 Yongshi Yang
Yongshi Yang Xiaotong Tian
Xiaotong Tian Congwei Jia3
Congwei Jia3 Taisheng Li
Taisheng Li Zhengyin Liu
Zhengyin Liu- 1Department of Allergy, Peking Union Medical College Hospital, Chinese Academy of Medical Science and Peking Union Medical College, Beijing, China
- 2Department of Infectious Disease, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
- 3Department of Pathology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
Background: Paragangliomas (PGLs) are rare neuroendocrine tumors originating from the extra-adrenal autonomic paraganglia with a strong genetic background. SDHB pathogenic variants are associated with the highest rate of malignancy in PGLs. Most head and neck paragangliomas (HNPGs) are asymptomatic and benign, and multiple metastases are rare.
Case presentation: A 37-year-old man presented at our hospital with a fever of unknown origin (FUO) without any other complaints except for mild consumption lasting over 6 months. Laboratory examinations showed elevated inflammatory markers (CRP, ESR, IL-6, and Ferritin) but no abnormalities in infection, immune, or tumor markers. Imaging examination found an oval-shaped space-occupying lesion in the right parapharyngeal space, with rare and unique vertebral imaging findings. Octreotide imaging and 68Ga-FAPI PET/CT scans indicated a potential for neuroendocrine tumors with lymph nodes, bone, and lung metastases. Pathology demonstrated metastatic paraganglioma. Whole-genome sequencing identified an SDHB pathogenic variant and a KIF1B variant of uncertain significance. Following multidisciplinary consultation, the patient opted for the cyclophosphamide-vincristine-dacarbazine (CVD) chemotherapy regimen and was subsequently transferred to a regional hospital for coordinated follow-up.
Conclusions: This case reported rare co-occurring variants in SDHB and KIF1B and unusual imaging findings of metastasis in paraganglioma. A multimodal imaging evaluation and whole-genome sequencing were instrumental in assessing paraganglioma patients. This case suggested that atypical imaging features should raise suspicion of malignant diseases and underscored the importance of interdisciplinary collaboration in guiding the diagnosis and treatment of complex and rare clinical cases.
Introduction
Paragangliomas (PGLs) are rare neuroendocrine tumors originating from the extra-adrenal autonomic paraganglia (1). Previous studies have shown that the incidence rates of pheochromocytoma (PCC) and sympathetic paraganglioma (sPGL) have increased significantly over the past two decades, with about 0.6 cases per 100,000 person-years (2). Clinically, the sPGL are characterized by catecholamine hypersecretion and predominantly localize to the sympathetic paravertebral ganglia of the thorax, abdomen, and pelvis. In contrast, most parasympathetic PGLs are nonfunctional, and the majority of parasympathetic PGLs arise in the head and neck (3).
With the development of next-generation sequencing, approximately 30%-50% of PGLs are associated with hereditary syndromes (4). To date, more than 20 susceptibility genes have been found to be associated with the development of hereditary PCC and PGL (PPGLs), such as VHL, SDHB, SDHD, SDHA, SDHC, NF1, and RET, which are considered the most common pathogenic genes. Other susceptibility genes, which have not yet undergone rigorous genotype–clinical outcome evaluation, include KIF1B, IDH1, HIF2A, MDH2, FH, SLC25A11, and DNMT3A (3, 5). Some hereditary paragangliomas, particularly head and neck PGLs (HNPGLs), are predominantly associated with the pseudohypoxia cluster and germline succinate dehydrogenase subunit SDHD and SDHB variants, indicating the need for genetic counseling and testing for all patients with PGLs (6). Many studies have shown that SDHB gene pathogenic variants are associated with the highest risk of malignancy and represent the most frequent cause of metastatic paragangliomas (7, 8). We herein present a diagnostically challenging case of fever of unknown origin (FUO), which was ultimately diagnosed with HNPGLs harboring an SDHB pathogenic variant and a KIF1B variant of uncertain significance (VUS), complicated by extensive bone and pulmonary metastases.
Case presentation
A 37-year-old man presented at our hospital with recurrent fever for over 6 months, without any other discomforts. Since March 2024, he had developed an intermittent fever in the afternoon, with a temperature max (Tmax) of 38.3°C, which could gradually return to normal without medication. The patient visited the local hospital, where laboratory tests showed a significant increase in inflammatory markers, including C-reactive protein (CRP) 154.5mg/L and erythrocyte sedimentation rate (ESR) 100mm/h, accompanied by mild anemia. A Computed Tomography (CT)-guided percutaneous puncture biopsy of the lumbar vertebral space-occupying lesions was performed, but pathology showed no clear evidence of malignant lesions. The local hospital was unable to provide a definitive diagnosis and attempted intermittent treatment with prednisone and broad-spectrum antibiotics therapy. However, the patient still had recurrent fevers.
Consequently, the patient was admitted to our hospital for investigation of FUO. Physical examination showed that the patient was emaciated, with a body mass index (BMI) of 15.78kg/m2, presenting an anemic appearance. There was no swelling of superficial lymph nodes. The spine showed no deformity and tenderness, and no obvious tenderness or percussion pain in the spinous process and paravertebral region. Limb activity was good, and limb muscle strength was normal. The patient has had anemia since childhood. He has a long-term smoking and drinking history. The patient reported no history of cancer or endocrine disorders in his parents, one older brother, one younger sister, and his son.
Laboratory tests showed the CRP 178.34mg/L, ESR 81mm/h, IL-6 42.2pg/mL, and Ferritin 753ng/mL, all significantly increased. Bone marrow culture, bone marrow tissue pathogen metagenomic next-generation sequencing (mNGS), as well as screening for Parasites, Brucella, Tuberculosis, and other infection-related indicators, and no suspected pathogenic pathogens were detected. The autoimmune antibodies spectrum, anti-neutrophil cytoplasmic antibodies, immunoglobulin, serum IgG subclasses, complement, and other immune disease-related indicators were all negative. There were no significant abnormal results in the examination of tumor-related indicators such as tumor serum markers, serum/urine protein electrophoresis, serum/urine immunofixation electrophoresis, angiotensin-converting enzyme (ACE), and the vascular endothelial growth factor (VEGF).
