 Jeeyeon Cha
Jeeyeon Cha Cristina Aguayo-Mazzucato
Cristina Aguayo-Mazzucato Peter J. Thompson
Peter J. Thompson- 1Division of Diabetes, Endocrinology and Metabolism, Vanderbilt University Medical Center, Nashville, TN, United States
- 2Joslin Diabetes Center, Harvard Medical School, Boston, MA, United States
- 3Diabetes Research Envisioned and Accomplished in Manitoba Theme, Children’s Hospital Research Institute of Manitoba, Winnipeg, MB, Canada
- 4Department of Physiology & Pathophysiology, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB, Canada
Cellular senescence is a response to a wide variety of stressors, including DNA damage, oncogene activation and physiologic aging, and pathologically accelerated senescence contributes to human disease, including diabetes mellitus. Indeed, recent work in this field has demonstrated a role for pancreatic β-cell senescence in the pathogenesis of Type 1 Diabetes, Type 2 Diabetes and monogenic diabetes. Small molecule or genetic targeting of senescent β-cells has shown promise as a novel therapeutic approach for preventing and treating diabetes. Despite these advances, major questions remain around the molecular mechanisms driving senescence in the β-cell, identification of molecular markers that distinguish senescent from non-senescent β-cell subpopulations, and translation of proof-of-concept therapies into novel treatments for diabetes in humans. Here, we summarize the current state of the field of β-cell senescence, highlighting insights from mouse models as well as studies on human islets and β-cells. We identify markers that have been used to detect β-cell senescence to unify future research efforts in this field. We discuss emerging concepts of the natural history of senescence in β-cells, heterogeneity of senescent β-cells subpopulations, role of sex differences in senescent responses, and the consequences of senescence on integrated islet function and microenvironment. As a young and developing field, there remain many open research questions which need to be addressed to move senescence-targeted approaches towards clinical investigation.
1 Introduction
Pancreatic islet β-cells are the primary insulin-producing cells in the body, and their cooperation with neighboring endocrine cells in the islets of Langerhans are essential for controlling metabolic responses at a whole organismal level. Malfunction and/or loss of β-cells is a signature of diabetes mellitus, a collection of conditions, such as in Type 1 Diabetes (T1D), Type 2 Diabetes (T2D) and Maturity Onset Diabetes of the Young (MODY), with diverse etiologies commonly marked by hyperglycemia. Recent work from our groups has now established that subpopulations of β-cells undergo senescence during the development and onset of diabetes across mouse models of T1D, T2D and MODY (1–3) and in human pancreas tissue from T1D and T2D donors (1, 2).
Senescence is generally considered to be a terminal stress response that occurs in many different cell types in physiological contexts including embryonic development (4, 5) and aging (6, 7), and in response a wide variety of stressors such as tissue damage (8–10), viral infection (11) and oncogene activation (12). Senescent cells often undergo a DNA damage response leading to cell cycle arrest and progressively acquire resistance to intrinsic apoptosis, chromatin remodeling of gene regulatory networks, metabolic alterations, and development of a complex Senescence Associated Secretory Phenotype, or SASP [reviewed extensively elsewhere (13, 14)]. DNA damage signaling during senescence involves activation of cytoplasmic DNA sensing pathway cyclic GMP-AMP Synthase-STimulator of InterferoN Genes (cGAS-STING), which is also essential for the SASP program (15–17). The SASP secretome consists of a tissue/cell-type specific milieu of growth factors, cytokines, chemokines, proteases, shed receptors and ligands and extracellular vesicles (18) and shows a temporal progression. At early stages SASP is TGF-β-rich and immunosuppressive while at later stages the SASP is highly pro-inflammatory and involves type I interferon signaling (19, 20).The evolution and functions of cellular senescence are complex and have been suggested to demonstrate antagonistic pleiotropy: early in life, senescent cells are beneficial by suppressing cancer and facilitating organogenesis, but paradoxically senescent cells are associated with aging-related disease later in life (21). Although the fundamental points of this theory have been challenged (22) recent evidence has now supported this view (23). The clearance of accumulated senescent cells by the immune system seems to be the primary way senescence is resolved in most tissues during aging and is necessary for tissue homeostasis (24). However, when immune surveillance fails during aging senescent cells accumulate and can impair tissue function and reduce longevity (25) [also reviewed in (26, 27)]. Accumulation of senescent cells, including β-cells, hepatocytes and preadipocytes contributes to the pathology of different forms of diabetes mellitus and metabolic syndrome, and pharmacologic and genetic strategies to mitigate senescence have shown beneficial effects in mouse models (1–3, 28–30), underscoring the pathogenic effects of senescent cells in these diseases.
The molecular signatures associated with senescence vary by cell type and context but recent work has established at least four common hallmarks of β-cell senescence across different models (1–3) (Figure 1). These include: [1] increased markers of DNA damage and upregulation of cyclin-dependent kinase inhibitors p21Cip1 and/or p16Ink4a; [2] increased activity of the lysosomal enzyme GLB1 also called senescence-associated βgalactosidase (SA-βgal); [3] activation of a prosurvival pathway, such as the anti-apoptotic BCL-2 family of proteins; and [4] the development of a functional SASP (each discussed in depth below). Moreover, senescent β-cells maintain insulin expression and do not seem to acquire expression of other islet hormones. Together these features and molecular markers clearly distinguish senescence from other stress responses in the β-cell, such as apoptosis or altered β-cell identity (31). Additional molecular markers of senescence vary by disease model and context; however, all of these common features are readily assayed at the gene expression, protein and cellular/phenotypic levels (Table 1). Despite these advances, our knowledge in this emerging area stems from a relatively small number of studies and thus there is much to be learned about the molecular mechanisms of senescence in the β-cell, heterogeneity of senescent signatures depending on context, and how specific signaling pathways in senescent β-cells may be exploited to develop therapies for diabetes subtypes. In this review, we summarize the current state of knowledge of β-cell senescence in T1D, T2D and MODY, and propose a set of best-practices for the field for evaluating β-cell senescence to help guide its potential for clinical translation.
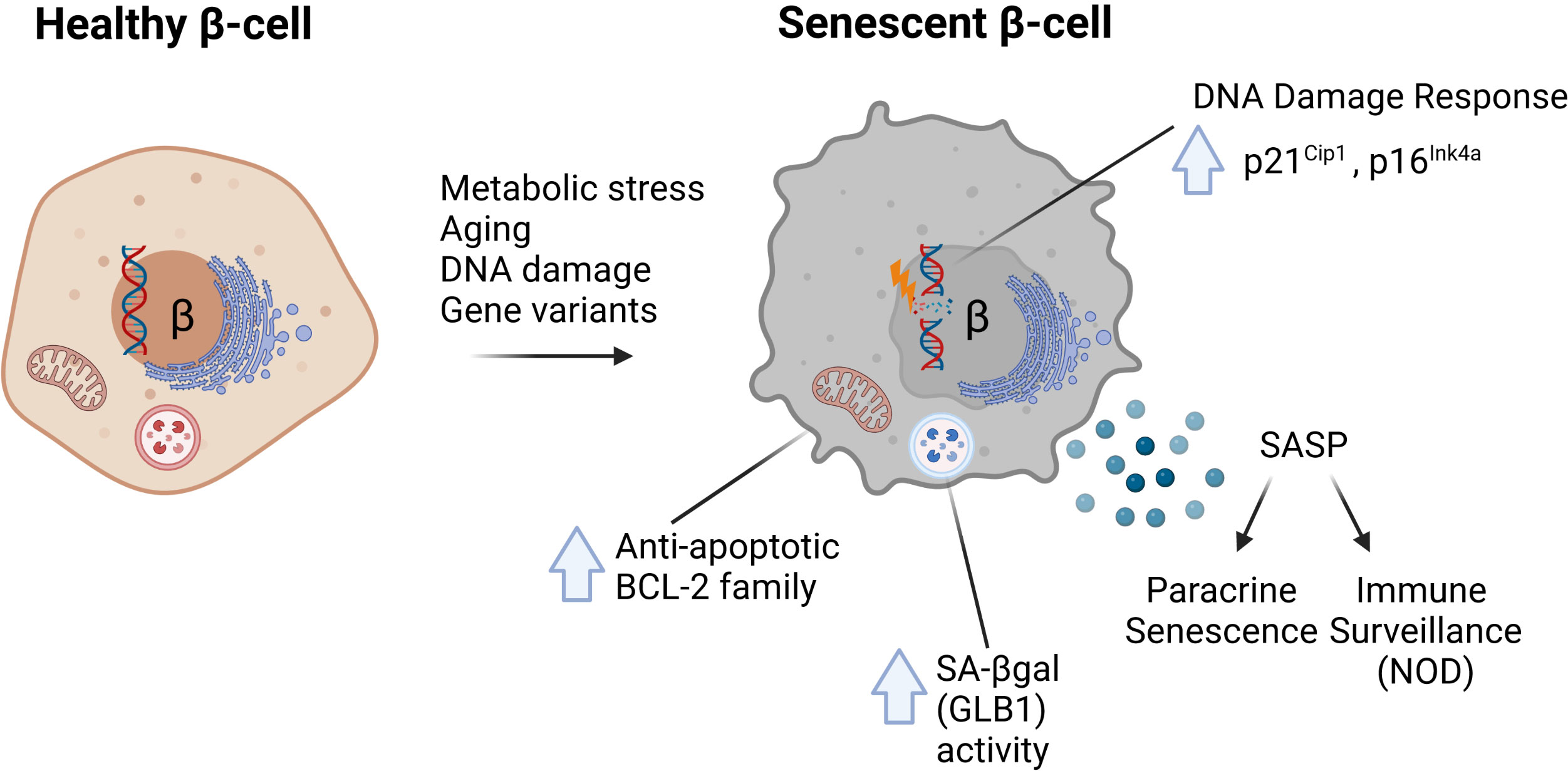
Figure 1 Common molecular features of senescent β-cells characterized in mouse models of diabetes. In response to metabolic stress, aging, direct DNA damage or the MafAS64F/+ gene variant, β-cells can develop senescence defined by several common features. These include (1) increased DNA damage and upregulation of Cdk inhibitors p21Cip1 and/or p16Ink4a; (2) increased activity of lysosomal enzyme GLB1, also referred to as senescence-associated βgalactosidase (SA-βgal); (3) increased expression of anti-apoptotic BCL-2 family proteins which mediate a prosurvival state; and (4) development of a Senescence-Associated Secretory Phenotype (SASP).
