 Henry H. Lindner
Henry H. Lindner- Private Practice, Tunkhannock, PA, United States
The 2012 American endocrine associations' guidelines on hypothyroidism were a reiteration of the TSH-T4 Paradigm from the 1970s. They likewise defined hypothyroidism as hypothyroxinemia, assumed that almost all hypothyroidism was primary, and relied upon the thyroid stimulating hormone (TSH) test and inactive prohormone thyroxine (T4) for diagnosis and treatment. The guidelines’ authors acknowledged many TSH and other “pitfalls” in the paradigm yet warned physicians against attending to patients’ signs and symptoms and relative free T4 (FT4) and free triiodothyronine (FT3) levels—the only means by which to identify and avoid all pitfalls and provide individualized diagnosis and treatment. This inadequate paradigm has distorted medical practice and research for 50 years, including laboratories’ FT4 and FT3 reference ranges. It produces overdiagnosis, underdiagnosis, inadequate treatment, and widespread patient dissatisfaction. Since the 1970s, our understanding of thyroid hormone production, transport, metabolism, reception, and signaling has increased greatly, as has our appreciation of the importance of optimal T3 effects for health and wellbeing. Hypothyroidism must be defined physiologically as insufficient T3 effect in some or all tissues. The best indicators of tissue T3 effect are the patient’s signs and symptoms, and the best serum tests are FT4 and FT3, considered together. The TSH level is not a reliable indicator of T3 status in the untreated state and is oversuppressed by the peak levels that occur with once-daily oral T4 and/or T3. Normalizing an elevated TSH or low FT4 with T4 usually does not produce sufficient, let alone optimal, T3 effect and can leave some patients markedly hypothyroid. T4/T3 combination therapy is more physiological and effective than T4 monotherapy and must be guided by clinical criteria, not the TSH. Some patients cannot tolerate more T3 effect due to hypocortisolism, inflammation, and other disorders. There is no substitute for the practice of fully informed clinical medicine.
The ultimate test of whether a patient is experiencing the effects of too much or too little thyroid hormone is not a measurement of hormone concentration in the blood or of the size and functional activity of the thyroid but of the effect of thyroid hormones on the peripheral tissues. Unfortunately, no simple, reproducible, and specific tests are available to do this (1).
1 Introduction
The biologically active human thyroid hormone is triiodothyronine (T3). It stimulates mitochondrial function and biogenesis (2, 3), gluconeogenesis (4), and glucose transport into cells, thereby increasing the energy available to every cell, tissue, and organ and improving their function. It also alters the expression of thousands of genes (5, 6). Optimal T3 effect is essential to every aspect of human development, health, and vitality, including the function of the rest of the endocrine system.
Under the influence of thyroid-stimulating hormone (TSH), secreted by the hypothalamic-pituitary (HP) system, the thyroid gland secretes thyroxine (T4) and T3. T4 is its dominant product but is only a prohormone—inactive until converted into T3. While the serum T4 level is largely determined by the HP system, the serum and intracellular T3 levels are largely determined by the intracellular deiodinases: D1, D2, and D3. Their amounts and activities vary from tissue to tissue. They locally increase or decrease T3 availability in a time-variable fashion (7, 8). Much of the T3 in the serum is derived from peripheral T4-to-T3 conversion (9). The serum FT4 level is three times greater than the FT3 level (in pmol/l). Different tissues obtain T3 by different mechanisms. Some preferentially take in T4 from the serum and convert it to T3 (e.g., brain, pituitary, heart, brown adipose tissue), whereas others are more reliant on serum T3 (e.g., liver, kidneys, adrenal glands) (10). We are still learning to appreciate the complexities and fallibilities of T3 transport, reception, and signaling (11). Therefore, the amount of T3 within the cells of any given tissue and the resultant T3 effect are dependent upon a complex chain of events, any link in which may be defective in any person, resulting in hypothyroidism in some or all tissues (12) (Table 1).

Table 1. Steps required to produce T3 effect.
Hypothyroidism is the clinical state resulting from insufficient T3 effect in some or all tissues of the body. It varies from mild to severe and can variably affect different tissues. Its clinical presentation varies with the person’s genetics, cortisol status, medical conditions, nutrition, and inflammation. Hypothyroidism causes a vast array of signs and symptoms and causes or contributes to a host of medical and psychiatric disorders and diseases. (See Tables 2, 3 for partial lists.) Indeed, nearly every functional complaint and disorder encountered in medical practice can either be caused by hypothyroidism in whole or in part or be affected by it. Therefore, physicians must know how to determine every patient’s thyroid status and how to effectively diagnose and treat every variety and degree of hypothyroidism.
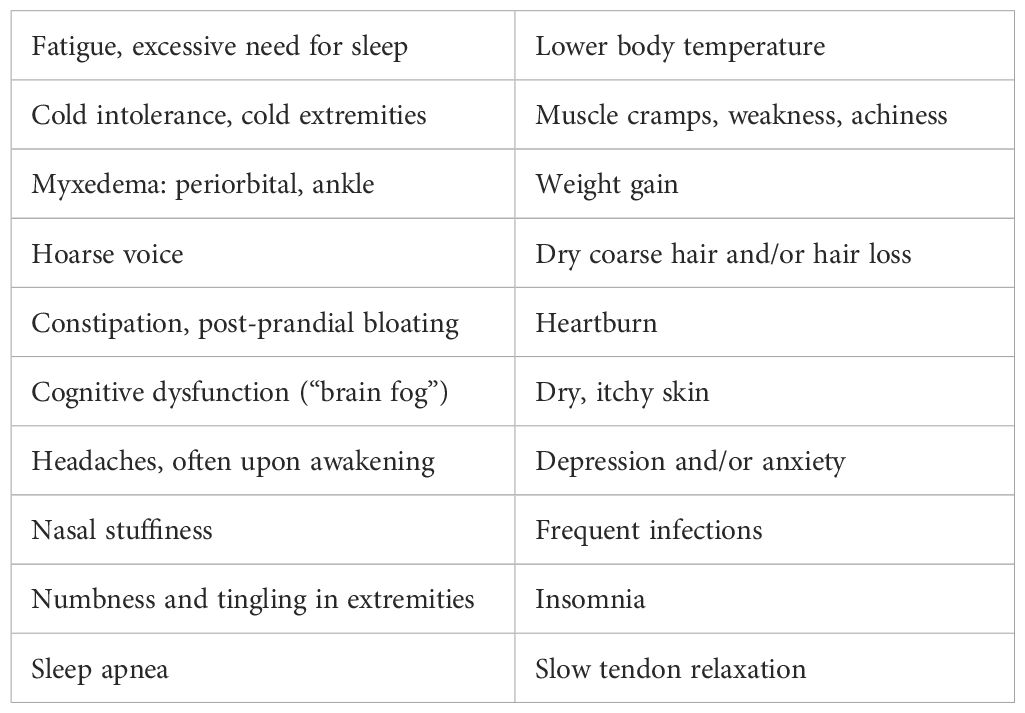
Table 2. Hypothyroidism: signs and symptoms.
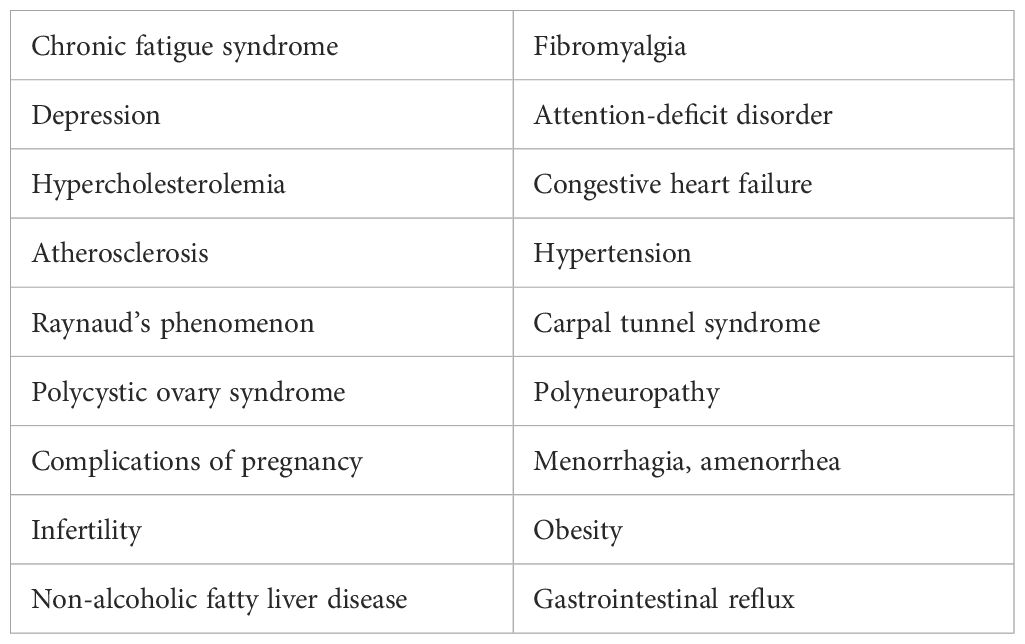
Table 3. Hypothyroidism: associated medical conditions.
2 The TSH-T4 Paradigm
How does a physician decide if a patient is suffering from insufficient T3 effect? How does a physician intervene to optimize T3 effect in all tissues while avoiding overdosing? For answers, physicians look to guidelines published by endocrine associations. In their most recent guidelines, the American Thyroid Association (ATA) and the American Association of Clinical Endocrinology (AACE) claim to produce “evidence-based clinical guidelines for the clinical management of hypothyroidism” (13). However, their guidelines are neither clinical nor evidence-based. Their assumptions, analyses, and recommendations are all products of an approach invented in the 1970s that I call the “TSH-T4 Paradigm”. A paradigm is a set of ideas (e.g., definitions and assumptions) according to which evidence is gathered and interpreted. Contradictory evidence is often ignored or recategorized—as in this case. False paradigms can dominate sciences for decades or even centuries before they are replaced (14).
2.1 Clinical thyroidology before the TSH-T4 Paradigm
In the late 19th and early 20th centuries, physicians linked the conditions of myxedema and cretinism to the thyroid gland—to its damage or disease. They discovered that they could correct hypothyroidism with extracts of mammalian thyroid glands. Desiccated thyroid extract (DTE) was initially obtained from the thyroid glands of sheep, cows, or pigs. It was standardized by organic iodine content and contained T4 and T3 in ratios ranging from 2:1 to 5:1 (15). By the 1930s, most DTE was branded porcine thyroid, Armour Thyroid® and Westhroid®, which had a reliable content and 4:1 T4/T3 ratio. DTE was the standard treatment for hypothyroidism from the 1890s to the 1970s. Physicians adjusted the DTE dose by clinical criteria—signs and symptoms. Experts affirmed the efficacy and safety of clinically adjusted DTE therapy (16). Although levothyroxine (T4) became available in 1955, most physicians continued to prescribe DTE because it was effective and normalized the serum protein-bound iodine (PBI). T4 monotherapy produced a high PBI and physicians feared that it would cause T3 deficiency. Restoring clinical euthyroidism required 2 to 3 grains (120 to 180mgs) of DTE, 200 to 400mcgs of T4, or 75 to 125mcgs of T3 (15, 17). T4/T3 ratios of 3.3:1 and 4:1 worked best to produce clinical euthyroidism and a normal PBI (15, 18).
2.2 Invention of the TSH-T4 Paradigm
In the early 1970s, a reliable test for TSH became widely available. Physicians discovered that most patients who were on clinically optimized DTE or T4 doses had low or suppressed TSH levels. They assumed that this indicated overtreatment, despite the lack of clinical evidence (19). Normalizing an elevated TSH required much lower DTE or T4 doses than recommended in the textbooks, often just one-half of the clinical dose (19–22). Research revealed that T4 was converted into T3 (23), alleviating the fear of T3 deficiency with T4 monotherapy. T4 produced more stable T4 and T3 levels, but higher FT4 and lower FT3 levels than seen in healthy persons.
A few endocrinologists believed that they could replace clinical diagnosis and DTE-T4/T3 treatment with a simple laboratory-based approach to diagnosis and treatment using T4 monotherapy to normalize the TSH: the TSH-T4 Paradigm (24, 25). They assumed that almost all hypothyroidism was primary. They cited studies that found that T4 doses that produced a low or suppressed TSH level and/or high FT4 levels were often associated with changes seen in hyperthyroidism, including increases in liver enzymes (26, 27), bone loss in women (28), and reduced cardiac systolic time intervals (29). An influential review concluded that “overzealous treatment” was producing “subclinical hyperthyroidism”, and that the goal of treatment should be a normal TSH (30). Some cited reports of thyrotoxicity with excessive doses of DTE (31) and T4/T3 (32), and the high T3 levels after doses to argue that DTE and T3 should not be prescribed.
From the start, it was clear that TSH-normalizing T4 therapy (TSHT4Rx) produced suboptimal clinical results. It produced Billewicz hypothyroidism scores (33) in the equivocal hypothyroid or, at best, the low euthyroid range (34). Adding 50mcgs of T4 to the TSH-normalizing dose raised Billewicz scores into the low euthyroid range and improved self-reported wellbeing (35) but was believed to be overtreatment. The paradigm’s architects ignored a T4 dosing study that found the TSH to be the least reliable, and the FT3 the most reliable serum indicator of clinical euthyroidism (36).
2.3 ATA/AACE’s TSH-T4 guidelines
The authors of the 2012 guidelines adhere to the TSH-T4 Paradigm but are conflicted. They similarly define hypothyroidism as an underactive thyroid gland indicated by a high TSH and/or low FT4. They state that the first goal of treatment is to resolve symptoms and admit that their guidelines cannot replace clinical judgment. However, they then warn physicians against attending to clinical criteria and using clinical judgment. After describing the euthyroid state as the “normalization of a variety of clinical and metabolic end points”, they thereafter equate euthyroidism with normal TSH and FT4 levels. They claim that neither clinical criteria nor relative FT4 and FT3 levels can be used to diagnose hypothyroidism, and “do not have sufficient specificity to serve as therapeutic endpoints…”. They provide no evidence or arguments for these beliefs; they are only assumptions of the paradigm (Table 4). They thus pay lip service to clinical thyroidology while promoting TSH normalization with T4 as the “standard treatment”.

Table 4. The TSH-T4 Paradigm’s assumptions.
The authors also assume that almost all hypothyroidism is primary—thyroid gland disease. They provide no evidence for this assumption because none exists. Such a study would require a definition of hypothyroidism that is not TSH-dependent and includes clinical and physiological criteria, FT4 and FT3 levels, and clinical response to a trial of effective T4/T3 treatment. (See below.) The authors frequently omit the adjective “primary” from “hypothyroidism”, thus reinforcing the idea that all hypothyroidism is thyroid gland disease evidenced by a high TSH, and thyroidology is just “TSH control” (37).
The authors consider central hypothyroidism (suboptimal TSH production) to be extremely rare, limited to cases of obvious HP disease or damage (~1 in 100,000 persons). They thus assume that every anatomically intact HP system functions perfectly. They neither state this assumption nor present any evidence to support it because it is false. (See TSH “pitfalls” below.) To this, the authors add the highly improbable assumption that the HP system responds to once-daily oral thyroid replacement therapy (TRT) exactly as it does to continuous endogenous thyroidal T4/T3 output. This too is contradicted by the evidence. They assert that a normal TSH level during the treatment of primary hypothyroidism with T4 is “euthyroidism”. To support this claim, they cite one old, small observational study (38), while almost all other studies refute it. (See below.)
The authors’ reliance on the TSH and T4 monotherapy requires the assumption that T4-to-T3 conversion is always perfect—sufficient to restore optimal FT3 levels and effects in all tissues if the FT4 is normal. They cite only one study to support this claim (39); while almost all other studies refute it. (See below.) Assuming perfect conversion, they see no reason to check a patient’s T3 level or prescribe T3. They state that a low FT3 should be ignored, as should persisting signs and symptoms of hypothyroidism, as long as the TSH and/or FT4 are normal. The authors discuss thyroid resistance separately because it is outside the paradigm, even though it is a cause of hypothyroidism as properly defined.
2.4 Acknowledged and unacknowledged TSH “pitfalls”
Reliance on the TSH test is illogical, as proven by the fact that the guidelines’ authors must warn physicians of 14 pitfalls to be avoided when attempting to rely on the TSH and its reference range (Table 5). Logically then, the TSH level is neither a sensitive nor specific indicator of thyroid (T3) status: It is the wrong test. The authors assume that physicians will somehow recognize and avoid all these pitfalls. How? There is only one way: by attending to the true indicators of T3 availability and effect—the relative FT4 and FT3 levels and, most importantly, the patient’s signs and symptoms. That is how each of the pitfalls was discovered and it is the only way to recognize them now. I will demonstrate that doing so results in the detection of more pitfalls (Table 6).
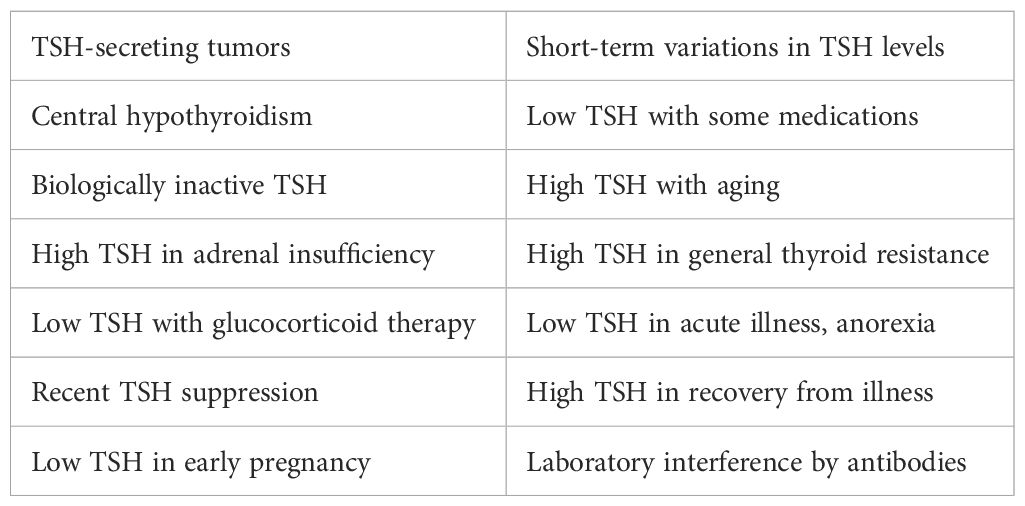
Table 5. ATA/AACE-acknowledged TSH pitfalls.

Table 6. Unacknowledged TSH pitfalls.
Because they know that the TSH can mislead, the guidelines’ authors state that hypothyroidism must be “biochemically confirmed” by a low FT4. Logically then, physicians should check the FT4 first to establish the diagnosis of hypothyroidism and then check the TSH to see if it is primary or central. However, they know that relying on a normal (within reference range) FT4 also has pitfalls. T4 is inactive and T4-to-T3 conversion may or may not be compensatory. T4/FT4 levels can be increased or decreased by various non-thyroidal causes (40). Patients can be clinically hypothyroid with a normal FT4, and merely normalizing a low FT4 with T4 therapy will usually be inadequate. So, they attempt to rely on the TSH level, despite its many pitfalls.
The TSH-T4 Paradigm still dominates the practice of medicine today. Most physicians follow the ATA/AACE’s primary message: The TSH level tells them everything they need to know about a patient’s thyroid status, untreated or treated. They typically order only a TSH, or TSH with reflex—the FT4 is checked only if the TSH is abnormal. If the TSH is elevated, physicians usually prescribe just enough levothyroxine to normalize it. If the TSH is low on TRT, they declare the patient “thyrotoxic” and reduce the dose, even though the patient is feeling well and has no signs or symptoms of hyperthyroidism. If the patient feels worse on the lower dose, they will not raise it. They do not check the FT3 level and typically refuse to prescribe DTE or any T3. For patients, the results of this anti-clinical thyroidology range from suboptimal to catastrophic.
2.5 False diagnostic scheme
Because the TSH-T4 Paradigm is arbitrary, illogical, and isolated from clinical reality, it necessarily produces diagnostic and therapeutic errors: underdiagnosis, overdiagnosis, unnecessary treatment, and undertreatment. It is immune to correction by its clinical failures. The paradigm implies, and physicians follow, a simplistic two-test diagnostic scheme for the diagnosis of hypo- and hyperthyroidism and determination of the cause (Table 7).
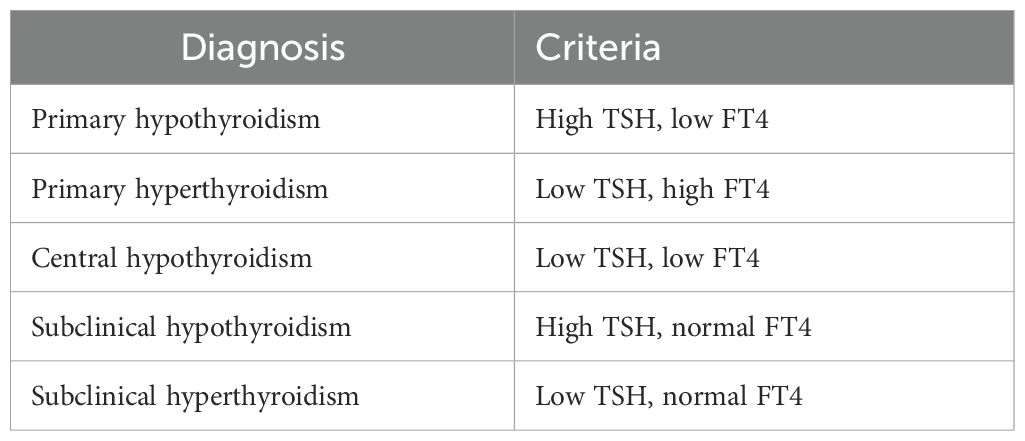
Table 7. The TSH-T4 Paradigm’s diagnostic scheme.
Since this algorithm does not include signs, symptoms, or relative FT4 and FT3 levels, it guarantees errors. It even creates two pseudo-diagnoses. A patient with a high TSH and normal FT4 is labeled “subclinical hypothyroidism”. However, the normal FT4 and/or FT3 levels may be low in-range, midrange, or high in-range. The patient may be clinically hypothyroid, euthyroid, or hyperthyroid, or may suffer from hypocortisolism. This is the bulk of the 15% of older adults who are diagnosed with hypothyroidism and given T4 (41). The low T4 doses usually produce no clinical improvement (42, 43) and can reduce T3 production, levels, and effects. (See below.) The scheme fails to diagnose partial central hypothyroidism and partial peripheral thyroid resistance because the TSH and FT4 are usually normal.
2.6 Patient dissatisfaction and physician disagreement
Since its invention, the TSH-T4 Paradigm has produced dissatisfaction among patients. Many testify that their health and vitality were restored only when they were diagnosed and treated clinically—according to their symptoms, not their TSH level. Most state that T4/T3 combination therapy, usually DTE with its 4:1 ratio, works better than T4 monotherapy, and that the doses they require produce a low or suppressed TSH (44–49). Patients with residual symptoms on T4 prefer T4/T3 and DTE (50–53). Published surveys have found prominent patient dissatisfaction with TSHT4Rx and greater satisfaction with DTE (54, 55). Patients reported that on DTE they experienced improved weight management, energy, mood, and memory (56). Many experienced physicians, including some prominent former advocates of the paradigm, have argued that hypothyroidism should be treated according to clinical criteria, not the TSH, and/or that treatment should include T3 (57–69).
