Abstract
Nuclear receptor subfamily 0 group B member 1 gene (NR0B1) encodes an orphan nuclear receptor that plays a critical role in the development and regulation of the adrenal gland and hypothalamic–pituitary–gonadal axis. In this study, we report a novel mutation in NR0B1 that led to adult-onset adrenal hypoplasia congenita (AHC) and pubertal development failure in a male adult. Clinical examinations revealed hyponatremia, elevated adrenocorticotropic hormone levels, reduced testosterone and gonadotropin levels, and hyper-responses to gonadotropin-releasing hormone and human chorionic gonadotropin stimulation tests. Whole-exome sequencing and Sanger sequencing were performed to identify the potential causes of AHC. Candidate variants were shortlisted based on the X-linked recessive models. Sequence analyses identified a novel hemizygous variant of c.1034delC in exon 1 of NR0B1 at Xp21.2, resulting in a frameshift mutation and premature stop codon formation. The c.1034delC/p.Pro345Argfs*27 in the NR0B1 gene was detected in the hemizygous state in affected males and in the heterozygous state in healthy female family carriers. These results expand the clinical features of AHC as well as the mutation profile of the causative gene NR0B1. Further studies are needed to elucidate the biological effects of the mutation on the development and function of the adrenal gland and the hypothalamic–pituitary–gonadal axis.
Introduction
The nuclear receptor subfamily 0 group B member 1 (NR0B1) gene encodes DAX1, an orphan nuclear receptor that plays a critical role in the development and differentiation of the adrenal gland and hypothalamus-pituitary-gonadal axis (1). It is predominantly expressed in the adrenal cortex, hypothalamus, pituitary gland, and gonads (testis and ovary). DAX1 has been proposed to serve as a transcription factor involved in the development of the adrenal cortex and pituitary gonadotropes (2). More than 200 mutations have been reported in NR0B1, most of which are nonsense or frameshift mutations that result in premature truncation of the protein (3). Defective NR0B1 leads to X-linked adrenal hypoplasia congenita (AHC), resulting in a failure to develop the permanent adult adrenal cortex (4). Patients with NR0B1 mutations may present with severe salt wasting in infancy or have a more insidious onset during childhood. AHC rarely presents with adrenal failure later in adulthood, and late-onset primary adrenal insufficiency (AI) is often accompanied by hypogonadotropic hypogonadism (HH) and azoospermia (5–7). In this study, we report a novel mutation in NR0B1 that leads to late-onset AHC and failure of pubertal development.
Materials and Methods
Subjects
The proband and all available family members were recruited from Wuhan Union Hospital (Figure 1). Medication history, disease symptoms, and progression of each family member was recorded by dictation as well as from medical records. Venous blood samples were collected from each family member after obtaining written informed consent. The present study was approved by the ethics committee of Wuhan Union Hospital.
Figure 1
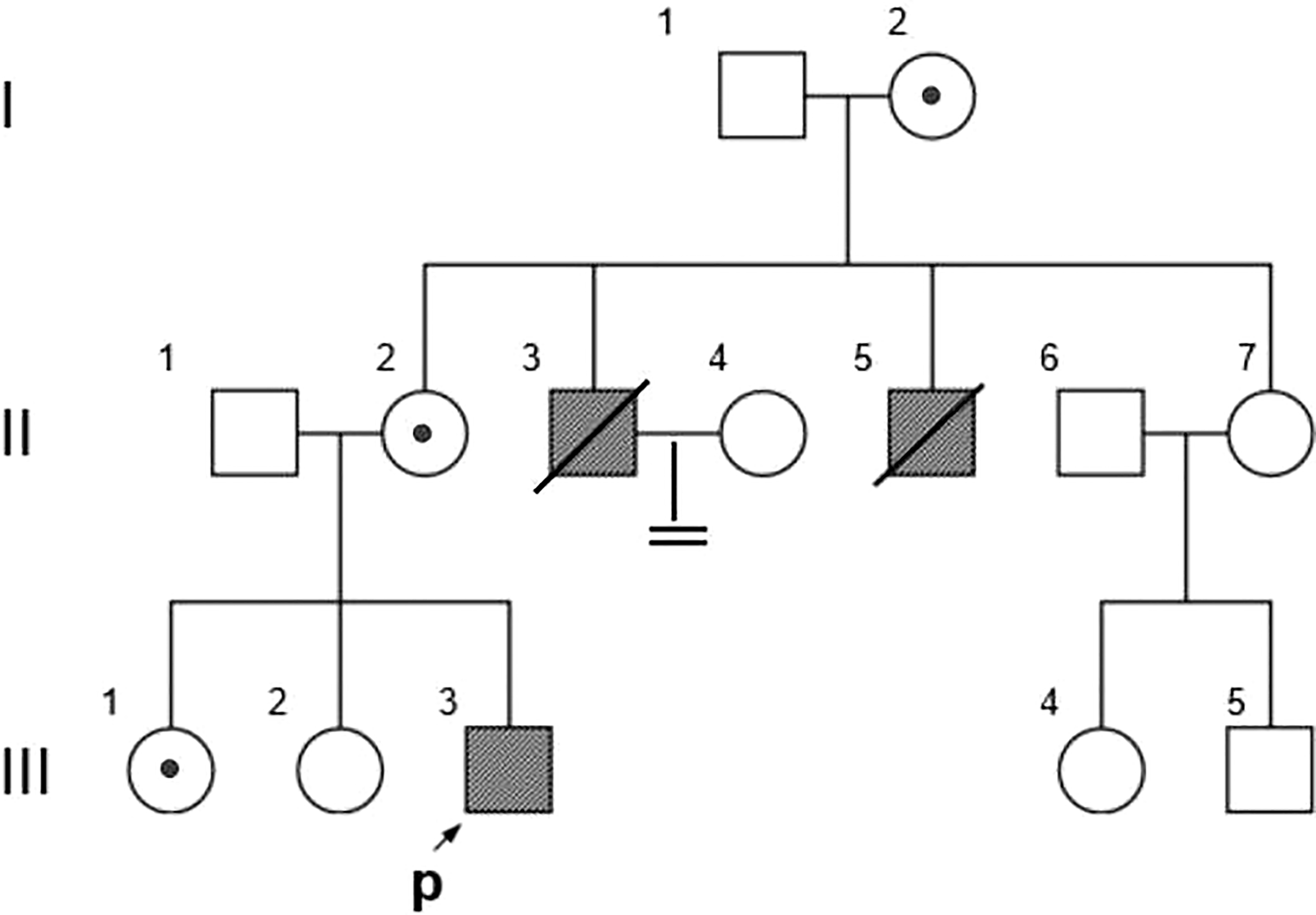
Family Pedigree. Symbols represent males (squares), females (circles), affected subjects (solid symbols), carriers (dotted symbols), and the proband (arrow). The propositus is indicated by an arrow.
