 Xueqian Wang1†
Xueqian Wang1† Shengzhuang Guan1,2†Yiqing Gao3†
Shengzhuang Guan1,2†Yiqing Gao3† Rongrong Xie2
Rongrong Xie2 Fengyun Wang2
Fengyun Wang2 Xiuli Chen2
Xiuli Chen2 Haiying Wu2Xiaohui Zhang4
Haiying Wu2Xiaohui Zhang4 Dandan Zhang1,2
Dandan Zhang1,2 Bingyu Yang1,2Qisang Fan1,2Qing Wang1,2
Bingyu Yang1,2Qisang Fan1,2Qing Wang1,2 Hongying Wang1Tao Feng1
Hongying Wang1Tao Feng1 Haitao Lv1
Haitao Lv1 Ting Chen1,2*
Ting Chen1,2*- 1Suzhou Clinical Center for Rare Diseases in Children, Children’s Hospital of Soochow University, Suzhou, Jiangsu, China
- 2Department of Endocrinology, Genetics and Metabolism, Children’s Hospital of Soochow University, Suzhou, Jiangsu, China
- 3Department of Endocrinology and Metabolism, Xuzhou Children’s Hospital, Xuzhou, Jiangsu, China
- 4State Key Laboratory of Common Mechanisms Research for Major Diseases, Suzhou Institute of Systems Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College, Suzhou, China
Background: Brachydactyly type E (BDE) is characterized by variable shortening of metacarpals or metatarsals, often involving phalanges. It may occur as an isolated anomaly or as part of congenital syndromes. With advancements in molecular diagnostic technologies, how genetic testing enhances the precise diagnosis of BDE remains unclear. Our aims were to establish an algorithm for molecular genetic diagnostics in Chinese children with BDE and to explore the phenotype-genotype correlations of Chinese patients with BDE.
Methods: We reviewed left-hand wrist X-rays from children visiting Children’s Hospital of Soochow University (Jun 2021–Dec 2023). From 60,650 films, 135 BDE cases were identified, and their comprehensive phenotypes were collected. Whole-exome sequencing (WES) with copy number variation (CNV) analysis was performed on 60 patients and their parents. Sanger sequencing was used to validate single nucleotide variants (SNV) and indels.
Results: Causative variants were found in 19 patients. SNVs and indels affecting 10 genes were identified in 15 patients, and CNVs in four. GNAS mutations were the leading cause (four cases), followed by EXT1 and ACAN defects. The diagnostic yield was 19.1% in patients with isolated brachydactyly; 75% in patients with brachydactyly combined with short stature; 77.8% in patients with brachydactyly combined with facial dysmorphism; 83.3% in patients with brachydactyly combined with intellectual disability.
Conclusion: Through comprehensive evaluation of genotype-phenotype correlations, we propose a diagnostic algorithm for precise molecular diagnosis in Chinese children with BDE.
1 Introduction
Brachydactyly (BD) refers to the shortening of the hands/feet caused by abnormal development of the metacarpals, metatarsals and/or phalanges. BD is classified into five types, A through E, based on anatomical characteristics (1). Most types of BD are rare, with the exception of types A3 (BDA3, OMIM 112700) and D (BDD, OMIM 113200) (2). Brachydactyly type E (BDE) involves variable shortening of the metacarpals/metatarsals, with frequent involvement of the phalanges. As a minor morphologic anomaly, BDE can occur in isolation or as a feature of various congenital syndromes (3, 4).
To date, mutations in two genes have been associated with isolated BDE. Brachydactyly type E1 (BDE1, OMIM 113300) is caused by a heterozygous mutation in the HOXD13 gene. Some patients with HOXD13 gene variants present not only with BDE, but also with overlapping features of BDD, characterized by the shortening and broadening of thumbs (5–7). Brachydactyly type E2 (BDE2, OMIM 613382) is caused by a heterozygous mutation in the PTHLH gene. Most, but not all, patients with PTHLH gene defect exibit short stature in addition to BDE (8–12).
The most common syndromic forms of BDE are inactivating PTH/PTHrP signaling disorders (iPPSD), such as pseudohypoparathyroidism 1A (PHP1A, OMIM 103580), pseudopseudohypoparathyroidism (PPHP, OMIM 612463), acrodysostosis with multihormonal resistance (ACRDYS1, OMIM 101800), acrodysostosis without multihormonal resistance (ACRDYS2, OMIM 614613), and hypertension with brachydacytly syndrome (HTNB, OMIM 112410) (13–15). Patients with these conditions often exhibit features of Albright Hereditary Osteodystrophy (AHO), which include BDE, short stature, obesity, round face, subcutaneous ossifications, and other skeletal anomalies (16–18). Syndromic BDE can also be related to chromosomal aberrations, with the most common disorder being Turner syndrome. BDE is also observed in 2q37 deletion syndrome, alternatively known as brachydactyly mental retardation syndrome (BDMR, OMIM 600430) (19, 20). The most common phenotypic features of BDMR comprise developmental delay, BDE, short stature, obesity, autistic features, and craniofacial or skeletal dysmorphism (21–23).
In this study, we reviewed the left-hand wrist X-ray film of 60,650 children and identified 135 children with BDE. Among these, 60 unrelated families consented to genetic testing. Our objectives are to establish an algorithm for molecular genetic diagnostics in Chinese children with BDE, and to explore the phenotype-genotype correlations of Chinese patients with BDE. This research may provide insights into the necessity of genetic investigations for BDE in future clinical practice.
2 Methods
2.1 Study design and participants
Our study retrospectively analyzed left-wrist radiographs from 60,650 children who did this test at Children’s Hospital of Soochow University between June 2021 and December 2023. Figure 1 shows the diagnostic imaging. Out of 60,650 children, we identified 135 patients with BDE. With fully informed consent, 60 patients and their parents agreed to participate in follow-up visits and underwent Trio-whole-exome sequencing (Trio-WES) (Figure 2). This study received approval from the Ethics Committee of Children’s Hospital of Soochow University.
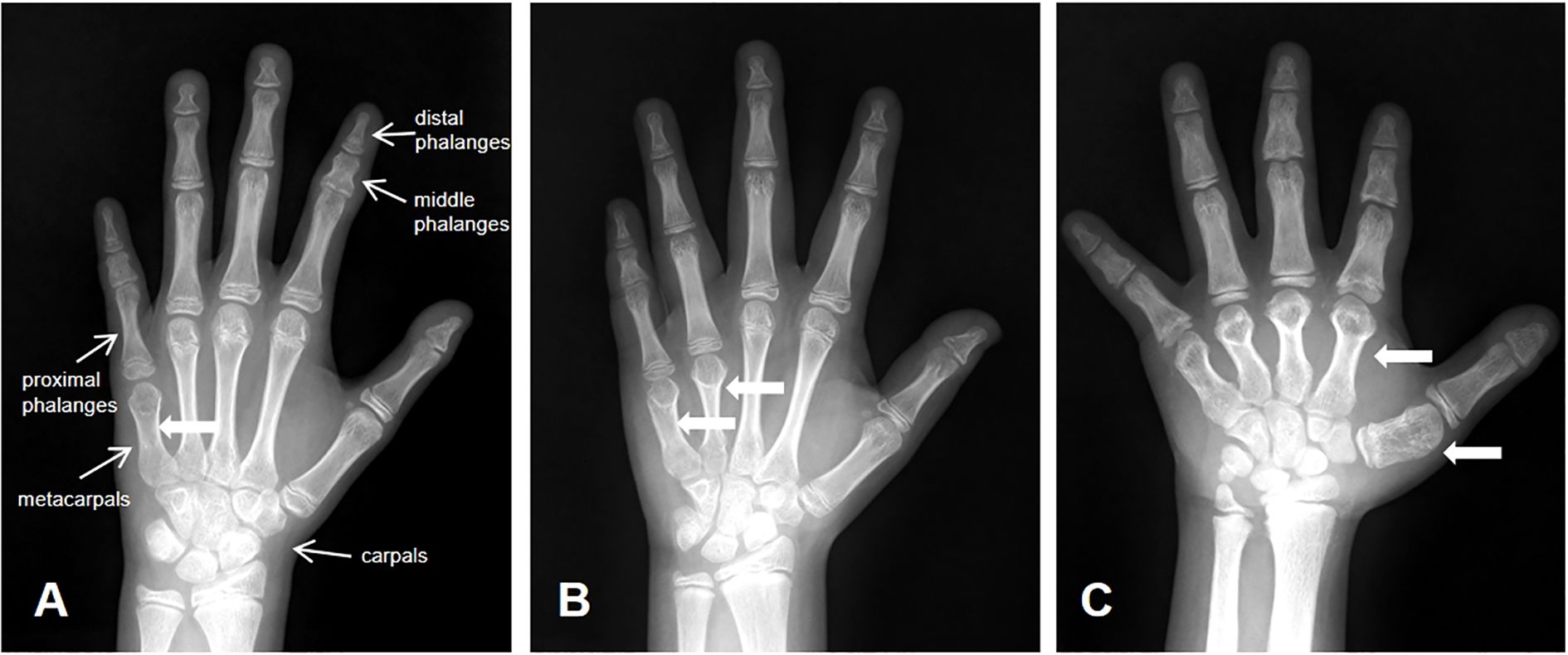
Figure 1. Diagnostic imaging. (A) BDE with shortened fifth metacarpal; (B) BDE with shortened fourth and fifth metacarpals; (C) BDE with shortened first to fifth metacarpls.
