 A. T. M. Emdadul Haque1
A. T. M. Emdadul Haque1 Mohammed S. Razzaque2*
Mohammed S. Razzaque2*- 1Preclinical Department, Universiti Kuala Lumpur Royal College of Medicine Perak (UniKL RCMP), Ipoh, Perak, Malaysia
- 2Department of Medical Education, School of Medicine, University of Texas Rio Grande Valley (UTRGV), Edinburg, TX, United States
Introduction
We are currently in an exciting time to advance the field of renal endocrinology. There has never been a more favorable time to study the molecular and cellular mechanisms underlying renal hormone functions in physiology, pharmacology, and pathophysiology. The availability of advanced research tools provides unique opportunities to dissect the complex regulatory roles of renal hormones. The kidneys are vital organs in the human body; each one is composed of approximately one million functional nephrons. Understanding the precise roles and regulation of kidney-derived hormones and hormones that exert endocrine effects on the kidneys will help in preventing or delaying various renal and systemic diseases, including hypertensive complications. The primary functions of the kidneys include filtration of blood, maintenance of fluid, electrolyte and acid–base balances, controlling blood pressure, and facilitating erythropoiesis by generating erythropoietin. In addition to erythropoietin, kidneys also produce renin and help generate functional vitamin D by converting 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D (1, 2). We briefly discuss the extrarenal functions of these kidney-derived factors below, with an emphasis on the clinical and functional aspects that remain to be elucidated.
Erythropoietin
Under normal physiological conditions, erythropoietin synthesis is tightly regulated; however, when oxygen levels are low, hypoxia-inducible factor (HIF) promotes the expression, synthesis and release of erythropoietin from the kidney (3), which then binds to erythropoietin receptors on erythroid progenitor cells to enhance their survival, proliferation, and differentiation (4). The molecular feedback mechanism that fine-tunes erythropoietin synthesis and subsequent signaling has not yet been clearly defined. Recombinant human erythropoietin is commonly used to treat renal anemia in patients with chronic kidney disease (CKD), cancer, or those undergoing chemotherapy (5, 6). Of therapeutic importance, exogenous erythropoietin and other erythropoiesis-stimulating agents have been linked to an increased risk of adverse cardiovascular events, including hypertension, stroke, and heart failure. Overcorrection of hemoglobin levels may cause hyperviscosity with increased thrombotic risk and endothelial dysfunction (7, 8). Additionally, the distribution of erythropoietin receptors in cells outside the hematopoietic system has been identified, and their physiological significance remains to be determined. For example, the exogenous use of erythropoietin has the potential to exert proliferative effects on tumor cells, as the expression of erythropoietin receptors is detected on tumor cells (usually at low levels) (9, 10). Careful monitoring is necessary when recombinant erythropoietin is used in tumor patients.
Renin
Renin is another important kidney-derived hormone and is secreted by juxtaglomerular cells in response to hypotension (11). It is an essential component of the renin–angiotensin–aldosterone system, which regulates blood pressure, fluid, and electrolyte balance. The secretion of renin is tightly controlled by baroreceptors, macula densa cells, and sympathetic nerve endings (12). Dysregulation of the renin–angiotensin–aldosterone system, especially excess renin or angiotensin II activity, has been implicated in the development of hypertension, heart failure, and CKD (13). Since the (pro)renin receptor binds both renin and prorenin independent of angiotensin II, the in vivo physiological role of the (pro)renin receptor beyond angiotensin generation is unclear. Importantly, renin primarily acts systemically through angiotensinogen cleavage in the circulation; however, the biological significance of tissue-specific renin–angiotensin systems (e.g., the brain, heart, adipose, and immune systems) is not yet clearly understood. It has been observed that tissue-specific renin-angiotensin systems can exert protective effects by activating an anti-inflammatory regulatory pathway, which is mediated through angiotensin II type 2 receptor (AT2R) signaling (14). Additionally, the underlying mechanisms of low-renin-mediated essential hypertension need further mechanistic clarification.
Calcitriol
Normal kidney structure and function are essential for the production of calcitriol (1,25-dihydroxyvitamin D) via renal 1α-hydroxylase activity. Vitamin D is a pleiotropic hormone that has both skeletal and extraskeletal functions. Vitamin D enables the intestinal absorption of calcium and phosphate to support the mineralization process of the bone and helps maintain the homeostatic balance of serum ionized calcium and phosphate. Vitamin D is also believed to exert immunomodulatory effects on both the innate and adaptive immune systems (15). The replacement of exogenous vitamin D is a common clinical practice in patients with CKD and skeletal disorders. Traditionally, calcitriol has been used to manage secondary hyperparathyroidism in patients with CKD. However, concerns about its association with cardiovascular calcification, along with the availability of selective vitamin D receptor activators (e.g., paricalcitol) and calcimimetic drugs, have introduced alternative treatment options. Additionally, the benefits of calcitriol for bone health, particularly osteoporosis and fracture prevention, are not conclusive, as some randomized trials have shown inconsistent effects on fracture risk and bone mineral density (BMD) (16–18). For updated information on vitamin D testing and dosing, readers are referred to a recent Endocrine Society Clinical Practice Guideline (19).
