 Antía Fernández-Pombo1,2
Antía Fernández-Pombo1,2 Sofía Sánchez-Iglesias1
Sofía Sánchez-Iglesias1 Ana I. Castro-Pais2,3
Ana I. Castro-Pais2,3 Maria José Ginzo-Villamayor4Silvia Cobelo-Gómez1Teresa Prado-Moraña1,2
Maria José Ginzo-Villamayor4Silvia Cobelo-Gómez1Teresa Prado-Moraña1,2 Everardo Josué Díaz-López1,2Felipe F. Casanueva3
Everardo Josué Díaz-López1,2Felipe F. Casanueva3 Lourdes Loidi5
Lourdes Loidi5 David Araújo-Vilar1,2*
David Araújo-Vilar1,2*- 1Department of Psychiatry, Radiology, Public Health, Nursing and Medicine, IDIS-CiMUS, University of Santiago de Compostela, Santiago de Compostela, Spain
- 2Division of Endocrinology and Nutrition, University Clinical Hospital of Santiago de Compostela, Santiago de Compostela, Spain
- 3CIBER Fisiopatología de la Obesidad y la Nutrición (CIBERobn), Madrid, Spain
- 4Department of Estatística, Análise Matemática e Optimización, University of Santiago de Compostela, Santiago de Compostela, Spain
- 5Galician Public Foundation for Genomic Medicine (SERGAS-Xunta de Galicia), Santiago de Compostela, Spain
The rarity of lipodystrophies implies that they are not well-known, leading to delays in diagnosis/misdiagnosis. The aim of this study was to assess the natural course and comorbidities of generalised and partial lipodystrophy in Spain to contribute to their understanding. Thus, a total of 140 patients were evaluated (77.1% with partial lipodystrophy and 22.9% with generalised lipodystrophy). Clinical data were collected in a longitudinal setting with a median follow-up of 4.7 (0.5-17.6) years. Anthropometry and body composition studies were carried out and analytical parameters were also recorded. The estimated prevalence of all lipodystrophies in Spain, excluding Köbberling syndrome, was 2.78 cases/million. The onset of phenotype occurred during childhood in generalised lipodystrophy and during adolescence-adulthood in partial lipodystrophy, with the delay in diagnosis being considerable for both cohorts. There are specific clinical findings that should be highlighted as useful features to take into account when making the differential diagnosis of these disorders. Patients with generalised lipodystrophy were found to develop their first metabolic abnormalities sooner and a different lipid profile has also been observed. Mean time to death was 83.8 ± 2.5 years, being shorter among patients with generalised lipodystrophy. These results provide an initial point of comparison for ongoing prospective studies such as the ECLip Registry study.
1 Introduction
Lipodystrophy syndromes are a heterogeneous group of rare disorders distinguished by the selective loss of adipose tissue. They may be generalised if the loss of adipose tissue affects the whole body, or partial if only part of the body is affected (1, 2). These generalised and partial lipodystrophy syndromes are also characterised by the ectopic accumulation of adipose tissue and the presence of insulin resistance and, consequently, by a variable degree of metabolic dysfunction, with diabetes mellitus (DM), dyslipidaemia, fatty liver disease, cardiovascular disease and reproductive dysfunction.
In 2017, the range of worldwide prevalence of all lipodystrophies (excluding human immunodeficiency virus (HIV)-related lipodystrophy) as determined by electronic medical record (EMR) database searches has been estimated at 1.3 to 4.7 cases per million inhabitants. However, it has also been estimated that for partial Mendelian lipodystrophies the estimated prevalence is higher (3, 4). The extreme rarity of lipodystrophy syndromes implies that they are not well-known. In addition, the clinical characterisation of these disorders is often deficient, there is a certain degree of phenotypical variability between the different subtypes of lipodystrophy and their aetiology is diverse, as are the pathogenetic mechanisms which lead to the alteration of adipose tissue. Thus, to date, they are considered to be underdiagnosed conditions, poorly understood by healthcare professionals, which often leads to long delays in diagnosis or misdiagnosis and to obstacles in the development of specific therapies.
The knowledge we currently have about the natural history, burden of the disease and mortality of these patients is based fundamentally on studies limited to small samples or studies focused on a specific lipodystrophy subtype (5–12). To the best of our knowledge, only one study, focused on five specific centres across three countries (Turkey, Brazil and United States) included patients with both generalised and partial lipodystrophy with a larger sample size (13).
In light of the previously-mentioned knowledge gap, the aim of this study was to assess the natural course and comorbidities of patients with generalised and partial lipodystrophies in Spain (both acquired and congenital) in an effort to contribute to the understanding of these syndromes.
2 Materials and methods
2.1 Study population
Of a total of 343 patients with rare adipose tissue disorders, 140 subjects were diagnosed with lipodystrophy and evaluated between 2001 and 2020 in the leading management and treatment centre for lipodystrophies in Spain (UETeM, CiMUS), which receives patients from the whole country.
Patients with localised lipodystrophy, lipomatosis and adiposis dolorosa were excluded from this analysis. Patients with HIV-related lipodystrophy are not evaluated at said centre and they were also not included in the analysis. Among patients with genetic lipodystrophy, subjects with familial partial lipodystrophy (FPLD) type 1 were also excluded taking into account the following reasons: its genetic origin is unknown and, therefore, its diagnosis is merely clinical; this specific FPLD subtype may be confused with android obesity in general practice and, therefore, it is rare for these patients to be referred for evaluation (80.6% of them come from the same geographic region in which the centre is located); the high number of patients with a diagnosis of FPLD type 1 would distort the prevalence results of the current study; most of the data on these patients have already been analysed and reported by our group (14). In addition, patients with progeroid syndromes associated with lipodystrophy and patients with a diagnosis of lipodystrophy but no signs of the disease taking into account their young age were only included for the demographic and epidemiological analysis, considering that their inclusion may also affect the prevalence results. More detailed data of the inclusion/exclusion of patients and the population evaluated can be seen in the flow diagram of Figure 1.
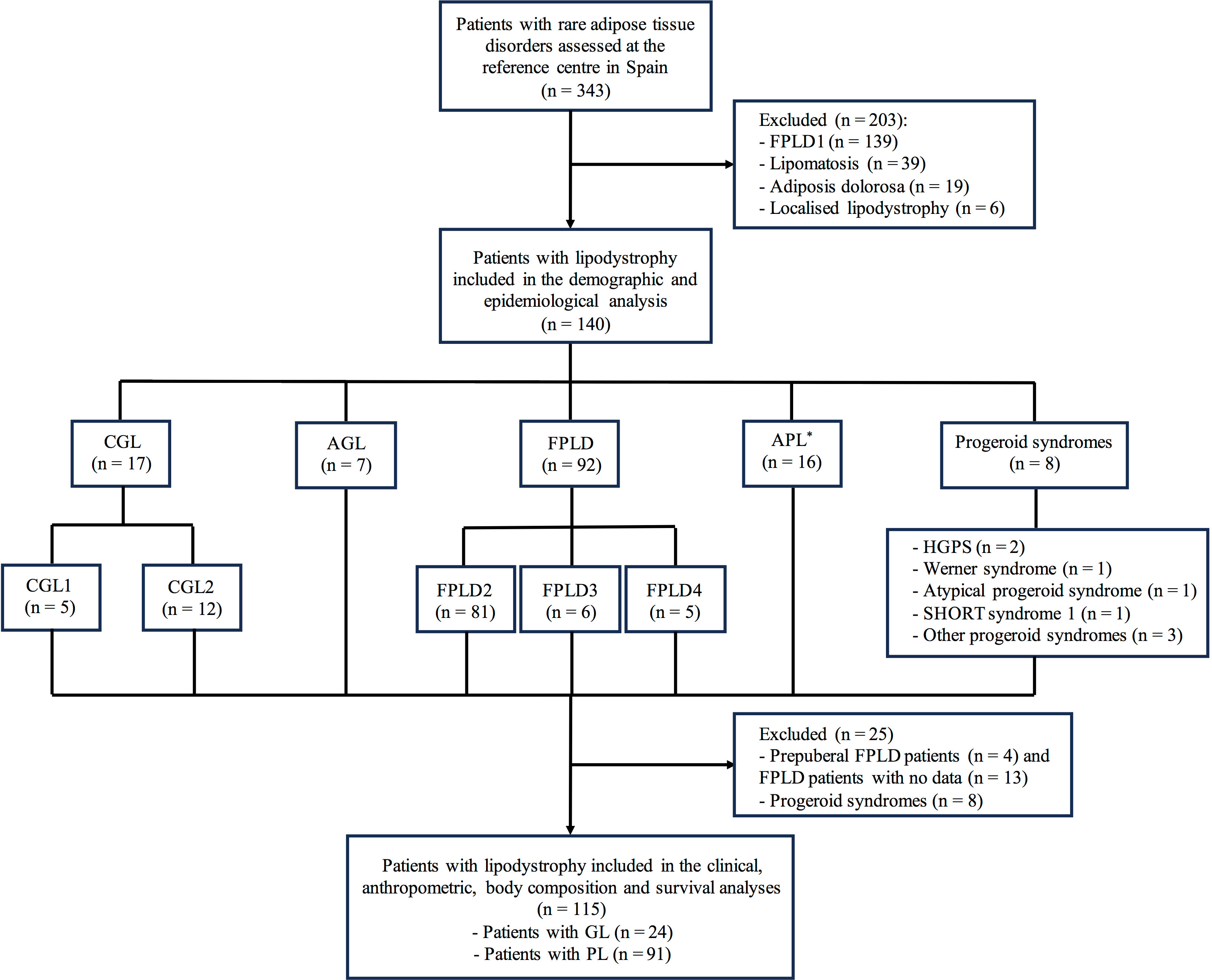
Figure 1 Flow diagram of patients with lipodystrophy evaluated at the reference centre in Spain. *The group of patients with acquired partial lipodystrophy included 15 subjects with Barraquer-Simons syndrome and one subject with partial lipodystrophy after bone marrow transplantation and radiotherapy. CGL, congenital generalised lipodystrophy; CGL1, congenital generalised lipodystrophy type 1; CGL2, congenital generalised lipodystrophy type 2; FPLD, familial partial lipodystrophy; FPLD1, familial partial lipodystrophy type 1; FPLD2, familial partial lipodystrophy type 2; FPLD3, familial partial lipodystrophy type 3; FPLD4, familial partial lipodystrophy type 4; AGL, acquired generalised lipodystrophy; APL, acquired partial lipodystrophy; HGPS, Hutchinson-Gilford Progeria Syndrome; SHORT, short stature, hyperextensibility, ocular depression, Rieger anomaly, and teething delay; GL, generalised lipodystrophy; PL, partial lipodystrophy.
On the other hand, it should be noted that twelve patients included in the analysis (11 with generalised lipodystrophy and one with partial lipodystrophy) started treatment with recombinant human leptin during follow-up.
2.2 Study design
This is a retrospective, observational, longitudinal study, carried out in a single centre attending patients with lipodystrophy from all over Spain. All the analyses were executed for the overall sample, as well as for generalised and partial lipodystrophy cohorts. Clinical data were recorded for analysis from birth until loss of follow-up, death or closure of the database.
This study was approved by the ethics review panel of the Red Gallega de Comités de Ética de la Investigación (approval code 2017/477) and carried out according to the ethical guidelines of the Helsinki Declaration.
