 Justyna Małgorzata Fercho1,2,3,4*†
Justyna Małgorzata Fercho1,2,3,4*† Oskar G. Chasles4†
Oskar G. Chasles4† Jakub Chamier-Gliszczyński4
Jakub Chamier-Gliszczyński4 Dominik Ryszard Płaza4Bogdan Jabłoński4
Dominik Ryszard Płaza4Bogdan Jabłoński4 Klaudia Kokot4Maciej Mielczarek5Małgorzata Małłek-Grabowska6,7Jacek Szypenbejl2,4Adrian Szkudlarek1Tomasz Szmuda8
Klaudia Kokot4Maciej Mielczarek5Małgorzata Małłek-Grabowska6,7Jacek Szypenbejl2,4Adrian Szkudlarek1Tomasz Szmuda8 Mariusz Siemiński2,4
Mariusz Siemiński2,4 Jacek Furtak1,7
Jacek Furtak1,7- 1Neurosurgery Department, 10th Military Research Hospital and PolyClinic SPZOZ, Bydgoszcz, Poland
- 2Department of Emergency Medicine, Medical University of Gdańsk, Gdańsk, Poland
- 3Lung Transplant Unit, Cardiac Surgery Department, Medical University of Gdańsk, Gdańsk, Poland
- 4Scientific Circle of Neurotraumatology, Department of Emergency Medicine, Medical University of Gdańsk, Gdańsk, Poland
- 5Neurosurgery Department, Stanisław Staszic Specialist Hospital, Piła, Poland
- 6Department of Anesthesiology and Intensive Therapy, 10th Military Research Hospital and Polyclinic SPZOZ, Bydgoszcz, Poland
- 7Faculty of Medicine, Bydgoszcz University of Science and Technology, Bydgoszcz, Poland
- 8Faculty of Medicine, Medical University of Gdańsk, Gdańsk, Poland
Background: Pheochromocytoma (PCC) is a rare neuroendocrine tumor, with 10–15% of cases showing malignant behavior defined by metastatic spread, including exceptionally rare central nervous system (CNS) involvement. Brain metastases present unique diagnostic and therapeutic challenges due to their potential to impair neurological function. This study reports a case of malignant PCC (mPCC) with CNS metastases and a systematic review to clarify the clinical patterns, management strategies, and prognostic factors.
Methods: We describe the surgically managed case of a 41-year-old man with right frontoparietal brain metastasis. A systematic review, adhering to the PRISMA 2020 guidelines, searched PubMed, Scopus, and Web of Science for peer-reviewed studies on mPCC with brain or spinal metastases confirmed by radiology or histopathology. Data on demographics, symptoms, imaging, treatments, and outcomes were extracted and descriptively analyzed using Python-generated graphics.
Results: This review identified 18 cranial (1948–2022) and 60 spinal (1977–2024) metastasis cases from 53 studies. Cranial metastases were present at a mean age of 46.6 years (SD 14.1), commonly with headaches (44.4%) and neurological deficits, such as weakness, presented in our case, with 72.2% surgically treated. Spinal metastases occurred at a mean age of 44.5 years (SD, 16.0), often with hypertension (51.7%) or pain, with a mean of 1.7 lesions (SD 1.5). The patient achieved short-term symptom relief post-resection, but incomplete follow-up (33.3% cranial) and reporting gaps (63.3% spinal laterality) limited the prognostic insights. MRI and PET improved the diagnostic accuracy over historical non-contrast CT use (41.7% spinal cases).
Interpretation: CNS mPCC metastases are exceedingly rare with distinct neurological (cranial) and structural (spinal) presentations. Advanced imaging, particularly magnetic resonance imaging (MRI) and positron emission tomography (PET), is critical for accurate diagnosis and surgical planning. Sparse data underscores the need for registries and prospective studies to standardize care and improve outcomes.
Introduction
Pheochromocytoma (PCC) is a rare neuroendocrine tumor arising from chromaffin cells, predominantly in the adrenal medulla, with an estimated incidence of 0.8 per 100,000 person-years (1). Alongside extra-adrenal paragangliomas, PCC forms a spectrum of catecholamine-secreting tumors that affect patients globally, with variations in prevalence across sporadic and hereditary contexts. Its pathophysiology centers on excessive catecholamine production, including epinephrine, norepinephrine, and dopamine, manifesting as paroxysmal hypertension, tachycardia, diaphoresis, and, in extreme cases, hypertensive crises that threaten cardiovascular stability (1). PCC occurs sporadically or within hereditary syndromes like von Hippel-Lindau disease (VHL), multiple endocrine neoplasia type 2 (MEN2), or neurofibromatosis type 1 (NF1), with mutations, particularly SDHB, increasing malignancy risk (2, 3).
Approximately 10–15% of PCCs are malignant, defined by metastatic spread, with a slight male predominance noted in some series (4). Common metastatic sites include the liver, lungs, and skeleton; however, central nervous system (CNS) involvement, encompassing brain and spinal metastases, is extraordinarily rare, with fewer than 100 cases reported over eight decades (5, 6). The scarcity of CNS metastases complicates diagnosis and management, as their neurological consequences (e.g., deficits and seizures) and structural impacts (e.g., spinal instability) require specialized intervention.
Diagnosis of malignant PCC (mPCC) integrates biochemical assays, imaging, and genetic profiling. Plasma-free metanephrines and urinary catecholamines offer high sensitivity, although false positives require careful interpretation (7, 8). Imaging modalities such as computed tomography (CT), magnetic resonance imaging (MRI), and 123I-metaiodobenzylguanidine (MIBG) scintigraphy localize tumors, with MRI excelling for CNS lesions and positron emission tomography (PET) aiding systemic staging (9). Genetic testing identifies high-risk mutations; however, global disparities limit access (2). Management hinges on preoperative alpha- and beta-adrenergic blockade to stabilize hemodynamics, followed by surgery for resectable metastases. CNS cases require additional consideration, balancing neurological preservation and oncological control (5, 10).
This study presents a rare case of CNS mPCC cerebral metastasis, illustrating distinct clinical and therapeutic complexities. The patient signed an informed consent form, allowing the authors to publish the results. Through a systematic review, we synthesized global data to elucidate the demographics, diagnostics, treatments, and gaps, aiming to inform clinical practice and future research on this understudied malignancy.
Case report
Cerebral metastasis
Patient background
A 41-year-old right-handed male was referred to the Neurosurgery Department for a right frontoparietal tumor identified by computed tomography (CT) at Głogów Hospital after two weeks of progressive left-sided weakness. His history included mPCC diagnosed post-laparoscopic adrenalectomy (September 2007) with open resection of a local recurrence (July 2008). Exploratory laparotomy (June 2015) revealed unresectable inferior vena cava invasion. He received stereotactic radiotherapy to the adrenal bed (30 Gy, May 2017), followed by 177Lu-DOTATATE for retroperitoneal lymph node metastases (first dose, November 2022), and sunitinib (initiated on March 9, 2023), achieving a partial response. The comorbidities included resistant hypertension, likely catecholamine-driven, and suspected autoimmune thyroiditis. To date, no allergies have been reported. The patient was later referred to the Neurosurgery Department at the 10th Military Research Hospital and Polyclinic in Bydgoszcz, Poland, due to the need for further treatment requiring greater neurosurgical capabilities. Enabling this treatment necessitated the patient’s transport across two voivodeships.
Clinical findings
On admission, the patient was alert and oriented, with a Glasgow Coma Scale (GCS) score of 15 and Karnofsky Performance Status (KPS) score of 80, reflecting preserved function despite systemic disease. Neurological examination revealed no motor deficits, although prior weakness prompted imaging.
