 Erika Messina1
Erika Messina1 Alessia Inglesi1,2
Alessia Inglesi1,2 Soraya Puglisi1*‡
Soraya Puglisi1*‡ Anja Barač Nekić3
Anja Barač Nekić3 Valentina Morelli4
Valentina Morelli4 Ylenia Alessi5
Ylenia Alessi5 Karin Zibar Tomsic3
Karin Zibar Tomsic3 Serena Palmieri6Pasquale Tomaiuolo2Enrico Grosso2
Serena Palmieri6Pasquale Tomaiuolo2Enrico Grosso2 Francesco Ferraù7
Francesco Ferraù7 Iacopo Chiodini8
Iacopo Chiodini8 Darko Kastelan3Anna Pia9Maria Scatolini2
Darko Kastelan3Anna Pia9Maria Scatolini2 Giuseppe Reimondo1†
Giuseppe Reimondo1† Massimo Terzolo1†
Massimo Terzolo1†- 1Department of Clinical and Biological Sciences, Internal Medicine, San Luigi Gonzaga Hospital, University of Turin, Orbassano, Italy
- 2Molecular Oncology Laboratory, Fondazione Edo ed Elvo Tempia, Ponderano, Italy
- 3Department of Endocrinology, University Hospital Zagreb, Zagreb, Croatia
- 4Unit for Bone Metabolism Diseases and Diabetes and Lab of Endocrine and Metabolic Research, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Istituto Auxologico Italiano, Milan, Italy
- 5Department of Biomedical, Dental and Morphological and Functional Imaging Sciences, University of Messina, Messina, Italy
- 6Endocrinology Unit, Fondazione Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Ca’ Granda-Ospedale Maggiore Policlinico, Milan, Italy
- 7Department of Human Pathology G. Barresi, Endocrine Unit, University Hospital G. Martino, University of Messina, Messina, Italy
- 8Department of Biotechnology and Translational Medicine, Unit of Endocrinology, Ospedale Niguarda Cà Granda, University of Milan, Milan, Italy
- 9Department of Endocrinology, Diabetes and Metabolism, Santa Croce and Carle Hospital, Cuneo, Italy
Objective: Adrenal incidentalomas are commonly detected in clinical practice. Despite growing interest in their molecular features, their germline genetic background remains largely unexplored. This study investigated the presence and potential pathogenic role of germline variants (GVs) in these patients using a targeted next-generation sequencing (NGS) approach, and explored possible genotype-phenotype correlations.
Design: This multicenter retrospective study included 191 patients with incidentally discovered adrenal masses from four European reference centers. Patients with adrenocortical carcinoma, pheochromocytoma and primary aldosteronism were excluded.
Methods: Germline DNA was extracted from peripheral blood and analyzed using a custom next-generation sequencing (NGS) panel targeting 21 genes potentially involved in adrenal tumorigenesis. Bioinformatic analysis was followed by variant classification using the ClinVar and VarSome databases, in accordance with ACMG guidelines.
Results: GVs were identified in 12 of 191 patients (6.3%), affecting 7 different genes: ZNRF3, ARMC5, APC, CACNA1H, SCNN1B, PDE11A, and KCNJ5. Most of the detected variants were classified as variants of uncertain significance (VUS). Genotype-phenotype analysis revealed that some patients with GVs had bilateral adrenal lesions and/or mild autonomous cortisol secretion (MACS). No variants were classified as clearly pathogenic.
Conclusion: This study provides new insights into the germline genetic landscape of adrenal incidentalomas. Although GVs were identified in a minority of patients, their clinical significance remains unclear due to the predominance of VUS. These findings do not currently support widespread germline testing in routine clinical management of adrenal incidentalomas. Nevertheless, the detection of potentially pathogenic variants may inform future studies exploring their possible role in adrenal tumorigenesis.
Introduction
Adrenal tumors are a heterogeneous group of neoplasms in terms of frequency, clinical presentation, and outcomes. The group includes a wide spectrum of pathological entities, from the rare and aggressive adrenocortical carcinoma (ACC) to the frequent and benign adrenal adenoma (1). Currently, the majority of adrenal adenomas are serendipitously detected by imaging tests performed for unrelated reasons in up to 5-7% of people, and therefore they are called “adrenal incidentalomas” (2, 3).
Adrenal incidentalomas are most commonly non-functioning; however, overt cortisol or aldosterone hypersecretion occurs in approximately 15% of cases, while mild autonomous cortisol secretion (MACS) is observed in up to 30–50% of cases (4, 5). Malignant tumors are infrequent among adrenal incidentalomas but should not be underestimated since they accounted for 8.6% of all cases according to population-based data (6).
The molecular mechanisms underlying the occurrence of adrenocortical tumors are complex and not yet fully elucidated. Nevertheless, several key oncogenic pathways have been identified for adrenocortical carcinoma, such as IGF, TP53 and the Wnt/β-catenin pathways (7–11). In benign adrenocortical adenomas, the activation of the cAMP/PKA signaling cascade has been frequently reported: somatic mutations in the catalytic alpha subunit of the protein kinase A (PRKACA) and the stimulatory G-protein alpha subunit (GNAS) are the most frequently observed alterations (12–14). These are driving factors in up to 35–60% of cortisol producing adenoma associated with overt Cushing syndrome, and are associated with a clinical phenotype characterized by more severe hormonal hypersecretion (15, 16). Conversely, these genetic variants are less frequently found in non-functioning adrenocortical adenoma or adenomas associated with MACS (12, 17–19). Indeed, the overall prevalence of PRKACA mutations in adenomas associated with MACS is less than 4.5%, while the most common genetic alteration is a somatic mutation of CTNNB1 (encoding β-catenin) with a prevalence similar to that observed in non-functioning adenomas (17).
An increasing number of molecular events with pathogenetic role have been identified in aldosterone-secreting adrenocortical adenomas: almost all (90%) are due to somatic variants in genes encoding ion channels or transporters. Somatic variants of KCNJ5 (potassium inwardly rectifying channel subfamily J member 5) are present in approximately 40% of aldosterone-producing adenomas (20). Other genes (CACNA1D, ATP1A1, ATP2B3, CTNNB1) are less frequently involved (21–23), and familial forms of primary aldosteronism due to germline mutations are uncommon.
The availability of next-generation sequencing (NGS) has boosted the study of genetic mechanisms and has facilitated the identification of both germline and somatic variants in human diseases, including adrenal disorders (21, 24). Although the genomic classification of benign adrenal lesions has been increasingly explored (21, 25–27), most studies have focused on somatic mutations in tumoral tissue (28), leaving the genetic background of adrenal incidentalomas still largely uncharted. This study aimed to identify germline mutations-associated with adrenal incidentalomas using a targeted sequencing approach and to explore potential genotype-phenotype correlations.
Materials and methods
Patient cohort
This multicenter study was conducted at four reference institutes which are part of the European Network for the Study of Adrenal Tumors (ENS@T): Orbassano (Turin), Milan, Messina (Italy) and Zagreb (Croatia). The study was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all participants, and the study was approved by the Local Ethics Committees. Patients with incidentally detected adrenal masses referred to the centers between 1999 and 2024 were retrospectively included in the study.