DNA Sequencing
A library of genomic DNA was created from the proband’s blood sample and enriched for exon targets according to the protocols of the Agilent SureSelect Human All Exon V6 Kit. After validation for size distribution using the Agilent 2100 Bioanalyzer, the enriched library was sequenced on the Illumina HiSeq X Ten platform. Raw files were generated and mapped to the human reference genome (GRCh37/hg19) using the Burrows-Wheeler alignment tool (version 0.7.8-r455). SAMtools was then applied to call single-nucleotide variants (SNVs) and indels (deletions and insertions, < 50 bp). Subsequently, ANNOVAR, accompanied by several prediction tools, was used to annotate SNVs and indels. Variants were annotated using genomic coordinates, referent nucleotides, variant nucleotides, mutation types, alleles, allele frequencies, gene names, amino acids variants and their evolutionary conservation. For allele frequency, each variant was compared against public population genetic databases. Based on the variant annotations, we focused on nonsynonymous coding substitutions, frameshifts, and splicing site mutations on chromosome X. Post these filtering steps, a list of candidate variants and related genes was created. To prioritize the most likely candidate disease-causing genes, all candidate genes were ranked using Phenolyzer (8). To validate the suspected variant in the proband, PCR and Sanger sequencing were performed on DNA samples of all available family members (II-2, II-3, II-6, III-1, III-2, III-3, III-4, and III-5) (Figure 1). The primer pairs used for PCR amplification of the region encompassing the suspected variant were forward 5′-GCTTTTAAAGAGCACCCGCC-3′ and reverse 5′-TTTCTTCACCTTTGCCCCGAC-3′.
Case Presentation
Clinical Presentations
The pedigree in Figure 1 shows the inheritance pattern of the X-linked recessive disorder. The proband (III-3), a 26-year-old male patient (height: 5 feet; weight: 75 kg; BMI 25.95), was admitted to the hospital with suspected adrenocortical failure. Medical history revealed that the patient had not exhibited secondary sexual characteristics by 17 years of age and had initial symptoms of skin hyperpigmentation; he had initiated prednisone oral tablet 5 mg per day along with some unknown Chinese herbs to help grow pubic hair. Up until the age of 24 years, there had been no incidence of fatigue, weight loss, dehydration, anorexia, nausea, or vomiting; however, in the following 2 years, he had developed worsening skin hyperpigmentation and experienced nausea and weakness following occasional treatment interruptions. Physical examination revealed generalized hyperpigmentation (Figures 2A, B) and delayed puberty, with the absence of secondary sexual characteristics (Figures 2C–F).
Figure 2
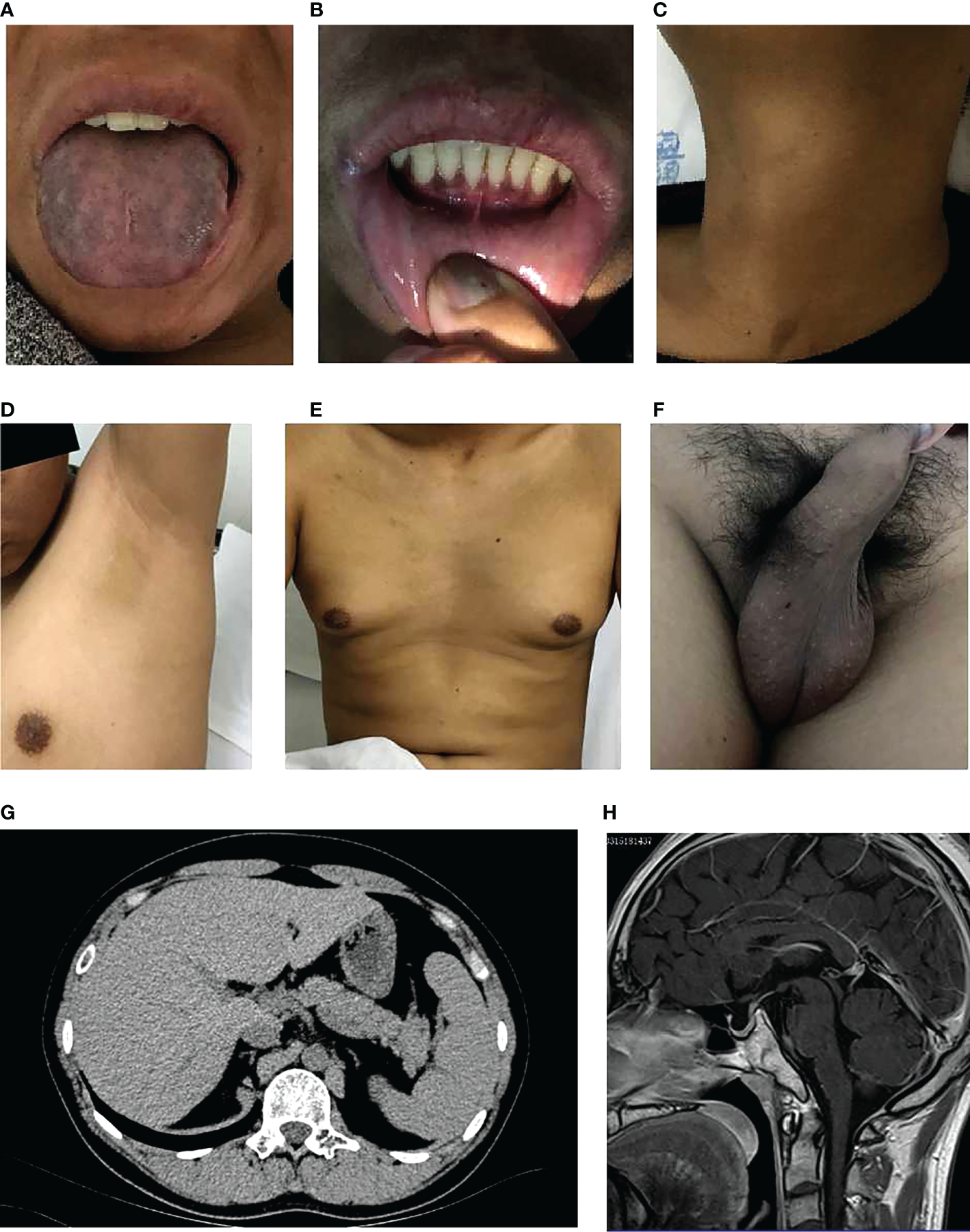
