 Polina Tsabai*
Polina Tsabai* Zaira KumykovaVictoria Averkova
Zaira KumykovaVictoria Averkova Nadezhda PavlovaDmitry MaslennikovAnna BolshakovaZalina BatyrovaTamara KolpakovaAndrey BystritskiyNatalia Karetnikova
Nadezhda PavlovaDmitry MaslennikovAnna BolshakovaZalina BatyrovaTamara KolpakovaAndrey BystritskiyNatalia Karetnikova Alexey EkimovAndrey Goltsov
Alexey EkimovAndrey Goltsov Maria KuznetsovaAnna TurchinetsIrina MukoseyTaisiya KochetkovaIgor Sadelov
Maria KuznetsovaAnna TurchinetsIrina MukoseyTaisiya KochetkovaIgor Sadelov Jekaterina ShubinaElena Uvarova
Jekaterina ShubinaElena Uvarova Svetlana YurenevaDmitry TrofimovGennady Sukhikh
Svetlana YurenevaDmitry TrofimovGennady Sukhikh- National Medical Research Center for Obstetrics, Gynecology and Perinatology named after Academician V.I.Kulakov of Ministry of Health of Russian Federation, Moscow, Russia
Introduction: While variants in hundreds of genes have been linked to premature ovarian insufficiency (POI), monogenic disorders account for fewer than half of idiopathic POI cases in adolescents with 46,XX karyotype. This highlights the need for the further genetic investigation across diverse populations.
Patients and methods: We recruited 63 Russian patients diagnosed with 46,XX POI before age 18. All underwent FMR1 premutation testing and whole-exome sequencing (WES). Copy number variation (CNV) analysis was conducted on WES data. Segregation studies by Sanger sequencing were performed where samples from the patients’ relatives were available.
Results: We identified variants in 15 genes in 38% of the cohort, including 13 causative genes (FMR1, DCAF17, FOXL2, STAG3, TP63, BNC1, CPEB1, NOBOX, LMNA, FSHR, SPIDR, MCM8, EIF2B2) and 2 candidate genes (MYRF, LATS1). 3.2% of patients carried an FMR1 premutation. WES detected causative single nucleotide variants (SNVs) in 15 patients (17.5% of the cohort). CNV analysis increased the diagnostic yield to 20.6%, identifying 15q25.2 microdeletions (BNC1, CPEB1) in two patients and FSHR exon 2 deletion in one patient with resistant ovary syndrome. Overall, the combination of molecular genetic approaches established a diagnosis of monogenic POI (pathogenic or likely pathogenic variants) in 23.8% of adolescents with normal female karyotype. 5 patients (7.9%) carried variants of unknown significance in FSHR, LMNA, NOBOX, SPIDR, LATS1 genes, warranting further investigation.
Discussion: Our findings demonstrate that WES is an effective diagnostic tool for adolescents with POI and should supplement standard karyotyping and FMR1 testing in routine clinical practice. We report several novel variants in POI-associated genes and propose new gene-disease association.
Introduction
Premature ovarian insufficiency (POI) is a condition defined as the loss of ovarian function before the age of 40, affecting 1-3.7% of women and representing a common cause of female infertility (1). The actual definition of POI includes clinical entities named by terms ovarian/gonadal agenesis/dysgenesis, primary ovarian insufficiency, premature/primary ovarian failure, premature menopause, hypergonadotropic amenorrhea (2). POI can develop due to genetic abnormalities, autoimmune diseases, infections, or medical interventions. The mechanisms of POI include abnormal gonadal development, diminished number of primordial follicles at birth, accelerated depletion or atresia of follicles, destruction of ovarian tissue, and resistance to gonadotropins. Depending on the extent and period of the pathological process in ovaries, it manifests as primary or secondary amenorrhea (3).
In adolescents, chromosomal abnormalities (primarily Turner syndrome) account for approximately 20% of POI cases (4, 5). Non-Turner POI is rare in this age group, with an estimated prevalence of 1 in 10,000 females under 18 (6, 7). To date, variants in hundreds of genes involved in different cellular and organ processes (gonadogenesis, meiosis, germ cells differentiation, folliculogenesis and postnatal maintenance of ovaries, hormone biosynthesis, mitochondrial function, DNA damage response and repair, etc.) have been implicated in POI (4). Targeted or whole exome sequencing (WES) enables precise diagnostics of POI, prevention of comorbidities due to syndromic POI or genetic susceptibility, and prediction of residual ovarian reserve (8–11). Genetic testing has become an essential tool for identifying causative variants and providing genetic counseling for affected individuals and their families (2). Due to the advancements in diagnostics, the proportion of idiopathic POI has decreased twofold (12).
Beyong its reproductive implications, POI has detrimental effect on overall health, increasing the risk of metabolic, cardiovascular, cognitive, and psychological issues (2). It is also significantly associated with complex genetic disorders and congenital malformations (5). Furthermore, syndromic forms of POI can be inherited, potentially leading to reproductive or extragenital disorders in offspring. Since fertility is retained to some extent in POI-affected individuals, understanding the underlying genetic features is particularly important. Ovarian dysfunction may be the only symptom of multiorganic genetic disease (10). Additionally, some forms of POI (e.g. caused by variants in meiosis-related genes) are associated with negative prognosis for euploid oocyte retrieval or increased risk of miscarriage, making assisted reproductive technologies (ART) attempts with patient’s oocytes ineffective (13, 14). Therefore, molecular diagnostics can enhance genetic counselling and improve prognosis for ART in these patients (15).
Despite the abovementioned progress on genetics of POI, 36%-67% of cases remain unexplained after thorough evaluation and require further investigation (2). Several studies including patients of mixed ancestry were published and discovered novel genetic variants related to POI via WES (9, 10, 16). This highlights the need for further research on the genetics of POI in different national cohorts, especially as menstrual irregularities are common in adolescence, and observational tactics often leads to delay in POI diagnosis (17).
Here, we describe the cohort of Russian individuals diagnosed with 46,XX POI before the age of 18 and present the results of FMR1 premutation testing and whole exome sequencing (WES).
Patients and methods
Ethics review committee approval
The institutional review board of the National Medical Research Centre for Obstetrics, Gynecology and Perinatology named after Academician V.I. Kulakov approved this study. This study was conducted according to the World Medical Association International Code of Medical Ethics (Declaration of Helsinki). All participants provided a written informed consent for the use of their data for scientific purposes.
Patients
Between January 2021 and April of 2025, patients with onset of POI before 18 years old and normal female karyotype 46,XX were recruited in the study. POI was defined as primary or secondary amenorrhea/spaniomenorrhea for more than 4 months accompanied by follicle-stimulating hormone (FSH) levels ≥25 IU/l measured on at least two separate occasions, more than four weeks apart (2). Patients with known etiology of POI, such as systemic chemotherapy, radiotherapy, autoimmune disorders or extensive ovarian surgery, established chromosomal causes of POI, were excluded from the study. The following clinical data were registered for each patient: age at the time of diagnosis, type of menstrual disorder, results of hormonal studies (FSH, E2, AMH), ovarian volume and antral follicle count (AFC), anamnesis, extragenital symptoms, family history with special concern of POI, early menopause, female and male infertility, miscarriage, ethnic origin, and consanguinity. Characteristics of the cohort (age at diagnosis, levels of hormones and volumes of the right and left ovaries) are presented as median and median absolute deviation (MAD).
Sampling and genetic testing
Samples of venous blood from the included patients were collected in anticoagulant tubes with EDTA. DNA was isolated using the PREP-MB MAX DNA Extraction Kit (DNA-Technology, Moscow, Russia) according to the manufacturer’s protocol.
FMR1 premutation testing
The number of CGG-repeats in the FMR1 gene was determined by PCR amplification using specific primers, one of which was labelled with the fluorescent dye FAM. The primers targeted the promoter CGG-repeats-containing region of the gene. Following PCR, fragment analysis was conducted with DNA samples using the Nanophor 05 genetic analyzer (Syntol, Russia) to measure the length of PCR products for each allele. The number of CGG repeats in each allele was calculated based on the fragment length.
Whole exome sequencing and chromosomal microarray analysis
WES was performed using a NovaSeq 6000 Illumina sequencer (San Diego, CA), sequencing libraries were prepared using DNA Prep (S) Tagmentation, IDT Illumina DNA/RNA UD Indexes (both Illumina), and xGen Exome Research Panel version 2 enrichment kit according to the manufacturer’s protocol. All samples were sequenced with 70×–100× coverage depth, and 10× coverage width was at least 0.95. The data were analyzed using in-house software, which included sequence alignment to the reference GRCh38 (hg38) genome, variant calling (GATK v4.5.0.0), and quality filtering. The Ensembl Variant Effect Predictor v113.3, a number of variant significance prediction algorithms (SIFT, PolyPhen-2, SpliceAI, CADD), along with OMIM, Human Gene Mutation Database (HGMD), and ClinVar were used for the annotation of variants. LOVD and other specialized databases (if present for a particular gene) were used for variant interpretation, along with the MASTERMIND genomic search engine. The genome aggregation population database (gnomAD v4.1.0) and our internal database were used to estimate the population frequencies of the identified variants. CNVs were searched using an algorithm developed by the laboratory, which is based on the application of ExomeDepth v1.1.17. The evaluation of the pathogenicity of identified CNVs is based on technical standards for interpretation and reporting of constitutional copy number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen) (18).