3 Why the TSH-T4 Paradigm fails to diagnose most hypothyroidism
3.1 Misunderstanding and misuse of endocrine reference ranges
Most endocrine tests are reported with broad population ranges, not with physician-adjudicated ranges. Their limits are just population statistics: two standard deviations (SDs) from the mean of the tested group. This includes the middle 95% of the tested population and so defines almost everyone as “normal”. Only the highest and lowest 2.5% are reported as “high” or “low”. To be used as diagnostic ranges, the persons tested to produce the ranges should be very carefully screened for any signs or symptoms of deficient or excessive hormone effect. They should also have ideal-optimal health and vitality. However, the subjects are not screened for symptoms; laboratory guidelines do not call for it (70). In addition, all subjects would need to have the same hormone sensitivity.
To produce a suggested reference range, a test kit manufacturer usually samples 120 or so “apparently healthy” persons. They are screened only for diseases and medications. The lack of symptom screening and the use of 2SDs to define “normal” explain the breadth of endocrine ranges. The upper limits are typically 2 to 5 times greater than the lower limits, and up to 30 times greater (Table 8). The ranges are far from representing “the most normal of normals”, as often claimed.
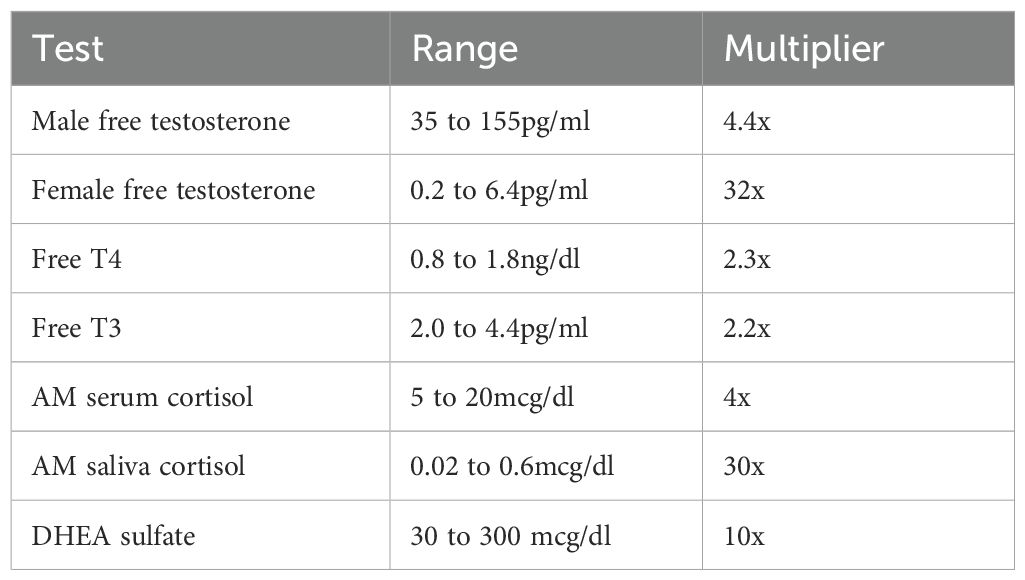
Table 8. Breadth of endocrine reference ranges.
A large problem in the interpretation of hormone levels is their greater degree of individuality compared to most of the tests that physicians perform every day—tests that have narrow ranges and low degrees of individuality (e.g., serum sodium levels). Physicians carry their training and experience with narrow-ranged, low-individuality tests over to the broad-ranged, high-individuality endocrine tests and so think that a hormone level anywhere within the laboratory’s range is “sufficient”. They will often ignore a hormone level that is “a little low”. However, a healthy person’s hormone levels vary much less with time than the breadth of the population range (high degree of individuality) (71). Hormone effects also exist on a continuum from lower to higher levels. Consider that patients could have FT4 and FT3 levels that are one-half of their previous levels yet still test as “normal”. Even if we had perfectly accurate tests and all persons had the same hormone sensitivity and milieu, the placement of dividing lines between “deficient”, “sufficient”, and “excessive” would still be an arbitrary act. Reference ranges only provide a statistical description of the population studied. They do not define what is optimal for the human species, let alone for any individual.
3.2 Distortion of FT4 and FT3 reference ranges
The FT4 and FT3 ranges reported by laboratories are distorted by a practice that is unique in laboratory science. As a result of the TSH-T4 Paradigm, laboratory scientists believe that a normal TSH equals euthyroidism. Therefore, they think it appropriate to include FT4 and FT3 levels from physician-ordered thyroid panels in their reference range determinations—if the TSH was normal. Their ranges thus include symptomatic untreated and T4-treated clinic and hospital patients. The result is evident in their ranges’ increased breadth compared to those found in studies of non-patients. With kits that yield similar values, laboratories report FT4 ranges with lower limits of only 0.6 to 0.8ng/dl and upper limits of 1.8 to 2.2ng/dl. The upper limits are 2 to 3 times greater than the lower limits. In contrast, studies of non-patient populations like blood donors and soldiers, without symptom screening, consistently yield narrower 2SD FT4 ranges of around 1.0 to 1.65ng/dl (72–78). The results of this misuse of the TSH test were documented in a test kit manufacturer’s detailed report (Table 9). The advised reference ranges included patient data (79). The large differences in the lower limits indicate that dysfunctional TSH secretion and T4-to-T3 conversion are much more common than primary hypothyroidism, which affects only 0.3 to 0.4% of the population (80, 81).
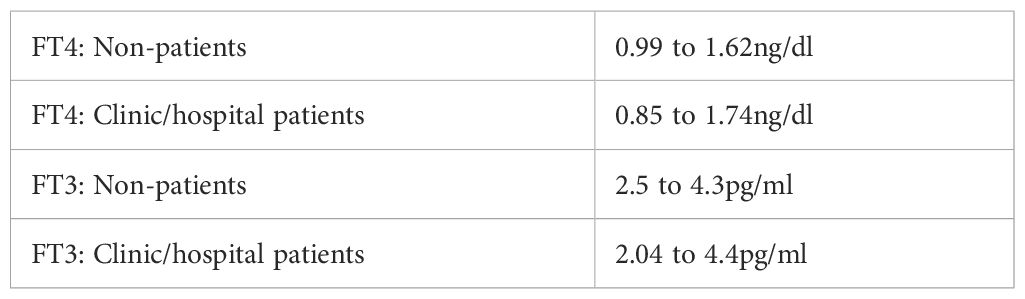
Table 9. Non-patient vs. patient ranges.
Laboratories using similar kits should report FT4s with a lower limit of 1.0ng/dl (range: 1.0 to 1.65) and FT3s with a lower limit of 2.5pg/ml (range: 2.5 to 4.3). However, these are still symptom-unscreened population ranges, and diagnosis should never be made by a laboratory test alone. FT4 and FT3 test results can also be inaccurate (82–84) and patients vary greatly in their need for, and response to T4 and T3 levels. There is no laboratory substitute for the physician’s clinical judgment.
3.3 FT4 and FT3 levels matter—within their reference ranges
The insensitivity of the TSH test and FT4/FT3 population ranges is illustrated by many published studies that associate indices of better well-being and health in untreated persons with higher compared to lower FT4 and/or FT3 levels within their ranges. For instance, higher T4 levels are associated with better cognitive function (85, 86) and lower risk of cognitive decline (87). Lower maternal FT4 levels in the first trimester of pregnancy are associated with lower neonatal neurobehavioral scales (88), impaired psychomotor development (89), and autism (90). Lower T4 and T3 levels are associated with depression or a worse prognosis for remission of depression (91–94). Persons with lower FT4 levels complain more of myalgias and weakness and have lower muscle strength on testing (95). Higher FT4 levels are associated with lower all-cause mortality and higher FT3 levels with lower cancer mortality (96). Higher FT4 levels are associated with less arterial stiffness, lower diastolic blood pressure, and less insulin resistance (97). Carotid artery intimal thickness is inversely associated with FT4 levels (98, 99). Persons undergoing cardiac catheterization who have FT3s in the highest tertile have half the incidence of severe coronary atherosclerosis as those in the lowest tertile (100). Lower FT4 levels are associated with hypercoagulability (101), higher body mass index and weight gain (102), subcutaneous fat (103), metabolic syndrome (104–106), and non-alcoholic fatty liver disease (107). Lower FT3 levels are also associated with a lower metabolic rate and weight gain (108). One can find studies that do not show these correlations or, in some cases, show the opposite. A sufficient explanation is that a FT4 or FT3 level by itself is insufficient to assess the thyroid status of any individual. They must be considered together to obtain the best serum estimate. (See below.)
3.4 Nature of the TSH-FT4 population correlation
The ATA/AACE’s emphasis on the TSH test encourages physicians to think that the higher the patient’s TSH level, the lower their thyroid (T3) status, and vice versa, even within the TSH range. This is an example of the ecological fallacy, i.e., drawing an inference about an individual from a correlation seen in the group (109). The best-fit lines in Figure 1 show the general inverse correlation between TSH and FT4 levels in a population of untreated (b) and untreated plus T4-treated patients (B) referred to a tertiary center (110). Patients with HP disease/damage were excluded. Reliance on the TSH might be defensible if every data point were close to the best-fit line. However, the data points are widely scattered—very high and low TSH levels coexist with normal FT4 levels and vice versa. The scatter is greater in the T4-treated patients. The group correlation clearly has no relevance to any individual. The general group correlation is also absent in the middle of the FT4 reference range, where the slope is almost flat. The TSH-FT4 correlation has strength only at higher and lower FT4 levels due to the presence of a subgroup with the correlation: patients with primary hypothyroidism and hyperthyroidism. The large scatter in the TSH levels attests to the high degree of individuality in HP-thyroidal axis function. The TSH level is clearly an unreliable inverse indicator of any patient’s FT4 level.
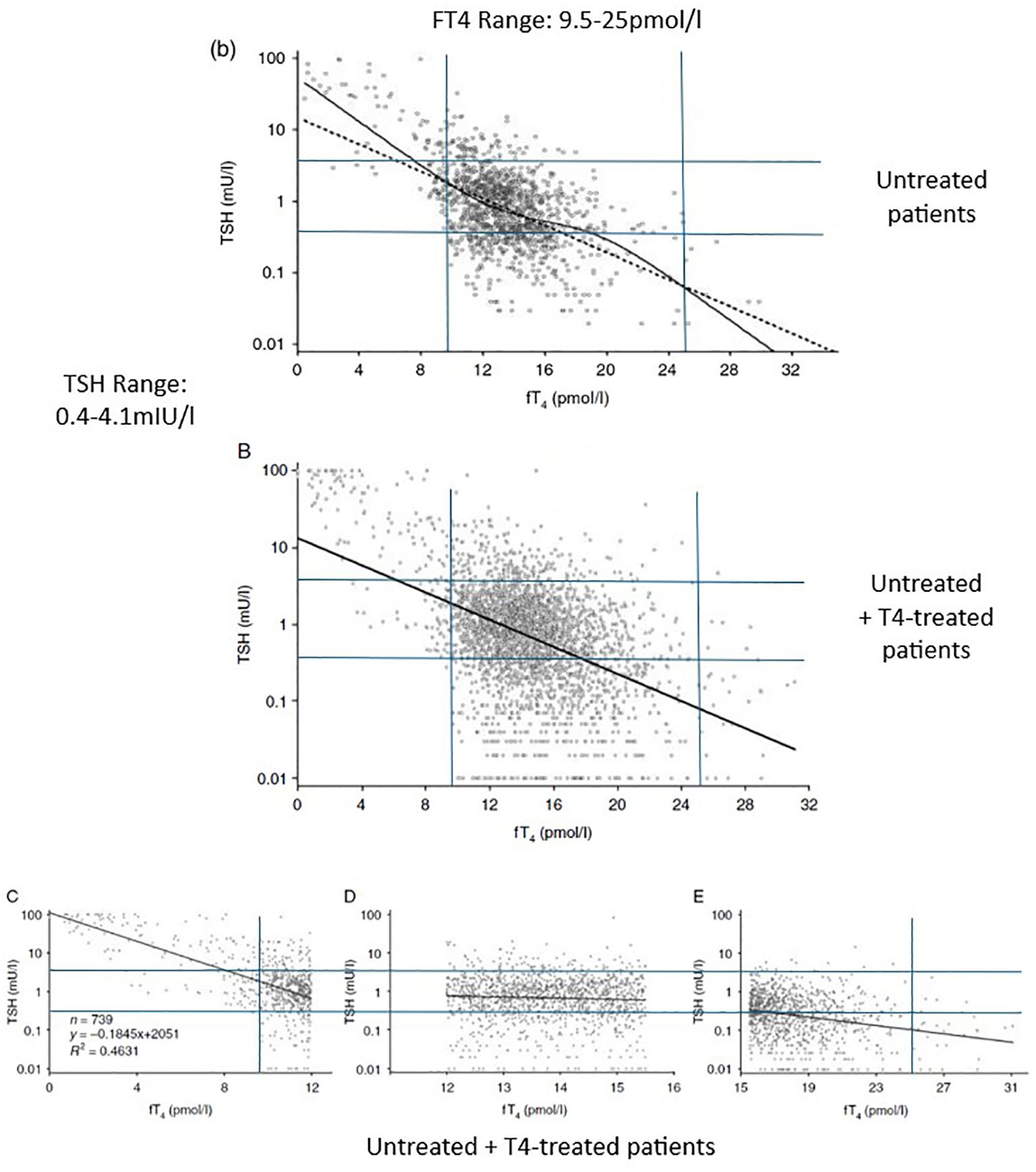
Figure 1. The TSH-FT4 correlation seen in untreated (b) and untreated plus T4-treated patients (B–E). The TSH and FT4 reference ranges (in blue) were added by the author (by permission of Oxford University Press). Adapted with permission from ‘Complex relationship between free thyroxine and TSH in the regulation of thyroid function’ by Hoermann R, Eckl W, Hoermann C, Larisch R, license number 6043140018466, Oxford University Press.
3.5 Why TSH is not a reliable indicator of T3 levels or effects
The TSH’s many pitfalls arise from the attempt to use the level of a stimulatory pituitary hormone as a surrogate indicator of end-hormone levels and effects. It is for good reasons that physicians do not attempt to do so with the gonadal and adrenal systems. It is equivalent to claiming that one’s home heating thermostat is working perfectly even as the house continues to get colder. Absent disease of the primary gland, the HP system controls serum hormone levels, and it is neither perfect nor passive. The hypothalamus and pituitary gland are parts of the brain and are affected by inputs from many regions of the brain and by many neurotransmitters. The HP system is also part of the larger neuro-endocrine-immune system and is affected by genetic variants, cytokines, illnesses, nutritional factors, chemicals, drugs, stress, etc. Because the HP system is much more complex than the primary gland, it is much more likely to be dysfunctional. It should be so judged when the clinical and laboratory evidence so indicates.
TSH is produced and secreted as a mixture of isoforms, the majority of which have differences in their oligosaccharide structure and possess different bioactivity (111, 112). Dysfunctional central hypothyroidism has been associated with relatively inactive forms of TSH, genetic variants, and other molecular disorders (113–115). There are published reports of patients with central hypothyroidism who had normal HP imaging studies (116–119). Most patients with central hypothyroidism due to known HP damage/disease have normal TSH levels and often have normal, but low-in-range FT4 levels (120–122). This “euthyroid” central hypothyroidism is another pitfall in the paradigm. The HP-thyroidal system also becomes dysfunctional with age. Between the ages of 20 and 80, the TSH response to low FT4 levels declines by 75% (123).
The HP system necessarily responds differently to serum T4 and T3 levels than other tissues, in some persons pathologically so. Whereas other tissues have separate membrane transporters for T4, T3, and reverse T3 (rT3), the pituitary has one transporter for all three (124) and it is not energy-dependent (T3-dependent) as are those in other tissues (125, 126). There are also four different T3 receptors (TRs) with various tissue distributions. The HP system primarily expresses TRβ2 (127). The function of any of these tissue-specific proteins can be altered by single nucleotide polymorphisms (SNPs) in any individual.
In mild thyroid gland failure, the elevated TSH may be perfectly compensatory—maintaining T3 effect in the periphery by assuring sufficient thyroidal T4/T3 output and peripheral T4-to-T3 conversion. (See below.) Other persons with the same higher TSH may be hypothyroid. Some may be hypothyroid with a normal or low TSH due to dysfunctional TSH hyposecretion or partial peripheral thyroid resistance (e.g., reduced T4-to-T3 conversion), or both.
Physicians defend the paradigm’s reliance on the TSH test by touting its “sensitivity”. The TSH level does respond in a logarithmically amplified way to changes in the serum FT4 level in any given person. In addition, the latest-generation TSH test is highly precise; it can measure the TSH to within 1/1000th of one mIU/l. However, neither of these facts implies diagnostic sensitivity or specificity. In fact, a TSH level alone implies nothing certain about any individual’s T3 status. It has no meaning apart from the FT4 and FT3 levels and the clinical context.
3.6 Why FT4 is not a reliable indicator of T3 levels or effects
The FT4 level indicates the serum availability of the more abundant prohormone. Compared to the TSH, it has a more reliable causal relationship with T3 availability and therefore T3 effect. The ATA/AACE argue that one can rely on the FT4 level because when “sufficient” T4 is available the deiodinases will adapt and assure optimal intracellular T3 availability and effect in every tissue. Some have proposed that the FT4 replace the TSH as the primary measure of thyroid status (128).
However, the FT4 level is still an indirect indicator. While the serum T4 level is controlled by the HP system, the production and elimination of T3 are largely controlled by intracellular deiodinases: D1, D2, and D3. Their concentrations and activities vary from tissue to tissue (8). They are affected not only by serum T4, T3, and TSH concentrations but also by many other factors: e.g., genetic variants, aging, inflammation, cytokines, nutrients, chemicals, cortisol, testosterone, growth hormone, etc.
In untreated primary hypothyroidism and less so in central hypothyroidism, deiodinases do attempt to compensate for the relative lack of T4 by increasing their conversion of T4 to T3. Patients with primary hypothyroidism are much less symptomatic if their FT3 is within range rather than low (129). When conversion is vigorous and compensatory, the FT3 can be above mid-range or even high. However, people with a mildly elevated TSH who lack compensatory conversion can have FT4 and FT3 levels that are “normal”, but if both are low in their ranges can suffer from hypothyroidism—even myxedema coma (130). T4-to-T3 conversion declines with aging. TSH and FT4 tend to rise while FT3 steadily declines, with deleterious effects (131).
The deiodinases are altered by genetic variants in many persons—another anomaly for the paradigm. A large percentage of the population has D1 and/or D2 variants that reduce T4-to-T3 conversion. The C allele of locus rs225014 of D2 (a.k.a. Thr92Ala) has a frequency of 39% in the population, so 15% of persons are homozygous. On T4 monotherapy post-thyroidectomy, homozygotes have lower FT3 levels (132). They have lower quality of life scores and experience significant improvement on T4/T3 combination therapy (133). The T allele of locus rs11206244 of D1 (a.k.a. C785T or D1a C/T) has a frequency of 34%, so 12% of the population is homozygous. Homozygotes have reduced T4-to-T3 conversion and reduced rT3 elimination. They have higher rT3/T4 ratios and lower T3/rT3 ratios (134). rT3 is not just inactive; it inhibits D2 activity and thereby T4-to-T3 conversion (135–138). Homozygotes have a greater tendency to major depression (139) that responds to T3 supplementation (140). Given these alleles’ frequencies, 13% of the population is heterozygous for both D1 and D2 variants, and 2% is homozygous for both.
Heterozygosity for either one of these variants has been associated with hypothyroid symptoms and various health problems, while having little effect on serum TSH, FT4, or FT3 levels (141). Homozygosity further impairs T4-to-T3 conversion. Homozygosity for one or even both of these known variants (i.e., partial peripheral hypothyroidism) is much more common than primary hypothyroidism. Many persons with deiodinase variants will be sufficiently symptomatic that they require T4/T3 therapy. A few require T3 monotherapy (142).
Other forms of genetic and acquired peripheral thyroid resistance are more common than currently acknowledged (143). Any one of the many steps involved in producing T3 effect (Table 1) can be defective in some or all tissues (144–146). Many of these proteins vary among the tissues, so peripheral thyroid resistance can be tissue-specific. There are at least 10 active T4 and T3 transport systems with variable tissue distributions (147, 148). There are four known T3 receptors and resistance syndromes have been associated with variants of each (149), some of which produce hypothyroidism while TSH, FT4 and FT3 levels remain normal (150, 151). There are at least 13 thyroid receptor-interacting proteins (TRIPs) that modulate T3 signaling and effect in various tissues (152). Each of these proteins’ functions can be affected by SNPs, nutritional deficiencies, toxins, and other factors. Each protein is also a possible target of autoimmunity. Unlike generalized thyroid resistance that also affects the HP system, in peripheral resistance the TSH is usually normal as are the FT4 and FT3 levels (134).
3.7 Most persons suffering from hypothyroidism have normal “thyroid function” tests
The preceding discussion explains why normal “thyroid function tests” (TSH and FT4) do not imply that a person has sufficient, let alone optimal T3 levels and effects in all tissues. Of course, population studies find that hypothyroid symptoms are more common in persons with low FT4 levels than in those with normal FT4 levels (153, 154). This is an expected population correlation; it does not justify defining hypothyroidism as a “low FT4”. What matters is T3-related physiology—of which symptoms are the most sensitive indicator. The ATA/AACE attempt to dismiss symptoms as guides to diagnosis by citing a study that found that hypothyroid symptoms were prevalent in persons with normal TSH and FT4 levels (81). However, if one defines hypothyroidism as insufficient T3 effect one draws a very different conclusion.
In that study of persons attending a health fair, of those who were not taking thyroid medication 8.5% had “subclinical hypothyroidism” and only 0.4% had “overt hypothyroidism”. However, “euthyroid” (TSH-T4 normal) persons were nearly as likely to have hypothyroid signs and symptoms as “subclinical hypothyroid” persons. Using the Billewicz score, “euthyroid” persons were also nearly as likely as “overt hypothyroid” persons to have moderate hypothyroidism (48% vs. 52%). Among those with Billewicz scores representing severe hypothyroidism, “euthyroid” persons outnumbered “overt hypothyroid” persons by a 50-to-1 margin (20% of 22,842 vs. 80% of 114).