In addition to the aforementioned factors, prostaglandins (e.g., PGE2), kallikrein, and urodilatin also exert autocrine and paracrine effects within the kidney, influencing renal blood flow, sodium excretion (natriuresis), and urine formation (diuresis) (Figure 1) (20–22). Normally functioning kidneys are also important for the endocrine effects of hormones produced elsewhere but act on the kidneys. Below, we briefly highlight several key examples, with an emphasis on the unresolved clinical and biological challenges.
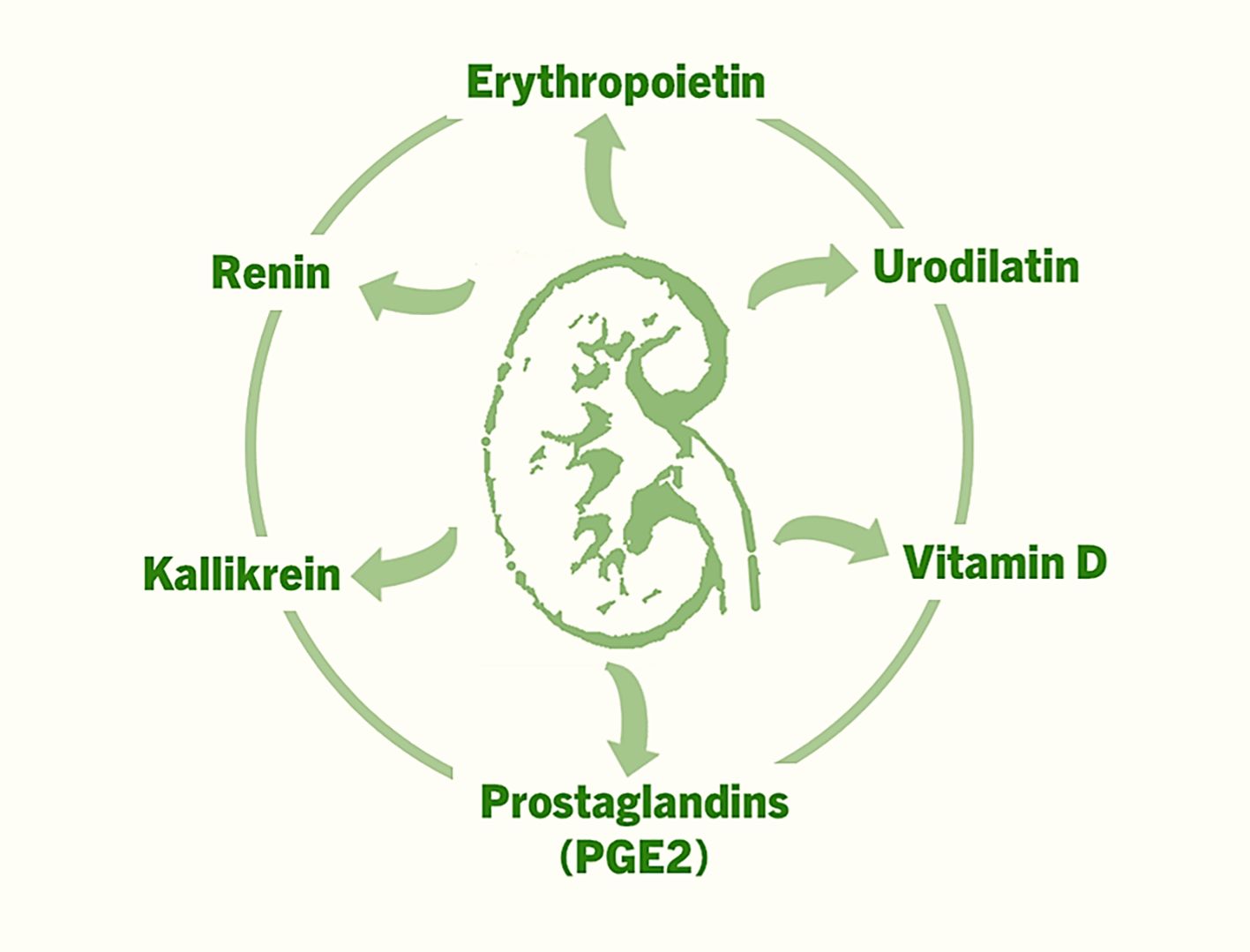
Figure 1. The factors that are produced in the kidneys, and exert autocrine, paracrine and endocrine effects to regulate various functions ranging from controlling blood pressure to facilitating erythropoiesis to musculoskeletal functions.