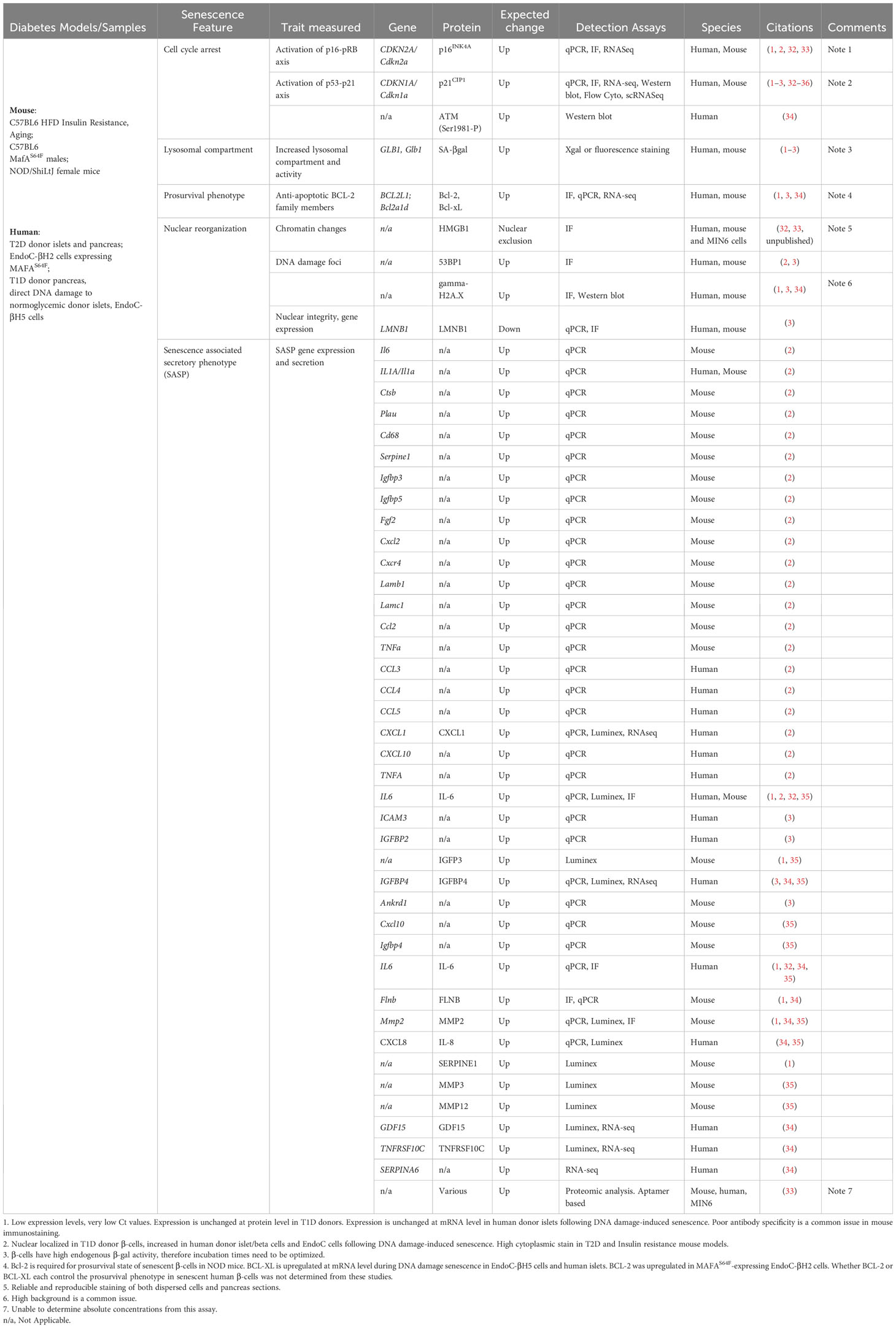
Table 1 Published markers of β-cell senescence in mouse models of diabetes and human cells/samples.
2 Strategies to evaluate β-cell senescence
A universal senescence signature that defines all senescent cells across tissues and aging has not been identified (37), underscoring the complexity of this cellular state. Therefore, it is critical to avoid ambiguity and make an accurate determination of whether senescence is involved in particular model. It is recommended that several independent markers representing distinct features of senescence are tested in parallel to establish the presence of senescent β-cells. As discussed earlier, senescence is inherently heterogeneous, and markers of β-cell senescence are likely to vary based on species (mouse vs. human), stressors involved, disease state, and cellular environment (in vitro versus in vivo). Published approaches which have been used to identify mouse and human β-cell senescence are summarized in Table 1. In this section we briefly discuss approaches for assaying these features and their utility for characterizing senescent β-cells in mice and humans.
2.1 SA-βgal activity
The lysosomal SA-βgal enzyme activity levels increase dramatically during senescence in many cell types and can be detected using X-gal staining (38) or C12FDG fluorescence staining (39). Mouse and human β-cells with high SA-βgal activity accumulate during aging and exhibit other features of senescence (2, 40) (Table 1), making this a useful approach for scoring or sorting presumed senescent β-cells. However, it should be kept in mind that lysosomal SA-βgal activity is not required for senescence (41) nor it is specific to senescence, as it can occur during other cellular responses such as autophagy or with high cell culture confluency (42, 43). Therefore, as a stand-alone marker, it is insufficient to prove that a β-cell is senescent, and additional molecular testing is recommended.
2.2 Expression of Cdk inhibitors
Senescent β-cells in both mouse and human frequently upregulate p21Cip1 and/or p16Ink4a and both should be monitored at the mRNA and protein levels. Validated primer sets are available to detect mRNA levels of Cdkn1a (encoding p21Cip1) or Cdkn2a (specific to exons encoding p16ink4a). Specific and validated antibodies for p21Cip1 are commercially available, however, available antibodies to detect p16Ink4a in mouse samples are often nonspecific [e.g. its detection in non-senescent macrophages (43)], and claims of specificity must be empirically established using appropriate controls. Also, care should be taken to note the localization patterns of each of these proteins in immunohistochemistry studies, as cytoplasmic localization for either one could indicate non-canonical functions. Furthermore, results in immortalized cell culture models (i.e. MIN6, HeLa) should be carefully interpreted as these cells have dysregulation of cell cycle regulators at baseline.
2.3 Loss of nuclear LaminB1 and HMGB1
Another common feature of senescence across different cell types is loss of nuclear integrity and reduced expression of nuclear LaminB1 (44) and HMGB1 (45). Loss of nuclear integrity impacts transcriptional activity as LaminB1 and HMGB1 proteins dynamically interact with active versus repressive chromatin to establish distinct topological domains, thus influencing the production of SASP (46, 47). Loss of LaminB1 and HMGB1 alters the spatial repositioning of heterochromatin to facilitate the formation of senescence-associated heterochromatin foci (SAHF) (reviewed in (48). Detection and localization of these markers can be assessed using immunocytochemistry or immunohistochemistry (Table 1). While loss of LaminB1 and HMGB1 have been found in some contexts of β-cell senescence in mice and cultured human β-cells (3, 32, 33), it is currently unclear whether this occurs across all models in which senescent β-cells are present and whether it occurs in resident senescent human β-cells in clinical disease.
2.4 Markers of DNA damage response
Markers of DNA damage are often seen in senescent cells, particularly with DNA damage-induced senescence (49). Enriched, punctate nuclear staining Trp53 binding protein 1 (53BP1) and phosphorylated H2AX (gH2AX) by immunostaining protocols mark double stranded DNA breaks implicated in driving cellular senescence (reviewed in (50). Lack of specificity of commercially available gH2AX antibodies have been noted across islet studies and therefore appropriate controls should always be included and results interpretated with caution.
2.5 SASP
The culture medium conditioned by senescent cells is enriched with secreted proteins which can promote the transformation of premalignant cells or induce phenotypic changes in neighboring cells, including paracrine senescence (51, 52) or tissue regeneration (10). Such enhanced secretory activity can also activate the adaptive Unfolded Protein Response (UPR) (19) although whether this occurs in β-cell senescence requires investigation. β-cell SASP repertoire consist of a variety of secreted molecules, including cytokines, chemokines, matrix metalloproteases, and growth factors (1–3, 33, 34), and those factors secreted from senescent β-cells across diabetes contexts have been identified using candidate and discovery approaches (Table 1) and show considerable differences between mouse and human β-cells (33, 34). SASP can be monitored at multiple levels, including mRNA, protein expression and secretion. To establish that senescent β-cells exhibit SASP, the SASP factors in question should, at the least, be monitored at the secretory level, as this is the only way to confirm that the factor is part of their secretome. In models involving the immune system, care should be taken to rule out contributions from immune cells, since some immune cell types secrete factors that overlap with common SASP constituents, such as IL-6 and IL-8. A bona fide SASP also should possess biological activity, which is best demonstrated by paracrine assays using the conditioned media from islets containing senescent β-cells and culturing this media with either naïve islets/cell lines or with immune cells (1, 2). A biologically relevant SASP can be confirmed by paracrine assays, demonstrating that the senescent β-cell conditioned media induces phenotypic changes in the naïve cells, such as paracrine senescence (1–3), or chemotaxis from immune cells in culture (1).
2.6 Prosurvival phenotype
Senescent cells activate anti-apoptotic pathways to resist normal programmed death signals [reviewed in (27)]. Senescent β-cells in mouse models of diabetes have thus far been shown to upregulate anti-apoptotic members of the BCL-2 family (1–3), although other pathways may also contribute. Key family members include Bcl-2, Bcl-xL, Bcl-w and Mcl-1 (1–3). As has been found in other senescent cell types, these changes can be assayed at the mRNA and protein levels (53). To confirm that the upregulation of one or more of these factors confers a prosurvival phenotype, small molecule BH3 mimetic compounds (such as ABT-263, ABT-737 and/or ABT-199) often used in cancer therapy (reviewed in (54) and which have been shown to possess senolytic activity (1, 53, 55), can be used to inhibit these proteins. Senescent cells are preferentially induced to undergo apoptosis in the presence of these compounds if the prosurvival mechanism employs anti-apoptotic BCL-2 family proteins (53, 55). It should be noted that there are a variety of other prosurvival mechanisms co-opted by senescent cells (56, 57), and these alternative pathways can also be explored if an upregulation of BCL-2 family members is not detected.
3 Molecular mechanisms of β-cell senescence
While cell cycle arrest can be mediated by several proteins in the Cyclin-dependent kinase (Cdk) inhibitor family, p21Cip1 and p16Ink4a have emerged as the major drivers of senescence in a broad range of cell types. This conclusion was established from key early studies on human fibroblast senescence models, genetic knockout mice and cancer (58–61) and has been extensively confirmed in subsequent studies [reviewed in (62)]. Activation of p21Cip1 (encoded by Cdkn1a) and p16Ink4a (an alternative splice product encoded by the Cdkn2a locus) tumor suppressor pathways are often upregulated by different cellular stressors, including (but not limited to): oncogene activation, telomere shortening, DNA damage, protein aggregation and reactive oxygen species (ROS) [reviewed in (13)]. This section will address the role of p21Cip1 and p16Ink4a in β-cells in the context of function, proliferation and senescence.