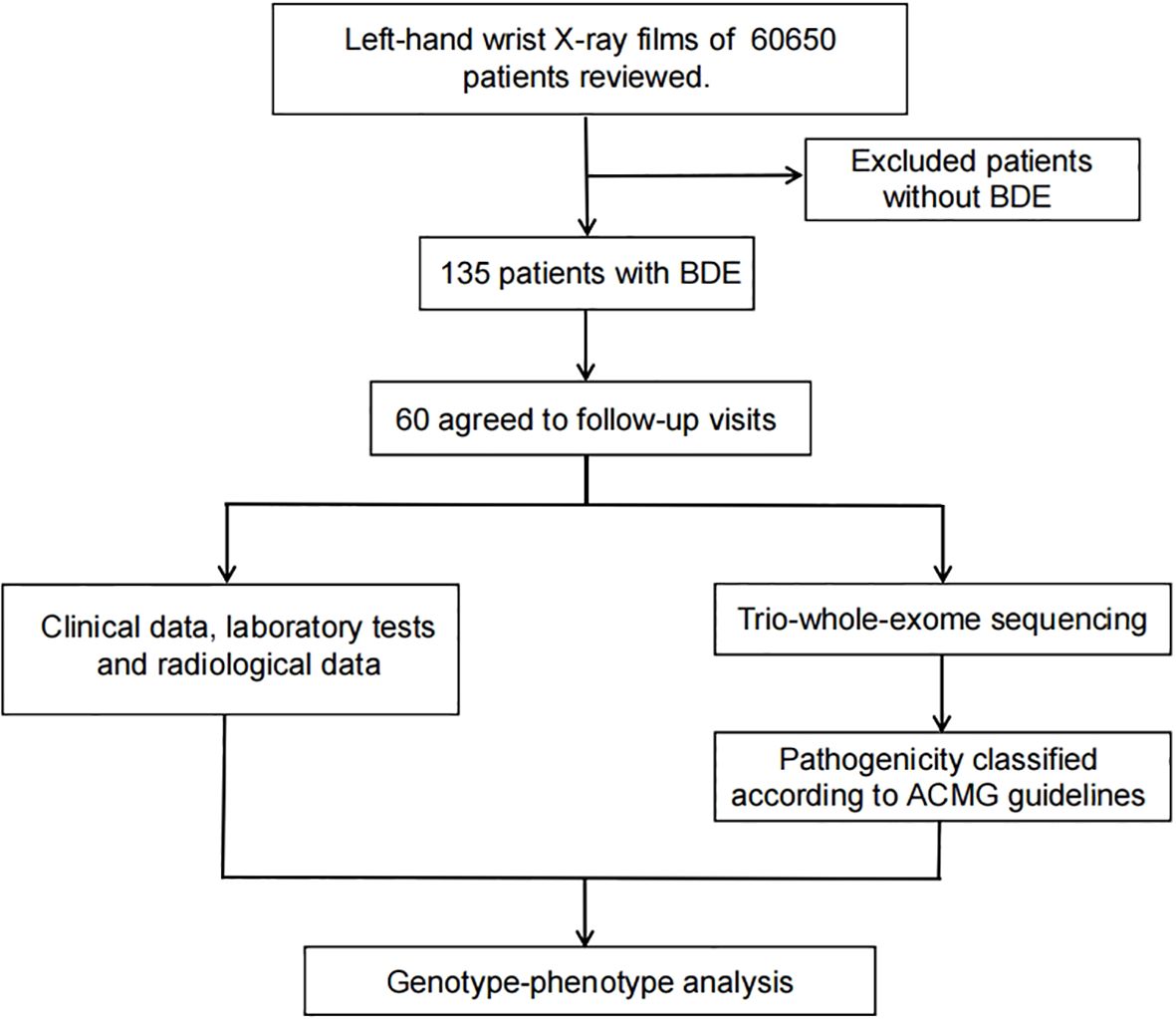
Figure 2. Flowchart depicting patient enrollment and follow-up process.
2.2 Data collection
We retrieved clinical data from the medical charts of 135 patients with BDE. Additionally, we collected detailed phenotypic information from the 60 children and their family members who agreed to follow-up visits. Height was measured using a wall-mounted stadiometer, and weight was measured using a digital floor scale. Each measurement was repeated three times, and the average values were calculated. Height standard deviation scores (SDS) were calculated based on the normal range for Chinese children (24, 25).
The left-hand wrist X-ray films were reviewed by two well-trained pediatric endocrinologists and one radiologist. Bone age was assessed by radiograph using the Tanner-Whitehouse III method (26). Advanced bone age was defined as bone age exceeding the chronological age by one year or more, while delayed bone age was defined as bone age behind the chronological age by one year or more.
2.3 Genetic testing
Peripheral blood samples were collected from the patients and their parents after obtaining written informed consent. Genomic DNA was extracted from these samples. WES was performed using the Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) with an average sequencing depth 120X. The sequences were aligned to the reference human genome (hg19). Candidate variants were validated by Sanger sequencing, and their pathogenicity was classified following the guidelines of the American College of Medical Genetics and Genomics (ACMG) (27). For SNV classification, population data were obtained from gnomAD, literature-based variant information was sourced from HGMD Professional (HGMDpro), and REVEL was used as an in silico prediction tool for missense variants. For CNV classification, the DECIPHER website (https://www.deciphergenomics.org/) was used to identify genes involved in the CNV, and dosage sensitivity information was obtained from the ClinGen Dosage Sensitivity Map.
2.4 Statistical analysis
Statistical analyses were performed using SPSS Statistics version 26. Continuous variables are presented as mean ± standard deviation, while categorical variables are reported as frequencies and percentages. The chi-square test was used to assess intergroup differences for categorical variables. A p-value of < 0.05 was considered statistically significant. To evaluate the performance of genetic testing across different clinical symptom profiles, we calculated diagnostic yield, sensitivity, and specificity using a 2×2 contingency table, comparing genetic test results against clinical diagnostic criteria.
