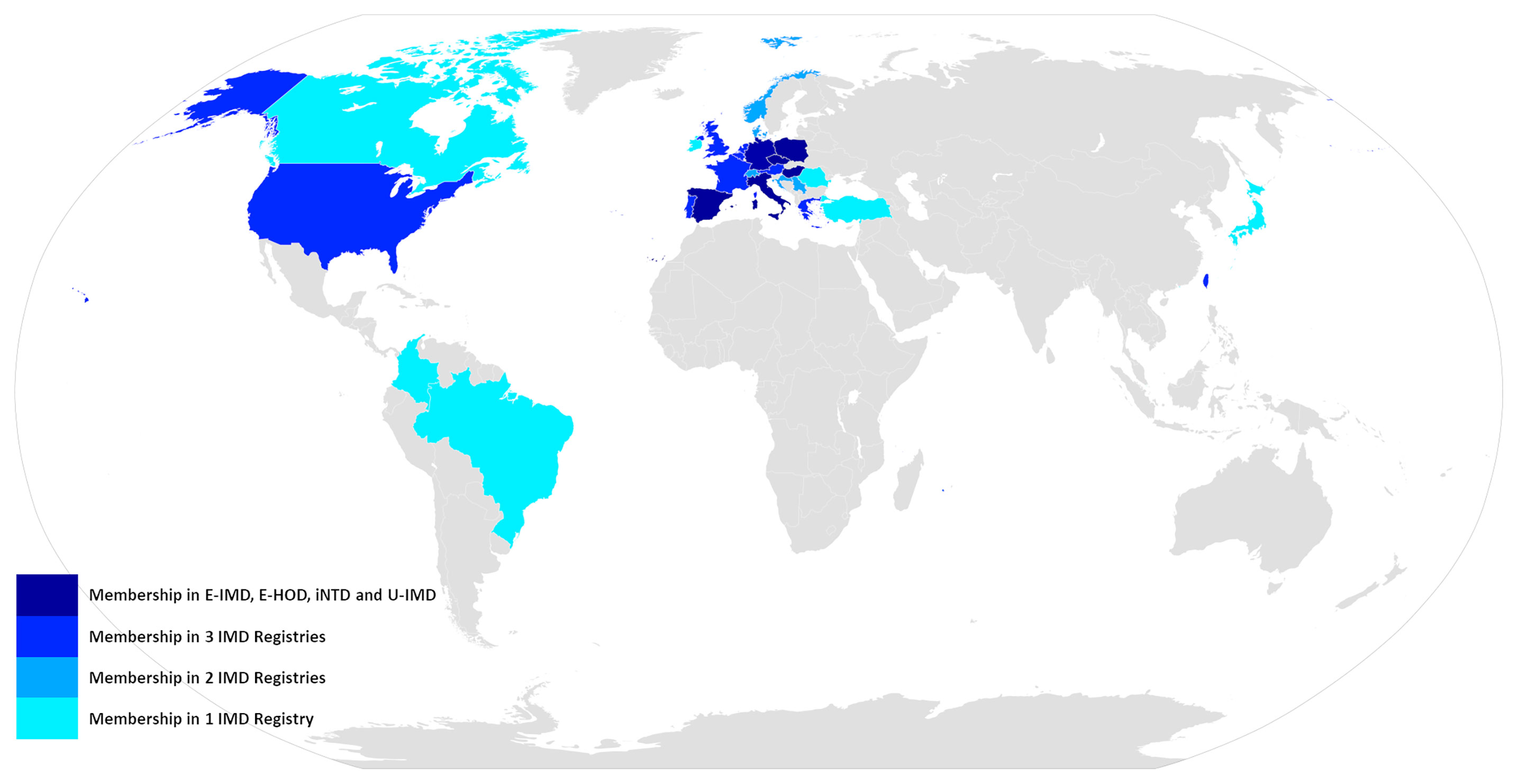
Figure 2 Global outreach of the E-IMD, E-HOD, iNTD and U-IMD registries.
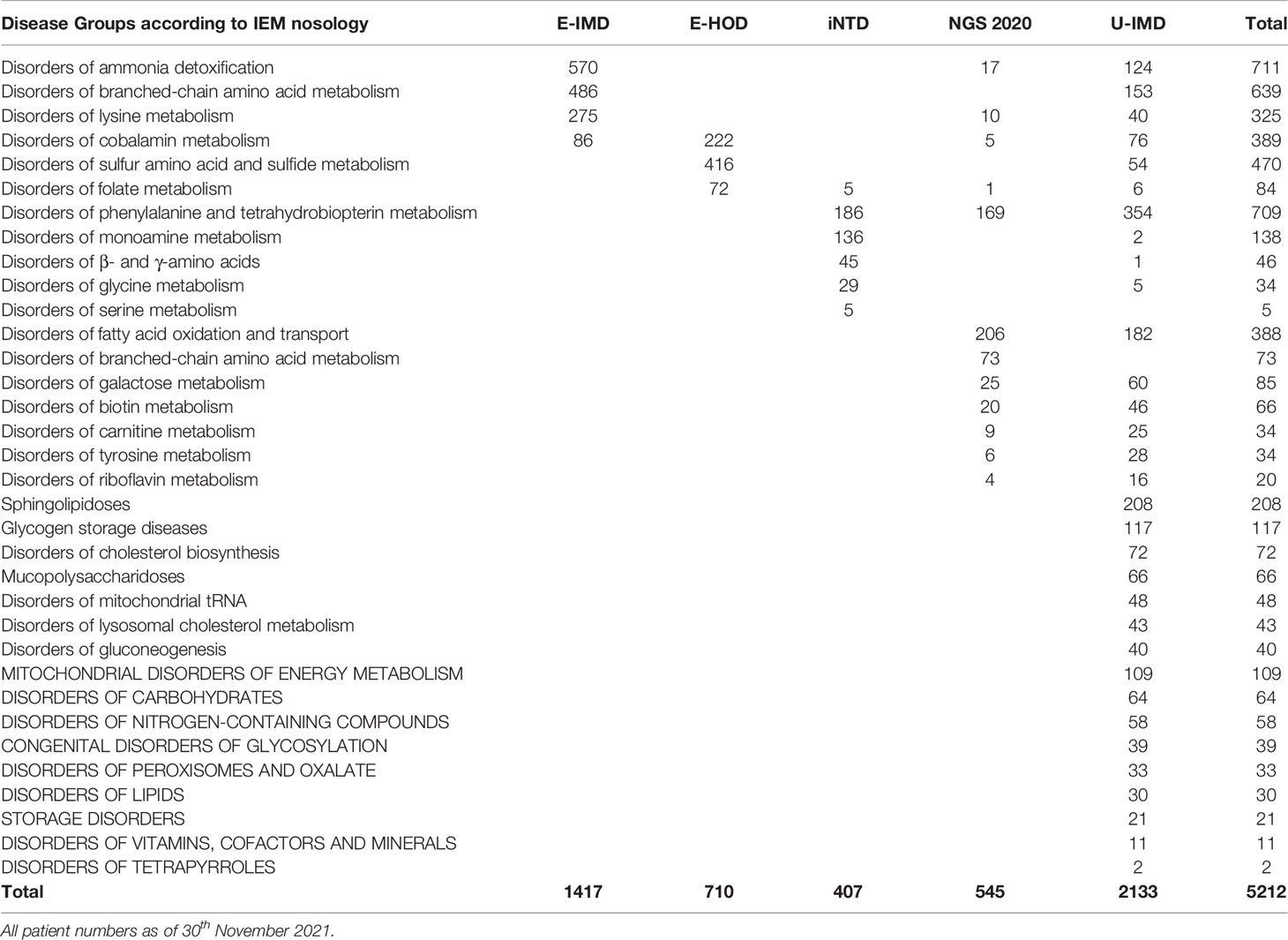
Table 1 Enrolment of active patients for the E-IMD, E-HOD, iNTD and U-IMD registries and for the German newborn screening outcome study (NGS2020, DRKS00013329).