Antidiuretic hormone
ADH (vasopressin) is a hormone synthesized in the hypothalamus that is released from the posterior pituitary gland and acts on the kidneys (23). When plasma osmolality is increased or blood volume is reduced, ADH acts on the collecting ducts of the kidneys to increase water reabsorption, concentrates the urine, and reduces water loss to restore the fluid balance (24). ADH can induce vasoconstriction by interacting with V1 receptors on vascular smooth muscles to maintain blood pressure during hypovolemia (25). The different subtypes of ADH receptors (V1a, V1b, and V2) have distinct tissue distributions and signaling pathways (26, 27). The specific roles of the receptor subtypes in various physiological and pharmacological processes remain poorly understood and require further study to elucidate the delicate functions of the receptor subtypes. Furthermore, ADH and oxytocin are structurally similar peptide hormones, and their receptors have cross-reactivity, indicating that ADH can activate the receptors of oxytocin and vice versa (28). This receptor crosstalk further complicates the interpretation of the behavioral and pharmacological effects of both ADH and oxytocin. Diabetes insipidus (too little or resistance to ADH) and syndrome of inappropriate antidiuretic hormone secretion (SIADH; too much ADH) are the two most commonly encountered clinical conditions that are associated with dysregulation of ADH functions (29). In SIADH, excessive ADH leads to excessive water retention, causing hyponatremia, and in clinical practice, V2 receptor antagonists are used to reduce water reabsorption and promote free water excretion to correct hyponatremia (30). However, the long-term safety of V2 receptor antagonists and their efficacy need further evaluation.
Aldosterone
Aldosterone is produced by the adrenal gland and exerts its functionality by acting on the distal tubules and collecting ducts of the kidneys. Aldosterone plays a key role in the regulation of blood pressure, electrolyte balance, and fluid homeostasis by promoting sodium retention and potassium excretion in the kidneys (31). On the distal tubules and collecting ducts, aldosterone binds with mineralocorticoid receptors to induce the expression of sodium–potassium ATPase and epithelial sodium channels (ENaCs), increasing sodium reabsorption, potassium secretion, and water retention (32). Collectively, these water and ion redistributions help maintain blood volume and blood pressure. Although the renal effects of aldosterone via mineralocorticoid receptors are well documented, its effects on the cardiovascular and immune systems, as well as neuronal and adipose tissues, need further study, as these receptors are also present in nonrenal tissues, including heart, brain, and fat tissues (33). In addition to aldosterone, cortisol can also activate mineralocorticoid receptors in nonrenal tissues and may exert adverse effects on the heart under stressful conditions or during aging (34). The clinical use of mineralocorticoid receptor antagonists is expanding in conditions such as diabetic nephropathy, heart failure, primary aldosteronism, and liver cirrhosis with ascites. However, further studies are needed to explain why not all patients respond equally to these treatments. Additionally, the regulation of CYP11B2, the gene encoding aldosterone synthase (an enzyme that is necessary for the biosynthesis of aldosterone from precursors such as corticosterone), has yet to be clearly defined in certain clinical conditions, including primary aldosteronism or stress conditions (35, 36). Importantly, angiotensin II and elevated plasma potassium levels are major stimulators of aldosterone release, whereas atrial natriuretic peptide (ANP) inhibits aldosterone secretion (37).
Atrial natriuretic peptide
ANP is another hormone that is secreted by the atrial myocytes of the heart and affects the kidneys (distal nephrons). In response to hypertension, ANP is released and binds to its receptor-A (NPR-A) to induce vasodilation, an increased glomerular filtration rate (GFR), and sodium excretion to control blood pressure, sodium balance, and fluid volume (38, 39). Furthermore, ANP can suppress the activity of the renin–angiotensin–aldosterone system by reducing the secretion of renin and aldosterone and suppressing sympathetic nervous system activity (40). Recombinant ANP has shown natriuretic, vasodilatory, and neurohormonal-suppressing effects in clinical studies in Japan (41); patients with acute myocardial infarction who received ANP had a smaller infarct size, fewer reperfusion injuries, and better outcomes than controls did. The investigators concluded that ANP could be an adjunctive therapy for patients with acute myocardial infarction who receive percutaneous coronary intervention (41). However, some of the follow-up trials failed to demonstrate such protective benefits (42).
Parathyroid hormone
PTH plays an essential role in regulating calcium and phosphate homeostasis. PTH is secreted by the parathyroid glands in response to hypocalcemia, hyperphosphatemia and reduced levels of calcitriol (43). PTH acts on distal tubular epithelial cells to increase calcium reabsorption and thereby reduce urinary calcium loss to increase serum calcium levels. PTH inhibits sodium–phosphate cotransporters in proximal tubular epithelial cells to increase urinary phosphate excretion to lower serum phosphate levels. PTH also increases the activity of 1α-hydroxylase in proximal tubular epithelial cells to increase the production of calcitriol, which eventually increases the intestinal absorption of calcium and phosphate (44). In patients with CKD, persistent stimulation of PTH leads to secondary hyperparathyroidism. Although the Kidney Disease Improvement Global Outcomes (KDIGO) working group guidelines recommend maintaining certain levels of PTH in CKD patients, in clinical practice, maintaining such levels is not always easy (45). Additionally, from a biological standpoint, PTH binds to the PTH1 receptor (PTH1R) to exert its bioactivities, and what determines whether PTH1R signals via cAMP pathway or calcium pathway in different tissues is not yet clearly defined (46).