3 Results
3.1 Demographic and clinical features
We reviewed left-hand wrist X-ray films from 60,650 children (female to male ratio 1.2), and identified 135 children with BDE (female to male ratio 6.1). The overall prevalence of BDE was 0.22%, with a higher prevalence of 0.35% in females and a lower prevalence of 0.07% in males. The average chronological age and bone age of the 135 BDE patients were 9.4 ± 2.3 years and 9.9 ± 2.6 years, respectively. Among these patients, 31.1% exhibited advanced bone age, and 12.6% showed delayed bone age. Most BDE patients had near-normal height (height SDS between -1 and 0), while approximately 6.7% patients were of short stature (height SDS<-2). About 80.0% BDE patients had shortening of metacarpals limited to the fourth or fifth metacarpals, 14.8% had shortening involving both the fourth and fifth metacarpals, and 5.9% had shortening of all metacarpals. A further 7.4% of patients had shortening of metatarsals. About 4.4% had shortening of metatarsals limited to the fourth or fifth metatarsals, 3.0% had shortening involving both the fourth and fifth metatarsals. Additionally, 32.0% of patients also had a shorter middle phalanx of fifth finger, known as BDA3; 5.0% BDE patients had a shorter middle phalanx of both the second and fifth finger, known as brachydactyly type A4 (BDA4, OMIM 112800); and 10.0% had short and broad thumbs, known as BDD.
With fully informed consent, 60 patients and their parents were recruited. Of these 60 patients, 50 were females and 10 were males. The average age was 9.1 ± 2.2 years, and their average bone age was at 10.0 ± 2.5 years. Similar to the overall statistics of the 135 BDE patients, among these 60 patients, 31.7% presented with advanced bone age, and 6.7% showed delayed bone age. Most patients had near-normal stature, while 6.7% had short stature. In addition to the shorting of metacarpal/metatarsal, these patients exhibited other skeletal abnormalities: three patients (5.0%) had multiple exostoses, three patients (5.0%) had pectus carinatum, four patients (6.7%) had elbow valgus, and one patient (1.7%) had knee valgus. Ten patients (16.7%) had a family history of brachydactyly. Nine patients (15.0%) exhibited facial dysmorphism, and six patients (10.0%) had intellectual disability.
3.2 Genetic findings
We identified causative single nucleotide variants (SNV) or small indels from ten different genes in 15 patients (Table 1) (28). The defect of the GNAS gene is identified as the most prevalent genetic cause, observed in 26.7% of patients with BDE. Defects of EXT1 and ACAN were both detected in two patients, respectively. Other genetic abnormalities include variants of IHH, PRKAR1A, PTHLH, PRMT7, POGZ, NPR2, and FBXW11 genes.
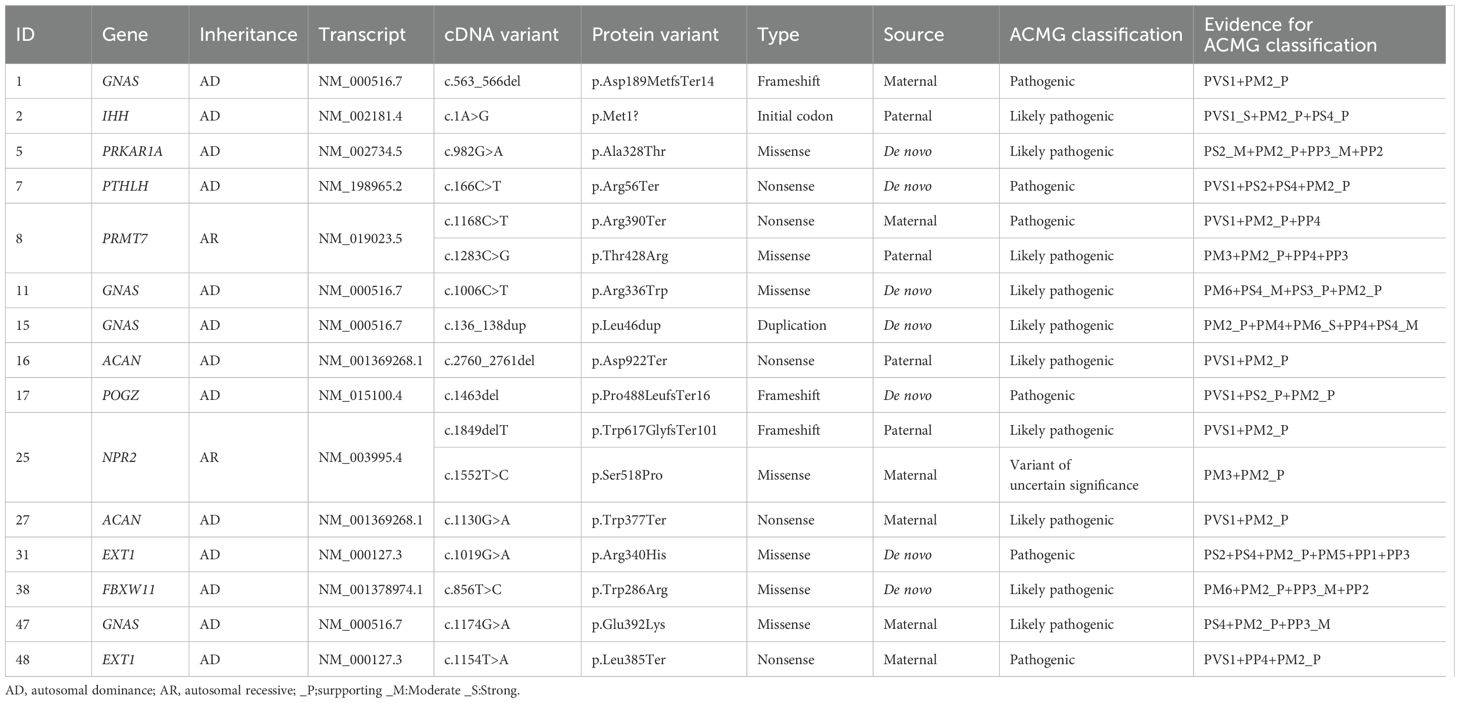
Table 1. Pathogenic and likely pathogenic genetic variants.
Pathogenic copy number variations (CNV) were identified in four patients (Table 2) (28). Patient 12 exhibited a duplication on 16q22.3-24.3, encompassing 132 protein coding genes. Patient 35 presented with a deletion on 7q11.21-11.22, including the AUTS2 gene, which overlaps with Mental Retardation, Autosomal Dominant 26 (MRD26, OMIM 615834). Patient 44 had a deletion on 2q37.2-37.3 containing HDAC4 gene, known to be chromosome 2q37 deletion syndrome (2q37DS, OMIM 600430). Lastly, patient 56 was found to have a deletion of approximately 950kb on chromosome 17, affecting the NF1 and adjacent genes, which is known as NF1 microdeletion syndrome (OMIM 613675).
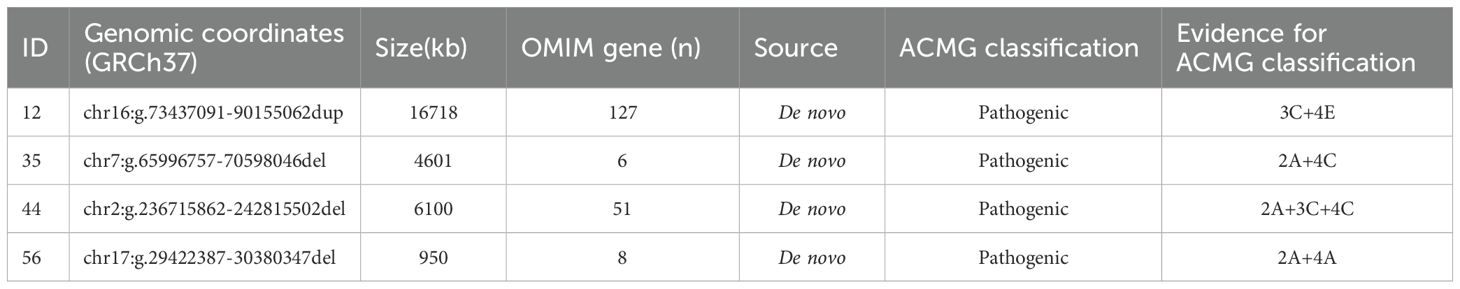
Table 2. Pathogenic and likely pathogenic CNVs.