The clinical significance of identified variants was assessed according to the ACMG criteria (19). To prioritize potentially causative SNVs and CNVs, HPO terms (HP:0008209 Premature ovarian insufficiency, HP:0000141 Amenorrhea, HP:0000786 Primary amenorrhea, HP:0000869 Secondary amenorrhea, HP:0008232 Elevated circulating follicle stimulating hormone level) were used; variants in genes from the OMIM phenotypic series were also analyzed: PS311360, Premature ovarian failure; PS233300, Ovarian dysgenesis. For an additional search for possible causes of POI, a panel of genes from the FeRGI Database was used, specifically filtered for the POI phenotype. In the presence of additional phenotypic features (syndromic forms of POI), additional HPO terms were applied. Pathogenic, likely pathogenic variants and variants of uncertain significance (VUS) relevant to the patient’s phenotype were reported. Variants in genes of uncertain significance were considered VUS by default. The presence of clinically significant DNA copy number variations (CNVs) was confirmed by chromosomal microarray analysis (CMA; ThermoFisher CytoScan™ Optima Suite, Thermo Fisher Scientific, Waltham, MA, USA).
Validation of the variants found by WES and segregation studies in parents and siblings of the proband were performed by Sanger sequencing when possible.
Results
Patients’ characteristics
The characteristics of patients recruited in this study are summarized in Table 1 and Figure 1, the detailed information on patients’ clinical, hormonal and anamnestic data is presented in the Supplementary Table 1.
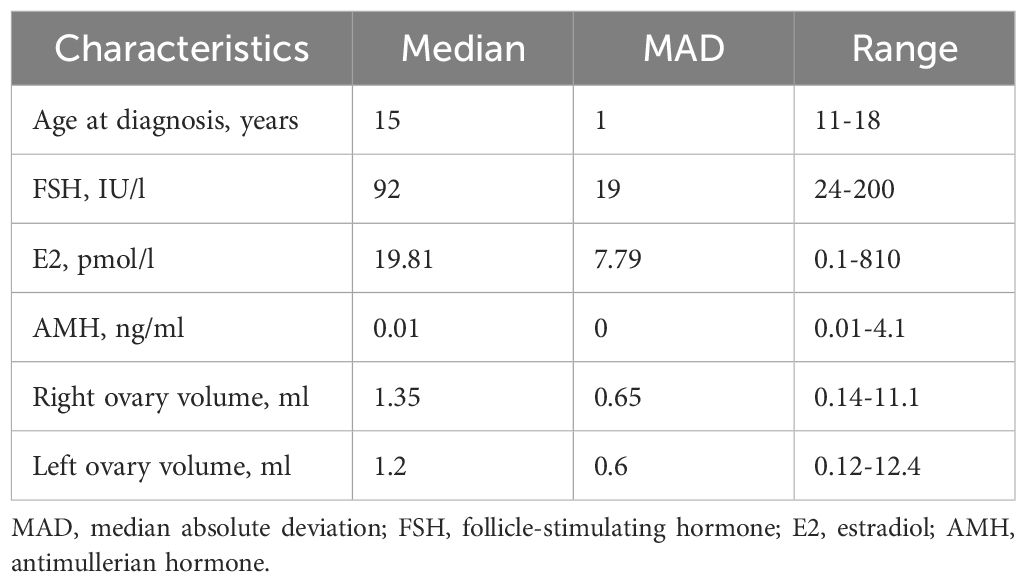
Table 1. Characteristics of the cohort of patients with idiopathic adolescent-onset POI (n=63).
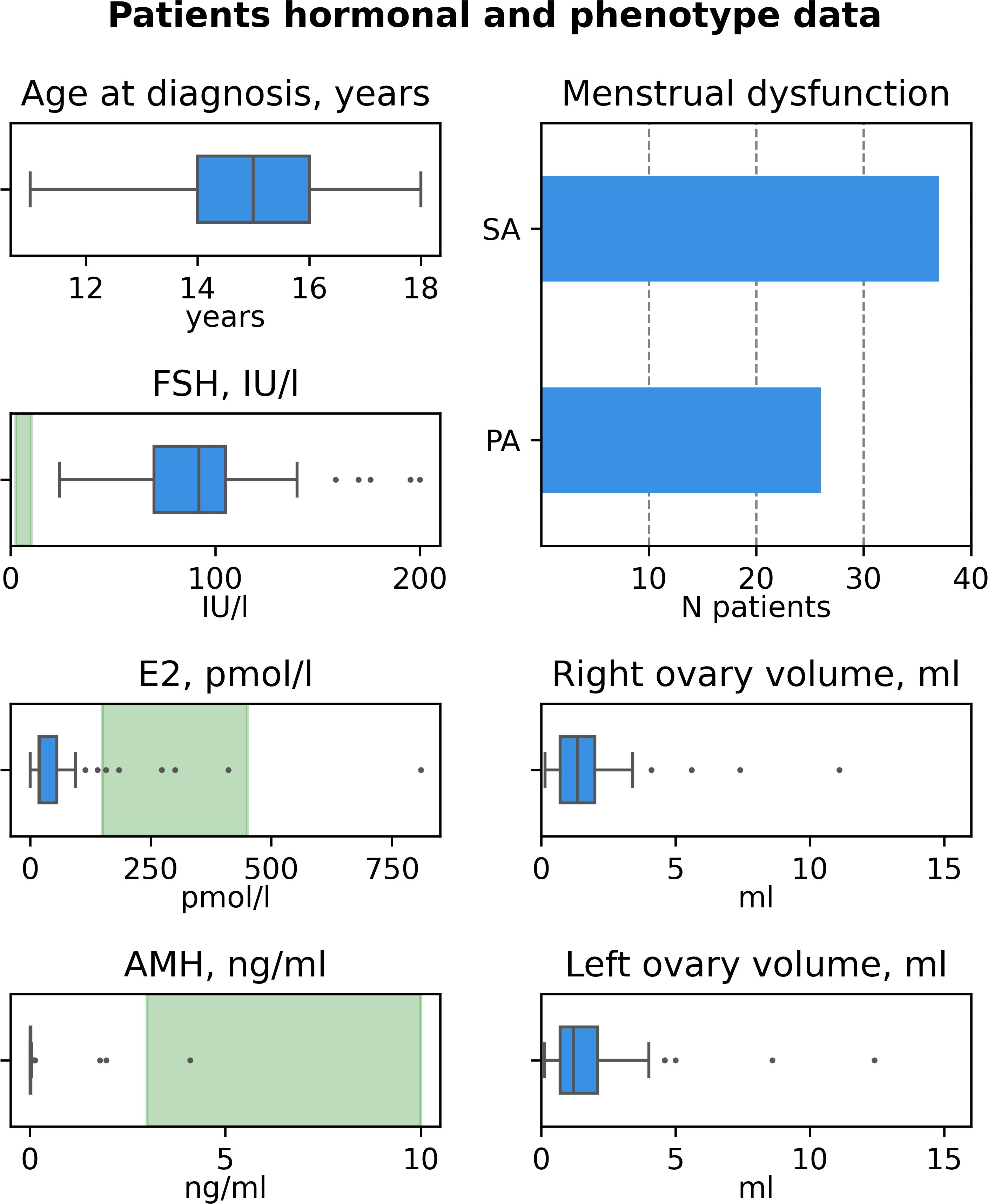
Figure 1. Characteristics of the cohort of patients with idiopathic adolescent-onset POI (n=63).
The cohort comprised 63 patients from 61 families. Median age at diagnosis was 15 years old (MAD 1, range 11-18, Figure 1A). In 26 (41.3%) of patients POI manifested as primary amenorrhea (PA), in 37 (58.7%) – as secondary amenorrhea (SA) or spaniomenorrhea (SP) (Figure 1B). Median hormone concentrations were 92 IU/l for FSH (MAD 19, Figure 1C), 19.81 pmol/l for E2 (MAD 7.79, Figure 1D), 0.01 ng/ml for AMH (MAD 0, Figure 1E). AMH levels fell within normal range in 3 patients (P31, P49, P54). P31 and P54 also had normal AFC suggesting resistant ovaries syndrome (ROS) as a cause of POI (20). In 17 cases, ovarian tissue was visualized only on one side (27%). In 21 cases, both ovaries were not visualized (33.3%). Median volume of the right ovary was 1.35 ml (MAD 0.65, Figure 1F), median volume of the left ovary was 1.2 ml (MAD 0.6, Figure 1G). 62/63 patients (98%) were Caucasians.
In 17/61 (27.9%) families proband’s relatives also faced reproductive issues. POI or early menopause were reported in relatives in 12 families (19.7%, for pedigrees see Figure 2). In 5 families, sisters were diagnosed with POI (two pairs of sisters were included in study – P6 and P7, P41 and P42). Consanguinity was reported in two families (F6 and F22), where parents were first cousins.