The ATA/AACE guidelines’ authors dismiss the possibility of hypothyroidism in symptomatic persons with normal TSH and FT4/FT3 levels by citing one study in which such patients and healthy controls were given 100mcgs of T4 daily or placebo (155). T4 treatment caused both groups to have lower SF-36 quality of life scores, while scores improved on placebo. This subreplacement T4 dose lowered TSH and raised FT4 and FT3 levels. The timing of the blood test relative to the last dose was not stated. The FT3 rise was far less in the patient group, suggesting reduced T4-to-T3 conversion. The poor clinical result suggests that this T4 dose reduced T3 effect in some tissues. It was not an effective trial of thyroid optimization (See below). In contrast, when patients with normal TSH and FT4 levels but long-standing hypothyroid symptoms were treated with up to 200mcgs of T4 according to clinical criteria, most benefitted dramatically (156). In a case series of “euthyroid” patients, T3 and T4 prescribed to optimize the T3/rT3 ratio and maintain a low-normal FT4 produced dramatic improvements in symptoms and many disorders (66). I have seen similar dramatic improvements with prescribing T4/T3 therapy to symptomatic “euthyroid” persons. Most had low-in-range FT4 and/or FT3 levels but a few had mid-range levels.
4 Why the TSH-T4 Paradigm fails to produce euthyroidism
Consider that even if TSH production and T4-to-T3 conversion were perfectly vigorous in a patient, the many checks and balances in the complex thyroidal system make it highly improbable that an unphysiological intervention like once-daily oral T4 or even T4/T3 therapy could produce optimal tissue T3 levels and effects in all tissues while producing the same serum TSH, FT4, and FT3 levels seen in euthyroid controls. In fact, there is only one way to do so. In a landmark study involving thyroidectomized rats, investigators measured the TSH and the serum and tissue levels of T4 and T3 in animals given various amounts of T4 and/or T3 by continuous infusion. A T3 infusion that produced normal serum T3 levels failed to restore T3 levels in many tissues (157, 158), as expected in the absence of T4. A T4 infusion failed to restore T3 levels in the serum and some tissues until T4 levels were supraphysiological and the TSH suppressed (159). Only a continuous infusion of T4 and T3 in a 6:1 ratio restored both serum and tissue levels of T4 and T3 to those of controls without suppressing the TSH (10). In another study of thyroidectomized rats, normalizing the TSH with T4 delivered continuously by subcutaneous pellets also failed to restore T3 levels in the serum and some tissues; to do so required the addition of T3 (160).
4.1 HP system is uniquely sensitive to T4
The inability of T4 monotherapy, even by continuous infusion, to restore serum TSH, T4, and T3 levels and T3 levels in many tissues to those of controls can be explained. To start, the thyroid gland secretes T3 in addition to T4. Also, as the thyroid controller, the HP system cannot attempt to maintain stable intracellular T3 levels in the face of changing serum FT4 and FT3 levels. It must react directly and proportionally to changes in FT4 and FT3, with more or less TSH production, in order to stabilize those levels. Since T4 is the dominant product of the thyroid gland, it is understandable that the HP system is especially sensitive to serum T4. In the hypothalamus, D2 is not ubiquitinated and thereby not deactivated by its interaction with T4 as in other tissues. As T4 levels rise, the HP system maintains its D2 expression and activity and thereby its T4-to-T3 conversion (161, 162) to reduce TSH secretion and avoid thyrotoxicosis (163). Therefore, on T4 monotherapy the degree of TSH suppression is disproportionate to the amount of T3 effect in other tissues (160). The TSH level only reflects the T3 effect in the HP system.
4.2 Oral T4 and/or T3 peak levels over-suppress the TSH for 24 hours
Unlike the rat studies mentioned above, we administer TRT by mouth, and usually once daily. This dumps the entire day’s T4 or T4/T3 into the circulation within a few hours, producing unphysiological peak levels. Four hours after a daily T4 dose, FT4 levels are 13% to 36% higher, and FT3 levels 8% higher than at the 24-hr trough (164–166). Three hours after a large daily T3 dose, FT3 levels are 100 to 300% higher than at the 24-hr trough. Even with these FT4 and FT3 peaks and troughs, the TSH level varies little over 24hrs (167). A single large T3 dose (50mcgs) given to controls reduced the TSH to a nadir of 0.5mIU/l at 12hrs. At 24hrs, the TSH was still near its nadir (0.55mIU/l) even though the FT3 had nearly returned to baseline (168). In rats, a rapid T4 infusion suppressed TSH production for over 22 hours (169). The over-suppression of the TSH by once-daily T4 therapy in patients can also be seen in Figure 1. by comparing graph (b) (untreated) to graph B (untreated and treated patients).
If the peak levels are reduced by splitting the daily dose, the TSH is less suppressed. A medical statistician determined that each additional splitting of his daily T4/T3 dose increased his TSH by ~1.0mIU/l (Supplementary 1). Compared to adding 6mcgs of immediate-release T3 to patients’ daily T4 dose, adding 6mcgs of slow-release T3 produced lower peak T3 levels and higher trough TSH levels (170). Since the peak levels after once-daily oral TRT oversuppress the TSH, neither the population TSH reference range nor the patient’s own premorbid TSH level can be the goal of therapy. The treated TSH will usually need to be lower than the non-treated TSH.
4.3 TSH- and FT4-normalizing T4 therapies fail to restore T3 levels and effects
When there is thyroid gland dysfunction, the higher TSH is compensatory, stimulating additional thyroidal T4/T3 production and additional D1- and D2-mediated T4-to-T3 conversion not only in the thyroid gland but in many non-thyroidal tissues as well (171–173). In both thyroidectomized dogs and humans on T4 replacement therapy, TSH injections raised serum T3 levels by approximately 50% and lowered rT3 levels (174, 175). Higher serum T3 levels stimulate D1-related T4-to-T3 conversion and degradation of rT3 (7): a positive feedback loop that further increases T3 levels and effects. This is evident in primary hypothyroidism where the high TSH level produces abnormally high serum T3/T4 ratios, 2x higher than in central hypothyroidism (176).
This positive feedback loop is interrupted by T4 monotherapy since it over-suppresses the TSH and thereby over-reduces T4-to-T3 conversion in the thyroid gland and throughout the body. In addition, the higher FT4 levels reduce T3 production by a direct effect upon the deiodinases. Higher FT4 levels suppress D2 activity in tissues like the skeletal muscles (177) which are a major source of circulating T3 (178). The lower TSH and FT3 levels on T4 monotherapy reduce D1 activity, thereby reducing T4-to-T3 conversion and the degradation of rT3. Higher T4 and/or T3 levels induce D3 production (179). D3 reduces T3 levels and effects by catalyzing the conversion of T4 to rT3 and the degradation of intracellular T3 to inactive T2. On T4 therapy serum rT3 levels are higher than in controls, often above the population range (180). They are 50% higher than on DTE titrated to the same normal TSH level (181).
In summary, once-daily T4 monotherapy is an unnatural intervention into a highly complex, tightly regulated system (172). For the reasons discussed, when T4 is dosed to normalize an elevated TSH, it fails to restore serum T3 levels or T3 effects in the tissues of most persons (Table 10).

Table 10. Why TSHT4Rx does not produce euthyroidism.
TSHT4Rx does, of course, rescue a patient from severe primary hypothyroidism and may produce optimal results for some patients. It can even be beneficial for some symptomatic “subclinical hypothyroidism” patients who have vigorous T4-to-T3 conversion—when the T4 dose produces significantly higher 24-hour trough FT3 levels (182). Symptom relief with higher T4 doses is strongly correlated with greater global deiodinase activity as evidenced by higher FT3 levels (183). However, T4 doses that produce low-normal, high-normal, or mildly elevated TSH levels usually do not restore symptom scores to those of healthy persons, and can leave FT3 levels low, even when the TSH is low-normal (184).
TSHT4Rx for primary hypothyroidism therefore produces higher FT4 levels and lower FT3 levels than seen in healthy controls (185–193). As a result of this T3 deficit, TSHT4Rx does not eliminate most hypothyroid symptoms and metabolic abnormalities in most patients at any TSH or FT4 levels within or near their reference ranges. TSHT4Rx can leave hypothyroid symptoms as severe and 24-hour urine T3 levels as low as in untreated hypothyroid patients (62). Most patients on TSHT4Rx continue to suffer significant impairment of their psychological wellbeing, health status, and cognitive function compared to healthy persons (194–197). The higher the TSH level, even within its reference range, the worse the symptoms (198).
Patients on TSHT4Rx have a lower quality of life and a higher rate of comorbidities that further degrade their quality of life (199). They have more depression and anxiety, especially when TSH levels are higher in range (200). They are more likely to be taking antidepressants (201). Compared to carefully matched controls, they have higher body mass indices (BMIs) despite reporting lower calorie intake corrected for body weight. They report lower physical activity levels and are more likely to be taking statins, beta-blockers, and antidepressants (202). They have a lower resting energy expenditure (190) and a 21% greater fat mass (189). They have a larger waist circumference, higher BMI, higher total and LDL cholesterol levels, and lower HDL levels (203). They have lower sex hormone-binding globulin levels (193)—a sign of less T3 effect in the liver. They have higher cholesterol levels (also less T3 effect in the liver) unless they are given T4 doses that suppress the TSH (204). They have persistent endothelial dysfunction (205) and higher cardiovascular morbidity (206). Higher T4 doses that lower the TSH to under 2.0mIU/l produce lower cholesterol, homocysteine, and C-reactive protein levels (207), but produce little improvement in hypothyroid symptoms (208). Therefore, the oft-repeated statement that “almost all” patients receiving TSHT4Rx are “well-replaced” is contradicted by almost all of the evidence. It is a myth; a product of the TSH-T4 Paradigm.
4.4 Athyreotic patients fare worse
TSHT4Rx produces worse results in patients without thyroid tissue (65). They have lower FT3 levels (209) due to the loss of the gland’s abundant deiodinases and therefore of intraglandular T3 production (172). Post-thyroidectomy, TSHT4Rx leaves them with much lower FT3 levels than controls (210). To restore their serum T3 to preoperative levels requires FT4 levels that are 40% higher than before surgery (39). Restoring both FT3 levels and metabolic markers to preoperative levels requires T4 doses that suppress the TSH to 0.3 to 0.03mIU/l (211). Alleviating hypothyroid symptoms requires T4 doses that produce a low TSH and an above-mid-range FT3 (212).
Patients who were euthyroid prior to thyroidectomy gain 2.2kgs more than controls in the first year (213). Patients who were hyperthyroid gain 10kgs in the first 18 months on TSHT4Rx, whereas those given TSH-suppressive T4 therapy do not gain weight (214). The failure of TSHT4Rx is obvious when a patient’s health and quality of life deteriorate dramatically after thyroidectomy and are restored with T4/T3 therapy (215). I have seen athyreotic patients who were nearly incapacitated by TSHT4Rx. I was able to restore their health and quality of life with DTE titrated by clinical criteria, without regard for the low or suppressed TSH.
4.5 Inadequate treatment of central hypothyroidism
The TSH-T4 Paradigm produces even worse results for patients with central hypothyroidism. The TSH should be ignored, but because physicians reflexively associate a low TSH with thyrotoxicity, they often prescribe only enough T4 to produce a low-in-range TSH (216, personal experience). This necessarily leaves the FT4 much lower than in T4-treated primary hypothyroidism (217). Most physicians will just normalize the FT4, leaving the FT3 low in nearly 50% of patients (120). The ATA/AACE recognize this pitfall and recommend keeping the FT4 in the upper half of its reference range (tweaking the paradigm). However, because the TSH is suppressed, thyroidal and peripheral T4-to-T3 conversion are reduced. At the same FT4 level, central hypothyroid patients will produce less T3. Some experts have recommended a T4 dose that raises the FT4 to the top of its reference range and the FT3 to the upper half of its range (216). Others have recommended ignoring the paradigm altogether and attending to the relative FT4 and FT3 levels and indicators of peripheral T3 effect—i.e., signs and symptoms (218).
In T4-treated central hypothyroidism weight gain is common (“hypothalamic obesity”), as are listlessness and other hypothyroid symptoms. The cause is inadequate treatment. The addition of 30 to 60mcgs of T3 to such patients’ “appropriate” T4 doses produced weight loss and alleviated hypothyroid symptoms (219). Increasing endocrinologists’ “empirical” 1mcg/kg T4 doses by 60% improved patients’ metabolic indicators and symptoms. Increasing their T4 doses by 44% and adding a body-weight-adjusted 8 to 12mcgs of T3 produced greater improvements (216).
5 Clinical diagnosis of hypothyroidism
5.1 Clinical medicine
The alternative to the TSH-T4 Paradigm is clinical thyroidology. Clinical medicine is “the study and practice of medicine in relation to the actual patient; the art of medicine as distinguished from laboratory science” (220). To practice clinical medicine is to acknowledge that the human body is more complex than we understand and that individuals vary greatly in their response to any given serum level, medical condition, or therapy. Our medical knowledge remains rudimentary, our tests limited, and our ideas frequently inadequate or false. There are hundreds of symptoms, disorders and diseases whose causes we do not know and for which we have no cure. We must admit our ignorance and treat patients—not reference ranges, not population correlations, not randomized controlled studies, and not guidelines. Population studies and trials can only inform, they can never replace clinical medicine. To practice clinical medicine is to practice mindfully within the context of all that is known about human physiology, its disorders and their treatment. It requires attention to the patient’s history, symptoms, physical signs, and test results. It requires gathering additional evidence as required, interpreting that evidence, and creating and testing theories of the cause. It may require trials of diagnostic and/or therapeutic interventions. It requires clinical judgment (221).
5.2 Best indicators of T3 effect: signs and symptoms
Signs are objective indicators—they can be observed and/or measured by the physician. Signs can be quantitative or qualitative. Some quantitative signs of T3 effect are body temperature, pulse, basal metabolic rate, and tendon reflex relaxation time. Others are serum tests including sex hormone binding globulin, total and LDL cholesterol, lipoprotein(a), and apolipoprotein B (222). Qualitative signs of suboptimal T3 effect include facial and/or ankle myxedema, dry skin, coarse scalp hair, loss of body hair, slow movement, and cold extremities.
Physicians must also attend to the patient’s symptoms. Symptoms are much more sensitive indicators of tissue T3 effect than signs. They are usually present long before signs appear, and often exist in the absence of signs. It is true that, considered individually, many hypothyroid symptoms are non-specific—they can have other causes. However, the greater the number of symptoms that are more specific for hypometabolism/hypothyroidism (e.g., cold intolerance, dry skin, hoarseness, constipation, weight gain, and muscle cramps), the greater the likelihood that hypothyroidism is the cause. The likelihood of hypothyroidism is further increased if the FT4 and/or FT3 levels are low in their ranges or low. It is also increased when the physician cannot find any other explanation for the symptoms. Non-anemic iron deficiency can mimic hypothyroidism and should not be missed (223). It reduces FT4 and FT3 levels and iron supplementation increases them (224, 225). A serum ferritin of <100ng/ml suggests iron deficiency and a ferritin of less than 60ng/ml is a common cause of inadequate improvement with TRT (226).
Because the TSH-T4 Paradigm has caused physicians to dismiss the signs and symptoms of hypothyroidism for decades, their awareness of them has declined. To diagnose hypothyroidism clinically, physicians must familiarize themselves with its protean manifestations. They must not only attend to the patient’s complaints but must actively search for signs and symptoms that could be caused by hypothyroidism. Patients are less aware of symptoms that have come on gradually over time (153) because they have accepted them as their “new normal”. Physicians must therefore tease out chronic symptoms through direct and indirect inquiries, conducted in a neutral manner to avoid leading the patient. Often a patient will present with one complaint, but careful questioning will reveal other hypothyroid symptoms. Many symptoms can be discovered only by inquiring about coping mechanisms (e.g., extra heaters, blankets, laxatives, skin lotions, etc.). Sometimes the physician will learn of a symptom only when the patient reports that it improved or disappeared with treatment.
5.3 Best serum indicator of T3 effect: FT4 and FT3 considered together
There is a need for a more reliable serum indicator of T3 status for untreated persons, both for population studies and clinical practice. The TSH is manifestly unfit for this purpose. An FT4 or FT3 level alone provides only part of the picture. The FT4 indicates the availability of the more abundant prohormone, while the FT3 indicates both the amount of T4-to-T3 conversion that is occurring in the tissues and the availability of the active hormone to those tissues that rely upon serum T3 (227). Both must be considered to obtain the most accurate serum estimate of intracellular T3 levels.
To judge whether a patient’s symptoms could be caused by hypothyroidism requires clinical judgment informed by the FT4 and FT3 levels, the exclusion of other causes, and ultimately, a trial thyroid optimization therapy that includes both T4 and T3. (See below.) Persisting improvement on clinically optimized T4/T3 therapy strongly supports the diagnosis.
6 Clinical treatment of hypothyroidism
6.1 Clinical reference ranges for T4 therapy in primary hypothyroidism
The treatment of hypothyroidism must proceed by clinical criteria first and the FT4 and FT3 levels second. TSH secretion evolved to control the thyroid gland and peripheral deiodinase activity; not to tell doctors what dose of T4 or T4/T3 a person should swallow every morning. Even the most carefully produced untreated TSH, FT4, and FT3 population reference ranges do not apply to treated patients. The timing of the blood draw relative to the T4 or T4/T3 dose strongly affects the results.
In 1986, four experienced clinicians adjusted the once-daily T4 doses of 148 long-term patients based upon clinical criteria alone—signs and symptoms—using a modified Wayne clinical index (36). For those patients judged to be clinically euthyroid, the 2SD reference ranges were as shown in Table 11. The TSH level was the least reliable indicator of clinical euthyroidism. The FT4 treatment range was 50% higher than the population range. In contrast, the FT3 treatment range was nearly identical to the conventional range. Therefore, if T4 monotherapy is to be guided by any test it should be the FT3. The breadth of the clinical treatment FT4 and FT3 ranges attests to their high individuality. Laboratories should report similar ranges for T4-treated persons.
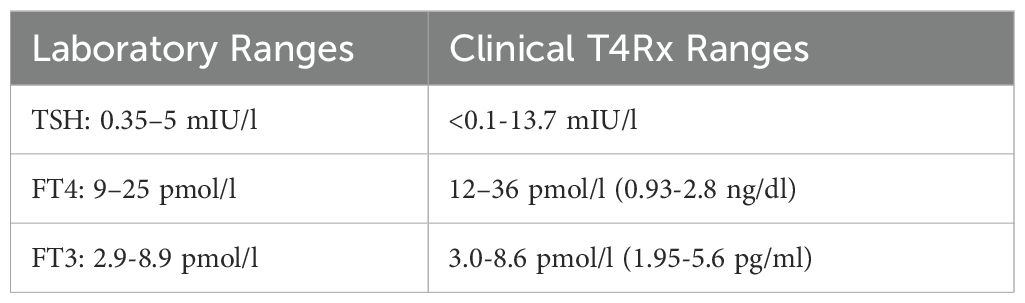
Table 11. Laboratory vs. clinical T4Rx ranges.
6.2 Low or undetectable TSH on TRT does not indicate thyrotoxicosis
The primary impediment to the effective clinical diagnosis and treatment of hypothyroidism is the ATA/AACE’s illogical reliance upon the TSH test and the resulting fear of a low TSH. Almost all physicians believe that a low TSH on replacement therapy has the same implications as does a low TSH in untreated persons in population studies—that it indicates thyrotoxicosis and portends bone loss, cardiac arrhythmias, heart attack, and stroke. Thus, they will not diagnose dysfunctional central hypothyroidism or peripheral resistance when the TSH is normal because effective treatment will suppress the TSH. They also will not try to alleviate a treated patient’s persisting hypothyroid symptoms by raising the T4 dose or adding T3. If the TSH is low on TRT, they reflexively reduce the dose, giving no thought to the nature of the hypothyroidism, the actual FT4 and FT3 levels, or the patient’s signs and symptoms.
TSH deficiency, of itself, has no known pathological effects. It simply reduces intrathyroidal T4/T3 and peripheral T3 production. The fear of a low TSH on TRT arises from population correlations seen in untreated persons, in whom a low TSH is correlated with thyrotoxicosis and its sequelae. However, population correlations have no relevance to any individual, unless proven so, and untreated TSH-population correlations have no relevance to T4- or T4/T3-treated populations. As mentioned, the TSH is oversuppressed by the serum peaks with once-daily oral therapy. Many studies, including many cited in this paper, have found that many patients require T4 doses that produce a low or suppressed TSH to restore T3 levels and effects—to restore clinical euthyroidism.
A treated TSH is not the same as an untreated TSH. Patients with true endogenous “subclinical hyperthyroidism” (i.e., Graves’ disease, toxic multinodular goiter, etc.) can have signs and symptoms of thyroid excess even though their TSH is only slightly low (avg. 0.15mIU/l). Their FT4 and FT3 levels are typically both in the upper thirds of their ranges (228). In endogenous “overt hyperthyroidism,” the TSH is often only partially suppressed when the FT4 and FT3 levels are both high 24hrs/day. The FT3 is usually disproportionately high relative to the FT4. Neither these clinical findings, nor high FT4 and FT3 levels, nor high FT3/FT4 ratios are seen with clinically adjusted T4 therapy that happens to produce a low or undetectable TSH. In thyroidectomized patients, extensive testing found no evidence of thyrotoxicity with T4 and T4/T3 doses that produced an average TSH of 0.1 to 0.3mIU/l (229).
What is true is that T4 or T4/T3 dosing that is excessive for the patient will usually produce a low or suppressed TSH. So, among persons with low or undetectable TSH levels on TRT, there will be more persons who are actually overtreated, an expected population correlation. In a retrospective cohort study of T4-treated patients, very low (<0.1mIU/l) or undetectable TSH levels were associated with a small increase in mortality (230). However, a low or undetectable TSH does not imply overtreatment in any given individual. On the other hand, some patients have medical conditions that make them intolerant of any increase in T3 effect, even when they have relatively low FT4 and FT3 levels and hypothyroid symptoms. (See below.) Some cannot tolerate the normalization of their TSH with T4 or T4/T3 therapy.
The benignity of a low TSH on once-daily T4 therapy for primary hypothyroidism has been acknowledged by some authorities. The UK’s Royal College of Physicians states that a correct dose of T4 may produce a “normal or below normal serum thyroid stimulating hormone concentration” (231). Others have stated, “Some patients achieve a sense of wellbeing only if free T4 is slightly elevated and TSH low or undetectable … it is not unreasonable to allow these patients to take a higher dose if T3 is unequivocally normal” (232). There is also a great deal of data from patients receiving TSH-suppressive T4 therapy (TSHSupT4Rx) for thyroid cancer. They are not usually thyrotoxic, either by symptoms or objective measures. Calorimetry shows no increase in metabolism compared to their pre-surgical state (233). Many still have worse SF-35 mental scores than healthy controls and some have low FT3 levels (234).
I am not arguing that a low or undetectable TSH is a goal of thyroid optimization therapy. It is best if the TSH does not need to be suppressed as it retains thyroid gland activity and improves peripheral T4-to-T3 conversion. It may be possible to avoid TSH suppression in many persons who have vigorous TSH secretion and no peripheral thyroid resistance, especially with multiple daily T4/T3 doses or slow-release T4/T3 tablets that produced more stable levels 24hrs/day. What I am arguing is that the TSH cannot be used to determine the treatment of hypothyroidism. In my experience, many persons on DTE or T4/T3 therapy require doses that completely suppress their TSH level, yet they have moderate FT4/FT3 levels and no signs or symptoms of thyrotoxicosis.