The inclusion criteria were as follows: age ≥18 years, incidentally detected tumor, presumed benign cortical adenoma, availability of a peripheral whole blood sample, and complete follow-up information. Diagnosis of adrenal adenoma was based on computed tomography (CT) features according to the guidelines of the time and later revised considering the current clinical practice guidelines (5, 29). In all patients, CT was repeated after 6 months to document unchanged CT features. Patients with radiological features suggesting ACC or with known extra-adrenal malignancies were excluded from the study. In agreement with the definition of adrenal incidentaloma, patients with severe or resistant hypertension, or hypokalemia or overt clinical signs of hypercortisolism were excluded. Catecholamine excess was excluded in all patients by measuring urinary fractionated metanephrines, while primary aldosteronism (PA) was excluded using the aldosterone-to-renin ratio (ARR), calculated from the pair plasma aldosterone concentration and plasma renin activity. Patients were considered negative for PA when their ARR values were below 30, in accordance with the guideline-recommended cutoff available at the time of study design (30).
Clinical and hormonal data were collected at diagnosis and at last follow-up. Clinical signs and symptoms of adrenal hormone overproduction were ruled out based on medical history and physical examination. The following data were obtained for further evaluation: age at the time of adrenal incidentaloma diagnosis, sex, family history of adrenal tumors, presence of arterial hypertension and glucose metabolism impairment, of any level. Hypertension and diabetes were either reported by the patient in the past medical history or diagnosed during the follow-up according to the guidelines available at the time (31, 32).
The measurement of urinary free cortisol (UFC), adrenocorticotropic hormone (ACTH), serum cortisol after overnight 1 mg dexamethasone suppression test (DST) were performed at baseline and at last follow-up. An incomplete cortisol suppression was defined by a post-DST cortisol level >1.8 μg/dL (50 nmol/L). Mild autonomous cortisol secretion (MACS) was considered for patients with an incomplete cortisol suppression following the DST without signs and symptoms of overt Cushing syndrome (5).
Next-Generation Sequencing (NGS) and bioinformatics analyses were conducted at the Molecular Oncology Laboratory, Edo and Elvo Tempia Foundation (Biella, Italy).
NGS custom panel design
Targeted NGS was conducted using a custom panel (Illumina, San Diego, CA) targeting 21 genes potentially associated with benign adrenal tumors (APC, AXIN1, ZNRF3, ARMC5, PRKAR1A, GNAS, CTNNB1, PRKACA, PRKACB, CACNA1H, SCNN1B, ATP1A1, KCNJ5, ATP2B3, CLCN2, DOT1L, HDAC9, NR3C1, PDE8B, PDE11A, and CYP11B2) (Supplementary Table S1). The study protocol was conceived in 2019, when the targeted NGS panel was defined to include 21 genes selected based on the evidence available at the time and in accordance with the scope of the study that was approved by the local Ethics Committee. Given the subsequent delays in patient enrollment and laboratory processing (partly attributable to restrictions and work disruptions related to the COVID-19 pandemic) the initial gene panel was retained without modification to ensure consistency with the original design and Ethical clearance. Briefly, we included oncosuppressor genes involved in the regulation of the Wnt/B-catenin pathway in adrenal tumors and primary bilateral macronodular adrenal hyperplasia (PBMAH), as well as the cAMP-protein kinase A pathway (APC, AXIN1, ZNRF3, ARMC5, and PRKAR1A) (9, 33, 34). Additionally, genes associated with cancer-predisposition (GNAS, CTNNB1, PRKACA, PRKACB) (9, 35, 36) were added. Genes encoding proteins of membrane transport that are involved in the pathogenesis of aldosterone-producing adenomas (CACNA1H, KCNJ5, SCNN1B, ATP1A1, ATP2B3, CLCN2) (21–23) were also selected. Further, we included additional 6 genes encoding membrane receptors (NR3C1) (37, 38), enzymes (PDE8B, PDE11A, CYP11B2) (9, 21, 39) and proteins involved in the histone modification (DOT1L, HDAC9) (40). The custom panel was designed using the web-platform DesignStudio v.8.2.0.78 (https://www.illumina.com/products/by-type/informatics-products/designstudio.html) (Details in Supplementary Table S2). The selection of these genes was based on literature evidence highlighting their possible role in the pathogenesis of adrenal tumors (9, 25, 33, 34).
DNA extraction from blood samples
Germline DNA was isolated from peripheral blood leukocytes in EDTA using QIAamp® DNA Blood Mini-Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. DNA quantity of each sample was quantified by DeNovix DS-11 spectrophotometer (DeNovix Inc., Wilmington, DE, USA).
NGS library preparation and sequencing for genomic germline DNA
DNA library preparation was conducted using the AmpliSeq Library Plus (Illumina, San Diego, CA, USA) following manufacturer’s instructions. Briefly, a total amount of 30 ng of genomic DNA per sample was used to generate amplicon libraries in a multiplex PCR amplification with two primer pools. The amplicons were partially digested and phosphorylated with FuPa reagent (Thermo Fisher Scientific), ligated to AmpliSeq CD indexes and purified with Agencourt® AMPure® XP beads (Beckman Coulter, Brea, CA, USA). The libraries were then amplified, purified with beads, and quantified on a Qubit® 3.0 Fluorometer (Invitrogen, Waltham, MA, USA) using the dsDNA High Sensitivity kit. The pooled libraries were loaded for sequencing by synthesis on the Illumina MiSeq™ platform (Illumina, San Diego, CA, USA).
NGS analysis and bioinformatics interpretation
A semi-automated bioinformatics pipeline was used. Bioinformatics analysis was performed with the DNA amplicon Analysis Module v2.1.0 workflow in Local Run Manager (Illumina, San Diego, CA, USA) through the following steps of analysis: demultiplexing and FASTQ generation, alignment to a reference genome and variant calling. The ANNOVAR tool (www.openbioinformatics.org/annovar/) was used to annotate all variants. Variant prioritization was obtained with a cascade of filtering. In particular, single-nucleotide polymorphisms (SNPs) ≥ 1% were excluded using data from the 1000Genome Project Data MAF (www.internationalgenome.org/) and subsequently with greater stringency based on the Non-Finnish European (NFE) category of the Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org/). Furthermore, we removed synonymous variants that do not result in changes at the amino acid level. The identified variants were verified by the IGV (Integrative Genomics Viewer 92 v.2.5) (UC San Diego, Broad Institute).
Polymorphisms located in intronic regions were excluded from analysis. The pathogenic role of germline variants (GV) was assessed using the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) and the VarSome Premium tool (https://sso.varsome.com/) based on the American College of Medical Genetics and Genomics (ACMG) guidelines (41–43). Pathogenic (P), likely pathogenic (LP), variants of uncertain significance (VUS) or increased risk alleles variants were studied. Selected variants were validated through a Sanger sequencing (See Supplementary Table S3).
Statistical analysis
Descriptive statistics were used to analyze the clinical variables. Continuous data are expressed as median and interquartile range (IQR). Categorical variables are indicated as numbers and percentages. Statistical analyses were performed with Jamovi - version 2.6.25.0.
Results
Patient characteristics
We evaluated 191 subjects, 127 females and 64 males (median age at the blood sample 58.2 years, IQR 33–82 years). None of them had a family history of adrenal diseases, thus all cases were apparently sporadic.
At diagnosis, 141 patients (74%) had a unilateral adrenal lesion, and 50 patients (26%) had bilateral masses. The average tumor size was 28.2 mm (IQR 20-33.5 mm). At diagnosis, 116 patients (61%) were hypertensive and 19 (10%) were diabetic. MACS, according to the criteria defined above, was detected in 46.6% (n=89) of patients, while non-functioning tumors were detected in 53.4% (n=102) of patients. The median follow-up was 8.5 years (IQR 3–15 years).