Clinical characteristics of the proband. (A) Significant melanosis could be seen in the wrinkles of the face, gums, tongue, and limbs. (B) A few hyperpigmented macules were noted on the lips and oral mucosa. (C-F) Inconspicuous male secondary sexual characteristics: no beard, axillary hair, no obvious laryngeal knot, inverted triangle distributed pubic hair, significantly reduced bilateral testicles. (G, H). Scanning results (adrenal hypoplasia, hypogonadotropic hypogonadism, small bilateral testes, olfactory sulci and pituitary, mild fatty liver).
Laboratory examinations revealed a serum sodium level of 123.6 mmol/L at admission, which confirmed hyponatremia; however, it elevated to 136.6 mmol/L following hydrocortisone treatment. The serum potassium levels were normal. The serum triglyceride level was extraordinarily high (29.57 mmol/L) at admission but dropped to 3.91 mmol/L following 5 days of lipid-lowering treatment. The serum cholesterol level was high (7.18 mmol/L). The plasma cortisol level (11.017 μg/L) was low. The adrenocorticotropic hormone level was extremely high, above the upper limit of detection (> 2000 pg/ml), which was consistent with primary AI. The VLCFA (very long-chain fatty acids) and 17-Hydroxyprogesterone concentrations were normal (Table 1). Initial hormone examinations showed normal levels of luteinizing hormone (2.49 IU/L) and follicle-stimulating hormone (3.08 IU/L) but a low level of testosterone (2.14 nmol/L). Thereafter, both gonadotropin-releasing hormone (GnRH) and human chorionic gonadotropin (hCG) stimulation tests were performed to diagnose the cause of hypotestosteronemia. As depicted in Table 2, in response to GnRH, testosterone levels increased from 1.96 IU/I to 10.27 IU/I at 90 s, but then decreased to 9.67 IU/I at 120 s. In response to hCG, testosterone levels increased by more than nine-fold, from 2.14 nmol/L to 18.33 nmol/L, after 48 h of stimulation, indicating that the testicular tissue was capable of achieving normal secretory function upon stimulation.
Table 1
| Measures | At admission | After treatment | Normal value |
|---|---|---|---|
| Na/K | 123.6/4.3 | 136.6/4.5 | [135–150]/[3.5–5.5] Eq/L |
| ACTH | >2000 | NA | [9–40] pg/mL |
| Cortisol | 11.017 | NA | [37–194] μg/L |
| Triglyceride | 29.57 | 5.59 | [<1.7] mmol/L |
| Cholesterol | 7.18 | 5.28 | [2.9-6.0] mmol/L |
| 17-OHG | 2.5 | NA | [0.7–3.6ng/mL] ng/mL |
| VLCFA | 0.30 | NA | [<0.50] μg/ml |
| LH | 2.49 | NA | [1.7–8.6] IU/L |
| FSH | 3.08 | NA | [1.5-12.4] IU/L |
| Testosterone | 2.14 | NA | [8.6-29.0] nmol/L |
| Glucose | 6.0 | NA | [3.9-6.1] mmol/L |
Basal biochemical and hormone measurements.
NA, not available.
Table 2
| GnRH | |
|---|---|
| Time | LH(IU/L) |
| 0’ | 1.96 |
| 30’ | 6.85 |
| 60’ | 9.95 |
| 90’ | 10.27 |
| 120’ | 9.67 |
| hCG | |
| Time | Testosterone(nmol/L) |
| 0 hr | 2.14 |
| 48 hr | 18.33 |
| 72 hr | 19.55 |
GnRH stimulation test and hCG stimulation test.