3.3 Genotype-phenotype correlations
The comparison of clinical characteristics between genetically positive cases and negative cases was depicted in Table 3. The most recurrent phenotype among positive cases was two or more shortened metacarpals/metatarsals (n=15, 78.9%). Most patients had near-normal stature, while four patients (6.7%) had short stature, within whom three carried genetic abnormalities. Nine patients (15.0%) exhibited facial dysmorphism, within whom seven carried genetic abnormalities. Six patients (10.0%) had intellectual disability, and five of them carried genetic abnormalities. Clinical characteristics of 19 patients carrying causative variants are shown in Table 4.
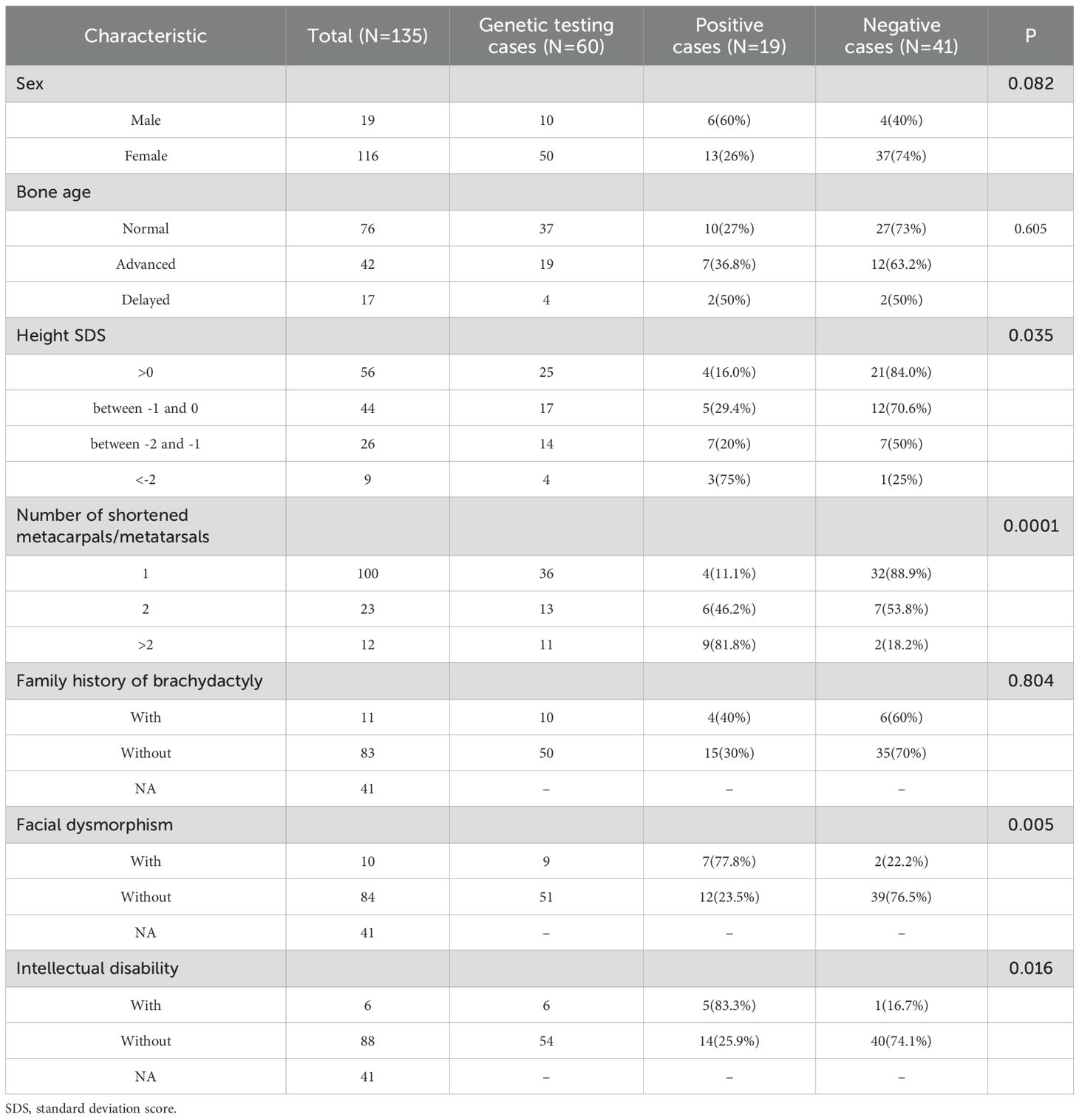
Table 3. Summary of the clinical characteristics of the 135 patients.
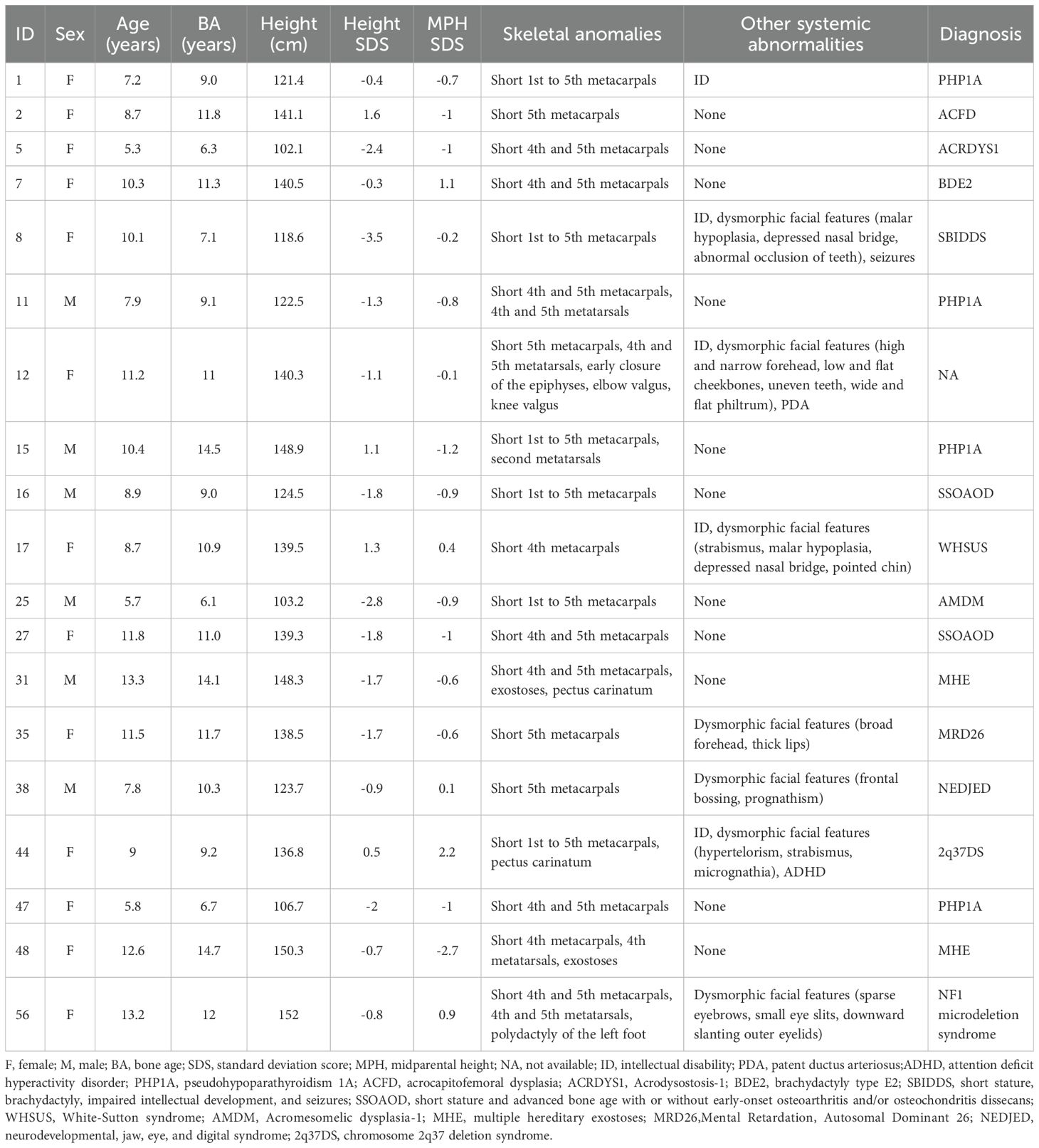
Table 4. Clinical characteristics of 19 patients with pathogenic/likely pathogenic variants.