Patient Registries Are Multi-Purpose Instruments for Rare Disease Research
Natural History Studies
The most common application for patient registries is natural history studies, e.g., ultra-rare diseases such as mevalonic aciduria (33), to raise awareness. The precise description of the initial presentation helps to reduce diagnostic delay and to guide the diagnostic process. Noteworthy, phenotypic diversity can be extreme such as in urea cycle disorders, ranging from life-threatening sepsis-like hyperammonemic encephalopathy in newborns with severe disease onset to variable fluctuating episodes of neurological, gastrointestinal and psychiatric symptoms in patients with attenuated forms (34, 35). Phenotypic diversity of variant disease courses can be easily mistaken as discrepant diseases with overlapping biochemical and clinical presentations. This is exemplified by genotype phenotype correlation studies in neuroblastoma-amplified sequence (NBAS)-associated disease which identified three different but partially overlapping subgroups with complex phenotypes which are directly related to the affected region of the NBAS protein (36). In order to distinguish the initial presentation and the evolving clinical phenotypes of rare diseases, comparative phenotyping within and between disease groups is a helpful strategy. Recently, this approach was successfully applied to elucidate clinical similarities and differences between disorders of biogenic amine and tetrahydrobiopterin (BH4) metabolism (37), urea cycle disorders, and organic acidurias (24). This knowledge is of relevance for the optimization of the diagnostic process, patient clinical paths, healthcare planning, and counselling of parents having a child with a rare disease. For example, comparative phenotyping helped to identify the need to carefully monitor renal function not only in individuals with isolated methylmalonic acidurias but also in individuals with other organic acidurias such as propionic aciduria and glutaric aciduria type 1 (24, 38) since chronic kidney dysfunction was already well known in methylmalonic aciduria (39, 40), but was underestimated in propionic aciduria and unknown in glutaric aciduria type 1 before. Patient registries also allow comparison between different diagnostic journeys and related health outcomes on an international level, especially if there are known rigid differences in diagnostic approaches between countries, that may be evaluated against each other, as is the case with differing national newborn screening panels. Results from such comparisons may help in harmonizing newborn screening programs by illustrating different needs for long term care resulting from different diagnostic journeys in the light of health economics.
Patient registries are powerful tools to describe and compare disease variants and to challenge existing case definition, disease classification, and indication to treat. Recently, careful biochemical and clinical evaluation of more than 300 individuals with confirmed cystathionine β-synthase deficiency resulted in the development of comprehensive criteria for the classification of pyridoxine responsiveness (41). This reclassification is likely to have enormous practical consequences on the diagnostic process, treatment and management of affected individuals with this disease since pyridoxine responsiveness is the major predictor of clinical severity and outcome. Furthermore, patient registries can help to elucidate the clinical relevance of newly identified disease variants with yet uncertain clinical significance. A good example is so-called mild isovaleric aciduria, an attenuated and putatively benign disease variant, which has been increasingly recognized since the inclusion of isovaleric aciduria into newborn screening panels. In contrast to individuals with the severe, so-called classic, disease variant with high risk of neonatal mortality, poor neurocognitive outcome in surviving individuals, and a clear indication to treat, individuals with the mild disease variant are not confronted with acute metabolic decompensations and increased neonatal mortality and showed excellent neurocognitive outcomes. Noteworthy, excellent health outcomes in individuals with mild isovaleric aciduria did not appear to be impacted by metabolic maintenance therapy (42), questioning the overall benefit from NBS and the indication to treat.
A solid description of the disease course with its age-dependent manifestations and variations is also an important source for the identification of meaningful endpoints for clinical trials instead of surrogate parameters with doubtful clinical relevance for affected individuals. Furthermore, it can guide the inclusion of individuals with well-defined clinical severity to clinical trials in order to avoid apparent (non-) effectiveness of the study drug due to case mix differences of study groups. For individuals with urea cycle disorders, the severity of the initial presentation, represented by age at disease onset, initial peak ammonium concentration in plasma, and coma, has a major impact on survival and neurocognitive outcomes in urea cycle disorders (43, 44). In addition to mortality as a hard clinical end point, collaborative evaluation of 300 individuals with ornithine transcarbamylase deficiency identified intelligence as another suitable clinical endpoint since it was shown to be the most distinguished indicator for cognitive function and to correlate highly with the initial peak ammonium concentration in plasma (45, 46).