The scanning results provided additional evidence of adrenal hypoplasia and HH. Chromosomal analysis confirmed a normal male 46, XY karyotype. Bilateral testicular volume reduction was detected using B-ultrasound (left: 25.7*19.4*12.7 mm, right: 27.4*18.2*12.8 mm). Abdominal computed tomography revealed bilateral adrenal gland hypoplasia (Figure 2G). Magnetic resonance imaging of the olfactory sulci and pituitary gland was normal (Figure 2H). In addition, ultrasonography of the upper abdomen showed a mildly fatty liver.
Steroid supplementation with hydrocortisone (15-35 mg/d) and fludrocortisone (100 μg/d) resulted in a rapid improvement in the clinical conditions of the proband; the laboratory results following treatment are listed in Table 1. For personal reasons, in addition to androgen replacement, patient refused other treatments such as spermatogenesis therapy. During the follow-up, the patient refused to discuss his condition. His sibling mentioned that the proband’s uncle died in a car accident, and the proband himself was bitter regarding not being able to have children, causing his depression and gloominess. He had abandoned his family plans.
Mutation Detection
Based on the reads aligned to the human reference genome (GRCh37/hg19), a total of 65,535 variants, including 58,704 SNVs and 6,831 indels, were identified. There were 965 variants located on chromosome X. Among them, 167 were nonsynonymous coding substitutions, and eight were frameshift variants. Prioritization with phenotype “adrenal hypoplasia” in Phenolyzer revealed a novel hemizygous frameshift deletion c.1034delC in the first exon of NR0B1 at Xp21.2. A cytosine deletion at c.1034 caused a frameshift change, with proline-345 being the first affected amino acid by becoming arginine and forming a new reading frame resulting in a premature stop codon 27 amino acids further down (Figure 3). As a result, the putative ligand-binding domain (LBD) of the DAX protein was 20 amino acids shorter than the wild type. To validate the suspected variant in the proband, PCR and Sanger sequencing were performed on DNA samples of all available family members (II-2, II-3, II-6, III-1, III-2, III-3, III-4, and III-5), which revealed a hemizygous state in affected males (II-3 and III-3) and a heterozygous state in healthy female carriers (II-2 and III-1) (Figure 1).
Figure 3
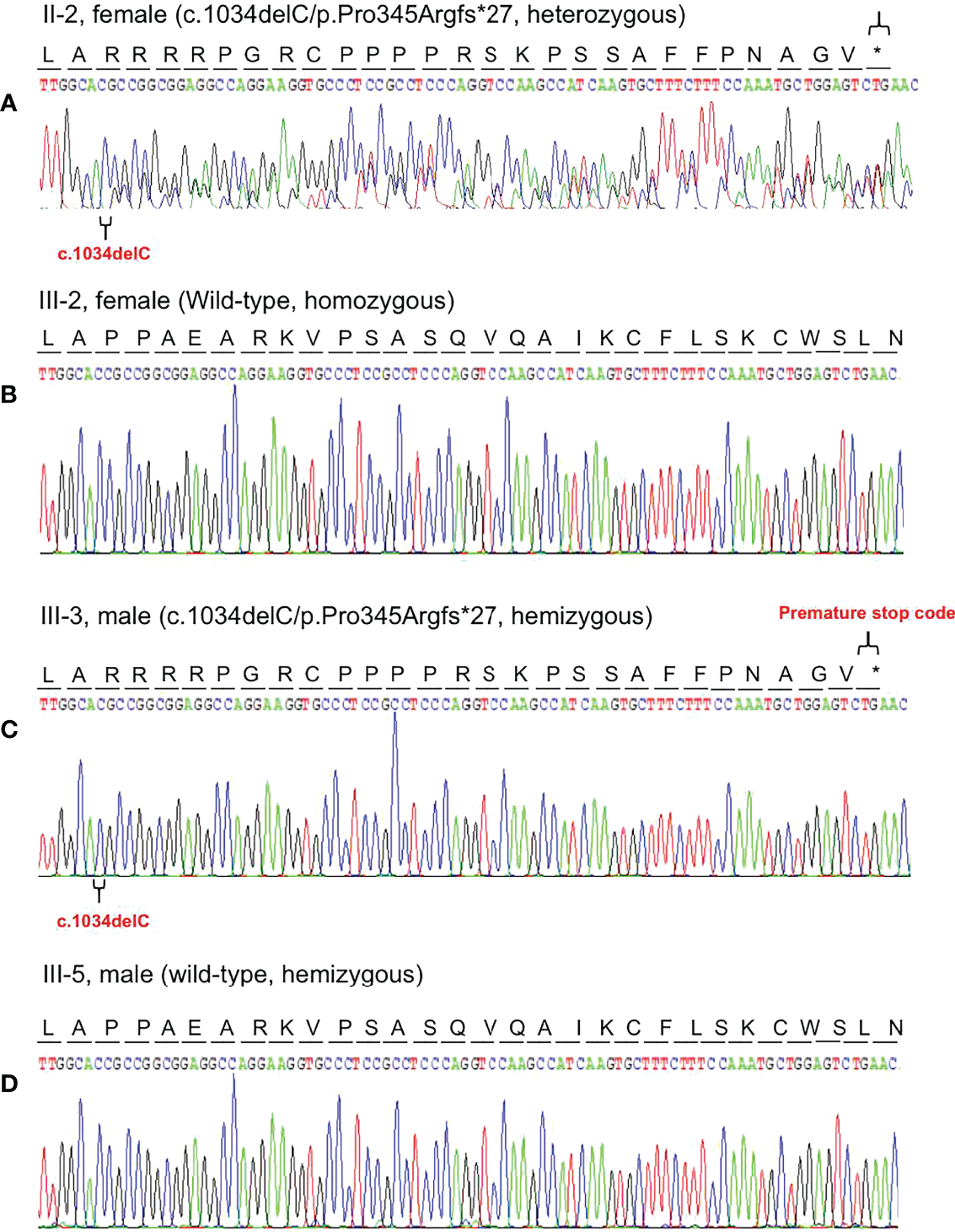
Representative chromatogram of the NR0B1 gene. The position of the mutation is indicated by special symbols (“ ” for the mutation, and “*” for Premature stop code). (A) The proband’s mother (II-2) inherited the c.1034delC variant in heterozygous status. (B) His health sister (III-2) presented with homozygous wild-type of the NR0B1 gene. (C) The proband was hemizygous for the c.1034delC variant in the NROB1 gene. (D) His maternal cousin (III-5) showed hemizygosity for wild-type of the NROB1gene.