Patients with shorter stature were significantly more likely to have genetic abnormalities (P=0.035). The number of shortened metacarpals and metatarsals was notably higher in positive cases compared to negative cases (p=0.0001). Additionally, the frequency of dysmorphic facial features was significantly greater in positive cases than in negative cases (P=0.005). Intellectual disability was also more frequent in positive cases, with a statistically significant difference (P=0.016).
3.4 Diagnostic yield in patients with different phenotypes
In patients with isolated brachydactyly, the diagnostic yield was 19.1%, sensitivity was 47.4%, and specificity was 7.3%. In patients with additional clinical manifestations, the diagnosis yield, sensitivity, and specificity increased, as shown in Table 5. Among them, the highest diagnosis yield of 83.3% was observed in patients with intellectual disability; the highest sensitivity of 78.9% was seen in patients with multiple metacarpal/metatarsal shortening; and the highest specificity of 97.6% was found in patients with short stature and intellectual disability.
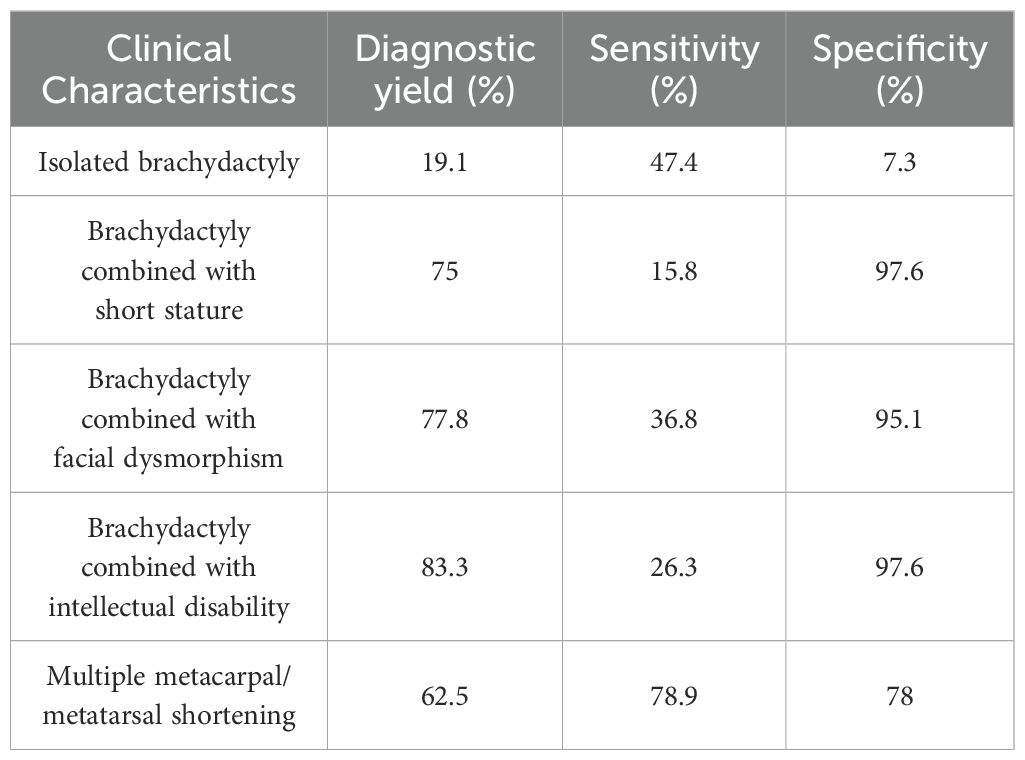
Table 5. Performance of the prediction rule for diagnosing brachydactyly.
4 Discussion
The aim of the present study was to investigate the prevalence of BDE and to assess the necessity of genetic testing for BDE within a pediatric medical center setting. We reviewed left-hand wrist X-ray films from 60650 children and identified 135 cases with BDE, which was more common in females than in males. Genetic testing was performed on 60 patients, revealing identified genetic etiology in 31.7% of them. In total, 15 variants from ten genes and four CNVs were identified in 19 patients. The genetic diagnostic yield was significantly higher in patients presenting with short stature, multiple shortened metacarpals or metatarsals, facial dysmorphism, and intellectual disability compared to those without these phenotypes.
The pronounced female predominance in our cohort (female-to-male ratio 6:1) significantly exceeds both our screened population baseline (1.2:1) and general population expectations. While BDE gender ratios remain unreported, our findings align with documented female predominance in subtypes of brachydactyly type A (BDA). A study in Chinese children found that out of 174 patients with BDA3, 111 were females. Even after excluding females with Turner syndrome, the prevalence of BDA3 in females remained significantly higher than in males (29). Similarly, surveys conducted in Native Americans, Mexico, and Pacific islander populations have also reported a higher frequency of BDA3 in females (30, 31). This disparity suggests either ethnic-specific biological factors, estrogen-mediated modulation of skeletal development pathways, or influence by genes on X-chromosome. To further validate this observation, we recommend collaborative meta-analyses of gender-stratified data from multinational consortia to distinguish true biological sex differences from sampling artifacts.
In this study, we found that the genetic etiology of BDE can be roughly divided into three categories. Approximately one-third of BDE patients have defects in PTH/PTHrP signaling (13–15), with GNAS gene defects being the most common within this group. Patients with iPPSDs, including PHP1A, PHP1C, ACRDYS1 and ACRDYS2, commonly present with BDE. Another one-third of BDE patients exhibit defects in growth plate or cartilage signaling, which include deficiencies in genes such as ACAN, IHH and EXT1. Defects in these genes typically result in short stature, although the presence of BDE is not consistent, with variations observed even among family members carrying the same mutation. The last one-third of patients display diverse genetic profiles, often accompanied by neurological manifestations in addition to BDE. Notably, patient 8 was found to have biallelic variants in the PRMT7 gene, causing short stature, brachydactyly, intellectual disability, dysmorphic facial features, and seizures syndrome (SBIDDS) (32–34), which aligns with her clinical presentation. Patient 17 had a de novo frameshift mutation in the POGZ gene, which is associated with White-Sutton syndrome (WHSUS, OMIM 616364) (35, 36). WHSUS is a neurodevelopmental disorder characterized by a spectrum of intellectual disabilities, global developmental delay and behavioral problems, short stature, dysmorphic facial features and brachydactyly (37–40).
In the present study, approximately one-third of patients who underwent genetic testing were found to have a genetic cause for their condition. Notably, individuals with isolated BDE, without accompanying symptoms did not exhibit detectable genetic defects. The key risk factors for identifying a genetic etiology in BED include the severity of short stature, multiple shortened metacarpals/metatarsals, the extent of facial dysmorphism, and the presence of intellectual disability.
We describe three instructive cases highlighting the genetic complexity of BDE. Patient 12 harbored a rare 16q22.3-24.3 duplication, exhibiting intellectual disability, patent ductus arteriosus (PDA), and skeletal anomalies including BDE—expanding the phenotypic spectrum of distal 16q duplications beyond the six previously reported cases with the duplicated region spanning from 6Mb to 17Mb (41–44). Patient 35 carried a deletion in the chromosomal region 7q11.21-11.22, which includes the AUTS2 gene, a key regulator of neuronal gene expression. This deletion is associated with MRD26 (45). Patient 35 exhibited BDE and short stature but normal cognition, challenging the classic MRD26 paradigm (46, 47). Patient 56 had a deletion of approximately 950kb on chromosome 17. This deletion, affecting NF1 and contiguous genes, is associated with NF1 microdeletion syndrome (48–50). Besides common symptoms of NF1, patient 56 exhibited BDE and polydactyly, reinforcing the skeletal involvement in NF1 microdeletion syndrome (51, 52).