Preoperative diagnosis
Suspected mPCC metastasis to the right frontoparietal region based on CT and oncological history.
Surgical management
On August 1, 2024, neuronavigation-assisted craniectomy was performed under general anesthesia. The patient was supine, and the head was secured using the Z-touch neuronavigation system to map the tumor. A parietal incision enabled a skin-fascia flap and craniectomy near the sagittal sinus, which was controlled with hemostatic sutures. Dural incision revealed no initial motor cortex response upon stimulation. A 5-mm cortical incision exposed a grayish-cream, moderately vascular tumor resected via a cavitron ultrasonic surgical aspirator (CUSA). A second brownish, highly vascular segment extended into the right lateral ventricle, requiring meticulous resection. Fluorescent residual tissue (pink under UV light) was cleared. Stimulation (5 mA) at the anteromedial margin elicited left extremity responses, indicating motor cortex proximity, prompting careful closure. Hemostasis was performed using Surgicel; the dura was sealed with sutures and TachoSil, and the bone flap was secured with CranioFix, followed by cranioplasty with bone cement. The wound was closed in layers and the patient was transferred to the ICU. Preoperative alpha- and beta-blockers ensured hemodynamic stability with no intraoperative complications.
Histopathology
Two samples confirmed metastatic mPCC, showing a nested (zellballen) architecture typical of pheochromocytoma. Immunohistochemistry revealed the following findings:
● Positive: Vimentin (++, strong), CD10 (+, weak), Chromogranin A (++, strong), and synaptophysin (++, strong).
● Negative: GATA3, Melan A, CAM 5.2, TTF1, CK-PAN, and S-100.
Strong positivity (++) for Chromogranin A and Synaptophysin, with weaker CD10, confirmed a neuroendocrine origin, ruling out carcinoma or melanoma. The absence of GATA3 and TTF1 excluded thyroid or lung primaries, aligning with the patient’s history of mPCC.
Postoperative imaging
Non-contrast CT (August 2, 2024) showed a 48 × 23 mm fluid-filled resection cavity, consistent with postoperative changes, with successful cranioplasty. Brain parenchyma, ventricles, and midline were unremarkable (Figure 1).
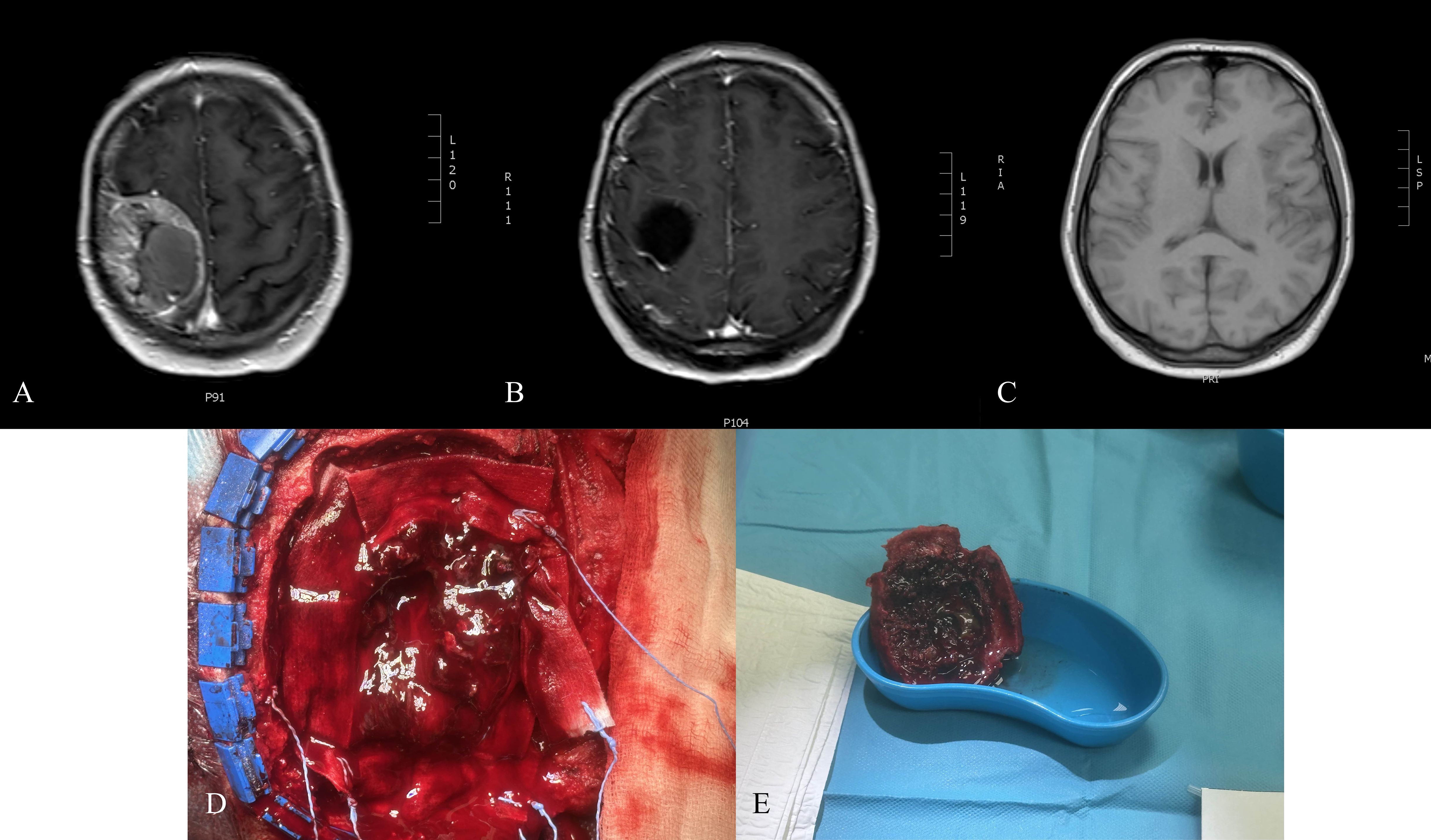
Figure 1. (A) Preoperative T1-weighted MRI with contrast showing cranial metastasis of the pheochromocytoma. (B) Non-contrast CT scan performed on the first postoperative day, demonstrating postsurgical changes. (C) T1-weighted MRI with contrast at 3 months postoperatively, illustrating the postoperative outcomes. (D) Intraoperative photograph of resected pheochromocytoma metastasis. (E) Intraoperative photograph obtained during the resection of a cranial metastatic tumor.
Postoperative diagnosis
Resected right frontoparietal mPCC metastasis with cranioplasty, histologically verified.
Outcome
Short-term neurological preservation was achieved with no immediate deficits, although systemic disease indicated a guarded prognosis.
Methodology
Search strategy and protocol
This systematic review was conducted in accordance with PRISMA 2020 guidelines. A comprehensive search strategy was developed and implemented across three electronic databases, PubMed, Scopus, and Web of Science. The search utilized the following terms: (“pheochromocytoma”) AND ((“malignant”) OR (“metastasis”)) AND ((“brain” OR “cerebrum” OR “intracranial” OR “cranium” OR “skull”) OR (“spine” OR “vertebra”)). No date or language restrictions were applied, resulting in 1426 records from 1948 to 2024. After removing 187 duplicates and 140 results owing to insufficient metadata for screening, 1099 unique records were retained for screening (Figure 2).