6.3 Common concerns about TSH-suppression with TRT
Physicians believe that a low TSH heralds bone loss that may lead to a fracture. TSHSupT4Rx has been associated with a faster loss of bone density in some studies, but not all (235). What matters is the cause. Greater T3 effect increases the metabolic activity of every tissue, including bone. Bone is constantly being remodeled. More T3 effect increases the rate of bone turnover (236, 237). If a person is in a bone-catabolic state, actively losing bone mass, then more T3 effect increases the rate of loss. After age 30 most persons are in a bone-catabolic state largely due to the age-related loss of bone-anabolic hormones; women more so than men (238). When women over age 30 develop primary hypothyroidism, just normalizing their TSH with T4 therapy increases their rate of bone loss for the first 6 months (239). Menopausal women are in a bone-catabolic state due to estradiol deficiency, so they are most likely to lose bone density faster on TSHSupT4Rx (240). Estrogen replacement therapy prevents this excess bone loss (241). The contrary is also true: if a person is in a bone-anabolic state, more T3 effect speeds up the increase in bone density. Adolescent females on TSHSupT4Rx gain bone mass faster than controls (242). A young hypopituitary woman experienced a 20% increase in bone density over 2 years when 50mcgs of T3 was added to her existing T4 and estradiol regimen (219).
Physicians also fear TSH suppression because hyperthyroidism is associated with liver pathology. An early study claimed that T4 doses that produced low TSH or high FT4 levels caused “hepatic damage” indicated by high glutathione-S-transferase levels (26). However, with T4 therapy glutathione-S-transferase and some transaminase levels are far less elevated than in endogenous hyperthyroidism (27). Again, what matters is the cause. Due to the first-pass effect, oral TRT exposes the liver to higher T4/T3 levels than other tissues, causing changes in liver function tests that resemble hyperthyroidism. The liver contains abundant D1 and converts portal T4 to T3 (243). These liver effects are apparently benign. There were no reports of liver pathology in the many decades when patients received higher, clinically optimized T4 and T4/T3 doses.
Physicians also fear TSH suppression because endogenous hyperthyroidism is associated with an increased risk of myocardial infarction, stroke, and thromboembolic disease (244). However, these patients have high FT4 and FT3 levels 24hrs/day; with proportionately higher FT3 levels. They have signs and symptoms of thyrotoxicosis and may have other abnormalities related to the cause of their hyperthyroidism. Such levels, signs, and symptoms are not present in patients on clinically adjusted T4 or T4/T3 doses that happen to suppress the TSH, for the reasons given above. Again, in the pre-TSH era, there was no data suggesting that treated patients had an increased risk of cardiovascular events. A recent review found that T4-treated primary hypothyroidism patients with low TSH levels (0.04-0.4mIU/l) had no increase in cardiovascular events or mortality compared to those with normal TSH levels. Only those with high TSH levels or the lowest TSH levels (<0.04mIU/l) had a statistical increase in morbidity (245). This is, again, an expected population correlation. Overtreatment can be avoided by careful attention to symptoms, signs, and FT4/FT3 levels.
More relevant to thyroid optimization therapy (TOT) is the fact that moderately excessive T3 effect in the heart, as in mild endogenous hyperthyroidism, produces increased heart rate, excessive contractility, impaired diastolic relaxation, and thickening of the walls. These changes are seen in endogenous “subclinical hyperthyroidism” with an average TSH of 0.15mIU/l and higher-in-range FT4 and FT3 levels than controls. Such patients usually have thyrotoxic symptoms (228). These changes can be seen in patients on TSHSupT4Rx when the T4 dose is excessive (246) and there are thyrotoxic signs and symptoms (247). Lowering the T4 dose eliminates the thyrotoxic signs/symptoms and cardiac abnormalities, even though the TSH remains low (avg: 0.1mIU/l) (248). Athyreotic patients on TSHSupT4Rx can have no cardiac symptoms and normal cardiovascular studies even though their average TSH is just 0.03mIU/l and their FT4 is 50% higher than controls. Their FT3 is similar to controls (249). On the other hand, persons with significant atherosclerotic heart disease may not tolerate any increase in T3 levels/effects due to increased cardiac work and resultant worsening of angina or triggering of arrhythmias.
6.4 Risk of atrial fibrillation with thyroid optimization therapy
The best reason to fear a low TSH with treatment is also the best reason to fear all TRT, particularly TOT: the risk of atrial fibrillation (AF). This too must be understood in context. Most persons who develop AF are not on TRT or TOT. AF is common; the lifetime risk of AF is 1 in 5 for persons with no risk factors and rises to over 1 in 3 for persons with one or more risk factors (250). The incidence of AF rises with age, affecting 1 in 25 persons over 60 and 1 in 10 persons over 80 (251). Risk factors for AF are common in the population.
AF risk is clearly related to T3 status. It has been reported with hypothyroidism (252) but is much more frequently seen with higher FT4 and FT3 levels and hyperthyroidism. More T3 effect increases automaticity and trigger activity in the pulmonary vein myocytes that initiate AF (253). In untreated persons, the risk of AF rises with higher FT4 levels within the population range (254, 255) and with lower-in-range or low TSH levels (256). Therefore, any increase T3 effect with T4 or T4/T3 therapy entails a risk of triggering AF in susceptible patients. It does not require overtreatment but is more likely with overtreatment. AF is more likely to occur with a suppressed TSH—an expected population correlation.
Clinically optimized T4 or T4/T3 therapy thus necessarily entails a higher risk of AF than less effective TSH- or FT4-normalizing therapy. The TSH-T4 Paradigm minimizes the risk of triggering AF in susceptible persons, but at the costs of widespread underdiagnosis and undertreatment. This is neither scientifically nor ethically justifiable. Mild-to-moderate hypothyroidism has its own medical risks, and patients want to feel and function as well as possible. The physician can resolve the ethical and legal dilemmas surrounding AF by obtaining informed consent for TOT. The physician and patient should together weigh the patient’s risk of AF against the potential health and quality-of-life benefits of optimal T3 effect. The patient should be given the right to choose. Fortunately, for most persons under age 60, the risk of AF is low, even with overtreatment. In persons over 60 and/or with risk factors, the physician can minimize the risk of AF by going “low and slow” with T4 or T4/T3 dosing. Fortunately, AF that is induced by TRT usually resolves with reducing the dose, except in older patients with significant underlying heart disease (257).
7 Hypocortisolism and inflammation
It is important to emphasize that none of the tests [for hypocortisolism] will be perfect and therefore, clinical judgment should prevail in patients with significant symptoms and apparently normal or equivocal biochemical data (258).
It is very convenient for physicians to limit thyroidology to one test, the TSH, and ignore the patients’ symptoms and all other aspects of their endocrine system, physiology, and medical conditions. However, that is not possible. The thyroidal system is part of an integrated neuro-endocrine-immune system. T3 has especially powerful interactions with cortisol. These interactions affect how hypothyroidism and hyperthyroidism present and can cause anomalous positive and negative reactions to T4, T4/T3, and T3 treatment. Knowledge of these interactions is necessary to treat patients and interpret studies, including T4/T3 combination therapy studies.
7.1 T4/T3 therapy can unmask hypocortisolism
One interaction between T3 and cortisol is well known: TRT can unmask or worsen “adrenal insufficiency” (AI). However, relative hypocortisolism, without adrenal gland or HP damage, can arise from many causes and cannot be ruled out by a normal AM serum cortisol (ref. range: 5 to 20mcg/dl (259). It also cannot be ruled out by a normal adrenocorticotropic hormone (ACTH) stimulation test (260–264). Relative hypocortisolism and/or inflammation is suggested when TRT rapidly causes worse fatigue, achiness, and/or brain fog, or worsening of an existing inflammatory disease. As with hypothyroidism, diagnosing relative hypocortisolism requires attention to the patient’s history, signs and symptoms, relative serum and saliva cortisol levels, responses to TRT, and ultimately, response to a trial of hydrocortisone.
7.2 Cortisol-T3 interactions
Cortisol and T3 are the most powerful hormones in the endocrine system, and they counteract and support each other’s levels and effects in various ways (Table 12). Their interactions are so powerful that the balance between them is as important as the absolute status of either. Together their actions and interactions play a large role in our mental and physical stamina and well-being. Both hormones affect the secretion and/or effectiveness of other major hormones (e.g., the gonadal steroids and growth hormone). Both have large roles in glucose production, availability, and utilization. Mitochondria have both T3 and cortisol receptors (265, 266); both hormones are required to ensure optimal mitochondrial energy production (267). A relative deficiency of one or both produces a hypometabolic state accompanied by fatigue, depression, muscle/joint pain, and cognitive dysfunction.

Table 12. T3-Cortisol interactions.
Most of T3’s effects are genomic and occur on a time scale of weeks and even months, but higher T3 levels/effects immediately stimulate more ACTH and cortisol production (268, 269) while also increasing cortisol utilization and metabolism (270). Oral T3 has greater effects than endogenous T3 on ACTH-cortisol production due to its high peak levels and on cortisol utilization and metabolism due to its strong first-pass effects in the liver. Almost all positive and negative clinical responses that occur within hours or days from the initiation of T4 and/or T3 supplementation are due to alterations in cortisol levels and effects or T3’s rapid immune system stimulating effects. Persons with relative hypocortisolism may improve rapidly when oral T3 succeeds in raising cortisol levels and effects. On the contrary, if T4 or T4/T3 increases cortisol metabolism more than its production, they may be unable to tolerate necessary thyroid supplementation.
In the absence of cortisol, the affinity of thyroid receptors for T3 is reduced by 50% (271). In marked hypocortisolism, endogenous T4 and T3 fail to suppress TSH production, resulting in an elevated TSH (“subclinical hypothyroidism”). On the other hand, higher cortisol levels and effects reduce T3 levels and effects by reducing TSH secretion (272) and T4-to-T3 conversion (273). In hypothyroidism ACTH and cortisol production are reduced (274), sometimes so much as to produce symptomatic central hypocortisolism (275).
7.3 Hypocortisolism and the female sex
Most persons who suffer from a relative hypocortisolism are female. Women have lower cortisol levels and effects, and lower cortisol responses to stress than men (276–283). This is a sufficient explanation for their greater resistance to infections and their much higher incidences of disorders and diseases that are associated with hypocortisolism (allergies, autoimmune diseases, chemical sensitivities, anxiety, achiness, etc.).
Women’s relative hypocortisolism is a sufficient explanation for their much higher incidences of Hashimoto’s and Graves’ diseases (15:1 and 7:1 respectively). Autoimmune thyroid disease (ATD) has been correlated with hypocortisolism (259, 284–286) and other autoimmune diseases (287). Those with Hashimoto’s thyroiditis suffer from constitutional symptoms caused by the autoimmune process and its accompanying inflammation, as shown by the improvements that they experience with thyroidectomy (288, 289). Relative hypocortisolism is therefore common among women with ATD—who comprise most of the subjects in studies of TRT, including the T4/T3 studies. Women’s relative hypocortisolism is a sufficient explanation for the fact that women are the majority of persons who suffer from unexplained fatigue, pain, cognitive dysfunction, anxiety, gastrointestinal dysfunction, and other constitutional symptoms, have mild TSH elevations, are diagnosed with hypothyroidism, have anomalous responses to TRT, and are dissatisfied with TRT.
8 Safety and efficacy of T4/T3
Producing optimal clinical results for patients with TRT is a very different exercise than just normalizing serum TSH, FT4, or FT3 levels. Without a trial of TOT, physicians cannot rule out hypothyroidism as a contributor to the patient’s ongoing symptoms. If TOT does not help or makes the patient feel worse, then physicians know to search for another cause: hypocortisolism, inflammation, autoimmune disease, chronic infection, etc. For patients with persisting hypothyroid symptoms on TSHT4Rx, physicians can increase the T4 dose, without regard for a low or suppressed TSH. However, studies show that this will usually fail to restore euthyroidism in all tissues and may cause hyperthyroidism in tissues with high D2 activity like the heart (290).
8.1 The need for T3
The thyroid gland secretes T3 and T4, so to replicate normal physiology physicians must include T3 (291). T4/T3 therapy is logically and scientifically necessary to restore optimal T3 effects in all tissues. Providing T3 with T4 keeps FT4 levels lower and thus avoids T4-mediated downregulation of D2 and upregulation of D3. Providing T3 induces greater D1 activity and therefore T4-to-T3 conversion. It compensates for the over-reduction or suppression of TSH secretion due to peak levels and resultant over-reduction in TSH-induced T4-to-T3 conversion. It compensates for the lack of thyroidal deiodinases in athyreotic patients and the lack of TSH in central hypothyroidism. Providing T3 keeps the T4/T3 ratio lower and so improves sensitivity to both T4 and T3, i.e., prevents and treats genetic and acquired forms of thyroid resistance including common deiodinase variants. T3 also helps to stimulate ACTH/cortisol secretion and so can prevent or treat relative hypocortisolism. Unlike low doses of T4 that can reduce T3 effect, providing T3 in a 4:1 T4/T3 ratio guarantees that even low doses increase T3 effect long term, providing reliable information to the physician and patient. T4/T3 therapy has other practical advantages. It improves compliance because patients experience therapeutic effects more quickly and quickly feel a loss of effect if they omit doses (18). In summary, it makes sense to give all patients T4/T3 combination therapy from the start. There will be some patients, however, who will not tolerate oral T3 and/or for whom T4 will work best.
The ATA/AACE guidelines’ authors suggest that oral T3 supplementation is unsafe due to the supraphysiological peak T3 levels after each dose. They provide no evidence. In fact, the T3 peaks have never been shown to produce any health problems. In my experience, the T3 peaks with once daily 4:1 T4/T3 therapy cause problems only for a few persons with hypocortisolism, inflammation, or cardiac disease. The guidelines’ authors also express concern that T3 levels vary throughout the day and “cannot be easily monitored”. In fact, they do not need to be monitored. Oral T3’s pharmacokinetics are well known. T3 levels peak 2.5hrs after an oral dose and have a half-life of around 22 hours (168). With a substantial T3 dose, FT3 levels are usually high at their peak and fall gradually to midrange or low in range at the 24-hr trough. The ATA/AACE authors also assert that we cannot give patients T3 because we do not have slow-release T3 tablets or capsules that can perfectly replicate the more stable 24-hr TSH, FT4, and FT3 levels of a normal person. It may be best to replicate such levels, however, the fact that we cannot do so does not imply that we cannot prescribe any T3.
T3 is our thyroid hormone. T3 supplementation can cause problems only if its dose is excessive (16). T3 is feared precisely because it is efficacious—providing T3 with T4 assures greater thyroid effect, and therefore a greater risk of overdosing and/or of causing problems in susceptible persons (e.g., bone loss, AF). Overdosing is what should be avoided, not T3. A 17-year observational study found that, compared to T4 therapy, patients receiving T4/T3 and T3-only therapies had no increased risk of cardiovascular disease, atrial fibrillation, or fractures (292). A more recent retrospective study found that DTE and T4/T3 treatment, adjusted to a normal TSH, produced no adverse effects or events (293).
8.2 Human thyroidal T4/T3 ratio
What is the ideal T4/T3 ratio for TRT for most persons? It is commonly stated that the human thyroidal T4/T3 output ratio is a high 17:1. This belief is based upon a single study in 1990 that involved injections of radio-labeled T4 and T3 in euthyroid persons who were given Lugol’s solution twice daily (294). It is known that such an acute excess of iodine substrate causes the thyroid gland to preferentially synthesize T4 over T3. It inhibits thyroidal D1 activity and D1 mRNA expression, reducing T3 output (295). The human ratio is certainly much lower than this. A 9:1 T4/T3 ratio given to thyroidectomized patients produced a high PBI—i.e., contained an excessive amount of T4. A 3.3:1 ratio produced better clinical results and a normal PBI (18). DTE’s 4:1 ratio normalizes rT3 levels but produces lower FT4 levels than controls with once-daily dosing. The human thyroid T4/T3 output ratio is probably between 6:1 and 8:1. The ratio can be determined as it was in rats, by giving thyroidectomized persons continuous infusions of various ratios of T4 and T3. However, the ideal ratio for once-daily oral dosing will likely be lower than the thyroidal output ratio due to the over-reduction of the TSH by peak levels. It will also vary among patients.
8.3 Existing studies support T4/T3 therapy
In the past 54 years, many studies have compared T4 monotherapy with T4/T3 combination therapy. These studies provide abundant detailed information about the serum levels, symptom scores and physiological parameters of persons on T4 monotherapy and various combinations of T4 and T3. Most studies involved arbitrary substitutions of some amount of T3 for some amount of the patient’s usual T4 dose, without regard for the patient’s symptoms or serum FT4 and FT3 levels. Arbitrary substitutions are problematic. They will result in overtreatment or undertreatment for some persons. While oral T3 (as monotherapy) is generally about 3.5 times more potent than T4 by weight in its ability to lower the TSH (296), their relative oral potency can differ by a factor of 3 in different persons and with different doses (297); i.e., from ~1:1 to 10:1. Using the 3.5:1 ratio, the arbitrary substitutions ranged from 1-for-1mcg oversubstitution to 1-for-5mcg undersubstitution—the latter being most common and often produced an effective reduction in dose as evidenced by a higher TSH. In some studies, T4/T3 was allowed to produce different TSH levels; in a few the T4 doses were adjusted to maintain a similar TSH.
Randomized trials with arbitrary substitutions of T3 for T4 cannot override our knowledge of thyroid physiology or the need for individualized treatment based upon clinical judgment (183). Consider that if one-half of the patients in a study improve and one-half deteriorate, the net result is no change. To obtain useful information one must separate the two groups and determine why each reacted as they did. Due to individual variations in T4-to-T3 conversion, cortisol status, autoimmune disease, and other factors, the same FT4/FT3 levels that produce benefits in some persons will produce negative symptoms in others. Furthermore, hypocortisolism and/or inflammation are more common in women with ATD—the majority of subjects in most T4/T3 studies. They can be sensitive to changes in their TRT.
Reviews and meta-analyses of the T4/T3 studies have concluded that they do not support replacing T4 monotherapy with any form of T4/T3 combination therapy (298–302). However, the metanalyses failed to account for the flaws and heterogeneity of the studies. To assess the T4/T3 studies’ implications, each must be analyzed individually, and the analysis must not be based upon the TSH-T4 Paradigm. Both significant and non-significant changes must be considered, along with patient preference. I provide such an analysis of each study here, informed by my clinical experience with attempting to optimize T3 status clinically with T4/T3 therapy (mostly DTE) in more than a thousand patients over 20 years. See Table 13 for a brief description of each study and its results. See Supplementary 2 for detailed summaries of and commentaries on each study.
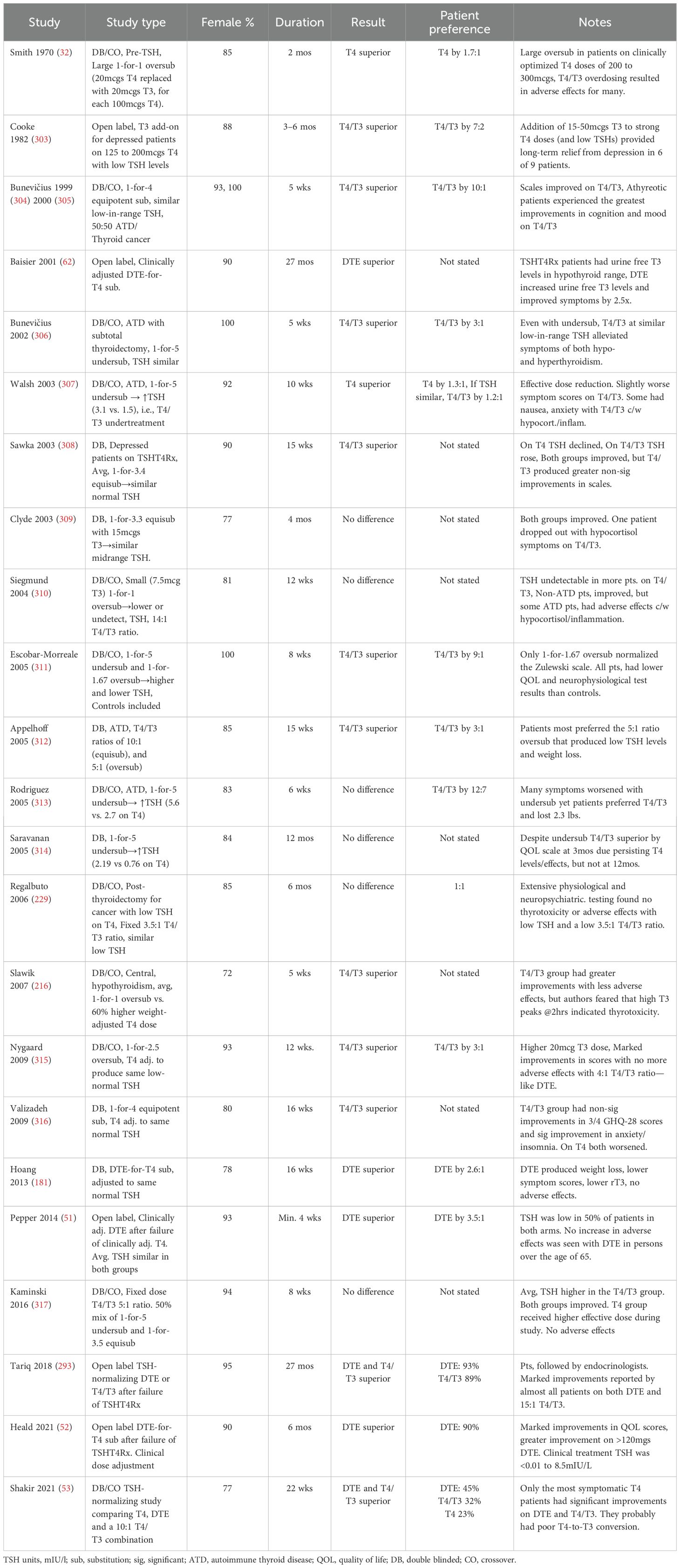
Table 13. Studies of T4/T3 combination therapy vs. T4 monotherapy.
Considering both the statistically significant and non-significant trends and patient preference, the 23 T4/T3 combination studies strongly favor combination therapy. This is despite frequent T4/T3 underdosing as evidenced by higher TSH levels, and occasional T4/T3 overdosing. Fifteen of the 23 studies favored T4/T3 therapy when all results are considered, and only two favored T4—a 7.5:1 ratio in favor of T4/T3. One study that favored T4 involved 1-for-1 oversubstitution and obvious overdosing in patients already receiving high, pre-TSH, clinically optimized T4 doses (Smith). The other involved 1-for-5 undersubstitution and obvious underdosing, indicated by a 2x higher average TSH. Even in that study, if the TSH rose by <0.99miU/L, patients preferred T4/T3 by a small margin (Walsh).
In 6 of 23 studies there was no overall difference in the results. Three of the six neutral studies used 1-for-5 undersubstitution (Rodriquez, Saravanan, Kaminski) and one used 1-for-1 oversubstitution (Siegmund). In the 15 studies in which subjects’ preference was assessed, they preferred T4/T3 in 12 (80%). They sometimes preferred T4/T3 even with 1-for-5 undersubstitution and higher TSH levels. For certain, the T4/T3 studies show that including some arbitrary amount of T3 is not a magic bullet; it doesn’t guarantee better results in all cases. However, it usually produces better results, especially with equipotent 3.5:1 substitutions and lower T4/T3 ratios.