Germline variants at NGS
Of the 191 patients, 12 (6.3%) had germline variants (GVs) in the panel of analyzed genes. A total of 12 unique GVs were identified in our patient cohort, including two identical variants in the ARMC5 gene in two distinct carriers. One subject was carrier of two different variants (in PDE11A and KCNJ5 genes respectively). These unique variants were identified in seven different genes: ZNRF3 (n=2), ARMC5 (n=1), APC (n=2), CACNA1H (n=4), SCNN1B (n=1), PDE11A (n=1), KCNJ5 (n=1) (Table 1). Variants were interpreted following ClinVar classifications as follows: a) one (8%) variant of potential pathogenicity, classified as variant of uncertain significance/likely pathogenic (VUS/LP) in the APC gene; b) six (50%) VUS in ARMC5 (n=1), CACNA1H (n=3), PDE11A (n=1), SCNN1B (n=1). Five out of 12 variants (42%) lack clinical evidence in ClinVar; therefore, the classification was implemented using the VarSome Premium tool, which applies the ACMG guidelines, resulting in: two VUS/LP variants in ZNRF3 and KCNJ5; two VUS in APC and CACNA1H; and one variant in ZNRF3 classified as benign (B). None GV was classified as pathogenic (P).
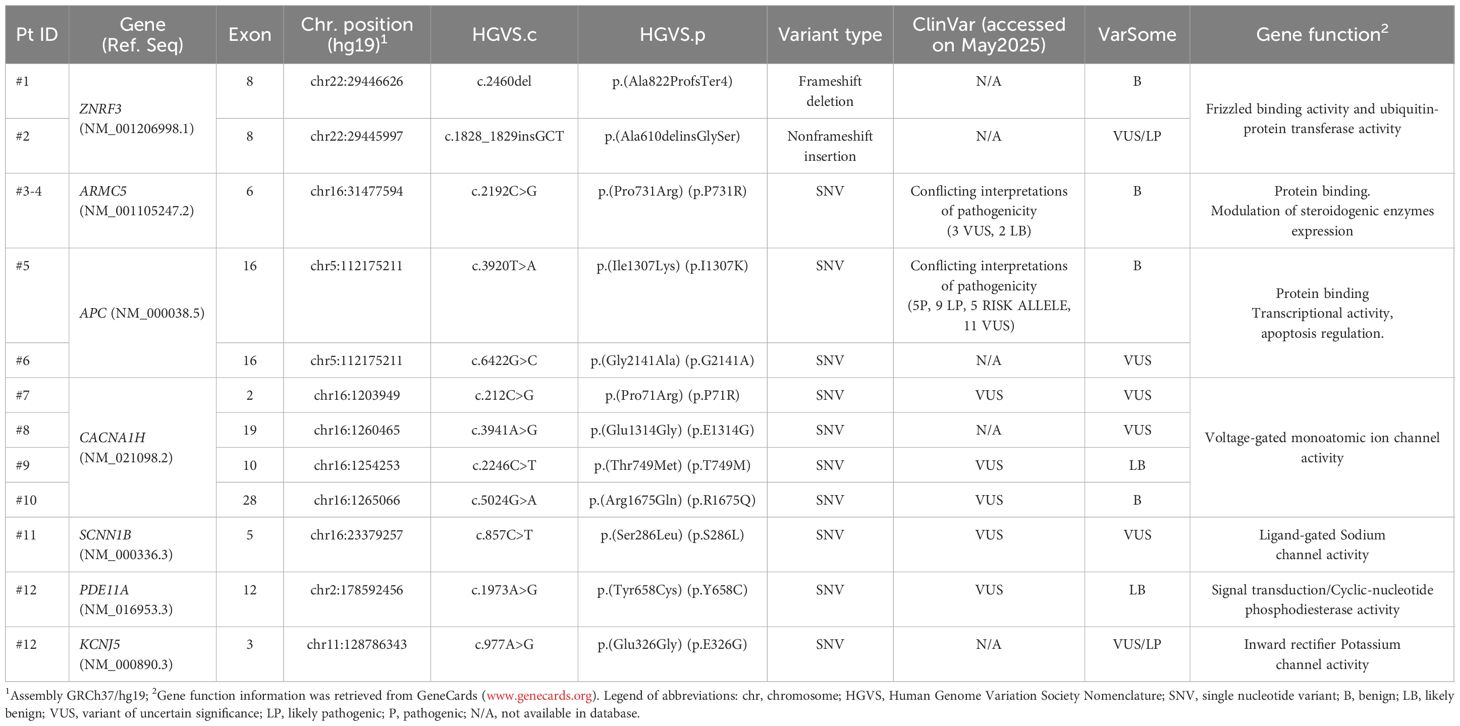
Table 1. Germline variants detected by NGS in a cohort of 191 patients with adrenal incidentalomas.
The complete list of germinal mutations including all the information about the type and localization of genetic alterations and the degree of pathogenicity classification is summarized in Table 1.
Genotype-phenotype analysis
Clinical features of patients are detailed in Table 2.
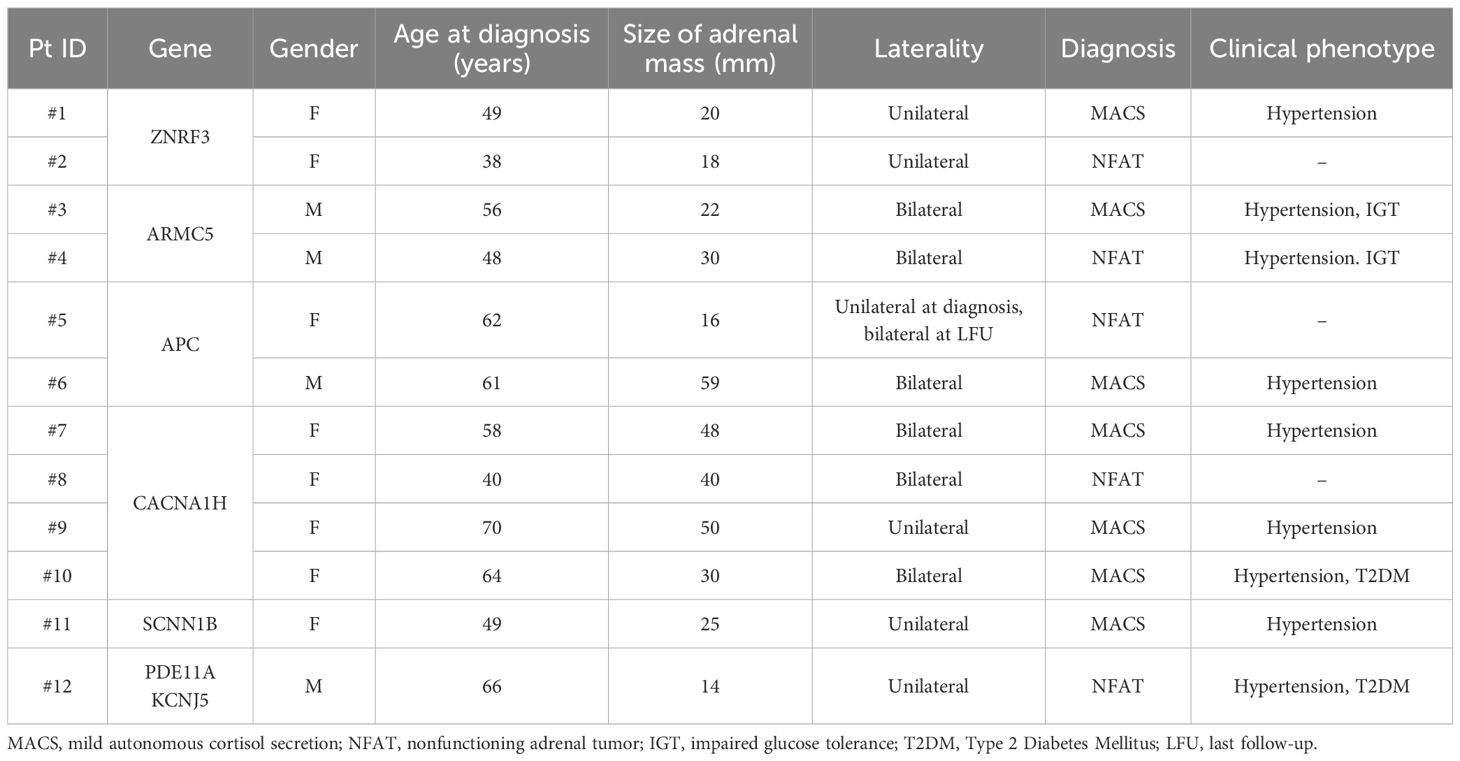
Table 2. Clinical characteristics of patients with GVs.