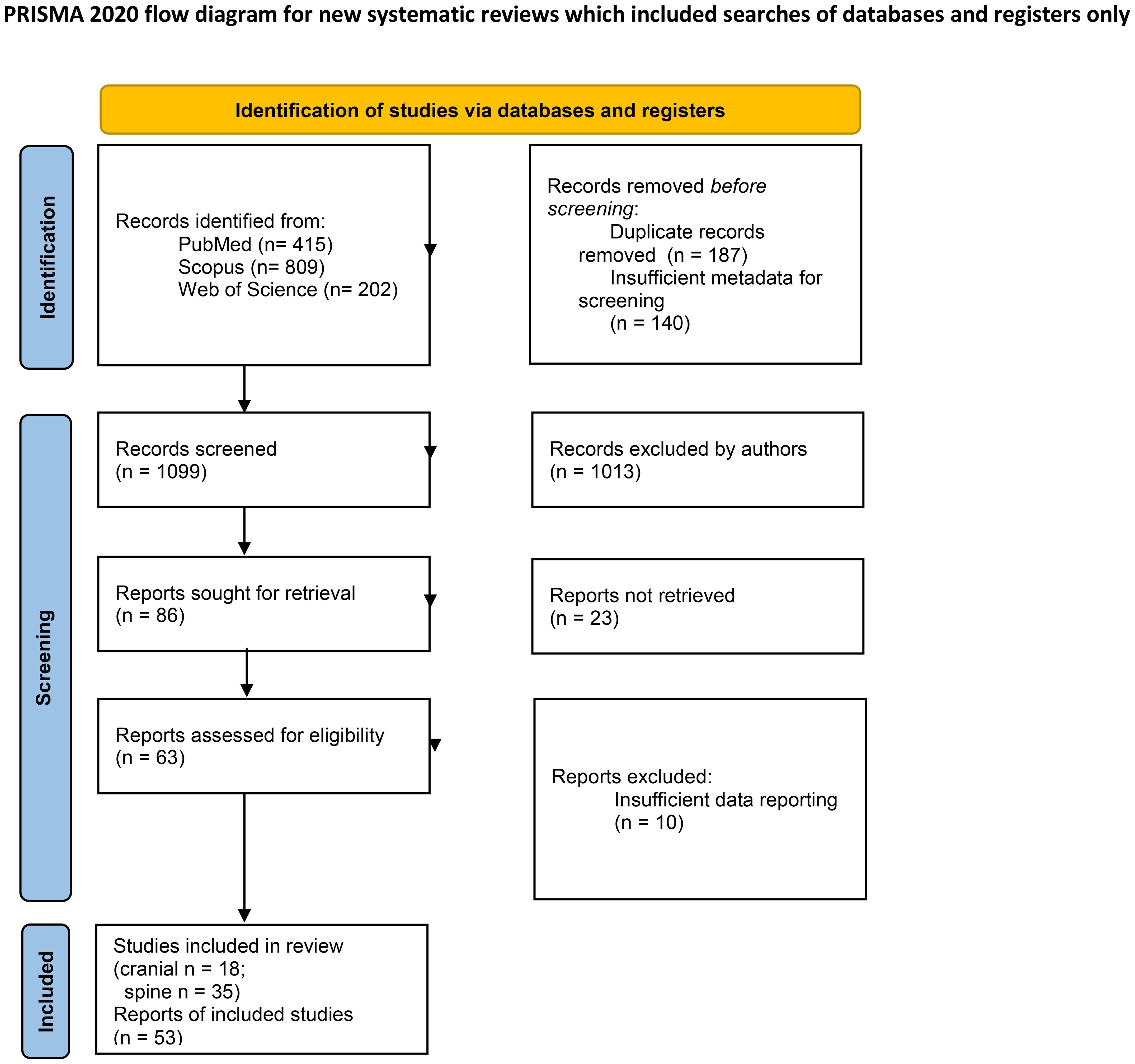
Figure 2. PRISMA Flowchart.
Selection criteria
The eligibility criteria were peer-reviewed case reports and case series documenting patients with primary adrenal pheochromocytoma (PCC) and metastatic lesions to the central nervous system or related bony structures (cranium or spine), who granted consent for publication to the authors of included studies. Metastatic disease requires confirmation via radiological (e.g., CT, MRI) or histopathological evidence. The exclusion criteria included studies with insufficient data (n = 10), studies unrelated to the specified anatomical regions, and studies with irretrievable full texts (n = 23). A two-stage screening process was conducted by four authors (OC, JG, DP, and BJ): initial title and abstract screening, followed by full-text analysis. Discrepancies during screening were resolved through consensus discussion or adjudication by senior authors (JF, MM, and TS). Manual screening was used exclusively, without artificial intelligence or automation tools, to ensure rigorous human oversight during the study selection.
Statistical analysis
Fifty-three peer-reviewed studies, comprising 78 individual cases of metastatic adrenal PCC (18 cranial, 60 spinal), were included in this retrospective analysis. Extracted clinical data included patient factors (age at diagnosis, sex, genetic predisposition, von Hippel-Lindau disease, tumor laterality, number and location of metastases, admission symptoms, urinary metanephrine/normetanephrine levels, and immunohistochemical markers) and treatment factors (treatment method, follow-up duration, and outcomes). Descriptive statistics were calculated as follows: continuous data (e.g., age and number of metastases) were reported as means, standard deviations (SD), medians, and ranges; categorical data (e.g., genetic causes, imaging modalities) were expressed as proportions. Missing data (“not specified” [NS]) were noted as percentages where applicable. Data were tabulated and synthesized descriptively, providing extractable insights into the clinical course of the patients. No artificial intelligence methods were employed in the data extraction or analysis; all graphical presentations, including histograms, bar charts, and pie charts, were generated using Python with the matplotlib and seaborn libraries to ensure a clear and accurate representation of the findings.
Results
Cranial metastases
Eighteen studies (1948–2022) reported 18 cases (10 males [55.6%] and 8 females [44.4%]) (Table 1). The mean age at PCC diagnosis was 35.0 years (SD 14.4, median 34, range 10–53); metastases occurred at 46.6 years (SD 14.1, median 48, range 23–73) (Figure 3), with an 11.6-year latency. Genetic predisposition was 16.7% (n=3, no VHL). The tumors were unilateral (72.2%, n=13), bilateral (5.6%, n=1), or unspecified (22.2%, n=4) (Figure 4). The metastases were solitary (n=10), dual (n=1), numerous (n=5), or unspecified (n=2). Symptoms included headache (44.4%, n=8), hypertension (27.8%, n=5), and sweating (5.6%, n=1). Treatments included resection (72.2%, n=13), radiotherapy (38.9%, n=7, including one MIBG, one stereotactic), or chemotherapy (11.1%, n=2); one case lacked data. Follow-up (12/18 cases, 33.3% missing) revealed 7 deaths, 1 relapse, and 4 no relapses. Immunohistochemistry revealed Chromogranin A (n=10), synaptophysin (n=7), and neuron-specific enolase (n=5).