3.1 p21Cip1 in β-cell proliferation and function
In vitro, induction of DNA damage leads to stable p21Cip1 expression in mouse β-cell lines MIN6 and NIT-1 concomitant with a senescent-like growth arrest (2, 33, 34). Inhibition of p21Cip1 during acute DNA damage in MIN6 cells led to apoptosis (63), suggesting a key role of this Cdk inhibitor in promoting survival of β-cells following DNA damage. Given their role as cell-cycle inhibitors, several reports studying p21Cip1 and p16Ink4a first elucidated their roles as inhibitors of β-cell proliferation (64, 65). Interestingly, in a model of constitutively active Akt, mouse β-cells showed increased β-cell mass and proliferation via cyclin D1 and cyclin D2 with increases of p21Cip1 and p16Ink4a (66), however, another study demonstrates that mice with reduced p21Cip1 in the setting of constitutively active Akt signaling showed fasting and fed hypoglycemia, hyperinsulinemia and improved glucose tolerance (65). In contrast, opposite effects of p21Cip1 in β-cell function were observed in mice deficient in the cell cycle regulator Sei1. Sei1 promotes proliferation through the assembly of Cdk4-cyclin D and transcription factor E2F1, and mice deficient in this regulator with increased p21Cip1 are glucose intolerant with impaired insulin secretion and reduced β-cell mass (67). These findings suggests that the molecular context in which p21Cip1 is increased affects β-cell responses.
The subcellular localization of p21Cip1 also plays an important role in its regulation of β-cell proliferation. Using INS-1E cells with increased expression of protein kinase PKCδ, it was shown that p21Cip1 was excluded from the nucleus favoring its cytosolic localization, which led to increased proliferation (64). Other work has shown an anti-apoptotic role for cytoplasmic p21Cip1 independent of its cell cycle inhibitor activities (68). Additional experiments to understand the role of cytosolic versus nuclear p21Cip1 in models of β-cell senescence will be important since there are studies that report both its nuclear localization (1, 32) in models of T1D and cytoplasmic localization (2, 69) in models of T2D. Unfortunately, none of these studies addressed β-cell senescence, and Cdkn1a knockout (KO) mice show normal islet mass, β-cell replication rates, and function (70). Therefore, further studies are required in models of β-cell-specific deletion and overexpression that focus on the roles of these regulators in terms of spatial localization, temporality, and duration of expression.
3.2 p16Ink4a in β-cell proliferation and function
p16Ink4a was also identified as an inhibitor of β-cell proliferation through its action on kinases Cdk4 and Cdk6, which are critical for β-cell proliferation in both mouse and humans, respectively (71, 72). Importantly, a second tumor suppressor is encoded at the Cdkn2a locus (p19Arf in mouse, p14Arf in humans) which can also play a role in cell cycle inhibition by altering the Mdm2/p53 axis (62). Most studies have focused on p16Ink4a and have not selectively altered expression of p19Arf without an impact on p16Ink4a. Therefore, comparatively less is known about specific functions of p19/p14Arf in β-cells. Here we focus on the roles of p16Ink4a in β-cell senescence.
Several studies have demonstrated that expression of p16Ink4a at the protein and mRNA levels across tissues increases with age in humans and mice (1, 2, 40, 69, 73, 74). In mice, this age-dependent increase in expression was correlated with decreased methylation of the Cdkn2a gene locus (74) and reduced repressive histone modifications including Histone H3 Lys 27 trimethylation (H3K27me3) and Histone H2B Lys119 ubiquitination (H2BK119ub) (75, 76). Expression of p16Ink4a in human β-cells is also age-dependent: its expression was detected in 8% of prenatal β-cells and increased to about 63% of adult β-cells (77), however, absolute quantities of p16Ink4a protein need to be interpreted with care due to the challenges in the field with specificity of p16Ink4a antibodies, mostly in mouse, to detect it by immunohistochemistry (50).
The age-related increase of p16Ink4a in pancreatic β-cells has been suggested to be responsible for the age-related limited regeneration of this cell type (73). Supporting this concept, mice that overexpress p16Ink4a had decreased islet proliferation while proliferation was increased in aged p16Ink4a-deficient mice (73). The regulation of β-cell proliferation by p16Ink4a is dependent on PTEN status, as p16Ink4a was downregulated when PTEN was lost as a result of cyclin D1 induction and activation of E2F transcription factors (78). In turn, activation of E2F transcriptional-factors led to methylation of Cdkn2a promoter, thus inhibiting its expression. Furthermore, a study using an overexpression model of p16Ink4a driven by the Ins2 promoter has shown that β-cell function as assessed by glucose stimulated insulin secretion (GSIS) was improved while it was decreased in islets from p16Ink4a-deficient mice (40), suggesting that this Cdk-inhibitor is also important for β-cell function by inducing β-cell maturation.
However, other studies suggest that p16Ink4a expression has a deleterious effect on insulin secretion. Mice with telomerase haploinsufficiency with increased p16Ink4a expression in β-cells were insulin intolerant suggesting a primary defect on β-cell function (79). A knockdown of CDKN2A in the EndoC-βH1 human β-cell line increased insulin secretion (80). In fact, several noncoding genetic signals in the CDKN2A/B locus associate with high risk for human T2D (81) and loss of β-cell function (80) by GWAS analysis. At least four independent signals have been described at this locus (rs7856455, rs10757282, rs10811661, and rs1575972) (82). In particular, the risk allele (T) of the rs10811661 SNP correlates with reduced insulin secretory capacity (83, 84), higher HbA1c (p=2.15-10), prevalence of T2D and higher transcription of CDKN2A transcript in the human pancreas (while the other SNPs are not). In human studies, individuals from familial melanoma kindreds with a heterozygous loss of function of CDKN2A had increased insulin secretion supporting a deleterious effect of this Cdk-inhibitor on human β-cell function (80).
While these studies have advanced our understanding of the roles of p21Cip1 and p16Ink4a in β-cells, studies addressing their distinct roles in physiologic β-cell senescence (and functional maturity) versus pathologic senescence in disease remain open. Sequential activation of p21Cip1 and p16Ink4a during senescence progression has been recently reported in primary human fibroblasts in response to three well-established senescence-inducing stimuli: replication, oncogene-induction and irradiation (20). Under these conditions, cellular senescence proceeded through an early DNA damage-response phase characterized by p21Cip1 upregulation followed by a SASP response which coincided with an increase in p16Ink4a (20). Indeed, in β-cells from adult mice treated for two weeks with insulin receptor antagonist S961 modeling insulin resistance, expression of Cdkn1a increased transiently followed by a peak of Cdkn2a expression (33). Interestingly, this duration of S961 treatment also induces hyperinsulinemia in the setting of hyperglycemia with proliferation of a subset of β-cells (85). scRNASeq of islet cells from S961-treated mice suggest distinct subpopulation of proliferative and senescent β-cells (33). Whether the proportion of β-cells entering these cell fates (i.e. proliferation and senescence) depends on the dose and/or duration of S961 exposure remains to be discerned. In contrast, in T1D donor pancreas tissue and in a human islet model of DNA damage-induced senescence, CDKN1A and p21Cip1 upregulation occurs without any increase in CDKN2A or p16Ink4a (1, 34, 35), suggesting that the response of p21Cip1 and p16Ink4a in β-cells depends on the stressor. In some models, these results support a temporal progression of β-cell senescence during metabolic stress in which Cdkn1a is expressed in early stages, whereas Cdkn2a persists as a marker of established cellular senescence (33). Additional work has shown that the senescence growth arrest in fibroblasts enforced by p16Ink4a and p21Cip1 can be decoupled from SASP (86), the latter of which being driven by other mechanisms occurring during senescence, such as persistent DNA damage response signaling (49), chromatin remodelling (87, 88) and activation of the p38/MAPK pathway (89). Whether this is also true in the context of senescence in β-cells remains to be determined. Overall, questions remain regarding the circumstances in which p21Cip1 and p16Ink4a are upregulated with respect to the other, whether their activation is sequential or occur in parallel and whether they can enforce different forms of senescence to drive heterogeneous senescent β-cell populations.
3.3 Heterogeneity in β-cell senescence: Do p21Cip1 and p16Ink4a mark different subpopulations of senescent β-cells?
Senescence is being recognized as a highly heterogeneous phenomenon, both in vitro (90, 91) and in vivo (14, 37), and there is accumulating evidence for distinctions between senescent cell populations expressing p21Cip1 versus p16Ink4a. Whereas early work viewed p16Ink4a as a near-universal marker for senescent cells in vivo and developed a suite of molecular and genetic tools based around the detection and ablation of p16Ink4a+ cells, a shift in the field has occurred in recent years to focus on p21Cip1-expressing senescent cells. Notably, a recent study showed that p21Cip1 and p16 Ink4a expression elicit differences in SASP, where the p21Cip1- but not p16 Ink4a -driven SASP is sufficient to trigger immune surveillance and removal of senescent cells in vivo (92). Similarly, selective deletion of p21High cells in adipose tissue improves metabolic health in the HFD mouse model (93) and depletion of p21Cip1+ but not p16Ink4a+ senescent cells improves bone health following radiation exposure (94). In contrast with early work showing benefits of ablating p16Ink4a+ cells on healthspan in mice, a recent study showed that p16High cells are critical for healthy aging (95), thus challenging the notion that all p16Ink4a-expressing senescent cells are deleterious.
Evidence for heterogeneity of senescent β-cell subpopulations expressing predominantly p21Cip1 or p16Ink4a is apparent in mouse and human models relevant to T1D. In the NOD mouse model, the percentage of β-cells expressing p21Cip1 is around 5-10% by 14-15 weeks of age, whereas the percentage expressing p16Ink4a is ~30-40% (1). Indeed, the frequency of p21Cip1-expressing β-cells in late-stage euglycemic NOD mice most closely matches that of β-cells expressing other later senescence markers, such as Bcl-2 and SASP factors Mmp2, IL-6 and Igfbp3. While both p21Cip1 and p16Ink4a-expressing β-cell subpopulations are sensitive to BH3-only mimetic inhibitors with senolytic activity [i.e. ABT-737 and ABT-199, which mimic binding of the BH3-only initiator proteins to anti-apoptotic members (54)], the differences in the depletion of these populations is notable. When islets isolated from NOD mice at 14 weeks of age are cultured in the presence of senolytic ABT-737, nearly all the p21Cip1 expression is depleted, whereas only about 50% of the islet p16Ink4a expression is lost (1). In contrast, in aged human donor islets, senolytic compound ABT-263 mainly depletes CDKN2A expression (p16Ink4a mRNA) rather than CDKN1A (2). In human T1D donor pancreas, the subset of p21Cip1+ β-cells is significantly higher than in healthy controls with autoantibody-positive donors (1 or 2 AAbs) and in contrast, p16Ink4a+ β-cells were no different among the donor groups (1). p16Ink4a expression was already quite high even in the youngest pediatric donors examined in that study (aged 12-14), consistent with the age-related β-cell maturation and cell cycle exit. Replicating this observation in a culture model, DNA damage-induced senescence in islets from healthy donors leads to stable activation of p21Cip1 but not p16Ink4a (1, 34, 35) and more recently p21Cip1 expression was confirmed in β-cells in this model (36). Additional evidence for distinctions between p21Cip1 and p16Ink4a in senescent β-cells was recently shown with the INK-ATTAC mouse model, which revealed that ablation of p16Ink4a-expressing β-cells left a remaining senescent β-cell population expressing SASP factor genes (96). In future work, it will be important to investigate the molecular features that distinguish p16Ink4a -expressing β-cells from p21Cip1-expressing β-cells in the context of T1D and T2D mouse models and human islets.