Both female patients with GVs in the ZNRF3 gene (c.2460delC, p.Ala822ProfsTer4 and c.1828_1829insGCT, p.Ala610delinsGlySer) had unilateral small adrenal lesions that did not change their CT characteristics during a mean follow-up of 7 years; however, the patients became hypertensive despite the absence of mineralocorticoid and glucocorticoid excess.
A unique GV c.2192C>G, p.(Pro731Arg) in exon 6 of the ARMC5 gene was found in two male subjects. Both had bilateral lesions at diagnosis (the largest size of 22 mm and 30 mm, respectively) that did not grow significantly over time. One of them had hypertension and MACS at diagnosis, while the other became hypertensive during follow-up despite the absence of cortisol autonomy.
A 62-year-old woman, carrier of a well-known GV in exon 16 of the APC gene c.3920T>A, (p.Ile1307Lys) (I1307K), presented with a non-secreting adrenal lesion of 16 mm. During follow-up she developed an adenoma with similar characteristics in the contralateral gland. Instead, a novel GV c.6422G>C, p.(Gly2141Ala) in the APC gene was found in a 61-year-old man with bilateral adrenal lesions (the largest of 59 mm) and MACS.
Three of four GVs in the CACNA1H gene were classified as VUS based on the ClinVar database (ID: #7, c.C212G; #9, c.2246C>T; #10, c.5024G>A) and were not associated with aldosterone hypersecretion. However, the three women who carried these VUS (#7, #9 and #10) were all hypertensive and diagnosed with MACS associated with bilateral adrenal lesions. The other patient (ID: 8#, c.3941A>G) displayed a unilateral adrenal mass with a size of 40 mm, which remained stable over time, with no occurrence of hypertension and no evidence of excess hormone secretion.
A 49-years old woman with a GV c.857C>T, p.(Ser286Leu) in the SCNN1B gene presented a single adrenal lesion with MACS and developed hypertension overtime.
GVs in both PDE11A and KCNJ5 genes were identified in a hypertensive, diabetic male patient presenting with a 14 mm non-secreting adrenal mass, which demonstrated no radiological progression over a 7-year follow-up period.
Discussion
Adrenal tumors occur sporadically, and only in rare instances in the context of familial genetic syndromes. This clinical observation supports the view that hereditable genetic factors contribute to their etiopathogenesis in a minority of cases (44). To date, genetic research has primarily focused on patients with primary aldosteronism (45, 46), or Cushing syndrome (47, 48) or ACC (49, 50). Clinically non-functioning adrenal incidentalomas received less attention. This multicenter study represents, to our knowledge, one of the few studies aimed to investigate the germline variants associated with adrenal incidentalomas using a targeted sequencing approach and to explore potential genotype-phenotype correlations.
Targeted NGS focused on 21 genes whose selection was based on literature data supporting their potential role in adrenal tumor formation. Our findings show that GVs in genes implicated in adrenal tumorigenesis are present in approximately 6.3% of patients with adrenal incidentaloma. Currently, the clinical significance of these variants remains uncertain, as the majority are classified as VUS according to the ClinVar database. Nonetheless, these VUS could be reclassified based on new evidence thanks to functional assays or emerging clinical data. Therefore, a potential pathogenic role for these variants cannot be ruled out at this time. In summary, we identified GVs in only 7 of the 21 genes analyzed in the panel, ZNRF3, ARMC5, APC, CACNA1H, SCNN1B, PDE11A, KCNJ5.
Somatic mutations of ZNRF3 are present in approximately 20% of patients with ACC (51), a finding that suggests an important role in tumor development through aberrant activation of the Wnt pathway (7, 8). In contrast, such mutations are rarely observed in adrenal adenomas (17, 25) suggesting a limited pathogenic role in benign lesions. In our series, we observed two GVs of the ZNRF3 gene in patients with adrenal adenomas lacking any suspicious features and remaining radiologically stable over time. Both identified tumor suppressor variants -a novel in-frame variant p.(Ala610delinsGlySer) and a frameshift variant p.(Ala822ProfsTer4)- have not been previously reported in the ClinVar database. While somatic mutations in ZNRF3 are commonly observed across various cancers, the implications of germline variations in this gene remain less well understood. Notably, the p.(Ala610delinsGlySer) variant is classified in the Varsome Premium tool as benign, based on its allele frequency in the gnomAD exome dataset (0.000164), and its presence primarily in healthy adult individuals. This alteration results in a change in the protein coding sequence length and does not occur within a known repeat region, suggesting that it may not arise from replication slippage or repeat-associated mutational mechanisms (52). Furthermore, we identified a novel null variant in the ZNRF3 gene (c.2460del), leading to a frameshift and premature stop codon. Although this variant has not yet been reported in ClinVar nor established as a germline contributor to adrenal incidentalomas, Varsome Premium classifies it as a variant of VUS/LP, due to a frameshift mutation presumed to cause nonsense-mediated decay. Importantly, ZNRF3 inactivation has been demonstrated to cause hyperactivation of Wnt/β-catenin signaling, a pathway critical for regulating cell growth, differentiation, and tumor development (51, 53). Nevertheless, to date, no studies have explored the clinical implications of these variants in patients with adrenal incidentalomas.
Inactivating mutations in ARMC5 are the most common genetic alterations in PBMAH, accounting for approximately 80% of familial and 20–25% of sporadic cases (10, 54, 55). Pathogenic GVs in ARMC5 have already been detected in 18.8% of patients with bilateral adrenal incidentalomas and MACS (56). The heterozygous p.(Pro731Arg) variant in ARMC5, currently classified as conflicting interpretation, has been reported in earlier studies, indicating that it represents a relatively frequent observation among reported cases (10, 50, 55, 56). We found this variant in two patients with bilateral adrenal lesions (and MACS in one of them). The relative frequency of non-pathogenic ARMC5 variants suggests that multiple molecular mechanisms are needed in PBMAH pathogenesis (57). This finding leads also to the speculation that a continuum may exist between bilateral adenomas and PBMAH and that what we currently interpret as bilateral adenomas might represent early stages of PBMAH. PBMAH frequently results from heterozygous germline inactivating mutations, often accompanied by a second somatic mutation, consistent with a ‘two-hit’ model of tumorigenesis (33). However, recent studies have identified somatic ARMC5 mutations occurring independently of germline alterations (56). A potential limitation of our study is the lack of genetic analysis on adrenal tissue, which precludes the assessment of somatic mutations role. Nonetheless, the recurrence of the ARMC5 variants in patients with bilateral adrenal tumors across different cohorts, including ours, supports the existence of a definitive genotype–phenotype correlation. In line with this, ARMC5 testing may be most informative in patients with bilateral adrenal lesions, especially when associated with MACS, as already proposed by Mariani et al. (2020) (58).