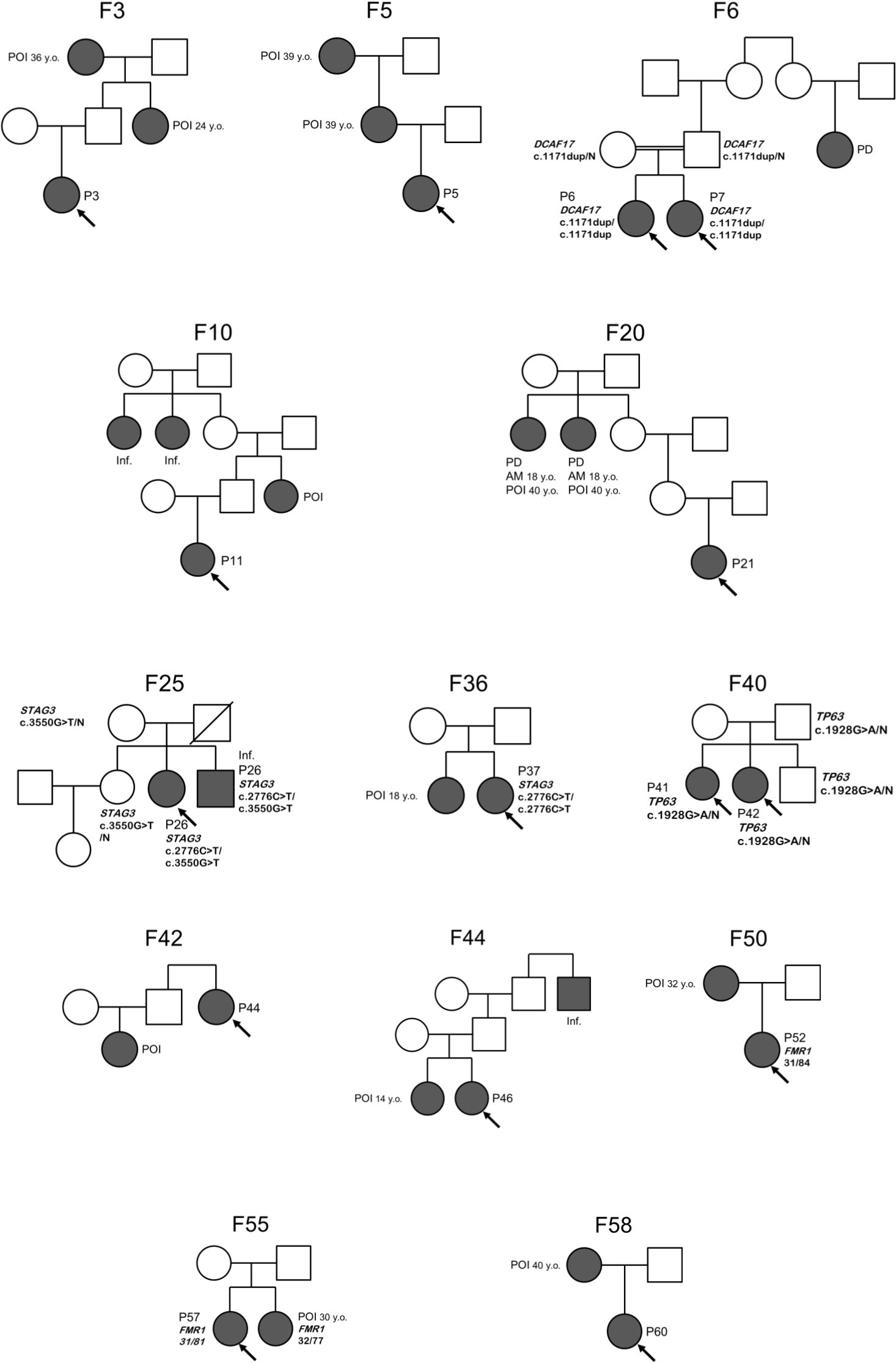
Figure 2. Pedigrees of familial POI cases. Proband is indicated by an arrow. Details of family history are presented in Supplementary Table 1. F, family; P, patient; POI, premature ovarian insufficiency; AM, age at menarche; PD, pubertal delay; Inf., infertility; N, wild type allele.
Molecular genetic testing
FMR1 alleles were normal in 60/63 (95.2%) patients. Two patients (P52 and P57, 3.2%) carried FMR1 premutations with expansion on one allele (84 and 81 CGG repeats, respectively). The patient P14 had one allele with 45 CGG repeats belonging to the grey zone (Supplementary Table 1).
All patients proceeded to the WES. In total, 18 single nucleotide variants (SNV) in 12 different genes were detected in 20 patients (31.7%), 14 of which were novel variants. Causative (likely) pathogenic SNVs were found in 11/63 patients (17.5%, Supplementary Tables 2, 3). 7 potentially causative variants of unknown significance (VUS) were found in 6 patients (9.5%) and corresponded to the suggested mode of inheritance. In 4 patients (6.3%) with non-syndromic POI, WES detected only single heterozygous variants in genes, associated with autosomal recessive POI (STAG3, SPIDR, MCM8, (21) EIF2B2, Supplementary Table 4). FMR1 premutation testing and CNV-analysis in these cases (P9, P25, P39, P40) were negative.
Segregation studies were performed in 14 cases. Both parents were tested in 10 cases, only mother was available for testing in 4 cases. In 7 cases, the origin of the variant could not be determined, as parent(s) and siblings were unavailable for testing. Segregation analysis confirmed that the variants were in trans for 5 genes (DCAF17, NOBOX, SPIDR, STAG3). In two cases, these studies prompted the reclassification of the variants’ pathogenicity. The variant p.Ser350ProfsTer55 in the MYRF gene was not inherited from parents of P63, thus, appeared de novo and was classified as pathogenic. In patient P28, we identified the heterozygous variants in STAG3: VUS p.Arg360Cys and pathogenic p.Arg926Ter. According to the ACMG guidelines, variant p.Arg360Cys initially met the criteria PM2, PP2 and PP3, thus classified as a variant of uncertain significance. Sanger sequencing of family members confirmed p.Arg360Cys to be in trans with the previously reported variant p.Arg926Ter (PM3). Taken together, these data support reclassification of p.Arg360Cys as likely pathogenic. In family F40, the father was an asymptomatic carrier of the pathogenic heterozygous variant p.Arg643Gln in TP63 gene. Both his daughters (patients P41 and P42) and son inherited the variants. Thus, the diagnostic value of WES after segregation analysis of SNVs was 17.5% (11/63 patients).
Furthermore, CNV analysis of WES data revealed pathogenic heterozygous microdeletions on chromosome 15q25.2, encompassing the BNC1 and CPEB1 genes, in two patients (P8, P10) with secondary amenorrhea. The deletions in P8 and P10 were verified with CMA (Supplementary Table 2). These patients had nonspecific phenotype (P8: mild eyelid ptosis, thin upper lip, smooth philtrum, streak of grey hair, P10: limited extension of elbows, hirsutism, astigmatism, tachycardia, speech delay, ADHD, pityriasis versicolor, mega cisterna magna on brain MRI). These findings increased the diagnostic performance of WES to 20.6% (13/63 patients).
Therefore, combined with FMR1 testing results, WES established the molecular diagnosis in 15/63 patients (23.8%). Nevertheless, 39 patients (61.9%) had no candidate variants detected by WES or FMR1 testing, including P46, who has sister with the same manifestations (ovarian dysgenesis).
It is worth noting that 9/15 (60%) patients with genetic POI suffered from autosomal recessive disorders. The consanguinity was reported only for three of them (sisters P6 and P7, P23). Among these 9 patients, there were 4 patients with STAG3- associated POI from unrelated families. Interestingly, all of them carried the same p.Arg926Ter variant in either homozygous or compound-heterozygous state.
The patients P6, P7, P15 and P63 had syndromic forms of POI. In these cases, clinical and molecular diagnosis was also based on specific phenotype (Supplementary Tables 2, 3), and in P15 clinical diagnosis of blepharophimosis-ptosis-inverse epicanthus syndrome precluded the molecular diagnosis. According to the patient’s mother, the father had a phenotypic appearance similar to that of P15, but he was not available for examination. We assume that he may have transmitted the pathogenic variant in FOXL2 to his daughter. Monozygotic twins P6 and P7 from a consanguineous family with a novel homozygous variant p.Glu391GlyfsTer4 in the DCAF17 gene manifested primary amenorrhea, ovarian agenesis, hypoplastic uterus, secondary hypothyroidism, short stature, global developmental delay, mild intellectual disability, broad chest, wide-set nipples, genu valgum, broad tip of the nose, high palate, widely spaced incisors, cone-shaped fingers, consistent with diagnosis of Woodhouse-Sakati syndrome. In patient P63, a novel heterozygous variant p.Ser350ProfsTer55 in the MYRF gene was identified. P63 also had a congenital heart defect (secundum atrial septal defect with an absent superior rim, partial anomalous pulmonary venous return draining to the right atrium, bicuspid aortic valve) and mild hypermetropia, both of which are distinctive of MYRF-related disorders.