Contrast-enhanced CT revealed multifocal micro- and small pulmonary nodules bilaterally, scattered vertebral body density abnormalities, and a right parapharyngeal space mass with heterogeneous prominent enhancement (CT value 65–118 HU), suspicious for paraganglioma. Figure 1 shows the patient’s MRI images, including head (A&B), pelvis (C), cervical (D), thoracic (E), and lumbar (F) vertebrae. These scans, with imaging features consistent with paraganglioma, further confirmed the diagnosis and suggested multiple metastases. Whole-body bone scintigraphy (Figure 2A) revealed cervical and thoracic vertebral abnormalities suggesting malignancy. Meta-iodobenzylguanidine (MIBG) scintigraphy (Figure 2B) showed no abnormal tracer uptake in adrenal regions or elsewhere. Octreotide imaging (Figure 2C) demonstrated somatostatin receptor overexpression in the right parapharyngeal tumor, spine, and bilateral lung nodules, indicating metastatic disease. 68Ga-FAPI PET/CT (Figure 2D) confirmed a malignant right parapharyngeal tumor and revealed FAPI-expressing metastases in cervical lymph nodes, spine, left ilium, and bilateral lungs. Based on the imaging findings, we performed additional bone metabolism and endocrine function tests. Results revealed no significant abnormalities in these parameters (Supplementary Table S1).
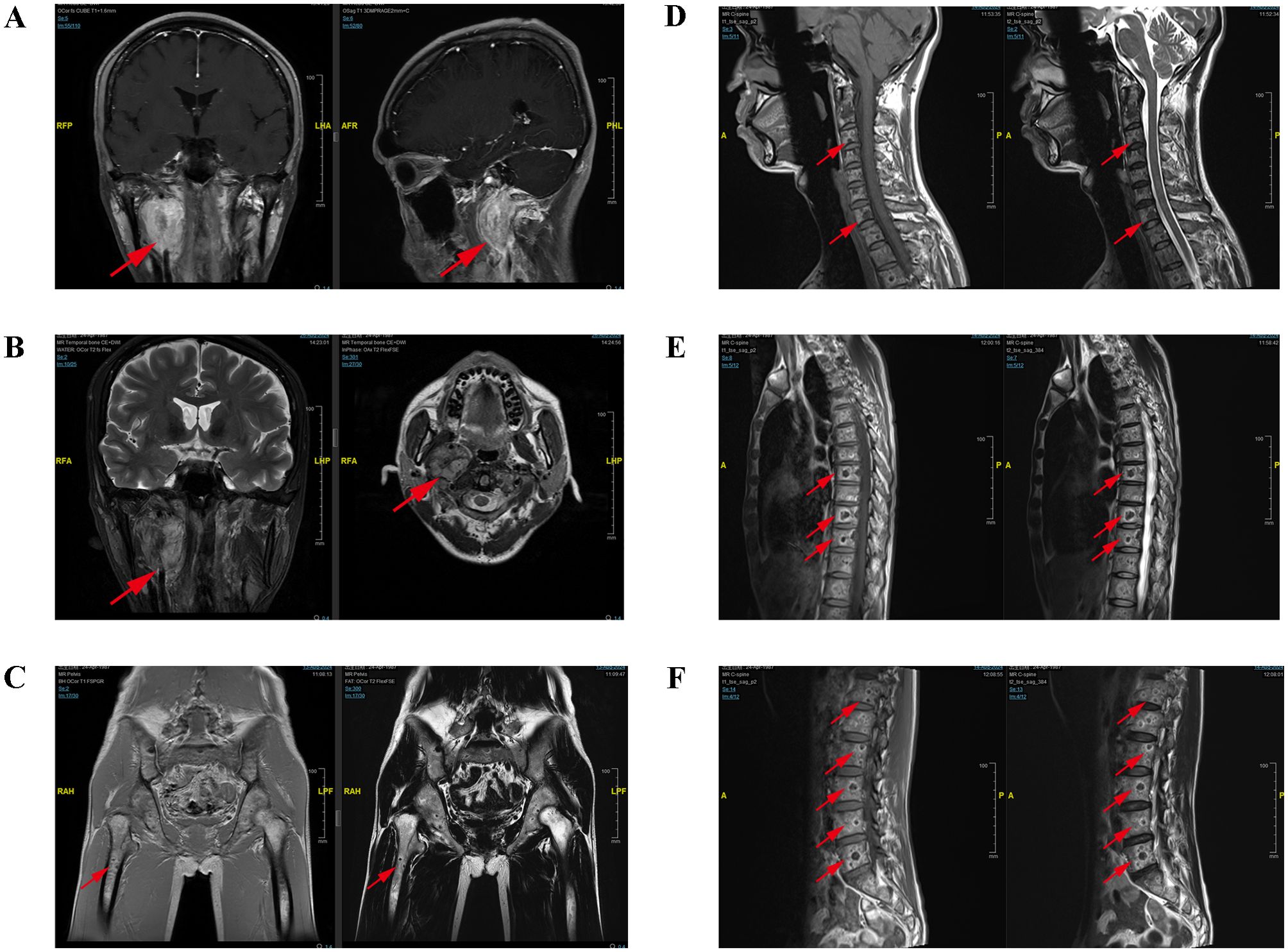
Figure 1. The MRI examination images of the patient. The MRI of the head (A, B) demonstrated an oval-shaped space-occupying lesion between the internal carotid artery and vein in the right parapharyngeal space, exhibiting uneven signal intensity and heterogeneous enhancement, with a larger section measured approximately 38mm×29mm×43mm. The MRI of the pelvis (C), cervical vertebra (D), thoracic vertebra (E) and lumbar vertebra (F) indicated the presence of multiple flaky and quasi-circular abnormal signal shadows in the pelvic bones and vertebral bodies. The MRI showed multiple patchy, quasi-circular abnormal signal shadows, with low signal intensity on T1WI and T2WI accompanied by surrounding ring-shaped high signal intensity shadows and slightly high signal intensity on fat-suppressed T2WI accompanied by peripheral annular low signal intensity shadows, which showed special ring signs.