Benefits and Limitations of Current Diagnostic and Therapeutic Strategies
It is a well-known fact that individuals with rare diseases are often confronted with a diagnostic odyssey and diagnostic delay, putting high burden on the affected individual and her/his family and being associated with an increased risk of losing therapeutic effectiveness and developing irreversible organ dysfunction. To shorten the path to diagnosis newborn screening programs for rare diseases, such as inherited metabolic diseases, have been continuously developed for more than 50 years following Robert Guthrie’s pioneering work (47). With technological developments, such as tandem mass spectrometry, and concomitant extensions of the disease panel, endogenous intoxication-type metabolic diseases with acute neonatal manifestations have been introduced to these programs (48, 49). As a consequence, some individuals affected by these diseases may become symptomatic in the first days of life, even before newborn screening results are available, questioning the benefit of newborn screening. Comparison of screened and unscreened cohorts followed by patient registries can help to estimate the proportion of individuals who can be reliably identified pre-symptomatically depending on the time point of sample collection (44, 50). Such studies can also evaluate the diagnostic process quality of newborn screening programs, identifying current strengths and weaknesses of its team-players, i.e. senders, mail services, and laboratories, evaluate promising new biomarkers, such as 3-O-methyldopa in dried blood spots for aromatic ʟ-amino-acid decarboxylase deficiency, and thus can highlight the potential for improvement (33, 51). Longitudinal outcome studies of newborn screening cohorts, which provide formal evidence of the clinical effectiveness and the long-term health benefits of screened individuals, have remained the neglected part of newborn screening programs. A long-term observational study in Germany recently demonstrated that newborn screening for inherited metabolic diseases resulted in overall normal development (96%), normal cognitive outcome (mean IQ: 100), and regular attendance of screened individuals at kindergarten (95%) and primary school (95%) (51). At the same time, it identified disease-specific variations of health outcomes and diseases with less favorable health outcomes, particularly if recommended treatment was not adequately prescribed or adhered to (38, 52), as well as individuals with a putatively benign disease variants with risk of over-treatment (42). In addition, observational studies based on patient registries are helpful to evaluate the prevalence and putative health impact of early diagnosis and treatment for conditions that are not yet regularly included in national screening panels, such as neonatal vitamin B12 deficiency mostly due to hitherto undiagnosed maternal vitamin B12 deficiency (53).
Another important but sometimes difficult application of patient registries is the evaluation of therapeutic safety and effectiveness under real-world conditions. In contrast to clinical trials, inclusion and exclusion criteria are kept to a minimum in patient registries in order to study affected individuals with a broad phenotypic spectrum (1). Furthermore, patients are observed as they present for care instead of asking for strict adherence to protocol. Because of the resulting high variation, which is important to achieve generalizable results, evidence-based answers to therapy-related questions often depend on a large sample size and hence require the pooling of data from different sources (44, 46). Interoperability is key to the success of this effort. If these hurdles can be overcome, patient registries are valuable sources for the optimization of therapies and guideline development. For instance, long-term follow-up of the largest cohort of individuals with urea cycle disorders did not find evidence for superiority of any of the available nitrogen scavenger therapies but highlighted that early liver transplantation appears to be beneficial (46). Another example is glutaric aciduria type 1, which was thought to be an untreatable condition in the pre-screening era since treatment given to symptomatic patients did not improve the clinical outcome (54). Introduction of tandem mass spectrometry-based newborn screening programs challenged this view, proving that pre-symptomatic start of treatment is the major prerequisite for favorable neurological outcome (38, 52). Long-term follow-up of two large patient cohorts in the USA and Germany further elucidated that in screened individuals with glutaric aciduria type 1 therapeutic quality and adherence to evidence-based recommendations become major predictors of neurological outcome and survival (55). Furthermore, these studies showed that low lysine diet supplemented with fortified metabolic formula is superior to protein restriction and allows for normal anthropometric long-term development (38, 56, 57). In another large group of individuals with protein-dependent inherited metabolic disorders, study of the dietary impact on growth highlighted the need for careful monitoring of plasma amino acids, particularly branched-chain amino acids and L-arginine, and to adjust the protein-to-energy prescription of diet to support normal growth (58). In brief, besides precise evaluation of the natural history patient registries are also valuable instruments to assess the iatrogenic impact on the evolving clinical phenotype.
Guideline Development
Patient registries are important data sources for guideline development, helping to improve the evidence level and the grade of recommendation. Since guideline development for rare diseases is often difficult and based on literature with a low level of evidence, such as case reports, small case control or cohort studies, all of them having a high risk of bias and low probability that the reported causes and effects are causally linked. Furthermore, guideline development often identifies topics which have remained white spots on the scientific map, with no published evidence at all. Observational studies and health economic evaluations based on patient registries can specifically address these questions and topics to larger cohorts, seeking for evidence. Furthermore, patient registries can inform guideline developmental groups about the feasibility, safety, and effectiveness of guideline recommendations, paving the way for successful guideline revision. The high potential of this approach can be illustrated by guideline development for glutaric aciduria type 1, being initiated more than 15 years ago. While the first guideline mostly included grade D and a few grade C recommendations, reflecting significant uncertainty (59), the level of evidence and grade of recommendations have been improved continuously, demonstrated by the first (60) and second revision (61). This progress was much accelerated by careful long-term follow-up of national and international cohorts (38, 56, 57, 59, 61–69), demonstrating that (i) newborn screening is the prerequisite of favorable neurological outcome and survival and (ii) is highly cost-effective, and that (iii) adherence to recommended maintenance and emergency treatment is associated with the most favorable neurological outcome and (iv) normal growth. Furthermore, these studies described a so far unknown renal disease manifestation, unraveled similarities (risk of striatal necrosis with concomitant complex movement disorder with predominant dystonia) and discrepancies (cognitive function, white matter changes, subdural hematoma) between biochemically delineated subgroups (high versus low excreter phenotype), and evaluated which part of the complex clinical spectrum can be specifically targeted and changed by current therapy, highlighting the need for safer and more effective medicines. Similar approaches to continuous improvement of guidelines for rare diseases through long-term observational studies coordinated by international scientific consortia have also been chosen for other rare diseases, such as urea cycle disorders (70, 71), propionic and methylmalonic aciduria (72, 73), cobalamin-related remethylation disorders (74–76), cystathionine beta-synthase deficiency (41, 77), and neurotransmitter-related disorders (37, 78, 79).
Post-Authorization Safety Studies (PASS)
Large scale data collection whether in clinical research or other scientific fields, raises concerns regarding the principles of long-term data stewardship, having resulted in the formulation of the FAIR concept, signifying findability, accessibility, interoperability and reusability of data (80). The aspect of reutilization has a special ethical significance in clinical research for rare disease, since comparatively small patient cohorts imply an enhanced risk of putting the burden of participation in multiple studies on a small number of patients, often severely affected. For the same reason of small patient numbers, orphan drug designations and the resulting need for post-authorization safety studies (PASS) are common for rare diseases, which usually are implemented as industry-driven single purpose drug registries. There is an increasing awareness of regulating bodies like the European Medicine Agency (EMA) for the under-utilization of existing data sources like scientific disease registries, which often already gather the type of longitudinal data also required for certain regulatory actions like post-authorization measures (81, 82). Therefore, the collaboration between marketing authorization holders (MAH) and scientific patient registries, within the model of a public private partnership, is a feasible approach for achieving the goals of certain drug safety measures, while at the same time reducing data fragmentation and duplication and ensuring ongoing data stewardship by publicly funded scientific consortia, better capable and more committed to ensuring data FAIR-ness than industry lead efforts. In the past E-HOD, as well as currently E-IMD, successfully implemented the model as envisioned by the EMA, for the orphan drugs Cystadane® (betaine anhydrous, http://www.encepp.eu/encepp/viewResource.htm?id=40022) and Ravicti® (glycerol phenylbutyrate, EUPAS17267; URL: http://www.encepp.eu/encepp/viewResource.htm?id=30377), respectively. In both cases the PASS studies were implemented on a protocol drafted by the MAH together with the scientific registries and accepted by the Pharmacovigilance Risk Assessment Committee (PRAC) of the EMA. Patients already being enrolled in the respective registries could choose to also participate in the PASS, allowing their already available, purely observational data to be used for facilitating the surveillance measure, without increasing their burden or generating new data that would be only available in an industry owned data source.