Calcitonin
Calcitonin is secreted by thyroid parafollicular (C) cells and acts on distal tubules of the kidneys to promote renal calcium excretion by reducing calcium reabsorption in the tubules. Additionally, some reports have shown that kidney cells express both calcitonin and its receptors to perform autocrine and paracrine functions. One of the major issues with calcitonin is its exact in vivo role. Importantly, calcitonin deficiency in humans (e.g., post-thyroidectomy) often results in no major disturbances in calcium homeostasis (47–49). Additionally, high serum calcium and gastrin can induce calcitonin secretion, but its feedback control is not yet clear. The presence of calcitonin receptors in tissues not directly involved in calcium regulation, including central nervous system cells and immune cells, requires further studies to dissect the biological role of these receptor complexes. Furthermore, calcitonin secretion decreases with age in both men and postmenopausal women, and whether such a decrease is associated with skeletal age-related phenotypes needs further clarification. Elevated calcitonin levels are detected in the medullary carcinoma of the thyroid gland (50). Clinically, in Paget’s disease of the bone, calcitonin is used to suppress osteoclastic activities and thereby reduce abnormal bone remodeling. The precise role and regulatory effect of calcitonin on renal calcium handling in CKD-related mineral and bone disorders are not yet well defined. Calcitonin, particularly salmon calcitonin, has been used in clinical practice for several conditions related to bone metabolism and calcium regulation. However, in recent days, its clinical use has been limited by the availability of alternative therapies (bisphosphonates or denosumab) with concerns about long-term safety (51).
Fibroblast growth factor 23
FGF23 is a relatively recently identified factor that is produced by bone cells and acts on kidney tubules to regulate renal phosphate uptake (Figure 2) (52–54). FGF23 reduces renal phosphate reabsorption by suppressing the activities of sodium–phosphate cotransporters (NaPi–IIa and NaPi–IIc) in the proximal tubules, increasing urinary phosphate excretion and resulting in lower levels of serum phosphate. FGF23 also suppresses the production of calcitriol (1,25-dihydroxyvitamin D) by suppressing the renal expression of 1α-hydroxylase (CYP27B1). Phosphate and calcitriol stimulate skeletal FGF23 production, which in turn suppresses renal 1α-hydroxylase expression and phosphate reabsorption, forming a feedback loop that finetunes phosphate and vitamin D homeostasis (55). FGF23 regulates phosphate and vitamin D homeostasis by binding with its cognate FGF receptors (FGFRs) in the kidney, a process that also requires αKlotho and heparan sulfate (HS) as mandatory coreceptors (56). Importantly, αKlotho is synthesized primarily in the kidney. The FGF23-FGFR-αKlotho-HS asymmetric signal transduction unit is responsible for the endocrine regulation of phosphate and vitamin D homeostasis (56). The renal expression of αKlotho is markedly reduced in CKD patients, whereas bone-derived FGF23 expression is upregulated in these patients (57, 58). The exact role of high levels of FGF23 in patients with CKD is an active area of research (59). Additionally, long-term effects of phosphate toxicity because of FGF23 dysregulation need to be resolved (60–63). Clinical and preclinical studies are underway to determine the effects of restoring αKlotho and FGF23 balance in patients with CKD and other diseases with phosphate dysregulation. The identification of FGF23-FGFR-αKlotho interactions provides a structural blueprint for drug discovery to reduce FGF23-linked tissue and organ damage.