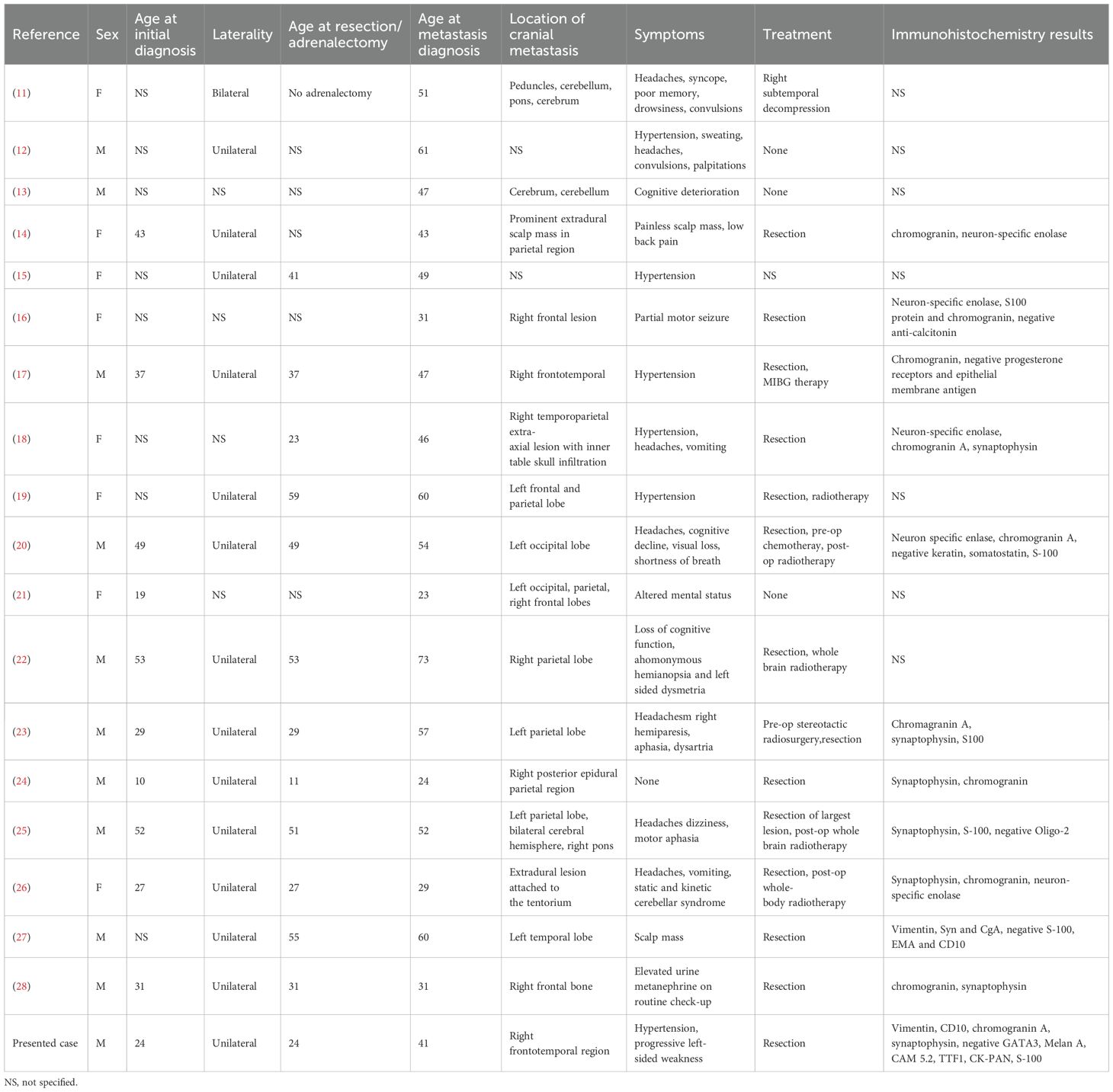
Table 1. summarizes the data from 18 studies (1948–2022, 18 patients) on cranial pheochromocytoma metastases, including demographics, symptoms, imaging, treatment, immunohistochemistry, and outcomes.
Spinal metastases
Thirty-five studies (1944–2024) reported 60 cases (Table 2). The mean age at PCC diagnosis was 40.5 years (SD, 15.4; median, 42; range, 11–68 years), and metastases occurred at 44.5 years (SD 16.0, median 44, range 9–74) (Figure 3), with a 4-year latency. Adrenalectomy occurred in 36.7% (n=22, mean age 42.1 years, SD 18.3), 13.3% (n=8) had none, and 50% (n=30) were unspecified. Genetics included an 8.3% predisposition (n=5, VHL 1.7% [n=1], NF1 3.3% [n=2]). The laterality was unspecified (63.3%, n=38), unilateral (31.7%, n=19), or bilateral (5%, n=3). The metastases were multiple (53.3%, n=32) or mean 1.7 lesions (SD 1.5, median 1, range 1–7). A total of 72 spinal metastases were recorded, nearly half of which were located in the thoracic spine (47.2%, n=34). No strong linear correlation was found between the co-occurrence of multiple tumors (Supplementary Correlation Matrix 1). The symptoms included hypertension (51.7%, n=31), headaches (13.3%, n=8), tachycardia (10%, n=6), and sweating/tremors (3.3%, n=2 each) (Figure 4). Imaging included non-contrast CT (41.7%, n=25), contrast CT (6.7%, n=4), non-contrast MRI (31.7%, n=19), contrast MRI (16.7%, n=10), and PET (21.7%, n=13), for a total of 71 modalities.

Table 2. details the data from 35 studies (1977–2024, 60 patients) on spinal pheochromocytoma metastases, including patient characteristics, symptoms, imaging findings, metastasis details, treatments, and outcomes.
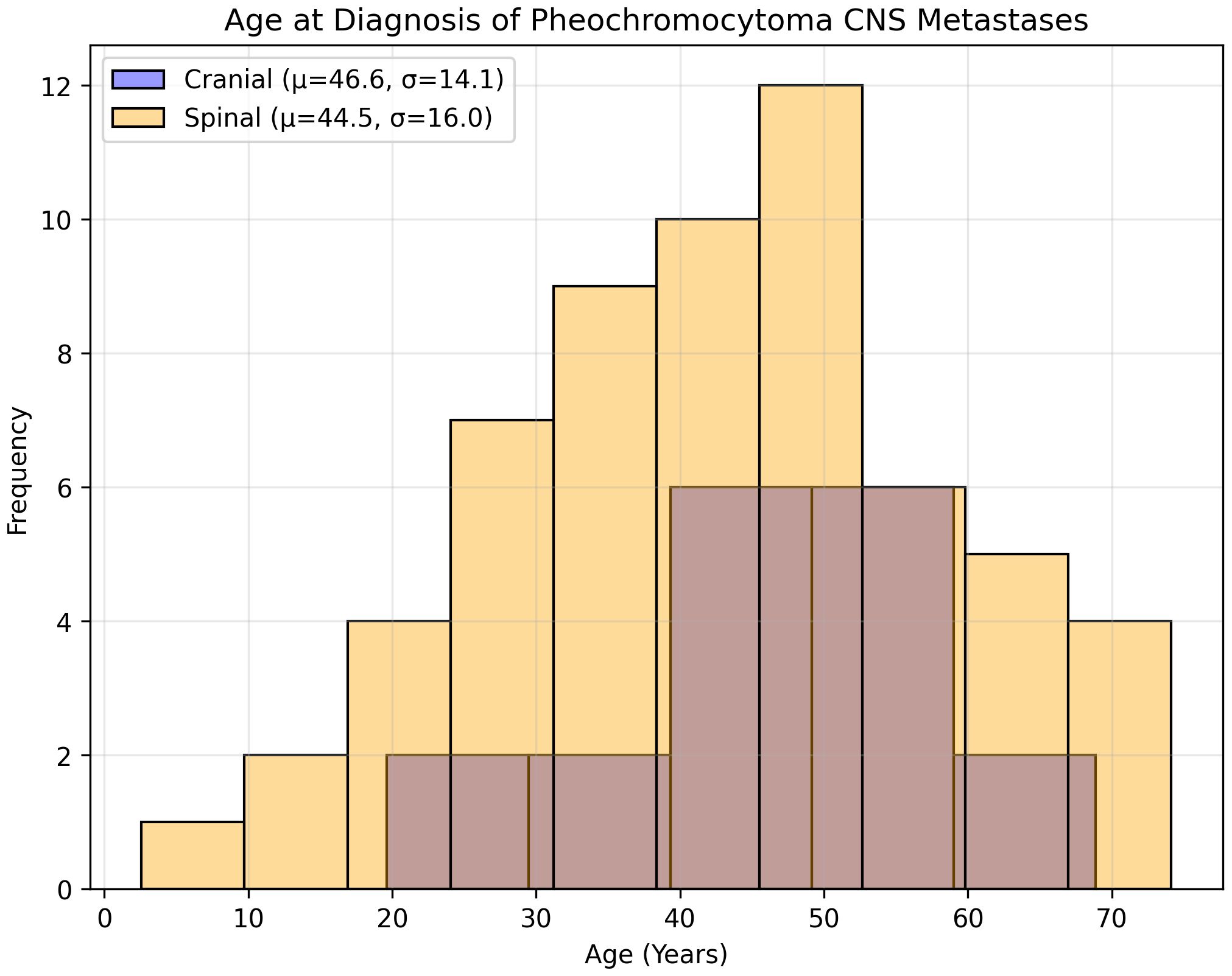
Figure 3. Age at diagnosis of pheochromocytoma CNS metastasis.