Discussion
Patient registries are key to personalized evidence-based medicine for individuals with rare diseases since they help to overcome the intrinsic obstacles of rare disease research through pooling of data and achievement of sufficient sample sizes. There is an increasing number of examples demonstrating that patient registries can fulfil multiple purposes for rare disease research, particularly (A) improving the knowledge about natural history and variant disease courses, (B) identifying meaningful endpoints for clinical trials, improving case definition, genotype phenotype correlation, risk stratification, and therapeutic decision, (C) evaluating the safety, effectiveness, and long-term health benefits as well as the health economical and societal benefits of preventive care programs, diagnostic strategies, and therapies, such as orphan drugs, and (D) increasing the evidence base and recommendation grade of guidelines.
However, successful establishment, sustainability, and usefulness of patient registries may be limited by (A) data fragmentation and duplication because of uncoordinated parallel activities with unharmonized data models, (B) lack of seeding and sustainability funding though industry-independent public sources, hampering projects via numerous funding dependent constraints like reimbursement of person hours invested by experts, (C) insufficient geographical coverage resulting in small cohorts, (D) non-adherence to data FAIRness, particularly syntactic and semantic interoperability, hampering data exchange and cross-site analysis, (E) non-compliance with data protection regulations resulting in the discontinuation of some older patient registries following the inception of the GDPR in 2018, and (F) non-involvement of patients and patient groups in the development of registries, leading to insufficient consideration of the patients’ view and experience. This last point is of special significance, since conservative treatment options for many IMDs can put a high psychosocial burden on patients and caregivers alike, often necessitating measures like permanent strict dietary management that conflict with many life choices. Adequately capturing these burdens with tailored patient reported outcome measures (PROMs) was often neglected in the past, but will become increasingly important going forward, since novel treatments directly addressing underlying disease causes will increasingly become available, with PROMs being an important measure for their efficacy (83).
In conclusion, the future success of patient registries for rare disease research in Europe critically depends on the compliance with formal requirements, particularly the FAIR data principles and the GDPR to ensure long-term data collection and data exchange in a protected environment. As indicated by a recent survey among 40 rare disease registries, there seems to be a high level of recognition within the community that adherence to formal aspects of governance, particularly FAIR principles, is a key criterion of successful implementation and administration of rare disease registries (84), although especially long-running projects might struggle to keep pace with adapting to recently introduced standards. Particular attention should be paid to facilitating the achievement of ethical approval and informed consent since these hurdles are importing rate-limiting steps (19, 24, 30). With the future development of safe and reliable IT tools for data extraction from electronic health records it is also hoped that the work- and cost-intensive process of gathering and entering personalized health data to registries can be significantly reduced. Finally, industry-independent sustainability of patient registries conducted by scientific consortia and European Reference Networks for Rare Diseases requires the establishment of long-term funding strategies on the level of the European Union and its Member States.
Author Contributions
All authors conceptualized the manuscript. SK and FG wrote the first draft of the manuscript. UM and TO edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Discussed studies, coordinated by some of the authors, received funding: SK received EU funding for the European Registry and Network for Intoxication-type Metabolic Diseases (E-IMD; CHAFEA agreement no. 2010 12 01), the European Network and Registry for Homocystinurias and Methylation Defects (E-HOD; CHAFEA agreement no. 2012 12 02), and the Unified Registry for Inherited Metabolic Diseases (U-IMD, CHAFEA agreement no. 777259), as well as funding from the Dietmar Hopp Foundation (Germany) for the observational study NGS 2020. TO received funding by Dietmar Hopp Foundation for conducting the International Working Group on Neurotransmitter-Related Disorders (iNTD).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are thankful to all patients with inherited metabolic diseases and their families for their trust and important contribution to the ongoing observational studies E-IMD, E-HOD, U-IMD, and iNTD. We thank all colleagues from the mentioned European and international consortia as well as the European Reference Network MetabERN for their valuable input and important scientific contribution. Representative for all contributors, we explicitly thank the executive board members of E-IMD (Matthias R. Baumgartner, Anupam Chakrapani, Kimberly Chapman, Dries Dobbelaere, Johannes Häberle, Monique Williams), E-HOD (Martina Huemer, Viktor Kozich, Andrew A. Morris, Henk Blom, Carlo Dionisi Vici, Matthias R. Baumgartner), iNTD (Angeles Garcia Cazorla, Kathrin Jeltsch, Manju Kurian, Vincenzo Leuzzi, Toni Pearson, Michel Willemsen, Roser Pons) and U-IMD (Carlo Dionisi Vici, Viktor Kozich, Maurizio Scarpa, Angeles Garcia Cazorla).
References
1. Gliklich RE, Dreyer NA, Leavy MB. AHRQ Methods for Effective Health Care. In: Gliklich RE, Dreyer NA, Leavy MB, editors. Registries for Evaluating Patient Outcomes: A User’s Guide. Rockville (MD): Agency for Healthcare Research and Quality (Us) (2014). Available at: http://www.effectivehealthcare.ahrq.gov/registries-guide-3.cfm.