We identified two different GVs of the APC gene. Other studies investigated the natural history of adrenal incidentalomas in familial adenomatous polyposis (FAP) and found that these lesions typically exhibit a non-aggressive behavior (59). Of particular interest, the APC p.(Ile1307Lys) variant is a missense polymorphism with an allele frequency of 0.002008 (gnomAD exome) in the general population, indicating that it is relatively common and should therefore be considered of limited clinical significance. In particular, this variant is a low-to-moderate penetrance alteration associated with a moderately increased risk of colorectal cancer exclusively in individuals of Ashkenazi Jewish origin (60–62). However, the InSight consortium has highlighted challenges in applying standard variant interpretation frameworks to this peculiar alteration, leading to inconsistencies in its classification (63). Currently, there is insufficient high-quality evidence to establish the p.(Ile1307Lys) variant as a significant risk factor for colorectal cancer in non-Ashkenazi Jewish populations or for extracolonic malignancies (64).
We identified another APC variant p.(Gly2164Ala) in a male patient presenting with bilateral adrenal lesions and MACS, but without evidence of colonic polyposis. This specific variant has not previously been documented in individuals with APC-related syndromes, nor is it reported in population databases such as the gnomAD, indicating that it is a rare or possibly novel alteration. Currently, there is insufficient data to clarify the pathogenicity or clinical significance of this variant. Mutations in the APC gene, particularly those affecting exon 16, are well-established contributors to colorectal carcinogenesis (65). Especially, exon 16 is the largest exon, encoding approximately 77% of the APC protein and encompassing multiple critical functional domains (63). It has been observed that the majority (~94%) of germline APC mutations result in truncated proteins, for instance caused by nonsense or frameshift mutations (66).
Literature data showed that both somatic and germline heterozygous mutations in CACNA1H, which encodes the alpha-1 subunit family of T-type calcium channel (CaV3.2), result in a gain-of-function effect, leading to enhanced aldosterone synthesis (22). In our cohort, primary aldosteronism was excluded at diagnosis; nonetheless, three of the four carriers of these variants of uncertain clinical significance in CACNA1H presented with hypertension and MACS. This finding is in line with the concept that autonomous aldosterone secretion extends beyond the traditional boundaries of primary aldosteronism (67) and reinforce the notion of a continuum, in which mild or subclinical forms may not be reliably captured by standard screening approaches. In the fourth carrier, CACNA1H SNV p.(Glu1314Gly) was identified, which has not yet been classified in the ClinVar database. According to Varsome, this variant is currently categorized as of uncertain significance while multiple in silico prediction tools suggest a likely deleterious effect (SIFT: 0.001; PolyPhen-2: 1.0; FATHMM: -4.55). This amino acid substitution occurs within the homologous domain III transmembrane α-helix, a region known to play a key role in regulating calcium ion flux through the CaV3.2 channel, thereby influencing cellular processes (68). The replacement of a negatively charged glutamic acid with a neutral glycine could disrupt the structural stability of the channel protein, potentially impairing its folding and function. Although these bioinformatic predictions raise concerns about its pathogenic potential, further functional validation is required to determine the true clinical significance of this variant in the context of adrenal incidentalomas.
Mutations in SCNN1B, belonging to the sodium channel genes, may cause a rare dominant form of monogenic hypertension. We found the p.(Ser286Leu) variant in a patient with MACS. However, this variant has been previously reported as benign (69). Functional studies on this variant, which is located in exon 5, suggest that it does not significantly alter the cellular sodium transport activity (70).
Defects in the PDE11A gene, encoding phosphodiesterase type 11, represent the main genetic cause of isolated primary pigmented nodular dysplasia of the adrenal cortex (iPPNAD), and are also detectable in PBMAH and adrenocortical adenomas (71). The p.(Tyr658Cys) variant, previously identified in a patient with ACTH-dependent macronodular adrenal hyperplasia (72), lies outside canonical functional domain. Although not located within the catalytic or regulatory regions, in silico algorithms SIFT and Polyphen-2 predict this missense substitution could be deleterious. Functional studies further support its significant impact on enzyme activity: transfection experiments in different cell lines demonstrated markedly reduced PDE activity associated with the Y658C variant, with the most pronounced inhibitory effect among the tested mutations (73). This reduction in enzyme activity likely inhibits intracellular cAMP and cGMP regulation, key pathways in adrenal cortex homeostasis. The study of Faucz et al. (2011) also reported that the observed differences in variant behavior between cell types suggest tissue-specific effects, which may underlie the clinical variability observed among patients carrying PDE11A mutations (73). Further in-depth functional and clinical studies are needed to definitively establish its pathogenic role.
GVs of KCNJ5 are implicated in rare syndromic forms of primary aldosteronism, unlike its somatic mutations which are often causative of aldosterone-producing adenomas (74). The p.(Glu326Gly) variant falls within a functional domain present in the third coding exon. The effect of amino acid substitution within the inward-rectifying potassium (K) channels domain is well studied, and the PA-associated mutations in KCNJ5 typically cause a marked reduction in K+ selectivity (75, 76). Hence the channels carry a significant inward Na+ current, which is thought to depolarize the zona glomerulosa cells triggering increased aldosterone synthesis and release. In silico predictors suggest that it could be deleterious; however, there are no studies reporting this specific mutation. Notably, our subject carrying both PDE11A and KCNJ5 variants presented with hypertension and a non-functioning adrenal tumor.
In conclusion, this study provides the first germline-level analysis in a large cohort of patients with adrenal incidentalomas to identify genetic variants in potential predisposition genes. In our cohort, no GV with a current clearly established pathogenic role was identified. Most of the variants found in our cohort were variants of uncertain significance (VUS), a classification that is likely due to the limited data available in the current literature. We disclose the limit that the use of a targeted panel in this study could be associated with a limited diagnostic yield, highlighting the need for broader genomic strategies in future cohorts. Larger, adrenal-disease–inclusive panels or whole-exome sequencing could help uncover rare or unexpected predisposition genes, provide a more comprehensive framework to assess genotype–phenotype correlations, and facilitate a deeper understanding of the molecular basis of adrenal incidentalomas. To clarify the potential pathogenicity of the variants of currently unknown significance, further functional studies correlating genetic findings with clinical phenotypes will be necessary. Given the current status of knowledge, the present findings suggest that germline testing is not useful in the management of patients with adrenal incidentalomas, and widespread germline testing may therefore not be warranted.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by Comitato etico interaziendale AOU San Luigi Gonzaga di Orbassano. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
EM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. AI: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. SoP: Data curation, Investigation, Validation, Visualization, Writing – review & editing. AB: Data curation, Investigation, Validation, Visualization, Writing – review & editing. VM: Data curation, Investigation, Validation, Visualization, Writing – review & editing. YA: Data curation, Investigation, Validation, Visualization, Writing – review & editing. KZ: Data curation, Investigation, Validation, Visualization, Writing – review & editing. SeP: Data curation, Investigation, Validation, Visualization, Writing – review & editing. PT: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Writing – review & editing. EG: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Writing – review & editing. FF: Data curation, Investigation, Validation, Visualization, Writing – review & editing. IC: Data curation, Investigation, Validation, Visualization, Writing – review & editing. DK: Data curation, Investigation, Validation, Visualization, Writing – review & editing. AP: Data curation, Investigation, Validation, Visualization, Writing – review & editing. MS: Data curation, Formal analysis, Investigation, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. GR: Conceptualization, Investigation, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. MT: Conceptualization, Funding acquisition, Investigation, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. The research was supported by funding for Massimo Terzolo from the Italian Association for Cancer Research (Associazione Italiana per la Ricerca sul Cancro -AIRC), grant number IG2019-23069 and from Ministry of Education, University and Research (Ministero dell’Istruzione, dell’Università e della Ricerca -MIUR), PRIN number 20222KAYY5. We thank the AIRC and the MIUR for financial support.
