 Ya-Ping Fang1†‡
Ya-Ping Fang1†‡ Yu Zhao
Yu Zhao Xin Yang
Xin Yang Xiao-Liang Zhang
Xiao-Liang Zhang- 1Institute of Nephrology, Zhong Da Hospital, Southeast University School of Medicine, Nanjing, Jiangsu, China
- 2Department of Clinical Medicine, Southeast University School of Medicine, Nanjing, Jiangsu, China
Vascular calcification (VC) has emerged as a key predictor of cardiovascular events in patients with chronic kidney disease (CKD). In recent years, an expanding body of research has put forth the concept of accelerated vascular aging among CKD patients, highlighting the significance of vascular cells senescence in the process of VC. Within the milieu of uremia, senescent vascular endothelial cells (VECs) release extracellular microvesicles (MV) that promote vascular smooth muscle cells (VSMCs) senescence, thereby triggering the subsequent osteogenic phenotypic switch and ultimately contributing to the VC process. In addition, senescent vascular progenitor or stem cells with diminished ability to differentiate into VECs and VSMCS, compromise the repair of vascular integrity, on the other hand, release a cascade of molecules associated with senescence, collectively known as the senescence-associated secretory phenotype (SASP), perpetuating the senescence phenomenon. Furthermore, SASP triggers the recruitment of monocytes and macrophages, as well as adjacent VECs and VSMCs into a pro-adhesive and pro-inflammatory senescent state. This pro-inflammatory microenvironment niche not only impacts the functionality of immune cells but also influences the differentiation of myeloid immune cells, thereby amplifying the reduced ability to effectively clear senescent cells of senescent macrophages, promoted calcification of VSMCs. The objective of this paper is to provide a comprehensive review of the contribution of vascular cell senescence to the emergence and advancement of VC. Gaining a comprehensive understanding of the involvement of cellular senescence within the vessel wall is pivotal, especially when it comes to its intersection with VC. This knowledge is essential for advancing groundbreaking anti-aging therapies, aiming to effectively mitigate cardiovascular diseases.
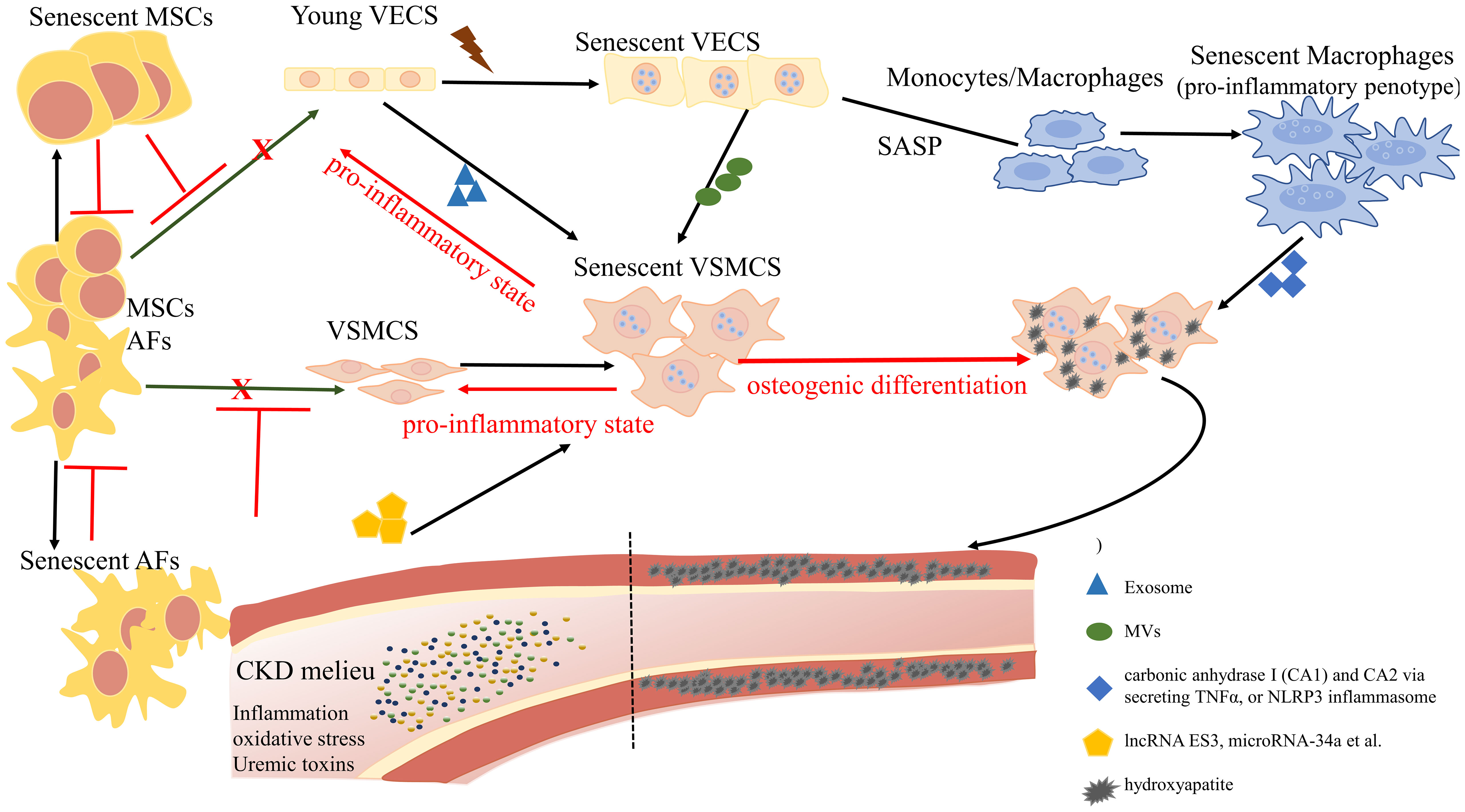
Graphical Abstract Overview of the role of cellular senescence in regulating vascular calcification. The complex landscape of CKD hastens the senescence of vascular endothelial cells (VECs) and undermines their functional integrity, exhibiting a distinctive profile characterized by the reduced expression of nitric oxide (NO) and an elevation in reactive oxygen species (ROS). Senescent VECs release microvesicles (MVs) to trigger senescence and calcification in VSMCs. On the other hand, senescent VECs attract monocytes to the endothelium and induce the proliferation and migration of VSMCs. Pro-inflammatory phenotypic macrophages subsequently promote VSMC calcification by stimulating carbonic anhydrase I (CA1) and CA2 via secreting TNFα, or NLRP3 inflammasome. Mesenchymal stromal cells (MSCs) and adventitial fibroblasts (AFs) exhibit a potential for differentiation, with the ability to transform into VECs and VSMCs, contributing to the replenishment of senescent cells. However, the proliferation and differentiation capacities of senescent MSCs and AFs are diminished.
1 Introduction
Cardiovascular disease is the leading cause of death in patients with chronic kidney disease (CKD), a reality that persists even during the initial CKD stages (CKD stages 1-3). Vascular calcification (VC) emerges as both an independent predictor and a pivotal driving force behind the onset and progression of cardiovascular disease in this patient demographic (1). Despite significant advancements in VC drug development, such as SNF472 and sodium thiosulfate, the incidence of cardiovascular mortality among CKD patients persists at elevated levels.
There is a growing focus on the role of senescent cells, particularly in the context of accelerated vascular aging among CKD patients, with VC emerging as a significant phenotype of this aging process (2). Accumulating data suggests that the transition of pro-calcificatory/osteoblastic phenotype of senescent vascular smooth muscle cells (VSMCs) play a vitol role. However, the deviation between vascular aging and chronological aging still need further invesatigated.
Improving our understanding of the interplay between aging and VC is of utmost importance. To gain a comprehensive understanding of the pathophysiological processes driving VC and to develop prospective therapies for CKD patients with VC, this review explores the current knowledge of VC in CKD, with a particular emphasis on the role of vascular cell senescence and cellular interactions in the progression of VC.