2.3 Diagnosis of lipodystrophy and clinical data collection
The loss of adipose tissue and the affected regions were clinically confirmed by the same expert evaluator in all cases. Clinical diagnosis of lipodystrophy was also supported by body composition imaging. The pattern of partial or total fat loss and its onset was taken into consideration for each of the lipodystrophy subtypes. The absence of family history was also taken into account in the acquired forms. Molecular analysis was carried out in all patients evaluated in the centre in order to confirm or discard a genetic origin of the disorder. Other causes associated with wasting or weight loss (such as cancer cachexia, malnutrition, malabsorption, anorexia nervosa, thyrotoxicosis or chronic infections) were ruled out.
Clinical data were obtained from the patients’ electronic medical records and were evaluated by the same two examiners following a standardised and homogeneous protocol. Of the patients evaluated, the proportion of subjects with more than three medical evaluations during follow-up was 62.1% in the overall sample (66.7% in the generalised lipodystrophy group and 50.4% in the partial lipodystrophy group).
Physical examination was performed on all participants in the study, establishing a routine in their evaluation to enable a homogeneous comparison between the lipodystrophy subgroups. Regarding comorbidities, data collection focused on capturing organ abnormalities mainly related with adipose tissue dysfunction, ectopic fat accumulation and insulin resistance characteristic of the disease. In addition, other global defects found in these patients, extending beyond the key organ systems, were also recorded. DM and gestational diabetes were defined according to the 2022 American Diabetes Association criteria (15). Foot examination for the diagnosis of diabetic peripheral neuropathy, starting at diagnosis of diabetes and annually thereafter, included visual inspection and pedal pulses, as well as the 10-g monofilament exam, determination of vibration with a 128-Hz tuning fork, temperature and/or pinprick sensation. Symptoms of diabetic autonomic neuropathy were also assessed (16). Causes of neuropathy other than diabetes were discarded. Active screening of atherosclerotic vascular disease (ASCVD) and other cardiac abnormalities (including rhythm disturbances, hypertrophic cardiomyopathy and valvulopathies) was made only if symptoms or signs of any of these disorders were present and not on a routine basis in asymptomatic patients. Polycystic Ovary Syndrome (PCOS) was diagnosed based on the 2006 proposal of the Androgen Excess and PCOS Society: the presence of hyperandrogenism (hirsutism and/or hyperandrogenaemia) and ovulatory dysfunction (oligo-anovulation and/or polycystic ovarian disease assessed by ultrasound) (17). Hepatic steatosis was measured using high resolution ultra-sound B-mode imaging with a convex transducer (frequency of 3.5–5 MHz). For the diagnosis of intellectual disability, cognitive evaluation tests, such as the Wechsler Adult Intelligence Scale (WAIS-IV), were carried out. The diagnosis of anxiety-depressive disorder was made according to the Diagnostic and Statistical Manual of Mental Disorders, fifth edition (DSM-5) and the 10th revision of the International Statistical Classification of Diseases and Related Health Problems (ICD-10) criteria (18). The ultimate diagnosis of the latter two conditions was made after referral to a psychiatry specialist in the centre. Progressive encephalopathy with or without lipodystrophy (PELD) was diagnosed in patients with pathogenic variants on chromosome 11q13 of the BSCL2 gene and developmental regression of motor and cognitive skills in the first years of life, leading to severe progressive neurodegeneration and death in the first decade of life (19).
2.4 Anthropometry and body composition analysis
Height and weight were verified with digital scales and a stadiometer after 12 h overnight fasting and body mass index (BMI) was calculated. Waist and hip circumferences were determined using a soft tape measure by a single examiner. Waist circumference was calculated taking as a reference the superior border of the iliac crest, and hip circumference taking as a reference the greater trochanter. The patients’ skinfolds were measured using a Lange skinfold calliper (Cambridge Scientific Industries, Cambridge, MD, USA) in the same hemibody by a single examiner. The mean of three consecutive determinations was obtained.
Determination of fat mass and fat-free mass (FFM), both total and segmental, was performed using Dual-energy X-ray absorptiometry (DXA) with a Lunar DPX model (GE Healthcare Lunar, Madison, WI, USA) between 8:30-10:30 a.m. after 12 h of overnight fasting, avoiding previous excessive physical effort.
2.5 Analytical measurements
Laboratory results associated to metabolic complications were documented. Blood samples were taken between 8:00-9:00 a.m. after 12 h overnight fasting. Glucose, creatinine, creatinine kinase, triglycerides and total and fractionated cholesterol levels were measured by standardised methods with appropriate quality control and quality assurance procedures. Aspartate aminotransferase (AST), alanine aminotransferase (ALT) and γ-glutamyl transpeptidase (GGT) were determined with enzymatic methods on an ADVIA analyser (Siemens, Bayer Diagnostics, Tarrytown, NY, USA). Glycated haemoglobin (HbA1c) was measured with ion-exchange high-performance liquid chromatography (Bio-Rad Laboratories Inc., Hercules, CA, USA). Plasma insulin concentrations were determined in duplicate by chemiluminescence using a commercial kit (Nichols Institute, San Juan Capistrano, CA, USA). The homeostatic model assessment for insulin resistance (HOMA-IR) was calculated using the following formula: (fasting glucose [mg/dL]/fasting insulin [mIU/L])/405. Plasma leptin levels and C-peptide were determined by enzyme-linked immunosorbent assay (ELISA) (DRG International Inc., Springfield, NJ, USA).
2.6 Molecular analysis
The search for variants in 26 genes involved in the aetiology of congenital lipodystrophies was made in all the patients evaluated in the centre by next-generation sequencing (NGS) (Ion torrent System, Thermo Fisher Scientific, Waltham, MA, USA) of the entire coding region of the genes and the flanking intronic regions (ADRA2A, AGPAT2, AKT2, BANF1, BLM, BSCL2, CAV1, CIDEC, ERCC6, ERCC8, FBN1, KCNJ6, LIPE, LMNA, MFN2, PCYT1A, PIK3R1, PLIN1, POLD1, POLR3A, PPARG, PSMB8, PTRF, SPRTN, WRN, ZMPSTE24). Patients with benign variants were discarded using databases such as Varsome, ClinVar, gnomAD and dbSNP. In cases in which no results were obtained, SIFT and Polyphen2 were used. If no reported damaging/deleterious variants were found in the databases evaluated, the characteristic phenotype of the disease was taken into consideration. Supplementary Table 1 lists the different variants in the genes that cause the disease in the patients with genetic lipodystrophy included in the study.
2.7 Statistical analysis
Data are expressed as the mean ± standard deviation (SD), median and interquartile range (IQR) or as n (%) values. For each continuous variable, the hypothesis of a normal distribution was verified by the Kolmogorov-Smirnov test. The χ2 test and Fisher’s exact test were used to compare qualitative variables in two groups. The t test or Mann-Whitney test were used to compare a quantitative variable in two groups. The analysis of covariance was also conducted to eliminate the possible modifying effect of certain covariates such as age and gender. Time to the onset of phenotype, time to the diagnosis of lipodystrophy, DM and hypertriglyceridaemia, and overall survival were described using a Kaplan-Meier curve. Log-rank tests were conducted to make the comparisons between patients with generalised and partial lipodystrophy. The level of significance was set at p < 0.05. All statistical analyses were performed with the SPSS 22.0 program (Chicago, IL, USA).
3 Results
3.1 Demographic and epidemiological data
A total of 140 patients were evaluated in the reference centre for lipodystrophies in Spain, with a median follow-up of 4.7 (0.5-17.6) years. Of these patients, 102 (72.9%) were women and 38 (27.1%) men (3:1 ratio). Age at first visit ranged from 5 months to 81.9 years of age.
A total of 117 (83.4%) patients presented genetic lipodystrophy and 23 (16.4%) acquired. According to the pedigree analysis, 62 index cases led to the detection of the rest of their affected relatives after cascade testing. As far as fat distribution is concerned, 108 (77.1%) patients presented partial lipodystrophy and 32 (22.9%) generalised lipodystrophy. As for the different lipodystrophy subtypes, 17 (12.1%) patients had congenital generalised lipodystrophy (CGL), 92 (65.7%) FPLD, 7 (5.0%) acquired generalised lipodystrophy (AGL), 16 (11.4%) acquired partial lipodystrophy (APL) and 8 (5.7%) patients had a progeroid syndrome. The most frequent disease was FPLD type 2 (57.9%), followed by Barraquer-Simons syndrome (10.7%) and CGL type 2 (8.6%). More detailed data on the distribution of the different lipodystrophy subtypes can be seen in Figure 1.
The estimated prevalence of all lipodystrophy cases in Spain was 2.95 cases/million. When separated into generalised and partial lipodystrophy, the estimated prevalence was 0.51 and 2.28 cases/million, respectively (1.94 cases/million for FPLD, 0.34 cases/million for APL, 0.36 cases/million for CGL and 0.15 cases/million for AGL) (Figure 2).
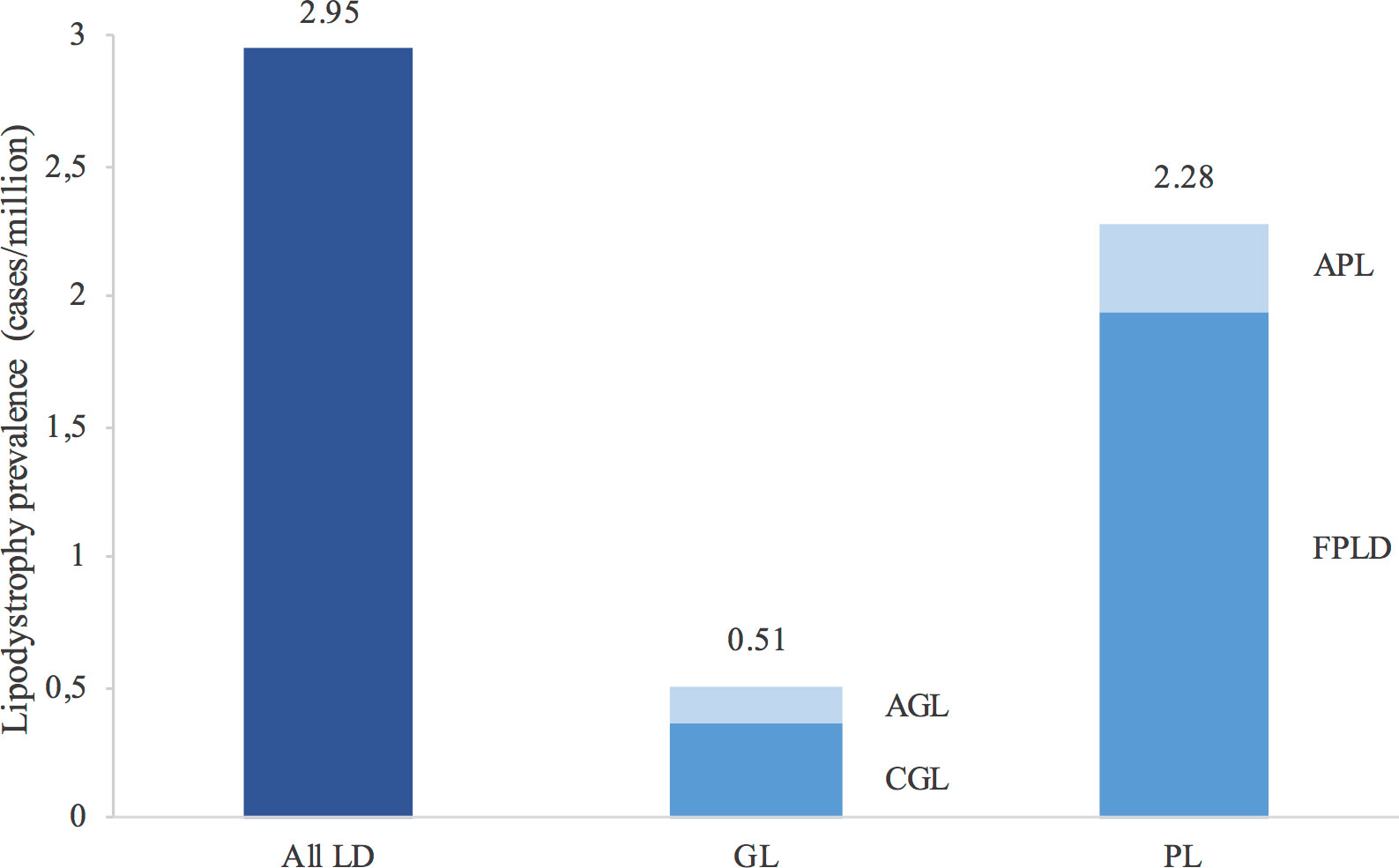
Figure 2 Estimated prevalence of lipodystrophies in Spain. Values above the bars report the prevalence. The estimated prevalence was calculated taking into account the population in Spain in 2020 according to the Instituto Nacional de Estadística (Spanish National Statistics Institute). Familial partial lipodystrophy type 1 has not been taken into account in the calculation of prevalence. LD, lipodystrophies; GL, generalised lipodystrophy; PL, partial lipodystrophy; AGL, acquired generalised lipodystrophy; CGL, congenital generalised lipodystrophy; APL, acquired partial lipodystrophy; FPLD, familial partial lipodystrophy.