2. Lanpher B, Brunetti-Pierri N, Lee B. Inborn Errors of Metabolism: The Flux From Mendelian to Complex Diseases. Nat Rev Genet (2006) 7(6):449–60. doi: 10.1038/nrg1880
3. Dimitrov B, Molema F, Williams M, Schmiesing J, Mühlhausen C, Baumgartner MR, et al. Organic Acidurias: Major Gaps, New Challenges, and a Yet Unfulfilled Promise. J Inherit Metab Dis (2021) 44(1):9–21. doi: 10.1002/jimd.12254
4. Kölker S, Burgard P, Sauer SW, Okun JG. Current Concepts in Organic Acidurias: Understanding Intra- and Extracerebral Disease Manifestation. J Inherit Metab Dis (2013) 36(4):635–44. doi: 10.1007/s10545-013-9600-8
5. Ravaglia S, Malovini A, Cirio S, Danesino C, de Filippi P, Moggio M, et al. Polymorphism in Exercise Genes and Respiratory Function in Late Onset Pompe Disease (LOPD). J Appl Physiol (Bethesda Md: 1985) (2021) 131(6):1762–71. doi: 10.1152/japplphysiol.00154.2020
6. Mecklenburg N, Kowalczyk I, Witte F, Görne J, Laier A, Mamo TM, et al. Identification of Disease-Relevant Modulators of the SHH Pathway in the Developing Brain. Development (2021) 148(17):1–17. doi: 10.1242/dev.199307
7. Bezerra GA, Foster WR, Bailey HJ, Hicks KG, Sauer SW, Dimitrov B, et al. Crystal Structure and Interaction Studies of Human DHTKD1 Provide Insight Into a Mitochondrial Megacomplex in Lysine Catabolism. IUCrJ (2020) 7(Pt 4):693–706. doi: 10.1107/S205225252000696X
8. Biagosch C, Ediga RD, Hensler SV, Faerberboeck M, Kuehn R, Wurst W, et al. Elevated Glutaric Acid Levels in Dhtkd1-/Gcdh- Double Knockout Mice Challenge Our Current Understanding of Lysine Metabolism. Biochim Biophys Acta Mol Basis Dis (2017) 1863(9):2220–8. doi: 10.1016/j.bbadis.2017.05.018
9. Leandro J, Khamrui S, Wang H, Suebsuwong C, Nemeria NS, Huynh K, et al. Inhibition and Crystal Structure of the Human DHTKD1-Thiamin Diphosphate Complex. ACS Chem Biol (2020) 15(8):2041–7. doi: 10.1021/acschembio.0c00114
10. Van Schaftingen E, Veiga-da-Cunha M, Linster CL. Enzyme Complexity in Intermediary Metabolism. J Inherit Metab Dis (2015) 38(4):721–7. doi: 10.1007/s10545-015-9821-0
11. Veiga-da-Cunha M, Van Schaftingen E, Bommer GT. Inborn Errors of Metabolite Repair. J Inherit Metab Dis (2020) 43(1):14–24. doi: 10.1002/jimd.12187
12. Kawase T, Nagasawa M, Ikeda H, Yasuo S, Koga Y, Furuse M. Gut Microbiota of Mice Putatively Modifies Amino Acid Metabolism in the Host Brain. Br J Nutr (2017) 117(6):775–83. doi: 10.1017/S0007114517000678
13. Krumsiek J, Mittelstrass K, Do KT, Stückler F, Ried J, Adamski J, et al. Gender-Specific Pathway Differences in the Human Serum Metabolome. Metabolomics (2015) 11(6):1815–33. doi: 10.1007/s11306-015-0829-0
14. Shin SY, Fauman EB, Petersen AK, Krumsiek J, Santos R, Huang J, et al. An Atlas of Genetic Influences on Human Blood Metabolites. Nat Genet (2014) 46(6):543–50. doi: 10.1038/ng.2982
15. Howard EJ, Lam TKT, Duca FA. The Gut Microbiome: Connecting Diet, Glucose Homeostasis, and Disease. Annu Rev Med (2021) 73:469–81. doi: 10.1146/annurev-med-042220-012821
16. Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal Microbiota Metabolism of L-Carnitine, a Nutrient in Red Meat, Promotes Atherosclerosis. Nat Med (2013) 19(5):576–85. doi: 10.1038/nm.3145
17. Matthijs G. Research Network: EUROGLYCANET: A European Network Focused on Congenital Disorders of Glycosylation. Eur J Hum Genet (2005) 13(4):395–7. doi: 10.1038/sj.ejhg.5201359
18. Bassanese G, Wlodkowski T, Servais A, Heidet L, Roccatello D, Emma F, et al. The European Rare Kidney Disease Registry (ERKReg): Objectives, Design and Initial Results. Orphanet J Rare Dis (2021) 16(1):251. doi: 10.1186/s13023-021-01872-8
19. Batshaw ML, Tuchman M, Summar M, Seminara J. A Longitudinal Study of Urea Cycle Disorders. Mol Genet Metab (2014) 113(1-2):127–30. doi: 10.1016/j.ymgme.2014.08.001
20. Orphanet. Rare Disease Registries in Europe (2000). Available at: https://www.orpha.net/orphacom/cahiers/docs/GB/Registries.pdfhttps://www.orpha.net/orphacom/cahiers/docs/GB/Registries.pdf (Accessed November 19, 2021).
21. European Commission. PROGRAMME OF COMMUNITY ACTION ON RARE DISEASES (1999 – 2003) (1999). Available at: https://ec.europa.eu/health/archive/ph_overview/previous_programme/rare_diseases/raredis_wpgm99_en.pdfhttps://ec.europa.eu/health/archive/ph_overview/previous_programme/rare_diseases/raredis_wpgm99_en.pdf (Accessed November 11, 2021).
22. European Commission. Commission Communication Rare Diseases: Europe’s Challenge (2008). Available at: https://ec.europa.eu/health/ph_threats/non_com/docs/rare_com_en.pdfhttps://ec.europa.eu/health/ph_threats/non_com/docs/rare_com_en.pdf (Accessed November 06, 2021).
23. European Commission. COMMISSION DECISION of 27 February 2008 on the Adoption of the Work Plan for 2008 for the Implementation of the Second Programme of Community Action in the Field of Health (2008-2013), and on the Selection, Award and Other Criteria for Financial Contributions to the Actions of This Programme (2008). Available at: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ%3AL%3A2008%3A056%3A0036%3A0062%3AEN%3APDFhttps://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ%3AL%3A2008%3A056%3A0036%3A0062%3AEN%3APDF (Accessed November 06, 2021).
24. Kölker S, Dobbelaere D, Häberle J, Burgard P, Gleich F, Summar ML, et al. Networking Across Borders for Individuals With Organic Acidurias and Urea Cycle Disorders: The E-IMD Consortium. JIMD Rep (2015) 22:29–38. doi: 10.1007/8904_2015_408
25. Opladen T, Cortès-Saladelafont E, Mastrangelo M, Horvath G, Pons R, Lopez-Laso E, et al. The International Working Group on Neurotransmitter Related Disorders (iNTD): A Worldwide Research Project Focused on Primary and Secondary Neurotransmitter Disorders. Mol Genet Metab Rep (2016) 9:61–6. doi: 10.1016/j.ymgmr.2016.09.006
26. European Commission. EU Health Programme: 2012 Abstracts of the Actions Selected for EU Co-Funding Call for Proposals (2014). Available at: https://op.europa.eu/en/publication-detail/-/publication/941eb798-ceb7-4269-bea7-c39405677ad1/language-en/format-PDF/source-240262429https://op.europa.eu/en/publication-detail/-/publication/941eb798-ceb7-4269-bea7-c39405677ad1/language-en/format-PDF/source-240262429 (Accessed November 05, 2021).