” for the mutation, and “*” for Premature stop code). (A) The proband’s mother (II-2) inherited the c.1034delC variant in heterozygous status. (B) His health sister (III-2) presented with homozygous wild-type of the NR0B1 gene. (C) The proband was hemizygous for the c.1034delC variant in the NROB1 gene. (D) His maternal cousin (III-5) showed hemizygosity for wild-type of the NROB1gene.
Discussion
X-linked AHC is an X-linked recessive inherited disease that typically affects males and presents as primary AI in early infancy or childhood, and HH at puberty with impaired spermatogenesis. Approximately 60% of male patients with AHC develop AI within 2 months after birth, whereas 40% do so between the age of 1 to 9 years. Recently, various cases of late-onset forms of X-linked AHC have been reported. In this study, we report a case of late-onset X-linked AHC and HH caused by a novel variant of NROB1. The proband presented with AI at 17 years of age and a lack of development of secondary sexual characteristics during puberty. Sequencing of NR0B1 revealed a c.1034delC frameshift variant in two affected males in the kindred. The diagnosis of X-linked AHC and HH was established based on the clinical presentation, laboratory tests, and molecular analysis. According to the family history, two other male members of the family had presented with similar manifestations; one (II-3) was diagnosed with Addison’s disease (AD) and male infertility at the age of 30 years and had been treated with prednisone, whereas the other (II-5), who had also been diagnosed with AD at the age of 33 years, succumbed to adrenal crisis at the age of 35 years. This indicates an X-linked mode of inheritance, as was confirmed by the results of the genetic analysis presented herein. In addition, no evident secondary causes of AD were identified, thus allowing us to exclude its presence.
NR0B1 consists of two exons and spans a 4.96 kb region at Xp21.2. It encodes a 470-amino-acid member of the nuclear hormone receptor superfamily. Most of the coding sequence is found in exon one (9, 10), which encodes the N-terminal domain and part of the C-terminal domain of the protein NR0B1. Exon two encodes the remaining part of the C-terminal domain. According to the statistical data of the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php, accessed on 20180630), a total of 252 mutations have been found in NR0B1. Truncating mutations such as nonsense or frameshift mutations have been identified throughout the gene. In contrast, missense changes have tended to cluster in the carboxyl terminus of the protein, corresponding to the putative LBD. Several cases of changes in the domain such as p.Ser259Pro, p.Pro279Leu, p.Ile439Ser, p.Tyr380Asp, and p.Gln305Hisfs*67 have been associated with a milder or late-onset phenotype (11). In functional studies of Tyr380Asp and Ile439Ser mutants, the partial loss of NR0B1 transcriptional repression was consistent with the late-onset phenotype (12). The p.Pro345Argfs*27 mutation is a novel frameshift mutation within the LBD that introduces premature termination of the NR0B1 protein, thus resulting in a truncated one. All the affected males in the family exhibited a late-onset phenotype. These findings suggest that the truncated NR0B1 protein produced by this mutation may retain part of its function. Further studies are needed to elucidate the function of p.Pro345Argfs*27.
The same variant was identified in another male family member (II-5), who had presented with a dysfunction of the adrenal cortex and had deceased owing to adrenal crisis. Although the dysfunction of his gonadal axis had occurred at an older age compared to that of the proband, it was more severe. His impairment involved the hypothalamus and pituitary, whereas the impairment of the proband primarily involved the hypothalamus. This suggests that, despite possessing the same genetic variant, there are differences in clinical manifestations, even in patients of the same pedigree. Liu also reported a nonsense variant change in exon 1 (c.192C>G) in two male siblings presenting with precocious puberty and late-onset HH but exhibiting different clinical manifestations (13).