In order to properly evaluate the natural course and comorbidities of lipodystrophies, subjects with progeroid syndrome, four prepuberal subjects with FPLD and 13 patients with FPLD and no data were discarded from the following analysis and, therefore, a total of 115 patients (91 with partial lipodystrophy and 24 with generalised lipodystrophy, 77.4% women, 45.2 ± 20.0 years of age) were subsequently analysed. The presence of women was predominant in the diagnosis of partial lipodystrophy (82.4%) in comparison with generalised lipodystrophy (58.3%) (p = 0.012), and patients with partial lipodystrophy were generally older (49.8 ± 18.4 vs 27.1 ± 15.5 years of age, p <0.0001), which is why the following non-demographic data were calculated after age and gender adjustment.
3.2 Clinical features
The onset of phenotype occurred during childhood in 95.6% of patients with generalised lipodystrophy (4.7 ± 11.5 years of age; 47.8% at birth), and during adolescence-adulthood in 91.7% of patients with partial lipodystrophy (16.5 ± 10.8 years of age), p < 0.0001. The delay in diagnosis was 7.4 ± 8.1 years for patients with generalised lipodystrophy and 23.8 ± 17.1 years for patients with partial lipodystrophy (p < 0.0001) (Figure 3).
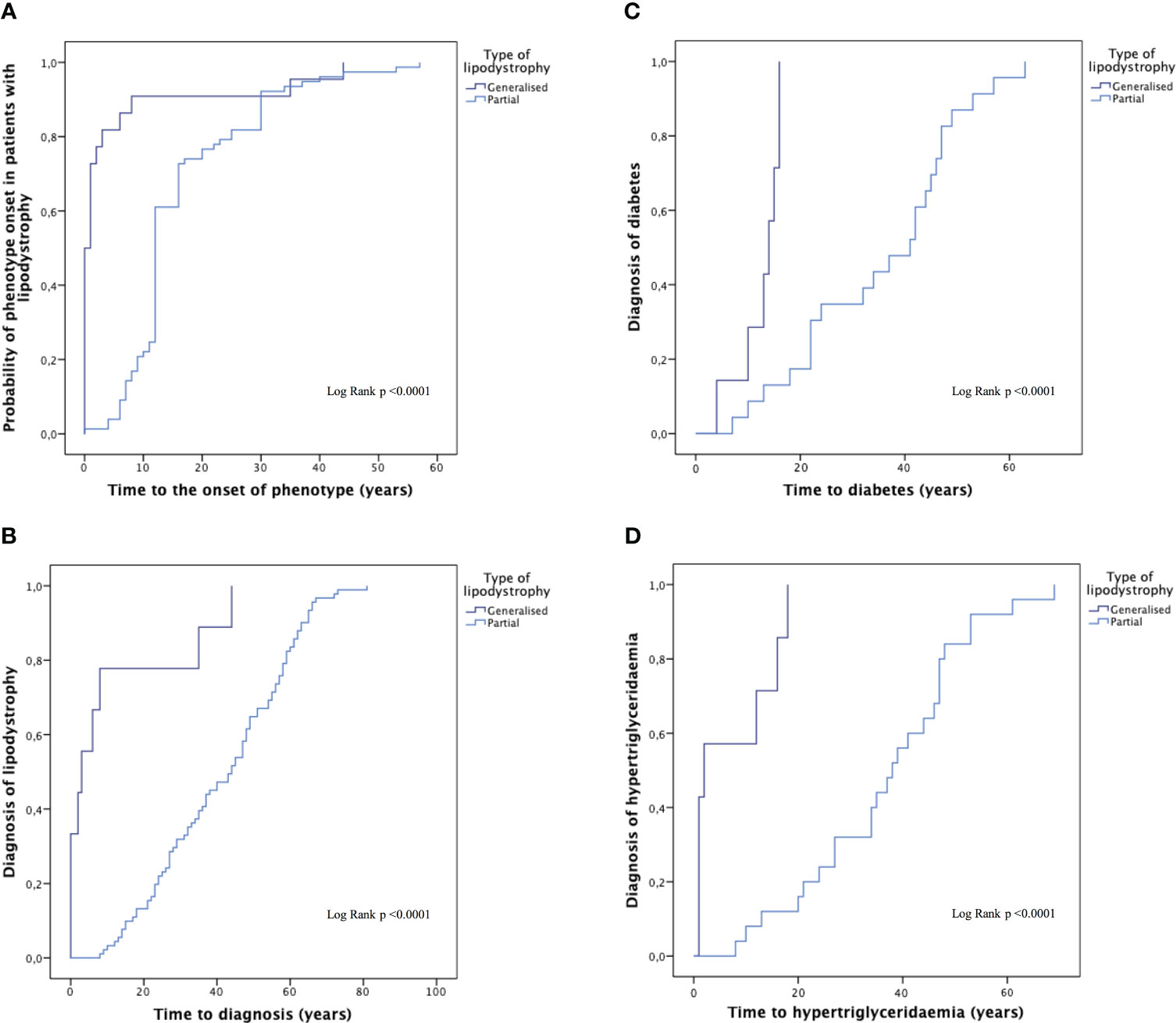
Figure 3 Time to the onset of phenotype, the diagnosis of the disease and the diagnosis of comorbidities in generalised and partial lipodystrophy. Time to the onset of phenotype (A), time to diagnosis of the disease (B) and time to the diagnosis of diabetes mellitus (C) and hypertriglyceridaemia (D) in generalised and partial lipodystrophy. Subjects with progeroid syndrome, prepuberal subjects with familial partial lipodystrophy and patients with familial partial lipodystrophy and no data were excluded from this analysis.
Table 1 summarises the lipodystrophy-associated clinical features of the patients evaluated. Consanguinity was present in 4 out of 24 (16.7%) patients with generalised lipodystrophy and in only one case (1.1%) with partial lipodystrophy. The most frequent clinical features in the overall sample were phlebomegaly (70 patients; 60.9%), followed by muscle hypertrophy (62 patients; 53.9%) and then by signs of insulin resistance such as acanthosis nigricans (48 patients; 41.7%). Among patients with generalised lipodystrophy, phlebomegaly and umbilical protrusion/hernia were the most commonly reported clinical features (70.8% and 62.5%, respectively), whereas in the partial lipodystrophy cohort, phlebomegaly and muscle hypertrophy were the most frequent clinical characteristics reported (58.2% and 59.3%, respectively). Furthermore, there were several distinctive features of the generalised lipodystrophy group in comparison with the partial lipodystrophy group, such as acromegaloid features (7/24 patients [29.2%] with generalised lipodystrophy vs 4/91 patients [4.4%], p = 0.004), prognathism (4/24 patients [16.7%] with generalised lipodystrophy vs 2/91 patients [2.2%] with partial lipodystrophy, p = 0.043) and the presence of umbilical protrusion/hernia (15/24 patients [62.5%] with generalised lipodystrophy vs 4/91 [4.4%] patients with partial lipodystrophy, p <0.0001).
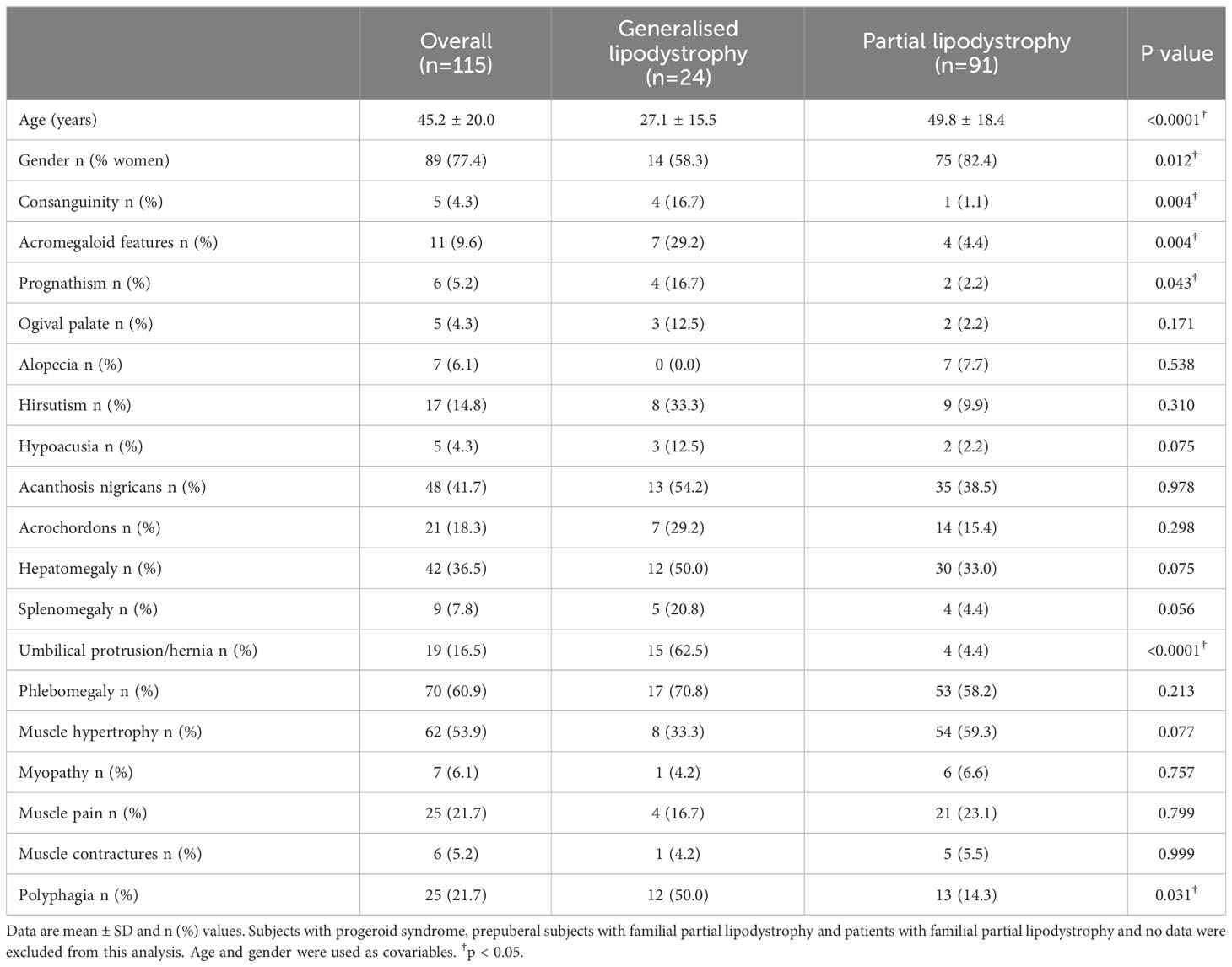
Table 1 Clinical features of patients with generalised and partial lipodystrophy.