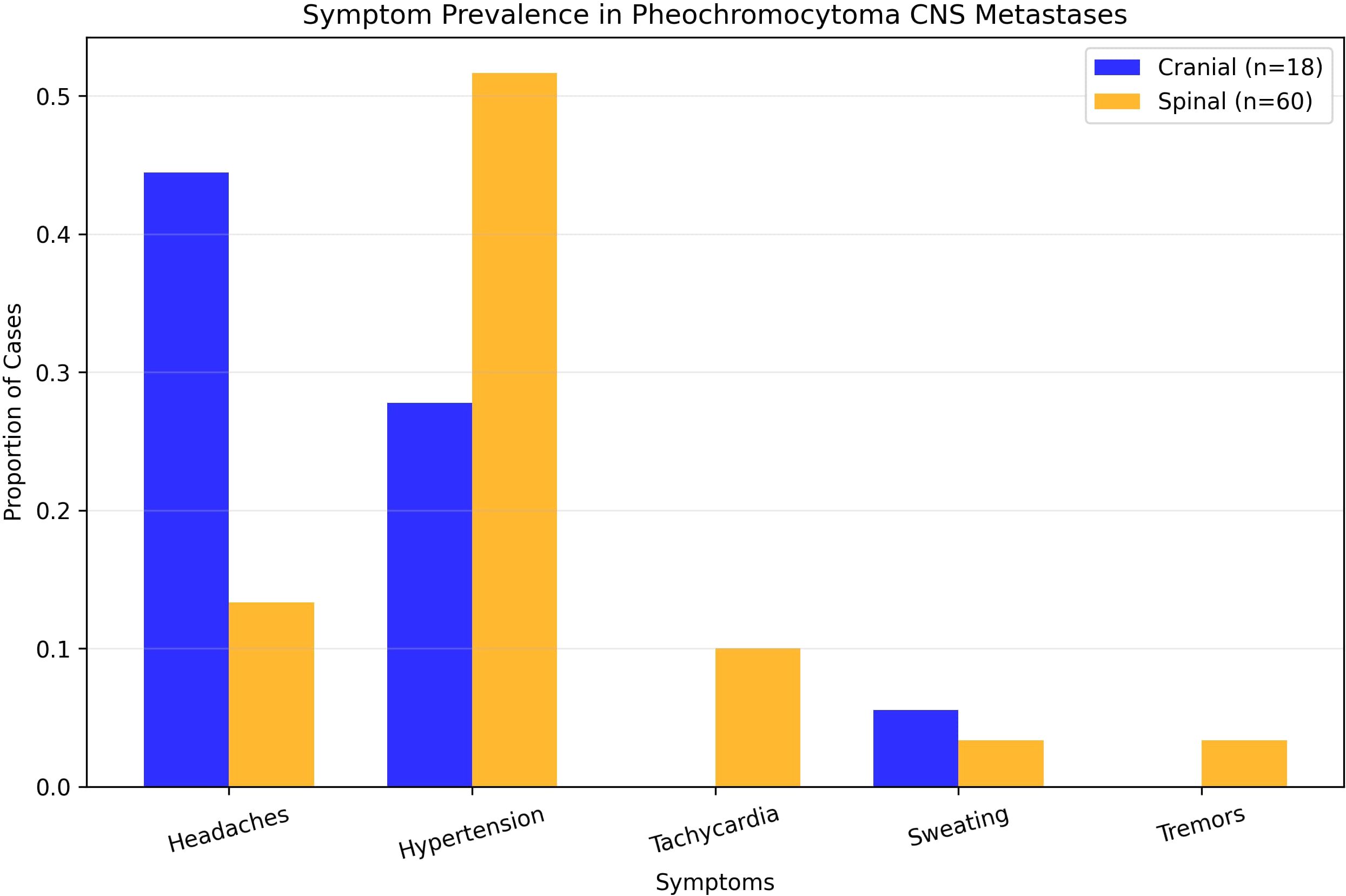
Figure 4. Prevalence of symptoms in pheochromocytoma CNS metastases.
Discussion
Pheochromocytoma metastases to the brain and spine are exceptionally rare, representing uncommon destinations for the malignant spread of NETs. This systematic review encompassed 35 studies from 1977 to 2024 (60 spinal cases) and 18 studies from 1948 to 2022 (18 cranial cases), along with one case report, consolidating sparse data to highlight distinct clinical, diagnostic, and therapeutic challenges of CNS involvement in malignant (mPCC) (1). Unlike more frequent metastatic sites, such as the liver or non-spinal skeleton, brain and spinal metastases pose unique complexities due to their potential to impair neurological function, disrupt spinal stability, or both (1). These challenges are exemplified by the case of a 41-year-old male with right frontoparietal brain metastasis causing progressive left-sided weakness. This case underscores the need for heightened diagnostic vigilance and tailored management strategies to address this rare metastatic site (1).
Clinical presentation and symptom variability
The clinical presentation of mPCC metastases varies significantly by site, reflecting the local anatomical and physiological impacts. Cranial metastases, as seen in our case and reported in 18 patients, predominantly manifest with neurological symptoms such as headaches (44.4%, n=8) and focal deficits (e.g., described case’s weakness), with hypertension present in only 27.8% (n=5) and no reports of tachycardia or tremors. This neurological predominance, often associated with solitary lesions (55.6%, n=10), contrasts with the systemic catecholamine-driven symptoms typical of classical PCC. Spinal metastases, analyzed in 60 patients, more frequently present with hypertension (51.7%, n=31), headaches (13.3%, n=8), and tachycardia (10%, n=6), consistent with catecholamine excess (10). This dichotomy—neurological deficits in cranial cases versus structural and pain-related issues in spinal cases—emphasizes the need for clinicians to consider mPCC in PCC patients presenting with neurological symptoms or intractable back pain, even in the absence of classical features (10).