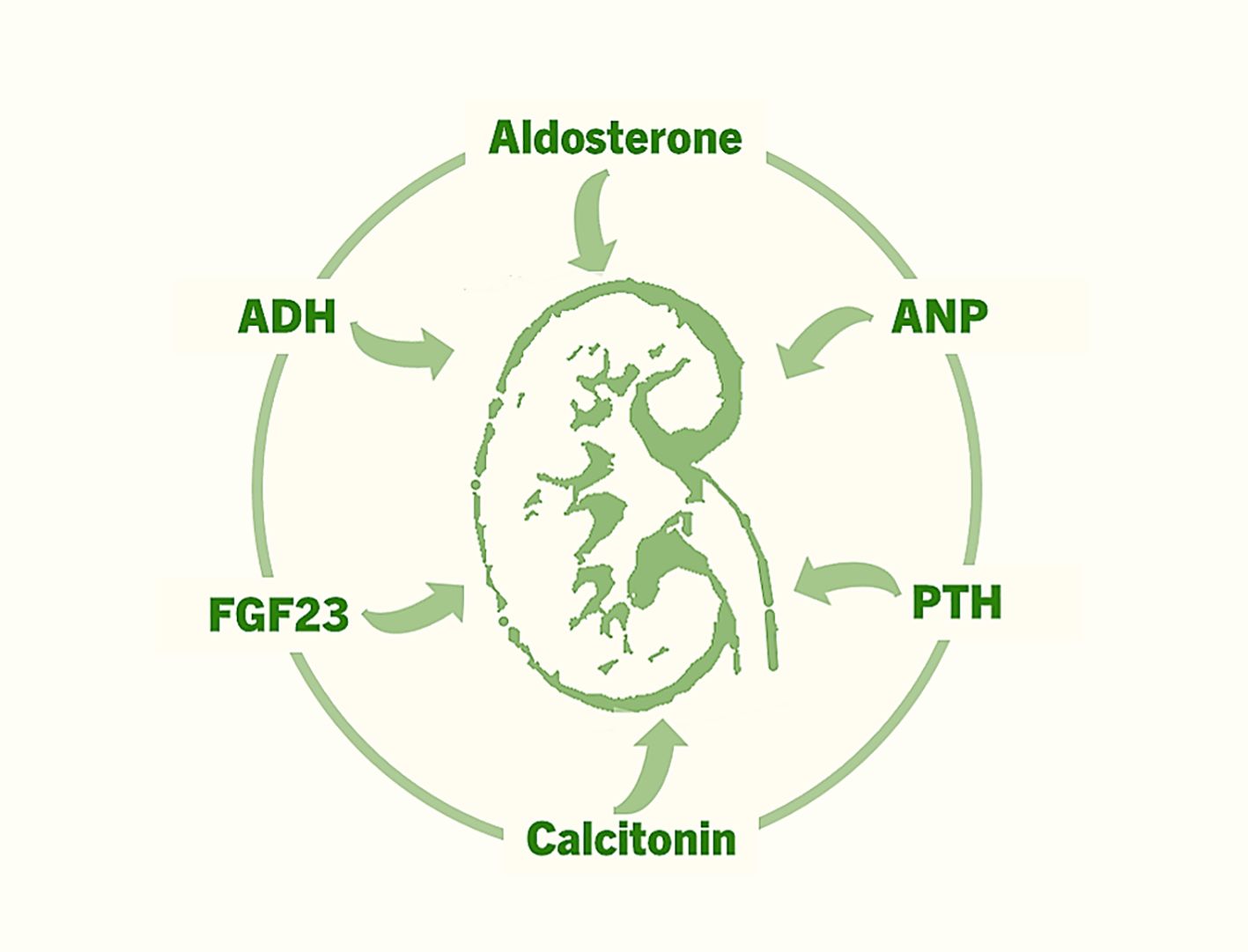
Figure 2. The major hormones that exert various effects on the kidneys to regulate fluid and electrolyte homeostasis, mineral ion balance, acid–base equilibrium, blood pressure, and erythropoiesis.
Conclusion
With access to a growing array of molecular and cellular manipulation techniques, including CRISPR-based gene editing, proteomics, genomics, in vitro and in vivo model systems, we are now better equipped to study the autocrine, paracrine, and endocrine functions of renal hormones, as well as those of hormones that affect the kidney. This presents a unique opportunity to expand our understanding of the mechanisms by which the kidney regulates fluid and electrolyte homeostasis, mineral ion balance, acid–base equilibrium, blood pressure, and erythropoiesis. While the challenges ahead are substantial, we can expect breakthrough research with applied potentials in this rapidly evolving field.
Author contributions
EH: Writing – original draft. MR: Conceptualization, Writing – review & editing, Visualization.
Acknowledgments
We want to thank Drs. Rahnuma Ahmad and Peace Uwambaye for providing useful suggestions. M.S.R. is a visiting professor at the University Kuala Lumpur Royal College of Medicine Perak, Ipoh, Malaysia.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Some of the information has been collected from online sources, including OpenAI GPT-3.5 and Google Scholar.
Correction note
This article has been corrected with minor changes. These changes do not impact the scientific content of the article.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Uwitonze AM and Razzaque MS. Role of magnesium in vitamin D activation and function. J Am Osteopath Assoc. (2018) 118:181–9. doi: 10.7556/jaoa.2018.037
2. Santoro D, Caccamo D, Lucisano S, Buemi M, Sebekova K, Teta D, et al. Interplay of vitamin D, erythropoiesis, and the renin-angiotensin system. BioMed Res Int. (2015) 2015:145828 2015. doi: 10.1155/2015/145828
3. Haase VH. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. (2013) 27:41–53. doi: 10.1016/j.blre.2012.12.003
5. Dwitanto K, Angginy N, and Sutandar W. Comparative study of recombinant human erythropoietin (rhEPO) products on CKD (Chronic kidney disease) patients. Drug Res (Stuttg). (2023) 73:271–8. doi: 10.1055/a-1982-381110.1055/a-1982-3811
6. Abdelrazik N and Fouda M. Once weekly recombinant human erythropoietin treatment for cancer-induced anemia in children with acute lymphoblastic leukemia receiving maintenance chemotherapy: a randomized case-controlled study. Hematology. (2007) 12:533–41. doi: 10.1080/10245330701521572
7. Peckham JD, Rizzo JA, April MD, Maddry JK, Meledeo MA, Stewart IJ, et al. A narrative review of erythropoietin stimulating agents for trauma. Mil Med. (2025). doi: 10.1093/milmed/usaf289
8. Schoener B and Borger J. Erythropoietin Stimulating Agents. In: StatPearls. Treasure Island (FL: StatPearls Publishing (2025).