3.3 Anthropometric measurements and body composition analysis
After age and gender adjustment, anthropometric parameters such as height, weight, waist and hip circumferences, as well as the skinfolds in the triceps, biceps, subscapular area, suprailiac area and thigh, were significantly lower in the generalised lipodystrophy cohort compared to the partial lipodystrophy cohort. However, no significant differences were found when measuring the skinfold of the calf in both groups. Regarding the DXA measurement of fat content, as expected, it was observed that, in patients with generalised lipodystrophy, this was significantly reduced both in total and in the limbs and trunk, compared to the partial lipodystrophy group. Patients with generalised lipodystrophy also presented higher FFM in the trunk, with no differences in the global analysis or in the upper and lower limbs (Table 2).
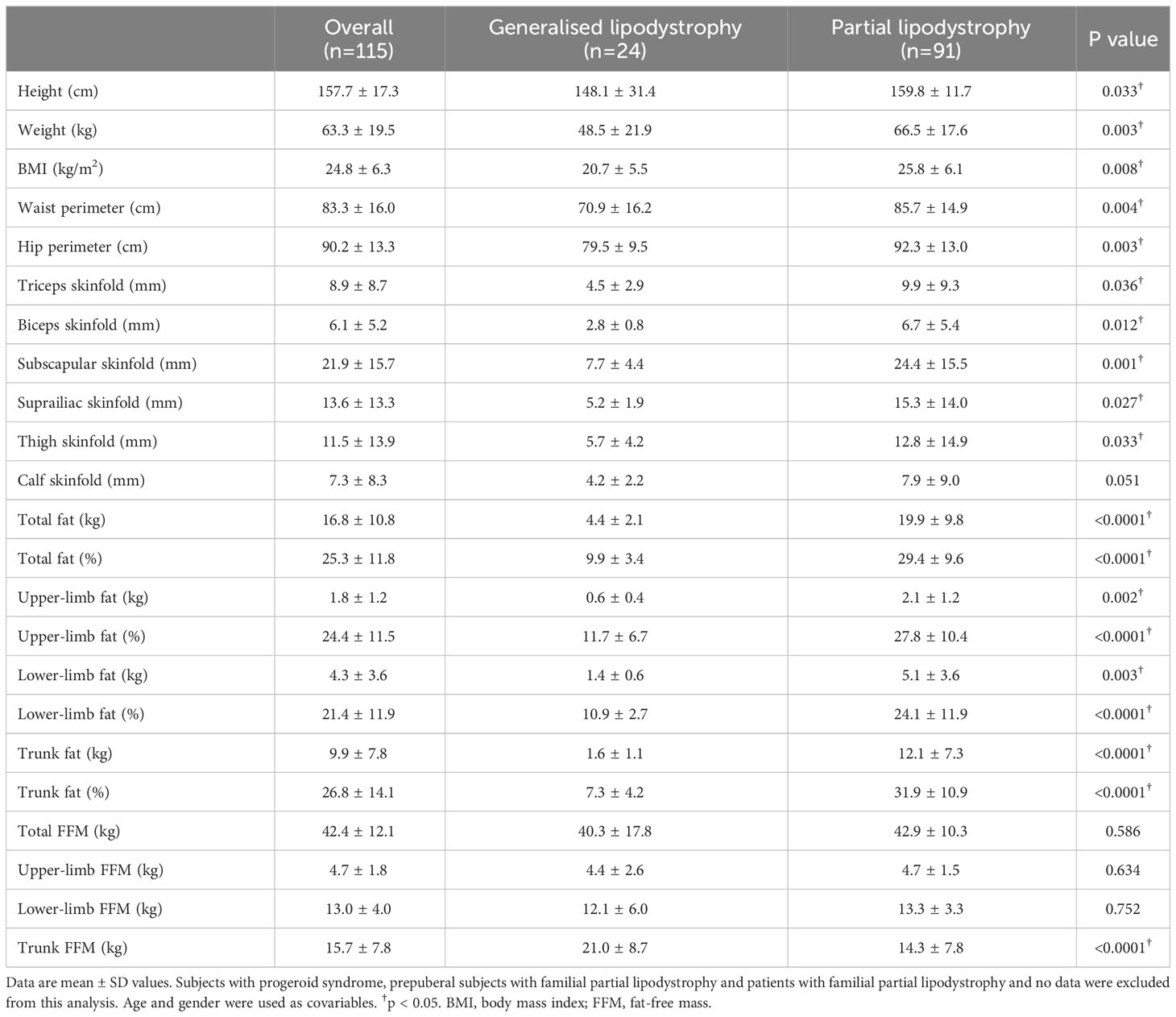
Table 2 Adjusted anthropometric and body composition data determined by Dual-energy X-ray absorptiometry (DXA) of lipodystrophy syndromes.
3.4 Prevalence of main comorbidities
Data on the prevalence of lipodystrophy-associated comorbidities are shown in Table 3. Throughout the follow-up, DM was reported in 50 individuals (43.5%) in the overall sample, with no differences among the groups. Prevalence of DM complications was of 36.0% in the case of nephropathy (16.0% presenting renal failure and the rest of them isolated albuminuria, one patient with partial lipodystrophy required dialysis) and 20.0% in the case of retinopathy and peripheral neuropathy. Mean age at diagnosis of DM (Figure 3) was 30.2 ± 3.1 years in the overall sample (12.6 ± 1.6 years in patients with generalised lipodystrophy and 35.5 ± 3.2 years in patients with partial lipodystrophy; p <0.0001). As regards cardiovascular disease (Table 3), ischemic cardiopathy was present in 14 patients (15.4%) with partial lipodystrophy. Valvulopathy and hypertrophic cardiomyopathy were distinctive comorbidities associated to the generalised lipodystrophy group (eight patients [33.3%] and seven patients [29.2%], respectively) in comparison with the partial lipodystrophy cohort (p <0.0001). As far as neurological disease is concerned, intellectual disability was also more frequent among subjects with generalised lipodystrophy (11 patients [45.8%] vs one patient [1.1%] in the partial lipodystrophy group, p<0.0001). In addition, four (16.7%) patients with generalised lipodystrophy were diagnosed with PELD and, therefore, presented encephalopathy and gait disturbances. Hepatic steatosis was the most common liver abnormality identified, affecting 49 patients (42.6%) in the overall sample, with no differences among the groups. Episodes of acute pancreatitis were reported in three patients (12.5%) with generalised lipodystrophy and seven patients (7.7%) with partial lipodystrophy. As far as gynaecological disorders are concerned, 20 women (24.4%) manifested oligo/amenorrhea (four women [20.0%] in the generalised group and 16 [22.2%] in the partial lipodystrophy group) and 13 (15.9%) were diagnosed with PCOS. Anxiety-depressive disorder was diagnosed in 18 subjects (15.7%) of the overall sample, with 10 of them receiving chronic antidepressant and/or anxiolytic treatment. In all these cases patients related psychological distress due to their physical appearance as one of the causes. Other disorders detected with relative frequency in these patients are also shown in Table 3.
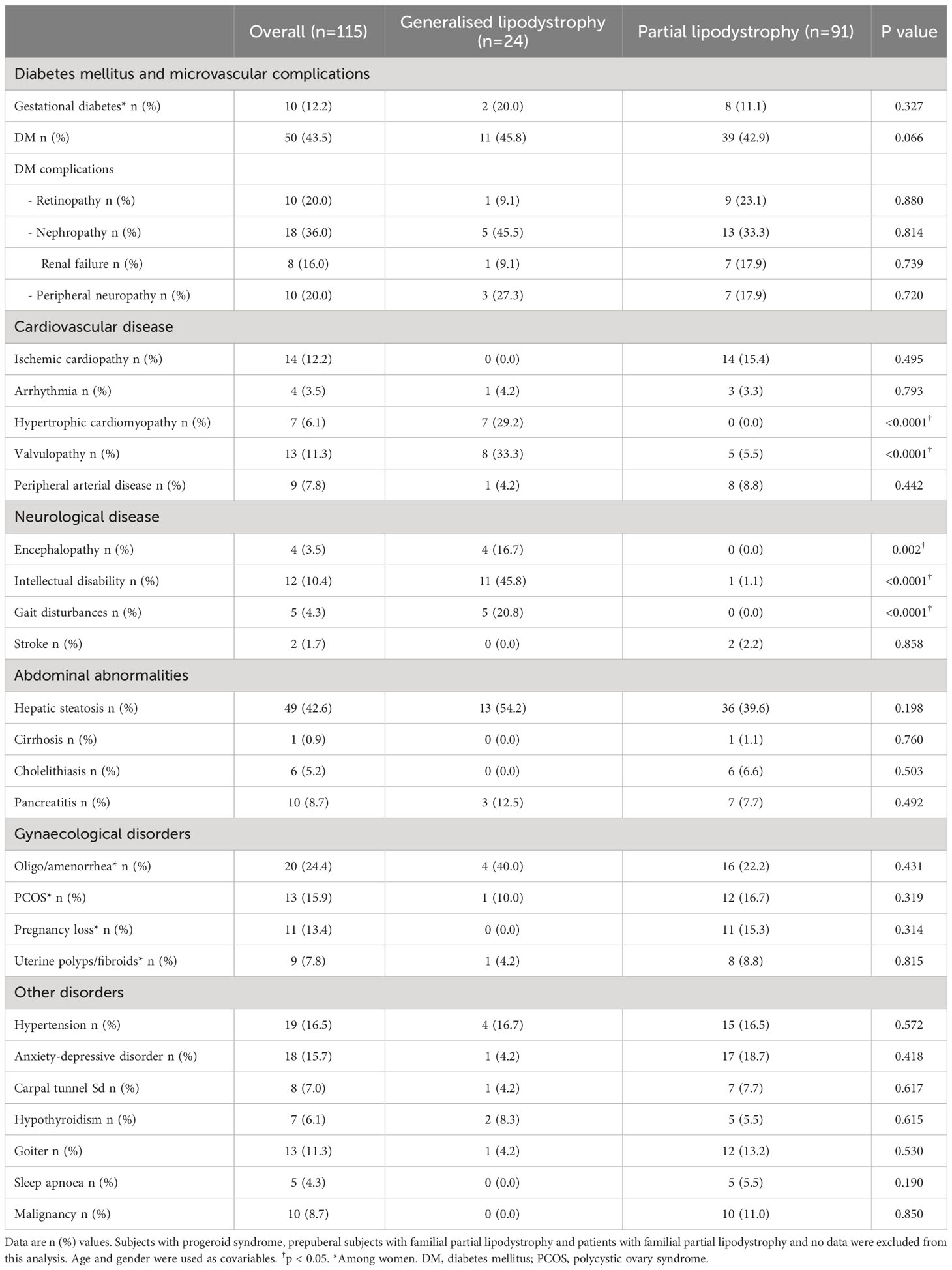
Table 3 Lifetime prevalence of organ abnormalities and comorbidities.