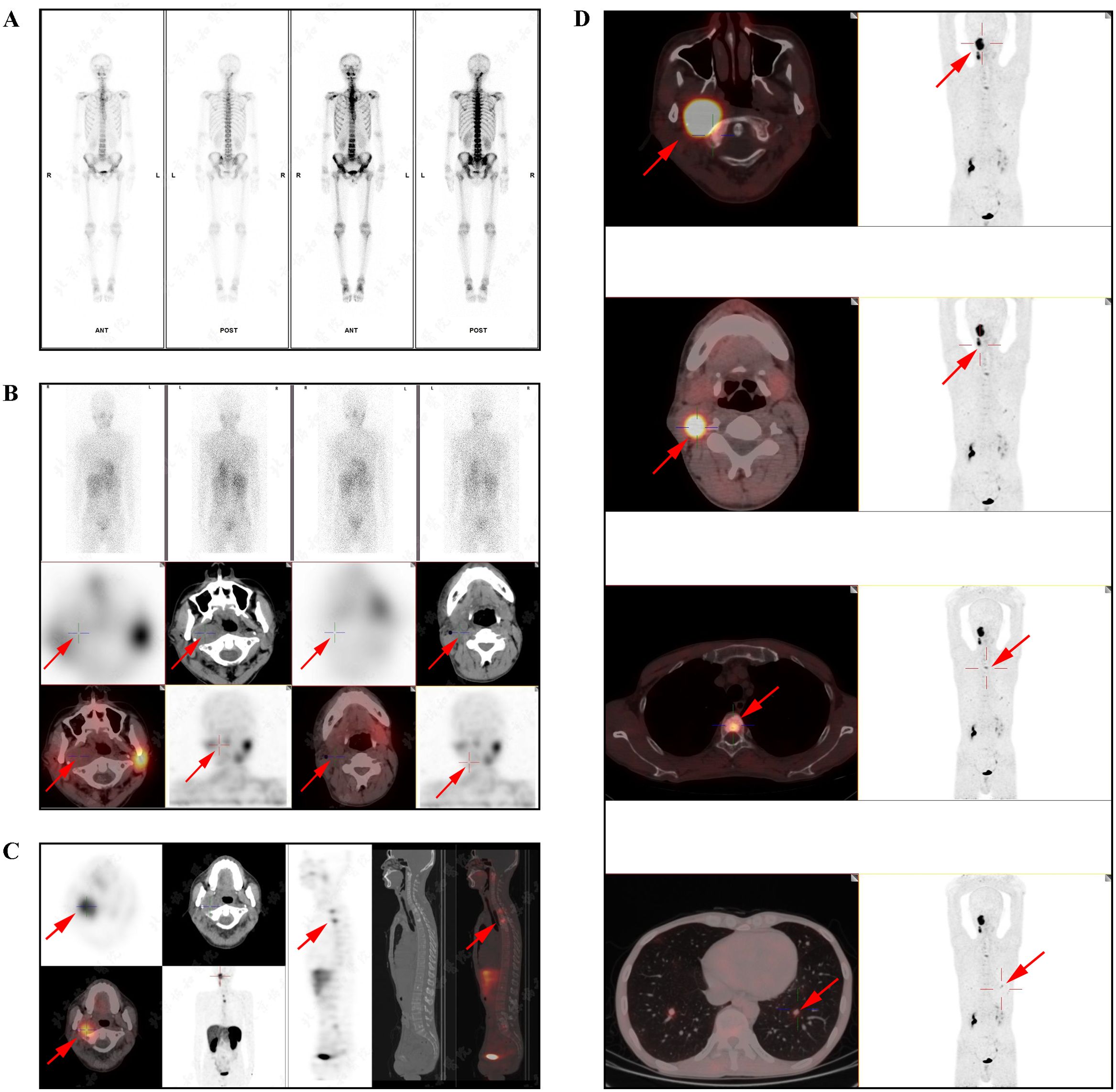
Figure 2. The nuclear medicine examination images of the patient. (A) The whole-body bone imaging indicated an abnormal increase in radioactive uptake in the right skull base bone and uneven distribution of radioactivity in some cervical and thoracic vertebrae. (B) SPECT/CT fusion imaging (MIBG): swollen lymph nodes in the right parapharyngeal space and right region II, multiple high-density nodules in the cervical and thoracic vertebrae, and the left second posterior rib, without radioactive uptake. (C) SPECT/CT fusion imaging (Octreotide): a tumor with heightened somatostatin receptor expression was observed in the right parapharyngeal space, measuring approximately 2.8×2.3cm; multiple high-density shadows were detected in the spine, pelvis, and left clavicle, bilateral scapulae, left humerus, and left femur, particularly prominent in the spine, among which larger lesions (T1 and T3 vertebral lesions) showed significantly increased radioactive uptake. (D) The PET/CT scans revealed a tumor in the right parapharyngeal space with heightened FAPI expression, measuring approximately 4.1×2.7cm in size and exhibiting a maximum standardized uptake value (SUVmax) of 7.9. Several lymph nodes in the deep cervical region (zone II) showed increased expression of somatostatin receptors. Diffuse multiple high-density shadows were observed in the cervical, thoracic, and lumbar vertebrae, and the left iliac bone, with some areas showing heightened FAPI expression (SUVmax of 3.8). Multiple micro and small nodules were detected in both lungs, with the larger one located in the lower lobe of the left lung, measuring approximately 1.0cm in diameter, with heightened FAPI expression (SUVmax of 2.7).