2 Vascular calcification
The type of VC that develops depends on its location, and it commonly takes the following forms: intimal calcification, media calcification, and valve calcification. Intimal calcification often presents as atherosclerosis, primarily impacting larger and medium-sized arteries along with their branches. This condition arises from chronic vascular inflammation and involves the infiltration of vascular smooth muscle cells (VSMCs) and macrophages in the lipid-rich regions of atherosclerotic plaques. Ischemic heart disease stems from the formation, rupture, and obstruction of these plaques within arteries (3). Monckeberg’s sclerosis, also known as medial arterial calcification (MAC), is an age-related degenerative aiflment first proposed by Johann Georg Monckeb in 1903 (4). The hallmark of MAC is the diffuse and continuous deposition of hydroxyapatite in the medial layer (5). The presence of MAC can be observed in the walls of vessels with varying internal diameters, which may experience stiffness. This stiffness often leads to increased pulse wave velocity (PWV), reduced blood flow to the heart (cardiac hypoperfusion), elevated systolic blood pressure, left ventricular hypertrophy, and eventually heart failure (4–6). MAC is frequently associated with abnormalities in calcium and phosphorus metabolism, as well as mineral and bone metabolism, the severity of CKD, dialysis vintage, aging (7), diabetes (5), and rapid progression of medial calcification in patients with end-stage renal disease who are undergoing hemodialysis (8).
In contemporary medical practices, conventional X-rays, ultrasonography, and computed tomography (CT) scans have been used for detecting and monitoring calcification. Additionally, circulating biomarkers, which reflect pathophysiological changes, thus contribute valuable insights. However, despite these advancements, the clinical challenge in the context of CKD persists due to the limited sensitivity of VC diagnosis (9). Previously, it was believed that VC occurred passively, involving the deposition of calcium and phosphate as hydroxyapatite in the arterial wall. However, mounting evidence now suggests that VC is an active, highly regulated, and resembling the bone-forming process known as osteogenesis. This active process is driven by a osteogenic-like phenotypic transformation of VSMCs (5). Beyond, a range of factors contribute to the complex process of VSMCs calcification, including imbalances in the regulation of calcium and phosphate levels, cell death (apoptosis, ferroptosis), diminished presence of calcification inhibitors, the release of vesicles containing mineral-forming components (mineralizing matrix vesicles or MVs), and crosstalk between VSMCs and VECs (10–12). In an environment with high calcium levels, the quantity of calcium-sensing receptors (CaSRs) on the surface of VSMCs decreases, resulting in an increase in intracellular calcium content. This heightened calcium concentration triggers apoptosis in VSMCs, leading to the formation of hydroxyapatite deposits sites from these apoptotic cells. The release of calcium from dying cells perpetuates apoptosis, thus accelerating the vicious cycle of VC (13, 14). Elevated levels of phosphorus have a direct effect, triggering monocytes to synthesize and release tumor necrosis factor-alpha (TNF-α) via the PiT-1 pathway. This initiates an inflammatory state, furthering the transformation of VSMCs into an osteogenic phenotype and the subsequent calcification process (15). As a result, VSMCs shift toward a cell phenotype resembling that of osteoblasts, leading to an increase in the expression of regulatory proteins like Runx2, Osterix, Msx2, and Sox9. Additionally, these cells release mineralized MVs while reducing the presence of endogenous calcification inhibitors within these vesicles, such as MGP (16–18). Moreover, VSMCs produce alkaline phosphatase (ALP), which deactivates the mineralization-inhibiting effects of pyrophosphate, releasing free phosphate that serves as a building block for VC (19).
Recent advancements highlight abnormalities in epigenetic modifications (such as DNA methylation, histone modifications, and noncoding RNAs), ferroptosis of VSMCs, autophagy dysfunction, and senescence as active participants in the cellular processes leading to VC (12, 20, 21). Extensive studies have delved into the involvement of senescent vascular cells, including VECs, VSMCs, and macrophages, in propagating senescence through paracrine mechanisms (e.g., SASP and MV) (22–24). Through immunostaining and analysis of protein expression in abdominal aortic samples from 61 kidney transplant recipients, it was disclosed that CKD patients exhibiting coronary artery calcification, showed notably elevated levels of p16. Remarkably, the count of cells displaying both p16 and SA-β-Gal-positivity exhibited a positive correlation with the severity of medial calcification in CKD (25). Furthermore, eliminating senescent cells yielded significant reductions in aortic aging markers and osteogenic marker, osterix (26). The burden of senescent cells may drive the VC process.
3 Cellular senescence
Cellular senescence forms the cornerstone of organismal aging. Professor Leonard Hayflick’s breakthrough discovery in the 1960s revealed that human diploid cells, when cultured in vitro, possess a finite capacity for proliferation, eventually culminating in an irreversible state of growth arrest irrespective of culturing conditions. This phenomenon, termed cellular senescence, defines the maximum number of cellular divisions, referred to as the Hayflick limit (27, 28). Cellular senescence entails the irreversible and enduring cessation of cell division, accompanied by observable morphological alterations such as cell flattening, expanded cytoplasm, and nucleus changes. Cellular senescence is currently divided into two main categories: replicative senescence (RS) and stress-induced premature senescence (SIPS). Replicative senescence occurs during normal physiological processes when cells continuously replicate, leading to telomere shortening and cell exit from the cell cycle, which also represents a cause for the Hayflick limit. However, replication is not the only factor causing telomere shortening, environmental stress can indeed lead to telomere shortening (29). SIPS refers to aging that occurs in cells before the Hayflick limit in response to various environmental stimuli. Stimulating factors include excessive activation of oncogenes, loss of tumor suppressor factors, damage to DNA or chromosome structures, mitochondrial dysfunction, oxidative stress, wound healing, various cytokines, and intercellular signal transduction (30). This process is also marked by the expression of the senescence-associated secretory phenotype (SASP), which bridges cellular senescence with tissue dysfunction and organismal aging (27, 31). The SASP components exhibit strong autocrine activity and can also transmit senescence via paracrine signaling to adjacent cells. This characterization creates an pro-inflammatory microenvironment, resulting in a persistent low-level chronic inflammation, a state known as inflammaging, and causing age-related diseases (32). Indeed, senescent cells play a dual role: they not only generate a pro-inflammatory microenvironment primarily through the senescence-associated SASP but also impede stem cell proliferation and regeneration. This dual action serves to blunt the tissue regeneration and repair processes, further complicating the overall health and recovery mechanisms (33). On one hand, senescent cells display anti-proliferative attributes that prevent the propagation of damaged cells, thus averting the development of potential malignant cells. Additionally, senescent cells release SASP factors that extend senescence to neighboring healthy cells while recruiting immune cells for removal (34). On the other hand, senescent cells express BCL-2 family proteins that confer resistance to apoptosis (35), allowing them to amass within the body as age advances, and ultimately leading to the progression of age-related diseases.
Targeting the elimination of senescent cells as a strategy to alleviate age-related diseases has been a focal point for scientists. The use of senolytics, a cocktail designed to selectively clear senescent cells, has shown promising outcomes. By reducing the number of senescent cells, this approach not only ameliorates physical dysfunction but also decreases mortality hazard and enhances healthspan, providing direct evidence that cellular senescence is a causative factor in age-related diseases (36). In the field of Geroscience, researchers advocate for a shift in perspective by focusing on the aging process itself rather than individual diseases. This approach holds the potential to delay or even halt the development of age-related diseases (37). However, the complexity of cellular senescence poses challenges, particularly in identifying specific biomarkers for senescent cell detection. Researchers have made strides in this regard, developing tools such as DNAm PhenoAge, inflammatory aging clock (iAge), and others, to predict physiological age. Since the introduction of the epigenetic clock by Prof. Horvath (38–40), various methods combining multiple markers have been employed to confirm the occurrence of cellular senescence at the cellular level. These markers encompass senescence-associated β-galactosidase (SA-β-gal), cell cycle-dependent protein kinase inhibitors (p16, p21, p53), cell proliferation indicators (Ki67), senescence-associated heterochromatin foci (SAHF), DNA damage response, and SASP (41–43). The cell cycle arrest that characterizes cellular senescence is primarily governed by the p53/p21 and p16/RB pathways (44, 45). Notably, p53 functions upstream of p21, while p16 and p21 belong to the cyclin-dependent kinase inhibitor (CDKI) family, inhibiting cyclinD-CDK4/6 and cyclinE/A-CDK2, respectively. This action binds minimally phosphorylated RB closely to E2F, halting cells in the G1 phase (46) (Figure 1). It is important to acknowledge that p21 is mainly expressed in the early stages of senescence induction and does not persist, while p16 is not highly expressed in all senescent cells (49–51). While SASP is a prevalent feature of senescent cells, its composition is not entirely fixed. Components of SASP differ due to variations in cell characteristics, stimulus methods, and senescence duration (52). Nevertheless, the significance of employing a combination of multiple markers to identify cellular senescence cannot be emphasized enough.