4 Lessons on β-cell senescence from genetic models
In studying the molecular programs and effectors of senescence, much can be learned from the relationships with apoptosis. Senescence and apoptosis have been proposed as complementary cell fates (97), where senescence is often favored when the stress or damage is irremediable but not as severe or extensive as compared with apoptosis (98). Yet how these distinct stress responses in β-cells relate to one another during the development and progression of diabetes is poorly understood. In mouse models for T1D (e.g. NOD mouse) and T2D (e.g. HFD mouse), only a minority of β-cells develop senescence whereas others undergo apoptosis or (mal)adaptive stress responses. Thus, it is important to consider that β-cell senescence in the context of diabetes does not occur in isolation from other stress responses such as apoptosis.
Studies from whole-body mouse genetic models provide instructive examples of the intricate balance between senescence and apoptosis in β-cells. Mutations altering the p53 pathway or mutations affecting β-cell replication and DNA repair result in a heterogeneous population of β-cells, where some undergo senescence and others apoptosis (99–101). Notably, α-cells, though harboring the same mutation are not generally affected in these models (Table 2). For instance, mice harboring knock-out of the DNA ligase Lig4 (leading to increased endogenous unrepaired DNA strand breaks) and a point mutation in Tp53 (R172P) that mitigates p53-induced apoptosis, leads to very early onset of diabetes, involving both senescent β-cells and progressive loss of β-cell mass due to p53-independent apoptosis (100). In line with these findings, selective ablation of Tp53 failed to ameliorate dysglycemia induced by genetic modifications, diet, or pharmacologic agents (103), however β-cell senescence was not addressed. Similarly, deletion of the chromosome licensing factor Pttg1 (encoding the protein Securin), leads to a mixture of both senescent and apoptotic β-cells (99). In both the p53R172P;Lig4-/- mutants and the Pttg1-/- mutants, senescent and apoptotic β-cells were present, and progressive loss of β-cell mass and diabetes occurred at relatively young ages (5-10 weeks). Furthermore, mice harboring a whole-body point mutation that reduces expression of the DNA repair gene Ercc1, show accelerated aging, glucose intolerance and reduced β-cell mass with the accumulation of β-cells expressing senescence markers (101). Ercc1 hypomorphic mutant mice also exhibit senescent cell accumulation in other organs which may contribute to the metabolic dysfunction observed, nevertheless, this study affirms the importance of efficient repair of endogenous DNA damage for β-cell health. What causes some β-cells to activate a senescence program and others to undergo apoptosis in the same islets, in constitutive genetic models (where all β-cells have the same genotype), is not clear.
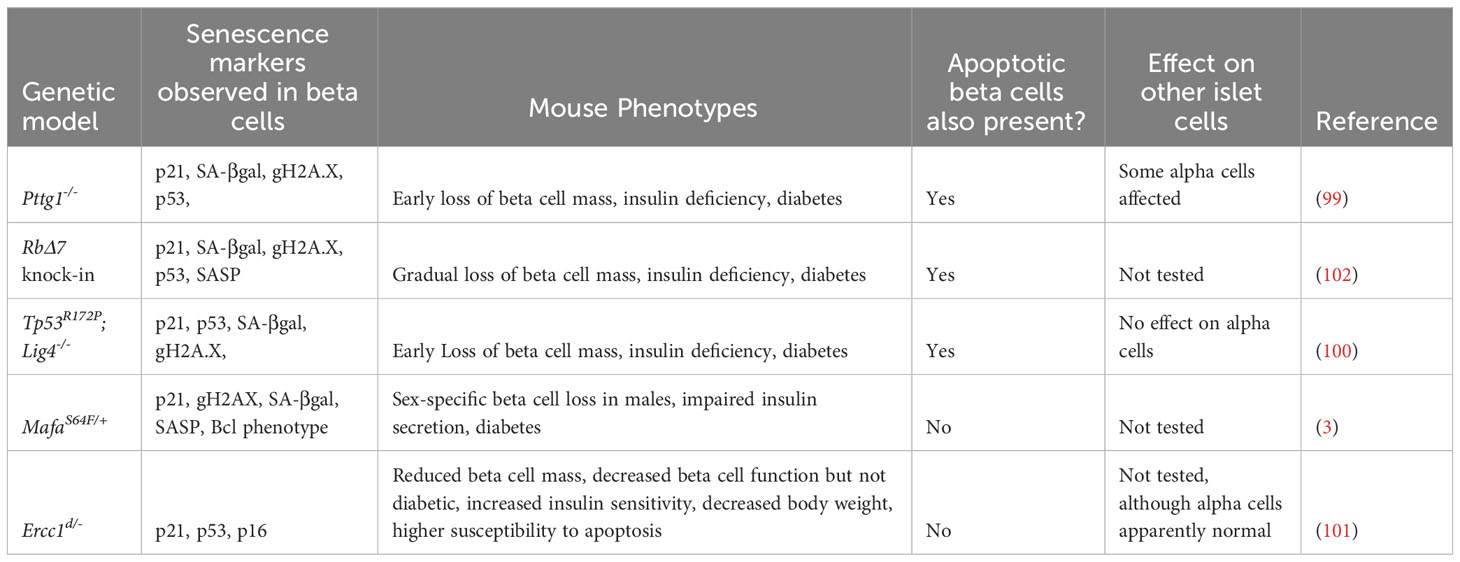
Table 2 Genetic mouse models with phenotypes of β-cell senescence and apoptosis.
In contrast with the above models that lead to a heterogeneous population of senescent and apoptotic β-cells, there are models that show predominantly one or the other. A recent study showed that point mutations in Rb that abrogate its phosphorylation sites (and hence prevent cell cycle progression), inhibit post-natal expansion of β-cell mass, resulting in early onset senescence and insufficient β-cell mass to maintain glucose homeostasis (102). A further example is illustrated with MafAS64F/+ mutant mice, in which males exhibit diabetes involving the accumulation of senescent β-cells, whereas females are protected from this phenotype (3), suggesting discrete functional β-cell populations. On the other hand, deletion of the transcription factor Yy1 leads to profound accumulation of β-cell DNA damage and extreme loss of β-cell mass by apoptosis by 5 weeks of age, without evidence of senescent β-cell accumulation (104). A recently published mouse model might be useful to further understand cellular fate after DNA damage. In the Inducible Changes to the Epigenome (ICE) mice, the endonuclease I-PpoI is used to create double stranded breakages in cells without causing mutations, allowing induction of non-mutagenic cuts in vivo (105). Interestingly, during and immediately after I-PpoI induction, there were no detectable changes in cell-cycle profile, apoptosis, or senescence. However, as the mice age, their tissues accumulated senescent cells at a faster rate than control mice (105), suggesting that DNA damage in non-coding regions might preferentially drive senescence versus apoptosis.
Together these genetic models reveal that senescence is finely balanced with apoptosis in β-cells and suggest that the fate choice between these different responses may be dependent on a variety of factors, including not only the type and extent of the damage, but also the age/maturation stage, sex and genetic background.
5 Sex differences in β-cell senescence
Sexual dimorphism in disease risk and manifestation has been described across metabolic tissues regulating glucose homeostasis, such as adipose (106), liver (107), and pancreas (108–110). While multiple factors can contribute to (and confound) intrinsic sex differences in diabetes such as age (in relation to puberty and menopause), sex hormone status, ethnicity, obesity, and diet, clinical reports have shown that adult premenopausal women have lower fasting glucose levels and higher insulin sensitivity than men (111, 112). In addition, in those exhibiting prediabetes (defined as HbA1C 5.7-6.4), women tend to manifest with glucose intolerance while men show impaired fasting glucose (reviewed in 113). In general, men are reported to have increased risk of T2D and ketosis-prone diabetes compared to premenopausal women (114), and reports of equivocal rates of T1D in men and women are paradoxical given female susceptibility to autoimmune conditions. Sex differences are also evident at the level of the human pancreatic islet, with women having increased insulin secretion after a glucose load, enhanced GSIS from isolated islets, and increased basal β-cell mass (reviewed in (109). Recent work notes sex differences in not only islet gene expression (115) but also magnitude of transcriptional responses (110). In whole, these reports support intrinsic differences in glucose metabolism and risk for diabetes between men and women.
Biologic sex differences are attributed to a complex interplay of sex chromosomes, physiological sex hormone production, sex hormone receptor status and environmental exposures (including endocrine disruptors) (116). Interpretations from studies using animal and culture models used are often limited by exposure to non-physiologic doses of sex hormone treatment and sex chromosome aneuploidy with progressive passages in cultured cells. Historically, animal research in metabolism were largely performed on male subjects (117). Recent efforts to incorporate sex as a biological variable (SABV) have facilitated the discovery of sex-dependent differences in β-cell function. For example, female mouse islets demonstrate improved resilience to ER stress, with larger capacity for protein production and folding than male islets (110). In line with this, many mouse models of diabetes show a relative protection from disease by female sex, including HFD-fed mice, insulin receptor antagonist (S961) treatment, STZ toxin treatment, and transgenic mice overexpressing hIAPP (reviewed in (109); a notable exception is the NOD mouse model, which manifests a T1D at a higher penetrance in female mice as compared with males (118, 119).
Aging itself is a predominant risk factor in diabetes, and declining levels of sex hormones is tightly linked to advanced tissue aging in natural andropause and menopause in men and women, respectively. Estradiol 17β (E2) demonstrated protective effects from DNA damage, by promoting telomerase expression in skin (120) and reducing p21Cip1 and senescence in a breast cancer cell line (121), while testosterone ameliorated vascular senescence (122) and DNA-damage induced senescence in cardiomyocytes (123). In fact, both exogenous E2 and testosterone treatment have been shown to improve β-cell function (124, 125). Chromosomal sex also modifies the senescence program triggered by the DNA damage-inducing agent bleomycin in human donor islets ex vivo. While isolated human islets of both sexes showed increases in p21Cip1, the extent of p21Cip1 induction varied dramatically (1.5-fold in a male donor versus 12-fold in a female donor) (34, 35), and male islets also showed more robust and sustained activation of DNA damage sensor ATM (34). Further studies addressing sex differences in β-cell senescent signatures and the magnitude of these changes in more human islet donors is warranted.