27. Aymé S, Rodwell C. The European Union Committee of Experts on Rare Diseases: Three Productive Years at the Service of the Rare Disease Community. Orphanet J Rare Dis (2014) 9:30. doi: 10.1186/1750-1172-9-30
28. European Union Committee of Experts on Rare Diseases. Eucerd Core Recommendations on Rare Disease Patient Registration and Data Collection (2013). Available at: https://rarediseases.org/wp-content/uploads/2015/12/EUCERD_Recommendations_RDRegistryDataCollection_adopted.pdfhttps://rarediseases.org/wp-content/uploads/2015/12/EUCERD_Recommendations_RDRegistryDataCollection_adopted.pdf (Accessed November 05, 2021).
29. European Commission Joint Research Centre. Set of Common Data Elements for Rare Diseases Registration (2017). Available at: https://eu-rd-platform.jrc.ec.europa.eu/sites/default/files/CDS/EU_RD_Platform_CDS_Final.pdfhttps://eu-rd-platform.jrc.ec.europa.eu/sites/default/files/CDS/EU_RD_Platform_CDS_Final.pdf (Accessed November 05, 2021).
30. Opladen T, Gleich F, Kozich V, Scarpa M, Martinelli D, Schaefer F, et al. U-IMD: The First Unified European Registry for Inherited Metabolic Diseases. Orphanet J Rare Dis (2021) 16(1):95. doi: 10.1186/s13023-021-01726-3
31. Posset R, Garbade SF, Boy N, Burlina AB, Dionisi-Vici C, Dobbelaere D, et al. Transatlantic Combined and Comparative Data Analysis of 1095 Patients With Urea Cycle Disorders-A Successful Strategy for Clinical Research of Rare Diseases. J Inherit Metab Dis (2019) 42(1):93–106. doi: 10.1002/jimd.12031
32. Dhombres F, Bodenreider O. Interoperability Between Phenotypes in Research and Healthcare Terminologies–Investigating Partial Mappings Between HPO and SNOMED Ct. J BioMed Semantics (2016) 7:3. doi: 10.1186/s13326-016-0047-3
33. Brennenstuhl H, Kohlmüller D, Gramer G, Garbade SF, Syrbe S, Feyh P, et al. High Throughput Newborn Screening for Aromatic ʟ-Amino-Acid Decarboxylase Deficiency by Analysis of Concentrations of 3-O-Methyldopa From Dried Blood Spots. J Inherit Metab Dis (2020) 43(3):602–10. doi: 10.1002/jimd.12208
34. Kölker S, Garcia-Cazorla A, Valayannopoulos V, Lund AM, Burlina AB, Sykut-Cegielska J, et al. The Phenotypic Spectrum of Organic Acidurias and Urea Cycle Disorders. Part 1: The Initial Presentation. J Inherit Metab Dis (2015) 38(6):1041–57. doi: 10.1007/s10545-015-9839-3
35. Rüegger CM, Lindner M, Ballhausen D, Baumgartner MR, Beblo S, Das A, et al. Cross-Sectional Observational Study of 208 Patients With Non-Classical Urea Cycle Disorders. J Inherit Metab Dis (2014) 37(1):21–30. doi: 10.1007/s10545-013-9624-0
36. Staufner C, Peters B, Wagner M, Alameer S, Barić I, Broué P, et al. Defining Clinical Subgroups and Genotype-Phenotype Correlations in NBAS-Associated Disease Across 110 Patients. Genet Med (2020) 22(3):610–21. doi: 10.1038/s41436-019-0698-4
37. Kuseyri Hübschmann O, Horvath G, Cortès-Saladelafont E, Yıldız Y, Mastrangelo M, Pons R, et al. Insights Into the Expanding Phenotypic Spectrum of Inherited Disorders of Biogenic Amines. Nat Commun (2021) 12(1):5529. doi: 10.1038/s41467-021-25515-5
38. Boy N, Mengler K, Thimm E, Schiergens KA, Marquardt T, Weinhold N, et al. Newborn Screening: A Disease-Changing Intervention for Glutaric Aciduria Type 1. Ann Neurol (2018) 83(5):970–9. doi: 10.1002/ana.25233
39. Hörster F, Baumgartner MR, Viardot C, Suormala T, Burgard P, Fowler B, et al. Long-Term Outcome in Methylmalonic Acidurias Is Influenced by the Underlying Defect (Mut0, Mut-, Cbla, Cblb). Pediatr Res (2007) 62(2):225–30. doi: 10.1203/PDR.0b013e3180a0325f
40. Hörster F, Tuncel AT, Gleich F, Plessl T, Froese SD, Garbade SF, et al. Delineating the Clinical Spectrum of Isolated Methylmalonic Acidurias: cblA and Mut. J Inherit Metab Dis (2021) 44(1):193–214. doi: 10.1002/jimd.12297
41. Kožich V, Sokolová J, Morris AAM, Pavlíková M, Gleich F, Kölker S, et al. Cystathionine β-Synthase Deficiency in the E-HOD Registry-Part I: Pyridoxine Responsiveness as a Determinant of Biochemical and Clinical Phenotype at Diagnosis. J Inherit Metab Dis (2021) 44(3):677–92. doi: 10.1002/jimd.12338
42. Mütze U, Henze L, Gleich F, Lindner M, Grünert SC, Spiekerkoetter U, et al. Newborn Screening and Disease Variants Predict Neurological Outcome in Isovaleric Aciduria. J Inherit Metab Dis (2021) 44(4):857–70. doi: 10.1002/jimd.12364
43. Enns GM, Berry SA, Berry GT, Rhead WJ, Brusilow SW, Hamosh A. Survival After Treatment With Phenylacetate and Benzoate for Urea-Cycle Disorders. N Engl J Med (2007) 356(22):2282–92. doi: 10.1056/NEJMoa066596
44. Posset R, Garcia-Cazorla A, Valayannopoulos V, Teles EL, Dionisi-Vici C, Brassier A, et al. Age at Disease Onset and Peak Ammonium Level Rather Than Interventional Variables Predict the Neurological Outcome in Urea Cycle Disorders. J Inherit Metab Dis (2016) 39(5):661–72. doi: 10.1007/s10545-016-9938-9
45. Buerger C, Garbade SF, Dietrich Alber F, Waisbren SE, McCarter R, Kölker S, et al. Impairment of Cognitive Function in Ornithine Transcarbamylase Deficiency Is Global Rather Than Domain-Specific and Is Associated With Disease Onset, Sex, Maximum Ammonium, and Number of Hyperammonemic Events. J Inherit Metab Dis (2019) 42(2):243–53. doi: 10.1002/jimd.12013