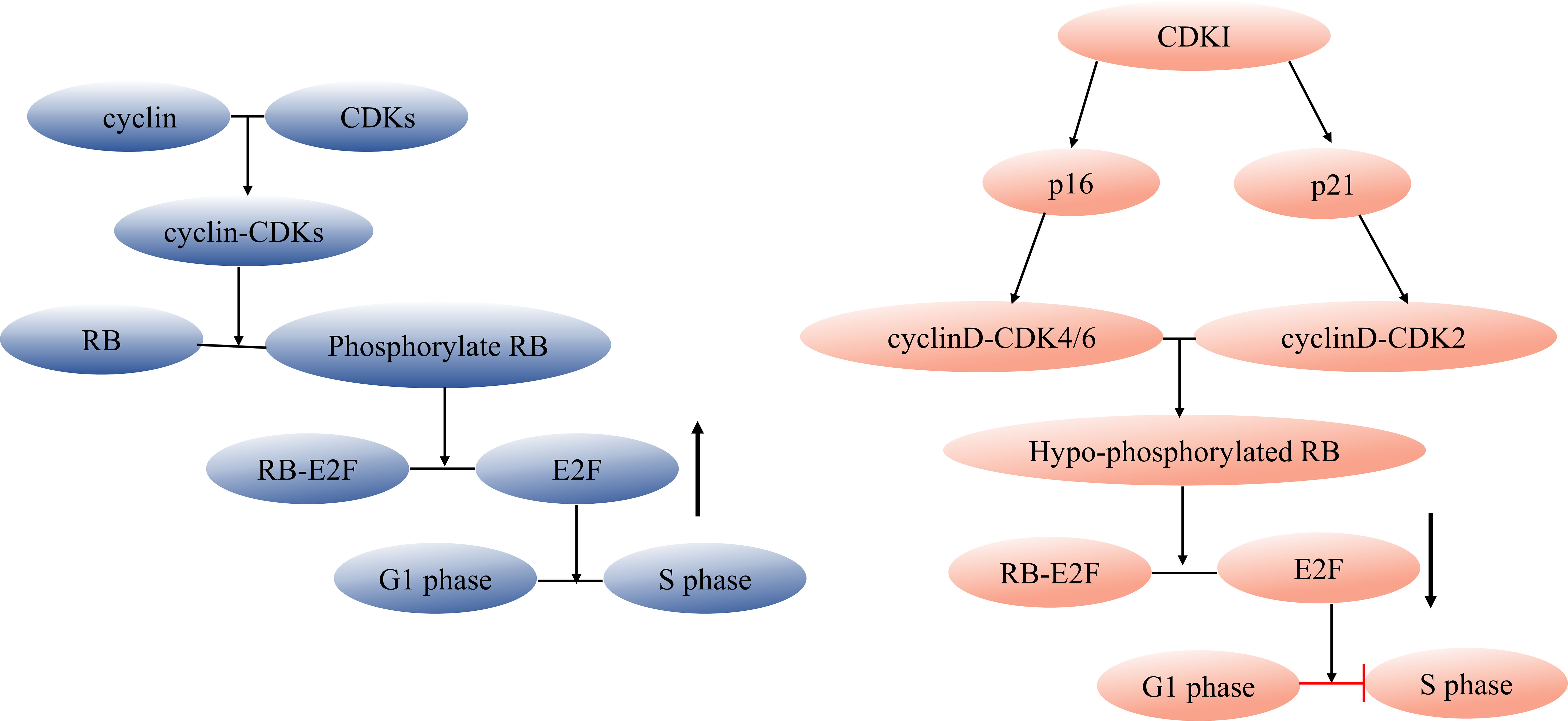
Figure 1 Regulation of cell cycle and senescence pathways. During cell division, cyclin and CDK combine forming a cyclin-CDK complex that initiates the downstream phosphorylation of RB. The phosphorylated RB then disengages the transcription factor E2F from the RB-E2F complex, facilitating DNA transcription and propelling the cell cycle into the S phase. CDKI can inhibit this process. Cell cycle arrest during cellular senescence primarily hinges on the p53/p21 and p16/RB pathways (47, 48), p53 acts as an upstream regulator of p21, while both p16 and p21 belong to the CDKI family. These members inhibit cyclinD-CDK4/6 and cyclinE/A-CDK2, respectively, tightly tethering low-phosphorylated RB to E2F and halting cells in the G1 phase. CDK, cyclin-dependent kinase, CDKI, cyclin-dependent kinase inhibitor.
4 Role of cell senescence in vascular calcification: insights from chronic kidney disease
In 1989, Professor Virchow first introduced a captivating discovery: individuals suffering from CKD manifest the same pathological transformations of medial calcification commonly observed in the elderly population (53). Foley et al. made a comparable observation regarding vascular traits among young hemodialysis patients, likening them to those found in individuals in their eighties and nineties (54). A study featured in NEJM in 2000 disclosed that the occurrence of VC among young hemodialysis patients aged 20 to 30 years reached a staggering 87.5% (14 out of 16), markedly surpassing the corresponding age group in the general population (3.3%, 2 out of 60) (55). Furthermore, Girndt et al. proposed that the extent of VC could serve as an indicator of an individual’s biological age (2). Collectively, these findings indicate an accelerated vascular aging progress in CKD patients.
In the realm of CKD, a milieu defined by the presence of uremic toxins, inflammatory processes, and oxidative stress persists, impervious even to renal transplantation. This environment fosters a state of premature aging, a condition further exacerbated by hemodialysis, which accelerates telomere attrition (56, 57). Within this complex scenario, the burden of senescent cells emerges as a substantial factor contributing to VC in the CKD milieu. Notably, interventions aimed at eliminating senescent cells using genetic approaches or dasatinib plus quercetin (D+Q) yielded significant reductions in aortic aging markers and calcification in aging and atherosclerotic mice. This was accompanied by a downregulation of the osteogenic marker, osterix (26). Furthermore, the pro-inflammatory secretome (SASP) derived from senescent cells has been found to elevate in both uremic mouse models and patients (58, 59). However, the direct impact of SASP on vascular cells remains underexplored, with potential mechanisms possibly involving the propagation of senescence or amplification of localized inflammation. However, numerous studies have shown that sirtuins, histone deacetylases, or known as the family of longevity proteins, play a protective role in VC. For example, sirtuin 6 mediated the deacetylation of Runx2, which subsequently facilitated its nuclear export via exportin 1 (XPO1). This event eventually triggered the degradation of Runx2 through the ubiquitin-proteasome system, thereby inhibiting the osteogenic transdifferentiation of VSMCs (60). Additionally, sirtuin 1 was upregulated by spermidine, thus alleviating VC in CKD (61). Moreover, compelling evidence suggests a decline in nicotinamide adenine dinucleotide (oxidized form, NAD+) levels in both CKD and acute kidney injury (AKI). This molecule plays a pivotal role as a coenzyme in crucial cellular metabolic activities. The supplementation of its precursor, nicotinamide mononucleotide (NMN), has demonstrated positive effects on physical endurance and the maintenance of biological age (62). The popularity of NMN is significant due to its potential to activate longevity proteins, improve mitochondrial metabolism, and contribute to the repair of damaged DNA. In the near future, there is hope that NMN could become a promising choice for extending healthy lifespan. Furthermore, nuclear factor erythroid 2-related factor 2 (NRF2) has emerged as a proposed protective factor against calcification and aging. Its transcriptional suppression of pro-inflammatory genes and antioxidant effects contribute to its role in mitigating these processes (63).
Senescent cells are not quiescent, nonfunctional and may be continuously releasing inflammatory mediators, microparticles, etc., affecting the level of transcription, post-transcriptional regulation, translation, and metabolism by autocrine/paracrine means, which in turn affects phenotypic transformation and microenvironmental niche on itself or adjacent cells, resulting acceleration of calcification. The intricate association between aging and VC implies that delving deeper into their interaction could potentially pave the way for novel avenues in diagnosing and treating VC.