Diagnostic challenges and imaging evolution
Diagnosing CNS mPCC metastases is challenging because of their rarity, nonspecific symptoms, and evolution of imaging modalities (5). Historically, spinal cases have often utilized non-contrast CT (41.7%, n=25) for its accessibility, despite its limited soft tissue resolution, compared to contrast-enhanced MRI, which accurately delineates epidural spinal cord compression and instability, guiding surgical planning (5). MRI (16.7%, n=10) and PET (21.7%, n=13) were underutilized in spinal cases but offered superior detailed and metabolic insights, as recommended by the current guidelines (60). For cranial metastases, MRI was critical in our case, although the review’s imaging data were incomplete, variably mentioning MRI and CT findings. The prolonged latency between initial PCC diagnosis and metastasis—11.6 years for cranial (mean age 46.6 years, SD 14.1) and 4 years for spinal (mean age 44.5 years, SD 16)—is reflected in the 17-year interval post-adrenalectomy (5). These extended intervals underscore the importance of long-term surveillance, prioritizing MRI for suspected CNS metastases to assess neurological and structural involvement, supplemented by CT for spinal bony anatomy, and PET for systemic staging (60). The historical reliance on CT and underutilization of advanced imaging highlights the diagnostic gap that modern protocols must address (9).
Surgical management and outcomes
Surgical management remains the cornerstone of both cranial and spinal mPCC and is tailored to address site-specific deficits (6). The neuronavigation-assisted frontoparietal tumor resection with cranioplasty preserved neurological function, aligning with the review’s high surgical prevalence for cranial metastases (72.2%, n=13), often supplemented by radiotherapy (38.9%, n=7) or chemotherapy (11.1%, n=2) (6). The focal nature of spinal lesions (mean 1.7, SD 1.5) suggests amenability to targeted intervention, but our case’s prior recurrences emphasize the need for meticulous preoperative optimization to mitigate catecholamine surges, which is a critical consideration in CNS mPCC surgeries (6). Systemic therapies, such as 177Lu-DOTATATE in the described Case, and radiotherapy show promise for unresectable or recurrent disease, warranting further exploration (6).
Histopathological insights and genetic context
Histopathologically, CNS metastatic pheochromocytoma (mPCC) consistently exhibits neuroendocrine marker expression. Described Case’s cranial lesion (Chromogranin A++, Synaptophysin++, Vimentin++) aligns with the systematic review’s findings, which reported Chromogranin A in 10 cranial cases and Synaptophysin in 7, confirming diagnostic consistency (2). However, the lack of comparative proliferation data, such as Ki67 levels, limits its prognostic utility. Genetically, hereditary syndromes are rare in cranial cases, with only 16.7% (n=3) showing predisposition and none linked to von Hippel-Lindau syndrome (2). Described in this study Case exhibited no genetic predisposition, consistent with most cases. The absence of SDHB mutation testing, a known malignancy risk factor, reflects a common gap in the historical data, restricting risk stratification (2, 3). Routine genetic profiling, particularly for SDHB, could identify patients at a higher risk of CNS metastases, thereby enhancing surveillance and management (3).
Limitations and future directions
The rarity of cranial metastatic pheochromocytoma (mPCC) poses significant limitations, with a small sample size (18 cranial cases) and extended study periods (1944–2024), leading to incomplete reporting, including 33% of missing follow-up data (61). The historical preference for non-contrast CT over MRI or PET reduces diagnostic precision, whereas inconsistent genetic testing (e.g., SDHB) limits comprehensive risk assessment (9). These gaps underscore the need for prospective studies to standardize diagnostic and therapeutic protocols (61). Interdisciplinary collaboration—integrating neurosurgery, endocrinology, oncology, radiotherapy, and potentially plastic surgery for soft tissue reconstruction after spinal procedures—could optimize functional outcomes and quality of life (61). International registries and collaborative research are essential to address these uncommon metastatic sites and improve the detection, management, and survival in this rare malignancy.
Conclusions
This systematic review and case report highlights the rarity of pheochromocytoma metastases to the brain and spine, emphasizing distinct clinical presentations (neurological for cranial and pain-driven for spinal), the critical role of MRI and PET in diagnosis, and the necessity of tailored surgical interventions. Despite diagnostic and data limitations, the findings advocate for long-term surveillance, advanced imaging, and multidisciplinary management to optimize outcomes in this challenging malignancy. Prospective studies and international registries are needed to standardize care and improve prognoses.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
JFe: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing – original draft, Writing – review & editing. OC: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. JC-G: Data curation, Writing – review & editing. DP: Data curation, Writing – review & editing. BJ: Data curation, Writing – review & editing. KK: Writing – review & editing. MM: Project administration, Writing – review & editing. MM-G: Writing – original draft, Writing – review & editing. JS: Writing – review & editing. AS: Writing – review & editing. TS: Supervision, Writing – review & editing. MS: Supervision, Writing – review & editing. JFu: Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fendo.2025.1694463.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2025.1633411/full#supplementary-material.
References
1. Lenders JWM, Eisenhofer G, Mannelli M, and Pacak K. Phaeochromocytoma. Lancet. (2005) 366:665–75. doi: 10.1016/S0140-6736(05)67139-5, PMID: 16112304
2. Amar L, Bertherat J, Baudin E, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. (2005) 23:8812–8. doi: 10.1200/JCO.2005.03.1484, PMID: 16314641
3. Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. (2017) 31:181–93. doi: 10.1016/j.ccell.2017.01.001, PMID: 28162975
4. Plouin P-F, Chatellier G, Fofol I, and Corvol P. Tumor recurrence and hypertension persistence after successful pheochromocytoma operation. Hypertension. (1997) 29:1133–9. doi: 10.1161/01.HYP.29.5.1133, PMID: 9149678
5. Kaloostian PE, Zadnik PL, Kim JE, Groves ML, Wolinsky JP, Gokaslan ZL, et al. High incidence of morbidity following resection of metastatic pheochromocytoma in the spine: Report of 5 cases. J Neurosurg Spine. (2014) 20:726–33. doi: 10.3171/2014.3.SPINE13535, PMID: 24725182
6. Singh A, Santangelo G, Ellens N, Kohli G, Pranaat R, and Bender MT. Preoperative embolization and en bloc resection of a metastatic pheochromocytoma of the cervical spine. J Cerebrovasc Endovasc Neurosurg. (2024) 26:331–7. doi: 10.7461/jcen.2024.e2023.04.005, PMID: 38897596
7. Schürfeld R, Pamporaki C, Peitzsch M, Fliedner A, Timmers HJLM, Lenders JWM, et al. False-positive results for pheochromocytoma associated with norepinephrine reuptake blockade. Endocr Relat Cancer. (2023) 31. doi: 10.1530/ERC-23-0063, PMID: 37955319
8. Kline GA, Boyd J, Leung AA, Tang A, and Sadrzadeh HM. Very high rate of false positive biochemical results when screening for pheochromocytoma in a large, undifferentiated population with variable indications for testing. Clin Biochem. (2020) 77:26–31. doi: 10.1016/j.clinbiochem.2020.01.005, PMID: 31978379
9. Taïeb D, Hicks RJ, Hindié E, Guillet BA, Avram A, Gheduzzi D, et al. European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. (2019) 46:2112–37. doi: 10.1007/s00259-019-04398-1, PMID: 31254038