46. Posset R, Gropman AL, Nagamani SCS, Burrage LC, Bedoyan JK, Wong D, et al. Impact of Diagnosis and Therapy on Cognitive Function in Urea Cycle Disorders. Ann Neurol (2019) 86(1):116–28. doi: 10.1002/ana.25492
47. Guthrie R, Susi A. A Simple Phenylalanine Method for Detecting Phenylketonuria in Large Populations of Newborn Infants. Pediatrics (1963) 32:338–43. doi: 10.1542/peds.32.3.338
48. Therrell BL, Padilla CD, Loeber JG, Kneisser I, Saadallah A, Borrajo GJ, et al. Current Status of Newborn Screening Worldwide: 2015. Semin Perinatol (2015) 39(3):171–87. doi: 10.1053/j.semperi.2015.03.002
49. Loeber JG, Platis D, Zetterström RH, Almashanu S, Boemer F, Bonham JR, et al. Neonatal Screening in Europe Revisited: An ISNS Perspective on the Current State and Developments Since 2010. Int J Neonatal Screen (2021) 7(1):1–21. doi: 10.3390/ijns7010015
50. Heringer J, Valayannopoulos V, Lund AM, Wijburg FA, Freisinger P, Barić I, et al. Impact of Age at Onset and Newborn Screening on Outcome in Organic Acidurias. J Inherit Metab Dis (2016) 39(3):341–53. doi: 10.1007/s10545-015-9907-8
51. Mütze U, Garbade SF, Gramer G, Lindner M, Freisinger P, Grunert SC, et al. Long-Term Outcomes of Individuals With Metabolic Diseases Identified Through Newborn Screening. Pediatrics (2020) 146(5):1–11. doi: 10.1542/peds.2020-0444
52. Boy N, Mengler K, Heringer-Seifert J, Hoffmann GF, Garbade SF, Kölker S. Impact of Newborn Screening and Quality of Therapy on the Neurological Outcome in Glutaric Aciduria Type 1: A Meta-Analysis. Genet Med (2021) 23(1):13–21. doi: 10.1038/s41436-020-00971-4
53. Mütze U, Walter M, Keller M, Gramer G, Garbade SF, Gleich F, et al. Health Outcomes of Infants With Vitamin B(12) Deficiency Identified by Newborn Screening and Early Treated. J Pediatr (2021) 235:42–8. doi: 10.1016/j.jpeds.2021.02.009
54. Bjugstad KB, Goodman SI, Freed CR. Age at Symptom Onset Predicts Severity of Motor Impairment and Clinical Outcome of Glutaric Acidemia Type 1. J Pediatr (2000) 137(5):681–6. doi: 10.1067/mpd.2000.108954
55. Boy N, Mühlhausen C, Maier EM, Heringer J, Assmann B, Burgard P, et al. Proposed Recommendations for Diagnosing and Managing Individuals With Glutaric Aciduria Type I: Second Revision. J Inherit Metab Dis (2017) 40(1):75–101. doi: 10.1007/s10545-016-9999-9
56. Märtner EMC, Maier EM, Mengler K, Thimm E, Schiergens KA, Marquardt T, et al. Impact of Interventional and Non-Interventional Variables on Anthropometric Long-Term Development in Glutaric Aciduria Type 1: A National Prospective Multi-Centre Study. J Inherit Metab Dis (2021) 44(3):629–38. doi: 10.1002/jimd.12335
57. Strauss KA, Williams KB, Carson VJ, Poskitt L, Bowser LE, Young M, et al. Glutaric Acidemia Type 1: Treatment and Outcome of 168 Patients Over Three Decades. Mol Genet Metab (2020) 131(3):325–40. doi: 10.1016/j.ymgme.2020.09.007
58. Molema F, Gleich F, Burgard P, van der Ploeg AT, Summar ML, Chapman KA, et al. Evaluation of Dietary Treatment and Amino Acid Supplementation in Organic Acidurias and Urea-Cycle Disorders: On the Basis of Information From a European Multicenter Registry. J Inherit Metab Dis (2019) 42(6):1162–75. doi: 10.1002/jimd.12066
59. Kölker S, Christensen E, Leonard JV, Greenberg CR, Burlina AB, Burlina AP, et al. Guideline for the Diagnosis and Management of Glutaryl-CoA Dehydrogenase Deficiency (Glutaric Aciduria Type I). J Inherit Metab Dis (2007) 30(1):5–22. doi: 10.1007/s10545-006-0451-4
60. Kölker S, Christensen E, Leonard JV, Greenberg CR, Boneh A, Burlina AB, et al. Diagnosis and Management of Glutaric Aciduria Type I–revised Recommendations. J Inherit Metab Dis (2011) 34(3):677–94. doi: 10.1007/s10545-011-9289-5
61. Boy N, Heringer J, Brackmann R, Bodamer O, Seitz A, Kölker S, et al. Extrastriatal Changes in Patients With Late-Onset Glutaric Aciduria Type I Highlight the Risk of Long-Term Neurotoxicity. Orphanet J Rare Dis (2017) 12(1):77. doi: 10.1186/s13023-017-0612-6
62. Kölker S, Garbade SF, Greenberg CR, Leonard JV, Saudubray JM, Ribes A, et al. Natural History, Outcome, and Treatment Efficacy in Children and Adults With Glutaryl-CoA Dehydrogenase Deficiency. Pediatr Res (2006) 59(6):840–7. doi: 10.1203/01.pdr.0000219387.79887.86
63. Heringer J, Boy SP, Ensenauer R, Assmann B, Zschocke J, Harting I, et al. Use of Guidelines Improves the Neurological Outcome in Glutaric Aciduria Type I. Ann Neurol (2010) 68(5):743–52. doi: 10.1002/ana.22095
64. Boy N, Mohr A, Garbade SF, Freisinger P, Heringer-Seifert J, Seitz A, et al. Subdural Hematoma in Glutaric Aciduria Type 1: High Excreters Are Prone to Incidental SDH Despite Newborn Screening. J Inherit Metab Dis (2021) 44(6):1343–52. doi: 10.1002/jimd.12436
65. Pfeil J, Listl S, Hoffmann GF, Kölker S, Lindner M, Burgard P. Newborn Screening by Tandem Mass Spectrometry for Glutaric Aciduria Type 1: A Cost-Effectiveness Analysis. Orphanet J Rare Dis (2013) 8:167. doi: 10.1186/1750-1172-8-167
66. Märtner EMC, Thimm E, Guder P, Schiergens KA, Rutsch F, Roloff S, et al. The Biochemical Subtype Is a Predictor for Cognitive Function in Glutaric Aciduria Type 1: A National Prospective Follow-Up Study. Sci Rep (2021) 11(1):19300. doi: 10.1038/s41598-021-98809-9
67. Strauss KA, Puffenberger EG, Robinson DL, Morton DH. Type I Glutaric Aciduria, Part 1: Natural History of 77 Patients. Am J Med Genet C Semin Med Genet (2003) 121c(1):38–52. doi: 10.1002/ajmg.c.20007