As shown in Supplementary Tables 2 and 3, 5 patients (7.9%) carried VUSes in known human POI-causing genes (LMNA, NOBOX, SPIDR, FSHR) or in genes, which disruption leads to ovarian dysgenesis in mice (LATS1). A heterozygous VUS p.Ala491Gly in the LMNA gene was identified in patient P17. The origin of the variant could not be determined, as her father died at age 52 from heart failure, and her mother was not a carrier. Further segregation studies were not possible. The homozygous in-frame deletion p.Cys310_Glu313del in the RAD51-binding domain of the SPIDR gene was identified via WES in patient P25. Sanger sequencing revealed that the patients’ elder fertile sister, younger brother, and both parents were heterozygous carriers of the variant. Patient P23, from a consanguineous family, harbored a homozygous missense variant p.Leu287Val in the NOBOX gene. The family declined further segregation studies in the patients’ elder fertile sisters. In patient P49, diagnosed with resistant ovaries syndrome (ROS) and PA, we identified a compound heterozygous SNV and CNV in the FSHR gene: a novel hemizygous variant p.Phe66Cys and a heterozygous deletion involving exon 2 of the gene. We could not define the precise boundaries of the intragenic deletion. P49 also carried a novel likely pathogenic heterozygous variant p.Pro2002Arg in the FBN1 gene, which likely accounts for her ophthalmological features (OU mild myopia, OS peripheral vitreochorioretinal dystrophy).
In patient P47, we identified two heterozygous missense variants in LATS1 gene, which is not associated with any monogenic disease, according to OMIM: c.1334C>G (p.Pro445Arg) and c.234G>T (p.Leu78Phe), which were classified as VUS. Sanger sequencing confirmed that variants are compound-heterozygous and parents are heterozygous carriers. The family history of patient P47 was unremarkable, she had no siblings. Her extragenital features included inferior vermian hypoplasia, hypermetropic astigmatism, and migraine.
Genetic POI was diagnosed more often in patients with PA (9/26, 34.6%) than with SA/SP (6/37, 16.2%). Vice versa, patients with genetic POI were more likely to manifest PA (9/15, 60%) in comparison to patients with no molecular diagnosis (17/48, 35.4%). Among 15 patients with established genetic POI, three had congenital anomalies (20%), 4 had dysmorphic features (26.7%) and 2 had developmental delay (13.3%). Among patients without established genetic cause of POI, one had congenital anomalies (2%, P59 born prematurely from complicated pregnancy had Klippel-Feil syndrome), 9 had non-specific dysmorphic features (18.7%), such as ocular hypertelorism, short palpebral fissures, high forehead, hemifacial hypoplasia, upslanting palpebral fissures, long lashes, low-set columella, smooth philtrum, thin upper lip, mandibular prognathism, short broad neck, connective tissue disorders, etc. 4 had either intellectual disability (P36) or neurological problems, e.g. migraine, diffuse muscular hypotonia, syncope (10.4%, P19, P31, P36, P54).
In 12 cases with familial POI or early menopause, the rate of genetic diagnosis (pathogenic or likely pathogenic variants) was 41.7%. This included 2 families with autosomal recessive POI (F6 with DCAF17 variants and F36 with STAG3 variants), one family with autosomal dominant POI (F40, paternally inherited TP63 variant), and 2 families with FMR1 premutation (F50, F55). Still, family history of POI or early menopause was a predictor of genetic POI diagnosis only if the proband had an affected sister [4/5 cases (80%)], but not when other relatives, e.g. mother, grandmother, aunt, were affected (1/7 cases – P52 with FMR1 premutation has an affected mother, 14.3%). Thus, in families with intergenerational transmission, the prevalence of genetic POI was compatible to that in sporadic cases (8/49, 16.3%). Thus, in most families with intergenerational transmission of POI we did not find a molecular explanation for disorder in affected relatives.
Discussion
In our cohort of adolescents with 46,XX POI, whole-exome sequencing yielded a diagnostic rate of 17.5%, consistent with other WES studies (9, 22, 23). Combining WES with FMR1 testing, CNV analysis, and segregation studies established a molecular diagnosis in 15 of 63 adolescents (23.8%). Recent study by Cosette et al. using array-CGH and next-generation sequencing resulted in very similar rate of causative variants detection of 28.6%, although the average age of diagnosis was more than in our study (27.7 years vs. 15 years) (24). In our cohort 9 out of 63 cases (14.3%) were caused by variants in genes associated with autosomal recessive POI. 3 of these cases had consanguinity in family history and carried homozygous likely pathogenic variants or VUS. For STAG3-related POI, which showed high prevalence in our cohort, we did not find correlation with consanguinity or specific ethnic groups.
Among genetic findings, we diagnosed 3 patients with SA with fragile X-related POI (FXPOI). Early-onset secondary amenorrhea is not typical for FXPOI, and median age of amenorrhea in premutation carriers is around 38 years (25). It is estimated that approximately 3% of FMR1 premutation carriers have irregular menstrual cycles in adolescence and only 1% of them experience final menstruation before the age of 18 (26). We performed WES in these patients, but did not find alternative monogenic cause of POI.
The etiology of POI remains unknown for 48 of 63 patients (76.2%) in whom no candidate variants were found or who carried only VUS. The possibility of other undetected variants cannot be excluded. Studies show that besides variants in single genes, many POI patients have oligogenic or multigenic contribution to phenotype (9, 27, 28). Other undetectable on WES causes may include variants in mitochondrial DNA (29), epigenetic factors (30), mosaic chromosomal aneuploidy in ovarian tissue (31). In this study, we could not establish the clinical significance of variants in FSHR, LMNA, NOBOX, SPIDR, LATS1 genes, mainly due to unavailability of blood samples of proband’s relatives. Further studies and animal modelling are required to determine the real causative relationship between these variants and ovarian dysfunction that is beyond the scope of this article.
Notably, in familial cases with intergenerational inheritance of ovarian dysfunction, we identified no shared variants among affected relatives aside from the FMR1 premutation. Rouen et al. studied 36 familial cases of non-syndromic POI and found causative variants in 50%, both with inter- and intragenerational transmission (32). Thus, within families POI and early menopause can be of different origin reflecting polyetiological nature of ovarian dysfunction.
Below we discuss the genetic variants found in our patients.
Variants in established syndromic POI genes
Monozygotic twins P6 and P7 from a consanguineous family with a novel homozygous variant p.Glu391GlyfsTer4 in the DCAF17 gene had a Turner syndrome-like phenotype and intellectual disability. The DCAF17 (Ddb1- and Cul4-associated factor 17) protein acts as a substrate receptor for the CUL4-DDB1 E3 ubiquitin ligase complex. The loss of its function results in Woodhouse-Sakati syndrome (WSS), a rare autosomal recessive disorder presenting with hypogonadism in both sexes, lack of secondary sex characteristics, partial alopecia, diabetes mellitus, intellectual disability, deafness, electrocardiographic abnormalities, and extrapyramidal disorders (33). Hypogonadism is a constant feature of WSS: in females, there are streak or hypoplastic ovaries with no oocytes on biopsy and hypergonadotropic hypogonadism (34, 35), while males have azoospermia and hypogonadotropic hypogonadism (36). In females, FSH can be elevated more or less significantly (37), despite there are no documented cases of normal ovarian function. All patients with WSS show ovarian dysgenesis and hypoplastic mullerian derivatives (38). Murine models support the role of this gene in ovariogenesis. Dcaf17-knockout mice are subfertile and exhibit follicular depletion at all stages (39). At presentation, our patients did not exhibit diabetes mellitus, alopecia, hearing loss, or extrapyramidal disorders. Still, different symptoms of WSS may manifest at older age: for example, neurological disorders appear in adolescence, and diabetes mellitus usually develops up to the 25 years of age (40). Thus, the results of WES suggest a risk of developing the abovementioned issues in future, so we recommended patients to assess their endocrine (screening for glucose intolerance and thyroid dysfunction), cardiological and neurological status (assessment for dystonia, dysarthria, dysphagia and hearing loss) annually.
Patient P15 presented with the facial phenotype of blepharophimosis-ptosis-inverse epicanthus syndrome and primary amenorrhea resulting from ovarian dysgenesis. WES identified the previously reported in POI heterozygous variant p.Lys193SerfsTer78 in the FOXL2 (forkhead box L2) gene (41).
A heterozygous VUS p.Ala491Gly in the LMNA gene was identified in patient P17. The LMNA gene is associated with a wide spectrum of disorders, including premature aging syndromes. In 2003, Chen et al. described three female patients with atypical Werner syndrome and hypogonadism who carried heterozygous variants Ala57Pro and Arg133Leu in LMNA (42). In 2008, McPherson et al. proposed an association between a heterozygous missense variant p.Leu59Arg in LMNA and Malouf syndrome (dilated cardiomyopathy with hypergonadotropic hypogonadism), based on two unrelated patients with dysgenetic ovaries and onset of cardiomyopathy at ages 12 and 17 (43). In contrast, P17 showed no signs of premature aging, lipodystrophy, or cardiomyopathy. Her growth and intelligence were normal. The same variant has been reported in ClinVar in a patient with primary dilated cardiomyopathy (RCV004013487.2). Considering this, p.Ala491Gly variant was classified as VUS, and regular echocardiography monitoring was recommended for the patient. The causative relationship between LMNA variants and POI remains questionable, as most patients with a clinical diagnosis of Malouf syndrome have no identifiable variants in the coding regions of LMNA (44–46). The pathogenetic mechanism of ovarian dysgenesis in carriers of heterozygous LMNA variants remains unknown. Although mice lacking lamin A showed no abnormalities in ovarian morphology, spermatogenesis is severely disrupted at the pachytene stage (47). Dominant-negative variants in LMNA may cause Malouf syndrome through a mechanism different from the one studied by Alsheimer et al.