Only one T4/T3 study included healthy controls and it found that most of the T4 and T4/T3 treated patients were more hypothyroid than controls by subjective and objective scales. Scales improved to those of controls only with T3 oversubstitution that suppressed the TSH (Escobar-Morreale). No other studies included controls, so this raises the probability that most patients with normal or even low TSH levels in both the T4 and T4/T3 arms of most of the studies were undertreated. In only three studies was the T4/T3 regimen (DTE) adjusted according to clinical criteria without regard for the TSH (Baisier, Pepper, Heald). Two of those studies compared clinically adjusted T4 and DTE therapies (Baisier, Pepper) and both found DTE to be superior.
8.4 Conclusions from the T4/T3 studies
Most of the T4/T3 studies’ designs and interpretations were vitiated by errors, many of which are encountered in other studies of TRT performed within the TSH-T4 Paradigm. Some of these are discussed in Supplementary 2. From the standpoint of the clinical paradigm and the scientific evidence here presented, I submit that the following conclusions can be drawn:
● T4/T3 treatment is both efficacious and safe, except with overdosing or underdosing. The T4/T3 studies have disproved the idea that including T3 is somehow dangerous.
● Supraphysiological peak T3 levels after T3 doses do not produce any deleterious effects or negative symptoms in most patients.
● T4/T3 treatment usually produces superior results except when the T4/T3 dose is clearly excessive or inadequate; the latter often indicated by a much higher TSH. Even at significantly higher in-range TSH levels, T4/T3 sometimes produces the same clinical benefits as T4 monotherapy (Saravanan), attesting to its physiological benefits.
● Primary hypothyroid patients usually feel best, and/or obtain the best symptom or physiological test scores on T4 and/or T4/T3 regimens that produce a low or suppressed TSH (Cooke, Escobar-Morreale, Appelhof). They can have no signs or symptoms of thyrotoxicosis, even with extensive testing (Regalbuto).
● Athyreotic and central hypothyroid patients are most likely to experience benefits with T4/T3 combination therapy (Bunevičius, Slawik; Regalbuto is the outlier).
● Studies that recruited patients who are dissatisfied with TSHT4Rx tended to show more positive effects with T4/T3 therapy (Baisier, Pepper, Heald, Tariq).
● The best clinical results were obtained with DTE adjusted by clinical criteria (Baisier, Pepper, Heald). Clinically optimized DTE doses frequently result in a suppressed TSH (Pepper, Heald).
● Persons with ATD tend to have higher FT3 levels on treatment, and some do not tolerate greater thyroid effect on T4/T3, even though clearly not overtreated (Clyde, Siegmund).
In sum, the existing studies indicate that T4/T3 therapy is not harmful and is superior to T4 monotherapy for most patients. Because T4/T3 therapy is more physiological and effective, it should be considered the preferred treatment for hypothyroidism. There is no rational justification for more arbitrary substitution or TSH-normalizing T4/T3 studies. The only meaningful study of T4/T3 vs. T4 monotherapy is a comparison of clinically adjusted dosing of each by experienced clinicians and/or according to hypothyroid symptom scales and patient preference, without regard for the TSH level.
9 Conclusion
The 2012 ATA/AACE guidelines for the treatment of hypothyroidism were a reiteration of the TSH-T4 Paradigm from the 1970s. This old, arbitrary, ineffective, laboratory-based paradigm must be replaced by a clinical, patient-centered paradigm that incorporates all current knowledge. Hypothyroidism must be properly defined as insufficient T3 effect in some or all tissues. Diagnosis and treatment must be guided by the most sensitive criteria: the patient’s signs and symptoms first and the relative FT4 and FT3 levels second. T4/T3 therapy is superior and should be favored. DTE’s 4:1 ratio worked well for many decades and still appears to work better than higher T4:T3 ratios for most people. If a person has hypothyroid symptoms for which there is no other medical explanation and has relatively low FT4 and/or FT3 levels, then he/she deserves a trial of gradually increasing T4/T3 doses with careful monitoring for both improvements and evidence of overdosing. If the latter occurs, or if the FT4 and FT3 levels are rather high with treatment, the dose should be reduced and the result monitored. The optimal T4/T3 dose is the lowest dose that eliminates hypothyroid signs and symptoms without producing any signs or symptoms of excess T3 effect. The TSH level provides some information but cannot be relied upon for either diagnosis or treatment. A suppressed TSH with treatment is not proof of thyrotoxicosis.
Hypothyroidism can cause a wide variety of symptoms and disorders and so must be ruled out in all cases of unexplained symptoms. Physicians’ ability to understand hypothyroidism and willingness to provide a trial of T4/T3 thyroid optimization therapy will improve the lives of countless patients around the world. It will also open the door to a greater understanding of the underlying causes of fatigue, achiness, cognitive dysfunction and other “functional” disorders. If a trial of T4/T3 thyroid optimization eliminates patients’ symptoms indefinitely, then they were indeed suffering only from a lack of T3 effect in their tissues—for whatever reason. If their symptoms improve only temporarily or are worsened by T4/T3 optimization therapy, then physicians gain additional clues regarding the cause. They must consider hypocortisolism, chronic infection, autoimmune disease, iron deficiency, toxins, genetic disorders, etc. By performing additional tests and/or therapeutic trials for these other conditions, they will be able to help more suffering people. An effective thyroidology therefore will not only optimize the diagnosis and treatment of all forms of hypothyroidism but will open the door to greater understanding of endocrinology and of the causes of many currently unexplained disorders.
Author contributions
HHL: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author declares that no financial support was received for the research and/or publication of this article.
Acknowledgments
The author thanks Mel Rowe, thyroid patient advocate, for his help with editing this paper.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fendo.2025.1656676.
Generative AI statement
The author declares that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the author and do not necessarily represent those of his affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2025.1529791/full#supplementary-material
Abbreviations
AACE, American Association of Clinical Endocrinologists; ATA, American Thyroid Association; D1-D3, deiodinase types 1-3; FT3, free T3; FT4, free T4; HP, hypothalamic-pituitary; DTE, desiccated thyroid extract; rT3, reverse T3; SD, standard deviation; SNP, single nucleotide polymorphism; T3, 3,5,3’-triiodothyronine, liothyronine; T4, thyroxine, levothyroxine; TRT, thyroid replacement therapy; TOT, thyroid optimization therapy; TSH, thyroid-stimulating hormone, thyrotropin.
References
1. Greenspan FS ed. Basic and Clinical Endocrinology. 3rd edition. Norwalk, CT: Appleton & Lange (1991). p. 211.
2. Weitzel JM, Iwen KA, and Seitz HJ. Regulation of mitochondrial biogenesis by thyroid hormone. Exp Physiol. (2003) 88:121–8. doi: 10.1113/eph8802506
3. Vaitkus JA, Farrar JS, and Celi FS. Thyroid hormone mediated modulation of energy expenditure. Int J Mol Sci. (2015) 16:16158–75. doi: 10.3390/ijms160716158
4. Sandler MP, Robinson RP, Rabin D, Lacy WW, and Abumrad NN. The effect of thyroid hormones on gluconeogenesis and forearm metabolism in man. J Clin Endocrinol Metab. (1983) 56:479–85. doi: 10.1210/jcem-56-3-479
5. Wu Y and Koenig RJ. Gene regulation by thyroid hormone. Trends Endocrinol Metab. (2000) 11:207–11. doi: 10.1016/s1043-2760(00)00263-0
6. Chatonnet F, Flamant F, and Morte B. A temporary compendium of thyroid hormone target genes in brain. Biochim Biophys Acta. (2015) 1849:122–9. doi: 10.1016/j.bbagrm.2014.05.023
7. Bianco AC, Salvatore D, Gereben B, Berry MJ, and Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev. (2002) 23:38–89. doi: 10.1210/edrv.23.1.0455
8. Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, et al. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev. (2008) 29:898–938. doi: 10.1210/edrv.23.1.0455
9. Peeters RP, Visser TJ, and Feingold KR. Metabolism of thyroid hormone. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al, editors. Endotext. MDText.com, Inc, South Dartmouth (MA) (2000).
10. Escobar-Morreale HF, del Rey FE, Obregón MJ, and de Escobar GM. Only the combined treatment with thyroxine and triiodothyronine ensures euthyroidism in all tissues of the thyroidectomized rat. Endocrinology. (1996) 137:2490–502. doi: 10.1210/endo.137.6.8641203
11. Flamant F, Gauthier K, and Samarut J. Thyroid hormones signaling is getting more complex: STORMs are coming. Mol Endocrinol. (2007) 21:321–33. doi: 10.1210/me.2006-0035
12. Dumitrescu AM, Korwutthikulrangsri M, and Refetoff S. Impaired Sensitivity to Thyroid Hormone: Defects of Transport, Metabolism and Action. In: Feingold KR, Ahmed SF, Anawalt B, Blackman MR, Boyce A, Chrousos G, et al editors. Endotext [Internet]. South Dartmouth, MA: MDText.com, Inc. (2023).
13. Garber JR, Cobin RH, Gharib H, Hennessey JV, Klein I, Mechanick JI, et al. American association of clinical endocrinologists and american thyroid association taskforce on hypothyroidism in adults 2012 clinical practice guidelines for hypothyroidism in adults: co-sponsored by american association of clinical endocrinologists and the American thyroid association. Endocr Pract. (2012) 18:988–1028. doi: 10.4158/ep12280.gl
15. Selenkow HA and Wool MS. A new synthetic thyroid hormone combination for clinical therapy. Ann Intern Med. (1967) 67:90–9. doi: 10.7326/0003-4819-67-1-90
16. Idrees T, Palmer S, Maciel RMB, and Bianco AC. Liothyronine and desiccated thyroid extract in the treatment of hypothyroidism. Thyroid. (2020) 10:1399–413. doi: 10.1089/thy.2020.0153
17. McGavack TH and Reckendorf HK. Therapeutic activity of desiccated thyroid substance, sodium L-thyroxine and D, L-triiodothyronine; a comparative study. Am J Med. (1956) 20:774–7. doi: 10.1016/0002-9343(56)90159-0
18. Taylor S, Kapur M, and Adie R. Combined thyroxine and triiodothyronine for thyroid replacement therapy. Br Med J. (1970) 2:270–1. doi: 10.1136/bmj.2.5704.270
19. Cotton GE, Gorman CA, and Mayberry WE. Suppression of thyrotropin (h-TSH) in serums of patients with myxedema of varying etiology treated with thyroid hormones. N Engl J Med. (1971) 285:529–33. doi: 10.1056/NEJM197109022851001
20. Pearce CJ and Himsworth RL. Total and free thyroid hormone concentrations in patients receiving maintenance replacement treatment with thyroxine. Br Med J (Clin Res Ed). (1984) 288:693–5. doi: 10.1136/bmj.288.6418.693
21. Helfand M and Crapo LM. Monitoring therapy in patients taking levothyroxine. Ann Intern Med. (1990) 113:450–4. doi: 10.7326/0003-4819-113-6-450
22. McAninch EA and Bianco AC. The history and future of treatment of hypothyroidism. Ann Intern Med. (2016) 164:50–6. doi: 10.7326/M15-1799
23. Braverman LE, Ingbar SH, and Sterling K. Conversion of thyroxine (T4) to triiodothyronine (T3) in athyreotic human subjects. J Clin Invest. (1970) 49:855–64. doi: 10.1172/JCI106304
25. Stock JM, Surks MI, and Oppenheimer JH. Replacement dosage of L-thyroxine in hypothyroidism. A re-evaluation. N Engl J Med. (1974) 290:529–33. doi: 10.1056/nejm197403072901001
26. Beckett GJ, Kellett HA, Gow SM, Hussey AJ, Hayes JD, and Toft AD. Raised plasma glutathione S-transferase values in hyperthyroidism and in hypothyroid patients receiving thyroxine replacement: evidence for hepatic damage. Br Med J (Clin Res Ed). (1985) 291:427–31. doi: 10.1136/bmj.291.6493.427
27. Gow SM, Caldwell G, Toft AD, Seth J, Hussey AJ, Sweeting VM, et al. Relationship between pituitary and other target organ responsiveness in hypothyroid patients receiving thyroxine replacement. J Clin Endocrinol Metab. (1987) 64:364–70. doi: 10.1210/jcem-64-2-364
28. Ross DS, Neer RM, Ridgway EC, and Daniels GH. Subclinical hyperthyroidism and reduced bone density as a possible result of prolonged suppression of the pituitary-thyroid axis with L-thyroxine. Am J Med. (1987) 82:1167–70. doi: 10.1016/0002-9343(87)90219-1
29. Banovac K, Papic M, Bilsker MS, Zakarija M, and McKenzie JM. Evidence of hyperthyroidism in apparently euthyroid patients treated with levothyroxine. Arch Intern Med. (1989) 149:809–12. doi: 10.1001/archinte.1989.00390040043008
30. Ross DS. Subclinical hyperthyroidism: possible danger of overzealous thyroxine replacement therapy. Mayo Clin Proc. (1988) 63:1223–9. doi: 10.1016/s0025-6196(12)65409-3
31. Jackson IM and Cobb WE. Why does anyone still use desiccated thyroid USP? Am J Med. (1978) 64:284–8. doi: 10.1016/0002-9343(78)90057-8
32. Smith RN, Taylor SA, and Massey JC. Controlled clinical trial of combined triiodothyronine and thyroxine in the treatment of hypothyroidism. Br Med J. (1970) 4:145–8. doi: 10.1136/bmj.4.5728.145
33. Billewicz WZ, Chapman RS, Crooks J, Day ME, Gossage J, Wayne E, et al. Statistical methods applied to the diagnosis of hypothyroidism. Q J Med. (1969) 38:255–66.
34. Evered D, Young ET, Ormston BJ, Menzies R, Smith PA, and Hall R. Treatment of hypothyroidism: a reappraisal of thyroxine therapy. Br Med J (1973). (5872) 3:131–4. doi: 10.1136/bmj.3.5872.131
35. Carr D, McLeod DT, Parry G, and Thornes HM. Fine adjustment of thyroxine replacement dosage: comparison of the thyrotrophin releasing hormone test using a sensitive thyrotrophin assay with measurement of free thyroid hormones and clinical assessment. Clin Endocrinol (Oxf). (1988) 28:325–33. doi: 10.1111/j.1365-2265.1988.tb01219.x
36. Fraser WD, Biggart EM, O’Reilly DS, Gray HW, McKillop JH, and Thomson JA. Are biochemical tests of thyroid function of any value in monitoring patients receiving thyroxine replacement? Br Med J (Clin Res Ed). (1986) 293:808–10. doi: 10.1136/bmj.293.6550.808
37. Lee SL, Haugen BR, and Singer PA. New Dimensions in TSH Control, Summaries of 3 lectures presented at the Annual Meeting of the American Association of Clinical Endocrinologists in San Diego, California (2003), originally posted at ThyroidToday.com.
38. Petersen K, Bengtsson C, Lapidus L, Lindstedt G, and Nyström E. Morbidity, mortality, and quality of life for patients treated with levothyroxine. Arch Intern Med. (1990) 150:2077–81. doi: 10.1001/archinte.1990.00390210063015
39. Jonklaas J, Davidson B, Bhagat S, and Soldin SJ. Triiodothyronine levels in athyreotic individuals during levothyroxine therapy. JAMA. (2008) 299:769–77. doi: 10.1001/jama.299.7.769
40. Ross DS, Burch HB, Cooper DS, Greenlee MC, Laurberg P, Maia AL, et al. American thyroid association guidelines for diagnosis and management of hyperthyroidism and other causes of thyrotoxicosis. Thyroid. (2016) 10:1343–421. doi: 10.1089/thy.2016.0229
41. Qato DM, Wilder J, Schumm LP, Gillet V, and Alexander GC. Changes in prescription and over-the-counter medication and dietary supplement use among older adults in the United States, 2005 vs 2011. JAMA Intern Med. (2016) 176:473–82. doi: 10.1001/jamainternmed.2015.8581
42. Stott DJ, Rodondi N, Kearney PM, Ford I, Westendorp RGJ, Mooijaart SP, et al. Thyroid hormone therapy for older adults with subclinical hypothyroidism. N Engl J Med. (2017) 376:2534–44. doi: 10.1056/NEJMoa1603825
43. Teixeira PF, Reuters VS, Ferreira MM, Almeida CP, Reis FA, Melo BA, et al. Treatment of subclinical hypothyroidism reduces atherogenic lipid levels in a placebo-controlled double-blind clinical trial. Horm Metab Res. (2008) 40:50–5. doi: 10.1055/s-2007-993216
44. Bowthorpe JA. Stop the Thyroid Madness (2008). Fredericksburg, TX: Laughing Grape. Available online at: http://www.stopthethyroidmadness.com/ (Accessed April 6, 2025).
45. Thyroid UK. Available online at: https://thyroiduk.org/ (Accessed April 6, 2025).
46. Thyroid Change. Available online at: https://www.facebook.com/ThyroidChange (Accessed April 6, 2025).
47. Thyroid Patient Advocacy. Available online at: http://tpauk.com/main/ (Accessed April 6, 2025).
49. Robinson P. The Thyroid Patient’s Handbook (2018). North Somerset, UK: Elephant in the Room Books. Available online at: http://recoveringwitht3.com/ (Accessed April 6, 2025).
50. Michaelsson LF, Medici BB, la Cour JL, Selmer C, Røder M, Perrild H, et al. Treating hypothyroidism with thyroxine/triiodothyronine combination therapy in Denmark: following guidelines or following trends? Eur Thyroid J. (2015) 4:174–80. doi: 10.1159/000437262
51. Pepper GM and Casanova-Romero PY. Conversion to Armour Thyroid from levothyroxine improved patient satisfaction in the treatment of hypothyroidism. J Endocrinol Diabetes Obes. (2014) 2:1055–9. doi: 10.47739/2333-6692/105
52. Heald AH, Premawardhana L, Taylor P, Okosieme O, Bangi T, Devine H, et al. Is there a role for natural desiccated thyroid in the treatment of levothyroxine unresponsive hypothyroidism? Results from a consecutive case series. Int J Clin Pract. (2021) 75:e14967. doi: 10.1111/ijcp.14967
53. Shakir MKM, Brooks DI, McAninch EA, Fonseca TL, Mai VQ, Bianco AC, et al. Comparative effectiveness of levothyroxine, desiccated thyroid extract, and levothyroxine+Liothyronine in hypothyroidism. J Clin Endocrinol Metab. (2021) 106:e4400–13. doi: 10.1210/clinem/dgab478
54. Peterson SJ, Cappola AR, Castro MR, Dayan CM, Farwell AP, Hennessey JV, et al. An online survey of hypothyroid patients demonstrates prominent dissatisfaction. Thyroid. (2018) 28:707–21. doi: 10.1089/thy.2017.0681
55. Mitchell AL, Hegedüs L, Žarković M, Hickey JL, and Perros P. Patient satisfaction and quality of life in hypothyroidism: An online survey by the british thyroid foundation. Clin Endocrinol (Oxf). (2021) 94:513–20. doi: 10.1111/cen.14340
56. Toloza FJK, Espinoza Suarez NR, El Kawkgi O, Golembiewski EH, Ponce OJ, Yao L, et al. Patient experiences and perceptions associated with the use of desiccated thyroid extract. Medicina (Kaunas). (2020) 56:161. doi: 10.3390/medicina56040161
57. O’Reilly DS. Thyroid hormone replacement: an iatrogenic problem. Int J Clin Pract. (2010) 64:991–4. doi: 10.1111/j.1742-1241.2009.02317.x
58. Barnes BO and Galton L. Hypothyroidism: The Unsuspected Illness. New York: HarperCollins (1976).
59. Holtorf K, Brownstein D, Wilson D, Freidman M, and Shomon M. National Academy of Hypothyroidism. Available online at: http://www.nahypothyroidism.org/ (Accessed April 6, 2025).
62. Baisier WV, Hertoghe J, and Eeckhaut W. Thyroid insufficiency. Is thyroxine the only valuable drug? J Nutr Environ Med. (2001) 11:159–66. doi: 10.1080/13590840120083376
63. Derry D. Interview with Shomon M. Available online at: https://web.archive.org/web/20190525200844/http:/www.thyroid-info.com/articles/david-derry.htm (Accessed April 6, 2025).
65. Toft AD. Thyroid hormone replacement - a counterblast to guidelines. J R Coll Physicians Edinb. (2017) 47:307–9. doi: 10.4997/JRCPE.2017.401
66. Levine DT. The definition of optimal metabolism and its association with large reductions in chronic diseases. Trends Med. (2018) 18:5–5. doi: 10.15761/TiM.1000122
67. Midgley JEM, Toft AD, Larisch R, Dietrich JW, and Hoermann R. Time for a reassessment of the treatment of hypothyroidism. BMC Endocr Disord. (2019) 19:37. doi: 10.1186/s12902-019-0365-4
68. Hoermann R, Midgley JEM, Larisch R, and Dietrich JW. Individualised requirements for optimum treatment of hypothyroidism: complex needs, limited options. Drugs Context. (2019) 8:212597. doi: 10.7573/dic.212597
69. Bianco AC. Rethinking Hypothyroidism: Why Treatment Must Change and What Patients Can Do. Chicago IL: Univ. of Chicago Press (2022).
70. Clinical and Laboratory Standards Institute. Defining, Establishing, and Verifying Reference Intervals in the Clinical Laboratory: Approved Guideline. 3rd ed. Vol. 28. . Wayne, PA: CSLI document (2008) p. C28–A3.
71. Maes M, Mommen K, Hendrickx D, Peeters D, D’Hodte P, Ranjan R, et al. Components of biological variation, including seasonality, in blood concentrations of TSH, TT3, FT4, PRL, cortisol and testosterone in healthy volunteers. Clin Endocrinol (Oxf). (1997) 46:587–98. doi: 10.1046/j.1365-2265.1997.1881002.x
72. Walter Reed Army Medical Center Laboratory. Reported FT4 range 1.01-1.79ng/dl based upon 120 healthy soldiers—per telephone communication with Ms. King, head laboratory scientist. (2009).