3.5 Analytical parameters
As for analytical measurements at first visit (Table 4), mean HbA1c among individuals with DM was 7.9 ± 2.2% in the overall sample, with HbA1c levels >7% occurring in around half of the patients (26 patients [52.0%]). Analytical parameters such as HOMA-IR showed results related with significant insulin resistance in these subjects. In terms of lipid profile, hypertriglyceridaemia was present both in patients with generalised and partial lipodystrophy. However, severe hypertriglyceridaemia was observed more frequently in the generalised lipodystrophy cohort, with triglyceride levels >500 mg/dl occurring in 7 out of 24 (29.2%) subjects with generalised lipodystrophy vs 5 out of 91 (5.5%) subjects with partial lipodystrophy, p = 0.011. In addition, mean age at diagnosis of hypertriglyceridaemia was 30.4 ± 3.3 in the overall sample (7.3 ± 2.9 years in patients with generalised lipodystrophy and 36.9 ± 3.1 years in patients with partial lipodystrophy; p <0.0001) (Figure 3). On the other hand, low-density lipoprotein cholesterol (LDL-C) and high-density lipoprotein cholesterol (HDL-C) levels were also significantly lower in the generalised group of subjects, with no differences in non-HDL-C or total cholesterol. As expected, lower leptin levels were observed in patients with generalised lipodystrophy in comparison with partial lipodystrophy. No differences were found regarding transaminase levels in both groups. The rest of the metabolic parameters analysed are shown in Table 4.

Table 4 Metabolic parameters of lipodystrophy syndromes at first visit.
3.6 Comparison between the two main subtypes of partial lipodystrophy
Taking into account the differences in the adipose tissue phenotype between the two main partial lipodystrophy subtypes (FPLD and APL), a subanalysis was made in order to compare body composition analysis and metabolic abnormalities (Table 5). Although there were no statistically significant differences in leptin levels or in the percentage of total fat, as expected, fat distribution differed between the two partial lipodystrophy subtypes. Thus, while the percentage of lower-limb fat was lower in the patients with FPLD, patients with APL showed lower trunk fat percentage. Consistent with this, subjects with FPLD also showed lower thigh and calf skinfolds and subjects with APL showed lower subscapular and suprailiac skinfolds (data not shown). A greater BMI was observed in the FPLD population, mainly based on greater FFM (both in total and in the limbs). The presence of phlebomegaly and muscle hypertrophy were also especially characteristic of the FPLD cohort (observed in 64.0% and 70.7% of these subjects, respectively). While DM was diagnosed in 37 (49.3%) patients with FPLD, it was only diagnosed in two (12.5%) patients with APL (p = 0.006), with worse metabolic control in the FPLD population (HbA1c 7.7 ± 2.2 vs 6.9 ± 0.0 in subjects with APL, p = 0.029). Although no statistically significant differences were found regarding organ abnormalities, peripheral arterial disease was only diagnosed in the FPLD cohort (10.7%) and a tendency towards greater triglyceride levels and towards a greater prevalence of hepatic steatosis was also observed for the FPLD subjects (44.0% vs 18.8% in comparison with patients with APL, p = 0.052). In addition, PCOS was likewise only diagnosed in women with FPLD (20.0%).
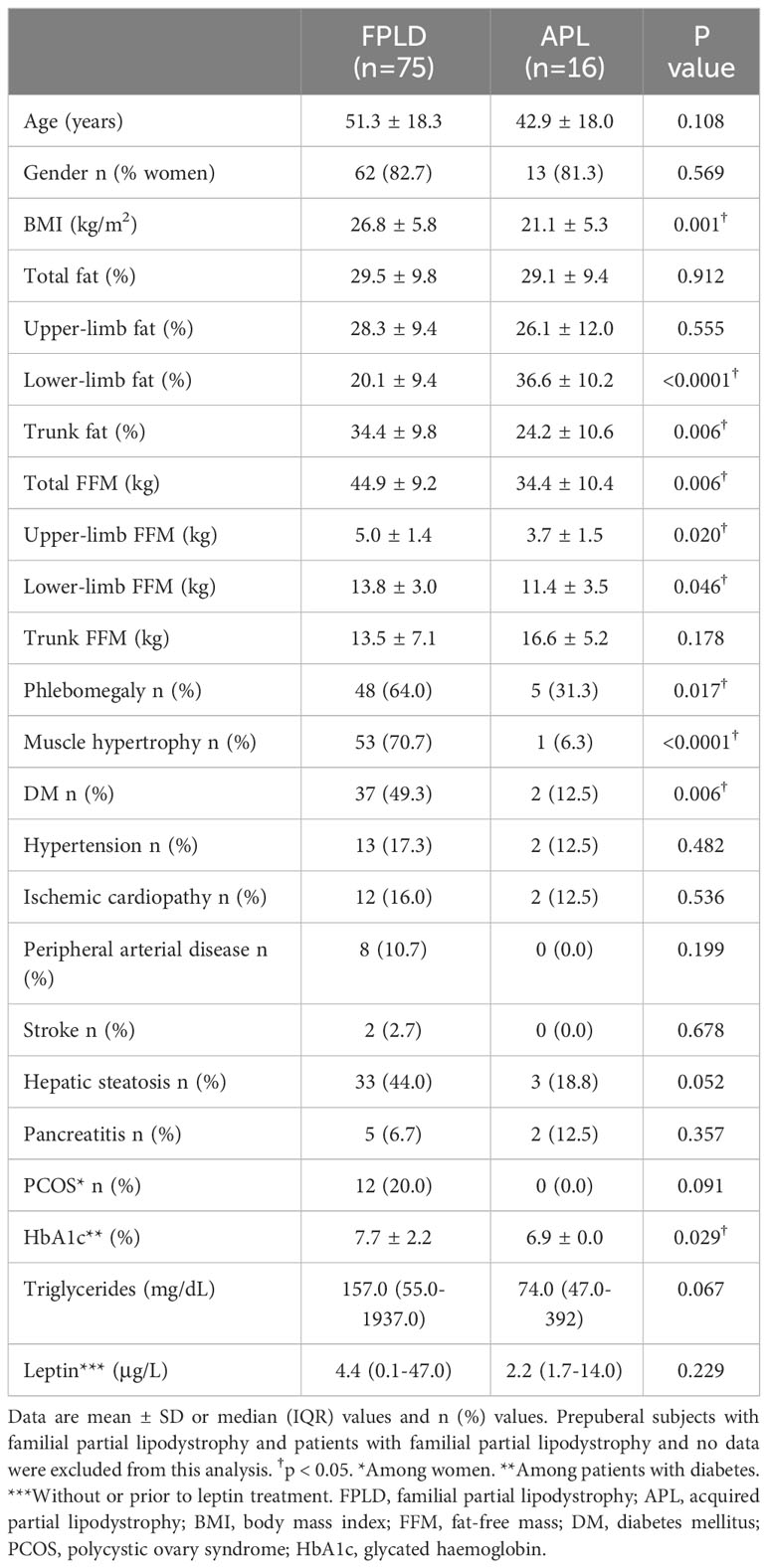
Table 5 Comparison of body composition and metabolic abnormalities between patients with familial partial lipodystrophy and acquired partial lipodystrophy.
3.7 Overall survival
During the follow-up, there were a total of eight deaths (6.8%); four (3.4%) in patients with generalised lipodystrophy and four (3.4%) in patients with partial lipodystrophy. Mortality status was unknown for 15 patients. Overall mean time to death was 83.8 ± 2.5 years (Figure 4). Furthermore, mean time to death was shorter among patients with generalised lipodystrophy in comparison to patients with partial lipodystrophy (55.3 ± 3.8 vs 86.2 ± 2.3 years, respectively; p <0.0001).
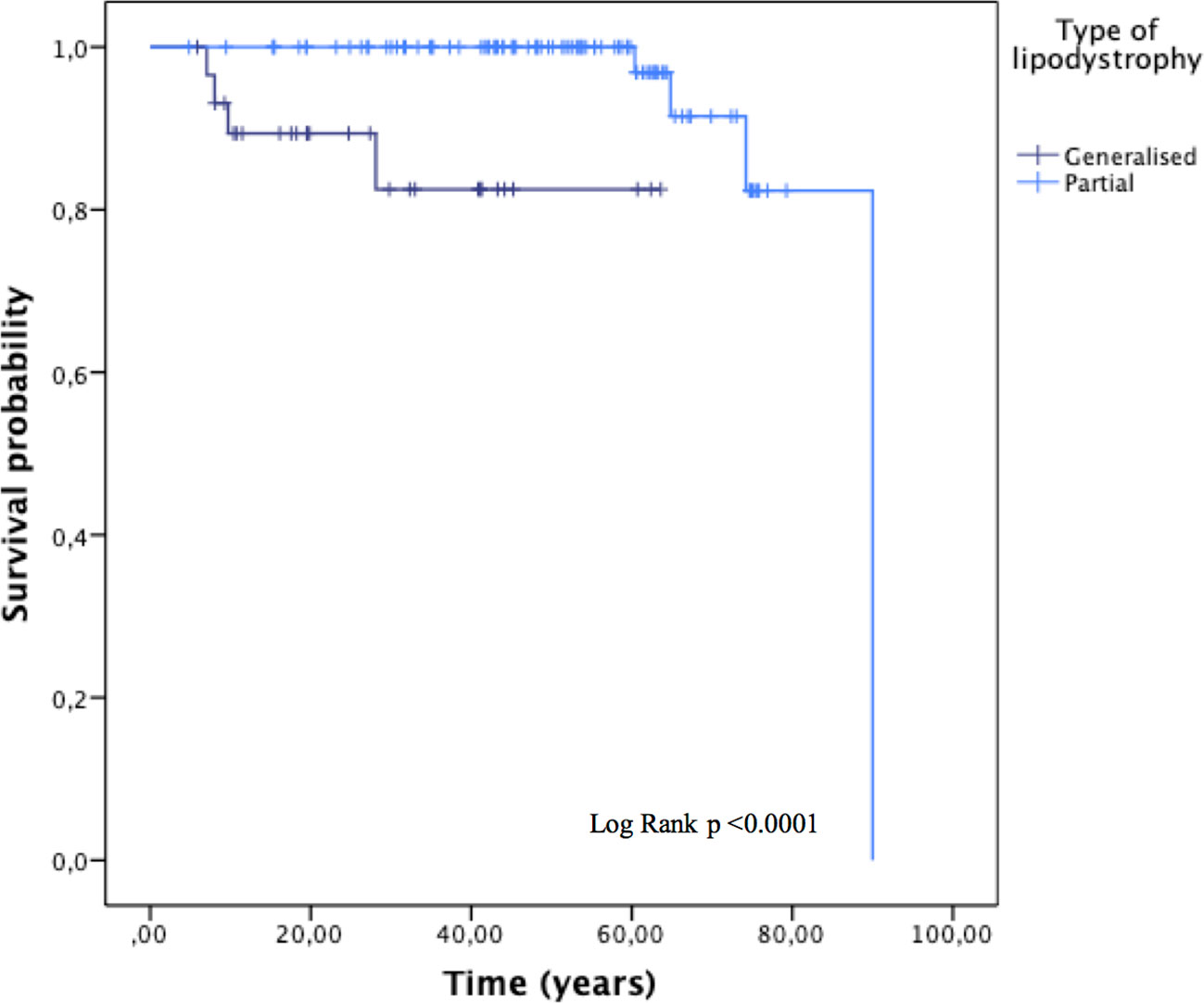
Figure 4 Overall survival, stratified by the type of lipodystrophy (generalised or partial). Subjects with progeroid syndrome, 13 patients with familial partial lipodystrophy and no data and four prepuberal subjects with familial partial lipodystrophy according to molecular analysis in whom the disease phenotype has not yet been developed, were excluded from this analysis.