9. Diensthuber M, Ilner T, Rodt T, Samii M, Brandis A, Lenarz T, et al. Erythropoietin and erythropoietin receptor expression in vestibular schwannoma: potential role in tumor progression. Otol Neurotol. (2007) 28:559–65. doi: 10.1097/mao.0b013e3180423b05
10. Arcasoy MO, Amin K, Chou SC, Haroon ZA, Varia M, and Raleigh JA. Erythropoietin and erythropoietin receptor expression in head and neck cancer: relationship to tumor hypoxia. Clin Cancer Res. (2005) 11:20–7. doi: 10.1158/1078-0432.20.11.1
11. Kobori H, Nangaku M, Navar LG, and Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. (2007) 59:251–87. doi: 10.1124/pr.59.3.3
12. Fountain JH, Kaur J, and Lappin SL. Physiology, Renin Angiotensin System. In: StatPearls. Treasure Island (FL: StatPearls Publishing (2025).
13. Chang TI and Lerma EV. Optimizing renin-Angiotensin system inhibitor use in CKD. Clin J Am Soc Nephrol. (2022) 17:131–3. doi: 10.2215/CJN.12950921
14. Saravi B, Li Z, Lang CN, Schmid B, Lang FK, Grad S, et al. The tissue renin-angiotensin system and its role in the pathogenesis of major human diseases: quo vadis? Cells. (2021) 10(3):650. doi: 10.3390/cells10030650
15. Adorini L. Immunomodulatory effects of vitamin D receptor ligands in autoimmune diseases. Int Immunopharmacol. (2002) 2:1017–28. doi: 10.1016/S1567-5769(02)00049-8
16. O’Donnell S, Moher D, Thomas K, Hanley DA, and Cranney A. Systematic review of the benefits and harms of calcitriol and alfacalcidol for fractures and falls. J Bone Miner Metab. (2008) 26:531–42. doi: 10.1007/s00774-008-0868-y
17. Tilyard MW, Spears GF, Thomson J, and Dovey S. Treatment of postmenopausal osteoporosis with calcitriol or calcium. N Engl J Med. (1992) 326:357–62. doi: 10.1056/NEJM199202063260601
18. Ebeling PR, Wark JD, Yeung S, Poon C, Salehi N, Nicholson GC, et al. Effects of calcitriol or calcium on bone mineral density, bone turnover, and fractures in men with primary osteoporosis: a two-year randomized, double blind, double placebo study. J Clin Endocrinol Metab. (2001) 86:4098–103. doi: 10.1210/jcem.86.9.7847
19. Demay MB, Pittas AG, Bikle DD, Diab DL, Kiely ME, Lazaretti-Castro M, et al. Vitamin D for the prevention of disease: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2024) 109:1907–47. doi: 10.1210/clinem/dgae290
20. Breyer MD and Breyer RM. Prostaglandin E receptors and the kidney. Am J Physiol Renal Physiol. (2000) 279:F12–23. doi: 10.1152/ajprenal.2000.279.1.F12
21. Suzuki T, Katori M, Fujita T, Kumagai Y, and Majima M. Involvement of the renal kallikrein-kinin system in K(+)-induced diuresis and natriuresis in anesthetized rats. Eur J Pharmacol. (2000) 399:223–7. doi: 10.1016/S0014-2999(00)00382-4
22. Drummer C, Fiedler F, König A, and Gerzer R. Urodilatin, a kidney-derived natriuretic factor, is excreted with a circadian rhythm and is stimulated by saline infusion in man. J Am Soc Nephrol. (1991) 1:1109–13. doi: 10.1681/ASN.V191109
23. Robertson GL. Regulation of arginine vasopressin in the syndrome of inappropriate antidiuresis. Am J Med. (2006) 119:S36–42. doi: 10.1016/j.amjmed.2006.05.006
24. Bankir L, Bichet DG, and Morgenthaler NG. Vasopressin: physiology, assessment and osmosensation. J Intern Med. (2017) 282:284–97. doi: 10.1111/joim.12645
25. Verbalis JG. Disorders of body water homeostasis. Best Pract Res Clin Endocrinol Metab. (2003) 17:471–503. doi: 10.1016/S1521-690X(03)00049-6
26. Ostrowski NL, Young WS 3rd, Knepper MA, and Lolait SJ. Expression of vasopressin V1a and V2 receptor messenger ribonucleic acid in the liver and kidney of embryonic, developing, and adult rats. Endocrinology. (1993) 133:1849–59. doi: 10.1210/endo.133.4.8404628
27. Sparapani S, Millet-Boureima C, Oliver J, Mu K, Hadavi P, Kalostian T, et al. The biology of vasopressin. Biomedicines. (2021) 9(1):89. doi: 10.3390/biomedicines9010089