Variants in established non-syndromic POI genes
The pathogenic effect of heterozygous POI-causing variants in the TP63 gene is sex-limited. Paternal inheritance of the variant p.Arg643Gln found in patients P41 and P42 has been previously reported (48, 49), while p.Arg594Ter variant detected in P45 was described as de novo (50). Variants in TP63 are associated with broad spectrum of conditions related to ectodermal dysplasia. Recently, a case series on TP63-associated POI showed that same variant may cause POI or isolated cleft palate in relatives, but also that non-syndromic POI may be caused by variant associated with ADULT syndrome (51), which makes the resulting phenotype prediction uncertain. Knowledge of the molecular diagnosis allows family F40 to make informed reproductive choices and to consider options such as PGT-M or prenatal diagnostics for family planning.
In contrast to the TP63 gene, variants in genes involved in meiosis often cause infertility in both females and males (52). The STAG3 gene encodes for the stromal antigen 3 protein, which is involved in the formation of the cohesion complex. Inactivation of Stag3 in mice results in the absence of axial elements and synaptonemal complex formation, leading to gonadal dysgenesis in both sexes (53). In the family of patient P26, who carried compound-heterozygous variants in the STAG3 gene, segregation studies led to the identification of an affected brother with severe oligoasthenoteratozoospermia (54). The p.Arg926Ter variant in the STAG3 gene was the most common causative variant in non-syndromic POI, found in 4 unrelated patients in our cohort, proposing p.Arg926Ter as an enriched variant in our cohort. Bergant et al. (55) have reported this variant in homozygous state in patient with PA (55). For the non-Russian population, it is the only case with this variant described to our current knowledge, which may indicate a higher prevalence in our population. However, the possibility of random fluctuation due to the limited sample size cannot be excluded, especially given that monogenic causes of POI are understudied. The variant frequency according to gnomAD v.4.0 is 0.000065, and its frequency is yet to be investigated in Russia to reveal whether or not it is a recurrent variant or just reflects chance variation due to the limited sample size. We also report a novel likely pathogenic missense variant, p.Arg360Cys, located in the armadillo domain of the STAG3 gene, found in compound-heterozygote with p.Arg926Ter variant in patient P28. Several causative missense variants within armadillo (56–58) or STAG (57) domains of the protein have been described in publications on POI.
Similar to the variants in the STAG3 gene, biallelic variants in the SPIDR gene cause both female and male infertility (59). The SPIDR gene encodes for a scaffold protein involved in DNA repair which is an essential factor in meiotic homologous recombination (60). The homozygous in-frame deletion p.Cys310_Glu313del in the RAD51-binding domain of SPIDR was identified via WES in patient P25. This case is of particular interest due to the rarity of SPIDR-associated POI: only three infertile females with biallelic SPIDR variants have been described up to date (61, 62). Further studies (e.g. sister chromatide exchanges) are required to determine the pathogenicity of this novel in-frame deletion.
In patients P8 and P10, CNV-analysis of WES data suggested a deletion on chromosome 15q25.2, encompassing the BNC1 and CPEB1 genes. Complete deletions of both genes have been reported to cause non-syndromic POI. Hyon et al. described three women with a microdeletion in the 15q25.2 region encompassing the BNC1 and CPEB1 genes; two of these patients exhibited primary amenorrhea (63). Similarly, in a study by Bestetti et al., microdeletions of 15q25.2 including both genes were found in two out of 67 women with POI (64). Chen et al. described a 14-year-old girl with POI and a deletion of approximately 0.447 Mb in the 15q25.2 region that, interestingly, did not include the CPEB1 gene, emphasizing the role of BNC1 haploinsufficiency in POI development (65). The BNC1 (basonuclin 1) gene regulates transcription in germ cells and affects follicle development and survival. Zhang et al. reported a familial case of POI in which a heterozygous frameshift variant in BNC1 was identified. A mouse model carrying this mutation in Bnc1 demonstrated infertility, elevated FSH levels, decreased ovarian size, and reduced follicle count (66). Later, they showed that BNC1 deficiency triggers oocyte ferroptosis leading to premature follicular activation and excessive follicular atresia (67). The CPEB1 gene encodes cytoplasmic polyadenylation element-binding protein 1, which regulates the polyadenylation and translation of several mRNAs important for oocyte reentry into the meiotic cell cycle. Takahashi et al. demonstrated that Cpeb1 mRNA translation and protein levels decrease with age, resulting in altered translation in oocytes and aberrant progression through the meiotic cell cycle. They observed that Cpeb1 haploinsufficiency caused similar changes in proteostasis in young oocytes, while increasing CPEB1 protein levels in aged oocytes rescued the translation phenotype (68). Thus, haploinsufficiency of both BNC1 and CPEB1 should be considered a potential mechanism for POI development; however, BNC1 may contribute more significantly. Although the deletions found in patients P8 and P10 included other haploinsufficiency-sensitive genes, like RPS17, HOMER2, the patients exhibited no extragenital symptoms similar to those described in syndromic patients with 15q25.2 microdeletions (69).
In patient P49, diagnosed with resistant ovaries syndrome (ROS), we identified a compound heterozygous SNV and CNV in the FSHR gene: a novel hemizygous variant p.Phe66Cys and a heterozygous deletion involving exon 2 of the gene. Biallelic missense and truncating variants in FSHR causing POI with ROS have been reported, as well as several exon deletions in this gene (70–72). ROS usually leads to primary amenorrhea, while rare cases of secondary amenorrhea were reported (73). Interestingly, in two other patients (P31 and P54) with secondary amenorrhea clinically diagnosed with ROS, we did not identify any FSHR variants. Also, in P49 we found likely pathogenic heterozygous variant p.Pro2002Arg in the FBN1 gene located outside exons 65–66 of FBN1, which encode asprosin, a C-terminal cleavage product of fibrillin 1 that may influence ovarian function (74), thus, it is not considered to contribute to the development of POI in this patient.
Patient P23, from a consanguineous family, harbored a homozygous missense variant p.Leu287Val located in the homeobox domain of the NOBOX gene. NOBOX (newborn ovary homeobox) is a gene involved in the earliest stages of folliculogenesis. Most reported variants are located within the homeodomain, which is responsible for nuclear localization of NOBOX protein (75). Biallelic variants in this gene have been reported as the most frequent cause of monogenic POI (76), though they were uncommon in our cohort (1/63, 1.6%).
Potential novel gene-phenotype associations in POI
A novel heterozygous variant p.Ser350ProfsTer55 in the MYRF gene was identified in patient P63. MYRF (myelin regulatory factor) is a transcription factor essential for oligodendrocyte development and coelomic epithelium-derived cells proliferation and migration. Heterozygous variants in MYRF are associated with cardiac-urogenital syndrome, nanophthalmos and mild encephalopathy with reversible myelin vacuolization. Cardiac-urogenital syndrome is a 46,XY and 46,XX disorder of sex development caused via dysregulation of gonadogenesis and, possibly, upregulation of CITED2 gene (77, 78). Some of the several reported 46,XX patients had ovarian and/or müllerian agenesis or hypoplasia and often were severely affected by multiple congenital anomalies leading to premature death (79, 80). Recently, Ding and Tian (81) reported a patient with hypergonadotropic hypogonadism, primary amenorrhea and mullerian aplasia. Exome sequencing revealed a de novo variant c.1468C>G (p.Arg490Gly) in MYRF gene. Importantly, this patient had no significant extragenital anomalies, except possible renal duplication. Also, we described patient who had ovarian hypoplasia with preserved ovarian function and extragenital issues, and carried a heterozygous de novo p.Ala440ThrfsTer2 variant in MYRF (82). These two cases illustrate pleiotropic effect of MYRF variants and high variability of associated phenotype. These cases show that precise molecular diagnosis is important in syndromic ovarian dysfunction as long as fertility management of these patients must include preimplantation genetic testing of embryos for monogenic condition (PGT-M) to prevent severe malformations in children.
In patient P47, we identified two heterozygous missense variants in LATS1, c.1334C>G (p.Pro445Arg) and c.234G>T (p.Leu78Phe), which were classified as VUS. LATS1 (large tumor suppressor kinase 1) encodes for a regulator of the Hippo pathway (83) and has not previously been associated with POI in humans, while the deleterious consequences of LATS1 deficiency on ovariogenesis were demonstrated in murine models. The ablation of LATS1 in mice causes increased germ cell apoptosis with subsequent primordial follicle loss, development of ovarian cysts and stromal tumors, and lack of mammary glands (84). The Lats1−/− mice exhibit a high perinatal mortality (85). In mice with deletions of both Lats1 and Lats2, ovarian enlargement was observed along with transdifferentiation of granulosa cells into seminiferous tubule-like structures and bone tissue (86). Germline variants in the LATS1 gene are not associated with any monogenic disease in humans (OMIM: 603473), but Kim et al. proposed that heterozygous missense variant p.Arg96Trp may be a cause of familial schwannomatosis (87). The data in the current manuscript is not sufficient for suggesting LATS1 as a potential POI-associated gene as functional studies could not be performed, nevertheless we consider this observation to be of an interest of future studies.