Acknowledgments
Author SP has been supported by COST Action CA20122 Harmonization. We thank the COST Action CA20122 Harmonization for supportive networking.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2025.1685220/full#supplementary-material
References
1. Mete O, Erickson LA, Juhlin CC, de Krijger RR, Sasano H, Volante M, et al. Overview of the 2022 WHO classification of adrenal cortical tumors. Endocr Pathol. (2022) 33:155–96. doi: 10.1007/s12022-022-09710-8
2. Reimondo G, Castellano E, Grosso M, Priotto R, Puglisi S, Pia A, et al. Adrenal incidentalomas are tied to increased risk of diabetes: findings from a prospective study. J Clin Endocrinol Metab. (2020) 105:e973–81. doi: 10.1210/clinem/dgz284
3. Ebbehoj A, Li D, Kaur RJ, Zhang C, Singh S, Li T, et al. Epidemiology of adrenal tumours in Olmsted County, Minnesota, USA: a population-based cohort study. Lancet Diabetes Endocrinol. (2020) 8:894–902. doi: 10.1016/S2213-8587(20)30314-4
4. Prete A and Bancos I. Mild autonomous cortisol secretion: pathophysiology, comorbidities and management approaches. Nat Rev Endocrinol. (2024) 20:460–73. doi: 10.1038/s41574-024-00984-y
5. Fassnacht M, Tsagarakis S, Terzolo M, Tabarin A, Sahdev A, Newell-Price J, et al. European Society of Endocrinology clinical practice guidelines on the management of adrenal incidentalomas, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol. (2023) 189:G1–G42. doi: 10.1093/ejendo/lvad066
6. Bancos I and Prete A. Approach to the patient with adrenal incidentaloma. J Clin Endocrinol Metab. (2021) 106:3331–53. doi: 10.1210/clinem/dgab512
7. Juhlin CC, Goh G, Healy JM, Fonseca AL, Scholl UI, Stenman A, et al. Whole-exome sequencing characterizes the landscape of somatic mutations and copy number alterations in adrenocortical carcinoma. J Clin Endocrinol Metab. (2015) 100:E493–502. doi: 10.1210/jc.2014-3282
8. Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell. (2016) 29:723–36. doi: 10.1016/j.ccell.2016.04.002
9. Kamilaris CDC, Hannah-Shmouni F, and Stratakis CA. Adrenocortical tumorigenesis: Lessons from genetics. Best Pract Res Clin Endocrinol Metab. (2020) 34:101428. doi: 10.1016/j.beem.2020.101428
10. Espiard S and Bertherat J. The genetics of adrenocortical tumors. Endocrinol Metab Clin North Am. (2015) 44:311–34. doi: 10.1016/j.ecl.2015.02.004
11. Catalano R, Nozza E, Altieri B, Esposito E, Croci GA, Barbieri AM, et al. Emerging role of IGF1R and IR expression and localisation in adrenocortical carcinomas. Cell Commun Signal. (2025) 23:119. doi: 10.1186/s12964-025-02115-0
12. Beuschlein F, Fassnacht M, Assie G, Calebiro D, Stratakis CA, Osswald A, et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N Engl J Med. (2014) 370:1019–28. doi: 10.1056/NEJMoa1310359
13. Calebiro D, Di Dalmazi G, Bathon K, Ronchi CL, and Beuschlein F. cAMP signaling in cortisol-producing adrenal adenoma. Eur J Endocrinol. (2015) 173:M99–106. doi: 10.1530/EJE-15-0353
14. Drougat L, Omeiri H, Lefevre L, and Ragazzon B. Novel insights into the genetics and pathophysiology of adrenocortical tumors. Front Endocrinol (Lausanne). (2015) 6:96. doi: 10.3389/fendo.2015.00096
15. Dalmazi GD and Beuschlein F. PRKACA mutations in adrenal adenomas: genotype/phenotype correlations. Horm Metab Res. (2017) 49:301–6. doi: 10.1055/s-0042-120416
16. Ronchi CL. cAMP/protein kinase A signalling pathway and adrenocortical adenomas. Curr Opin Endocr Metab Res. (2019) 8:15–21. doi: 10.1016/j.coemr.2019.06.003
17. Ronchi CL, Di Dalmazi G, Faillot S, Sbiera S, Assie G, Weigand I, et al. Genetic landscape of sporadic unilateral adrenocortical adenomas without PRKACA p.Leu206Arg mutation. J Clin Endocrinol Metab. (2016) 101:3526–38. doi: 10.1210/jc.2016-1586
18. Di Dalmazi G, Kisker C, Calebiro D, Mannelli M, Canu L, Arnaldi G, et al. Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: a European multicentric study. J Clin Endocrinol Metab. (2014) 99:E2093–100. doi: 10.1210/jc.2014-2152
19. Thiel A, Reis AC, Haase M, Goh G, Schott M, Willenberg HS, et al. PRKACA mutations in cortisol-producing adenomas and adrenal hyperplasia: a single-center study of 60 cases. Eur J Endocrinol. (2015) 172:677–85. doi: 10.1530/EJE-14-1113
20. Lenzini L, Rossitto G, Maiolino G, Letizia C, Funder JW, and Rossi GP. A meta-analysis of somatic KCNJ5 K(+) channel mutations in 1636 patients with an aldosterone-producing adenoma. J Clin Endocrinol Metab. (2015) 100:E1089–95. doi: 10.1210/jc.2015-2149
21. Pitsava G, Maria AG, and Faucz FR. Disorders of the adrenal cortex: Genetic and molecular aspects. Front Endocrinol (Lausanne). (2022) 13:931389. doi: 10.3389/fendo.2022.931389
22. Scholl UI. Genetics of primary aldosteronism. Hypertension. (2022) 79:887–97. doi: 10.1161/HYPERTENSIONAHA.121.16498
23. Akerstrom T, Maharjan R, Sven Willenberg H, Cupisti K, Ip J, Moser A, et al. Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci Rep. (2016) 6:19546. doi: 10.1038/srep19546
24. Patocs A, Nagy P, Papp J, Bozsik A, Antal B, Grolmusz VK, et al. Cost-effectiveness of genetic testing of endocrine tumor patients using a comprehensive hereditary cancer gene panel. J Clin Endocrinol Metab. (2024) 109:3220–33. doi: 10.1210/clinem/dgae300