73. Kratzsch J, Fiedler GM, Leichtle A, Brügel M, Buchbinder S, Otto L, et al. New reference intervals for thyrotropin and thyroid hormones based on National Academy of Clinical Biochemistry criteria and regular ultrasonography of the thyroid. Clin Chem. (2005) 51:1480–6. doi: 10.1373/clinchem.2004.047399
74. Takeda K, Mishiba M, Sugiura H, Nakajima A, Kohama M, and Hiramatsu S. Evaluated reference intervals for serum free thyroxine and thyrotropin using the conventional outliner rejection test without regard to presence of thyroid antibodies and prevalence of thyroid dysfunction in Japanese subjects. Endocr J. (2009) 56:1059–66. doi: 10.1507/endocrj.k09e-123
75. González-Sagrado M and Martín-Gil FJ. Population-specific reference values for thyroid hormones on the Abbott ARCHITECT i2000 analyzer. Clin Chem Lab Med. (2004) 42:540–2. doi: 10.1515/CCLM.2004.091
76. Valeix P, Dos Santos C, Castetbon K, Bertrais S, Cousty C, and Hercberg S. Thyroid hormone levels and thyroid dysfunction of French adults participating in the SU.VI.MAX study. Ann Endocrinol (Paris). (2004) 65:477–86. doi: 10.1016/s0003-4266(04)95956-2
77. Builes-Barrera CA, Marquez-Fernandez JM, Gomez-Baena R, and Cardenas-Gomez M. TSH and free T4 serum values in an adult population in Medellin, Columbia. Presented at the 85th Annual Meeting of the American Thyroid Association, Lake Buena Vista, FL. Poster no. 184. Thyroid. (2015) 25:A76. doi: 10.1089/thy.2015.29004.abstracts
78. Beckman Coulter, Inc. Access FT4, ref. 33880:5 (2010). (316 non-patients: FT4 range: 0.61-1.12ng/dl).
79. Roche Diagnostics. Reference Intervals for Children and Adults. Mannheim, Germany: Elecsys Thyroid Tests (2009).
80. Hollowell JG, Staehling NW, Flanders WD, Hannon WH, Gunter EW, Spencer CA, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. (2002) 87:489–99. doi: 10.1210/jcem.87.2.8182
81. Canaris GJ, Manowitz NR, Mayor G, and Ridgway EC. The Colorado thyroid disease prevalence study. Arch Intern Med. (2000) 160:526–34. doi: 10.1001/archinte.160.4.526
82. Gurnell M, Halsall DJ, and Chatterjee VK. What should be done when thyroid function tests do not make sense? Clin Endocrinol (Oxf). (2011) 74:673–8. doi: 10.1111/j.1365-2265.2011.04023.x
83. Koulouri O, Moran C, Halsall D, Chatterjee K, and Gurnell M. Pitfalls in the measurement and interpretation of thyroid function tests. Best Pract Res Clin Endocrinol Metab. (2013) 27:745–62. doi: 10.1016/j.beem.2013.10.003
84. Soldin OP and Soldin SJ. Thyroid hormone testing by tandem mass spectrometry. Clin Biochem. (2011) 44:89–94. doi: 10.1016/j.clinbiochem.2010.07.020
85. Prinz PN, Scanlan JM, Vitaliano PP, Moe KE, Borson S, Toivola B, et al. Thyroid hormones: positive relationships with cognition in healthy, euthyroid older men. J Gerontol A Biol Sci Med Sci. (1999) 54:M111–6. doi: 10.1093/gerona/54.3.m111
86. Beydoun MA, Beydoun HA, Kitner-Triolo MH, Kaufman JS, Evans MK, and Zonderman AB. Thyroid hormones are associated with cognitive function: moderation by sex, race and depressive symptoms. J Clin Endocrinol Metab. (2013) 98:3470–81. doi: 10.1210/jc.2013-1813
87. Volpato S, Guralnik JM, Fried LP, Remaley AT, Cappola AR, and Launer LJ. Serum thyroxine level and cognitive decline in euthyroid older women. Neurology. (2002) 58:1055–61. doi: 10.1212/wnl.58.7.1055
88. Kooistra L, Crawford S, van Baar AL, Brouwers EP, and Pop VJ. Neonatal effects of maternal hypothyroxinemia during early pregnancy. Pediatrics. (2006) 117:161–7. doi: 10.1542/peds.2005-0227
89. Pop VJ, Kuijpens JL, van Baar AL, Verkerk G, van Son MM, de Vijlder JJ, et al. Low maternal free thyroxine concentrations during early pregnancy are associated with impaired psychomotor development in infancy. Clin Endocrinol (Oxf). (1999) 50:149–55. doi: 10.1046/j.1365-2265.1999.00639.x
90. Román GC, Ghassabian A, Bongers-Schokking JJ, Jaddoe VW, Hofman A, de Rijke YB, et al. Association of gestational maternal hypothyroxinemia and increased autism risk. Ann Neurol. (2013) 74:733–42. doi: 10.1002/ana.23976
91. Berent D, Zboralski K, Orzechowska A, and Gałecki P. Thyroid hormones association with depression severity and clinical outcome in patients with major depressive disorder. Mol Biol Rep. (2014) 41:2419–25. doi: 10.1007/s11033-014-3097-6
92. Cole DP, Thase ME, Mallinger AG, Soares JC, Luther JF, Kupfer DJ, et al. Slower treatment response in bipolar depression predicted by lower pretreatment thyroid function. Am J Psychiatry. (2002) 159:116–21. doi: 10.1176/appi.ajp.159.1.116
93. Gitlin M, Altshuler LL, Frye MA, Suri R, Huynh EL, Fairbanks L, et al. Peripheral thyroid hormones and response to selective serotonin reuptake inhibitors. J Psychiatry Neurosci. (2004) 29:383–6.
94. Boral GC, Ghosh AB, Pal SK, Ghosh KK, and Nandi DN. Thyroid function in different psychiatric disorders. Indian J Psychiatry. (1980) 22:200–2.
95. Reuters VS, Buescu A, Reis FA, Almeida CP, Teixeira PF, Costa AJ, et al. Clinical and muscular evaluation in patients with subclinical hypothyroidism. Arq Bras Endocrinol Metabol. (2006) 50:523–31. doi: 10.1590/s0004-27302006000300016
96. Zhang Y, Chang Y, Ryu S, Cho J, Lee WY, Rhee EJ, et al. Thyroid hormones and mortality risk in euthyroid individuals: the Kangbuk Samsung health study. J Clin Endocrinol Metab. (2014) 99:2467–76. doi: 10.1210/jc.2013-3832
97. Wang J, Zheng X, Sun M, Wang Z, Fu Q, Shi Y, et al. Low serum free thyroxine concentrations associate with increased arterial stiffness in euthyroid subjects: a population-based cross-sectional study. Endocrine. (2015) 50:465–73. doi: 10.1007/s12020-015-0602-1
98. Dullaart RP, de Vries R, Roozendaal C, Kobold AC, and Sluiter WJ. Carotid artery intima media thickness is inversely related to serum free thyroxine in euthyroid subjects. Clin Endocrinol (Oxf). (2007) 67:668–73. doi: 10.1111/j.1365-2265.2007.02943.x
99. Valentina VN, Marijan B, Chedo D, and Branka K. Subclinical hypothyroidism and risk to carotid atherosclerosis. Arq Bras Endocrinol Metabol. (2011) 55:475–80. doi: 10.1590/s0004-27302011000700007
100. Auer J, Berent R, Weber T, Lassnig E, and Eber B. Thyroid function is associated with presence and severity of coronary atherosclerosis. Clin Cardiol. (2003) 26:569–73. doi: 10.1002/clc.4960261205
101. Chadarevian R, Bruckert E, Ankri A, Beucler I, Giral P, and Turpin G. Relationship between thyroid hormones and plasma D-dimer levels. Thromb Haemost. (1998) 79:99–103.
102. Knudsen N, Laurberg P, Rasmussen LB, Bülow I, Perrild H, Ovesen L, et al. Small differences in thyroid function may be important for body mass index and the occurrence of obesity in the population. J Clin Endocrinol Metab. (2005) 90:4019–24. doi: 10.1210/jc.2004-2225
103. Alevizaki M, Saltiki K, Voidonikola P, Mantzou E, Papamichael C, and Stamatelopoulos K. Free thyroxine is an independent predictor of subcutaneous fat in euthyroid individuals. Eur J Endocrinol. (2009) 161:459–65. doi: 10.1530/EJE-09-0441
104. Roos A, Bakker SJ, Links TP, Gans RO, and Wolffenbuttel BH. Thyroid function is associated with components of the metabolic syndrome in euthyroid subjects. J Clin Endocrinol Metab. (2007) 92:491–6. doi: 10.1210/jc.2006-1718
105. Inoue K, Tsujimoto T, Saito J, and Sugiyama T. Association between low normal thyroid function and mortality. In: Abstract presented at the 85th Annual Meeting of the American Thyroid Association, vol. 25. Poster 164) Thyroid, Lake Buena Vista, FL (2015). p. A–68.
106. Mehran L, Amouzegar A, Bakhtiyari M, Mansournia MA, Rahimabad PK, Tohidi M, et al. Variations in serum free thyroxine concentration within the reference range predicts the incidence of metabolic syndrome in non-obese adults: A cohort study. Thyroid. (2017) 27:886–93. doi: 10.1089/thy.2016.0557
107. Tao Y, Gu H, Wu J, and Sui J. Thyroid function is associated with non-alcoholic fatty liver disease in euthyroid subjects. Endocr Res. (2015) 40:74–8. doi: 10.3109/07435800.2014.952014
108. Ortega E, Pannacciulli N, Bogardus C, and Krakoff J. Plasma concentrations of free triiodothyronine predict weight change in euthyroid persons. Am J Clin Nutr. (2007) 85:440–5. doi: 10.1093/ajcn/85.2.440
109. Fisher AJ, Medaglia JD, and Jeronimus BF. Lack of group-to-individual generalizability is a threat to human subjects research. Proc Natl Acad Sci U.S.A. (2018) 115:E6106–15. doi: 10.1073/pnas.1711978115
110. Hoermann R, Eckl W, Hoermann C, and Larisch R. Complex relationship between free thyroxine and TSH in the regulation of thyroid function. Eur J Endocrinol. (2010) 162:1123–9. doi: 10.1530/EJE-10-0106
111. Persani L, Borgato S, Romoli R, Asteria C, Pizzocaro A, and Beck-Peccoz P. Changes in the degree of sialylation of carbohydrate chains modify the biological properties of circulating thyrotropin isoforms in various physiological and pathological states. J Clin Endocrinol Metab. (1998) 83:2486–92. doi: 10.1210/jcem.83.7.4970
112. Beck-Peccoz P and Persani L. Variable biological activity of thyroid-stimulating hormone. Eur J Endocrinol. (1994) 131:331–40. doi: 10.1530/eje.0.1310331
113. Yamada M and Mori M. Mechanisms related to the pathophysiology and management of central hypothyroidism. Nat Clin Pract Endocrinol Metab. (2008) 4:683–94. doi: 10.1038/ncpendmet0995
114. Beck-Peccoz P, Amr S, Menezes-Ferreira MM, Faglia G, and Weintraub BD. Decreased receptor binding of biologically inactive thyrotropin in central hypothyroidism. Effect of treatment with thyrotropin-releasing hormone. N Engl J Med. (1985) 312:1085–90. doi: 10.1056/NEJM198504253121703
115. Faglia G, Bitensky L, Pinchera A, Ferrari C, Paracchi A, Beck-Peccoz P, et al. Thyrotropin secretion in patients with central hypothyroidism: evidence for reduced biological activity of immunoreactive thyrotropin. J Clin Endocrinol Metab. (1979) 48:989–98. doi: 10.1210/jcem-48-6-989
116. Prieto-Tenreiro A and Diaz-Guardiola P. Isolated idiopathic central hypothyroidism in an adult, possibly caused by thyrotropin releasing hormone (TRH) deficiency. Hormones (Athens). (2010) 9:176–80. doi: 10.14310/horm.2002.1268
117. Gharib H and Abboud CF. Primary idiopathic hypothalamic hypothyroidism. Report of four cases. Am J Med. (1987) 83:171–4. doi: 10.1016/0002-9343(87)90515-8
118. Preiss D, Todd L, and Panarelli M. Diagnosing unsuspected hypopituitarism in adults from suggestive thyroid function test results. Ann Clin Biochem. (2008) 45:70–5. doi: 10.1258/acb.2007.007100
119. Tanaka Y, Sawa H, Inden M, Kosaka Y, and Takezawa H. A case of idiopathic hypothalamic hypothyroidism. Jpn J Med. (1981) 20:222–6. doi: 10.2169/internalmedicine1962.20.222
120. Alexopoulou O, Beguin C, De Nayer P, and Maiter D. Clinical and hormonal characteristics of central hypothyroidism at diagnosis and during follow-up in adult patients. Eur J Endocrinol. (2004) 150:1–8. doi: 10.1530/eje.0.1500001
121. Shimon I, Cohen O, Lubetsky A, and Olchovsky D. Thyrotropin suppression by thyroid hormone replacement is correlated with thyroxine level normalization in central hypothyroidism. Thyroid. (2002) 12:823–7. doi: 10.1089/105072502760339406
122. Larsen PR and Davies TF. Hypothyroidism and thyroiditis. In: Larsen PR, Kronenberg HM, Melmed S, and Polonsky KS, editors. Williams Textbook of Endocrinology, 10th edition. Saunders, Philadelphia, PA (2003). p. 430.
123. Carlé A, Laurberg P, Pedersen IB, Perrild H, Ovesen L, Rasmussen LB, et al. Age modifies the pituitary TSH response to thyroid failure. Thyroid. (2007) 17:139–44. doi: 10.1089/thy.2006.0191
124. Hennemann G and Krenning EF. The kinetics of thyroid hormone transporters and their role in non-thyroidal illness and starvation. Best Pract Res Clin Endocrinol Metab. (2007) 21:323–38. doi: 10.1016/j.beem.2007.03.007
125. Wassen FW, Moerings EP, van Toor H, Hennemann G, and Everts ME. Thyroid hormone uptake in cultured rat anterior pituitary cells: effects of energy status and bilirubin. J Endocrinol. (2000) 165:599–606. doi: 10.1677/joe.0.1650599
126. Krenning E, Docter R, Bernard B, Visser T, and Hennemann G. Characteristics of active transport of thyroid hormone into rat hepatocytes. Biochim Biophys Acta. (1981) 676:314–20. doi: 10.1016/0304-4165(81)90165-3
127. Lazar MA. Thyroid hormone receptors: multiple forms, multiple possibilities. Endocr Rev. (1993) 14:184–93. doi: 10.1210/edrv-14-2-184
128. Fitzgerald SP and Falhammar H. Redefinition of successful treatment of patients with hypothyroidism. Is TSH the best biomarker of euthyroidism? Front Endocrinol (Lausanne). (2022) 13:920854. doi: 10.3389/fendo.2022.920854
129. Zulewski H, Müller B, Exer P, Miserez AR, and Staub JJ. Estimation of tissue hypothyroidism by a new clinical score: evaluation of patients with various grades of hypothyroidism and controls. J Clin Endocrinol Metab. (1997) 82:771–6. doi: 10.1210/jcem.82.3.3810
130. Mallipedhi A, Vali H, and Okosieme O. Myxedema coma in a patient with subclinical hypothyroidism. Thyroid. (2011) 21:87–9. doi: 10.1089/thy.2010.0175
131. Lawton RI, Sabatini BL, and Hochbaum DR. Longevity, demographic characteristics, and socio-economic status are linked to triiodothyronine levels in the general population. Proc Natl Acad Sci U.S.A. (2024) 121:e2308652121. doi: 10.1073/pnas.2308652121
132. Castagna MG, Dentice M, Cantara S, Ambrosio R, Maino F, Porcelli T, et al. DIO2 Thr92Ala reduces deiodinase-2 activity and serum-T3 levels in thyroid-deficient patients. J Clin Endocrinol Metab. (2017) 102:1623–30. doi: 10.1210/jc.2016-2587
133. Panicker V, Saravanan P, Vaidya B, Evans J, Hattersley AT, Frayling TM, et al. Common variation in the DIO2 gene predicts baseline psychological well-being and response to combination thyroxine plus triiodothyronine therapy in hypothyroid patients. J Clin Endocrinol Metab. (2009) 94:1623–9. doi: 10.1210/jc.2008-1301
134. Peeters RP, van Toor H, Klootwijk W, de Rijke YB, Kuiper GG, Uitterlinden AG, et al. Polymorphisms in thyroid hormone pathway genes are associated with plasma TSH and iodothyronine levels in healthy subjects. J Clin Endocrinol Metab. (2003) 88:2880–8. doi: 10.1210/jc.2002-021592
135. Silva JE, Gordon MB, Crantz FR, Leonard JL, and Larsen PR. Qualitative and quantitative differences in the pathways of extrathyroidal triiodothyronine generation between euthyroid and hypothyroid rats. J Clin Invest. (1984) 73:898–907. doi: 10.1172/JCI111313
136. Chopra IJ. A study of extrathyroidal conversion of thyroxine (T4) to 3,3’,5-triiodothyronine (T3) in vitro. Endocrinology. (1977) 101:453–63. doi: 10.1210/endo-101-2-453
137. Kim SW, Harney JW, and Larsen PR. Studies of the hormonal regulation of type 2 5’-iodothyronine deiodinase messenger ribonucleic acid in pituitary tumor cells using semiquantitative reverse transcription-polymerase chain reaction. Endocrinology. (1998) 139:4895–905. doi: 10.1210/endo.139.12.6334
138. Cettour-Rose P, Visser TJ, Burger AG, and Rohner-Jeanrenaud F. Inhibition of pituitary type 2 deiodinase by reverse triiodothyronine does not alter thyroxine-induced inhibition of thyrotropin secretion in hypothyroid rats. Eur J Endocrinol. (2005) 153:429–34. doi: 10.1530/eje.1.01984
139. Philibert RA, Beach SR, Gunter TD, Todorov AA, Brody GH, Vijayendran M, et al. The relationship of deiodinase 1 genotype and thyroid function to lifetime history of major depression in three independent populations. Am J Med Genet B Neuropsychiatr Genet. (2011) 156B:593–9. doi: 10.1002/ajmg.b.31200
140. Cooper-Kazaz R, van der Deure WM, Medici M, Visser TJ, Alkelai A, Glaser B, et al. Preliminary evidence that a functional polymorphism in type 1 deiodinase is associated with enhanced potentiation of the antidepressant effect of sertraline by triiodothyronine. J Affect Disord. (2009) 116:113–6. doi: 10.1016/j.jad.2008.10.019
141. Verloop H, Dekkers OM, Peeters RP, Schoones JW, and Smit JW. Genetics in endocrinology: genetic variation in deiodinases: a systematic review of potential clinical effects in humans. Eur J Endocrinol. (2014) 171:R123–35. doi: 10.1530/EJE-14-0302
142. Robinson P. Recovering with T3 (2018). North Somerset, UK: The Elephant in the Room Books. Available online at: http://recoveringwitht3.com/ (Accessed November 16, 2024).
143. Tjørve E, Tjørve KM, Olsen JO, Senum R, and Oftebro H. On commonness and rarity of thyroid hormone resistance: A discussion based on mechanisms of reduced sensitivity in peripheral tissues. Med Hypotheses. (2007) 69:913–21. doi: 10.1016/j.mehy.2006.12.056
144. Brent GA. Mechanisms of thyroid hormone action. J Clin Invest. (2012) 122:3035–43. doi: 10.1172/JCI60047
145. van der Deure WM, Appelhof BC, Peeters RP, Wiersinga WM, Wekking EM, Huyser J, et al. Polymorphisms in the brain-specific thyroid hormone transporter OATP1C1 are associated with fatigue and depression in hypothyroid patients. Clin Endocrinol (Oxf). (2008) 69:804–11. doi: 10.1111/j.1365-2265.2008.03267.x
146. Medici M, van der Deure WM, Verbiest M, Vermeulen SH, Hansen PS, Kiemeney LA, et al. A large-scale association analysis of 68 thyroid hormone pathway genes with serum TSH and FT4 levels. Eur J Endocrinol. (2011) 164:781–8. doi: 10.1530/EJE-10-1130
147. Hennemann G, Docter R, Friesema EC, de Jong M, Krenning EP, and Visser TJ. Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr Rev. (2001) 22:451–76. doi: 10.1210/edrv.22.4.0435
148. Everts ME, de Jong M, Lim CF, Docter R, Krenning EP, Visser TJ, et al. Different regulation of thyroid hormone transport in liver and pituitary: its possible role in the maintenance of low T3 production during nonthyroidal illness and fasting in man. Thyroid. (1996) 6:359–68. doi: 10.1089/thy.1996.6.359
149. Ortiga-Carvalho TM, Sidhaye AR, and Wondisford FE. Thyroid hormone receptors and resistance to thyroid hormone disorders. Nat Rev Endocrinol. (2014) 10:582–91. doi: 10.1038/nrendo.2014.143
150. Moran C, Agostini M, Visser WE, Schoenmakers E, Schoenmakers N, Offiah AC, et al. Resistance to thyroid hormone caused by a mutation in thyroid hormone receptor (TR)α1 and TRα2: clinical, biochemical, and genetic analyses of three related patients. Lancet Diabetes Endocrinol. (2014) 2:619–26. doi: 10.1016/S2213-8587(14)70111-1
151. van Gucht AL, Meima ME, Zwaveling-Soonawala N, Visser WE, Fliers E, Wennink JM, et al. Resistance to thyroid hormone alpha in an 18-month-old girl: clinical, therapeutic, and molecular characteristics. Thyroid. (2016) 26:338–46. doi: 10.1089/thy.2015.0463
152. Moore JM and Guy RK. Coregulator interactions with the thyroid hormone receptor. Mol Cell Proteomics. (2005) 4:475–82. doi: 10.1074/mcp.R500001-MCP200
153. Canaris GJ, Steiner JF, and Ridgway EC. Do traditional symptoms of hypothyroidism correlate with biochemical disease? J Gen Intern Med. (1997) 12:544–50. doi: 10.1046/j.1525-1497.1997.07109.x
154. Carlé A, Pedersen IB, Knudsen N, Perrild H, Ovesen L, and Laurberg P. Hypothyroid symptoms and the likelihood of overt thyroid failure: a population-based case-control study. Eur J Endocrinol. (2014) 171:593–602. doi: 10.1530/EJE-14-0481
155. Pollock MA, Sturrock A, Marshall K, Davidson KM, Kelly CJ, McMahon AD, et al. Thyroxine treatment in patients with symptoms of hypothyroidism but thyroid function tests within the reference range: randomised double blind placebo controlled crossover trial. BMJ. (2001) 323:891–5. doi: 10.1136/bmj.323.7318.891
156. Skinner GRB, Holmes D, Ahmad A, Davies JA, and Benitez J. Clinical response to thyroxine sodium in clinically hypothyroid but biochemically euthyroid patients. J Nutr Environ Med. (2009) 10:115–24. doi: 10.1080/13590840050043530
157. Escobar-Morreale HF, Obregon MJ, Calvo R, and Escobar del Rey F. Continuous infusion of different doses of T4 or T3 in thyroidectomized rats: circulating and tissue levels of T4 and T3. Presented at the 67th Annual Meeting of the American Thyroid Association, Tampa, Florida, Abstract no. T49 (1993).