In several mouse models of diabetes involving β-cell senescence, the metabolic phenotypes show marked sex differences (1, 2), however it is unclear whether the prevalence of senescent β-cells is primarily a function of dysglycemia or biologic sex. The most dramatic example of sexual dimorphism of β-cell function and cellular senescence is in human carriers of a pathogenic variant in the transcription factor MAFA (serine to phenylalanine at position 64, or MAFAS64F) (3, 126). This variant promotes MAFA protein stability and predisposes subjects in their late 30’s to either adult-onset diabetes or insulinomatosis (i.e. non-syndromic insulin-producing β-cell tumors). MAFAS64F-associated diabetes was much more prevalent (3:1) in males while females presented more often with insulinomatosis (4:1) (126). A mouse model harboring this point mutation in the endogenous Mafa locus showed that islet β-cell dysfunction is principally in males, mimicking the findings in human subjects. Significantly, dysfunction in male mice was associated with premature cellular aging and senescence, whereas females had improved glucose clearance without accelerated senescence. In sum, an interplay between risk for β-cell dysfunction, cellular senescence, and biologic sex is likely.
6 Translation of therapies targeting β-cell senescence
A long-term goal is to move senescence-targeting therapies towards the clinic as tools to treat and prevent diabetes (56, 57). Preclinical studies in mouse models have validated the use of both genetic and pharmacologic approaches for either targeted removal of proinflammatory senescent β-cells or mitigation of their proinflammatory SASP. Given that pharmacologic approaches targeting senescence are currently undergoing clinical trials, their long term safety and efficacy in other chronic diseases remains to be determined (27, 127). However, it is likely that with further refinement, such approaches could also be tested in trials to improve clinical outcomes for those with or at risk for developing different forms of diabetes. Here we review the literature of preclinical studies targeting β-cell senescence.
6.1 Genetic approaches for mitigating β-cell senescence
In the context of T2D, senescent β-cells with SASP can be genetically targeted in mouse models of insulin resistance and HFD using the INK-ATTAC transgene. This model incorporates the promoter region of p16Ink4a to drive expression of a drug-activated apoptosis gene (FKBP8-Caspase8) selective in p16Ink4a-expressing senescent cells (6) and has been widely used as an in vivo reporter and genetic tool to deplete p16Ink4a-expressing senescent cells. Upon drug-mediated dimerization, the reporter selectively triggers Caspase8-mediated apoptosis in p16Ink4a-expressing cells. Removal of senescent cells using this mouse model on adult HFD mice (7-8 months) or mice with insulin resistance induced by S961 treatment, mitigates glucose intolerance, restores insulin sensitivity concomitant with a reduction in the markers of proinflammatory senescent β-cells in mouse islets (2). More recent work on this model indicates that although it leads to ablation of p16Ink4a-expressing senescent β-cells, overall β-cell mass and replication are not affected (96). Similarly, populations of p21Cip1-expressing senescent β-cells with SASP are were not affected by genetic deletion of p16Ink4a-expressing senescent β-cells (96). Whereas genetic tools have recently been developed to target p21Cip1-expressing senescent cells in mice (94, 128), it will be of great interest to apply these to interrogate the roles of p21High senescent β-cells in various diabetes models.
6.2 Pharmacologic approaches for mitigating β-cell senescence
Several different pharmacologic approaches have been employed to selectively mitigate the accumulation or phenotypes of senescent β-cells in diabetes models, including senolytics and senomorphics (56, 57). While senolytics are agents that preferentially disarm the anti-apoptosis pathways in senescent cells towards their demise, senomorphics are agents that can modify the biological properties of senescent cells in favor of mitigating their deleterious effects, such as inhibiting/altering the SASP. Several senolytic compounds have been tested in vivo to clear senescent cells [reviewed in (129)], such as FOXO4 inhibiting peptide that disrupts binding to p53 (130), the broad spectrum senolytics cardiac glycosides (131), finestin (132) and Heat-shock protein 90 inhibitors (133). More recently, FOXO-DRI (a selective FOXO4 inhibitor) also showed senolytic properties in senescent Leydig testicular cells (134) and in non-small cell lung cancer (135).
Senescent β-cells in different mouse models of diabetes upregulate different anti-apoptotic programs, and thus are sensitive to different types of senolytic agents. For instance, senescent β-cells that accumulate in NOD mice preferentially upregulate Bcl-2 and become sensitive to Bcl-2 family senolytic agents, such as ABT-737 or ABT-199 (1). SA-βgal+ cells from 7-8 month old C57BL6 male mice, also are sensitive to ABT-263 (Navitoclax), leading to improved β-cell function and identity (2). Dasatinib + Quercetin (D+Q), a potent combination with broad senolytic activity towards cells using the PI3K/Akt pathway, also elicits senolysis in senescent β-cells of HFD mice (2). Bromodomain ExtraTerminal (BET) domain inhibitors such as JQ1 or iBET-762 act as senomorphics on senescent β-cells in NOD mice by inhibiting these epigenetic readers and thus blocking the SASP at the transcriptional level (35). Administration of endogenously occurring lipids known as palmitic acid hydroxy stearic acids (PAHSAs) can prevent or reverse early stages of β-cell senescence in NOD mice by interfering with the Mdm2/p53 pathway (32).
Sex dimorphism in β-cell senescence responses can impact outcomes to senolytic therapy. Senolytic agents either targeting BCL2 family members or treating with PAHSAs, in female NOD mice improved β-cell function and delayed and/or prevent diabetes progression (1, 32). Notably, only NOD females were used in these studies due to their higher penetrance for diabetes. Senescent β-cells produced in male mice after acute exposure to insulin receptor antagonist S961 were cleared with ABT-263 coincident with improved glycemic responses (2). Similarly, S961-treated males treated with the senolytic cocktail D+Q also improved glucose tolerance and reduced rates of SA-βgal+ cells in the final four days of treatment (2). HFD treatment also showed overt diabetes with increases in senescence/aging indices in males and females. In contrast to HFD-treated males, HFD-treated females treated with ABT263 did not show an improvement in glucose tolerance despite reductions in SA-βgal+ β-cell population, and Aging and SASP indices, suggesting sex differences in response to senolytics depends on the metabolic disturbance provoking dysglycemia (2). Intriguingly, males and female human islets from non-diabetic donors or those with T2D treated with ABT263 similarly showed increased clearance of senescent β-cells.
However, a significant drawback to the use of these systemically administered agents (senolytics, senomorphics) is the possibility of off-target effects: while senescent cells are preferentially sensitive to these compounds, the drug targets are also expressed in healthy β-cells as well as other cell types. Current efforts are being made to preferentially target senescent cells in a tissue specific manner. Along this vein, preventative senolytic/senomorphic therapy for diabetes not only will deplete senescent β-cells but will be safe to use in patients that ultimately would not develop diabetes (57). For example, senolytic therapy in the NOD model have been focused on female mice with evidence of dysglycemia, however full metabolic profiling of euglycemic females and males receiving similar preventative treatment have not been assessed. Wild-type euglycemic mice pretreated with the senolytic compounds Fisetin (F) or D+Q showed sexually dimorphic glycemic responses (136). In both treatment groups, adult male mice over time showed mild, but not statistically significant, improvement in glucose tolerance to an i.p. glucose load, while female mice did not show any impact. Notably, while adult mice treated with D+Q had minimal effects in males, females showed increased SASP expression and accumulation of white adipose tissue depots, which may signal detrimental metabolic effects long term, suggesting that such preventative therapy is not benign in all populations.
7 Impact of senescence on β-cell on islet function and microenvironment
Senescence can alter β-cell function in many ways that are modified by genetic background and may exhibit differences between T1D and T2D. For instance, in the NOD mouse model for T1D, depletion of senescent β-cells in euglycemic female mice did not lead to a significant improvement in glucose tolerance or GSIS in isolated islets (1) suggesting that senescent β-cells in this model are not overtly deleterious to islet insulin secretion. Similarly, during DNA damage-induced senescence in human donor islets in culture, the extent of GSIS in static assays is remarkably preserved compared to controls (34). Significant differences were apparent in DNA damage-induced senescence in mouse MIN6 (C57BL6 background) and NIT1 (NOD background) β-cell lines, where senescence led to increased GSIS relative to controls (34). In contrast, DNA damage-induced senescence in the human fetal-derived female EndoC-βH5 cell line impaired GSIS (34). These findings suggest that DNA damage-associated senescence can differentially impact β-cell function depending on maturation stage, genetic background, culture model and species, and islet donor characteristics.
In T2D models of HFD or acute S961 insulin resistance, senescence results in an accelerated aging phenotype in β-cells and dramatically impairs insulin secretion. Senescent β-cells show increased expression of disallowed genes (2), which may contribute to their diminished function. In both T1D and T2D, senescent β-cells develop a SASP and it remains unclear how SASP alters or may interfere with the natural secretory machinery of the β-cell. Although upregulation of some of the age-dependent factors on their own do not drive age-related deterioration of β-cells (40, 74), in the long-term, aging results in lower transcriptional fidelity and worse β-cell function (137, 138). Senescence has major effects on cellular metabolism, with changes in mitochondrial function and reactive oxygen species (14), which have yet to be elucidated in the context of senescent β-cells. Further studies are needed to explore the specific metabolic and functional changes that occur in senescent β-cells using more sophisticated techniques, such as Patch-seq (139).
In addition to the impact of senescence directly on the metabolism and secretory function of β-cells, senescent β-cells could also exert paracrine effects on neighboring islet cells by virtue of SASP. Although this idea has yet to be tested, culture model systems of senescence support this concept. Conditioned media transfer experiments show that SASP factors secreted by β-cells are capable of triggering phenotypic changes in naïve recipient cells (1–3, 35). Moreover, induction of SASP in human islets in culture leads to altered glucagon secretion from α-cells, suggesting that SASP from β-cells can affect α-cell function (36). BET inhibitor iBET-762, which reduces SASP, can partially rescue the glucagon secretion phenotype (36), lending further evidence implicating SASP in impaired α-cell function. Given recent work implicating α-cell dysfunction in T1D (139–141), further investigation into how the accumulation of senescent β-cells in the islet may affect α-cell function is warranted. Since α-cell function is also impaired in T2D (142), it will be of great interest to explore how senescent β-cell accumulation in T2D is related to alterations in glucagon secretion.