28. Song Z and Albers HE. Cross-talk among oxytocin and arginine-vasopressin receptors: Relevance for basic and clinical studies of the brain and periphery. Front Neuroendocrinol. (2018) 51:14–24. doi: 10.1016/j.yfrne.2017.10.004
29. Spasovski G, Vanholder R, Allolio B, Annane D, Ball S, Bichet D, et al. Clinical practice guideline on diagnosis and treatment of hyponatraemia. Nephrol Dial Transplant. (2014) 29 Suppl 2:i1–i39. doi: 10.1093/ndt/gfu040
30. Al Zaman K, Alhebsi AA, Almheiri A, Alhosani H, Alshehhi NT, and Alwheibi ES. Syndrome of inappropriate antidiuretic hormone (SIADH) in chronic respiratory diseases: A comprehensive review. Cureus. (2025) 17:e77407. doi: 10.7759/cureus.77407
31. Young MJ. Mechanisms of mineralocorticoid receptor-mediated cardiac fibrosis and vascular inflammation. Curr Opin Nephrol Hypertens. (2008) 17:174–80. doi: 10.1097/MNH.0b013e3282f56854
32. Garty H and Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. (1997) 77:359–96. doi: 10.1152/physrev.1997.77.2.359
33. Ekman N, Grossman AB, Nieckarz A, Jedrzejewski L, Wolf J, and Dworakowska D. Non-hypertensive effects of aldosterone. Int J Mol Sci. (2025) 26(2):540. doi: 10.3390/ijms26020540
34. de Kloet ER. Brain mineralocorticoid and glucocorticoid receptor balance in neuroendocrine regulation and stress-related psychiatric etiopathologies. Curr Opin Endocr Metab Res. (2022) 24:100352. doi: 10.1016/j.coemr.2022.100352
35. Takeda Y, Demura M, Kometani M, Karashima S, Yoneda T, and Takeda Y. Molecular and epigenetic control of aldosterone synthase, CYP11B2 and 11-hydroxylase, CYP11B1. Int J Mol Sci. (2023) 24(6):5782. doi: 10.3390/ijms24065782
36. Nanba K, Vaidya A, and Rainey WE. Aging and adrenal aldosterone production. Hypertension. (2018) 71:218–23. doi: 10.1161/HYPERTENSIONAHA.117.10391
37. Funder JW. Aldosterone and mineralocorticoid receptors: a personal reflection. Mol Cell Endocrinol. (2012) 350:146–50. doi: 10.1016/j.mce.2011.11.026
38. Kishimoto I, Tokudome T, Nakao K, and Kangawa K. Natriuretic peptide system: an overview of studies using genetically engineered animal models. FEBS J. (2011) 278:1830–41. doi: 10.1111/j.1742-4658.2011.08116.x
39. Potter LR, Abbey-Hosch S, and Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev. (2006) 27:47–72. doi: 10.1210/er.2005-0014
40. Gardner DG, Chen S, Glenn DJ, and Grigsby CL. Molecular biology of the natriuretic peptide system: implications for physiology and hypertension. Hypertension. (2007) 49:419–26. doi: 10.1161/01.HYP.0000258532.07418.fa
41. Kitakaze M, Asakura M, Kim J, Shintani Y, Asanuma H, Hamasaki T, et al. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): two randomised trials. Lancet. (2007) 370:1483–93. doi: 10.1016/S0140-6736(07)61634-1
42. Tholén M, Kolsrud O, Dellgren G, Karason K, Lannemyr L, and Ricksten SE. Atrial natriuretic peptide in the prevention of acute renal dysfunction after heart transplantation-a randomized placebo-controlled double-blind trial. Acta Anaesthesiol Scand. (2023) 67:738–45. doi: 10.1111/aas.14241
43. Muff R, Fischer JA, Biber J, and Murer H. Parathyroid hormone receptors in control of proximal tubule function. Annu Rev Physiol. (1992) 54:67–79. doi: 10.1146/annurev.ph.54.030192.000435
44. Alexander RT and Dimke H. Effects of parathyroid hormone on renal tubular calcium and phosphate handling. Acta Physiol (Oxf). (2023) 238:e13959. doi: 10.1111/apha.13959
45. Ketteler M, Block GA, Evenepoel P, Fukagawa M, Herzog CA, McCann L, et al. Executive summary of the 2017 KDIGO Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Guideline Update: what’s changed and why it matters. Kidney Int. (2017) 92:26–36. doi: 10.1016/j.kint.2017.04.006