158. Escobar-Morreale HF, Obregón MJ, Escobar del Rey F, and Morreale de Escobar G. Tissue-specific patterns of changes in 3,5,3’-triiodo-L-thyronine concentrations in thyroidectomized rats infused with increasing doses of the hormone. Which are the regulatory mechanisms? Biochimie. (1999) 81:453–62. doi: 10.1016/s0300-9084(99)80095-9
159. Escobar-Morreale HF, Obregón MJ, Escobar del Rey F, and Morreale de Escobar G. Replacement therapy for hypothyroidism with thyroxine alone does not ensure euthyroidism in all tissues, as studied in thyroidectomized rats. J Clin Invest. (1995) 96:2828–38. doi: 10.1172/JCI118353
160. Werneck de Castro JP, Fonseca TL, Ueta CB, McAninch EA, Abdalla S, Wittmann G, et al. Differences in hypothalamic type 2 deiodinase ubiquitination explain localized sensitivity to thyroxine. J Clin Invest. (2015) 125:769–81. doi: 10.1172/JCI77588
161. Larsen PR. Thyroid-pituitary interaction: feedback regulation of thyrotropin secretion by thyroid hormones. N Engl J Med. (1982) 306:23–32. doi: 10.1056/NEJM198201073060107
162. Batistuzzo A, Salas-Lucia F, Gereben B, Ribeiro MO, and Bianco AC. Sustained pituitary T3 production explains the T4-mediated TSH feedback mechanism. Endocrinology. (2023) 164:bqad155. doi: 10.1210/endocr/bqad155
163. Christoffolete MA, Ribeiro R, Singru P, Fekete C, da Silva WS, Gordon DF, et al. Atypical expression of type 2 iodothyronine deiodinase in thyrotrophs explains the thyroxine-mediated pituitary thyrotropin feedback mechanism. Endocrinology. (2006) 147:1735–43. doi: 10.1210/en.2005-1300
164. Ain KB, Pucino F, Shiver TM, and Banks SM. Thyroid hormone levels affected by time of blood sampling in thyroxine-treated patients. Thyroid. (1993) 3:81–5. doi: 10.1089/thy.1993.3.81
165. Wennlund A. Variation in serum levels of T3, T4, FT4 and TSH during thyroxine replacement therapy. Acta Endocrinol (Copenh). (1986) 113:47–9. doi: 10.1530/acta.0.1130047
166. Carpi A, Toni MG, and De Gaudio C. Effect of a single oral dose of L-thyroxine (150 micrograms) on serum thyroid hormone and TSH concentrations in clinically euthyroid goitrous patients. Thyroidology. (1992) 4:69–73.
167. Saberi M and Utiger RD. Serum thyroid hormone and thyrotropin concentrations during thyroxine and triiodothyronine therapy. J Clin Endocrinol Metab. (1974) 39:923–7. doi: 10.1210/jcem-39-5-923
168. Jonklaas J, Burman KD, Wang H, and Latham KR. Single-dose T3 administration: kinetics and effects on biochemical and physiological parameters. Ther Drug Monit. (2015) 37:110–8. doi: 10.1097/FTD.0000000000000113
169. Larsen PR and Frumess RD. Comparison of the biological effects of thyroxine and triiodothyronine in the rat. Endocrinology. (1977) 100:980–8. doi: 10.1210/endo-100-4-980
170. Hennemann G, Docter R, Visser TJ, Postema PT, and Krenning EP. Thyroxine plus low-dose, slow-release triiodothyronine replacement in hypothyroidism: proof of principle. Thyroid. (2004) 14:271–5. doi: 10.1089/105072504323030924
171. Ishii H, Inada M, Tanaka K, Mashio Y, Naito K, Nishikawa M, et al. Induction of outer and inner ring monodeiodinases in human thyroid gland by thyrotropin. J Clin Endocrinol Metab. (1983) 57:500–5. doi: 10.1210/jcem-57-3-500
172. Hoermann R, Midgley JE, Larisch R, and Dietrich JW. Integration of peripheral and glandular regulation of triiodothyronine production by thyrotropin in untreated and thyroxine-treated subjects. Horm Metab Res. (2015) 47:674–80. doi: 10.1055/s-0034-1398616
173. Citterio CE, Veluswamy B, Morgan SJ, Galton VA, Banga JP, Atkins S, et al. De novo triiodothyronine formation from thyrocytes activated by thyroid-stimulating hormone. J Biol Chem. (2017) 292:15434–44. doi: 10.1074/jbc.M117.784447
174. Kabadi UM. Role of thyrotropin in metabolism of thyroid hormones in nonthyroidal tissues. Metabolism. (2006) 55:748–50. doi: 10.1016/j.metabol.2006.01.010
175. Kabadi UM. T4 to T3 Conversion by TSH in Extra Thyroidal Tissues. Abstract presented at the 100th Annual Meeting of the Endocrine Society, Chicago, IL. Abstract no. OR34-3. Endocr Rev. (2018) 39:Supplement:1296. doi: 10.1093/edrv/39.supp.1
176. Kabadi UM. Role of thyrotropin in triiodothyronine generation in hypothyroidism. Thyroidology. (1993) 5:41–7.
177. Larsen PR, Davies TF, Schlumberger MJ, and Hay ID. Thyroid physiology and diagnostic evaluation of patients with thyroid disorders. In: Larsen PR, Kronenberg HM, Melmed S, and Polonsky KS, editors. Williams Textbook of Endocrinology, 10th edition. Philadelphia: Saunders (2003). p. 341–3.
178. Maia AL, Kim BW, Huang SA, Harney JW, and Larsen PR. Type 2 iodothyronine deiodinase is the major source of plasma T3 in euthyroid humans. J Clin Invest. (2005) 115:2524–33. doi: 10.1172/JCI25083
179. Esfandiari A, Courtin F, Lennon AM, Gavaret JM, and Pierre M. Induction of type III deiodinase activity in astroglial cells by thyroid hormones. Endocrinology. (1992) 131:1682–8. doi: 10.1210/endo.131.4.1396314
180. Jennings PE, O’Malley BP, Griffin KE, Northover B, and Rosenthal FD. Relevance of increased serum thyroxine concentrations associated with normal serum triiodothyronine values in hypothyroid patients receiving thyroxine: a case for “tissue thyrotoxicosis. Br Med J (Clin Res Ed). (1984) 289:1645–7. doi: 10.1136/bmj.289.6459.1645
181. Hoang TD, Olsen CH, Mai VQ, Clyde PW, and Shakir MK. Desiccated thyroid extract compared with levothyroxine in the treatment of hypothyroidism: A randomized, double-blind, crossover study. J Clin Endocrinol Metab. (2013) 98:1982–90. doi: 10.1210/jc.2012-4107
182. Monzani F, Caraccio N, Del Guerra P, Casolaro A, and Ferrannini E. Neuromuscular symptoms and dysfunction in subclinical hypothyroid patients: beneficial effect of L-T4 replacement therapy. Clin Endocrinol (Oxf). (1999) 51:237–42. doi: 10.1046/j.1365-2265.1999.00790.x
183. Hoermann R, Midgley JEM, Larisch R, and Dietrich JW. Lessons from randomised clinical trials for triiodothyronine treatment of hypothyroidism: have they achieved their objectives? J Thyroid Res. (2018) 2018:3239197. doi: 10.1155/2018/3239197
184. Samuels MH, Kolobova I, Niederhausen M, Janowsky JS, and Schuff KG. Effects of altering levothyroxine (L-T4) doses on quality of life, mood, and cognition in L-T4 treated subjects. J Clin Endocrinol Metab. (2018) 103:1997–2008. doi: 10.1210/jc.2017-02668
185. Woeber KA. Levothyroxine therapy and serum free thyroxine and free triiodothyronine concentrations. J Endocrinol Invest. (2002) 25:106–9. doi: 10.1007/BF03343972
186. Mortoglou A and Candiloros H. The serum triiodothyronine to thyroxine (T3/T4) ratio in various thyroid disorders and after Levothyroxine replacement therapy. Hormones (Athens). (2004) 3:120–6. doi: 10.14310/horm.2002.11120
187. Ito M, Miyauchi A, Morita S, Kudo T, Nishihara E, Kihara M, et al. TSH-suppressive doses of levothyroxine are required to achieve preoperative native serum triiodothyronine levels in patients who have undergone total thyroidectomy. Eur J Endocrinol. (2012) 167:373–8. doi: 10.1530/EJE-11-1029
188. Hoermann R, Midgley JE, Larisch R, and Dietrich JW. Is pituitary TSH an adequate measure of thyroid hormone-controlled homoeostasis during thyroxine treatment? Eur J Endocrinol. (2013) 168:271–80. doi: 10.1530/EJE-12-0819
189. Langdahl BL, Loft AG, Eriksen EF, Mosekilde L, and Charles P. Bone mass, bone turnover and body composition in former hypothyroid patients receiving replacement therapy. Eur J Endocrinol. (1996) 134:702–9. doi: 10.1530/eje.0.1340702
190. Samuels MH, Kolobova I, Smeraglio A, Peters D, Purnell JQ, and Schuff KG. Effects of levothyroxine replacement or suppressive therapy on energy expenditure and body composition. Thyroid. (2016) 26:347–55. doi: 10.1089/thy.2015.0345
191. Liewendahl K, Helenius T, Lamberg BA, Mähönen H, and Wägar G. Free thyroxine, free triiodothyronine, and thyrotropin concentrations in hypothyroid and thyroid carcinoma patients receiving thyroxine therapy. Acta Endocrinol (Copenh). (1987) 116:418–24. doi: 10.1530/acta.0.1160418
192. Fish LH, Schwartz HL, Cavanaugh J, Steffes MW, Bantle JP, and Oppenheimer JH. Replacement dose, metabolism, and bioavailability of levothyroxine in the treatment of hypothyroidism. Role of triiodothyronine in pituitary feedback in humans. N Engl J Med. (1987) 316:764–70. doi: 10.1056/NEJM198703263161302
193. Alevizaki M, Mantzou E, Cimponeriu AT, Alevizaki CC, and Koutras DA. TSH may not be a good marker for adequate thyroid hormone replacement therapy. Wien Klin Wochenschr. (2005) 117:636–40. doi: 10.1007/s00508-005-0421-0
194. Saravanan P, Chau WF, Roberts N, Vedhara K, Greenwood R, and Dayan CM. Psychological well-being in patients on ‘adequate’ doses of l-thyroxine: results of a large, controlled community-based questionnaire study. Clin Endocrinol (Oxf). (2002) 57:577–85. doi: 10.1046/j.1365-2265.2002.01654.x
195. Samuels MH, Schuff KG, Carlson NE, Carello P, and Janowsky JS. Health status, psychological symptoms, mood, and cognition in L-thyroxine-treated hypothyroid subjects. Thyroid. (2007) 17:249–58. doi: 10.1089/thy.2006.0252
196. Wekking EM, Appelhof BC, Fliers E, Schene AH, Huyser J, Tijssen JG, et al. Cognitive functioning and well-being in euthyroid patients on thyroxine replacement therapy for primary hypothyroidism. Eur J Endocrinol. (2005) 153:747–53. doi: 10.1530/eje.1.02025
197. Winther KH, Cramon P, Watt T, Bjorner JB, Ekholm O, Feldt-Rasmussen U, et al. Disease-specific as well as generic quality of life is widely impacted in autoimmune hypothyroidism and improves during the first six months of levothyroxine therapy. PloS One. (2016) 11:e0156925. doi: 10.1371/journal.pone.0156925
198. Morón-Díaz M, Saavedra P, Alberiche-Ruano MP, Rodríguez-Pérez CA, López-Plasencia Y, Marrero-Arencibia D, et al. Correlation between TSH levels and quality of life among subjects with well-controlled primary hypothyroidism. Endocrine. (2021) 72:190–7. doi: 10.1007/s12020-020-02449-4
199. Wouters H, Kobold AM, Slagter S, Links T, van der Klauw M, and Wolffenbuttel B. The Presence of Comorbidity Has More Impact on Health-Related Quality of Life in Individuals Using Levothyroxine. Presented at the 100th Annual Meeting of the Endocrine Society, Chicago, IL. Abstract no. OR34-1. Endocr Rev. (2018) 39:Supplement:1295. doi: 10.1093/edrv/39.supp.1
200. Panicker V, Evans J, Bjøro T, Asvold BO, Dayan CM, and Bjerkeset O. A paradoxical difference in relationship between anxiety, depression and thyroid function in subjects on and not on T4: findings from the HUNT study. Clin Endocrinol (Oxf). (2009) 71:574–80. doi: 10.1111/j.1365-2265.2008.03521.x
201. Kramer CK, von Mühlen D, Kritz-Silverstein D, and Barrett-Connor E. Treated hypothyroidism, cognitive function, and depressed mood in old age: the Rancho Bernardo Study. Eur J Endocrinol. (2009) 161:917–21. doi: 10.1530/EJE-09-0606
202. Peterson SJ, McAninch EA, and Bianco AC. Is a normal TSH synonymous with “Euthyroidism” in levothyroxine monotherapy? J Clin Endocrinol Metab. (2016) 101:4964–73. doi: 10.1210/jc.2016-2660
203. Azizi F, Amouzegar A, Tohidi M, Hedayati M, and Cheraghi L. Does Levothyroxine monotherapy mimic the effects of endogenous thyroid hormones? Presented at the 87th Annual Meeting of the American Thyroid Association, Victoria, BC, Canada. Poster no. 205. Thyroid. (2017) 27:A–66. doi: 10.1089/thy.2017.29046.abstracts
204. Franklyn JA, Daykin J, Betteridge J, Hughes EA, Holder R, Jones SR, et al. Thyroxine replacement therapy and circulating lipid concentrations. Clin Endocrinol (Oxf). (1993) 38:453–9. doi: 10.1111/j.1365-2265.1993.tb00339.x
205. Clausen P, Mersebach H, Nielsen B, Feldt-Rasmussen B, and Feldt-Rasmussen U. Hypothyroidism is associated with signs of endothelial dysfunction despite 1-year replacement therapy with levothyroxine. Clin Endocrinol (Oxf). (2009) 70:932–7. doi: 10.1111/j.1365-2265.2008.03410.x
206. Flynn RW, Macdonald TM, Jung RT, Morris AD, and Leese GP. Mortality and vascular outcomes in patients treated for thyroid dysfunction. J Clin Endocrinol Metab. (1997) 91:2159–64. doi: 10.1210/jc.2005-1833
207. Gursoy A, Ozduman Cin M, Kamel N, and Gullu S. Which thyroid-stimulating hormone level should be sought in hypothyroid patients under L-thyroxine replacement therapy? Int J Clin Pract. (2006) 60:655–9. doi: 10.1111/j.1368-5031.2006.00822.x
208. Samuels MH, Kolobova I, Smeraglio A, Niederhausen M, Janowsky JS, and Schuff KG. Effect of thyroid function variations within the laboratory reference range on health status, mood, and cognition in levothyroxine-treated subjects. Thyroid. (2016) 26:1173–84. doi: 10.1089/thy.2016.0141
209. Ito M, Miyauchi A, Kang S, Hisakado M, Yoshioka W, Ide A, et al. Effect of the presence of remnant thyroid tissue on the serum thyroid hormone balance in thyroidectomized patients. Eur J Endocrinol. (2015) 173:333–40. doi: 10.1530/EJE-15-0138
210. Gullo D, Latina A, Frasca F, Le Moli R, Pellegriti G, and Vigneri R. Levothyroxine monotherapy cannot guarantee euthyroidism in all athyreotic patients. PloS One. (2011) 6:e22552. doi: 10.1371/journal.pone.0022552
211. Ito M, Miyauchi A, Hisakado M, Yoshioka W, Ide A, Kudo T, et al. Biochemical markers reflecting thyroid function in athyreotic patients on levothyroxine monotherapy. Thyroid. (2017) 27:484–90. doi: 10.1089/thy.2016.0426
212. Larisch R, Midgley JEM, Dietrich JW, and Hoermann R. Symptomatic relief is related to serum free triiodothyronine concentrations during follow-up in levothyroxine-treated patients with differentiated thyroid cancer. Exp Clin Endocrinol Diabetes. (2018) 126:546–52. doi: 10.1055/s-0043-125064
213. Jonklaas J and Nsouli-Maktabi H. Weight changes in euthyroid patients undergoing thyroidectomy. Thyroid. (2011) 21:1343–51. doi: 10.1089/thy.2011.0054
214. Tigas S, Idiculla J, Beckett G, and Toft A. Is excessive weight gain after ablative treatment of hyperthyroidism due to inadequate thyroid hormone therapy? Thyroid. (2000) 10:1107–11. doi: 10.1089/thy.2000.10.1107
215. Ivkovic S. Polyneuropathy after radioactive iodine treatment of hyperthyroidism and beneficial effect of combined T4/T3 therapy of hypothyroidism. Eur Thyroid J. (2012) 1:129–31. doi: 10.1159/000337977
216. Slawik M, Klawitter B, Meiser E, Schories M, Zwermann O, Borm K, et al. Thyroid hormone replacement for central hypothyroidism: a randomized controlled trial comparing two doses of thyroxine (T4) with a combination of T4 and triiodothyronine. J Clin Endocrinol Metab. (2007) 92:4115–22. doi: 10.1210/jc.2007-0297
217. Koulouri O, Auldin MA, Agarwal R, Kieffer V, Robertson C, Falconer Smith J, et al. Diagnosis and treatment of hypothyroidism in TSH deficiency compared to primary thyroid disease: pituitary patients are at risk of under-replacement with levothyroxine. Clin Endocrinol (Oxf). (2011) 74:744–9. doi: 10.1111/j.1365-2265.2011.03984.x
218. Ferretti E, Persani L, Jaffrain-Rea ML, Giambona S, Tamburrano G, and Beck-Peccoz P. Evaluation of the adequacy of levothyroxine replacement therapy in patients with central hypothyroidism. J Clin Endocrinol Metab. (1999) 84:924–9. doi: 10.1210/jcem.84.3.5553
219. Fernandes JK, Klein MJ, Ater JL, Kuttesch JF, and Vassilopoulou-Sellin R. Triiodothyronine supplementation for hypothalamic obesity. Metabolism. (2002) 51:1381–3. doi: 10.1053/meta.2002.35591
221. Kienle GS and Kiene H. Clinical judgement and the medical profession. J Eval Clin Pract. (2011) 17:621–7. doi: 10.1111/j.1365-2753.2010.01560.x
222. de Bruin TW, van Barlingen H, van Linde-Sibenius Trip M, van Vuurst de Vries AR, Akveld MJ, and Erkelens DW. Lipoprotein(a) and apolipoprotein B plasma concentrations in hypothyroid, euthyroid, and hyperthyroid subjects. J Clin Endocrinol Metab. (1993) 76:121–6. doi: 10.1210/jcem.76.1.8421075
223. Soppi ET. Iron deficiency without anemia - a clinical challenge. Clin Case Rep. (2018) 6:1082–6. doi: 10.1002/ccr3.1529
224. Garofalo V, Condorelli RA, Cannarella R, Aversa A, Calogero AE, and La Vignera S. Relationship between iron deficiency and thyroid function: A systematic review and meta-analysis. Nutrients. (2023) 15:4790. doi: 10.3390/nu15224790
225. Eftekhari MH, Simondon KB, Jalali M, Keshavarz SA, Elguero E, Eshraghian MR, et al. Effects of administration of iron, iodine and simultaneous iron-plus-iodine on the thyroid hormone profile in iron-deficient adolescent Iranian girls. Eur J Clin Nutr. (2006) 60:545–52. doi: 10.1038/sj.ejcn.1602349
226. Soppi ET. Iron deficiency is the main cause of symptom persistence in patients treated for hypothyroidism. Abstract presented at the 15th International Thyroid Congress, Lake Buena Vista, FL, Poster no. 179. Thyroid. (2015) 25:A–74. doi: 10.1089/thy.2015.29004.abstracts
227. Salas-Lucia F and Bianco AC. T3 levels and thyroid hormone signaling. Front Endocrinol (Lausanne). (2022) 13:1044691. doi: 10.3389/fendo.2022.1044691
228. Biondi B, Palmieri EA, Fazio S, Cosco C, Nocera M, Saccà L, et al. Endogenous subclinical hyperthyroidism affects quality of life and cardiac morphology and function in young and middle-aged patients. J Clin Endocrinol Metab. (2000) 85:4701–5. doi: 10.1210/jcem.85.12.7085
229. Regalbuto C, Maiorana R, Alagona C, Paola RD, Cianci M, Alagona G, et al. Effects of either LT4 monotherapy or LT4/LT3 combined therapy in patients totally thyroidectomized for thyroid cancer. Thyroid. (2007) 17:323–31. doi: 10.1089/thy.2006.0084
230. Thayakaran R, Adderley NJ, Sainsbury C, Torlinska B, Boelaert K, Šumilo D, et al. Thyroid replacement therapy, thyroid stimulating hormone concentrations, and long term health outcomes in patients with hypothyroidism: longitudinal study. BMJ. (2019) 366:l4892. doi: 10.1136/bmj.l4892
231. Vanderpump MPJ, Ahlquist JAO, Franklyn JA, and Clayton RN. Consensus statement for good practice and audit measures in the management of hypothyroidism and hyperthyroidism. The Research Unit of the Royal College of Physicians of London, the Endocrinology and Diabetes Committee of the Royal College of Physicians of London, and the Society for Endocrinology. BMJ. (1996) 313:539–44. doi: 10.1136/bmj.313.7056.539
232. Toft AD and Beckett GJ. Thyroid function tests and hypothyroidism. BMJ. (2003) 326:295–6. doi: 10.1136/bmj.326.7384.295
233. Nozaki H, Funahashi H, Sato Y, Imai T, Oike E, Kato M, et al. Study of hormone replacement therapy following total thyroidectomy in thyroid cancer–with special reference to the analysis of thyroid hormone peripheral effects, using indirect calorimetry. Nippon Geka Gakkai Zasshi. (1991) 92:1700–7.
234. Samuels MH, Kolobova I, Smeraglio A, Peters D, Janowsky JS, and Schuff KG. The effects of levothyroxine replacement or suppressive therapy on health status, mood, and cognition. J Clin Endocrinol Metab. (2014) 99:843–51. doi: 10.1210/jc.2013-3686
235. Leese GP, Jung RT, Guthrie C, Waugh N, and Browning MC. Morbidity in patients on L-thyroxine: a comparison of those with a normal TSH to those with a suppressed TSH. Clin Endocrinol (Oxf). (1992) 37:500–3. doi: 10.1111/j.1365-2265.1992.tb01480.x
236. De Menis E, Da Rin G, Roiter I, Legovini P, Foscolo G, and Conte N. Bone turnover in overt and subclinical hyperthyroidism due to autonomous thyroid adenoma. Horm Res. (1992) 37:217–20. doi: 10.1159/000182315
237. Bassett JH, O’Shea PJ, Sriskantharajah S, Rabier B, Boyde A, Howell PG, et al. Thyroid hormone excess rather than thyrotropin deficiency induces osteoporosis in hyperthyroidism. Mol Endocrinol. (2007) 21:1095–107. doi: 10.1210/me.2007-0033
238. Speroff L and Fritz M. Clinical Gynecologic Endocrinology and Fertility. 7th edition. Philadelphia: Lippincott Williams & Wilkins (2005). p. 653.
239. Coindre JM, David JP, Rivière L, Goussot JF, Roger P, de Mascarel A, et al. Bone loss in hypothyroidism with hormone replacement. A histomorphometric study. Arch Intern Med. (1986) 146:48–53. doi: 10.1001/archinte.1986.00360130058007
240. Heemstra KA, Hamdy NA, Romijn JA, and Smit JW. The effects of thyrotropin-suppressive therapy on bone metabolism in patients with well-differentiated thyroid carcinoma. Thyroid. (2006) 16:583–91. doi: 10.1089/thy.2006.16.583
241. Ongphiphadhanakul B, Puavilai G, and Rajatanavin R. Effect of TSH-suppressive doses of levothyroxine on bone mineral density in Thai women. J Med Assoc Thai. (1996) 79:563–67.