25. Faillot S, Foulonneau T, Neou M, Espiard S, Garinet S, Vaczlavik A, et al. Genomic classification of benign adrenocortical lesions. Endocr Relat Cancer. (2021) 28:79–95. doi: 10.1530/ERC-20-0128
26. Vaduva P and Bertherat J. The molecular genetics of adrenal cushing. Hormones (Athens). (2024) 23:601–10. doi: 10.1007/s42000-024-00608-0
27. Jouinot A, Armignacco R, and Assie G. Genomics of benign adrenocortical tumors. J Steroid Biochem Mol Biol. (2019) 193:105414. doi: 10.1016/j.jsbmb.2019.105414
28. Wu L, Xie J, Qi Y, Su T, Jiang L, Zhou W, et al. Mutational landscape of non-functional adrenocortical adenomas. Endocr Relat Cancer. (2022) 29:521–32. doi: 10.1530/ERC-21-0410
29. Fassnacht M, Arlt W, Bancos I, Dralle H, Newell-Price J, Sahdev A, et al. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol. (2016) 175:G1–G34. doi: 10.1530/EJE-16-0467
30. Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, et al. The management of primary aldosteronism: case detection, diagnosis, and treatment: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2016) 101:1889–916. doi: 10.1210/jc.2015-4061
31. Unger T, Borghi C, Charchar F, Khan NA, Poulter NR, Prabhakaran D, et al. 2020 International society of hypertension global hypertension practice guidelines. Hypertension. (2020) 75:1334–57. doi: 10.1161/HYPERTENSIONAHA.120.15026
32. ElSayed NA, Aleppo G, Aroda VR, Bannuru RR, Brown FM, Bruemmer D, et al. Classification and diagnosis of diabetes: standards of care in diabetes-2023. Diabetes Care. (2023) 46:S19–40. doi: 10.2337/dc23-S002
33. Assie G, Libe R, Espiard S, Rizk-Rabin M, Guimier A, Luscap W, et al. ARMC5 mutations in macronodular adrenal hyperplasia with Cushing’s syndrome. N Engl J Med. (2013) 369:2105–14. doi: 10.1056/NEJMoa1304603
34. MacDonald BT, Tamai K, and He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. (2009) 17:9–26. doi: 10.1016/j.devcel.2009.06.016
35. Faillot S and Assie G. Endocrine Tumours: The genomics of adrenocortical tumors. Eur J Endocrinol. (2016) 174:R249–65. doi: 10.1530/EJE-15-1118
36. Gaujoux S, Tissier F, Groussin L, Libe R, Ragazzon B, Launay P, et al. Wnt/beta-catenin and 3’,5’-cyclic adenosine 5’-monophosphate/protein kinase A signaling pathways alterations and somatic beta-catenin gene mutations in the progression of adrenocortical tumors. J Clin Endocrinol Metab. (2008) 93:4135–40. doi: 10.1210/jc.2008-0631
37. Reimondo G, Chiodini I, Puglisi S, Pia A, Morelli V, Kastelan D, et al. Analysis of BCLI, N363S and ER22/23EK polymorphisms of the glucocorticoid receptor gene in adrenal incidentalomas. PloS One. (2016) 11:e0162437. doi: 10.1371/journal.pone.0162437
38. Vitellius G, Trabado S, Hoeffel C, Bouligand J, Bennet A, Castinetti F, et al. Significant prevalence of NR3C1 mutations in incidentally discovered bilateral adrenal hyperplasia: results of the French MUTA-GR Study. Eur J Endocrinol. (2018) 178:411–23. doi: 10.1530/EJE-17-1071
39. Nakamura Y, Yamazaki Y, Tezuka Y, Satoh F, and Sasano H. Expression of CYP11B2 in aldosterone-producing adrenocortical adenoma: regulatory mechanisms and clinical significance. Tohoku J Exp Med. (2016) 240:183–90. doi: 10.1620/tjem.240.183
40. Cao Y, He M, Gao Z, Peng Y, Li Y, Li L, et al. Activating hotspot L205R mutation in PRKACA and adrenal Cushing’s syndrome. Science. (2014) 344:913–7. doi: 10.1126/science.1249480
41. Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. (2018) 46:D1062–D7. doi: 10.1093/nar/gkx1153
42. Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: the human genomic variant search engine. Bioinformatics. (2019) 35:1978–80. doi: 10.1093/bioinformatics/bty897
43. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
44. Jeyaraman K, Concolino P, and Falhammar H. Adrenocortical tumors and hereditary syndromes. Expert Rev Endocrinol Metab. (2025) 20:1–19. doi: 10.1080/17446651.2024.2431748
45. Mourtzi N, Sertedaki A, Markou A, Piaditis GP, and Charmandari E. Unravelling the genetic basis of primary aldosteronism. Nutrients. (2021) 13:1–12. doi: 10.3390/nu13030875
46. Hacini I, De Sousa K, Boulkroun S, Meatchi T, Amar L, Zennaro MC, et al. Somatic mutations in adrenals from patients with primary aldosteronism not cured after adrenalectomy suggest common pathogenic mechanisms between unilateral and bilateral disease. Eur J Endocrinol. (2021) 185:405–12. doi: 10.1530/EJE-21-0338
47. Kamilaris CDC, Stratakis CA, and Hannah-Shmouni F. Molecular genetic and genomic alterations in cushing’s syndrome and primary aldosteronism. Front Endocrinol (Lausanne). (2021) 12:632543. doi: 10.3389/fendo.2021.632543
48. Ojha U, Ogunmwonyi I, Xiang J, and Ojha H. Gene mutations in cushing’s syndrome. Mol Syndromol. (2023) 13:459–70. doi: 10.1159/000524267
49. Cioppi F, Cantini G, Ercolino T, Chetta M, Zanatta L, Nesi G, et al. Targeted Next Generation Sequencing molecular profiling and its clinical application in adrenocortical cancer. Eur J Endocrinol. (2024) 191:17–30. doi: 10.1093/ejendo/lvae077
50. Scatolini M, Grisanti S, Tomaiuolo P, Grosso E, Basile V, Cosentini D, et al. Germline NGS targeted analysis in adult patients with sporadic adrenocortical carcinoma. Eur J Cancer. (2024) 205:114088. doi: 10.1016/j.ejca.2024.114088
51. Assie G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O, et al. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet. (2014) 46:607–12. doi: 10.1038/ng.2953