Before the biopsy, a comprehensive perioperative risk assessment was conducted to exclude major surgical contraindications, including infection, uncontrolled hypertension or hyperglycemia, hematological/coagulation abnormalities, anatomical risks, and anesthetic fitness. Preoperative evaluation was coordinated by multidisciplinary teams including otolaryngology, general surgery, interventional radiology, and anesthesiology. Finally, the otolaryngology-head and neck surgery team performed a lateral neck approach under general anesthesia for skull base tumor biopsy. No procedure-related adverse events were observed. The lymph nodes in zone II were enlarged and adhered. The lower pole of the tumor was located deep at the bifurcation of the internal and external carotid arteries, with a rich blood supply and hard texture. Part of the tumor was resected and sent for frozen section examination and pathological analysis. The pathological results are as follows: in the fibrous tissue of the tumor in the right parapharyngeal space, highly compressed dark-stained cells were observed. Immunohistochemical results (Figure 3) showed: AE1/AE3(-), CD20(-), CD3(-), Ki-67(index 5%), INSM-1(+), CgA(+), Syn(+); findings were consistent with neuroendocrine tumor. Tumor cells were observed in the lymph node tissue of the right II region. The immunohistochemical results were AE1/AE3(-), GATA3(-), INSM-1(+), CgA(+), Ki-67(index 5%), S-100(local scattered +), Syn(+), MGMT(partial +), SDHB(+), PIT-1(-), T-PIT(-), SF-1(-); EBER ISH(-); the diagnosis was considered as a metastatic neuroendocrine tumor, with a tendency towards paraganglioma.
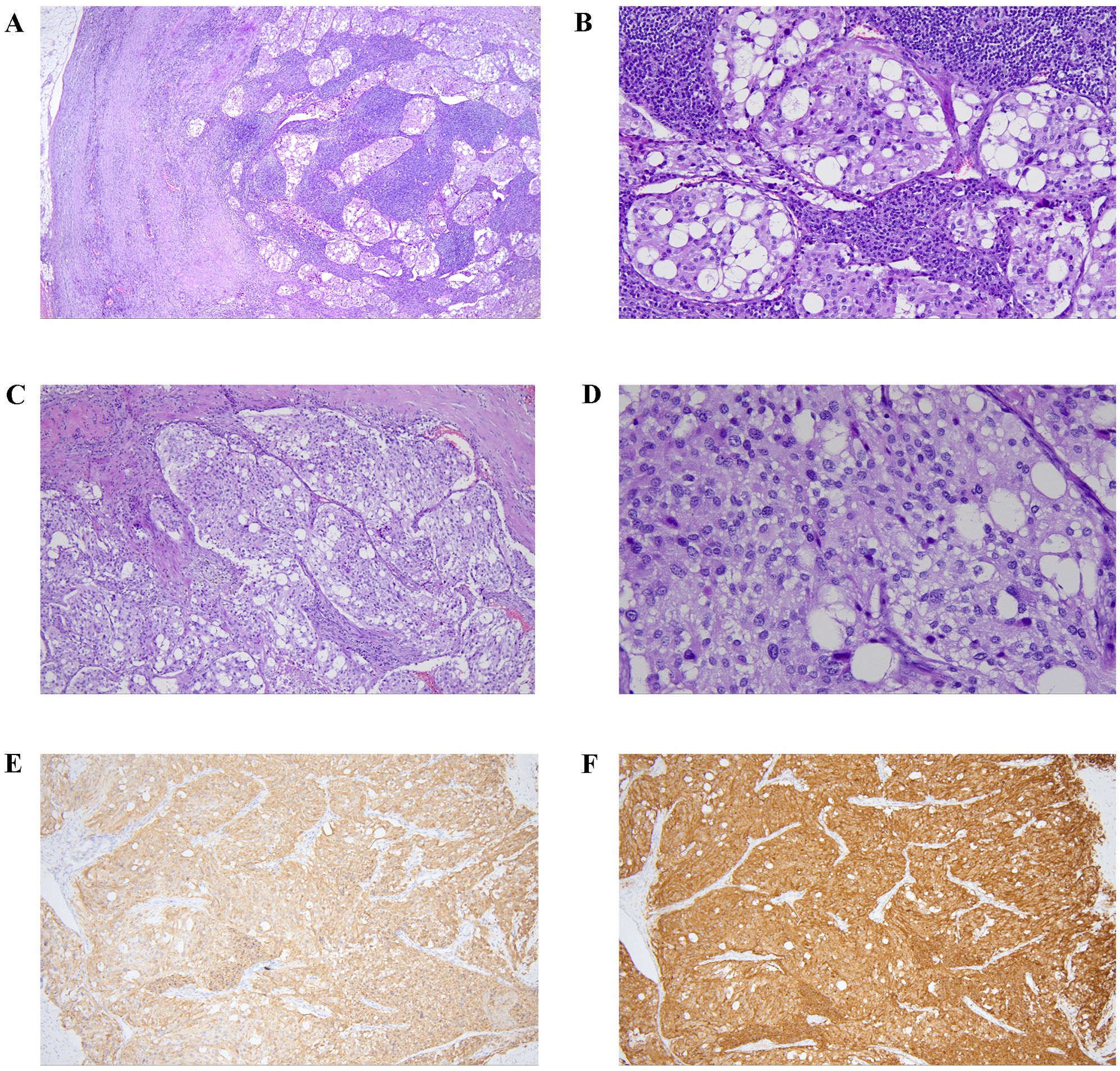
Figure 3. The pathological of the tumor tissue and the lymph node tissue. Tumor metastasis was observed in the sinus of lymph node (A, low power, HE ×40; B, medium power, HE ×200). Tumor cells were arranged in nest bulk pattern with abundant collagenous stroma (C, medium power, HE ×100), and had abundant pale cytoplasm and hyperchromatic nuclei (D, high power, HE ×400). Immunostaining showed tumor cells express chromogranin A (E, medium power, ×100) and synaptophysin (F, medium power, ×100).
Additionally, we performed whole-genome sequencing on the genomic DNA extracted from the patient’s peripheral blood. Molecular methods and bioinformatic analyses of whole-genome sequencing are summarized in the Supplementary Materials. The results (Figure 4) showed that the patient carried an SDHB gene substitution variant (NM_003000.2: c.725G>A; p.Arg242His), and a KIF1B gene deletion variant (NM_015074.3: c.1452 + 1293delG). Both variants were heterozygous. The chromosomal locations of these variants were at chr1:17349143 and chr1:10353473, respectively. According to ACMG (American College of Medical Genetics and Genomics) guidelines, the SDHB variant was referred to as pathogenic, while the KIF1B variant was classified as a VUS.