46. Prideaux M, Dallas SL, Zhao N, Johnsrud ED, Veno PA, Guo D, et al. Parathyroid hormone induces bone cell motility and loss of mature osteocyte phenotype through L-Calcium channel dependent and independent mechanisms. PloS One. (2015) 10:e0125731. doi: 10.1371/journal.pone.0125731
47. Chisthi MM, Nair RS, Kuttanchettiyar KG, and Yadev I. Mechanisms behind post-Thyroidectomy hypocalcemia: interplay of calcitonin, parathormone, and albumin-A prospective study. J Invest Surg. (2017) 30:217–25. doi: 10.1080/08941939.2016
48. Wilkin TJ, Isles TE, Paterson CR, Crooks J, and Beck J. Post-thyroidectomy hypocalcaemia: A feature of the operation or the thyroid disorder? Lancet. (1977) 1:621–3. doi: 10.1016/S0140-6736(77)92057-8
49. Demeester-Mirkine N, Hooghe L, Van Geertruyden J, and De Maertelaer V. Hypocalcemia after thyroidectomy. Arch Surg. (1992) 127:854–8. doi: 10.1001/archsurg.1992.01420070118021
50. Verbeek HH, de Groot JWB, Sluiter WJ, Muller Kobold AC, van den Heuvel ER, Plukker JT, et al. Calcitonin testing for detection of medullary thyroid cancer in people with thyroid nodules. Cochrane Database Syst Rev. (2020) 3:Cd010159. doi: 10.1002/14651858.CD010159.pub2
51. Evangelatos G, Fragoulis GE, and Iliopoulos A. Twenty-year scintigraphic follow-up of calcitonin versus zoledronic acid in Paget’s disease of bones. Rheumatol (Oxford). (2019) 58:1776. doi: 10.1093/rheumatology/kez070
52. Razzaque MS. FGF23-mediated regulation of systemic phosphate homeostasis: is Klotho an essential player? Am J Physiol Renal Physiol. (2009) 296:F470–476. doi: 10.1152/ajprenal.90538.2008
53. Razzaque MS and Mohammadi M. Can targeting the FGF23-alphaKlotho signaling system delay phosphate-driven organ damage? Expert Opin Ther Targets. (2025) 29:93–100. doi: 10.1080/14728222.2025.2482552
54. Razzaque MS. The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. (2009) 5:611–9. doi: 10.1038/nrendo.2009.196
55. Razzaque MS. Interactions between FGF23 and vitamin D. Endocr Connect. (2022) 11(10):e220239. doi: 10.1530/EC-22-0239
56. Chen L, Fu L, Sun J, Huang Z, Fang M, Zinkle A, et al. Structural basis for FGF hormone signalling. Nature. (2023) 618:862–70. doi: 10.1038/s41586-023-06155-9
57. Hu MC, Shi M, Zhang J, Quiñones H, Griffith C, Kuro-o M, et al. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. (2011) 22:124–36. doi: 10.1681/ASN.2009121311
58. Koh N, Fujimori T, Nishiguchi S, Tamori A, Shiomi S, Nakatani T, et al. Severely reduced production of klotho in human chronic renal failure kidney. Biochem Biophys Res Commun. (2001) 280:1015–20. doi: 10.1006/bbrc.2000.4226
59. Razzaque MS. Does FGF23 toxicity influence the outcome of chronic kidney disease? Nephrol Dial Transplant. (2009) 24:4–7. doi: 10.1093/ndt/gfn620
60. Alexander R, Debiec N, Razzaque MS, and He P. Inorganic phosphate-induced cytotoxicity. IUBMB Life. (2022) 74:117–24. doi: 10.1002/iub.2561
61. Brown RB and Razzaque MS. Phosphate toxicity and tumorigenesis. Biochim Biophys Acta Rev Cancer. (2018) 1869:303–9. doi: 10.1016/j.bbcan.2018.04.007
62. Ohnishi M, Kato S, and Razzaque MS. Genetic induction of phosphate toxicity significantly reduces the survival of hypercholesterolemic obese mice. Biochem Biophys Res Commun. (2011) 415:434–8. doi: 10.1016/j.bbrc.2011.10.076
Keywords: ADH, FGF23, kidney, endocrinology, phosphate
Citation: Haque ATME and Razzaque MS (2025) Specialty grand challenge: existing issues and emerging frontiers in renal endocrinology. Front. Endocrinol. 16:1651141. doi: 10.3389/fendo.2025.1651141
Received: 20 June 2025; Accepted: 30 June 2025;
Published: 17 July 2025; Corrected: 28 July 2025.
Edited and Reviewed by:
Jeff M P Holly, University of Bristol, United KingdomCopyright © 2025 Haque and Razzaque. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohammed S. Razzaque, bW9oYW1tZWQucmF6emFxdWVAdXRyZ3YuZWR1; bXNyLm5hZ2FzYWtpQGdtYWlsLmNvbQ==