Our comprehensive evaluation of 60 brachydactyly type E (BDE) patients reveals crucial insights into the diagnostic approach for this condition. In cases of isolated BDE, genetic testing demonstrates limited clinical utility. Routine molecular testing may not be justified for patients presenting with isolated digital shortening without additional clinical features. However, our data demonstrate dramatic improvements in diagnostic accuracy when BDE is accompanied by specific phenotypic red flags. The presence of even one additional clinical feature - particularly short stature (75% diagnostic yield), facial dysmorphism (77.8%), intellectual disability (83.3%), or multiple shortened metacarpals/metatarsals (62.5%) - increases the likelihood of identifying a genetic etiology by 3–4 fold compared to isolated cases. These clinical features serve as powerful predictors of underlying genetic pathology and should trigger more comprehensive evaluation. Based on these findings, we developed a phenotype-driven diagnostic algorithm (Figure 3) that optimizes testing efficiency while minimizing unnecessary procedures. In pediatric endocrinology practice, clinicians evaluating children with growth disorders should maintain particular vigilance for BDE, especially when assessing patients presenting with short stature, facial dysmorphism, or intellectual disability. A systematic examination of digital morphology should be performed, with particular attention to characteristic features such as shortened fourth/fifth metacarpals and positive Archibald’s sign. Upon identifying suspicious findings, clinicians are advised to implement our proposed diagnostic cascade beginning with standardized anthropometric documentation, followed by radiographic bone age assessment incorporating metacarpal pattern analysis, and culminating in targeted genetic testing.
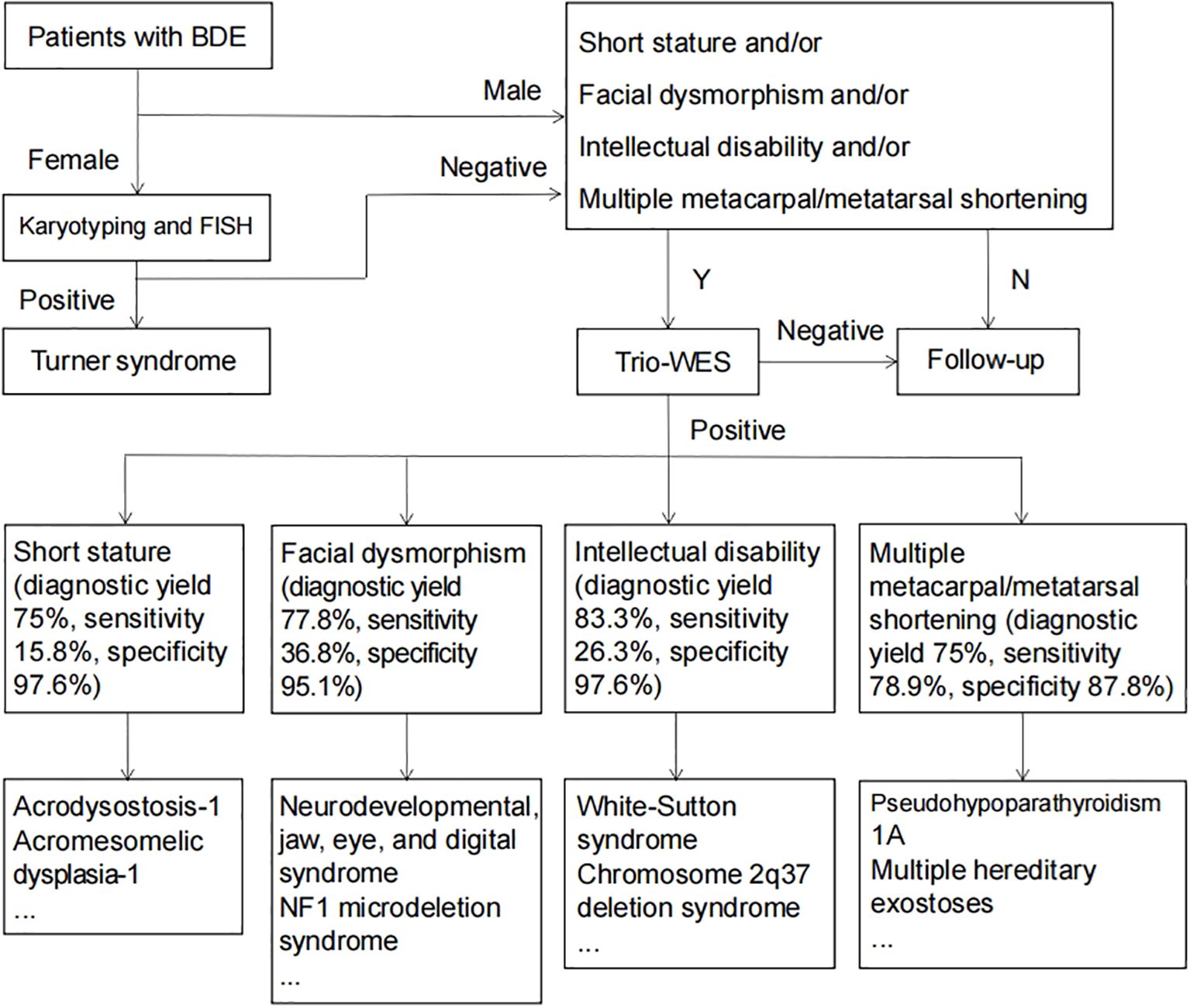
Figure 3. Diagnostic flowchart for patients with BDE.
While our study reveals significant genotype-phenotype correlations in BDE, several limitations should be noted. First, our selection approach of left-wrist radiographs may introduce bias toward children with growth abnormalities, potentially affecting prevalence estimates for incidental BDE findings. However, the large sample size helps mitigate this limitation for assessing overall BDE characteristics. Secondly, only a subset of BDE families consented to comprehensive genetic testing, resulting in a small sample size that may limit the generalizability of our findings and reduce statistical power for calculations. Besides, our study did not account for potential confounding factors such as epigenetic regulation, environmental influences, or genetic modifiers, which may contribute to phenotypic heterogeneity, especially for isolated brachydactyly. Furthermore, the pathogenic mechanisms of rare variants and the functional implications of variants of uncertain significance (VUS) remain unresolved. Lastly, while our study provides comprehensive data on left-wrist radiographs, cases of BDE with purely isolated right-hand involvement may have been underrepresented. These gaps highlight the need for larger, multicenter studies incorporating functional assays and population-level analyses to validate our findings and clarify disease mechanisms.
5 Conclusion
In conclusion, we conducted a comprehensive clinical and genetic analysis of Chinese patients with BDE. Our findings indicate that BDE, while considered a minor anomaly, is not uncommon among Chinese children and demonstrates a higher prevalence in females compared to males. The study underscores the importance of recognizing key phenotypic risk factors—such as pronounced short stature, involvement of multiple shortened metacarpals/metatarsals, distinct facial dysmorphism, and intellectual disability—for identifying an underlying genetic etiology. These insights provide a foundation for targeted genetic evaluations, which can enhance diagnostic accuracy and inform clinical management strategies for patients with BDE.