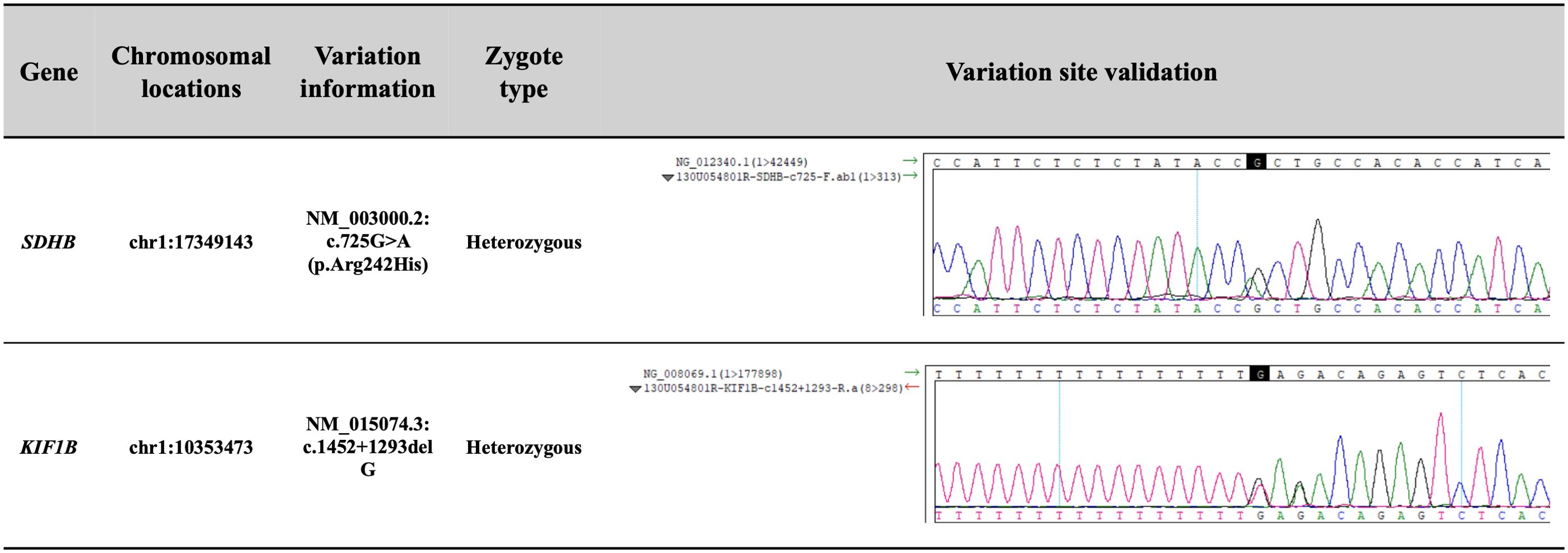
Figure 4. The results of the whole-genome sequencing on the genomic DNA from the patient’s peripheral blood. According to ACMG guidelines, the SDHB variant was referred to as pathogenic, supported by evidence codes: PP4 + PS3 + PP3 + PS4_Moderate + PM2. The KIF1B variant was classified as a variant of uncertain significance based solely on PP3.
Finally, after evaluation by the oncology department, it was recommended to choose anti-VEGFR TKI therapy or peptide receptor radionuclide therapy (PRRT), including Sunitinib or Cabozantinib, or CVD regimen (cyclophosphamide, vincristine, and dacarbazine) treatment. The patient ultimately chose the CVD regimen and returned to the local hospital for further treatment.
Discussion
Head and neck PGLs (HNPGLs) are extremely rare, comprising 3% of all PGLs and accounting for only 0.5% of all tumors in this region, which are very slow-growing and are often undetected for several years (9). Although most HNPGLs are benign, only 6% to 19% of HNPGLs may metastasize, which seriously affects survival (10). A review including 59 cases of HNPGLs extracted from the National Cancer Data Base showed five-year survival rates of 76.8% in patients with metastasis to regional lymph nodes and 11.8% in patients with distant metastasis (11). In this paper, we present a case that initially presented with FUO, and was ultimately diagnosed with HNPGLs with multiple metastases, accompanied by special imaging findings of bone metastasis, an SDHB pathogenic variant, and a KIF1B variant of uncertain significance.
The FUO is a challenging clinical syndrome with potential etiologies including infectious diseases, non-infectious inflammatory diseases, and neoplastic diseases, among others (12). It encompasses a broad differential diagnostic spectrum, and the causes exhibit significant geographical variations (13). In clinical practice, patients with PGLs with FUO as the main clinical manifestation are quite rare. Narechania S et al. reported a young healthy postpartum mother who presented with FUO and finally was found to have a retroperitoneal paraganglioma with metastases to the lungs and spine after appropriate diagnostic testing. And the genetic testing also identified a pathogenic variant in SDHB (14). Davies LL et al. also reported a patient with FUO and loin pain with elevated inflammatory markers and finally diagnosed with retroperitoneal paraganglioma confirmed by histological examination (15). Wang C et al. reported a 65-year-old man with retroperitoneal paraganglioma accompanied by elevated IL-6 levels and KIF1B single gene variant and without elevated levels of catecholamines and their derivatives, whose main symptoms were only fever and palpitations (16). Our patient was similar to this one with the IL-6 level increase and KIF1B gene variant. We also did not detect an increase in catecholamines and their metabolites in serum and urine. In addition, some studies have proposed that IL-6 secreting PCC should be considered in the differential diagnosis of FUO (17, 18). It is postulated that the IL-6 generated by the PCC is the culprit behind the fevers since the removal of the tumor, IL-6 levels declined, and the associated symptoms resolved (19, 20). However, it must be pointed out that compared to PCC, the relationship between IL-6 and PGLs still needs further confirmation.