8 Conclusions and future directions
In conclusion, it has become clear that β-cell senescence contributes to the pathophysiology of several forms of diabetes. Advancing our understanding of this stress response in the β-cell, its molecular control and heterogeneity, and how it is modified by sex, genetic background and maturation stage to impact other cells in the islet microenvironment holds promise for developing new therapies for treating diabetes. This field of investigation is still in its infancy and therefore establishing best-practices for characterizing senescence and developing accurate markers of senescence in β-cells is of utmost importance through collaborative efforts and concerted initiatives, such as the NIH SenNET [SenNet Consortium (143)]. The identification of such biomarkers will allow the translation of senotherapeutics into the clinic in the setting of diabetes. Care should be taken to accurately characterize senescence phenotypes at multiple levels (gene expression, protein and phenotypic) to avoid ambiguity and discriminate senescence from other β-cell stress responses. In addition, there is a need to continue identifying and testing therapeutic agents that target the vulnerabilities of senescent β-cells and neutralize their deleterious effects, while also minimizing the impact of such treatments on healthy β-cells. As the field matures, β-cell senescence will undoubtedly provide fruitful opportunities for advancing basic islet biology in addition to clinical translation efforts for diabetes.
Author contributions
JC, CA-M, and PT conceived the ideas together and jointly wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
JC is supported by K08 DK132507, Doris Duke Foundation Physician Scientist Fellowship, and Burroughs Wellcome Fund Career Award for Medical Scientists. Work in the lab of CA-M is supported by NIH grants R01DK132535 (to CA-M) and P30 DK036836 (to Joslin Diabetes Center Cores), and the Richard and Susan Smith Family Foundation Award. Work in the lab of PT is funded by a Project Grant from the Canadian Institutes of Health Research (Grant No. 202203PJT-479641).
Acknowledgments
We apologize to colleagues whose work was not cited due to space limitations. We are indebted to human islet donors and their families, without whom this research could not be possible.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, Bhushan A. Targeted elimination of senescent beta cells prevents type 1 diabetes. Cell Metab (2019) 29:1045–60. doi: 10.1016/j.cmet.2019.01.021
2. Aguayo-Mazzucato C, Andle J, Lee TB, Midha A, Talemal L, Chipashvili V, et al. Acceleration of β Cell aging determines diabetes and senolysis improves disease outcomes. Cell Metab (2019) 30:129–42.e4. doi: 10.1016/j.cmet.2019.05.006
3. Walker EM, Cha J, Tong X, Guo M, Liu J-H, Yu S, et al. Sex-biased islet β cell dysfunction is caused by the MODY MAFA S64F variant by inducing premature aging and senescence in males. Cell Rep (2021) 37:109813. doi: 10.1016/j.celrep.2021.109813
4. Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell (2013) 155:1119–30. doi: 10.1016/j.cell.2013.10.041
5. Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, et al. Programmed cell senescence during mamMalian embryonic development. Cell (2013) 155(5):1104–8. doi: 10.1016/j.cell.2013.10.019
6. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature (2011) 479:232–6. doi: 10.1038/nature10600
7. Yousefzadeh MJ, Zhao J, Bukata C, Wade EA, McGowan SJ, Angelini LA, et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell (2020) 19:e13094. doi: 10.1111/acel.13094
8. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell (2014) 31:722–33. doi: 10.1016/j.devcel.2014.11.012. é.
9. Mosteiro L, Pantoja C, Alcazar N, Marión RM, Chondronasiou D, Rovira M, et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science (2016) 354:aaf4445. doi: 10.1126/science.aaf4445
10. Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev (2017) 31:172–83. doi: 10.1101/gad.290635.116
11. Lee S, Yu Y, Trimpert J, Benthani F, Mairhofer M, Richter-Pechanska P, et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature (2021) 599:283–9. doi: 10.1038/s41586-021-03995-1
12. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell (1997) 88:593–602. doi: 10.1016/s0092-8674(00)81902-9
13. Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol (2021) 22:75–95. doi: 10.1038/s41580-020-00314-w
14. Wiley CD, Campisi J. The metabolic roots of senescence: mechanisms and opportunities for intervention. Nat Metab (2021) 3:1290–301. doi: 10.1038/s42255-021-00483-8
15. Glück S, Guey B, Gulen MF, Wolter K, Kang T-W, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol (2017) 19:1061–70. doi: 10.1038/ncb3586
16. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature (2017) 550:402–6. doi: 10.1038/nature24050
17. Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A (2017) 114:E4612–20. doi: 10.1073/pnas.1705499114
18. Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol (2020) 18:e3000599. doi: 10.1371/journal.pbio.3000599
19. Hoare M, Ito Y, Kang T-W, Weekes MP, Matheson NJ, Patten DA, et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol (2016) 18:979–92. doi: 10.1038/ncb3397
20. De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature (2019) 566:73–8. doi: 10.1038/s41586-018-0784-9
21. Campisi J. Aging and cancer: the double-edged sword of replicative senescence. J Am Geriatr Soc (1997) 45:482–8. doi: 10.1111/j.1532-5415.1997.tb05175.x
22. Giaimo S, d’Adda di Fagagna F. Is cellular senescence an example of antagonistic pleiotropy? Aging Cell (2012) 11:378–83. doi: 10.1111/j.1474-9726.2012.00807.x
23. Rodríguez JA, Marigorta UM, Hughes DA, Spataro N, Bosch E, Navarro A. Antagonistic pleiotropy and mutation accumulation influence human senescence and disease. Nat Ecol Evol (2017) 1:55. doi: 10.1038/s41559-016-0055
24. Ovadya Y, Landsberger T, Leins H, Vadai E, Gal H, BIran A, et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun (2018) 9:5435. doi: 10.1038/s41467-018-07825-3
25. Yousefzadeh MJ, Flores RR, Zhu Y, Schmiechen ZC, Brooks RW, Trussoni CE, et al. An aged immune system drives senescence and ageing of solid organs. Nature (2021) 594:100–5. doi: 10.1038/s41586-021-03547-7
26. Prata LGPL, Ovsyannikova IG, Tchkonia T, Kirkland JL. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin Immunol (2018) 40:101275. doi: 10.1016/j.smim.2019.04.003
27. Gasek NS, Kuchel GA, Kirkland JL, Xu M. Strategies for targeting senescent cells in human disease. Nat Aging (2021) 1:870–9. doi: 10.1038/s43587-021-00121-8
28. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med (2018) 24:1246–56. doi: 10.1038/s41591-018-0092-9
29. Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM, et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell (2019) 18:1–15. doi: 10.1111/acel.12950
30. Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun (2017) 8:15691 . doi: 10.1038/ncomms15691
31. Zhang J, Liu F. The De-, Re-, and trans-differentiation of β-cells: Regulation and function. Semin Cell Dev Biol (2020) 103:68–75. doi: 10.1016/j.semcdb.2020.01.003
32. Rubin de Celis MF, Garcia-Martin R, Syed I, Lee J, Aguayo-Mazzucato C, Bonner-Weir S, et al. PAHSAs reduce cellular senescence and protect pancreatic beta cells from metabolic stress through regulation of Mdm2/p53. Proc Natl Acad Sci U S A (2022) 119:e2206923119. doi: 10.1073/pnas.2206923119
33. Midha A, Pan H, Abarca C, Andle J, Carapeto P, Bonner-Weir S, et al. Unique human and mouse β-cell senescence-associated secretory phenotype (SASP) reveal conserved signaling pathways and heterogeneous factors. Diabetes (2021) 70:1098–116. doi: 10.2337/db20-0553
34. Brawerman G, Pipella J, Thompson PJ. DNA damage to β cells in culture recapitulates features of senescent β cells that accumulate in Type 1 Diabetes. Mol Metab (2022) 62:101524. doi: 10.1016/j.molmet.2022.101524
35. Thompson PJ, Shah A, Apostolopolou H, Bhushan A. BET proteins are required for transcriptional activation of the senescent islet cell secretome in type 1 diabetes. Int J Mol Sci (2019) 20:1–13. doi: 10.3390/ijms20194776
36. Brawerman G, Ntranos V, Thompson PJ. Alpha cell dysfunction in type 1 diabetes is independent of a senescence program. Front Endocrinol (Lausanne) (2022) 13:932516. doi: 10.3389/fendo.2022.932516
37. Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer (2015) 15:397–408. doi: 10.1038/nrc3960
38. Itahana K, Campisi J, Dimri GP. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol (2007) 371:21–31. doi: 10.1007/978-1-59745-361-5_3
39. Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc (2009) 4:1798–806. doi: 10.1038/nprot.2009.191
40. Helman A, Klochendler A, Azazmeh N, Gabai Y, Horwitz E, Anzi S, et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med (2016) 22:412–20. doi: 10.1038/nm.4054
41. Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, et al. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell (2006) 5:187–95. doi: 10.1111/j.1474-9726.2006.00199.x
42. Yang N-C, Hu M-L. The limitations and validities of senescence associated-beta-galactosidase activity as an aging marker for human foreskin fibroblast Hs68 cells. Exp Gerontol (2005) 40:813–9. doi: 10.1016/j.exger.2005.07.011
43. Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, et al. Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY) (2016) 8:1294–315. doi: 10.18632/aging.100991
44. Freund A, Laberge R-M, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell (2012) 23:2066–75. doi: 10.1091/mbc.E11-10-0884
45. Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, et al. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol (2013) 201:613–29. doi: 10.1083/jcb.201206006
46. Swanson EC, Manning B, Zhang H, Lawrence JB. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J Cell Biol (2013) 203:929–42. doi: 10.1083/jcb.201306073
47. Sofiadis K, Josipovic N, Nikolic M, Kargapolova Y, Übelmesser N, Varamogianni-Mamatsi V, et al. HMGB1 coordinates SASP-related chromatin folding and RNA homeostasis on the path to senescence. Mol Syst Biol (2021) 17:e9760. doi: 10.15252/msb.20209760
48. Rocha A, Dalgarno A, Neretti N. The functional impact of nuclear reorganization in cellular senescence. Brief Funct Genomics (2022) 21:24–34. doi: 10.1093/bfgp/elab012
49. Rodier F, Coppé JP, Patil CK, Hoeijmakers WAM, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol (2009) 11:973–9. doi: 10.1038/ncb1909
50. González-Gualda E, Baker AG, Fruk L, Muñoz-Espín D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J (2021) 288:56–80. doi: 10.1111/febs.15570
51. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol (2013) 15:978–90. doi: 10.1038/ncb2784
52. Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PloS Biol (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
53. Yosef R, Pilpel N, Tokarsky-Amiel R, BIran A, Ovadya Y, Cohen S, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun (2016) 7:11190. doi: 10.1038/ncomms11190
54. Delbridge ARD, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer (2016) 16:99–109. doi: 10.1038/nrc.2015.17
55. Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med (2016) 22:78–83. doi: 10.1038/nm.4010
56. Paez-Ribes M, González-Gualda E, Doherty GJ, Muñoz-Espín D. Targeting senescent cells in translational medicine. EMBO Mol Med (2019) 11:1–19. doi: 10.15252/emmm.201810234
57. Chaib S, Tchkonia T, Kirkland JL. Cellular senescence and senolytics: the path to the clinic. Nat Med (2022) 28:1556–68. doi: 10.1038/s41591-022-01923-y
58. Martín-Caballero J, Flores JM, García-Palencia P, Serrano M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res (2001) 61:6234–8.
59. Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, et al. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature (2001) 413:86–91. doi: 10.1038/35092592
60. Stein GH, Drullinger LF, Soulard A, Dulić V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol (1999) 19:2109–17. doi: 10.1128/MCB.19.3.2109
61. Okamoto A, Demetrick DJ, Spillare EA, Hagiwara K, Hussain SP, Bennett WP, et al. Mutations and altered expression of p16INK4 in human cancer. Proc Natl Acad Sci U S A (1994) 91:11045–9. doi: 10.1073/pnas.91.23.11045
62. Kumari R, Jat P. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol (2021) 9:645593. doi: 10.3389/fcell.2021.645593
63. Tay VSY, Devaraj S, Koh T, Ke G, Crasta KC, Ali Y. Increased double strand breaks in diabetic β-cells with a p21 response that limits apoptosis. Sci Rep (2019) 9:1–10. doi: 10.1038/s41598-019-54554-8
64. Ranta F, Leveringhaus J, Theilig D, Schulz-Raffelt G, Hennige AM, Hildebrand DG, et al. Protein kinase C delta (PKCδ) affects proliferation of insulin-secreting cells by promoting nuclear extrusion of the cell cycle inhibitor p21Cip1/WAF1. PLoS One (2011) 6:e28828. doi: 10.1371/journal.pone.0028828
65. Blandino-Rosano M, Alejandro EU, Sathyamurthy A, Scheys JO, Gregg B, Chen AY, et al. Enhanced beta cell proliferation in mice overexpressing a constitutively active form of Akt and one allele of p21Cip. Diabetologia (2012) 55:1380–9. doi: 10.1007/s00125-012-2465-9
66. Elghazi L, Balcazar N, Cras-Méneur C, Krits I, Kiyokawa H, Bernal-Mizrachi E. Akt induces beta-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes (2006) 55:318–25. doi: 10.2337/diabetes.55.02.06.db05-0757
67. Fernandez-Marcos PJ, Pantoja C, Gonzalez-Rodriguez A, Martin N, Flores JM, Valverde AM, et al. Normal proliferation and tumorigenesis but impaired pancreatic function in mice lacking the cell cycle regulator sei1. PloS One (2010) 5:e8744. doi: 10.1371/journal.pone.0008744
68. Jänicke RU, Sohn D, Essmann F, Schulze-Osthoff K. The multiple battles fought by anti-apoptotic p21. Cell Cycle (2007) 6:407–13. doi: 10.4161/cc.6.4.3855
69. Ebrahimi AG, Hollister-Lock J, Sullivan BA, Tsuchida R, Bonner-Weir S, Weir GC. Beta cell identity changes with mild hyperglycemia: Implications for function, growth, and vulnerability. Mol Metab (2020) 35:100959. doi: 10.1016/j.molmet.2020.02.002
70. Cozar-Castellano I, Haught M, Stewart AF. The cell cycle inhibitory protein p21cip is not essential for maintaining beta-cell cycle arrest or beta-cell function in vivo. Diabetes (2006) 55:3271–8. doi: 10.2337/db06-0627
71. Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet (1999) 22:44–52. doi: 10.1038/8751
72. Fiaschi-Taesch NM, Salim F, Kleinberger J, Troxell R, Cozar-Castellano I, Selk K, et al. Induction of human beta-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes (2010) 59:1926–36. doi: 10.2337/db09-1776
73. Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature (2006) 443:453–7. doi: 10.1038/nature05092
74. Avrahami D, Li C, Zhang J, Schug J, Avrahami R, Rao S, et al. Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved β cell function. Cell Metab (2015) 22:619–32. doi: 10.1016/j.cmet.2015.07.025
75. Dhawan S, Tschen SI, Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev (2009) 23:906–11. doi: 10.1101/gad.1742609
76. Chen H, Gu X, Su I, Bottino R, Contreras JL, Tarakhovsky A, et al. Polycomb protein Ezh2 regulates pancreatic β-cell Ink4a / Arf expression and regeneration in streptozotocin-induced diabetes mellitus. Genes Dev (2009) 23(8):975–85. doi: 10.1101/gad.1742509.regenerative
77. Köhler CU, Olewinski M, Tannapfel A, Schmidt WE, Fritsch H, Meier JJ. Cell cycle control of β-cell replication in the prenatal and postnatal human pancreas. Am J Physiol - Endocrinol Metab (2011) 300:E221–30. doi: 10.1152/ajpendo.00496.2010
78. Zeng N, Yang K-T, Bayan J-A, He L, Aggarwal R, Stiles JW, et al. PTEN controls β-cell regeneration in aged mice by regulating cell cycle inhibitor p16ink4a. Aging Cell (2013) 12:1000–11. doi: 10.1111/acel.12132
79. Guo N, Parry EM, Li L-S, Kembou F, Lauder N, Hussain MA, et al. Short telomeres compromise β-cell signaling and survival. PLoS One (2011) 6:e17858. doi: 10.1371/journal.pone.0017858
80. Pal A, Potjer TP, Thomsen SK, Ng HJ, Barrett A, Scharfmann R, et al. Loss-of-function mutations in the cell-cycle control gene CDKN2A impact on glucose homeostasis in humans. Diabetes (2016) 65:527–33. doi: 10.2337/db15-0602
81. Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PIW, Chen H, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science (2007) 316:1331–6. doi: 10.1126/science.1142358
82. Mahajan A, Spracklen CN, Zhang W, Ng MCY, Petty LE, Kitajima H, et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat Genet (2022) 54:560–72. doi: 10.1038/s41588-022-01058-3
83. Grarup N, Rose CS, Andersson EA, Andersen G, Nielsen AL, Albrechtsen A, et al. Studies of association of variants near the HHEX, CDKN2A/B, and IGF2BP2 genes with type 2 diabetes and impaired insulin release in 10,705 Danish subjects: validation and extension of genome-wide association studies. Diabetes (2007) 56:3105–11. doi: 10.2337/db07-0856
84. Hribal ML, Presta I, Procopio T, Marini MA, Stančáková A, Kuusisto J, et al. Glucose tolerance, insulin sensitivity and insulin release in European non-diabetic carriers of a polymorphism upstream of CDKN2A and CDKN2B. Diabetologia (2011) 54:795–802. doi: 10.1007/s00125-010-2038-8
85. Shirakawa J, Togashi Y, Basile G, Okuyama T, Inoue R, Fernandez M, et al. E2F1 transcription factor mediates a link between fat and islets to promote β cell proliferation in response to acute insulin resistance. Cell Rep (2022) 41:111436. doi: 10.1016/j.celrep.2022.111436
86. Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16 INK4a induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem (2011) 286:36396–403. doi: 10.1074/jbc.M111.257071
87. Chen H, Ruiz PD, McKimpson WM, Novikov L, Kitsis RN, Gamble MJ. MacroH2A1 and ATM play opposing roles in paracrine senescence and the senescence-associated secretory phenotype. Mol Cell (2015) 59:719–31. doi: 10.1016/j.molcel.2015.07.011
88. Aird KM, Iwasaki O, Kossenkov AV, Tanizawa H, Fatkhutdinov N, Bitler BG, et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J Cell Biol (2016) 215:325–34. doi: 10.1083/jcb.201608026
89. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J (2011) 30:1536–48. doi: 10.1038/emboj.2011.69
90. Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, Demaria M. Unmasking transcriptional heterogeneity in senescent cells. Curr Biol (2017) 27:2652–60.e4. doi: 10.1016/j.cub.2017.07.033
91. Wiley CD, Flynn JM, Morrissey C, Lebofsky R, Shuga J, Dong X, et al. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell (2017) 16:1043–50. doi: 10.1111/acel.12632
92. Sturmlechner I, Zhang C, Sine CC, van Deursen E-JJ, Jeganathan KB, Hamada N, et al. p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science (2021) 374:eabb3420. doi: 10.1126/science.abb3420
93. Wang L, Wang B, Gasek NS, Zhou Y, Cohn RL, Martin DE, et al. Targeting p21Cip1 highly expressing cells in adipose tissue alleviates insulin resistance in obesity. Cell Metab (2022) 34:75–89.e8. doi: 10.1016/j.cmet.2021.11.002
94. Chandra A, Lagnado AB, Farr JN, Doolittle M, Tchkonia T, Kirkland JL, et al. Targeted clearance of p21- but not p16-positive senescent cells prevents radiation-induced osteoporosis and increased marrow adiposity. Aging Cell (2022) 21:1–13. doi: 10.1111/acel.13602
95. Grosse L, Wagner N, Emelyanov A, Molina C, Lacas-Gervais S, Wagner KD, et al. Defined p16High senescent cell types are indispensable for mouse healthspan. Cell Metab (2020) 32:87–99.e6. doi: 10.1016/j.cmet.2020.05.002
96. Bahour N, Bleichmar L, Abarca C, Wilmann E, Sanjines S, Aguayo-mazzucato C. Clearance of p16 Ink4a -positive cells in a mouse transgenic model does not change β -cell mass and has limited effects on their proliferative capacity. Aging Cell (2023) 15:1–18. doi: 10.18632/aging.204483
97. Childs BG, Baker DJ, Kirkland JL, Campisi J, Deursen JM. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep (2014) 15:1139–53. doi: 10.15252/embr.201439245
98. Hsu CH, Altschuler SJ, Wu LF. Patterns of Early p21 Dynamics Determine Proliferation-Senescence Cell Fate after Chemotherapy. Cell (2019) 178:361–73.e12. doi: 10.1016/j.cell.2019.05.041
99. Chesnokova V, Wong C, Zonis S, Gruszka A, Wawrowsky K. Diminished pancreatic beta-cell mass in securin-null mice is caused by beta-cell apoptosis and senescence. Endocrinology (2009) 150:2603–10. doi: 10.1210/en.2008-0972
100. Tavana O, Puebla-osorio N, Sang M, Zhu C. Nonhomologous end-joining deficiency leads to a severe diabetic phenotype in mice. Diabetes (2010) 59(1):135–42. doi: 10.2337/db09-0792.O.T
101. Huerta Guevara AP, McGowan SJ, Kazantzis M, Stallons TR, Sano T, Mulder NL, et al. Increased insulin sensitivity and diminished pancreatic beta-cell function in DNA repair deficient Ercc1d/– mice. Metabolism (2021) 117:154711. doi: 10.1016/j.metabol.2021.154711