Data availability statement
The data presented in the study are deposited in the clinvar database https://www.ncbi.nlm.nih.gov/clinvar/, accession numbers: SCV005328512, SCV005328514, SCV005328517, and SCV005328539.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Children’s Hospital of Soochow University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
XW: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal Analysis. SG: Writing – original draft, Writing – review & editing, Data curation, Formal Analysis, Investigation. YG: Writing – original draft, Investigation, Writing – review & editing. RX: Data curation, Investigation, Writing – review & editing. FW: Writing – review & editing, Investigation, Methodology. XC: Writing – review & editing, Data curation, Validation. HWu: Resources, Writing – original draft. XZ: Data curation, Methodology, Writing – original draft. DZ: Investigation, Methodology, Writing – original draft. BY: Formal Analysis, Methodology, Writing – original draft. QF: Formal Analysis, Writing – original draft. QW: Writing – review & editing, Investigation. HWa: Writing – review & editing, Methodology. TF: Writing – review & editing, Data curation. HL: Writing – review & editing, Supervision. TC: Writing – review & editing, Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by a Suzhou Science and Technology Development Project (SKY2023007), a Research Project of Jiangsu Commission of Health (M2021082), and a Summit Project of Clinical Medicine (ML13100523) awarded to Ting Chen. It’s supported by the Jiangsu Health Innovation Team Program awarded to Xueqian Wang, and the Natural Science Foundation of Jiangsu Province (BK20220253) awarded to Bingyu Yang. It’s also supported by a Jiangsu Provincial Maternal and Child Health Care Program (F202119) and a Suzhou Science and Technology Development Project (MSXM2024014) awarded to Hongying Wang and the Suzhou Clinical Center for Rare Disease (Szlcyxzxj202105).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2025.1571136/full#supplementary-material
References
1. Temtamy SA and Aglan MS. Brachydactyly. Orphanet J Rare Dis. (2008) 3:15. doi: 10.1186/1750-1172-3-15
2. Temtamy SA and McKusick VA. The genetics of hand malformations. Birth Defects Orig Artic Ser. (1978) 14:i–xviii, 1–619.
3. Pereda A, Garin I, Garcia-Barcina M, Gener B, Beristain E, Ibañez AM, et al. Brachydactyly E: isolated or as a feature of a syndrome. Orphanet J Rare Dis. (2013) 8:141. doi: 10.1186/1750-1172-8-141
4. Hennekam RC, Biesecker LG, Allanson JE, Hall JG, Opitz JM, Temple IK, et al. Elements of morphology: General terms for congenital anomalies. Am J Med Genet A. (2013) 161:2726–33. doi: 10.1002/ajmg.a.36249
5. Johnson D, Kan S, Oldridge M, Trembath RC, Roche P, Esnouf RM, et al. Missense mutations in the homeodomain of HOXD13 are associated with brachydactyly types D and E. Am J Hum Genet. (2003) 72:984–97. doi: 10.1086/374721
6. Zhao X, Sun M, Zhao J, Leyva JA, Zhu H, Yang W, et al. Mutations in HOXD13 underlie syndactyly type V and a novel brachydactyly-syndactyly syndrome. Am J Hum Genet. (2007) 80:361–71. doi: 10.1086/511387
7. Jamsheer A, Sowińska A, Kaczmarek L, and Latos-Bieleńska A. Isolated brachydactyly type E caused by a HOXD13 nonsense mutation: a case report. BMC Med Genet. (2012) 13:4. doi: 10.1186/1471-2350-13-4
8. Klopocki E, Hennig BP, Dathe K, Koll R, de Ravel T, Baten E, et al. Deletion and point mutations of PTHLH cause brachydactyly type E. Am J Hum Genet. (2010) 86:434–9. doi: 10.1016/j.ajhg.2010.01.023
9. Scheffer-Rath MEA, Veenstra-Knol HE, and Boot AM. A novel mutation in PTHLH in a family with a variable phenotype with brachydactyly, short stature, oligodontia and developmental delay. Bone Rep. (2023) 19:101699. doi: 10.1016/j.bonr.2023.101699
10. Maass PG, Wirth J, Aydin A, Rump A, Stricker S, Tinschert S, et al. A cis-regulatory site downregulates PTHLH in translocation t (8,12)(q13;p11.2) and leads to Brachydactyly Type E. Hum Mol Genet. (2010) 19:848–60. doi: 10.1093/hmg/ddp553
11. Wang J, Wang Z, An Y, Wu C, Xu Y, Fu Q, et al. Exome sequencing reveals a novel PTHLH mutation in a Chinese pedigree with brachydactyly type E and short stature. Clin Chim Acta. (2015) 446:9–14. doi: 10.1016/j.cca.2015.03.019
12. Elli FM, Mattinzoli D, Lucca C, Piu M, Maffini MA, Costanza J, et al. Novel pathogenetic variants in PTHLH and TRPS1 genes causing syndromic brachydactyly. J Bone Miner Res. (2022) 37:465–74. doi: 10.1002/jbmr.4490
13. Elli FM, Pereda A, Linglart A, Perez de Nanclares G, and Mantovani G. Parathyroid hormone resistance syndromes – Inactivating PTH/PTHrP signaling disorders (iPPSDs). Best Pract Res Clin Endocrinol Metab. (2018) 32:941–54. doi: 10.1016/j.beem.2018.09.008
14. Thiele S, Mantovani G, Barlier A, Boldrin V, Bordogna P, De Sanctis L, et al. From pseudohypoparathyroidism to inactivating PTH/PTHrP signalling disorder (iPPSD), a novel classification proposed by the EuroPHP network. Eur J Endocrinol. (2016) 175:P1–P17. doi: 10.1530/EJE-16-0107
15. Pereda A, Elli FM, Thiele S, De Sanctis L, Rothenbuhler A, Hanna P, et al. Inactivating PTH/PTHrP signaling disorders (iPPSDs): evaluation of the new classification in a multicenter large series of 544 molecularly characterized patients. Eur J Endocrinol. (2021) 184:311–20. doi: 10.1530/EJE-20-0625
16. Jiang S, Yang Y, Song A, Jiang Y, Jiang Y, Li M, et al. Genotype-phenotype correlations in pseudohypoparathyroidism type 1a patients: a systemic review. Eur J Endocrinol. (2023) 189:S103–11. doi: 10.1093/ejendo/lvad142
17. Maduro AI, Pinto Saraiva A, Pimenta Rodrigues O, Marques M, B Sousa S, Malcata A, et al. Albright’s hereditary osteodystrophy: an entity to recognize. Rheumatol Oxf Engl. (2022) 61:e356–7. doi: 10.1093/rheumatology/keac277
18. Zhou Q, Liang B, Fu Q-X, Liu H, and Zou C-C. Different AHO phenotype in a Chinese family with a novel GNAS missense variant: a case report. Ital J Pediatr. (2022) 48:123. doi: 10.1186/s13052-022-01322-6
19. Gavril E-C, Nucă I, Pânzaru M-C, Ivanov AV, Mihai C-T, Antoci L-M, et al. Genotype-phenotype correlations in 2q37-deletion syndrome: an update of the clinical spectrum and literature review. Genes. (2023) 14:465. doi: 10.3390/genes14020465
20. Elli FM, de Sanctis L, Madeo B, Maffini MA, Bordogna P, Pirelli A, et al. 2q37 deletions in patients with an albright hereditary osteodystrophy phenotype and PTH resistance. Front Endocrinol. (2019) 10:604. doi: 10.3389/fendo.2019.00604
21. Le TN, Williams SR, Alaimo JT, and Elsea SH. Genotype and phenotype correlation in 103 individuals with 2q37 deletion syndrome reveals incomplete penetrance and supports HDAC4 as the primary genetic contributor. Am J Med Genet A. (2019) 179:782–91. doi: 10.1002/ajmg.a.61089
22. Wakeling E, McEntagart M, Bruccoleri M, Shaw-Smith C, Stals KL, Wakeling M, et al. Missense substitutions at a conserved 14-3–3 binding site in HDAC4 cause a novel intellectual disability syndrome. HGG Adv. (2021) 2:100015. doi: 10.1016/j.xhgg.2020.100015
24. Li H, Ji C-Y, Zong X-N, and Zhang Y-Q. Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years. Zhonghua Er Ke Za Zhi Chin J Pediatr. (2009) 47:487–92. doi: 10.3760/cma.j.issn.0578-1310.2009.07.003
25. Zong XN, Li H, Zhang YQ, and Wu HH. Updated growth standards for Chinese children under 7 years of age. Zhonghua Er Ke Za Zhi Chin J Pediatr. (2023) 61:1103–8. doi: 10.3760/cma.j.cn112140-20230925-00219
26. Malina RM and Beunen GP. Assessment of skeletal maturity and prediction of adult height (TW3 method). Am J Hum Biol. (2002) 14:788–9. doi: 10.1002/ajhb.10098
27. Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. (2020) 22:245–57. doi: 10.1038/s41436-019-0686-8
28. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med Off J Am Coll Med Genet. (2015) 17:405–24. doi: 10.1038/gim.2015.30
29. Wu H, Li Y, and Li H. Brachydactyly type A3 is more commonly seen in children with short stature but does not affect their height improvement by growth hormone therapy. Front Endocrinol. (2022) 13:824315. doi: 10.3389/fendo.2022.824315
30. Brown T, Lambert W, and Pinkerton SK. Brachymesophalangia-5 in a group of Australian aboriginals. Hum Biol. (1980) 52:651–9.