PGLs are rare neuroendocrine tumors characterized by a strong genetic determinism. A review described the evolution of the genetics of paragangliomas and pheochromocytomas (PPGLs) over the past 20 years (5). According to the whole genome molecular genetic testing, this patient had rare double gene variants of SDHB and KIF1B. SDHB is a mitochondrial gene that is related to the metabolic activities of cells and tissues. It has attracted more and more attention from researchers due to its important structural functions affecting the occurrence and progress of related diseases (21). Compared with other hereditary subtypes of PPGLs, PPGLs with SDHB have the highest rates of disease-specific morbidity and mortality. PPGLs with SDHB are usually less differentiated and do not produce large amounts of catecholamines, which allows these tumors to grow sub-clinically for a long time (22). Some scholars believe that germline variants in the SDHB gene predispose to metastatic PGLs (5, 23). A meta-analysis found that the SDHB mutation had the highest prevalence in PPGLs (23%-31%), similar to the highest risk of metastasis posed by SDHB mutation (12%-41%) (24). The KIF1B gene is related to the protein called kinesin family member 1B, which is essential for the transport of materials within cells (25). However, research on the KIF1B gene in PPGLs is far less abundant than reports on other gene variants. In the study of Evenepoel L, somatic variants were identified in 54% of the PPGLs patients, and the KIF1B (20.4%) and NF1 (20.8%) were the most frequently mutated genes (26). In 2020, a single-center study from our hospital included 314 PPGLs patients and found that 22.0% of patients had metastatic, 49.7% of patients were HNPGLs, and 107 variants were in 314 cases (34.1%). SDHB gene was the most frequently mutated gene (14.6%), followed by RET (3.8%), VHL (3.8%), SDHD (2.5%), SDHA (2.2%), MAX (1.9%), FH (0.3%), and KIF1B (0.3%) (27). Although some literatures have reported the identification of KIF1B gene variant in PPGLs patients, compared with SDHB, which is classified as a pathogenic variant gene, KIF1B gene variant can only be categorized as VUS. Further rigorous functional studies are still needed to confirm the clinical relevance. Over the last 20 years, significant progress has been made in the research of molecular biology and markers related to the aggressive behavior of PPGLs, but currently, there are still no clinical, biochemical, histopathological, or molecular markers that can be used to accurately predict the metastatic behavior of PPGLs at diagnosis (28).
It is reported that about 5-10% of cancer patients develop spinal metastasis during the disease process (29). The spine is the third most common site of metastatic disease, only following the lung and liver, and is also the most common site of bone metastasis (30). A retrospective study from our hospital included 250 patients with metastatic PPGLs and found that 85.6% had multisystem metastasis, with bone metastases being the most common site of metastasis (60.8%), following the lung (44.4%), lymph nodes (44.4%), and liver (30.8%). The incidence of germline variants, especially SDHB variants, was high in metastatic PPGLs patients, accounting for 76.5% (31). Using a large institutional database, Ayala-Ramirez M et al. conducted a retrospective study of 128 patients with pheochromocytoma or sympathetic paraganglioma. They found that 20% of the patients had metastatic disease located exclusively in the skeleton. The spine was the most common location of bone metastasis (81%), following the sacrum and pelvis (67%), proximal and distal long bones (49%), and skull (21%) (32). Further investigation is required to elucidate the roles of the SDHB pathogenic variant and the KIF1B variant of uncertain significance in driving the development of multiple metastases in this patient.
Another case report described a woman with SDHB variant and spinal metastatic PGL, presenting with multilevel lytic lesions at C1, C4-C7, L1-L3 (cervical/thoracic/lumbar spine) and compression fractures at C3 and T9 (33). Although they all have SDHB variant and extensive metastatic disease of the spine, this patient was significantly different from our reported patient in imaging and symptoms. In our report, the patient had no deformity of the spine, no tenderness, or percussion pain in the spinous process and paravertebral region, which seemed inconsistent with extensive imaging findings of vertebral metastasis. The patient’s vertebral MRI showed multiple patchy, quasi-circular abnormal signal shadows, with low signal intensity on T1WI and T2WI accompanied by surrounding ring-shaped high signal intensity shadows and slightly high signal intensity on fat-suppressed T2WI accompanied by peripheral annular low signal intensity shadows, which showed special ring signs. This was very similar to that reported by Mediouni A and his colleagues. They considered that the MRI of malignant HNPGLs had two types of lesions, namely nodular and expanding, and the nodular lesions showed specific imaging appearances, which were composed of low-intensity signals in the center, surrounded by a single fat-like halo or double halo (34). As reported by Yu and his colleagues, ring signs can be considered relatively specific signs in some other bone tumors, such as osteochondromyxoma in the Carney complex. They found the center of the ring signs had low or slightly equal signal intensities (compared with the muscle) on the T1WI and high or low signal intensities on the T2WI (35). The imaging findings of this patient also well remind us that when we find some special types of imaging appearance, we need to be alert to the possibility of malignant diseases.
The imaging localization examination of PGLs is beneficial for determining the selection of effective treatment methods. This patient underwent a very detailed imaging evaluation. Before surgery, temporal bone enhanced CT and MRI, as well as cervical CTA, were performed to clearly display the morphology, blood supply, and relationship with surrounding tissues of the tumor. The expert consensus recommends that PPGL patients with metastasis or inoperable conditions undergo 131I-MIBG nuclear imaging first and evaluate the possibility of 131I-MIBG treatment based on the functional and anatomical localization of the tumor (22). However, the 131I-MIBG radionuclide imaging of this patient was negative, so 131I-MIBG cannot be used for treatment. MIBG has low sensitivity in detecting metastatic, recurrent, located in the skull base, neck, chest, and bladder PGL, as well as PPGL genes associated with various subtypes of SDHx (especially SDHB); And the imaging effect is poor for low secretion function and small tumors, and false negatives may occur (36, 37). Somatostatin octreotide imaging (e.g., 99mTc HTOC) can complement the 131I-MIBG imaging of PPGL and assist in diagnosis (38).