There was no evident correlation between the NR0B1 variant type and the clinical presentation. Literature search revealed 11 reports of adult-onset AHC (6, 7, 11, 14–20). The reported cases of adult-onset AI and hypogonadotrophic hypogonadism associated with mutations of NR0B1 gene are summarized in Supplementary Table S1. Of the 11 reports, three (p.Gln37X in one, p.Trp39X in two kindreds) are amino-terminal nonsense mutations, whereas eight (p.Tyr378Cys, p.Leu386Phe, p.Ser259Pro in two kindreds, p.Pro279Leu, p.Gln305Hisfs*67 (c.915delG), p.Tyr380Asp, p.Ile439Ser) are mutations distributed throughout the carboxyl LBD. The mutation described in this study also falls in the “hotspot.” Most patients presented with AI as the initial symptom followed by HH (or not); however, two cases presented with HH first, and further endocrine investigations confirmed AI. Among these variants, p.W39X was identified in two kindreds; the proband reported by M. Guclu et al. (17) presented with delayed puberty and small size of testes as the initial symptoms, whereas the other proband, reported by M. L. Raffin-Sanson et al., presented with fatigue, sore throat, and dizziness, which are consistent with AI (6). Moreover, the p.Ser259Pro mutation was also found in two families (11, 18), wherein both probands presented with AI initially, however, hypogonadism was absent in the case reported by C. M. Oh et al. (18). The reproductive phenotypes were variable in these patients, ranging from azoospermia to spontaneous fertility, even within the same family, as shown by M. C. C. Vargas et al. (20), referring to a family with spontaneous fertility and a variable spectrum of reproductive phenotypes, in which all males presented a distinct degree of testosterone deficiency and fertility phenotypes, ranging from a variable degree of hypogonadism, oligoasthenoteratozoospermia to spontaneous fertility. Additionally, the same variant can cause diverse phenotypes within one kindred as reported in a family with the p.Trp39X variant; the proband was diagnosed with AI at the age of 19 years but exhibited a preserved hypothalamic–pituitary–gonadal axis; in contrast, his nephew experienced AI crisis at the age of 2 weeks (6). The underlying mechanism remains obscure. The mutation by itself as well as the repressive role of NR0B1 in steroidogenic factor-1 (SF1/Ad4BP, NR5A1) may be involved in this mechanism. DAX1 is expressed in tissues involved in steroid hormone production and reproductive function (21). This expression pattern overlaps significantly with that of another nuclear receptor, SF-1 (22). Previous studies have suggested that DAX1 is a negative regulator that represses SF-1-mediated transactivation of various genes involved in steroidogenesis (23). The loss of this inhibitory property in the LBD of DAX1 in the NR0B1 variant has been demonstrated to be responsible for the pathology of X-linked AHC and HH (24–30).
In addition, the proband’s serum triglyceride (TG) level was significantly high, which can be related to a low testosterone level. The plausible mechanism for the significant association between TG and testosterone levels in men is that low testosterone levels promote insulin resistance (31) by predisposing to visceral obesity, leading to a dysregulation of fatty acid metabolism, which in turn promotes insulin resistance (32). As reported previously, the relative risk of death in Swedish patients with AD is more than two-fold higher than that in the background population, primarily because of cardiovascular disease (CVD); elevated triglycerides are one of the CVD risk factors (33). Combining the dietary habits as well as no family history of hyperlipidemia makes it difficult to explain the common diseases of lipid metabolism. Therefore, whether or not the patient has a deletion of the glycerol kinase (GK) gene should be considered. The GK gene is located in Xp21.3, in a critical region of approximately 50–250 kb, close to the genomic coordinate of the AHC gene, and located distal to the Duchenne muscular dystrophy gene (34). Genomic analysis confirmed that the GK gene was unaffected, excluding combined glycerol kinase deficiency. Moreover, the serum VLCFA level was normal; therefore, adrenoleukodystrophy (ALD), another X-linked AI disorder, was also excluded. ALD is associated with primary testicular failure and characterized by primary AI and demyelination of the central or peripheral nervous system. The disease results from impaired transport of VLCFA into peroxisomes for beta-oxidation, which results in elevated levels of VLCFA in plasma and tissues (35).
Conclusion
In summary, we identified a novel frameshift variant of NROB1. This finding is important for expanding our knowledge of the phenotype and genotype of X-linked AHC. Functional studies of AHC patients are necessary to elucidate the biological effects of the identified mutation on the development and function of the adrenal gland and the hypothalamic–pituitary–gonadal axis.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81770736).
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://ngdc.cncb.ac.cn/, HRA002312.
Ethics statement
Ethical review and approval were not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the patient prior to his inclusion in the study.
Author contributions
JX designed and led this study. FZ and MZ searched the literature and drafted the manuscript. XD collected patient information, recruited the patient. YL analyzed the data. JX revised the manuscript. All authors read and approved the final manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.897069/full#supplementary-material
Abbreviations
ACTH, Adrenocorticotropic Hormone; AHC, Adrenal Hypoplasia Congenita; CAH, Congenital Adrenal Hyperplasia; DAX1, Dosage-sensitive sex reversal, adrenal hypoplasia congenital critical region on the X chromosome, gene 1; GnRH, Gonadotropin releasing Hormone; LH, luteinizing hormone; FSH, follicle-stimulating hormone; hCG, human chorionic gonadotropin; VLCFA, very long chain fatty acid, 17-OH, hydroxyprogesterone; CT, computed tomography; HH, hypogonadotropic hypogonadism.