102. Jiang Z, Li H, Schroer SA, Voisin V, Ju Y, Pacal M, et al. Hypophosphorylated pRb knock-in mice exhibit hallmarks of aging and vitamin C-preventable diabetes. EMBO J (2022) 41:1–23. doi: 10.15252/embj.2020106825
103. Uhlemeyer C, Müller N, Rieck M, Kuboth J, Schlegel C, Grieß K, et al. Selective ablation of P53 in pancreatic beta cells fails to ameliorate glucose metabolism in genetic, dietary and pharmacological models of diabetes mellitus. Mol Metab (2023) 67:101650. doi: 10.1016/j.molmet.2022.101650
104. Martins Peçanha FL, Jaafar R, Werneck-de-Castro JP, Apostolopolou CC, Bhushan A, Bernal-Mizrachi E. The transcription factor YY1 is essential for normal DNA repair and cell cycle in human and mouse β-cells. Diabetes (2022) 71:1694–705. doi: 10.2337/db21-0908
105. Yang J-H, Hayano M, Griffin PT, Amorim JA, Bonkowski MS, Apostolides JK, et al. Loss of epigenetic information as a cause of mamMalian aging. Cell (2023) 186:305–26.e27. doi: 10.1016/j.cell.2022.12.027
106. Reue K, Wiese CB. Illuminating the mechanisms underlying sex differences in cardiovascular disease. Circ Res (2022) 130:1747–62. doi: 10.1161/CIRCRESAHA.122.320259
107. Chen X, McClusky R, Itoh Y, Reue K, Arnold AP. X and Y chromosome complement influence adiposity and metabolism in mice. Endocrinology (2013) 154:1092–104. doi: 10.1210/en.2012-2098
108. Lefebvre P, Staels B. Hepatic sexual dimorphism - implications for non-alcoholic fatty liver disease. Nat Rev Endocrinol (2021) 17:662–70. doi: 10.1038/s41574-021-00538-6
109. Gannon M, Kulkarni RN, Tse HM, Mauvais-Jarvis F. Sex differences underlying pancreatic islet biology and its dysfunction. Mol Metab (2018) 15:82–91. doi: 10.1016/j.molmet.2018.05.017
110. Brownrigg GP, Xia YH, Jamie Chu CM, Wang S, Chao C, Zhang JA, et al. Sex differences in islet stress responses support female β cell resilience. Mol Metab (2023) 69:101678. doi: 10.1016/j.molmet.2023.101678
111. Mauvais-Jarvis F. Gender differences in glucose homeostasis and diabetes. Physiol Behav (2018) 187:20–3. doi: 10.1016/j.physbeh.2017.08.016
112. Sicree RA, Zimmet PZ, Dunstan DW, Cameron AJ, Welborn TA, Shaw JE. Differences in height explain gender differences in the response to the oral glucose tolerance test- the AusDiab study. Diabetes Med (2008) 25:296–302. doi: 10.1111/j.1464-5491.2007.02362.x
113. Kautzky-Willer A, Kosi L, Lin J, Mihaljevic R. Gender-based differences in glycaemic control and hypoglycaemia prevalence in patients with type 2 diabetes: results from patient-level pooled data of six randomized controlled trials. Diabetes Obes Metab (2015) 17:533–40. doi: 10.1111/dom.12449
114. Mauvais-Jarvis F, Sobngwi E, Porcher R, Riveline J-P, Kevorkian J-P, Vaisse C, et al. Ketosis-prone type 2 diabetes in patients of sub-Saharan African origin: clinical pathophysiology and natural history of beta-cell dysfunction and insulin resistance. Diabetes (2004) 53:645–53. doi: 10.2337/diabetes.53.3.645
115. Oliva M, Muñoz-Aguirre M, Kim-Hellmuth S, Wucher V, Gewirtz ADH, Cotter DJ, et al. The impact of sex on gene expression across human tissues. Science (2020) 369(6509):eaba3066. doi: 10.1126/science.aba3066
116. Mauvais-Jarvis F, Arnold AP, Reue K. A guide for the design of pre-clinical studies on sex differences in metabolism. Cell Metab (2017) 25:1216–30. doi: 10.1016/j.cmet.2017.04.033
117. Clayton JA, Collins FS. Policy: NIH to balance sex in cell and animal studies. Nature (2014) 509:282–3. doi: 10.1038/509282a
118. Pearson JA, Wong FS, Wen L. The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes. J Autoimmun (2016) 66:76–88. doi: 10.1016/j.jaut.2015.08.019
119. Chen YG, Mathews CE, Driver JP. The role of NOD mice in type 1 diabetes research: Lessons from the past and recommendations for the future. Front Endocrinol (Lausanne) (2018) 9:51. doi: 10.3389/fendo.2018.00051
120. Emmerson E, Hardman MJ. The role of estrogen deficiency in skin ageing and wound healing. Biogerontology (2012) 13:3–20. doi: 10.1007/s10522-011-9322-y
121. Pedram A, Razandi M, Evinger AJ, Lee E, Levin ER. Estrogen inhibits ATR signaling to cell cycle checkpoints and DNA repair. Mol Biol Cell (2009) 20:3374–89. doi: 10.1091/mbc.e09-01-0085
122. Chen Y-Q, Zhou H-M, Chen F-F, Liu Y-P, Han L, Song M, et al. Testosterone ameliorates vascular aging via the Gas6/Axl signaling pathway. Aging (Albany NY) (2020) 12:16111–25. doi: 10.18632/aging.103584
123. Altieri P, Barisione C, Lazzarini E, Garuti A, Bezante GP, Canepa M, et al. Testosterone antagonizes doxorubicin-induced senescence of cardiomyocytes. J Am Heart Assoc (2016) 5(1):e002383. doi: 10.1161/JAHA.115.002383
124. Xu W, Morford J, Mauvais-Jarvis F. Emerging role of testosterone in pancreatic β-cell function and insulin secretion. J Endocrinol (2019) 240(3):R97–R105. doi: 10.1530/JOE-18-0573
125. Xu B, Allard C, Alvarez-Mercado AI, Fuselier T, Kim JH, Coons LA, et al. Estrogens promote misfolded proinsulin degradation to protect insulin production and delay diabetes. Cell Rep (2018) 24:181–96. doi: 10.1016/j.celrep.2018.06.019
126. Iacovazzo D, Flanagan SE, Walker E, Quezado R, de Sousa Barros FA, Caswell R, et al. MAFA missense mutation causes familial insulinomatosis and diabetes mellitus. Proc Natl Acad Sci U S A (2018) 115:1027–32. doi: 10.1073/pnas.1712262115
127. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine (2019) 40:554–63. doi: 10.1016/j.ebiom.2018.12.052
128. Wang B, Wang L, Gasek NS, Zhou Y, Kim T, Guo C, et al. An inducible p21-Cre mouse model to monitor and manipulate p21-highly-expressing senescent cells in vivo. Nat Aging (2021) 1:962–73. doi: 10.1038/s43587-021-00107-6
129. Kirkland JL, Tchkonia T. Senolytic drugs: from discovery to translation. J Intern Med (2020) 288:518–36. doi: 10.1111/joim.13141
130. Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell (2017) 169:132–47.e16. doi: 10.1016/j.cell.2017.02.031
131. Triana-Martínez F, Picallos-Rabina P, Da Silva-Álvarez S, Pietrocola F, Llanos S, Rodilla V, et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat Commun (2019) 10:4731. doi: 10.1038/s41467-019-12888-x
132. Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine (2018) 36:18–28. doi: 10.1016/j.ebiom.2018.09.015
133. Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun (2017) 8:422. doi: 10.1038/s41467-017-00314-z
134. Zhang C, Xie Y, Chen H, Lv L, Yao J, Zhang M, et al. FOXO4-DRI alleviates age-related testosterone secretion insufficiency by targeting senescent Leydig cells in aged mice. Aging (Albany NY) (2020) 12:1272–84. doi: 10.18632/aging.102682
135. Meng J, Li Y, Wan C, Sun Y, Dai X, Huang J, et al. Targeting senescence-like fibroblasts radiosensitizes non-small cell lung cancer and reduces radiation-induced pulmonary fibrosis. JCI Insight (2021) 6(23):e146334. doi: 10.1172/jci.insight.146334
136. Fang Y, Medina D, Stockwell R, McFadden S, Quinn K, Peck MR, et al. Sexual dimorphic metabolic and cognitive responses of C57BL/6 mice to Fisetin or Dasatinib and quercetin cocktail oral treatment. GeroScience (2023). doi: 10.1007/s11357-023-00843-0
137. Shrestha S, Erikson G, Lyon J, Spigelman AF, Bautista A, Manning Fox JE, et al. Aging compromises human islet beta cell function and identity by decreasing transcription factor activity and inducing ER stress. Sci Adv (2022) 8:eabo3932. doi: 10.1126/sciadv.abo3932
138. Enge M, Arda HE, Mignardi M, Beausang J, Bottino R, Kim SK, et al. Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell (2017) 171:321–30.e14. doi: 10.1016/j.cell.2017.09.004
139. Camunas-Soler J, Dai XQ, Hang Y, Bautista A, Lyon J, Suzuki K, et al. Patch-seq links single-cell transcriptomes to human islet dysfunction in diabetes. Cell Metab (2020) 31:1017–31.e4. doi: 10.1016/j.cmet.2020.04.005
140. Brissova M, Haliyur R, Saunders D, Shrestha S, Dai C, Blodgett DM, et al. α Cell function and gene expression are compromised in type 1 diabetes. Cell Rep (2018) 22:2667–76. doi: 10.1016/j.celrep.2018.02.032
141. Doliba NM, Rozo AV, ROman J, Qin W, Traum D, Gao L, et al. α Cell dysfunction in islets from nondiabetic, glutamic acid decarboxylase autoantibody–positive individuals. J Clin Invest (2022) 132:e156243. doi: 10.1172/JCI156243
142. Martínez MS, Manzano A, Olivar LC, Nava M, Salazar J, D’marco L, et al. The role of the α cell in the pathogenesis of diabetes: A world beyond the mirror. Int J Mol Sci (2021) 22(17):9504. doi: 10.3390/ijms22179504
Keywords: pancreatic beta cells, cellular senescence, type 1 diabetes, type 2 diabetes, monogenic diabetes
Citation: Cha J, Aguayo-Mazzucato C and Thompson PJ (2023) Pancreatic β-cell senescence in diabetes: mechanisms, markers and therapies. Front. Endocrinol. 14:1212716. doi: 10.3389/fendo.2023.1212716
Received: 26 April 2023; Accepted: 15 August 2023;
Published: 31 August 2023.
Edited by:
Mathieu Ferron, Montreal Clinical Research Institute (IRCM), CanadaReviewed by:
Rohit B. Sharma, NewYork-Presbyterian, United StatesFrédérick A. Mallette, Montreal University, Canada
Copyright © 2023 Cha, Aguayo-Mazzucato and Thompson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeeyeon Cha, amVleWVvbi5jaGFAdnVtYy5vcmc=; Peter J. Thompson, cGV0ZXIudGhvbXBzb25AdW1hbml0b2JhLmNh