In summary, this case illustrated the detailed process of a fever of unknown origin and ultimately being diagnosed with paraganglioma and presented the rare imaging findings of vertebral metastasis in paraganglioma. This case highlighted the emphasis on interdisciplinary collaboration, including Otorhinolaryngology, Orthopedics, Oncology, Endocrinology, Pathology, Genetics, Anesthesiology, Nuclear Medicine, and Radiology, which formed a multidisciplinary team (MDT) to integrate clinical, imaging, pathological, and genetic insights for the comprehensive management of complex and rare clinical cases. Notably, this case identified rare co-occurring variants, an SDHB pathogenic variant and a KIF1B VUS, underscoring the critical need for comprehensive germline and somatic genetic testing in such cases, offering valuable insights and data for the diagnosis, management, and research of paraganglioma.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, and further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Peking Union Medical College Hospital (I-24PJ0680). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YY: Data curation, Investigation, Writing – original draft. XT: Data curation, Investigation, Writing – original draft. CJ: Data curation, Investigation, Writing – review & editing. HF: Data curation, Investigation, Writing – review & editing. TL: Writing – review & editing, Conceptualization. LZ: Funding acquisition, Writing – review & editing, Conceptualization. ZL: Writing – review & editing, Funding acquisition, Conceptualization.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was supported by the National High Level Hospital Clinical Research Funding (grant number 2022-PUMCH-B-043). Whole genome molecular genetic testing was supported by the Project of Enhancing Rare Disease Diagnosis and Treatment Capabilities Supported by the Central Government’s Public Welfare Lottery Fund (UPWARDS).
Acknowledgments
We acknowledge the collaboration of Otorhinolaryngology, Orthopedics, Oncology, Endocrinology, Pathology, Genetics, Anesthesiology, Nuclear Medicine, and Radiology.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2025.1612259/full#supplementary-material
References
1. Mete O, Asa SL, Gill AJ, Kimura N, de Krijger RR, and Tischler A. Overview of the 2022 WHO classification of paragangliomas and pheochromocytomas. Endocrine Pathol. (2022) 33:90–114. doi: 10.1007/s12022-022-09704-6
2. Berends AMA, Buitenwerf E, de Krijger RR, Veeger NJGM, van der Horst-Schrivers ANA, Links TP, et al. Incidence of pheochromocytoma and sympathetic paraganglioma in the Netherlands: A nationwide study and systematic review. Eur J Internal Med. (2018) 51:68–73. doi: 10.1016/j.ejim.2018.01.015
3. Neumann HPH, Young WF, and Eng C. Pheochromocytoma and paraganglioma. New Engl J Med. (2019) 381:552–65. doi: 10.1056/NEJMra1806651
4. Fishbein L, Merrill S, Fraker DL, Cohen DL, and Nathanson KL. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol. (2013) 20:1444–50. doi: 10.1245/s10434-013-2942-5
5. Buffet A, Burnichon N, Favier J, and Gimenez-Roqueplo AP. An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab. (2020) 34:101416. doi: 10.1016/j.beem.2020.101416
6. Lin EP, Chin BB, Fishbein L, Moritani T, Montoya SP, Ellika S, et al. Head and neck paragangliomas: an update on the molecular classification, state-of-the-art imaging, and management recommendations. Radiol Imaging Cancer. (2022) 4:e210088. doi: 10.1148/rycan.210088
7. King KS, Prodanov T, Kantorovich V, Fojo T, Hewitt JK, Zacharin M, et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol: Off J Am Soc Clin Oncol. (2011) 29:4137–42. doi: 10.1200/JCO.2011.34.6353
8. Martins R and Bugalho MJ. Paragangliomas/Pheochromocytomas: clinically oriented genetic testing. Int J Endocrinol. (2014) 12:794187. doi: 10.1155/2014/794187
9. Leonetti JP. Neurological manifestations of paragangliomas of the head and neck. Curr Neurol Neurosci Rep. (2022) 22:485–9. doi: 10.1007/s11910-022-01216-4
10. Sandow L, Thawani R, Kim MS, and Heinrich MC. Paraganglioma of the head and neck: A review. Endocrine Practice: Off J Am Coll Endocrinol Am Assoc Clin Endocrinol. (2023) 29:141–7. doi: 10.1016/j.eprac.2022.10.002
11. Lee JH, Barich F, Karnell LH, Robinson RA, Zhen WK, Gantz BJ, Hoffman HT, American College of Surgeons Commission on Cancer, and American Cancer Society. National Cancer Data Base report on Malignant paragangliomas of the head and neck. Cancer. (2002) 94:730–7. doi: 10.1002/cncr.10252
12. Haidar G and Singh N. Fever of unknown origin. New Engl J Med. (2022) 386:463–77. doi: 10.1056/NEJMra2111003
13. Ryan K. Fever of unknown origin. Med Clinics North America. (2024) 108:79–92. doi: 10.1016/j.mcna.2023.05.016
14. Narechania S, Bath A, Ghassemi L, Lokhande C, Haddad A, Yousuf AM, et al. Paraganglioma presenting as postpartum fever of unknown origin. Case Rep Endocrinol. (2015) p:864719. doi: 10.1155/2015/864719
15. Davies LL, Fitchett JM, and Jones D. Fever of unknown origin: a rare retroperitoneal cause. BMJ Case Rep. (2012) 2012. doi: 10.1136/bcr.03.2012.6122
16. Wang C, Guan M, Zhang S, and Cui C. Case Report: A 65-year-old man with paraganglioma accompanied by elevated interleukin-6 levels and KIF1B single gene mutation. Front Endocrinol. (2023) 14:1226468. doi: 10.3389/fendo.2023.1226468
17. Cheng X, Zhang M, Xiao Y, Li H, Zhang Y, and Ji Z. INTERLEUKIN-6-PRODUCING PHEOCHROMOCYTOMA AS A NEW REASON FOR FEVER OF UNKNOWN ORIGIN: A RETROSPECTIVE STUDY. Endocrine Practice: Off J Am Coll Endocrinol Am Assoc Clin Endocrinol. (2018) 24:507–11. doi: 10.4158/EP-2018-0048
18. Rai HK and Reddy K. Extensive fever of unknown origin (FUO) workup to unmask pheochromocytoma. Cureus. (2021) 13:e15218. doi: 10.7759/cureus.15218
19. Siddiqui UM, Matta S, Wessolossky MA, and Haas R. Fever of unknown origin: could it be a pheochromocytoma? A case report and review of the literature. Case Rep Endocrinol. (2018) p:3792691. doi: 10.1155/2018/3792691
20. Yarman S, Soyluk O, Altunoglu E, and Tanakol R. Interleukin-6-producing pheochromocytoma presenting with fever of unknown origin. Clinics (Sao Paulo Brazil). (2011) 66:1843–5. doi: 10.1590/S1807-59322011001000028
21. Liu C, Zhou D, Yang K, Xu N, Peng J, and Zhu Z. Research progress on the pathogenesis of the SDHB mutation and related diseases. Biomed Pharmacother Biomed Pharmacother. (2023) 167:115500. doi: 10.1016/j.biopha.2023.115500
22. Taïeb D, Nölting S, Perrier ND, Fassnacht M, Carrasquillo JA, Grossman AB, et al. Management of phaeochromocytoma and paraganglioma in patients with germline SDHB pathogenic variants: an international expert Consensus statement. Nature Reviews. Endocrinology. (2024) 20:168–84. doi: 10.1038/s41574-023-00926-0
23. Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, et al. Mutations in the SDHB gene are associated with extra-adrenal and/or Malignant phaeochromocytomas. Cancer Res. (2003) 63:5615–21.