The causes of death of the patients with generalised lipodystrophy (CGL and AGL) were fundamentally respiratory tract infections. The three patients with pathogenic variants in the BSCL2 gene who died presented PELD and died in the context of neurodegeneration, the deterioration of their general state due to respiratory sepsis or due to a status epilepticus not reversible with anticonvulsants. Among the patients with partial lipodystrophy, two of them died due to colorectal and gastric cancer and the other two due to myocardial infarction. More details are shown in Table 6.
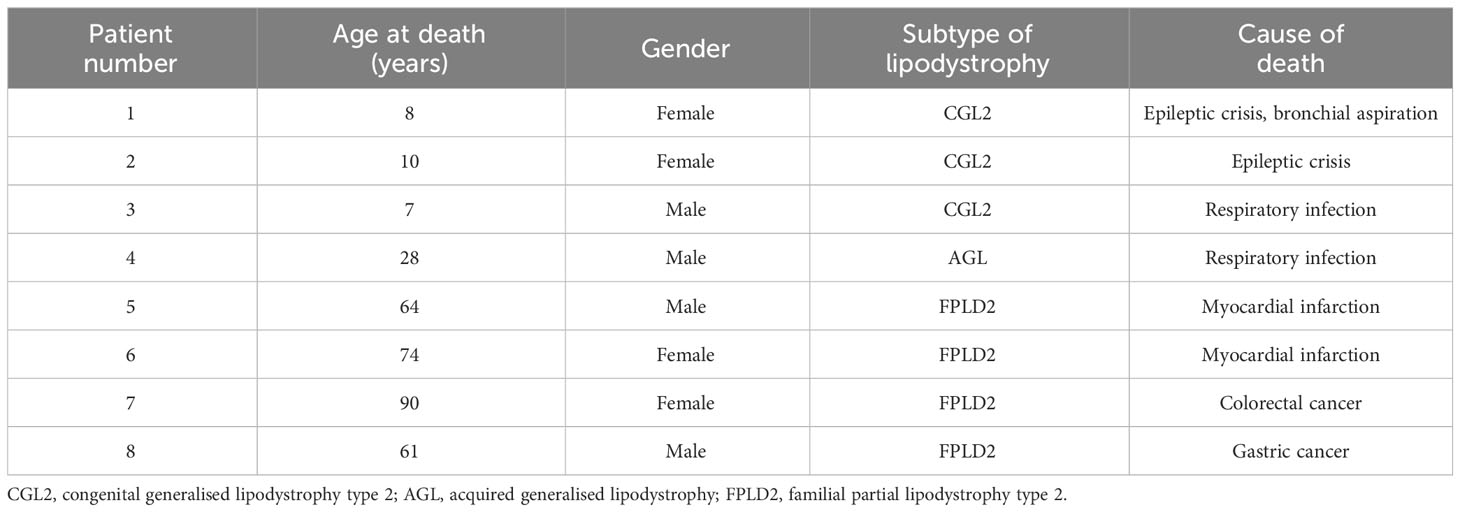
Table 6 Causes of mortality of lipodystrophy patients.
4 Discussion
The current study offers a comprehensive report of the status of lipodystrophy syndromes in Spain and adds more information about their natural course and comorbidities depending on fat distribution in order to contribute to increasing knowledge of these ultra-rare disorders.
In the current analysis, the estimated prevalence of lipodystrophies in Spain, according to the patients referred to its reference centre, was considered to be 2.95 cases/million (0.51 cases/million for generalised lipodystrophy [AGL and CGL] and 2.28 cases/million for partial lipodystrophy [APL and FPLD]). This is in line with a previous study in which, by evaluating the prevalence of lipodystrophy syndromes from five EMR database searches and four literature searches (also excluding HIV-related lipodystrophies), a range of prevalence for all lipodystrophies of 1.3–4.7 cases/million (for generalised lipodystrophy of 0.2–1.0 cases/million, and for partial lipodystrophy of 1.7–2.8 cases/million) was estimated (3). In a more recent analysis, using not only clinical but also molecular searches in the Geisinger Health System and MarketScan databases, a much higher prevalence for lipodystrophy syndromes was reported (47.3 cases/million in global and 1:7,588 for autosomal dominant FPLD) (4). One limitation of these database search analyses (in addition to probably including patients with FPLD type 1 without a firm diagnosis) is the possibility that cases of localised lipoatrophy or insulin lipoatrophy were also captured, which may likewise contribute to an increase in these estimated prevalences. There is definitely great difficulty in making a realistic estimate of a group of rare and heterogeneous diseases, and only large and prolonged registries will make it possible to present a more objective picture of the true prevalence.
Initial clinical characteristics were typically identified during childhood for patients with generalised lipodystrophy and during adolescence-adulthood for patients with partial lipodystrophy. In a systematic review, lower mean ages of onset for lipodystrophy have been reported. However, these results may not be directly comparable as the study population was limited to patients ≤ 18 years of age (20). The delay in the diagnosis in our sample was considerable for both groups of patients, but especially for subjects with partial lipodystrophy, taking into account that their particular phenotype may sometimes be subtle, making the diagnosis of the disease challenging (21), and probably due to the lack of recognition and understanding of these syndromes.
The most frequent clinical characteristics in the overall sample were phlebomegaly, muscle hypertrophy and acanthosis nigricans, common features in most lipodystrophy syndromes (1, 21–23). In addition, acromegaloid features, prognathism and the presence of umbilical protrusion/hernia were distinctive characteristics of patients with generalised loss of fat, as previously described in the literature (1, 24). Therefore, these specific clinical findings should be highlighted as important and useful features to take into account when making the clinical and differential diagnosis of these disorders. In addition, these lipodystrophy-associated changes in physical appearance can cause general physical discomfort, substantial psychological distress and, in consequence, have a negative impact on quality of life (25–27). In fact, anxiety-depressive disorder was diagnosed in 15.7% of our overall sample.
Beyond a physical examination, skinfold thickness measurement and DXA can provide information on the pattern of fat loss. As expected, total fat was significantly reduced in patients with generalised lipodystrophy compared to the partial lipodystrophy group. However, although significant differences were also found regarding fat loss in the lower limbs according to DXA (lower in the group of generalised lipodystrophy), skinfold thickness measurement showed no differences regarding calf skinfold in both groups. This could be due to the fact that DXA does not stratify the limbs by segments, but rather assesses the proximal and distal parts of lower limbs as a whole.
On the other hand, patients with generalised lipodystrophy developed their first metabolic abnormalities, such as DM, sooner than subjects with partial lipodystrophy. However, DM was finally reported in almost half of our sample with no differences among the groups. Dyslipidaemia in patients with lipodystrophy is characterised by hypertriglyceridaemia and low levels of HDL-C (28). Although high triglyceride levels were characteristic of both generalised and partial lipodystrophy in our study, severe hypetriglyceridaemia (triglycerides >500 mg/dl) was observed more frequently in the generalised lipodystrophy group and, in addition, it appeared earlier in these subjects, supporting the view that patients with generalised forms of lipodystrophy may have a greater severity in their metabolic complications (1, 5, 26). However, the manifestation of lipodystrophy syndromes is heterogeneous, and cases of partial lipodystrophy with comparable or more severe symptomology have also been documented (29, 30). In addition, not only HDL-C but also LDL-C levels were likewise significantly lower in patients with generalised lipodystrophy in our sample, with no differences in non-HDL-C or total cholesterol. This may suggest that other lipoproteins that are part of non-HDL-C, unlike LDL-C, are likely to be elevated in these patients and the fact that subjects with partial lipodystrophy had higher LDL-C levels could be due to increased intra-abdominal fat deposition. In relation with a greater severity of hypertriglyceridaemia in patients with generalised loss of fat, the prevalence of acute pancreatitis in this group of subjects was 12.5%, in comparison with 7.7% in partial lipodystrophy. Although no statistically significant differences were found, this was considerably higher than in the general population (31, 32) and in accordance with what has previously been reported in the literature, with an estimated prevalence of pancreatitis in generalised lipodystrophy of around 12% (6, 13).
Due to the increased prevalence of dyslipidaemia and DM, patients with lipodystrophy also appear to be at high risk of ASCVD (28). However, because these disorders are rare, only limited data from some cohorts of patients are available concerning the prevalence of atherosclerotic vascular complications (26). Thus, in our cohort, ischemic cardiopathy was present in 15.4% of cases with partial lipodystrophy and in none with generalised lipodystrophy. In this sense, although it is necessary to take into account the influence of the younger age of the generalised lipodystrophy group, a recent study conducted in 19 subjects with CGL types 1 and 2 quantifying coronary arterial calcification found that despite also evaluating a young CGL population, a quarter of them already presented an altered coronary calcium score, suggesting a potential increase in cardiovascular risk (33). On the other hand, while there are only a few anecdotal reports of ASCVD among CGL, AGL and APL patients, there is more substantial literature on ASCVD among patients with FPLD (26, 34, 35). This is consistent with the results observed in the current study. Thus, the prevalence of atherosclerotic cardiovascular events in these subjects ranged from 14% to 68% throughout the literature (12, 36–39), which, although variable, is also clearly higher than that reported for the general population (40). In addition, a higher frequency of cardiac events has likewise been observed in non-R482 FPLD type 2 carriers in comparison with FPLD type 2 subjects with variants at the 482nd codon (37, 41, 42). In this sense, in our cohort, 32/81 patients with FPLD type 2 harboured R482 pathogenic variants in the LMNA gene. On the contrary, while no differences were found regarding rhythm disturbances between both cohorts, hypertrophic cardiomyopathy and valvulopathy were distinctive comorbidities associated to the generalised lipodystrophy group in our sample, with a prevalence of 29.2% and 33.3%, respectively, which is higher than the previous estimates in the literature for these patients. In this sense, in a multicentre study evaluating a total of 230 subjects with these syndromes, cardiomyopathy was detected in 15.9% of patients with generalised lipodystrophy in comparison with 1.0% of patients with partial lipodystrophy (13). In another study focused on patients with CGL, cardiomyopathy was diagnosed in 2 out of 11 patients with CGL type 2 and in none with CGL type 1 or type 4 (6). Regarding liver abnormalities, hepatomegaly is commonly observed in patients with lipodystrophy (43). In a systematic review, it was shown that the rates of hepatomegaly are generally higher in generalised than in partial syndromes (7, 20, 44). However, no significant differences were found between both groups of patients in the present study. In addition, it has also been shown that the severity of metabolic liver disease (including hepatic steatosis) may depend on the type of lipodystrophy, being more severe in generalised lipodystrophy and presenting higher liver enzymes (44, 45). In our study, although hepatic steatosis was the most common liver abnormality identified, affecting 42.6% of the overall sample, its prevalence was lower than previously reported in other countries for both generalised and partial lipodystrophy (61.7%) (13). In addition, no differences were found between both groups of patients in our cohort. Neither were differences observed between generalised and partial lipodystrophy in another study in which the presence of hepatic steatosis was evaluated using magnetic resonance spectroscopy imaging, although in that study the possible influence of youth in the generalised lipodystrophy group on these results is emphasised (46), as is the case in our analysis. Another consideration that could explain the lack of differences between the generalised and partial lipodystrophy groups is the possible influence of the subjects with AGL, who usually have less severe hepatic steatosis than CGL patients (46). In addition, the metreleptin therapy initiated throughout follow-up in 11 patients with generalised lipodystrophy in our cohort should also be taken into consideration, as this treatment improves liver disease (25, 47). In fact, the previously-mentioned study reporting a greater overall prevalence of hepatic steatosis selected patients who have never received metreleptin therapy or other lipodystrophy-specific therapies, which could also explain these differences (13).