Conclusions
Our study demonstrates that whole exome sequencing combined with CNV-analysis is an effective diagnostic tool in the adolescent population with POI and should be as a supplement for standard karyotyping and FMR1 testing. Understanding the molecular mechanisms underlying POI is essential for improving clinical management and genetic counselling, including the evaluation of reproductive risks and extragenital symptoms.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by The Ethical Review Board of the Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
PT: Writing – original draft, Writing – review & editing, Investigation, Resources, Conceptualization. ZK: Resources, Investigation, Writing – original draft. VA: Resources, Writing – original draft, Investigation. NP: Formal analysis, Writing – original draft, Visualization. DM: Investigation, Writing – original draft. ABo: Writing – review & editing, Resources, Investigation. ZB: Resources, Writing – review & editing. TKol: Resources, Writing – original draft. ABy: Project administration, Writing – review & editing, Funding acquisition. NK: Conceptualization, Investigation, Writing – review & editing. AE: Investigation, Resources, Writing – review & editing. AG: Resources, Investigation, Writing – review & editing. MK: Writing – original draft, Investigation. AT: Resources, Writing – original draft. IM: Writing – original draft, Investigation. TKoc: Investigation, Writing – original draft. IS: Writing – original draft, Investigation. JS: Software, Methodology, Writing – review & editing. EU: Resources, Writing – review & editing. SY: Funding acquisition, Project administration, Writing – review & editing. DT: Funding acquisition, Writing – review & editing, Project administration. GS: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by a grant from the Russian Science Foundation (Grant No. 24-14-00460).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2025.1687148/full#supplementary-material
References
1. Golezar S, Ramezani Tehrani F, Khazaei S, Ebadi A, and Keshavarz Z. The global prevalence of primary ovarian insufficiency and early menopause: a meta-analysis. Climacteric. (2019) 22:403–11. doi: 10.1080/13697137.2019.1574738
2. Panay N, Anderson RA, Bennie A, Cedars M, Davies M, Ee C, et al. Evidence-based guideline: premature ovarian insufficiency. Hum Reprod Open. (2024) 2024):hoae065. doi: 10.1093/hropen/hoae065
3. Huang Y, Liu Z, Geng Y, Li F, Hu R, Song Y, et al. The risk factors, pathogenesis and treatment of premature ovarian insufficiency. J Ovarian Res. (2025) 18:134. doi: 10.1186/s13048-025-01714-2
4. Federici S, Rossetti R, Moleri S, Munari EV, Frixou M, Bonomi M, et al. Primary ovarian insufficiency: update on clinical and genetic findings. Front Endocrinol. (2024) 15:1464803. doi: 10.3389/fendo.2024.1464803
5. Silvén H, Savukoski SM, Pesonen P, Pukkala E, Ojaniemi M, Gissler M, et al. Association of genetic disorders and congenital malformations with premature ovarian insufficiency: a nationwide register-based study. Hum Reprod. (2023) 38:1224–30. doi: 10.1093/humrep/dead066
6. Gruber N, Kugler S, De Vries L, Brener A, Zung A, Eyal O, et al. Primary ovarian insufficiency nationwide incidence rate and etiology among Israeli adolescents. J Adolesc Health. (2020) 66:603–9. doi: 10.1016/j.jadohealth.2019.11.315
7. Silvén H, Savukoski SM, Pesonen P, Pukkala E, Gissler M, Suvanto E, et al. Incidence and familial risk of premature ovarian insufficiency in the Finnish female population. Hum Reprod. (2022) 37:1030–6. doi: 10.1093/humrep/deac014
8. Huhtaniemi I, Hovatta O, La Marca A, Livera G, Monniaux D, Persani L, et al. Advances in the molecular pathophysiology, genetics, and treatment of primary ovarian insufficiency. Trends Endocrinol Metab TEM. (2018) 29:400–19. doi: 10.1016/j.tem.2018.03.010
9. Ke H, Tang S, Guo T, Hou D, Jiao X, Li S, et al. Landscape of pathogenic mutations in premature ovarian insufficiency. Nat Med. (2023) 29:483–92. doi: 10.1038/s41591-022-02194-3
10. Heddar A, Ogur C, Da Costa S, Braham I, Billaud-Rist L, Findikli N, et al. Genetic landscape of a large cohort of Primary Ovarian Insufficiency: New genes and pathways and implications for personalized medicine. EBioMedicine. (2022) 84:104246. doi: 10.1016/j.ebiom.2022.104246
11. Jiao X, Ke H, Qin Y, and Chen Z-J. Molecular genetics of premature ovarian insufficiency. Trends Endocrinol Metab TEM. (2018) 29:795–807. doi: 10.1016/j.tem.2018.07.002
12. Csehely S, Kun A, Orbán E, Katona T, Orosz M, Krasznai ZT, et al. Changing etiological spectrum of premature ovarian insufficiency over the past decades: A comparative analysis of two cohorts from a single center. Diagnostics. (2025) 15:1724. doi: 10.3390/diagnostics15131724
13. Yi Y, Fu J, Xie S, Zhang Q, Xu B, Wang Y, et al. Association between ovarian reserve and spontaneous miscarriage and their shared genetic architecture. Hum Reprod. (2023) 38:2247–58. doi: 10.1093/humrep/dead180
14. Tang F, Gao Y, Li K, Tang D, Hao Y, Lv M, et al. Novel deleterious splicing variant in HFM1 causes gametogenesis defect and recurrent implantation failure: concerning the risk of chromosomal abnormalities in embryos. J Assist Reprod Genet. (2023) 40:1689–702. doi: 10.1007/s10815-023-02761-8
15. Xu Q, Ding H, Liu Y, Han D, Xia X, Li Y, et al. Genetic variants in diminished ovarian reserve and premature ovarian insufficiency: implications for assisted reproductive outcomes. J Assist Reprod Genet. (2025). doi: 10.1007/s10815-025-03663-7
16. McGlacken-Byrne SM, Suntharalingham JP, Ishida M, Buonocore F, Del Valle I, Cameron-Pimblett A, et al. A tiered approach to exome sequencing analysis in early-onset primary ovarian insufficiency. J Clin Endocrinol Metab. (2025) 110:3142–54. doi: 10.1210/clinem/dgaf124
17. Bakhsh H. An evidence-based approach to the management of primary ovarian insufficiency in adolescents and young women. Life. (2025) 15:1366. doi: 10.3390/life15091366
18. Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. (2020) 22:245–57. doi: 10.1038/s41436-019-0686-8
19. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
20. La Marca A, Spada E, Sighinolfi G, Argento C, Tirelli A, Giulini S, et al. Age-specific nomogram for the decline in antral follicle count throughout the reproductive period. Fertil Steril. (2011) 95:684–8. doi: 10.1016/j.fertnstert.2010.07.1069
21. Jolly A, Bayram Y, Turan S, Aycan Z, Tos T, Abali ZY, et al. Exome sequencing of a primary ovarian insufficiency cohort reveals common molecular etiologies for a spectrum of disease. J Clin Endocrinol Metab. (2019) 104:3049–67. doi: 10.1210/jc.2019-00248
22. Bestetti I, Barbieri C, Sironi A, Specchia V, Yatsenko SA, De Donno MD, et al. Targeted whole exome sequencing and Drosophila modelling to unveil the molecular basis of primary ovarian insufficiency. Hum Reprod. (2021) 36:2975–91. doi: 10.1093/humrep/deab192
23. Yang X, Touraine P, Desai S, Humphreys G, Jiang H, Yatsenko A, et al. Gene variants identified by whole-exome sequencing in 33 French women with premature ovarian insufficiency. J Assist Reprod Genet. (2019) 36:39–45. doi: 10.1007/s10815-018-1349-4
24. Cozette C, Pujalte M, Celton N, Bosquet D, Copin H, Cabry R, et al. Genetics investigation of idiopathic premature ovarian insufficiency: contribution of array-CGH and next-generation sequencing. Genes. (2025) 16:251. doi: 10.3390/genes16030251
25. Rodrigues B, Sousa V, Yrigollen CM, Tassone F, Villate O, Allen EG, et al. FMR1 allelic complexity in premutation carriers provides no evidence for a correlation with age at amenorrhea. Reprod Biol Endocrinol. (2024) 22:71. doi: 10.1186/s12958-024-01227-5