24. Lee H, Jeong S, Yu Y, Kang J, Sun H, Rhee JK, et al. Risk of metastatic pheochromocytoma and paraganglioma in SDHx mutation carriers: a systematic review and updated meta-analysis. J Med Genet. (2020) 57:217–25. doi: 10.1136/jmedgenet-2019-106324
25. Hirokawa N, Noda Y, Tanaka Y, and Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. (2009) 10:682–96. doi: 10.1038/nrm2774
26. Evenepoel L, Helaers R, Vroonen L, Aydin S, Hamoir M, Maiter D, et al. KIF1B and NF1 are the most frequently mutated genes in paraganglioma and pheochromocytoma tumors. Endocrine-related Cancer. (2017) 24:L57–61. doi: 10.1530/ERC-17-0061
27. Ma X, Li M, Tong A, Wang F, Cui Y, Zhang X, et al. Genetic and clinical profiles of pheochromocytoma and paraganglioma: A single center study. Front Endocrinol. (2020) 11:574662. doi: 10.3389/fendo.2020.574662
28. Torresan F, Iacobone C, Giorgino F, and Iacobone M. Genetic and molecular biomarkers in aggressive pheochromocytomas and paragangliomas. Int J Mol Sci. (2024) 25:7142. doi: 10.3390/ijms25137142
29. Miller R. MRI in metastatic spine disease. In: Eltorai AEM, Hyman CH, and Healey TT, editors. Essential Radiology Review: A Question and Answer Guide. Springer International Publishing, Cham (2019). p. 469–70.
30. Shah LM and Salzman KL. Imaging of spinal metastatic disease. Int J Surg Oncol. (2011) 2011:769753. doi: 10.1155/2011/769753
31. Cui YY, Zhou Y, Gao YJ, Li TY, Wang Y, Zhang WQ, et al. Clinical features of patients with metastatic pheochromocytoma/paraganglioma. Zhonghua Yi Xue Za Zhi. (2024) 104:1174–9. doi: 10.3760/cma.j.cn112137-20230820-00271
32. Ayala-Ramirez M, Palmer JL, Hofmann MC, de la Cruz M, Moon BS, Waguespack SG, et al. Bone metastases and skeletal-related events in patients with Malignant pheochromocytoma and sympathetic paraganglioma. J Clin Endocrinol Metab. (2013) 98:1492–7. doi: 10.1210/jc.2012-4231
33. Jabarkheel R, Pendharkar AV, Lavezo JL, Annes J, Desai K, Vogel H, et al. Metastatic paraganglioma of the spine with SDHB mutation: case report and review of the literature. Int J Spine Surg. (2021) 14:S37–45. doi: 10.14444/7163
34. Mediouni A, Ammari S, Wassef M, Gimenez-Roqueplo AP, Laredo JD, Duet M, et al. Malignant head/neck paragangliomas. Comparative study. Eur Ann Otorhinolaryngol Head Neck Dis. (2014) 131:159–66. doi: 10.1016/j.anorl.2013.05.003
35. Yu W, Zhang ZZ, Wang O, Huang MQ, Xia WB, and Guermazi A. Ring sign: an imaging sign for osteochondromyxoma in Carney complex. Quantitative Imaging Med Surg. (2019) 9:1958–65. doi: 10.21037/qims.2019.11.14
36. Timmers HJLM, Taïeb D, Pacak K, and Lenders JWM. Imaging of pheochromocytomas and paragangliomas. Endocrine Rev. (2024) 45:414–34. doi: 10.1210/endrev/bnae001
37. Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2014) 99:1915–42. doi: 10.1210/jc.2014-1498
Keywords: paraganglioma, metastasis, SDHB, case report, gene variant, FUO, KIF1B
Citation: Yang Y, Tian X, Jia C, Fan H, Li T, Zhang L and Liu Z (2025) Case Report: Rare multisystem metastasis in head and neck paraganglioma with SDHB pathogenic variant and KIF1B VUS manifested as FUO. Front. Endocrinol. 16:1612259. doi: 10.3389/fendo.2025.1612259
Received: 15 April 2025; Accepted: 29 July 2025;
Published: 21 August 2025.
Edited by:
Sergei Tevosian, University of Florida, United StatesReviewed by:
Felipe Freitas-Castro, Faculdade de Medicina da Universidade de São Paulo, BrazilTetsuro Tsumura, Daido Hospital, Japan
Sara Elena Campos Ramírez, Hospital Universitario Miguel Servet, Spain
Copyright © 2025 Yang, Tian, Jia, Fan, Li, Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Zhang, emhhbmdsaTM2QHB1bWNoLmNu; Zhengyin Liu, emhlbmd5aW5sQGhvdG1haWwuY29t
†These authors have contributed equally to this work