5 Role of vascular cell senescence in vascular calcification
Both VECs and VSMCs constitute essential cellular components residing within the intimal and medial layers of blood vessels, respectively. The adventitial layer, on the other hand, is comprised of a diverse array of cells, fibroblasts, lymphocytes, vascular progenitors/stem cells, pericytes, and primarily monocytes/macrophages-based immune cells. This rich composition could potentially open novel avenues for advancing the diagnosis and treatment of VC. A growing body of evidence is highlighting the pivotal role played by adventitial cells in blood vessel remodeling and the maintenance of vascular homeostasis (64, 65). Interestingly, adventitial fibroblasts (AFs) exhibit the remarkable ability to transdifferentiate into VSMCs in response to microenvironmental cytokines, subsequently migrating to the luminal side of the vessel (66). Recent advancements have shed light on the role of exosomes derived from AFs, predominant cells in the adventitia. These exosomes expedite VSMC calcification by delivering miR-21-5p, which, in turn, inhibits cysteine-rich motor neuron 1 (Crim1) (67). Conversely, adventitial pericytes (APC) counteract calcification by triggering an anti-calcific effect through the upregulation of microRNA-132-3p (68). Cells constitute the fundamental constituents of tissues, organs, and organisms. And it is the progression of cellular senescence that propels the aging process across these levels.
5.1 Role of vascular endothelial cell senescence in vascular calcification
VECs adeptly sense a myriad of stimuli coursing through the bloodstream and promptly respond, upholding vascular equilibrium via autocrine or paracrine mechanisms. These mechanisms play a role in regulating diverse functions, including blood pressure maintenance, promotion of angiogenesis, and coagulation regulation (69). In the complex landscape of CKD, characterized by heightened inflammation, oxidative stress, leukocyte migration, adhesion, cell death, and the emergence of a thrombotic phenotype, an array of circulating molecules holds sway over endothelial equilibrium These molecules exert their influence through frequently activated cellular signaling pathways, including ROS, MAPK/NF-κB, Aryl-Hydrocarbon Receptor, and RAGE pathways (70). A noteworthy phenomenon within this milieu is the endothelial-to-mesenchymal transition (EndMT) of VECs. This transition involves endothelial cells adopting a mesenchymal-like state, providing a conceptual link to the generation of osteogenic cells that contribute to VC. Wang et al. proposed that EndMT induced by Parathyroid hormone (PTH) plays a role in VC through the miR-29a-5p/GSAP/Notch1 pathway in CKD rats (71). Notably, indoxyl sulfate (IS) and sulfate paracresol emerge as significant players, hastening the senescence of endothelial cells and undermining their functional integrity (72). Senescent VECs exhibit a distinctive profile characterized by reduced expression of nitric oxide (NO) and an elevation in reactive oxygen species (ROS) (73). Moreover, the accumulation of senescent VECs suppresses the expression of adherens junction proteins and compromises the endothelial migration potential in non-senescent VECs (74). This intricate interplay underscores the impact of senescence on the functional and molecular dynamics within the endothelial environment. Given the spatial proximity of endothelial cells and VSMCs, an intriguing query arises: could endothelial cells, especially senescent ones, exert an impact on VSMC calcification? Intriguing in vitro findings add credence to this question. Under high glucose stimulation, endothelial cells have been observed to release exosomes housing Notch3 or versican proteins. This, in turn, triggers senescence and calcification in VSMCs (10, 11). Further studies reveal that senescent endothelial cells possess the ability to attract monocytes to the endothelium and induce the proliferation and migration of VSMCs (47). Additionally, the release of microvesicles (MVs) from senescent endothelial cells emerges as a key factor driving VSMC senescence and calcification. Noteworthy is the intervention using IS on human umbilical vein endothelial cells (HUVECs), which results in EC senescence and subsequent MV release. These MVs are identified as the primary instigators behind human aortic smooth muscle cells (HASMCs) calcification, coupled with the upregulation of pro-inflammatory factors (22). Furthermore, the influence of senescent endothelial cells and their derived MVs does not end there. Examination of plasma from elderly individuals and MVs sourced from senescent HUVECs underscores their promoting effect on HASMC calcification. These MVs are characterized by the upregulated expression of membrane-associated proteins A2, A6, and BMP2 (48).
5.2 Role of vascular smooth muscle cell senescence in vascular calcification
The transformation of VSMCs into osteogenic phenotypes stands as a pivotal cellular mechanism underlying VC (75). When VSMCs undergo senescence, they adopt a pro-calcification and osteogenic phenotype (76, 77). Microarray analysis of senescent VSMCs cultured in vitro unveiled distinct gene expression patterns related to VC including bone morphogenetic protein-2 (BMP-2), osteoprotegerin (OPG), osteopontin (OPN), secreted phosphoprotein 1 (SPP1), and matrix Gla protein (MGP) (76). Senescent VSMCs exhibit upregulation of RUNX2, while knockdown of RUNX2 resulted in a significant reduction of osteoblast markers, as well as in calcification (77). Furthermore, senescent VSMCs exhibit the expression of MMP9 and SASP, which in turn triggers adjacent ECs and VSMCs to assume a pro-adhesive and pro-inflammatory state. This cascade contributes to the establishment of chronic vascular inflammation (78).
Within the context of CKD, VSMCs encountered by patients face an array of pro-calcification environments. These include reactive oxygen species (ROS), elevated calcium and phosphate levels, and the influence of uremic toxins. This multifaceted assault can induce DNA damage in VSMCs. If left unresolved, persistent DNA damage prompts VSMCs to enter a state of senescence. The release of SASP, including BMP2, IL-6, and OPG, by senescent cells further spurs VSMCs toward osteogenic differentiation (23). Notably, the uremic toxin IS triggers upregulation of p21, p53, and nuclear lamin A in VSMCs through pathways driven by oxidative stress (79). The landscape of molecular mechanisms associated with VSMC senescence-induced calcification is intricate and multifaceted. For instance, the downregulation of microRNA-34a culminates in the inhibition of Sirtuin1 and the AXL receptor tyrosine kinase, thereby promoting both VSMC senescence and calcification (80, 81). Another intriguing facet is the involvement of long non-coding RNA (lncRNA) ES3, which binds to Bhlhe40. This interaction effectively silences multiple small interfering RNAs and subsequently ushers in the era of VSMC senescence and calcification (82).
5.3 Role of vascular progenitor/stem cell depletion in vascular calcification
The adventitia hosts an array of vascular progenitor/stem cells, encompassing mesenchymal stromal cells (MSCs), AFs, and pericytes. These vascular progenitor/stem cells exhibit a versatile potential for differentiation, capable of transforming into VECs and VSMCs. They play a pivotal role in vascular repair and remodeling following vascular injury and in the replenishment of senescent cells. The emergence of MSCs has ignited optimism within regenerative medicine, due to their remarkable attributes such as ease of procurement, isolation, immune regulation, and multi-lineage differentiation capabilities (83). Notably, MSC-derived extracellular vesicles (EVs) have demonstrated the ability to alleviate VEC senescence through microRNA-146a, thereby diminishing SASP expression while stimulating angiogenesis (84). Stem cell senescence is also a prominent theory of organismal aging (10). The depletion of vascular progenitor/stem cells can engender a decline in the self-renewal potential of VECs and VSMCs, potentially setting the stage for vascular aging and the jsubsequent advancement of age-associated vascular disorders (85).
Similar to senescent cells, senescent MSCs undergo distinct morphological alterations characterized by flattening and enlargement. Concomitantly, their proliferation and differentiation capacities wane (83). Senescent MSCs also express pro-inflammatory SASP, which inhibits the proliferation of hematopoietic stem cells, thus expediting their premature senescence (86). This, in turn, indirectly participates in the aging and calcification trajectory of blood vessels. Senescent AFs, akin to their cellular counterparts, release SASP laden with pro-inflammatory and pro-tissue degradation factors, thus perpetuating the senescence phenomenon (87). Aged AFs exhibit significant differential expression of pro-inflammatory and osteogenic genes via transcriptional analysis (88). Moreover, in patients with calcification associated with rheumatoid arthritis, the circulating osteogenic endothelial progenitor cells (EPC) are related to vascular aging, encountering a decline in both number and differentiation potential, culminating in hastened cellular senescence (89, 90). A distinct viewpoint is proposed by Wu et al., indicating that the proliferative and anti-apoptotic capabilities of aged adipose stem cells tend to outshine those of bone marrow stem cells (91). Consequently, adipose-derived stem cells may offer greater suitability for autsue and organismal aging, and chronic age-related conditions. Recent research underscores the potential of inhibiting the immune checkpoint protein programmed death-ligand 1 (PD-L1) in senescent cells. Such intervention appears to ameliorate the inflammatory state and mitigate age-related manifestations (92). Zhang et al. found that senescence characteristics of hematopoietic stem cells can be reversed and their long-term multilineage reconstitution capacity restored by harnessing matrix stiffness (93). In addition, senescent cells do not invariably induce detrimental effects, indeed, p16INK4a+ fibroblasts can actually foster the regeneration of epithelial stem cells (94). Although research on the role of vascular progenitor/stem cell senescence in VC remains limited, delving deeper into this avenue could yield strategies for anti-aging therapies aimed at postponing calcification processes. This unexplored territory holds promising potential for shedding light on the intricate intersections of senescence, vascular health, and aging-related outcomes.