Although, as previously mentioned, generalised lipodystrophy is typically associated to a more severe metabolic disease in proportion with the extent of fat loss or dysfunction, it has been demonstrated that it also depends on the regions affected. Thus, while the adipose tissue of the lower body does not correlate with insulin resistance and may even protect against metabolic dysfunction (48), the lipid accumulation within the abdominal adipose tissue has been linked with insulin resistance and metabolic disease (49, 50). In other words, it could also be said that upper-body fat loss is less prone to be associated with metabolic disease than gluteofemoral fat loss. Thus, in the case of APL, unlike FPLD, as can be observed in the current analysis, comorbidities associated with insulin resistance are not usually a feature, unless fat loss extends down to the gluteofemoral adipose depots or when affected patients gain weight and become obese (7).
As for survival, the estimates of mean time to death in this population correspond, surprisingly, to the mean life expectancy for the general population in Spain according to the Spanish National Statistics Institute. However, it was clearly shorter for patients with generalised lipodystrophy, causing a loss of around 30 years of life in these individuals. This is in agreement with a previous study focused on patients with CGL, for whom a life expectancy of 62.9 ± 4.8 years was also calculated (10). Although lower estimates have been found for the generalised lipodystrophy cohort presented in the current analysis, the influence of the presence of PELD in three cases must be taken into consideration in these results, which led to an early death during middle childhood and could bias the results. On the other hand, in contrast with the overall mean time to death reported in the current study (83.8 ± 2.5 years), in another multicentre study analysing survival in a cohort of 230 patients with lipodystrophy from the USA, Turkey and Brazil, mean time to death was 63.9 ± 1.2 years (13). Severe insulin resistance and associated comorbidities (DM, dyslipidaemia, liver abnormalities, kidney disease and cardiovascular disease) are considered to be the major contributors to the severity of the disease (51). However, deaths due to chronic complications of DM and dyslipidaemia were only observed in two patients with FPLD type 2, deceased due to myocardial infarction. Among the factors that may have contributed to these differences in the mean life expectancy and in the lower prevalence of metabolic comorbidities as a cause of death, it could be speculated that the influence of a different lifestyle and the priority use of drugs with demonstrated reduction in cardiovascular events in recent years among different countries may be included. Furthermore, it should be taken into account that of the total number of patients finally evaluated in the comorbidities and survival analyses, 61 were diagnosed through cascade testing after the detection of the index cases with a clearly developed phenotype and, therefore, the metabolic abnormalities leading to death may not yet have developed or evolved in some of these subjects during the follow-up time. In addition, the young age of the patients with generalised lipodystrophy in our cohort may also influence the lack of time to develop end-organ complications and makes it difficult to estimate the actual mean time to death of these patients. In fact, the main cause of early death in patients with generalised lipodystrophy was respiratory infection, which is not surprising considering that three of these patients had PELD (52). However, in our cohort of patients, respiratory tract infection was also the cause of death of a 28-year-old patient with AGL with no neurological involvement of the disease. In fact, beyond neurodegeneration, deterioration of the general state and bronchoaspiration in patients with PELD, a high frequency of deaths due to infections (especially of respiratory origin) has been reported throughout the literature in generalised lipodystrophy, mainly in patients with CGL type 2 (10, 16, 53–55), which leads to the belief that there must be other potential mechanisms involved.
One limitation of this study could be the inclusion of 13 patients treated with metreleptin throughout the follow-up. However, excluding these subjects could also have created a selection bias in our sample population toward patients with less severe disease and, therefore, could have underestimated the prevalence rates of metabolic comorbidities. Nevertheless, the vast majority of the patients evaluated in this analysis are drug naïve, and the sample reflects real-world data. On the other hand, due to the reasons previously mentioned, the decision was taken to dispense with the FPLD type 1 group, which can contribute to varying the estimated prevalence of comorbidities in the partial lipodystrophy group. In addition, the inclusion of patients with FPLD type 1 would also increase the global prevalence for all lipodystrophies in Spain and make it even higher than previous estimates. Thus, there is a compelling need to better differentiate FPLD type 1, as there are no clear diagnostic criteria and confusion can be created regarding the prevalence of lipodystrophy syndromes in general.
In conclusion, the current longitudinal study shows the natural history and burden of the disease of both generalised and partial lipodystrophies in Spain, which may contribute towards augmenting knowledge of these rare syndromes, providing an initial point of comparison for upcoming results from ongoing prospective studies, such as the ECLip Registry study, among others (56, 57).
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by Red Gallega de Comités de Ética de la Investigación. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
AF-P and DA-V were responsible for the construction and the design of the study and for setting up the methodology. AC-P, LL and FC provided the necessary resources to conduct the study. AF-P, TP-M and ED-L were responsible for the data acquisition. AF-P and MG-V were responsible for the analysis of data and interpretation. AF-P was responsible for drafting the article. DA-V, SS-I, and SC-G were responsible for the review and editing of the manuscript. DA-V was responsible for the final approval of the version submitted. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Instituto de Salud Carlos III (grant PI22/00514) and co-funded by the European Union, and an intramural grant from the Xunta de Galicia, ED431B 2020/37. AF-P receives funding from the Fundación Alfonso Martín Escudero. SS-I was awarded a Research Fellowship, granted by the Asociación Española de Familiares y Afectados de Lipodistrofias (AELIP).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1250203/full#supplementary-material
References
1. Brown RJ, Araujo-Vilar D, Cheung PT, Dunger D, Garg A, Jack M, et al. The diagnosis and management of lipodystrophy syndromes: A multi-society practice guideline. J Clin Endocrinol Metab (2016) 101(12):4500–11. doi: 10.1210/jc.2016-2466
2. Araújo-Vilar D, Santini F. Diagnosis and treatment of lipodystrophy: a step-by-step approach. J Endocrinol Invest (2019) 42(1):61–73. doi: 10.1007/s40618-018-0887-z
3. Chiquette E, Oral EA, Garg A, Araújo-Vilar D, Dhankhar P. Estimating the prevalence of generalized and partial lipodystrophy: findings and challenges. Diabetes Metab Syndr Obes (2017) 10:375–83. doi: 10.2147/DMSO.S130810
4. Gonzaga-Jauregui C, Ge W, Staples J, Van Hout C, Yadav A, Colonie R. et al; Geisinger-Regeneron DiscovEHR collaboration. Clin Mol Prevalence Lipodystrophy an Unascertained Large Clin Care Cohort Diabetes (2020) 69(2):249–58. doi: 10.2337/db19-0447
5. Akinci B, Oral E, Neidert A, Rus D, Cheng W, Thompson-Leduc P, et al. Burden of illness associated with generalized lipodystrophy in leptin replacement therapy-naïve patients: A longitudinal medical chart review study. In: 54th EASD annual meeting 2018. Berlin, Germany: Springer (2018). p. 296.
6. Akinci B, Onay H, Demir T, Ozen S, Kayserili H, Akinci G, et al. Natural history of congenital generalized lipodystrophy: A nationwide study from Turkey. J Clin Endocrinol Metab (2016) 101(7):2759–67. doi: 10.1210/jc.2016-1005
7. Misra A, Peethambaram A, Garg A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: report of 35 cases and review of the literature. Med (Baltimore) (2004) 83(1):18–34. doi: 10.1097/01.md.0000111061.69212.59
8. Ajluni N, Meral R, Neidert AH, Brady GF, Buras E, McKenna B, et al. Spectrum of disease associated with partial lipodystrophy: lessons from a trial cohort. Clin Endocrinol (Oxf) (2017) 86(5):698–707. doi: 10.1111/cen.13311
9. Akinci B, Onay H, Demir T, Savas-Erdeve Ş, Gen R, Simsir IY, et al. Clinical presentations, metabolic abnormalities and end-organ complications in patients with familial partial lipodystrophy. Metabolism (2017) 72:109–19. doi: 10.1016/j.metabol.2017.04.010
10. Lima JG, Nobrega LHC, Lima NN, Dos Santos MCF, Silva PHD, Baracho MFP, et al. Causes of death in patients with Berardinelli-Seip congenital generalized lipodystrophy. PloS One (2018) 13(6):e0199052. doi: 10.1371/journal.pone.0199052
11. Zhong ZX, Harris J, Wilber E, Gorman S, Savage DB, O’Rahilly S, et al. Describing the natural history of clinical, biochemical and radiological outcomes of children with familial partial lipodystrophy type 2 (FPLD2) from the United Kingdom: A retrospective case series. Clin Endocrinol (Oxf) (2022) 97(6):755–62. doi: 10.1111/cen.14806
12. Vasandani C, Li X, Sekizkardes H, Brown RJ, Garg A. Phenotypic differences among familial partial lipodystrophy due to LMNA or PPARG variants. J Endocr Soc (2022) 6(12):bvac155. doi: 10.1210/jendso/bvac155
13. Akinci B, Oral EA, Neidert A, Rus D, Cheng WY, Thompson-Leduc P, et al. Comorbidities and survival in patients with lipodystrophy: an international chart review study. J Clin Endocrinol Metab (2019) 104(11):5120–35. doi: 10.1210/jc.2018-02730
14. Guillín-Amarelle C, Sánchez-Iglesias S, Castro-Pais A, Rodriguez-Cañete L, Ordóñez-Mayán L, Pazos M, et al. Type 1 familial partial lipodystrophy: understanding the Köbberling syndrome. Endocrine (2016) 54(2):411–21. doi: 10.1007/s12020-016-1002-x
15. American Diabetes Association Professional Practice Committee. 2. Classification and diagnosis of diabetes: standards of medical care in diabetes-2022. In: Diabetes care, Diabetes Care vol. 45. (2022). p. S17–38. doi: 10.2337/dc22-S002
16. American Diabetes Association Professional Practice Committee. 12. Retinopathy, neuropathy, and foot care: standards of medical care in diabetes-2022. In: Diabetes care, Diabetes Care vol. 45. (2022). p. S185–94. doi: 10.2337/dc22-S012
17. Azziz R, Carmina E, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Futterweit W, et al. Androgen Excess Society. Positions statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society guideline. J Clin Endocrinol Metab (2006) 91(11):4237–45. doi: 10.1210/jc.2006-0178
18. Möller HJ, Bandelow B, Volz HP, Barnikol UB, Seifritz E, Kasper S. The relevance of ‘mixed anxiety and depression’ as a diagnostic category in clinical practice. Eur Arch Psychiatry Clin Neurosci (2016) 266(8):725–36. doi: 10.1007/s00406-016-0684-7
19. Guillén-Navarro E, Sánchez-Iglesias S, Domingo-Jiménez R, Victoria B, Ruiz-Riquelme A, Rábano A, et al. A new seipin-associated neurodegenerative syndrome. J Med Genet (2013) 50(6):401–9. doi: 10.1136/jmedgenet-2013-101525
20. Gupta N, Asi N, Farah W, Almasri J, Barrionuevo P, Alsawas M, et al. Clinical features and management of non-HIV-related lipodystrophy in children: A systematic review. J Clin Endocrinol Metab (2017) 102(2):363–74. doi: 10.1210/jc.2016-2271
21. Garg A. Acquired and inherited lipodystrophies. N Engl J Med (2004) 350(12):1220–34. doi: 10.1056/NEJMra025261
22. Araújo-Vilar D, Sánchez-Iglesias S, Guillín-Amarelle C, Fernández-Pombo A. Practical guidelines for the diagnosis and treatment of infrequent lipodystrophies. Murcia: AELIP (2020). 89 p.