26. De Caro JJ, Dominguez C, and Sherman SL. Reproductive health of adolescent girls who carry the FMR1 premutation: expected phenotype based on current knowledge of fragile X–associated primary ovarian insufficiency. Ann N Y Acad Sci. (2008) 1135:99–111. doi: 10.1196/annals.1429.029
27. Luo W, Ke H, Tang S, Jiao X, Li Z, Zhao S, et al. Next-generation sequencing of 500 POI patients identified novel responsible monogenic and oligogenic variants. J Ovarian Res. (2023) 16:39. doi: 10.1186/s13048-023-01104-6
28. Shekari S, Stankovic S, Gardner EJ, Hawkes G, Kentistou KA, Beaumont RN, et al. Penetrance of pathogenic genetic variants associated with premature ovarian insufficiency. Nat Med. (2023) 29:1692–9. doi: 10.1038/s41591-023-02405-5
29. Zhou Y, Jin Y, Wu T, Wang Y, Dong Y, Chen P, et al. New insights on mitochondrial heteroplasmy observed in ovarian diseases. J Adv Res. (2024) 65:211–26. doi: 10.1016/j.jare.2023.11.033
30. Li J, Liao Q, Guo Y, Zhang J, Zhang R, Liu Q, et al. Mechanism of crosstalk between DNA methylation and histone acetylation and related advances in diagnosis and treatment of premature ovarian failure. Epigenetics. (2025) 20:2528563. doi: 10.1080/15592294.2025.2528563
31. Nadesapillai S, van der Velden J, Smeets D, Van De Zande G, Braat D, Fleischer K, et al. Why are some patients with 45,X Turner syndrome fertile? A young girl with classical 45,X Turner syndrome and a cryptic mosaicism in the ovary. Fertil Steril. (2021) 115:1280–7. doi: 10.1016/j.fertnstert.2020.11.006
32. Rouen A, Rogers E, Kerlan V, Delemer B, Catteau-Jonard S, Reznik Y, et al. Whole exome sequencing in a cohort of familial premature ovarian insufficiency cases reveals a broad array of pathogenic or likely pathogenic variants in 50% of families. Fertil Steril. (2022) 117:843–53. doi: 10.1016/j.fertnstert.2021.12.023
33. Ali R, Al-Dewik N, Mohammed S, Elfituri M, Agouba S, Musa S, et al. Expanding on the phenotypic spectrum of Woodhouse-Sakati syndrome due to founder pathogenic variant in DCAF17 : Report of 58 additional patients from Qatar and literature review. Am J Med Genet A. (2022) 188:116–29. doi: 10.1002/ajmg.a.62501
34. Khalili Dehkordi A and Vakili R. DCAF17 mutation in woodhouse–sakati syndrome: A case report on a novel homozygous variant. Case Rep Pediatr. (2025) 2025:9913412. doi: 10.1155/crpe/9913412
35. Amosova M, Poluboyarinova I, Fadeev V, and Asanov A. Woodhouse-sakati syndrome: the new genetic variant of DCAF17 in 2 adult sisters. JCEM Case Rep. (2024) 2:luae130. doi: 10.1210/jcemcr/luae130
36. Alhuzaim ON, Ahmad MM, Sherbeeni SM, Almotawa F, Ali AS, and Alhejaily A-MG. Three siblings with woodhouse-sakati syndrome: A case report of A new Saudi family. Cureus. (2022) 14. doi: 10.7759/cureus.32225
37. Zhou M, Shi N, Zheng J, Chen Y, Wang S, Xiao K, et al. Case report: A Chinese family of woodhouse-sakati syndrome with diabetes mellitus, with a novel biallelic deletion mutation of the DCAF17 gene. Front Endocrinol. (2021) 12:770871. doi: 10.3389/fendo.2021.770871
38. Agopiantz M, Corbonnois P, Sorlin A, Bonnet C, Klein M, Hubert N, et al. Endocrine disorders in Woodhouse-Sakati syndrome: a systematic review of the literature. J Endocrinol Invest. (2014) 37:1–7. doi: 10.1007/s40618-013-0001-5
39. Gurbuz F, Desai S, Diao F, Turkkahraman D, Wranitz F, Wood-Trageser M, et al. Novel inactivating mutations of the DCAF17 gene in American and Turkish families cause male infertility and female subfertility in the mouse model. Clin Genet. (2018) 93:853–9. doi: 10.1111/cge.13183
40. Kohil A, Abdallah AM, Hussain K, and Al-Shafai M. Genetic epidemiology of Woodhouse-Sakati Syndrome in the Greater Middle East region and beyond: a systematic review. Orphanet J Rare Dis. (2023) 18:22. doi: 10.1186/s13023-023-02614-8
41. Meng T, Zhang W, Zhang R, Li J, Gao Y, Qin Y, et al. Ovarian reserve and ART outcomes in blepharophimosis-ptosis-epicanthus inversus syndrome patients with FOXL2 mutations. Front Endocrinol. (2022) 13:829153. doi: 10.3389/fendo.2022.829153
42. Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, Shafeghati Y, et al. LMNA mutations in atypical Werner’s syndrome. Lancet. (2003) 362:440–5. doi: 10.1016/s0140-6736(03)14069-x
43. McPherson E, Turner L, Zador I, Reynolds K, Macgregor D, and Giampietro PF. Ovarian failure and dilated cardiomyopathy due to a novel lamin mutation. Am J Med Genet A. (2009) 149A:567–72. doi: 10.1002/ajmg.a.32627
44. Flury G and Arquint F. A case report of non-lamin A/C dilated cardiomyopathy presenting in a patient with Najjar–Malouf syndrome. Eur Heart J - Case Rep. (2025) 9. doi: 10.1093/ehjcr/ytaf110
45. Gersak K, Strgulc M, Gorjup V, Dolenc-Strazar Z, Jurcic V, Penny DJ, et al. Dilated cardiomyopathy and ovarian dysgenesis in a patient with Malouf syndrome: A case report. Mol Med Rep. (2013) 8:1311–4. doi: 10.3892/mmr.2013.1669
46. Vaykhanskaya TG, Sivitskaya LN, Danilenko NG, Kurushko TV, Nizhnikova OG, and Davydenko OG. An orphan phenotype of cardiogenital laminopathy — Malouf syndrome. Russ J Cardiol. (2016) 90–4. doi: 10.15829/1560-4071-2016-11-90-94
47. Alsheimer M, Liebe B, Sewell L, Stewart CL, Scherthan H, and Benavente R. Disruption of spermatogenesis in mice lacking A-type lamins. J Cell Sci. (2004) 117:1173–8. doi: 10.1242/jcs.00975
48. Huang C, Zhao S, Yang Y, Guo T, Ke H, Mi X, et al. TP63 gain-of-function mutations cause premature ovarian insufficiency by inducing oocyte apoptosis. J Clin Invest. (2023) 133:e162315. doi: 10.1172/JCI162315
49. Ding L, Deng S, Zhang P, Zhang D, and Tian Q. Identification of novel variants and candidate genes in women with 46,XX complete gonadal dysgenesis. Reprod Biol Endocrinol. (2024) 22:140. doi: 10.1186/s12958-024-01309-4
50. Tucker EJ, Jaillard S, Grover SR, Den Bergen J, Robevska G, Bell KM, et al. TP63-truncating variants cause isolated premature ovarian insufficiency. Hum Mutat. (2019) 40:886–92. doi: 10.1002/humu.23744
51. Vanderschelden RK, Rodriguez-Escriba M, Chan SH, Berman AJ, Rajkovic A, and Yatsenko SA. Heterozygous TP63 pathogenic variants in isolated primary ovarian insufficiency. J Assist Reprod Genet. (2023) 40:2211–8. doi: 10.1007/s10815-023-02886-w
52. Adams IR and Davies OR. Meiotic chromosome structure, the synaptonemal complex, and infertility. Annu Rev Genomics Hum Genet. (2023) 24:35–61. doi: 10.1146/annurev-genom-110122-090239
53. Winters T, McNicoll F, and Jessberger R. Meiotic cohesin STAG3 is required for chromosome axis formation and sister chromatid cohesion. EMBO J. (2014) 33:1256–70. doi: 10.1002/embj.201387330
54. Tsabai PN, Pavlova NS, Shatylko TV, Kumykova Z, Stupko OK, Kochetkova TO, et al. Novel STAG3 variant causes oligoasthenoteratozoospermia with high sperm aneuploidy rate. J Assist Reprod Genet. (2025) 42:1239–45. doi: 10.1007/s10815-025-03417-5
55. Bergant G, Maver A, Lovrecic L, Čuturilo G, Hodzic A, and Peterlin B. Comprehensive use of extended exome analysis improves diagnostic yield in rare disease: a retrospective survey in 1,059 cases. Genet Med. (2018) 20:303–12. doi: 10.1038/gim.2017.142
56. Jaillard S, McElreavy K, Robevska G, Akloul L, Ghieh F, Sreenivasan R, et al. STAG3 homozygous missense variant causes primary ovarian insufficiency and male non-obstructive azoospermia. Mol Hum Reprod. (2020) 26:665–77. doi: 10.1093/molehr/gaaa050
57. Akbari A, Zoha Tabatabaei S, Salehi N, Padidar K, Almadani N, Ali Sadighi Gilani M, et al. Novel STAG3 variant associated with primary ovarian insufficiency and non-obstructive azoospermia in an Iranian consanguineous family. Gene. (2022) 821:146281. doi: 10.1016/j.gene.2022.146281
58. Heddar A, Dessen P, Flatters D, and Misrahi M. Novel STAG3 mutations in a Caucasian family with primary ovarian insufficiency. Mol Genet Genomics. (2019) 294:1527–34. doi: 10.1007/s00438-019-01594-4