References
1
Pereira BD Pereira I Portugal JR Goncalves J Raimundo L . X-Linked Adrenal Hypoplasia Congenita: Clinical and Follow-Up Findings of Two Kindreds, One With a Novel NR0B1 Mutation. Arch Endocrinol Metab (2015) 59(2):181–5. doi: 10.1590/2359-3997000000032
2
Swain A Narvaez S Burgoyne P Camerino G Lovell-Badge R . Dax1 Antagonizes Sry Action in Mammalian Sex Determination. Nature (1998) 391(6669):761–7. doi: 10.1038/35799
3
Stenson PD Mort M Ball EV Shaw K Phillips AD Cooper DN . The Human Gene Mutation Database: Building a Comprehensive Mutation Repository for Clinical and Molecular Genetics, Diagnostic Testing and Personalized Genomic Medicine. Hum Genet (2014) 133(1):1–9. doi: 10.1007/s00439-013-1358-4
4
Phelan JK McCabe ER . Mutations in NR0B1 (DAX1) and NR5A1 (SF1) Responsible for Adrenal Hypoplasia Congenita. Hum Mutat (2001) 18(6):472–87. doi: 10.1002/humu.1225
5
Mantovani G Ozisik G Achermann JC Romoli R Borretta G Persani L et al . Clinical Case Seminar - Hypogonadotropic Hypogonadism as a Presenting Feature of Late-Onset X-Linked Adrenal Hypoplasia Congenita. J Clin Endocrinol Metab (2002) 87(1):44–8. doi: 10.1210/jcem.87.1.8163
6
Raffin-Sanson ML Oudet B Salenave S Brailly-Tabard S Pehuet M Christin-Maitre S et al . A Man With a DAX1/NR0B1 Mutation, Normal Puberty, and an Intact Hypothalamic-Pituitary-Gonadal Axis But Deteriorating Oligospermia During Long-Term Follow-Up. Eur J Endocrinol (2013) 168(4):K45–50. doi: 10.1530/EJE-12-1055
7
Tabarin A Achermann JC Recan D Bex V Bertagna X Christin-Maitre S et al . A Novel Mutation in DAX1 Causes Delayed-Onset Adrenal Insufficiency and Incomplete Hypogonadotropic Hypogonadism. J Clin Invest (2000) 105(3):321–8. doi: 10.1172/JCI7212
8
Robinson PN Wang K . Phenolyzer: Phenotype-Based Prioritization of Candidate Genes for Human Diseases. Nat Methods (2015) 12(9):841–3. doi: 10.1038/nmeth.3484
9
Phelan JK McCabe ERB . Mutations in NROB1 (DAX1) and NR5A1 (SF1) Responsible for Adrenal Hypoplasia Congenita. Hum Mutat (2001) 18(6):472–87. doi: 10.1002/humu.1225
10
Guo WW Burris TP Zhang YH Huang BL Mason J Copeland KC et al . Genomic Sequence of the DAX1 Gene: An Orphan Nuclear Receptor Responsible for X-Linked Adrenal Hypoplasia Congenita and Hypogonadotropic Hypogonadism. J Clin Endocrinol Metab (1996) 81(7):2481–6. doi: 10.1210/jc.81.7.2481
11
Kyriakakis N Shonibare T Kyaw-Tun J Lynch J Lagos CF Achermann JC et al . Late-Onset X-Linked Adrenal Hypoplasia (DAX-1, NR0B1): Two New Adult-Onset Cases From a Single Center. Pituitary (2017) 20(5):585–93. doi: 10.1007/s11102-017-0822-x
12
Achermann JC Ito M Silverman BL Habiby RL Pang S Rosler A et al . Missense Mutations Cluster Within the Carboxyl-Terminal Region of DAX-1 and Impair Transcriptional Repression. J Clin Endocrinol Metab (2001) 86(7):3171–5. doi: 10.1210/jc.86.7.3171
13
Liu YX Yuan JL Zhang HJ Jiang YY Qin GJ . A Novel DAX-1 Mutation in Two Male Siblings Presenting With Precocious Puberty and Late-Onset Hypogonadotropic Hypogonadism. J Pediatr Endocrinol Metab (2017) 30(3):349–53. doi: 10.1515/jpem-2016-0228
14
Mantovani G Ozisik G Achermann JC Romoli R Borretta G Persani L et al . Hypogonadotropic Hypogonadism as a Presenting Feature of Late-Onset X-Linked Adrenal Hypoplasia Congenita. J Clin Endocrinol Metab (2002) 87(1):44–8. doi: 10.1210/jcem.87.1.8163
15
Ozisik G Mantovani G Achermann JC Persani L Spada A Weiss J et al . An Alternate Translation Initiation Site Circumvents an Amino-Terminal DAX1 Nonsense Mutation Leading to a Mild Form of X-Linked Adrenal Hypoplasia Congenita. J Clin Endocrinol Metab (2003) 88(1):417–23. doi: 10.1210/jc.2002-021034
16
Sekiguchi Y Hara Y Matsuoka H Hayashi Y Katsumata N Hirata Y . Sibling Cases of Addison’s Disease Caused by DAX-1 Gene Mutations. Intern Med (2007) 46(1):35–9. doi: 10.2169/internalmedicine.46.6082
17
Guclu M Lin L Erturk E Achermann JC Cangul H . Puberty, Stress, and Sudden Death. Lancet (2010) 376(9751):1512. doi: 10.1016/S0140-6736(10)61153-1
18
Oh CM Chun S Lee JE Lee JS Park S Gee HY et al . A Novel Missense Mutation in NR0B1 Causes Delayed-Onset Primary Adrenal Insufficiency in Adults. Clin Genet (2017) 92(3):344–6. doi: 10.1111/cge.12966
19
Suthiworachai C Tammachote R Srichomthong C Ittiwut R Suphapeetiporn K Sahakitrungruang T et al . Identification and Functional Analysis of Six DAX1 Mutations in Patients With X-Linked Adrenal Hypoplasia Congenita. J Endocr Soc (2019) 3(1):171–80. doi: 10.1210/js.2018-00270
20