5.4 Role of macrophage senescence in vascular calcification
Immunosenescence, the progressive decline in immune function with age, represents a causal contributor to systemic aging (95). This phenomenon is marked by a shift in immune cell differentiation, favoring myeloid lineage development while diminishing lymphoid lineage differentiation (96). As a result, immune cells experience a reduced ability to effectively clear senescent cells from the body, leading to the accumulation of these aged cells. Consequently, the role of immune cell senescence in age-related diseases has garnered substantial attention.
As mentioned above, specific components of the SASP are adept at recruiting immune cells. Significant contributors such as IL-6, IL-7, IL-8, IL-1α, IL-1β, TNF-α, macrophage colony-stimulating factor (M-CSF), G-CSF, and granulocyte-macrophage colony-stimulating factor (GM-CSF) exert their influence, drawing in a range of immune players including macrophages, granulocytes, neutrophils, and T lymphocytes (97). Immunosenescence results may lead to myeloid lineage bias, prompting us to consider that macrophages might indeed hold a central position in the realm of aging-related diseases. This notion finds its roots in the theory of inflamm-aging, which initially highlighted the pivotal role of macroph-aging – the gradual activation of macrophages as time advances (98). Macrophages, critical constituents of innate immunity, exhibit a tendency to shift from an anti-inflammatory M2 phenotype to a pro-inflammatory M1 phenotype with advancing age (99), and subsequently promote VSMCs calcification by sitimulating carbonic anhydrase I (CA1) and CA2 via secreting TNFα, or NLRP3 inflammasome (100, 101). Calcified smooth muscle cells induce the differentiation of macrophages into osteoblasts through RANKL, fostering a positive feedback loop that promotes VC (102). In a dystrophic muscle pathology model, the accumulation of pro-inflammatory M1 macrophages at ectopic calcification sites was evident. This accumulation was accompanied by the upregulation of senescence markers such as p21, C12FDG, and SASP (24). However, the specific molecular mechanisms by which senescent macrophages induce VC remain unclear, and require further investigation.
6 Summary and conclusions
Throughout the extensive trajectory of human evolution, cellular senescence emerges as the ultimate choice of cells when confronted with diverse physical and chemical stimuli, carefully weighing the pros and cons. While it acts as a brake on the proliferation of aberrant cells, it concurrently propels the advancement of age-related diseases. Notably, cardiovascular incidents and mortality rates maintain a distressingly high presence in patients with CKD. Within this context, the process of vascular premature senescence stands as a pivotal player in the intricate symphony of calcification. This article mainly focused on the multifaceted role of senescence across various vascular cell types, encompassing endothelial cells, vascular smooth muscle cells, vascular progenitor/stem cells, and macrophages. The ultimate aim is to distill the current corpus of literature, culminating in an informative synthesis that paves the way for prospective endeavors in the realm of anti-aging therapies, specifically designed to counteract and alleviate the menace of VC.
Senolytics, such as D+Q, fisetin (103), and procyanidin C1 (PCC1) (104), among others, have experienced continuous expansion and have even entered clinical trials since Professor Kirkland and colleagues discovered the anti-aging effects of D+Q in 2015 (105). Currently, D+Q appears to be the most promising agent (106, 107). However, it is noteworthy that many senolytics operate by suppressing antiapoptotic signal pathways in senescent cells, potentially increasing the risk of off-target effects on healthy tissues. To address this concern, chimeric antigen receptor (CAR) T cells have shown promise in selectively and effectively removing senescent cells by identifying proteins broadly induced in these cells (108, 109). Additionally, Senolytic vaccination, targeting glycoprotein nonmetastatic melanoma protein B (GPNMB), has successfully extended lifespan) in progeroid mice (about 20%) (110). The challenge lies in the variations in aging characteristics among species, individuals, organs, and even cells. Tailoring anti-aging regimens specifically to senescent cells is a future direction of exploration in this field. Despite these challenges, the development of a gene set (SenMayo) has made it possible to identify senescent cells in vivo. Moreover, the application of deep learning to discover senotherapeutics holds promise for the future (111, 112).
Author contributions
Y-PF: Data curation, Investigation, Visualization, Writing – original draft. YZ: Funding acquisition, Supervision, Writing – review & editing. J-YH: Data curation, Writing – review & editing. XY: Data curation, Writing – review & editing. YL: Data curation, Writing – review & editing. X-LZ: Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Natural Science Youth Foundation of China (Grant No. 82000650), and the National Natural Sciences Foundation of China (NO.81570612, NO.81870497).
Acknowledgments
The authors would like to express our gratitude to EditSprings (https://www.editsprings.cn) for the expert linguistic services provided.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
Afs, adventitial fibroblastsL AKI, acute kidney injury; ALP, alkaline phosphatase; APCs, adventitial pericytes; CaSRs, calcium-sensing receptors; CDKI, cyclin-dependent kinase inhibitor; CKD, chronic kidney disease; EndMT, endothelial-to-mesenchymal transition; MAC, medial arterial calcification; MSCs, mesenchymal stromal cells; MV, microvesicles; NMN, nicotinamide mononucleotide; NRF2, nuclear factor erythroid 2-related factor 2; PWV, pulse wave velocity; RS, replicative senescence; SASP, senescence-associated secretory phenotype; SAHF, senescence-associated heterochromatin foci; SIPS, stress-induced premature senescence; VC, Vascular calcification; VECs, vascular endothelial cells; VSMCs, vascular smooth muscle cells.