68. Strauss KA, Brumbaugh J, Duffy A, Wardley B, Robinson D, Hendrickson C, et al. Safety, Efficacy and Physiological Actions of a Lysine-Free, Arginine-Rich Formula to Treat Glutaryl-CoA Dehydrogenase Deficiency: Focus on Cerebral Amino Acid Influx. Mol Genet Metab (2011) 104(1-2):93–106. doi: 10.1016/j.ymgme.2011.07.003
69. Höliner I, Simma B, Reiter A, Sass JO, Zschocke J, Huemer M. Compliance to Clinical Guidelines Determines Outcome in Glutaric Aciduria Type I in the Era of Newborn Screening. Klin Padiatr (2010) 222(1):35–7. doi: 10.1055/s-0029-1239525
70. Häberle J, Burlina A, Chakrapani A, Dixon M, Karall D, Lindner M, et al. Suggested Guidelines for the Diagnosis and Management of Urea Cycle Disorders: First Revision. J Inherit Metab Dis (2019) 42(6):1192–230. doi: 10.1002/jimd.12100
71. Häberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, et al. Suggested Guidelines for the Diagnosis and Management of Urea Cycle Disorders. Orphanet J Rare Dis (2012) 7:32. doi: 10.1186/1750-1172-7-32
72. Baumgartner MR, Hörster F, Dionisi-Vici C, Haliloglu G, Karall D, Chapman KA, et al. Proposed Guidelines for the Diagnosis and Management of Methylmalonic and Propionic Acidemia. Orphanet J Rare Dis (2014) 9:130. doi: 10.1186/s13023-014-0130-8
73. Forny P, Hörster F, Ballhausen D, Chakrapani A, Chapman KA, Dionisi-Vici C, et al. Guidelines for the Diagnosis and Management of Methylmalonic Acidaemia and Propionic Acidaemia: First Revision. J Inherit Metab Dis (2021) 44(3):566–92. doi: 10.1002/jimd.12370
74. Huemer M, Diodato D, Schwahn B, Schiff M, Bandeira A, Benoist JF, et al. Guidelines for Diagnosis and Management of the Cobalamin-Related Remethylation Disorders Cblc, Cbld, Cble, Cblf, Cblg, cblJ and MTHFR Deficiency. J Inherit Metab Dis (2017) 40(1):21–48. doi: 10.1007/s10545-016-9991-4
75. Huemer M, Diodato D, Martinelli D, Olivieri G, Blom H, Gleich F, et al. Phenotype, Treatment Practice and Outcome in the Cobalamin-Dependent Remethylation Disorders and MTHFR Deficiency: Data From the E-HOD Registry. J Inherit Metab Dis (2019) 42(2):333–52. doi: 10.1002/jimd.12041
76. Keller R, Chrastina P, Pavlíková M, Gouveia S, Ribes A, Kölker S, et al. Newborn Screening for Homocystinurias: Recent Recommendations Versus Current Practice. J Inherit Metab Dis (2019) 42(1):128–39. doi: 10.1002/jimd.12034
77. Morris AA, Kožich V, Santra S, Andria G, Ben-Omran TI, Chakrapani AB, et al. Guidelines for the Diagnosis and Management of Cystathionine Beta-Synthase Deficiency. J Inherit Metab Dis (2017) 40(1):49–74. doi: 10.1007/s10545-016-9979-0
78. Opladen T, López-Laso E, Cortès-Saladelafont E, Pearson TS, Sivri HS, Yildiz Y, et al. Consensus Guideline for the Diagnosis and Treatment of Tetrahydrobiopterin (BH(4)) Deficiencies. Orphanet J Rare Dis (2020) 15(1):126. doi: 10.1186/s13023-020-01379-8
79. Wassenberg T, Molero-Luis M, Jeltsch K, Hoffmann GF, Assmann B, Blau N, et al. Consensus Guideline for the Diagnosis and Treatment of Aromatic L-Amino Acid Decarboxylase (AADC) Deficiency. Orphanet J Rare Dis (2017) 12(1):12. doi: 10.1186/s13023-016-0522-z
80. Wilkinson MD, Dumontier M, Aalbersberg IJ, Appleton G, Axton M, Baak A, et al. The FAIR Guiding Principles for Scientific Data Management and Stewardship. Sci Data (2016) 3:160018. doi: 10.1038/sdata.2016.18
81. European Medicines Agency. Patient Registry Initiative- Strategy and Mandate of the Cross-Committee Task Force (2017). Available at: https://www.ema.europa.eu/en/documents/other/patient-registry-initiative-strategy-mandate-cross-committee-task-force_en.pdfhttps://www.ema.europa.eu/en/documents/other/patient-registry-initiative-strategy-mandate-cross-committee-task-force_en.pdf (Accessed November 05, 2021).
82. European Medicines Agency. Initiative for Patient Registries Strategy and Pilot Phase (2015). Available at: https://www.ema.europa.eu/en/documents/other/initiative-patient-registries-strategy-pilot-phase_en.pdfhttps://www.ema.europa.eu/en/documents/other/initiative-patient-registries-strategy-pilot-phase_en.pdf (Accessed November 05, 2021).
83. Derks TGJ, Rodriguez-Buritica DF, Ahmad A, de Boer F, Couce ML, Grünert SC, et al. Glycogen Storage Disease Type Ia: Current Management Options, Burden and Unmet Needs. Nutrients (2021) 13(11):3828. doi: 10.3390/nu13113828
Keywords: rare disease (RD), inherited metabolic diseases, FAIR (findable, accessible, interoperable, and reusable) principles, personalized medicine, patient registry
Citation: Kölker S, Gleich F, Mütze U and Opladen T (2022) Rare Disease Registries Are Key to Evidence-Based Personalized Medicine: Highlighting the European Experience. Front. Endocrinol. 13:832063. doi: 10.3389/fendo.2022.832063
Received: 09 December 2021; Accepted: 31 January 2022;
Published: 04 March 2022.
Edited by:
Klaus Mohnike, University Hospital Magdeburg, GermanyReviewed by:
Maximilian Zeyda, Medical University of Vienna, AustriaChristiaan F. Mooij, Amsterdam University Medical Center, Netherlands
Alessandro Rossi, University of Naples Federico II, Italy
Copyright © 2022 Kölker, Gleich, Mütze and Opladen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan Kölker, U3RlZmFuLktvZWxrZXJAbWVkLnVuaS1oZWlkZWxiZXJnLmRl