Vargas MCC Moura FS Elias CP Carvalho SR Rassi N Kunii IS et al . Spontaneous Fertility and Variable Spectrum of Reproductive Phenotype in a Family With Adult-Onset X-Linked Adrenal Insufficiency Harboring a Novel DAX-1/NR0B1 Mutation. BMC Endocr Disord (2020) 20(1):21. doi: 10.1186/s12902-020-0500-2
21
Lalli E Sassone-Corsi P . DAX-1, an Unusual Orphan Receptor at the Crossroads of Steroidogenic Function and Sexual Differentiation. Mol Endocrinol (2003) 17(8):1445–53. doi: 10.1210/me.2003-0159
22
Ikeda Y Takeda Y Shikayama T Mukai T Hisano S Morohashi KI . Comparative Localization of Dax-1 and Ad4BP/SF-1 During Development of the Hypothalamic-Pituitary-Gonadal Axis Suggests Their Closely Related and Distinct Functions. Dev Dyn (2001) 220(4):363–76. doi: 10.1002/dvdy.1116
23
Suntharalingham JP Buonocore F Duncan AJ Achermann JC . DAX-1 (NR0B1) and Steroidogenic Factor-1 (SF-1, NR5A1) in Human Disease. Best Pract Res Clin Endoc Metab (2015) 29(4):607–19. doi: 10.1016/j.beem.2015.07.004
24
Muscatelli F Strom TM Walker AP Zanaria E Recan D Meindl A et al . Mutations In The Dax-1 Gene Give Rise To Both X-Linked Adrenal Hypoplasia Congenita And Hypogonadotropic Hypogonadism. Nature (1994) 372(6507):672–6. doi: 10.1038/372672a0
25
Zanaria E Muscatelli F Bardoni B Strom TM Guioli S Guo WW et al . An Unusual Member of the Nuclear Hormone-Receptor Superfamily Responsible for X-Linked Adrenal Hypoplasia Congenita. Nature (1994) 372(6507):635–41. doi: 10.1038/372635a0
26
Lalli E Bardoni B Zazopoulos E Wurtz JM Strom TM Moras D et al . A Transcriptional Silencing Domain in DAX-1 Whose Mutation Causes Adrenal Hypoplasia Congenita. Mol Endocrinol (1997) 11(13):1950–60. doi: 10.1210/mend.11.13.0038
27
Ito M Yu R Jameson JL . DAX-1 Inhibits SF-1-Mediated Transactivation via a Carboxy-Terminal Domain That Is Deleted in Adrenal Hypoplasia Congenita. Mol Cell Biol (1997) 17(3):1476–83. doi: 10.1128/MCB.17.3.1476
28
Crawford PA Dorn C Sadovsky Y Milbrandt J . Nuclear Receptor DAX-1 Recruits Nuclear Receptor Corepressor N-CoR to Steroidogenic Factor 1. Mol Cell Biol (1998) 18(5):2949–56. doi: 10.1128/MCB.18.5.2949
29
Altincicek B Tenbaum SP Dressel U Thormeyer D Renkawitz R Baniahmad A . Interaction of the Corepressor Alien With DAX-1 Is Abrogated by Mutations of DAX-1 Involved in Adrenal Hypoplasia Congenita. J Biol Chem (2000) 275(11):7662–7. doi: 10.1074/jbc.275.11.7662
30
Achermann JC Meeks JJ Jameson JL . Phenotypic Spectrum of Mutations in DAX-1 and SF-1. Mol Cell Endocrinol (2001) 185(1-2):17–25. doi: 10.1016/S0303-7207(01)00619-0
31
Pitteloud N Mootha VK Dwyer AA Hardin M Lee H Eriksson KF et al . Relationship Between Testosterone Levels, Insulin Sensitivity, and Mitochondrial Function in Men. Diabetes Care (2005) 28(7):1636–42. doi: 10.2337/diacare.28.7.1636
32
Boden G Jadali F White J Liang Y Mozzoli M Chen X et al . Effects of Fat on Insulin-Stimulated Carbohydrate Metabolism in Normal Men. J Clin Invest (1991) 88(3):960–6. doi: 10.1172/JCI115399
33
Bergthorsdottir R Leonsson-Zachrisson M Odén A Johannsson G . Premature Mortality in Patients With Addison’s Disease: A Population-Based Study. J Clin Endocrinol Metab (2006) 91(12):4849–53. doi: 10.1210/jc.2006-0076
34
Amato AA . Duchenne Muscular Dystrophy and Glycerol Kinase Deficiency: A Rare Contiguous Gene Syndrome. J Clin Neuromuscular Disease (2000) 1(4):191. doi: 10.1097/00131402-200006000-00006
35
Engelen M Kemp S de Visser M van Geel BM Wanders RJ Aubourg P et al . X-Linked Adrenoleukodystrophy (X-ALD): Clinical Presentation and Guidelines for Diagnosis, Follow-Up and Management. Orphanet J Rare Diseases (2012) 7:51. doi: 10.1186/1750-1172-7-51
Summary
Keywords
adrenal hypoplasia congenita, hypogonadotropic hypogonadism, NR0B1 gene, DAX1, X-linked recessive
Citation
Zhu F, Zhou M, Deng X, Li Y and Xiong J (2022) Case Report: A Novel Truncating Variant of NR0B1 Presented With X-Linked Late-Onset Adrenal Hypoplasia Congenita With Hypogonadotropic Hypogonadism. Front. Endocrinol. 13:897069. doi: 10.3389/fendo.2022.897069
Received
15 March 2022
Accepted
18 May 2022
Published
16 June 2022
Volume
13 - 2022
Edited by
Kunal Sharan, Central Food Technological Research Institute (CSIR), India
Reviewed by
Amalia Sertedaki, National and Kapodistrian University of Athens, Greece; Christina Bothou, University Hospital Zurich, Switzerland
Updates
Copyright
© 2022 Zhu, Zhou, Deng, Li and Xiong.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Xiong, jingxiong@hust.edu.cn
†These authors share first authorship
This article was submitted to Pediatric Endocrinology, a section of the journal Frontiers in Endocrinology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.