242. Poomthavorn P, Mahachoklertwattana P, Ongphiphadhanakul B, Preeyasombat C, and Rajatanavin R. Exogenous subclinical hyperthyroidism during adolescence: effect on peak bone mass. J Pediatr Endocrinol Metab. (2005) 18:463–9. doi: 10.1515/jpem.2005.18.5.463
243. Malik R and Hodgson H. The relationship between the thyroid gland and the liver. QJM. (2002) 95:559–69. doi: 10.1093/qjmed/95.9.559
244. Dekkers OM, Horváth-Puhó E, Cannegieter SC, Vandenbroucke JP, Sørensen HT, and Jørgensen JO. Acute cardiovascular events and all-cause mortality in patients with hyperthyroidism: a population-based cohort study. Eur J Endocrinol. (2017) 176:1–9. doi: 10.1530/EJE-16-0576
245. Flynn RW, Bonellie SR, Jung RT, MacDonald TM, Morris AD, and Leese GP. Serum thyroid-stimulating hormone concentration and morbidity from cardiovascular disease and fractures in patients on long-term thyroxine therapy. J Clin Endocrinol Metab. (2010) 95:186–93. doi: 10.1210/jc.2009-1625
246. Biondi B Fazio S, Carella C, Amato G, Cittadini A, Lupoli G, et al. Cardiac effects of long term thyrotropin-suppressive therapy with levothyroxine. J Clin Endocrinol Metab. (1993) 77:334–8. doi: 10.1210/jcem.77.2.8345037
247. Fazio S, Biondi B, Carella C, Sabatini D, Cittadini A, Panza N, et al. Diastolic dysfunction in patients on thyroid-stimulating hormone suppressive therapy with levothyroxine: beneficial effect of beta-blockade. J Clin Endocrinol Metab. (1995) 80:2222–6. doi: 10.1210/jcem.80.7.7608283
248. Mercuro G, Panzuto MG, Bina A, Leo M, Cabula R, Petrini L, et al. Cardiac function, physical exercise capacity, and quality of life during long-term thyrotropin-suppressive therapy with levothyroxine: effect of individual dose tailoring. J Clin Endocrinol Metab. (2000) 85:159–64. doi: 10.1210/jcem.85.1.6298
249. Shapiro LE, Sievert R, Ong L, Ocampo EL, Chance RA, Lee M, et al. Minimal cardiac effects in asymptomatic athyreotic patients chronically treated with thyrotropin-suppressive doses of L-thyroxine. J Clin Endocrinol Metab. (1997) 82:2592–5. doi: 10.1210/jcem.82.8.4155
250. Staerk L, Wang B, Preis SR, Larson MG, Lubitz SA, Ellinor PT, et al. Lifetime risk of atrial fibrillation according to optimal, borderline, or elevated levels of risk factors: cohort study based on longitudinal data from the Framingham Heart Study. BMJ. (2018) 361:k1453. doi: 10.1136/bmj.k1453
251. Go AS, Hylek EM, Phillips KA, Chang Y, Henault LE, Selby JV, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk factors In Atrial fibrillation (ATRIA) Study. JAMA. (2001) 285:2370–5. doi: 10.1001/jama.285.18.2370
252. Wong PSC, Hee FLLS, and Lip GYH. Atrial fibrillation and the thyroid. Heart. (1997) 78:623–4. doi: 10.1136/hrt.78.6.623a
253. Chen YC, Chen SA, Chen YJ, Chang MS, Chan P, and Lin CI. Effects of thyroid hormone on the arrhythmogenic activity of pulmonary vein cardiomyocytes. J Am Coll Cardiol. (2002) 39:366–72. doi: 10.1016/s0735-1097(01)01731-4
254. Gammage MD, Parle JV, Holder RL, Roberts LM, Hobbs FD, Wilson S, et al. Association between serum free thyroxine concentration and atrial fibrillation. Arch Intern Med. (2007) 167:928–34. doi: 10.1001/archinte.167.9.928
255. Cappola AR, Arnold AM, Wulczyn K, Carlson M, Robbins J, and Psaty BM. Thyroid function in the euthyroid range and adverse outcomes in older adults. J Clin Endocrinol Metab. (2015) 100:1088–96. doi: 10.1210/jc.2014-3586
256. Sawin CT, Geller A, Wolf PA, Belanger AJ, Baker E, Bacharach P, et al. Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons. N Engl J Med. (1994) 331:1249–52. doi: 10.1056/NEJM199411103311901
257. Woeber KA. Thyrotoxicosis and the heart. N Engl J Med. (1992) 327:94–8. doi: 10.1056/NEJM199207093270206
258. Al-Aridi R, Abdelmannan D, and Arafah BM. Biochemical diagnosis of adrenal insufficiency: the added value of dehydroepiandrosterone sulfate (DHEA-S) measurements. Endocr Pract. (2011) 17:261–70. doi: 10.4158/EP10262.RA
259. Greenfield JR and Samaras K. Evaluation of pituitary function in the fatigued patient: a review of 59 cases. Eur J Endocrinol. (2006) 154:147–57. doi: 10.1530/eje.1.02010
260. Streeten DH, Anderson GH Jr, and Bonaventura MM. The potential for serious consequences from misinterpreting normal responses to the rapid adrenocorticotropin test. J Clin Endocrinol Metab. (1996) 81:285–90. doi: 10.1210/jcem.81.1.8550765
261. Kamrath C and Boehles H. The low-dose ACTH test does not identify mild insufficiency of the hypothalamic-pituitary-adrenal axis in children with inadequate stress response. J Pediatr Endocrinol Metab. (2010) 23:1097–104. doi: 10.1515/jpem.2010.174
262. Soule S, Van Zyl Smit C, Parolis G, Attenborough S, Peter D, Kinvig S, et al. The low dose ACTH stimulation test is less sensitive than the overnight metyrapone test for the diagnosis of secondary hypoadrenalism. Clin Endocrinol (Oxf). (2000) 53:221–7. doi: 10.1046/j.1365-2265.2000.01057.x
263. Reimondo G, Bovio S, Allasino B, Terzolo M, and Angeli A. Secondary hypoadrenalism. Pituitary. (2008) 11:147–54. doi: 10.1007/s11102-008-0108-4
264. Ospina NS, Al Nofal A, Bancos I, Javed A, Benkhadra K, Kapoor E, et al. ACTH stimulation tests for the diagnosis of adrenal insufficiency: Systematic review and meta-analysis. J Clin Endocrinol Metab. (2016) 101:427–34. doi: 10.1210/jc.2015-1700
265. Psarra AM, Solakidi S, and Sekeris CE. The mitochondrion as a primary site of action of steroid and thyroid hormones: presence and action of steroid and thyroid hormone receptors in mitochondria of animal cells. Mol Cell Endocrinol. (2006) 246:21–33. doi: 10.1016/j.mce.2005.11.025
266. Lee SR, Kim HK, Song IS, Youm J, Dizon LA, Jeong SH, et al. Glucocorticoids and their receptors: insights into specific roles in mitochondria. Prog Biophys Mol Biol. (2013) 112:44–54. doi: 10.1016/j.pbiomolbio.2013.04.001
267. Du J, Wang Y, Hunter R, Wei Y, Blumenthal R, Falke C, et al. Dynamic regulation of mitochondrial function by glucocorticoids. Proc Natl Acad Sci U.S.A. (2009) 106:3543–8. doi: 10.1073/pnas.0812671106
268. Sánchez-Franco F, Fernández L, Fernández G, and Cacicedo L. Thyroid hormone action on ACTH secretion. Horm Metab Res. (1989) 21:550–2. doi: 10.1055/s-2007-1009285
269. Lizcano F and Rodríguez JS. Thyroid hormone therapy modulates hypothalamo-pituitary-adrenal axis. Endocr J. (2011) 58:137–42. doi: 10.1507/endocrj.k10e-369
270. Hoshiro M, Ohno Y, Masaki H, Iwase H, and Aoki N. Comprehensive study of urinary cortisol metabolites in hyperthyroid and hypothyroid patients. Clin Endocrinol (Oxf). (2006) 64:37–45. doi: 10.1111/j.1365-2265.2005
271. De Nayer P, Dozin B, Vandeput Y, Bottazzo FC, and Crabbe J. Altered interaction between triiodothyronine and its nuclear receptors in absence of cortisol: a proposed mechanism for increased thyrotropin secretion in corticosteroid deficiency states. Eur J Clin Invest. (1987) 17:106–10. doi: 10.1111/j.1365-2362.1987.tb02388.x
272. Samuels MH and McDaniel PA. Thyrotropin levels during hydrocortisone infusions that mimic fasting-induced cortisol elevations: a clinical research center study. J Clin Endocrinol Metab. (1997) 82:3700–4. doi: 10.1210/jcem.82.11.4376
273. Comtois R, Hébert J, and Soucy JP. Increase in T3 levels during hypocorticism in patients with chronic secondary adrenocortical insufficiency. Acta Endocrinol (Copenh). (1992) 126:319–24. doi: 10.1530/acta.0.1260319
274. Bigos ST, Ridgway EC, Kourides IA, and Maloof F. Spectrum of pituitary alterations with mild and severe thyroid impairment. J Clin Endocrinol Metab. (1978) 46:317–25. doi: 10.1210/jcem-46-2-317
275. Tunbridge WM, Marshall JC, and Burke CW. Primary hypothyroidism presenting as pituitary failure. Br Med J. (1973) 1:153–4. doi: 10.1136/bmj.1.5846.153
276. Purnell JQ, Brandon DD, Isabelle LM, Loriaux DL, and Samuels MH. Association of 24-hour cortisol production rates, cortisol-binding globulin, and plasma-free cortisol levels with body composition, leptin levels, and aging in adult men and women. J Clin Endocrinol Metab. (2004) 89:281–7. doi: 10.1210/jc.2003-030440
277. Weykamp CW, Penders TJ, Schmidt NA, Borburgh AJ, van de Calseyde JF, and Wolthers BJ. Steroid profile for urine: reference values. Clin Chem. (1989) 35:2281–4. doi: 10.1093/clinchem/35.12.2281
278. Vierhapper H, Nowotny P, and Waldhäusl W. Sex-specific differences in cortisol production rates in humans. Metabolism. (1998) 47:974–6. doi: 10.1016/s0026-0495(98)90353-5
279. Roca CA, Schmidt PJ, Deuster PA, Danaceau MA, Altemus M, Putnam K, et al. Sex-related differences in stimulated hypothalamic-pituitary-adrenal axis during induced gonadal suppression. J Clin Endocrinol Metab. (2005) 90:4224–31. doi: 10.1210/jc.2004-2525
280. Kudielka BM, Buske-Kirschbaum A, Hellhammer DH, and Kirschbaum C. HPA axis responses to laboratory psychosocial stress in healthy elderly adults, younger adults, and children: impact of age and gender. Psychoneuroendocrinology. (2004) 29:83–98. doi: 10.1016/s0306-4530(02)00146-4
281. Takai N, Yamaguchi M, Aragaki T, Eto K, Uchihashi K, and Nishikawa Y. Gender-specific differences in salivary biomarker responses to acute psychological stress. Ann N Y Acad Sci. (2007) 1098:510–5. doi: 10.1196/annals.1384.014
282. Van Cauter E, Leproult R, and Kupfer DJ. Effects of gender and age on the levels and circadian rhythmicity of plasma cortisol. J Clin Endocrinol Metab. (1996) 81:2468–73. doi: 10.1210/jcem.81.7.8675562
283. Kirschbaum C, Wüst S, and Hellhammer D. Consistent sex differences in cortisol responses to psychological stress. Psychosom Med. (1992) 54:648–57. doi: 10.1097/00006842-199211000-00004
284. Roosens B, Maes E, Van Steirteghem A, and Vanhaelst L. Primary hypothyroidism associated with secondary adrenocortical insufficiency. J Endocrinol Invest. (1982) 5:251–4. doi: 10.1007/BF03348331
285. Kasperlik-Załuska AA, Czarnocka B, Czech W, Walecki J, Makowska AM, Brzeziński J, et al. Secondary adrenal insufficiency associated with autoimmune disorders: a report of twenty-five cases. Clin Endocrinol (Oxf). (1998) 49:779–83. doi: 10.1046/j.1365-2265.1998.00611.x
286. Terzidis K, Panoutsopoulos A, Mantzou A, Tourli P, Papageorgiou G, Saltiki K, et al. Lower early morning plasma cortisol levels are associated with thyroid autoimmunity in the elderly. Eur J Endocrinol. (2010) 162:307–13. doi: 10.1530/EJE-09-0534
287. Boelaert K, Newby PR, Simmonds MJ, Holder RL, Carr-Smith JD, Heward JM, et al. Prevalence and relative risk of other autoimmune diseases in subjects with autoimmune thyroid disease. Am J Med. (2010) 123:183. doi: 10.1016/j.amjmed.2009.06.030
288. Ott J, Promberger R, Kober F, Neuhold N, Tea M, Huber JC, et al. Hashimoto’s thyroiditis affects symptom load and quality of life unrelated to hypothyroidism: a prospective case-control study in women undergoing thyroidectomy for benign goiter. Thyroid. (2011) 21:161–7. doi: 10.1089/thy.2010.0191
289. Guldvog I, Reitsma LC, Johnsen L, Lauzike A, Gibbs C, Carlsen E, et al. Thyroidectomy versus medical management for euthyroid patients with hashimoto disease and persisting symptoms: A randomized trial. Ann Intern Med. (2019) 170:453–64. doi: 10.7326/M18-0284
290. Dentice M, Morisco C, Vitale M, Rossi G, Fenzi G, and Salvatore D. The different cardiac expression of the type 2 iodothyronine deiodinase gene between human and rat is related to the differential response of the Dio2 genes to Nkx-2.5 and GATA-4 transcription factors. Mol Endocrinol. (2003) 17:1508–21. doi: 10.1210/me.2002-0348
291. Strich D, Chay C, Karavani G, Edri S, and Gillis D. Levothyroxine Therapy Achieves Physiological FT3/FT4 Ratios at Higher than Normal TSH Levels: A Novel Justification for T3 Supplementation? Horm Metab Res. (2018) 50:827–31. doi: 10.1055/a-0751-0498
292. Leese GP, Soto-Pedre E, and Donnelly LA. Liothyronine use in a 17 year observational population-based study - the tears study. Clin Endocrinol (Oxf). (2016) 85:918–25. doi: 10.1111/cen.13052
293. Tariq A, Wert Y, Cheriyath P, and Joshi R. Effects of long-term combination LT4 and LT3 therapy for improving hypothyroidism and overall quality of life. South Med J. (2018) 111:363–9. doi: 10.14423/SMJ.0000000000000823
294. Pilo A, Iervasi G, Vitek F, Ferdeghini M, Cazzuola F, and Bianchi R. Thyroidal and peripheral production of 3,5,3’-triiodothyronine in humans by multicompartmental analysis. Am J Physiol. (1990) 258:E715–726. doi: 10.1152/ajpendo.1990.258.4.E715
295. Wang K, Sun YN, Liu JY, Zhang L, Ye Y, Lin LX, et al. The impact of iodine excess on thyroid hormone biosynthesis and metabolism in rats. Biol Trace Elem Res. (2009) 130:72–85. doi: 10.1007/s12011-009-8315-z
296. Celi FS, Zemskova M, Linderman JD, Babar NI, Skarulis MC, Csako G, et al. The pharmacodynamic equivalence of levothyroxine and liothyronine: a randomized, double blind, cross-over study in thyroidectomized patients. Clin Endocrinol (Oxf). (2010) 72:709–15. doi: 10.1111/j.1365-2265.2009.03700.x
297. Sawin CT, Hershman JM, and Chopra IJ. The comparative effect of T4 and T3 on the TSH response to TRH in young adult men. J Clin Endocrinol Metab. (1977) 44:273–8. doi: 10.1210/jcem-44-2-273
298. Ma C, Xie J, Huang X, Wang G, Wang Y, Wang X, et al. Thyroxine alone or thyroxine plus triiodothyronine replacement therapy for hypothyroidism. Nucl Med Commun. (2009) 30:586–93. doi: 10.1097/MNM.0b013e32832c79e0
299. Grozinsky-Glasberg S, Fraser A, Nahshoni E, Weizman A, and Leibovici L. Thyroxine-triiodothyronine combination therapy versus thyroxine monotherapy for clinical hypothyroidism: meta-analysis of randomized controlled trials. J Clin Endocrinol Metab. (2006) 91:2592–9. doi: 10.1210/jc.2006-0448
300. Akirov A, Fazelzad R, Ezzat S, Thabane L, and Sawka AM. A systematic review and meta-analysis of patient preferences for combination thyroid hormone treatment for hypothyroidism. Front Endocrinol (Lausanne). (2019) 10:477. doi: 10.3389/fendo.2019.00477
301. Lan H, Wen J, Mao Y, Huang H, Chen G, and Lin W. Combined T4 + T3 therapy versus T4 monotherapy effect on psychological health in hypothyroidism: A systematic review and meta-analysis. Clin Endocrinol (Oxf). (2022) 97:13–25. doi: 10.1111/cen.14742
302. Nassar M, Hassan A, Ramadan S, Desouki MT, Hassan MA, and Chaudhuri A. Evaluating the effectiveness of combined T4 and T3 therapy or desiccated thyroid versus T4 monotherapy in hypothyroidism: a systematic review and meta-analysis. BMC Endocr Disord. (2024) 24:90. doi: 10.1186/s12902-024-01612-6
303. Cooke RG, Joffe RT, and Levitt AJ. T3 augmentation of antidepressant treatment in T4-replaced thyroid patients. J Clin Psychiatry. (1992) 53:16–8.
304. Bunevicius R, Kazanavicius G, Zalinkevicius R, and Prange AJ Jr. Effects of thyroxine as compared with thyroxine plus triiodothyronine in patients with hypothyroidism. N Engl J Med. (1999) 340:424–9. doi: 10.1056/NEJM199902113400603
305. Bunevicius R and Prange AJ. Mental improvement after replacement therapy with thyroxine plus triiodothyronine: relationship to cause of hypothyroidism. Int J Neuropsychopharmacol. (2000) 3:167–74. doi: 10.1017/S1461145700001826
306. Bunevicius R, Jakuboniene N, Jurkevicius R, Cernicat J, Lasas L, and Prange AJ Jr. Thyroxine vs thyroxine plus triiodothyronine in treatment of hypothyroidism after thyroidectomy for Graves’ disease. Endocrine. (2002) 18:129–33. doi: 10.1385/ENDO:18:2:129
307. Walsh JP, Shiels L, Lim EM, Bhagat CI, Ward LC, Stuckey BG, et al. Combined thyroxine/liothyronine treatment does not improve well-being, quality of life, or cognitive function compared to thyroxine alone: a randomized controlled trial in patients with primary hypothyroidism. J Clin Endocrinol Metab. (2003) 88:4543–50. doi: 10.1210/jc.2003-030249
308. Sawka AM, Gerstein HC, Marriott MJ, MacQueen GM, and Joffe RT. Does a combination regimen of thyroxine (T4) and 3,5,3’-triiodothyronine improve depressive symptoms better than T4 alone in patients with hypo-thyroidism? Results of a double-blind, randomized, controlled trial. J Clin Endocrinol Metab. (2003) 88:4551–5. doi: 10.1210/jc.2003-030139
309. Clyde PW, Harari AE, Getka EJ, and Shakir KM. Combined levothyroxine plus liothyronine compared with levothyroxine alone in primary hypothyroidism: a randomized controlled trial. JAMA. (2003) 290:2952–8. doi: 10.1001/jama.290.22.2952
310. Siegmund W, Spieker K, Weike AI, Giessmann T, Modess C, Dabers T, et al. Replacement therapy with levothyroxine plus triiodothyronine (bioavailable molar ratio 14:1) is not superior to thyroxine alone to improve well-being and cognitive performance in hypothyroidism. Clin Endocrinol (Oxf). (2004) 60:750–7. doi: 10.1111/j.1365-2265.2004.02050.x
311. Escobar-Morreale HF, Botella-Carretero JI, Gómez-Bueno M, Galán JM, and Barrios V. Sancho J Thyroid hormone replacement therapy in primary hypothyroidism: a randomized trial comparing L-thyroxine plus liothyronine with L-thyroxine alone. Ann Intern Med. (2005) 142:412–24. doi: 10.7326/0003-4819-142-6-200503150-00007
312. Appelhof BC, Fliers E, Wekking EM, Schene AH, Huyser J, Tijssen JG, et al. Combined therapy with levothyroxine and liothyronine in two ratios, compared with levothyroxine monotherapy in primary hypothyroidism: a double-blind, randomized, controlled clinical trial. J Clin Endocrinol Metab. (2005) 90:2666–74. doi: 10.1210/jc.2004-2111
313. Rodriguez T, Lavis VR, Meininger JC, Kapadia AS, and Stafford LF. Substitution of liothyronine at a 1:5 ratio for a portion of levothyroxine: effect on fatigue, symptoms of depression, and working memory versus treatment with levothyroxine alone. Endocr Pract. (2005) 11:223–33. doi: 10.4158/EP.11.4.223
314. Saravanan P, Simmons DJ, Greenwood R, Peters TJ, and Dayan CM. Partial substitution of thyroxine (T4) with tri-iodothyronine in patients on T4 replacement therapy: results of a large community-based randomized controlled trial. J Clin Endocrinol Metab. (2005) 90:805–12. doi: 10.1210/jc.2004-1672
315. Nygaard B, Jensen EW, Kvetny J, Jarløv A, and Faber J. Effect of combination therapy with thyroxine (T4) and 3,5,3’-triiodothyronine versus T4 monotherapy in patients with hypothyroidism, a double-blind, randomised cross-over study. Eur J Endocrinol. (2009) 161:895–902. doi: 10.1530/EJE-09-0542
316. Valizadeh M, Seyyed-Majidi MR, Hajibeigloo H, Momtazi S, Musavinasab N, and Hayatbakhsh MR. Efficacy of combined levothyroxine and liothyronine as compared with levothyroxine monotherapy in primary hypothyroidism: a randomized controlled trial. Endocr Res. (2009) 34:80–9. doi: 10.1080/07435800903156340
Keywords: clinical medicine, deiodinases, guidelines, hypocortisolism, hypothyroidism, paradigm, reference range, T4/T3 combination therapy
Citation: Lindner HH (2025) Clinical thyroidology: beyond the 1970s’ TSH-T4 Paradigm. Front. Endocrinol. 16:1529791. doi: 10.3389/fendo.2025.1529791
Received: 17 November 2024; Accepted: 08 April 2025;
Published: 24 June 2025; Corrected: 14 August 2025.
Edited by:
Iordanis Mourouzis, National and Kapodistrian University of Athens, GreeceReviewed by:
Ricardo V. Garcia-Mayor, Instituto de Investigación Sanitaria Galicia Sur (IISGS), SpainFrancesca Forini, National Research Council (CNR), Italy
Copyright © 2025 Lindner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Henry H. Lindner, aGxpbmRuZXIxQHlhaG9vLmNvbQ==