52. Sehn JK. Insertion and deltions (Indels). (Amsterdam: Elsevier Inc). (2015). pp. 129–50. pp. 129–50.
53. Basham KJ, Rodriguez S, Turcu AF, Lerario AM, Logan CY, Rysztak MR, et al. A ZNRF3-dependent Wnt/beta-catenin signaling gradient is required for adrenal homeostasis. Genes Dev. (2019) 33:209–20. doi: 10.1101/gad.317412.118
54. Bouys L, Vaczlavik A, Cavalcante IP, Violon F, Jouinot A, Berthon A, et al. The mutational landscape of ARMC5 in Primary Bilateral Macronodular Adrenal Hyperplasia: an update. Orphanet J Rare Dis. (2025) 20:51. doi: 10.1186/s13023-025-03554-1
55. Albiger NM, Regazzo D, Rubin B, Ferrara AM, Rizzati S, Taschin E, et al. A multicenter experience on the prevalence of ARMC5 mutations in patients with primary bilateral macronodular adrenal hyperplasia: from genetic characterization to clinical phenotype. Endocrine. (2017) 55:959–68. doi: 10.1007/s12020-016-0956-z
56. Morelli V, Elli FM, Frigerio S, Vena W, Palmieri S, Lucca C, et al. Prevalence and clinical features of armadillo repeat-containing 5 mutations carriers in a single center cohort of patients with bilateral adrenal incidentalomas. Eur J Endocrinol. (2023) 189:242–51. doi: 10.1093/ejendo/lvad088
57. Emms H, Tsirou I, Cranston T, Tsagarakis S, and Grossman AB. Do patients with incidentally discovered bilateral adrenal nodules represent an early form of ARMC5-mediated bilateral macronodular hyperplasia? Endocrine. (2016) 53:801–8. doi: 10.1007/s12020-016-0988-4
58. Mariani BMP, Nishi MY, Wanichi IQ, Brondani VB, Lacombe AMF, Charchar H, et al. Allelic variants of ARMC5 in patients with adrenal incidentalomas and in patients with cushing’s syndrome associated with bilateral adrenal nodules. Front Endocrinol (Lausanne). (2020) 11:36. doi: 10.3389/fendo.2020.00036
59. Will OC, Hansmann A, Phillips RK, Palazzo FF, Meeran K, Marshall M, et al. Adrenal incidentaloma in familial adenomatous polyposis: a long-term follow-up study and schema for management. Dis Colon Rectum. (2009) 52:1637–44. doi: 10.1007/DCR.0b013e3181a876d6
60. Schmidt RJ, Steeves M, Bayrak-Toydemir P, Benson KA, Coe BP, Conlin LK, et al. Recommendations for risk allele evidence curation, classification, and reporting from the ClinGen Low Penetrance/Risk Allele Working Group. Genet Med. (2024) 26:101036. doi: 10.1016/j.gim.2023.101036
61. Laken SJ, Petersen GM, Gruber SB, Oddoux C, Ostrer H, Giardiello FM, et al. Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC. Nat Genet. (1997) 17:79–83. doi: 10.1038/ng0997-79
62. Liang J, Lin C, Hu F, Wang F, Zhu L, Yao X, et al. APC polymorphisms and the risk of colorectal neoplasia: a HuGE review and meta-analysis. Am J Epidemiol. (2013) 177:1169–79. doi: 10.1093/aje/kws382
63. Spier I, Yin X, Richardson M, Pineda M, Laner A, Ritter D, et al. Gene-specific ACMG/AMP classification criteria for germline APC variants: Recommendations from the ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Variant Curation Expert Panel. Genet Med. (2024) 26:100992. doi: 10.1016/j.gim.2023.100992
64. Valle L, Katz LH, Latchford A, Mur P, Moreno V, Frayling IM, et al. Position statement of the International Society for Gastrointestinal Hereditary Tumours (InSiGHT) on APC I1307K and cancer risk. J Med Genet. (2023) 60:1035–43. doi: 10.1136/jmg-2022-108984
65. Al-Tameemi HK, Al-Husseini RM, Al-Mudhafer RH, Abid HA, Al-Gazali HR, Abdullah DAA, et al. Molecular and immunohistochemical study of APC exon 16 and its possible role in colorectal carcinoma development. Heliyon. (2024) 10:e23443. doi: 10.1016/j.heliyon.2023.e23443
66. Galiatsatos P and Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol. (2006) 101:385–98. doi: 10.1111/j.1572-0241.2006.00375.x
67. Vaidya A and Carey RM. Evolution of the primary aldosteronism syndrome: updating the approach. J Clin Endocrinol Metab. (2020) 105:3771–83. doi: 10.1210/clinem/dgaa606
68. Zhang Y, Jiang X, Snutch TP, and Tao J. Modulation of low-voltage-activated T-type Ca(2)(+) channels. Biochim Biophys Acta. (2013) 1828:1550–9. doi: 10.1016/j.bbamem.2012.08.032
69. Guan WJ, Li JC, Liu F, Zhou J, Liu YP, Ling C, et al. Next-generation sequencing for identifying genetic mutations in adults with bronchiectasis. J Thorac Dis. (2018) 10:2618–30. doi: 10.21037/jtd.2018.04.134
70. Ray EC, Chen J, Kelly TN, He J, Hamm LL, Gu D, et al. Human epithelial Na+ channel missense variants identified in the GenSalt study alter channel activity. Am J Physiol Renal Physiol. (2016) 311:F908–F14. doi: 10.1152/ajprenal.00426.2016
71. Libe R, Fratticci A, Coste J, Tissier F, Horvath A, Ragazzon B, et al. Phosphodiesterase 11A (PDE11A) and genetic predisposition to adrenocortical tumors. Clin Cancer Res. (2008) 14:4016–24. doi: 10.1158/1078-0432.CCR-08-0106
72. Vezzosi D, Libe R, Baudry C, Rizk-Rabin M, Horvath A, Levy I, et al. Phosphodiesterase 11A (PDE11A) gene defects in patients with acth-independent macronodular adrenal hyperplasia (AIMAH): functional variants may contribute to genetic susceptibility of bilateral adrenal tumors. J Clin Endocrinol Metab. (2012) 97:E2063–9. doi: 10.1210/jc.2012-2275
73. Faucz FR, Horvath A, Rothenbuhler A, Almeida MQ, Libe R, Raffin-Sanson ML, et al. Phosphodiesterase 11A (PDE11A) genetic variants may increase susceptibility to prostatic cancer. J Clin Endocrinol Metab. (2011) 96:E135–40. doi: 10.1210/jc.2010-1655
74. Murthy M, Xu S, Massimo G, Wolley M, Gordon RD, Stowasser M, et al. Role for germline mutations and a rare coding single nucleotide polymorphism within the KCNJ5 potassium channel in a large cohort of sporadic cases of primary aldosteronism. Hypertension. (2014) 63:783–9. doi: 10.1161/HYPERTENSIONAHA.113.02234
75. Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. (2011) 331:768–72. doi: 10.1126/science.1198785
Keywords: genetics, variant, mutation, genotype-phenotype correlation, cortisol, adrenal adenoma, primary bilateral macronodular adrenal hyperplasia
Citation: Messina E, Inglesi A, Puglisi S, Barač Nekić A, Morelli V, Alessi Y, Zibar Tomsic K, Palmieri S, Tomaiuolo P, Grosso E, Ferraù F, Chiodini I, Kastelan D, Pia A, Scatolini M, Reimondo G and Terzolo M (2025) Germline targeted next-generation sequencing in patients with adrenal incidentalomas. Front. Endocrinol. 16:1685220. doi: 10.3389/fendo.2025.1685220
Received: 13 August 2025; Accepted: 17 September 2025;
Published: 02 October 2025.
Edited by:
Piotr Glinicki, Centre of Postgraduate Medical Education, PolandReviewed by:
Raymond H. Kim, University of Toronto, CanadaMasanori Murakami, Tokyo Medical and Dental University, Japan
Copyright © 2025 Messina, Inglesi, Puglisi, Barač Nekić, Morelli, Alessi, Zibar Tomsic, Palmieri, Tomaiuolo, Grosso, Ferraù, Chiodini, Kastelan, Pia, Scatolini, Reimondo and Terzolo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Soraya Puglisi, c29yYXlhLnB1Z2xpc2lAdW5pdG8uaXQ=
†These authors have contributed equally to this work and share last authorship
‡ORCID: Soraya Puglisi, orcid.org/0000-0002-2883-6139