31. Abbie AA. Brachymesophalangy V in Australian aborigines. Med J Aust. (1970) 2:736–7. doi: 10.5694/j.1326-5377.1970.tb63148.x
32. Poquérusse J, Whitford W, Taylor J, Alburaiky S, Snell RG, Lehnert K, et al. Novel PRMT7 mutation in a rare case of dysmorphism and intellectual disability. J Hum Genet. (2022) 67:19–26. doi: 10.1038/s10038-021-00955-5
33. Rodari G, Villa R, Porro M, Gangi S, Iascone M, Elli F, et al. Short stature in PRMT7 Mutations: first evidence of response to growth hormone treatment. Eur J Hum Genet EJHG. (2023) 31:195–201. doi: 10.1038/s41431-022-01220-9
34. Agolini E, Dentici ML, Bellacchio E, Alesi V, Radio FC, Torella A, et al. Expanding the clinical and molecular spectrum of PRMT7 mutations: 3 additional patients and review. Clin Genet. (2018) 93:675–81. doi: 10.1111/cge.13137
35. Batzir NA, Posey JE, Song X, Akdemir ZC, Rosenfeld JA, Brown CW, et al. Phenotypic expansion of POGZ-related intellectual disability syndrome (White-Sutton syndrome). Am J Med Genet A. (2020) 182:38–52. doi: 10.1002/ajmg.a.61380
36. Heath J, Cheyou ES, Findlay S, Luo VM, Carpio EP, Lee J, et al. POGZ promotes homology-directed DNA repair in an HP1-dependent manner. EMBO Rep. (2022) 23:e51041. doi: 10.15252/embr.202051041
37. White J, Beck CR, Harel T, Posey JE, Jhangiani SN, Tang S, et al. POGZ truncating alleles cause syndromic intellectual disability. Genome Med. (2016) 8:3. doi: 10.1186/s13073-015-0253-0
38. Donnarumma B, Riccio MP, Terrone G, Palma M, Strisciuglio P, and Scala I. Expanding the neurological and behavioral phenotype of White-Sutton syndrome a case report. Ital J Pediatr. (2021) 47:148. doi: 10.1186/s13052-021-01101-9
39. Garde A, Cornaton J, Sorlin A, Moutton S, Nicolas C, Juif C, et al. Neuropsychological study in 19 French patients with White-Sutton syndrome and POGZ mutations. Clin Genet. (2021) 99:407–17. doi: 10.1111/cge.13894
40. Murch O, Jain V, Benneche A, Metcalfe K, Hobson E, Prescott K, et al. Further delineation of the clinical spectrum of White-Sutton syndrome: 12 new individuals and a review of the literature. Eur J Hum Genet EJHG. (2022) 30:95–100. doi: 10.1038/s41431-021-00961-3
41. Gunther K, Mowrey K, and Farach LS. Two new reported cases of 16q22.3q23.3 duplication syndrome highlight intrafamilial variability and potential sex expression differences within a rare duplication syndrome. Clin Case Rep. (2021) 9:1629–33. doi: 10.1002/ccr3.3862
42. Tokutomi T, Wada T, Nakagawa E, Saitoh S, and Sasaki MA. de novo direct duplication of 16q22.1 → q23.1 in a boy with midface hypoplasia and mental retardation. Am J Med Genet A. (2009) 149A:2560–3. doi: 10.1002/ajmg.a.33049
43. Nguyen HH, Pham VA, Barcia G, Malan V, Nguyen KLT, Ngo DN, et al. Distal duplication of chromosome 16q22.1q23.1 in a Vietnamese patient with midface hypoplasia and intellectual disability. Am J Med Genet A. (2018) 176:1981–4. doi: 10.1002/ajmg.a.40375
44. Rudd D, Axelsen M, Epping EA, Andreasen N, and Wassink T. Childhood-onset schizophrenia case with 2.2 Mb deletion at chromosome 3p12.2–p12.1 and two large chromosomal abnormalities at 16q22.3–q24.3 and Xq23–q28. Clin Case Rep. (2015) 3:201–7. doi: 10.1002/ccr3.192
45. Gao Z, Lee P, Stafford JM, von Schimmelmann M, Schaefer A, and Reinberg D. An AUTS2–Polycomb complex activates gene expression in the CNS. Nature. (2014) 516:349–54. doi: 10.1038/nature13921
46. Beunders G, Voorhoeve E, Golzio C, Pardo LM, Rosenfeld JA, Talkowski ME, et al. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am J Hum Genet. (2013) 92:210–20. doi: 10.1016/j.ajhg.2012.12.011
47. Biel A, Castanza AS, Rutherford R, Fair SR, Chifamba L, Wester JC, et al. AUTS2 syndrome: molecular mechanisms and model systems. Front Mol Neurosci. (2022) 15:858582. doi: 10.3389/fnmol.2022.858582
48. Kehrer-Sawatzki H, Bäzner U, Krämer J, Lewerenz J, and Pfeiffer C. The NF1 microdeletion syndrome: early genetic diagnosis facilitates the management of a clinically defined disease. J Dtsch Dermatol Ges J Ger Soc Dermatol JDDG. (2022) 20:273–7. doi: 10.1111/ddg.14707
49. Tritto V, Bettinaglio P, Mangano E, Cesaretti C, Marasca F, Castronovo C, et al. Genetic/epigenetic effects in NF1 microdeletion syndrome: beyond the haploinsufficiency, looking at the contribution of not deleted genes. Hum Genet. (2024) 143:775–95. doi: 10.1007/s00439-024-02683-0
50. Kehrer-Sawatzki H and Cooper DN. Classification of NF1 microdeletions and its importance for establishing genotype/phenotype correlations in patients with NF1 microdeletions. Hum Genet. (2021) 140:1635–49. doi: 10.1007/s00439-021-02363-3
51. Mensink KA, Ketterling RP, Flynn HC, Knudson RA, Lindor NM, Heese BA, et al. Connective tissue dysplasia in five new patients with NF1 microdeletions: further expansion of phenotype and review of the literature. J Med Genet. (2006) 43:e08–8. doi: 10.1136/jmg.2005.034256
Keywords: brachydactyly, genotype, phenotype, algorithm, children
Citation: Wang X, Guan S, Gao Y, Xie R, Wang F, Chen X, Wu H, Zhang X, Zhang D, Yang B, Fan Q, Wang Q, Wang H, Feng T, Lv H and Chen T (2025) Establishing an algorithm for molecular genetic diagnostics in Chinese children with brachydactyly type E. Front. Endocrinol. 16:1571136. doi: 10.3389/fendo.2025.1571136
Received: 05 February 2025; Accepted: 21 May 2025;
Published: 16 June 2025.
Edited by:
Semra Çaglar Çetinkaya, University of Health Sciences, TürkiyeReviewed by:
Jing Chen, Sichuan University, ChinaKarolina Gruca-Stryjak, Medical University Poznan, Poland
Mingjie Liu, University of South China, China
Copyright © 2025 Wang, Guan, Gao, Xie, Wang, Chen, Wu, Zhang, Zhang, Yang, Fan, Wang, Wang, Feng, Lv and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ting Chen, Y2hlbnRpbmc4ODhAc3VkYS5lZHUuY24=
†These authors have contributed equally to this work