10. Greenberg MS. Greenberg’s handbook of neurosurgery. 10. Auflage. Stuttgart: Thieme Medical Publishers (2023).
11. Spatt SD and Ghayzel DM. PHEOCHROMOCYTOMA OF THE ADRENAL MEDULLA: A clinicopathological study of five cases. Am J Med Sci. (1948) 216:39–50. doi: 10.1097/00000441-194807000-00007, PMID: 18877314
12. Melicow MM. One hundred cases of pheochromocytoma (107 tumors) at the columbia-presbyterian medical center, 1926-1976.A clinicopathological analysis. Cancer. (1977) 40:1987–2004. doi: 10.1002/1097-0142(197711)40:5<1987::AID-CNCR2820400502>3.0.CO;2-R, PMID: 922654
13. Ferrari G, Togni R, Pizzedaz C, and Aldovini D. Cerebral metastases in pheochromocytoma. Pathologica. (1979) 71:703–10., PMID: 551390
15. Mornex R, Badet C, and Peyrin L. Malignant pheochromocytoma: A series of 14 cases observed between 1966 and 1990. J Endocrinol Invest. (1992) 15:643–9. doi: 10.1007/BF03345808, PMID: 1479147
16. Gentile S, Rainero I, Savi L, Rivoiro C, and Pinessi L. Brain metastasis from pheochromocytoma in a patient with multiple endocrine neoplasia type 2A. Panminerva Med. (2001) 43:305–6., PMID: 11677427
17. Liel Y, Zucker G, Lantsberg S, Argov S, and Zirkin HJ. Malignant pheochromocytoma simulating meningioma: coexistence of recurrent meningioma and metastatic pheochromocytoma in the base of the skull. Isr Med Assoc J. (2002) 4:829–31., PMID: 12389355
18. Mercuri S, Gazzeri R, Galarza M, Esposito S, and Giordano M. Primary meningeal pheochromocytoma: Case report. J Neurooncol. (2005) 73:169–72. doi: 10.1007/s11060-004-4595-5, PMID: 15981108
19. Kowalska A and Lizis-Kolus K. Malignant pheochromocytoma with brain metastases and coexisting meningioma: case report. In: P316. Endocrine abstracts (2009). (Bioscientifica). Available online at: https://www.endocrine-abstracts.org/ea/0020/ea0020p316.
20. Schaefer I-M, Martinez R, Enders C, et al. Molecular cytogenetics of Malignant pheochromocytoma with cerebral metastasis. Cancer Genet Cytogenet. (2010) 200:194–7. doi: 10.1016/j.cancergencyto.2010.04.013, PMID: 20620607
21. Kharlip J, Teytelboym O, Kleinberg L, Purtell M, and Basaria S. Successful radiation therapy for cerebral metastatic lesions from a pheochromocytoma. Endocr Pract. (2010) 16:334–6. doi: 10.4158/ep.16.2.334, PMID: 20361415
22. Schulte DM, Faust M, Schmidt M, et al. Novel therapeutic approaches to CNS metastases in Malignant phaeochromocytomas – case report of the first patient with a large cystic CNS lesion. Clin Endocrinol (Oxf). (2012) 77:332–4. doi: 10.1111/j.1365-2265.2012.04354.x, PMID: 22288775
23. Miyahara M, Okamoto K, Noda R, Inoue M, Tanabe A, and Hara T. Cerebral metastasis of Malignant pheochromocytoma 28 years after of disease onset. Interdiscip Neurosurg. (2017) 10:130–4. doi: 10.1016/j.inat.2017.08.003
24. Boettcher LB, Abou-Al-Shaar H, Ravindra VM, Horn J, Palmer CA, and Menacho ST. Intracranial epidural metastases of adrenal pheochromocytoma: A rare entity. World Neurosurg. (2018) 114:235–40. doi: 10.1016/j.wneu.2018.03.126, PMID: 29588244
25. Cho YS, Ryu HJ, Kim SH, and Kang S-G. Pheochromocytoma with brain metastasis: A extremely rare case in worldwide. Brain Tumor Res Treat. (2018) 6:101. doi: 10.14791/btrt.2018.6.e18, PMID: 30381926
26. Kammoun B, Belmabrouk H, Kolsi F, Mnejja M, Gdoura Y, Hammami B, et al. Brain metastasis of pheochromocytoma: diagnostic and therapeutic challenge. World Neurosurg. (2019) 130:391–9. doi: 10.1016/j.wneu.2019.06.163, PMID: 31260853
27. Chen JC, Zhuang DZ, Luo C, and Chen WQ. Malignant pheochromocytoma with cerebral and skull metastasis: A case report and literature review. World J Clin cases. (2021) 9:2791–800. doi: 10.12998/wjcc.v9.i12.2791, PMID: 33969061
28. Hernandez MALU and Khu KJO. Calvarial metastasis from Malignant pheochromocytoma associated with multiple endocrine neoplasia. Surg Neurol Int. (2022) 13:444. doi: 10.25259/SNI_644_2022, PMID: 36324941
29. Ein SH, Weitzman S, Thorner P, Seagram CG, Filler RM, and Calgary A. Pediatric Malignant pheochromocytoma. J Pediatric Surg. (1994) 29:1197–201. doi: 10.1016/0022-3468(94)90799-4, PMID: 7807344
30. Miyamori I, Yamamoto I, Nakabayashi E, Takeda R, Okada Y, and Kitacawa M. Malignant pheochromocytoma with features suggesting the brown-sequard syndrome A Case Report. Cancer. (1977) 40:402–5., PMID: 880568
31. Dow CJ, Palmer MK, O’sullivan JP, et al. Malignant phaeochromocytoma: report of a case and a critical review. Br J Surg. (1982) 69:338–40. doi: 10.1002/bjs.1800690616, PMID: 7082961
32. Chiras J, Cognard C, Rose M, et al. Percutaneous injection of an alcoholic embolizing emulsion as an alternative preoperative embolization for spine tumor. AJNR. (1993) 14:1113–7., PMID: 8237690
33. Teno S, Tanabe A, Nomura K, and Demura H. Acutely exacerbated hypertension and increased inflammatory signs due to radiation treatment for metastatic pheochromocytoma. Endocr J. (1996) 43:511–6. doi: 10.1507/endocrj.43.511, PMID: 8980890
34. Mössner R and Keidel M. Malignant pheochromocytoma with progressive paraparesis in von Hippel–Lindau disease. Eur J Neurol. (2000) 7:439–42. doi: 10.1046/j.1468-1331.2000.00093.x, PMID: 10971605
35. Mori S, Okura T, Kitami Y, et al. A case of metastatic extra-adrenal pheochromocytoma 12 years after surgery case report first admission. Hypertens Res. (2002) 25(1):141–4. doi: 10.1291/hypres.25.141, PMID: 11924720
36. Yurt A, Nuri Arda M, and Vardar E. Metastatic pheochromocytoma of the thoracic spinal extradural space case report and review of the literature. Kobe J Med Sci. (2005) 51:49–53., PMID: 16444096
37. Kheir E, Pal D, Mohanlal P, Shivane A, Chakrabarty A, and Timothy J. Cervical spine metastasis from adrenal pheochromocytoma. Acta Neurochir (Wien). (2006) 148:1219–20. doi: 10.1007/s00701-006-0892-4, PMID: 16990988
38. Kasliwal MK, Sharma MS, Vaishya S, and Sharma BS. Metachronous pheochromocytoma metastasis to the upper dorsal spine-6-year survival. Spine J. (2008) 8:845–8. doi: 10.1016/j.spinee.2007.06.004, PMID: 18024223
39. Shinoto M, Hasuo K, Aibe H, et al. Percutaneous osteoplasty for hypervascular bone metastasis. Radiat Med - Med Imaging Radiat Oncol. (2008) 26:603–8. doi: 10.1007/s11604-008-0277-0, PMID: 19132491
40. Scalfani MT, Arnold PM, and Anderson KK. Metastatic adrenal pheochromocytoma to the thoracic spine Feocromocitoma adrenal metastático para a coluna torácica Feocromocitoma adrenal metastásico para la columna torácica. COLUNA/COLUMNA. (2010) 9(3):343–6. doi: 10.1590/S1808-18512010000300017