59. Alhathal N, Maddirevula S, Coskun S, Alali H, Assoum M, Morris T, et al. A genomics approach to male infertility. Genet Med. (2020) 22:1967–75. doi: 10.1038/s41436-020-0916-0
60. Huang T, Wu X, Wang S, Bao Z, Wan Y, Wang Z, et al. SPIDR is required for homologous recombination during mammalian meiosis. Nucleic Acids Res. (2023) 51:3855–68. doi: 10.1093/nar/gkad154
61. Smirin-Yosef P, Zuckerman-Levin N, Tzur S, Granot Y, Cohen L, Sachsenweger J, et al. A biallelic mutation in the homologous recombination repair gene SPIDR is associated with human gonadal dysgenesis. J Clin Endocrinol Metab. (2017) 102:681–8. doi: 10.1210/jc.2016-2714
62. Heddar A, Guichoux N, Auger N, and Misrahi MA. SPIDR homozygous nonsense pathogenic variant in isolated primary ovarian insufficiency with chromosomal instability. Clin Genet. (2022) 101:242–6. doi: 10.1111/cge.14080
63. Hyon C, Mansour-Hendili L, Chantot-Bastaraud S, Donadille B, Kerlan V, Dodé C, et al. Deletion of CPEB1 gene: A rare but recurrent cause of premature ovarian insufficiency. J Clin Endocrinol Metab. (2016) 101:2099–104. doi: 10.1210/jc.2016-1291
64. Bestetti I, Castronovo C, Sironi A, Caslini C, Sala C, Rossetti R, et al. High-resolution array-CGH analysis on 46,XX patients affected by early onset primary ovarian insufficiency discloses new genes involved in ovarian function. Hum Reprod. (2019) 34:574–83. doi: 10.1093/humrep/dey389
65. Chen Z, Chen H, Yuan K, and Wang C. A 15q25.2 microdeletion phenotype for premature ovarian failure in a Chinese girl: a case report and review of literature. BMC Med Genomics. (2020) 13:126. doi: 10.1186/s12920-020-00787-w
66. Zhang D, Liu Y, Zhang Z, Lv P, Liu Y, Li J, et al. Basonuclin 1 deficiency is a cause of primary ovarian insufficiency. Hum Mol Genet. (2018) 27:3787–800. doi: 10.1093/hmg/ddy261
67. Wang F, Liu Y, Ni F, Jin J, Wu Y, Huang Y, et al. BNC1 deficiency-triggered ferroptosis through the NF2-YAP pathway induces primary ovarian insufficiency. Nat Commun. (2022) 13. doi: 10.1038/s41467-022-33323-8
68. Takahashi N, Franciosi F, Daldello EM, Luong XG, Althoff P, Wang X, et al. CPEB1-dependent disruption of the mRNA translation program in oocytes during maternal aging. Nat Commun. (2023) 14. doi: 10.1038/s41467-023-35994-3
69. Wat MJ, Enciso VB, Wiszniewski W, Resnick T, Bader P, Roeder ER, et al. Recurrent microdeletions of 15q25.2 are associated with increased risk of congenital diaphragmatic hernia, cognitive deficits and possibly Diamond-Blackfan anaemia. J Med Genet. (2010) 47:777–81. doi: 10.1136/jmg.2009.075903
70. Chen X, Chen L, Wang Y, Shu C, Zhou Y, Wu R, et al. Identification and characterization of novel compound heterozygous variants in FSHR causing primary ovarian insufficiency with resistant ovary syndrome. Front Endocrinol. (2023) 13:1013894. doi: 10.3389/fendo.2022.1013894
71. Lokchine A, Bergougnoux A, Servant N, Akloul L, Launay E, Mary L, et al. Identification and characterization of novel FSHR copy number variations causing premature ovarian insufficiency. Am J Med Genet A. (2025) 197. doi: 10.1002/ajmg.a.63924
72. He W-B, Du J, Yang X-W, Li W, Tang W-L, Dai C, et al. Novel inactivating mutations in the FSH receptor cause premature ovarian insufficiency with resistant ovary syndrome. Reprod BioMed Online. (2019) 38:397–406. doi: 10.1016/j.rbmo.2018.11.011
73. Cooper OO, Quint EH, Smith YR, and Dendrinos ML. FSH receptor variant: an unusual cause of secondary amenorrhea. J Pediatr Adolesc Gynecol. (2025), S1083318825002748. doi: 10.1016/j.jpag.2025.06.002
74. Maylem ERS, Spicer LJ, Batalha I, and Schutz LF. Discovery of a possible role of asprosin in ovarian follicular function. J Mol Endocrinol. (2021) 66:35–44. doi: 10.1530/jme-20-0218
75. Jordan P, Verebi C, Perol S, Grotto S, Fouveaut C, Christin-Maitre S, et al. NOBOX gene variants in premature ovarian insufficiency: ethnicity-dependent insights. J Assist Reprod Genet. (2024) 41:135–46. doi: 10.1007/s10815-023-02981-y
76. Veitia RA, Cowles JD, and Caburet S. Reclassifying NOBOX variants in primary ovarian insufficiency cases with a corrected gene model and a novel quantitative framework. Hum Reprod. (2025) 40:1220–33. doi: 10.1093/humrep/deaf058
77. Hamanaka K, Takata A, Uchiyama Y, Miyatake S, Miyake N, Mitsuhashi S, et al. MYRF haploinsufficiency causes 46,XY and 46,XX disorders of sex development: bioinformatics consideration. Hum Mol Genet. (2019) 28:2319–29. doi: 10.1093/hmg/ddz066
78. Calonga-Solís V, Fabbri-Scallet H, Ott F, Al-Sharkawi M, Künstner A, Wünsch L, et al. MYRF: A new regulator of cardiac and early gonadal development—Insights from single cell RNA sequencing analysis. J Clin Med. (2022) 11:4858. doi: 10.3390/jcm11164858
79. Slaba K, Jezova M, Pokorna P, Palova H, Tuckova J, Papez J, et al. Two sisters with cardiac-urogenital syndrome secondary to pathogenic splicing variant in the MYRF gene with unaffected parents: A case of gonadal mosaicism? Mol Genet Genomic Med. (2023) 11:e2139. doi: 10.1002/mgg3.2139
80. Qi H, Yu L, Zhou X, Wynn J, Zhao H, Guo Y, et al. De novo variants in congenital diaphragmatic hernia identify MYRF as a new syndrome and reveal genetic overlaps with other developmental disorders. PloS Genet. (2018) 14:e1007822. doi: 10.1371/journal.pgen.1007822
81. Ding L and Tian Q. Identification of a novel variant in MYRF gene in a patient with 46, XX disorders of sex development. Gynecol Endocrinol. (2025) 41:2546985. doi: 10.1080/09513590.2025.2546985
82. Tsabai PN, Batyrova ZK, Kumykova Z, Kovalskaya VA, Sidorchuk MA, Goltsov A, et al. Uterus didelphys, longitudinal vaginal septum and diminished ovarian reserve in a patient with MYRF-associated disease. Akush Ginekol (Sofiia). (2025) 4_2025:171–7. doi: 10.18565/aig.2024.321
83. Clark KL, George JW, Przygrodzka E, Plewes MR, Hua G, Wang C, et al. Hippo signaling in the ovary: emerging roles in development, fertility, and disease. Endocr Rev. (2022) 43:1074–96. doi: 10.1210/endrev/bnac013
84. Sun T, Pepling ME, and Diaz FJ. Lats1 deletion causes increased germ cell apoptosis and follicular cysts in mouse ovaries1. Biol Reprod. (2015) 93:1–11. doi: 10.1095/biolreprod.114.118604
85. St John MAR, Tao W, Fei X, Fukumoto R, Carcangiu ML, Brownstein DG, et al. Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat Genet. (1999) 21:182–6. doi: 10.1038/5965
86. Tsoi M, Morin M, Rico C, Johnson RL, Paquet M, Gévry N, et al. Lats1andLats2are required for ovarian granulosa cell fate maintenance. FASEB J. (2019) 33:10819–32. doi: 10.1096/fj.201900609r
Keywords: premature ovarian insufficiency, ovarian dysgenesis, whole-exome sequencing (WES), FMR1, differences of sex development (DSD), adolescent, genetics
Citation: Tsabai P, Kumykova Z, Averkova V, Pavlova N, Maslennikov D, Bolshakova A, Batyrova Z, Kolpakova T, Bystritskiy A, Karetnikova N, Ekimov A, Goltsov A, Kuznetsova M, Turchinets A, Mukosey I, Kochetkova T, Sadelov I, Shubina J, Uvarova E, Yureneva S, Trofimov D and Sukhikh G (2025) Novel variants associated with premature ovarian insufficiency in Russian adolescents. Front. Endocrinol. 16:1687148. doi: 10.3389/fendo.2025.1687148
Received: 16 August 2025; Accepted: 05 November 2025; Revised: 05 November 2025;
Published: 27 November 2025.
Edited by:
Alfredo Ulloa-Aguirre, National Autonomous University of Mexico, MexicoReviewed by:
Xiaopan Chen, Zhejiang Provincial People’s Hospital, ChinaAbdelkader Heddar, Laboratoire National de Santé, Luxembourg
Copyright © 2025 Tsabai, Kumykova, Averkova, Pavlova, Maslennikov, Bolshakova, Batyrova, Kolpakova, Bystritskiy, Karetnikova, Ekimov, Goltsov, Kuznetsova, Turchinets, Mukosey, Kochetkova, Sadelov, Shubina, Uvarova, Yureneva, Trofimov and Sukhikh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Polina Tsabai, cG9saW5hdHNhYmFpQGdtYWlsLmNvbQ==