References
1. Jankowski J, Floege J, Fliser D, Böhm M, Marx N. Cardiovascular disease in chronic kidney disease: pathophysiological insights and therapeutic options. Circulation (2021) 143:1157–72. doi: 10.1161/circulationaha.120.050686
2. Shaw LJ, Raggi P, Berman DS, Callister TQ. Coronary artery calcium as a measure of biologic age. Atherosclerosis (2006) 188:112–9. doi: 10.1016/j.atherosclerosis.2005.10.010
3. Jebari-Benslaiman S, Galicia-García U, Larrea-Sebal A, Olaetxea JR, Alloza I, Vandenbroeck K, et al. Pathophysiology of atherosclerosis. Int J Mol Sci (2022) 23:3346. doi: 10.3390/ijms23063346
4. Lu CL, Liao MT, Hou YC, Fang YW, Zheng CM, Liu WC, et al. Sirtuin-1 and its relevance in vascular calcification. Int J Mol Sci (2020) 21(5):1593. doi: 10.3390/ijms21051593
5. Lanzer P, Hannan FM, Lanzer JD, Janzen J, Raggi P, Furniss D, et al. Medial arterial calcification: JACC state-of-the-art review. J Am Coll Cardiol (2021) 78:1145–65. doi: 10.1016/j.jacc.2021.06.049
6. Nelson AJ, Raggi P, Wolf M, Gold AM, Chertow GM, Roe MT. Targeting vascular calcification in chronic kidney disease. JACC Basic Transl Sci (2020) 5:398–412. doi: 10.1016/j.jacbts.2020.02.002
7. Pescatore LA, Gamarra LF, Liberman M. Multifaceted mechanisms of vascular calcification in aging. Arterioscler Thromb Vasc Biol (2019) 39:1307–16. doi: 10.1161/atvbaha.118.311576
8. Zhang H, Li G, Yu X, Yang J, Jiang A, Cheng H, et al. Progression of vascular calcification and clinical outcomes in patients receiving maintenance dialysis. JAMA Netw Open (2023) 6:e2310909. doi: 10.1001/jamanetworkopen.2023.10909
9. Marreiros C, Viegas C, Simes D. Targeting a silent disease: vascular calcification in chronic kidney disease. Int J Mol Sci (2022) 23:16114. doi: 10.3390/ijms232416114
10. Lin X, Li S, Wang YJ, Wang Y, Zhong JY, He JY, et al. Exosomal Notch3 from high glucose-stimulated endothelial cells regulates vascular smooth muscle cells calcification/aging. Life Sci (2019) 232(24):116582. doi: 10.1016/j.lfs.2019.116582
11. Li S, Zhan JK, Wang YJ, Lin X, Zhong JY, Wang Y, et al. Exosomes from hyperglycemia-stimulated vascular endothelial cells contain versican that regulate calcification/senescence in vascular smooth muscle cells. Cell Biosci (2019) 9:1. doi: 10.1186/s13578-018-0263-x
12. Ye Y, Chen A, Li L, Liang Q, Wang S, Dong Q, et al. Repression of the antiporter SLC7A11/glutathione/glutathione peroxidase 4 axis drives ferroptosis of vascular smooth muscle cells to facilitate vascular calcification. Kidney Int (2022) 102:1259–75. doi: 10.1016/j.kint.2022.07.034
13. Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res (2000) 87:1055–62. doi: 10.1161/01.res.87.11.1055
14. Olszak IT, Poznansky MC, Evans RH, Olson D, Kos C, Pollak MR, et al. Extracellular calcium elicits a chemokinetic response from monocytes in vitro and in vivo. . J Clin Invest (2000) 105:1299–305. doi: 10.1172/jci9799
15. Ding M, Zhang Q, Zhang M, Jiang X, Wang M, Ni L, et al. Phosphate overload stimulates inflammatory reaction via piT-1 and induces vascular calcification in uremia. J Ren Nutr (2022) 32:178–88. doi: 10.1053/j.jrn.2021.03.008
16. Shroff R, Long DA, Shanahan C. Mechanistic insights into vascular calcification in CKD. J Am Soc Nephrol (2013) 24:179–89. doi: 10.1681/ASN.2011121191
17. Tyson KL, Reynolds JL, Mcnair R, Zhang Q, Weissberg PL, Shanahan CM. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler Thromb Vasc Biol (2003) 23:489–94. doi: 10.1161/01.Atv.0000059406.92165.31
18. Kapustin AN, Davies JD, Reynolds JL, Mcnair R, Jones GT, Sidibe A, et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res (2011) 109:e1–12. doi: 10.1161/circresaha.110.238808
19. Lomashvili KA, Garg P, Narisawa S, Millan JL, O’neill WC. Upregulation of alkaline phosphatase and pyrophosphate hydrolysis: potential mechanism for uremic vascular calcification. Kidney Int (2008) 73:1024–30. doi: 10.1038/ki.2008.26
20. Ding N, Lv Y, Su H, Wang Z, Kong X, Zhen J, et al. Vascular calcification in CKD: New insights into its mechanisms. J Cell Physiol (2023) 238:1160–82. doi: 10.1002/jcp.31021
21. Liu Q, Luo Y, Zhao Y, Xiang P, Zhu J, Jing W, et al. Nano-hydroxyapatite accelerates vascular calcification via lysosome impairment and autophagy dysfunction in smooth muscle cells. Bioact Mater (2022) 8:478–93. doi: 10.1016/j.bioactmat.2021.06.004
22. Alique M, Bodega G, Corchete E, García-Menéndez E, De Sequera P, Luque R, et al. Microvesicles from indoxyl sulfate-treated endothelial cells induce vascular calcification. vitro. Comput Struct Biotechnol J (2020) 18:953–66. doi: 10.1016/j.csbj.2020.04.006
23. Shanahan CM. Mechanisms of vascular calcification in CKD-evidence for premature ageing? Nat Rev Nephrol (2013) 9:661–70. doi: 10.1038/nrneph.2013.176
24. Mu X, Lin CY, Hambright WS, Tang Y, Ravuri S, Lu A, et al. Aberrant RhoA activation in macrophages increases senescence-associated secretory phenotypes and ectopic calcification in muscular dystrophic mice. Aging (Albany NY) (2020) 12:24853–71. doi: 10.18632/aging.202413
25. Stenvinkel P, Luttropp K, Mcguinness D, Witasp A, Qureshi AR, Wernerson A, et al. CDKN2A/p16INK4(a) expression is associated with vascular progeria in chronic kidney disease. Aging (Albany NY) (2017) 9:494–507. doi: 10.18632/aging.101173
26. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell (2016) 15:973–7. doi: 10.1111/acel.12458
27. Karatza C, Stein WD, Shall S. Kinetics of in vitro ageing of mouse embryo fibroblasts. J Cell Sci (1984) 65:163–75. doi: 10.1242/jcs.65.1.163
28. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. (1965) 37:614–36. doi: 10.1016/0014-4827(65)90211-9
29. Karlseder J, Smogorzewska A, De Lange T. Senescence induced by altered telomere state, not telomere loss. Science (2002) 295:2446–9. doi: 10.1126/science.1069523
30. Martínez-Zamudio RI, Robinson L, Roux PF, Bischof O. SnapShot: cellular senescence pathways. Cell (2017) 170:816–816.e1. doi: 10.1016/j.cell.2017.07.049
31. Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev (2020) 34:1565–76. doi: 10.1101/gad.343129.120
32. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol (2014) 15:482–96. doi: 10.1038/nrm3823
33. Moiseeva V, Cisneros A, Sica V, Deryagin O, Lai Y, Jung S, et al. Senescence atlas reveals an aged-like inflamed niche that blunts muscle regeneration. Nature (2023) 613:169–78. doi: 10.1038/s41586-022-05535-x
34. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
35. Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun (2016) 7:11190. doi: 10.1038/ncomms11190
36. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med (2018) 24:1246–56. doi: 10.1038/s41591-018-0092-9
37. Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, et al. Geroscience: linking aging to chronic disease. Cell (2014) 159:709–13. doi: 10.1016/j.cell.2014.10.039
38. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol (2013) 14:R115. doi: 10.1186/gb-2013-14-10-r115
39. Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY) (2018) 10:573–91. doi: 10.18632/aging.101414
40. Sayed N, Huang Y, Nguyen K, Krejciova-Rajaniemi Z, Grawe AP, Gao T, et al. An inflammatory aging clock (iAge) based on deep learning tracks multimorbidity, immunosenescence, frailty and cardiovascular aging. Nat Aging (2021) 1:598–615. doi: 10.1038/s43587-021-00082-y
41. Mehdizadeh M, Aguilar M, Thorin E, Ferbeyre G, Nattel S. The role of cellular senescence in cardiac disease: basic biology and clinical relevance. Nat Rev Cardiol (2022) 19:250–64. doi: 10.1038/s41569-021-00624-2
42. González-Gualda E, Baker AG, Fruk L, Muñoz-Espín D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J (2021) 288:56–80. doi: 10.1111/febs.15570
43. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell (2019) 179:813–27. doi: 10.1016/j.cell.2019.10.005
44. Chen J, Huang X, Halicka D, Brodsky S, Avram A, Eskander J, et al. Contribution of p16INK4a and p21CIP1 pathways to induction of premature senescence of human endothelial cells: permissive role of p53. Am J Physiol Heart Circ Physiol (2006) 290:H1575–86. doi: 10.1152/ajpheart.00364.2005
45. Mijit M, Caracciolo V, Melillo A, Amicarelli F, Giordano A. Role of p53 in the regulation of cellular senescence. Biomolecules (2020) 10(3):420. doi: 10.3390/biom10030420
46. Roger L, Tomas F, Gire V. Mechanisms and regulation of cellular senescence. Int J Mol Sci (2021) 22(23):13173. doi: 10.3390/ijms222313173
47. Ding YN, Wang HY, Chen HZ, Liu DP. Targeting senescent cells for vascular aging and related diseases. J Mol Cell Cardiol (2022) 162:43–52. doi: 10.1016/j.yjmcc.2021.08.009
48. Alique M, Ruíz-Torres MP, Bodega G, Noci MV, Troyano N, Bohórquez L, et al. Microvesicles from the plasma of elderly subjects and from senescent endothelial cells promote vascular calcification. Aging (Albany NY) (2017) 9:778–89. doi: 10.18632/aging.101191
49. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol (2018) 28:436–53. doi: 10.1016/j.tcb.2018.02.001
50. Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol (2006) 7:667–77. doi: 10.1038/nrm1987
51. Stein GH, Drullinger LF, Soulard A, Dulić V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol (1999) 19:2109–17. doi: 10.1128/mcb.19.3.2109
52. Hernandez-Segura A, De Jong TV, Melov S, Guryev V, Campisi J, Demaria M. Unmasking transcriptional heterogeneity in senescent cells. Curr Biol (2017) 27:2652–2660.e4. doi: 10.1016/j.cub.2017.07.033
53. Virchow R. Cellular pathology. As based upon physiological and pathological histology. Lecture XVI–Atheromatous affection of arteries. 1858. Nutr Rev (1989) 47:23–5. doi: 10.1111/j.1753-4887.1989.tb02747.x
54. Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis (1998) 32:S112–9. doi: 10.1053/ajkd.1998.v32.pm9820470
55. Goodman WG, Goldin J, Kuizon BD, Yoon C, Gales B, Sider D, et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med (2000) 342:1478–83. doi: 10.1056/nejm200005183422003
56. Speer T, Dimmeler S, Schunk SJ, Fliser D, Ridker PM. Targeting innate immunity-driven inflammation in CKD and cardiovascular disease. Nat Rev Nephrol (2022) 18:762–78. doi: 10.1038/s41581-022-00621-9
57. Levstek T, Trebušak Podkrajšek K. Telomere attrition in chronic kidney diseases. Antioxidants (Basel) (2023) 12(3):579. doi: 10.3390/antiox12030579
58. Tölle M, Henkel C, Herrmann J, Daniel C, Babic M, Xia M, et al. Uremic mouse model to study vascular calcification and “inflamm-aging”. J Mol Med (Berl) (2022) 100:1321–30. doi: 10.1007/s00109-022-02234-y
59. Sanchis P, Ho CY, Liu Y, Beltran LE, Ahmad S, Jacob AP, et al. Arterial “inflammaging” drives vascular calcification in children on dialysis. Kidney Int (2019) 95:958–72. doi: 10.1016/j.kint.2018.12.014
60. Li W, Feng W, Su X, Luo D, Li Z, Zhou Y, et al. SIRT6 protects vascular smooth muscle cells from osteogenic transdifferentiation via Runx2 in chronic kidney disease. J Clin Invest (2022) 132(1):e150051. doi: 10.1172/jci150051
61. Liu X, Chen A, Liang Q, Yang X, Dong Q, Fu M, et al. Spermidine inhibits vascular calcification in chronic kidney disease through modulation of SIRT1 signaling pathway. Aging Cell (2021) 20:e13377. doi: 10.1111/acel.13377
62. Morevati M, Fang EF, Mace ML, Kanbay M, Gravesen E, Nordholm A, et al. Roles of NAD(+) in acute and chronic kidney diseases. Int J Mol Sci (2022) 24(1):137. doi: 10.3390/ijms24010137
63. Laget J, Hobson S, Muyor K, Duranton F, Cortijo I, Bartochowski P, et al. Implications of senescent cell burden and NRF2 pathway in uremic calcification: A translational study. Cells (2023) 12(4):643. doi: 10.3390/cells12040643
64. Ren XS, Tong Y, Qiu Y, Ye C, Wu N, Xiong XQ, et al. MiR155-5p in adventitial fibroblasts-derived extracellular vesicles inhibits vascular smooth muscle cell proliferation via suppressing angiotensin-converting enzyme expression. J Extracell Vesicles (2020) 9:1698795. doi: 10.1080/20013078.2019.1698795
65. Chen Y, Yang X, Kitajima S, Quan L, Wang Y, Zhu M, et al. Macrophage elastase derived from adventitial macrophages modulates aortic remodeling. Front Cell Dev Biol (2022) 10:1097137. doi: 10.3389/fcell.2022.1097137
66. Karamariti E, Zhai C, Yu B, Qiao L, Wang Z, Potter CMF, et al. DKK3 (Dickkopf 3) alters atherosclerotic plaque phenotype involving vascular progenitor and fibroblast differentiation into smooth muscle cells. Arterioscler Thromb Vasc Biol (2018) 38:425–37. doi: 10.1161/atvbaha.117.310079
67. Zheng MH, Shan SK, Lin X, Xu F, Wu F, Guo B, et al. Vascular wall microenvironment: exosomes secreted by adventitial fibroblasts induced vascular calcification. J Nanobiotechnology (2023) 21:315. doi: 10.1186/s12951-023-02000-3
68. Jover E, Fagnano M, Cathery W, Slater S, Pisanu E, Gu Y, et al. Human adventitial pericytes provide a unique source of anti-calcific cells for cardiac valve engineering: Role of microRNA-132-3p. Free Radic Biol Med (2021) 165:137–51. doi: 10.1016/j.freeradbiomed.2021.01.029
69. Trimm E, Red-Horse K. Vascular endothelial cell development and diversity. Nat Rev Cardiol (2023) 20:197–210. doi: 10.1038/s41569-022-00770-1
70. Harlacher E, Wollenhaupt J, Baaten C, Noels H. Impact of uremic toxins on endothelial dysfunction in chronic kidney disease: A systematic review. Int J Mol Sci (2022) 23(1):531. doi: 10.3390/ijms23010531
71. Wang L, Tang R, Zhang Y, Chen S, Guo Y, Wang X, et al. PTH-induced EndMT via miR-29a-5p/GSAP/Notch1 pathway contributed to valvular calcification in rats with CKD. Cell Prolif (2021) 54:e13018. doi: 10.1111/cpr.13018
72. Guerrero F, Carmona A, Jimenez MJ, Obrero T, Pulido V, Moreno JA, et al. Passage number-induced replicative senescence modulates the endothelial cell response to protein-bound uremic toxins. Toxins (Basel) (2021) 13(10):738. doi: 10.3390/toxins13100738
73. Ungvari Z, Labinskyy N, Gupte S, Chander PN, Edwards JG, Csiszar A. Dysregulation of mitochondrial biogenesis in vascular endothelial and smooth muscle cells of aged rats. Am J Physiol Heart Circ Physiol (2008) 294:H2121–8. doi: 10.1152/ajpheart.00012.2008
74. Wong PF, Tong KL, Jamal J, Khor ES, Lai SL, Mustafa MR. Senescent HUVECs-secreted exosomes trigger endothelial barrier dysfunction in young endothelial cells. Excli J (2019) 18:764–76. doi: 10.17179/excli2019-1505
75. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res (2018) 114:590–600. doi: 10.1093/cvr/cvy010
76. Burton DG, Giles PJ, Sheerin AN, Smith SK, Lawton JJ, Ostler EL, et al. Microarray analysis of senescent vascular smooth muscle cells: A link to atherosclerosis and vascular calcification. Exp Gerontol (2009) 44:659–65. doi: 10.1016/j.exger.2009.07.004
77. Nakano-Kurimoto R, Ikeda K, Uraoka M, Nakagawa Y, Yutaka K, Koide M, et al. Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am J Physiol Heart Circ Physiol (2009) 297:H1673–84. doi: 10.1152/ajpheart.00455.2009
78. Gardner SE, Humphry M, Bennett MR, Clarke MC. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1α-dependent senescence-associated secretory phenotype. Arterioscler Thromb Vasc Biol (2015) 35:1963–74. doi: 10.1161/atvbaha.115.305896
79. Muteliefu G, Shimizu H, Enomoto A, Nishijima F, Takahashi M, Niwa T. Indoxyl sulfate promotes vascular smooth muscle cell senescence with upregulation of p53, p21, and prelamin A through oxidative stress. Am J Physiol Cell Physiol (2012) 303:C126–34. doi: 10.1152/ajpcell.00329.2011