41. Rittirsch D, Battegay E, Zimmerli LU, et al. Cement-augmented dorsal instrumentation of the spine as a safe adjunct to the multimodal management of metastatic pheochromocytoma: A case report. Patient Saf Surg. (2012) 6. doi: 10.1186/1754-9493-6-1, PMID: 22222147
42. Kaloostian PE, Zadnik PL, Awad AJ, McCarthy E, Wolinsky JP, and Sciubba DM. En bloc resection of a pheochromocytoma metastatic to the spine for local tumor control and for treatment of chronic catecholamine release and related hypertension. J Neurosurg Spine. (2013) 18:611–6. doi: 10.3171/2013.3.SPINE12966, PMID: 23600583
43. Kapur. A case of Malignant metastatic pheochromocytoma after eight years of primary diagnosis. World J Oncol. (2014) 5(1):33–40. doi: 10.14740/wjon760w, PMID: 29147374
44. Chopko BW. Endovascular treatment of vertebral column metastases using intra-arterial cisplatin: Pilot experience. Case Rep Med. (2014) 2014. doi: 10.1155/2014/915904, PMID: 24963303
45. Asad S, Peters-Willke J, and Nott L. Malignant paraganglioma, a rare presentation with foot drop: a case report. J Spine Surg. (2015) 1:99–102. doi: 10.3978/j.issn.2414-469X.2015.10.01, PMID: 27683685
46. Liu S, Song A, Zhou X, Kong X, Li WA, Wang Y, et al. Malignant pheochromocytoma with multiple vertebral metastases causing acute incomplete paralysis during pregnancy: Literature review with one case report. Medicine. (2017) 96(44):e8535. doi: 10.1097/MD.0000000000008535, PMID: 29095319
47. Liu S, Zhou X, Song A, Li WA, Rastogi R, Wang Y, et al. Successful treatment of malignant pheochromocytoma with sacrum metastases: A case report. Medicine. (2018) 97(35):e12184. doi: 10.1097/MD.0000000000012184, PMID: 30170467
48. Fadiga L, Saraiva J, Paiva I, and Carrilho F. Thoracic spine metastasis presenting 18 years after complete resection of a phaeochromocytoma. BMJ Case Rep. (2019) 12. doi: 10.1136/bcr-2019-229621, PMID: 31439569
49. Liu S, Zhou X, Huo Z, Yao S, Wang Y, and Liu Y. Clinical features and prognosis analysis of metastatic spinal pheochromocytoma: A single center retrospective study. J Bone Oncol. (2020) 24. doi: 10.1016/j.jbo.2020.100312, PMID: 32793409
50. Jester G, Hassanein H, and El-Far A. Late diagnosis of metastatic pheochromocytoma in multiple endocrine neoplasia 2B with rapid clinical decline. BMJ Case Rep. (2021) 14. doi: 10.1136/bcr-2020-240488, PMID: 33541961
51. Colton MD, Tompkins K, O’donnell E, et al. Case of metastatic pheochromocytoma and meningiomas in a patient with lynch syndrome. JCO Precis Oncol. (2022) 6:e2100251. doi: 10.1200/PO.21, PMID: 35025617
52. Kurosawa S, Yamasaki H, Hasegawa W, and Mori T. Major intraoperative bleeding and drastic change in circulatory dynamics in a pregnant patient with metastatic pheochromocytoma: a case report. JA Clin Rep. (2022) 8. doi: 10.1186/s40981-022-00504-9, PMID: 35192091
53. Suresh S, Hrishi AP, Divakar G, and Sethuraman M. At the eye of the hurricane! Perioperative management of an unoptimized metastatic pheochromocytoma presenting for emergency neurosurgery. J Neurosci Rural Pract. (2022) 13:563–7. doi: 10.1055/s-0042-1749457, PMID: 35946015
54. Zeitler C, Fuderer L, Schmitz K, Arora R, and Dammerer D. Pheochromocytoma turned Malignant during pregnancy in a patient with neurofibromatosis type I - A case report and systematic review of the current literature. Anticancer Res. (2022) 42:4647–56. doi: 10.21873/anticanres.15969, PMID: 36039448
55. Du S, Hu P, Yang S, et al. Surgical treatment of spinal metastatic pheochromocytoma and paraganglioma: A single institutional cohort of 18 patients. Global Spine J. (2023) 13:2454–62. doi: 10.1177/21925682221087600, PMID: 35341356
56. Zhang Y, Li H, Wang W, Shan L, and Hao D. A novel technology for 3D-printing artificial vertebral bodies for treating lumbar spine adrenal pheochromocytoma metastases: A case report and review of the literature. Orthop Surg. (2023) 15:3335–41. doi: 10.1111/os.13899, PMID: 37771116
57. Ni X, Wang J, Cao J, Zhou Y, Zhang H, Zhang Z, et al. Surgical management and outcomes of spinal metastasis of Malignant adrenal tumor: A retrospective study of six cases and literature review. Front Oncol. (2023) 13:1110045. doi: 10.3389/fonc.2023.1110045, PMID: 36776311
58. Yunasan E, Ning X, Shaik MR, and Pennant M. Recurrent pheochromocytoma with bone metastasis eight years after bilateral adrenalectomies in a patient with neurofibromatosis type 1. AACE Clin Case Rep. (2024) 10:93–6. doi: 10.1016/j.aace.2024.02.006, PMID: 38799052
59. Segawa K, Yamamoto Y, Kato T, et al. A case of Malignant pheochromocytoma with neurofibromatosis type 1 having difficulty in differentiating spinal tumor. IJU Case Rep. (2024) 7:336–40. doi: 10.1002/iju5.12751, PMID: 38966763
60. Eisenhofer G, Bornstein SR, Brouwers FM, et al. Malignant pheochromocytoma: current status and initiatives for future progress. Endocr Relat Cancer. (2004) 11:423–36. doi: 10.1677/erc.1.00829, PMID: 15369446
61. Amar L, Lussey-Lepoutre C, Lenders JWM, Djadi-Prat J, Plouin P-F, and Steichen O. MANAGEMENT OF ENDOCRINE DISEASE: Recurrence or new tumors after complete resection of pheochromocytomas and paragangliomas: a systematic review and meta-analysis. Eur J Endocrinol. (2016) 175:R135–145. doi: 10.1530/EJE-16-0189, PMID: 27080352
Keywords: pheochromocytoma, malignant pheochromocytoma, spinal metastases, cerebral metastases, central nervous system
Citation: Fercho JM, Chasles OG, Chamier-Gliszczyński J, Płaza DR, Jabłoński B, Kokot K, Mielczarek M, Małłek-Grabowska M, Szypenbejl J, Szkudlarek A, Szmuda T, Siemiński M and Furtak J (2025) Pheochromocytoma metastasis to the central nervous system: a case report and systematic review. Front. Endocrinol. 16:1633411. doi: 10.3389/fendo.2025.1633411
Received: 22 May 2025; Accepted: 28 July 2025;
Published: 25 August 2025; Corrected: 10 September 2025.
Edited by:
Sergei Tevosian, University of Florida, United StatesReviewed by:
Vitaly Kantorovich, Hartford HealthCare, United StatesFederico Polverino, University of Naples Federico II, Italy
Copyright © 2025 Fercho, Chasles, Chamier-Gliszczyński, Płaza, Jabłoński, Kokot, Mielczarek, Małłek-Grabowska, Szypenbejl, Szkudlarek, Szmuda, Siemiński and Furtak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Justyna Małgorzata Fercho, amZlcmNob0BndW1lZC5lZHUucGw=
†These authors share first authorship