23. Garg A. Gender differences in the prevalence of metabolic complications in familial partial lipodystrophy (Dunnigan variety). J Clin Endocrinol Metab (2000) 85(5):1776–82. doi: 10.1210/jcem.85.5.6605
24. Garg A. Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab (2011) 96(11):3313–25. doi: 10.1210/jc.2011-1159
25. Leclercq P, Goujard C, Duracinsky M, Allaert F, L’henaff M, Hellet M, et al. High prevalence and impact on the quality of life of facial lipoatrophy and other abnormalities in fat tissue distribution in HIV-infected patients treated with antiretroviral therapy. AIDS Res Hum Retroviruses (2013) 29(5):761–8. doi: 10.1089/aid.2012.0214
26. Hussain I, Garg A. Lipodystrophy syndromes. Endocrinol Metab Clin North Am (2016) 45(4):783–97. doi: 10.1016/j.ecl.2016.06.012
27. Calabrò PF, Ceccarini G, Calderone A, Lippi C, Piaggi P, Ferrari F, et al. Psychopathological and psychiatric evaluation of patients affected by lipodystrophy. Eat Weight Disord (2020) 25(4):991–8. doi: 10.1007/s40519-019-00716-6
28. Hussain I, Patni N, Garg A. Lipodystrophies, dyslipidaemias and atherosclerotic cardiovascular disease. Pathology (2019) 51(2):202–12. doi: 10.1016/j.pathol.2018.11.004
29. Oral EA, Gorden P, Cochran E, Araújo-Vilar D, Savage DB, Long A, et al. Long-term effectiveness and safety of metreleptin in the treatment of patients with partial lipodystrophy. Endocrine (2019) 64(3):500–11. doi: 10.1007/s12020-019-01862-8
30. Diker-Cohen T, Cochran E, Gorden P, Brown RJ. Partial and generalized lipodystrophy: comparison of baseline characteristics and response to metreleptin. J Clin Endocrinol Metab (2015) 100(5):1802–10. doi: 10.1210/jc.2014-4491
31. Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology (2013) 144(6):1252–61. doi: 10.1053/j.gastro.2013.01.068
32. Machicado JD, Yadav D. Epidemiology of recurrent acute and chronic pancreatitis: similarities and differences. Dig Dis Sci (2017) 62(7):1683–91. doi: 10.1007/s10620-017-4510-5
33. Feijó BMXCRR, Mendonça RM, Egito EST, Lima DN, Campos JTAM, Lima JG. Coronary arterial calcification in patients with congenital generalised lipodystrophy: A case series. Clin Endocrinol (Oxf) (2022) 97(6):863–6. doi: 10.1111/cen.14800
34. Hoorntje ET, Bollen IA, Barge-Schaapveld DQ, van Tienen FH, Te Meerman GJ, Jansweijer JA, et al. Lamin A/C-related cardiac disease: late onset with a variable and mild phenotype in a large cohort of patients with the lamin A/C p.(Arg331Gln) founder mutation. Circ Cardiovasc Genet (2017) 10(4):e001631. doi: 10.1161/CIRCGENETICS.116.001631
35. Andre P, Schneebeli S, Vigouroux C, Lascols O, Schaaf M, Chevalier P. Metabolic and cardiac phenotype characterization in 37 atypical Dunnigan patients with nonfarnesylated mutated prelamin A. Am Heart J (2015) 169(4):587–93. doi: 10.1016/j.ahj.2014.12.021
36. Hegele RA. Premature atherosclerosis associated with monogenic insulin resistance. Circulation (2001) 103(18):2225–9. doi: 10.1161/01.cir.103.18.2225
37. Treiber G, Flaus Furmaniuk A, Guilleux A, Medjane S, Bonfanti O, Schneebeli S, et al. A recurrent familial partial lipodystrophy due to a monoallelic or biallelic LMNA founder variant highlights the multifaceted cardiac manifestations of metabolic laminopathies. Eur J Endocrinol (2021) 185(4):453–62. doi: 10.1530/EJE-21-0282
38. Bidault G, Garcia M, Vantyghem MC, Ducluzeau PH, Morichon R, Thiyagarajah K, et al. Lipodystrophy-linked LMNA p.R482W mutation induces clinical early atherosclerosis and in vitro endothelial dysfunction. Arterioscler Thromb Vasc Biol (2013) 33(9):2162–71. doi: 10.1161/ATVBAHA.113.301933
39. Kwapich M, Lacroix D, Espiard S, Ninni S, Brigadeau F, Kouakam C, et al. Diamenord–AEDNL Working Group. Cardiometabolic assessment of lamin A/C gene mutation carriers: a phenotype-genotype correlation. Diabetes Metab (2019) 45(4):382–9. doi: 10.1016/j.diabet.2018.09.006
40. Khan MA, Hashim MJ, Mustafa H, Baniyas MY, Al Suwaidi SKBM, AlKatheeri R, et al. Global epidemiology of ischemic heart disease: results from the global burden of disease study. Cureus (2020) 12(7):e9349. doi: 10.7759/cureus.9349
41. Eldin AJ, Akinci B, da Rocha AM, Meral R, Simsir IY, Adiyaman SC, et al. Cardiac phenotype in familial partial lipodystrophy. Clin Endocrinol (Oxf) (2021) 94(6):1043–53. doi: 10.1111/cen.14426
42. Mosbah H, Vatier C, Boccara F, Jéru I, Lascols O, Vantyghem MC, et al. Looking at new unexpected disease targets in LMNA-linked lipodystrophies in the light of complex cardiovascular phenotypes: implications for clinical practice. Cells (2020) 9(3):765. doi: 10.3390/cells9030765
43. Polyzos SA, Perakakis N, Mantzoros CS. Fatty liver in lipodystrophy: A review with a focus on therapeutic perspectives of adiponectin and/or leptin replacement. Metabolism (2019) 96:66–82. doi: 10.1016/j.metabol.2019.05.001
44. Handelsman Y, Oral EA, Bloomgarden ZT, Brown RJ, Chan JL, Einhorn D, et al. American Association of Clinical Endocrinologists. The clinical approach to the detection of lipodystrophy - an AACE consensus statement. Endocr Pract (2013) 19(1):107–16. doi: 10.4158/endp.19.1.v767575m65p5mr06
45. Misra A, Garg A. Clinical features and metabolic derangements in acquired generalized lipodystrophy: case reports and review of the literature. Med (Baltimore) (2003) 82(2):129–46. doi: 10.1097/00005792-200303000-00007
46. Altay C, Seçil M, Adıyaman SC, Özgen Saydam B, Demir T, Akıncı G, et al. Magnetic resonance spectroscopy to assess hepatic steatosis in patients with lipodystrophy. Turk J Gastroenterol (2020) 31(8):588–95. doi: 10.5152/tjg.2020.19114
47. Brown RJ, Oral EA, Cochran E, Araújo-Vilar D, Savage DB, Long A, et al. Long-term effectiveness and safety of metreleptin in the treatment of patients with generalized lipodystrophy. Endocrine (2018) 60(3):479–89. doi: 10.1007/s12020-018-1589-1
48. Zhang M, Hu T, Zhang S, Zhou L. Associations of different adipose tissue depots with insulin resistance: A systematic review and meta-analysis of observational studies. Sci Rep (2015) 5:18495. doi: 10.1038/srep18495
49. Karpe F, Pinnick KE. Biology of upper-body and lower-body adipose tissue–link to whole-body phenotypes. Nat Rev Endocrinol (2015) 11(2):90–100. doi: 10.1038/nrendo.2014.185
50. Mann JP, Savage DB. What lipodystrophies teach us about the metabolic syndrome. J Clin Invest (2019) 129(10):4009–21. doi: 10.1172/JCI129190
51. Cook K, Ali O, Akinci B, Foss de Freitas MC, Montenegro RM, Fernandes VO, et al. Effect of leptin therapy on survival in generalized and partial lipodystrophy: A matched cohort analysis. J Clin Endocrinol Metab (2021) 106(8):e2953–67. doi: 10.1210/clinem/dgab216
52. Sánchez-Iglesias S, Crocker M, O’Callaghan M, Darling A, García-Cazorla A, Domingo-Jiménez R, et al. Celia’s encephalopathy and c.974dupG in BSCL2 gene: a hidden change in a known variant. Neurogenetics (2019) 20(2):73–82. doi: 10.1007/s10048-019-00574-5
53. Van Maldergem L, Magré J, Khallouf TE, Gedde-Dahl T Jr, Delépine M, Trygstad O, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet (2002) 39(10):722–33. doi: 10.1136/jmg.39.10.722
54. Hsu RH, Lin WD, Chao MC, Hsiao HP, Wong SL, Chiu PC, et al. Congenital generalized lipodystrophy in Taiwan. J Formos Med Assoc (2019) 118(1 Pt 1):142–7. doi: 10.1016/j.jfma.2018.02.003
55. Seip M, Trygstad O. Generalized lipodystrophy, congenital and acquired (lipoatrophy). Acta Paediatr Suppl (1996) 413:2–28. doi: 10.1111/j.1651-2227.1996.tb14262.x
56. von Schnurbein J, Adams C, Akinci B, Ceccarini G, D’Apice MR, Gambineri A, et al. European lipodystrophy registry: background and structure. Orphanet J Rare Dis (2020) 15(1):17. doi: 10.1186/s13023-020-1295-y
Keywords: generalised lipodystrophy, partial lipodystrophy, body composition, diabetes mellitus, hypertriglyceridaemia, atherosclerotic cardiovascular disease, mortality
Citation: Fernández-Pombo A, Sánchez-Iglesias S, Castro-Pais AI, Ginzo-Villamayor MJ, Cobelo-Gómez S, Prado-Moraña T, Díaz-López EJ, Casanueva FF, Loidi L and Araújo-Vilar D (2023) Natural history and comorbidities of generalised and partial lipodystrophy syndromes in Spain. Front. Endocrinol. 14:1250203. doi: 10.3389/fendo.2023.1250203
Received: 29 June 2023; Accepted: 10 October 2023;
Published: 16 November 2023.
Edited by:
Milena Teles, University of São Paulo, BrazilReviewed by:
Renan Magalhães Montenegro Jr., Federal University of Ceara, BrazilCynthia Valerio, Instituto Estadual de Diabetes e Endocrinologia Luiz Capriglione, Brazil
Copyright © 2023 Fernández-Pombo, Sánchez-Iglesias, Castro-Pais, Ginzo-Villamayor, Cobelo-Gómez, Prado-Moraña, Díaz-López, Casanueva, Loidi and Araújo-Vilar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Araújo-Vilar, ZGF2aWQuYXJhdWpvQHVzYy5lcw==