80. Zuccolo E, Badi I, Scavello F, Gambuzza I, Mancinelli L, Macrì F, et al. The microRNA-34a-induced senescence-associated secretory phenotype (SASP) favors vascular smooth muscle cells calcification. Int J Mol Sci (2020) 21(12):4454. doi: 10.3390/ijms21124454
81. Badi I, Mancinelli L, Polizzotto A, Ferri D, Zeni F, Burba I, et al. miR-34a promotes vascular smooth muscle cell calcification by downregulating SIRT1 (Sirtuin 1) and axl (AXL receptor tyrosine kinase). Arterioscler Thromb Vasc Biol (2018) 38:2079–90. doi: 10.1161/atvbaha.118.311298
82. Zhong JY, Cui XJ, Zhan JK, Wang YJ, Li S, Lin X, et al. LncRNA-ES3 inhibition by Bhlhe40 is involved in high glucose-induced calcification/senescence of vascular smooth muscle cells. Ann N Y Acad Sci (2020) 1474:61–72. doi: 10.1111/nyas.14381
83. Weng Z, Wang Y, Ouchi T, Liu H, Qiao X, Wu C, et al. Mesenchymal stem/stromal cell senescence: hallmarks, mechanisms, and combating strategies. Stem Cells Transl Med (2022) 11:356–71. doi: 10.1093/stcltm/szac004
84. Xiao X, Xu M, Yu H, Wang L, Li X, Rak J, et al. Mesenchymal stem cell-derived small extracellular vesicles mitigate oxidative stress-induced senescence in endothelial cells via regulation of miR-146a/Src. Signal Transduct Target Ther (2021) 6:354. doi: 10.1038/s41392-021-00765-3
85. Kovacic JC, Moreno P, Hachinski V, Nabel EG, Fuster V. Cellular senescence, vascular disease, and aging: Part 1 of a 2-part review. Circulation (2011) 123:1650–60. doi: 10.1161/circulationaha.110.007021
86. Gnani D, Crippa S, Della Volpe L, Rossella V, Conti A, Lettera E, et al. An early-senescence state in aged mesenchymal stromal cells contributes to hematopoietic stem and progenitor cell clonogenic impairment through the activation of a pro-inflammatory program. Aging Cell (2019) 18:e12933. doi: 10.1111/acel.12933
87. Wlaschek M, Maity P, Makrantonaki E, Scharffetter-Kochanek K. Connective tissue and fibroblast senescence in skin aging. J Invest Dermatol (2021) 141:985–92. doi: 10.1016/j.jid.2020.11.010
88. Vidal R, Wagner JUG, Braeuning C, Fischer C, Patrick R, Tombor L, et al. Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart. JCI Insight (2019) 4(4):e131092. doi: 10.1172/jci.insight.131092
89. D’apolito M, Colia AL, Lasalvia M, Capozzi V, Falcone MP, Pettoello-Mantovani M, et al. Urea-induced ROS accelerate senescence in endothelial progenitor cells. Atherosclerosis (2017) 263:127–36. doi: 10.1016/j.atherosclerosis.2017.06.028
90. Chan YH, Ngai MC, Chen Y, Wu MZ, Yu YJ, Zhen Z, et al. Osteogenic circulating endothelial progenitor cells are associated with vascular aging of the large arteries in rheumatoid arthritis. Clin Interv Aging (2022) 17:287–94. doi: 10.2147/cia.S337118
91. Wu H, Li JZ, Xie BD, Tian H, Fang SH, Jiang SL, et al. Lower senescence of adipose-derived stem cells than donor-matched bone marrow stem cells for surgical ventricular restoration. Stem Cells Dev (2018) 27:612–23. doi: 10.1089/scd.2017.0271
92. Wang TW, Johmura Y, Suzuki N, Omori S, Migita T, Yamaguchi K, et al. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature (2022) 611:358–64. doi: 10.1038/s41586-022-05388-4
93. Zhang X, Cao D, Xu L, Xu Y, Gao Z, Pan Y, et al. Harnessing matrix stiffness to engineer a bone marrow niche for hematopoietic stem cell rejuvenation. Cell Stem Cell (2023) 30:378–395.e8. doi: 10.1016/j.stem.2023.03.005
94. Reyes NS, Krasilnikov M, Allen NC, Lee JY, Hyams B, Zhou M, et al. Sentinel p16(INK4a+) cells in the basement membrane form a reparative niche in the lung. Science (2022) 378:192–201. doi: 10.1126/science.abf3326
95. Yousefzadeh MJ, Flores RR, Zhu Y, Schmiechen ZC, Brooks RW, Trussoni CE, et al. An aged immune system drives senescence and ageing of solid organs. Nature (2021) 594:100–5. doi: 10.1038/s41586-021-03547-7
96. Kovtonyuk LV, Fritsch K, Feng X, Manz MG, Takizawa H. Inflamm-aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Front Immunol (2016) 7:502. doi: 10.3389/fimmu.2016.00502
97. Kale A, Sharma A, Stolzing A, Desprez PY, Campisi J. Role of immune cells in the removal of deleterious senescent cells. Immun Ageing (2020) 17:16. doi: 10.1186/s12979-020-00187-9
98. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
99. Becker L, Nguyen L, Gill J, Kulkarni S, Pasricha PJ, Habtezion A. Age-dependent shift in macrophage polarisation causes inflammation-mediated degeneration of enteric nervous system. Gut (2018) 67:827–36. doi: 10.1136/gutjnl-2016-312940
100. Song X, Song Y, Ma Q, Fang K, Chang X. M1-type macrophages secrete TNF-α to stimulate vascular calcification by upregulating CA1 and CA2 expression in VSMCs. J Inflammation Res (2023) 16:3019–32. doi: 10.2147/jir.S413358
101. Lu J, Xie S, Deng Y, Xie X, Liu Y. Blocking the NLRP3 inflammasome reduces osteogenic calcification and M1 macrophage polarization in a mouse model of calcified aortic valve stenosis. Atherosclerosis (2022) 347:28–38. doi: 10.1016/j.atherosclerosis.2022.03.005
102. Byon CH, Sun Y, Chen J, Yuan K, Mao X, Heath JM, et al. Runx2-upregulated receptor activator of nuclear factor κB ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler Thromb Vasc Biol (2011) 31:1387–96. doi: 10.1161/atvbaha.110.222547
103. Yousefzadeh MJ, Zhu Y, Mcgowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine (2018) 36:18–28. doi: 10.1016/j.ebiom.2018.09.015
104. Xu Q, Fu Q, Li Z, Liu H, Wang Y, Lin X, et al. The flavonoid procyanidin C1 has senotherapeutic activity and increases lifespan in mice. Nat Metab (2021) 3:1706–26. doi: 10.1038/s42255-021-00491-8
105. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell (2015) 14:644–58. doi: 10.1111/acel.12344
106. Justice JN, Nambiar AM, Tchkonia T, Lebrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine (2019) 40:554–63. doi: 10.1016/j.ebiom.2018.12.052
107. Gonzales MM, Garbarino VR, Marques Zilli E, Petersen RC, Kirkland JL, Tchkonia T, et al. Senolytic therapy to modulate the progression of Alzheimer’s disease (SToMP-AD): A pilot clinical trial. J Prev Alzheimers Dis (2022) 9:22–9. doi: 10.14283/jpad.2021.62
108. Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature (2020) 583:127–32. doi: 10.1038/s41586-020-2403-9
109. Yang D, Sun B, Li S, Wei W, Liu X, Cui X, et al. NKG2D-CAR T cells eliminate senescent cells in aged mice and nonhuman primates. Sci Transl Med (2023) 15:eadd1951. doi: 10.1126/scitranslmed.add1951
110. Suda M, Shimizu I, Katsuumi G, Yoshida Y, Hayashi Y, Ikegami R, et al. Senolytic vaccination improves normal and pathological age-related phenotypes and increases lifespan in progeroid mice. Nat Aging (2021) 1:1117–26. doi: 10.1038/s43587-021-00151-2
111. Saul D, Kosinsky RL, Atkinson EJ, Doolittle ML, Zhang X, Lebrasseur NK, et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat Commun (2022) 13:4827. doi: 10.1038/s41467-022-32552-1
Keywords: vascular calcification, senescence, chronic kidney disease, vascular smooth muscle cells, vascular endothelial cells, vascular progenitor/stem cells, macrophages
Citation: Fang Y-P, Zhao Y, Huang J-Y, Yang X, Liu Y and Zhang X-L (2024) The functional role of cellular senescence during vascular calcification in chronic kidney disease. Front. Endocrinol. 15:1330942. doi: 10.3389/fendo.2024.1330942
Received: 31 October 2023; Accepted: 03 January 2024;
Published: 22 January 2024.
Edited by:
James Harper, Sam Houston State University, United StatesReviewed by:
Nhat Tu Le, Houston Methodist Research Institute, United StatesPanagiotis Vasileiou, National and Kapodistrian University of Athens, Greece
Copyright © 2024 Fang, Zhao, Huang, Yang, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Zhao, ZG5keHp5QDEyNi5jb20=; Xiao-Liang Zhang, dG9ueXhsekAxNjMuY29t
†These authors have contributed equally to this work
‡ORCID: Ya-Ping Fang, orcid.org/0000-0002-7996-1001
Yu Zhao, orcid.org/0000-0003-3295-7379