


- 1 Medicinal Chemistry and Drug Action, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC, Australia
- 2 Centre for Immunology, Burnet Institute, Melbourne, VIC, Australia
- 3 Department of Surgery Austin Health, University of Melbourne, Heidelberg, VIC, Australia
- 4 Department of Immunology, Alfred Medical Research and Education Precinct, Monash University, Melbourne, VIC, Australia
Recognition of pathogen-associated carbohydrates by a broad range of carbohydrate-binding proteins is central to both adaptive and innate immunity. A large functionally diverse group of mammalian carbohydrate-binding proteins are lectins, which often display calcium-dependent carbohydrate interactions mediated by one or more carbohydrate recognition domains. We report here the application of molecular docking and site mapping to study carbohydrate recognition by several lectins involved in innate immunity or in modulating adaptive immune responses. It was found that molecular docking programs can identify the correct carbohydrate-binding mode, but often have difficulty in ranking it as the best pose. This is largely attributed to the broad and shallow nature of lectin binding sites, and the high flexibility of carbohydrates. Site mapping is very effective at identifying lectin residues involved in carbohydrate recognition, especially with cases that were found to be particularly difficult to characterize via molecular docking. This study highlights the need for alternative strategies to examine carbohydrate–lectin interactions, and specifically demonstrates the potential for mapping methods to extract additional and relevant information from the ensembles of binding poses generated by molecular docking.
Introduction
Recognition of pathogens by the innate immune system involves a multitude of cellular receptors and secreted proteins that can be broadly classified as pattern recognition receptors (PRRs), which have inherent specificities for pathogen-associated molecular patterns (PAMPs). Within the PAMPs there are a diversity of carbohydrates that are either unique to the pathogen or share similarities to autologous or “self” carbohydrates of the host (e.g., Lewis type histo-blood group carbohydrates; Yuriev et al., 2005). In humans, like other mammals, PRRs include the Toll-like receptors that often have specificity for both carbohydrates and non-carbohydrate PAMPs. There is also a diversity of carbohydrate-binding proteins (lectins) that have roles both in innate immunity as PRRs and in modulating adaptive immunity. Lectins functioning as PRRs often contain multiple carbohydrate recognition domains and are subdivided into the C-type lectins, galectins, and siglecs and are further grouped within these families according to their sequences, structures, molecular locations, and fine specificities (Kerrigan and Brown, 2009; Sato et al., 2009; Dam and Brewer, 2010).
As a group, lectins have been shown to be structurally diverse and adopt a wide range of folds (Gabius, 2008). Despite this, lectins from different species have been demonstrated to bind to the same carbohydrate, often utilizing similar mechanisms of recognition (Yuriev et al., 2009). However, lectins may recognize multiple carbohydrates either through the same binding site or by the assembly of closely related monomeric structures. As discussed later in this manuscript, several mammalian C-type lectins, such as DC-SIGN and human lung surfactant protein D (SP-D), have quite shallow binding sites which permit the recognition of multiple types of glycans. Galectins, of which there are at least 15 in mammals, bind β-galactose residues usually in the form of N-acetyllactosamine-containing glycans found in many pathogen and host glycoproteins and glycolipids (Rabinovich and Toscano, 2009). Siglecs bind to sialic acid residues often displayed at the termini of a range of complex glycan structures in pathogen and host molecules and play a multitude of roles in adaptive and innate immunity (Crocker et al., 2007).
While the role of lectins in innate immunity is to recognize and defend against invading pathogens, occasionally through adopting or mimicking carbohydrates of the host, pathogens can gain entry into cells and establish infection. For example, the high mannose shield of the HIV envelope glycoprotein binds to DC-SIGN as the first step in HIV invasion of dendritic cells and some macrophages (Hertje et al., 2010). The same type of interaction with langerin, a C-type lectin restricted to Langerhans cells, transfers HIV to the intracellular Birbeck granules for degradation and clearance (Van Der Vlist and Geijtenbeek, 2010). Similar to DC-SIGN and langerin, cell membrane-associated lectins are often involved in phagocytosis of pathogens for elimination or processing for antigen presentation (Kerrigan and Brown, 2009). In addition, secreted lectins (e.g., some of the galectins and SP-D) are typically involved as soluble PPRs in blocking pathogen invasion of the host and mediating signaling pathways, such as the lectin complement activation pathway (Wallis et al., 2010).
The large amount of carbohydrate–lectin functional and interaction data that is being generated by major international glycomics efforts has led us to pursue computational strategies to both augment the existing structural knowledge and to provide new insights into the structural aspects of carbohydrate recognition by lectins. In this regard, computational (in silico) techniques are particularly useful. Homology modeling can be used to generate the structure of a lectin in the absence of an experimentally determined structure. Molecular docking can be used to investigate carbohydrate recognition by a lectin where that particular carbohydrate–lectin complex is unavailable. Molecular dynamics can be used to study the movement of the protein over time, and how this movement affects ligand binding. We previously developed a site mapping technique to investigate carbohydrate–antibody recognition (Agostino et al., 2009b). The technique considers multiple binding modes obtained from molecular docking (thereby considering the dynamic behavior of the system) in order to identify the most likely interacting residues of the protein. We have also developed ligand-based mapping techniques, specific for carbohydrates, which, in combination with site mapping output, were used to determine likely carbohydrate–antibody complexes (Agostino et al., 2010). The combination of site- and ligand-based mapping procedures allows us to “score” individual carbohydrate poses, and therefore determine the likelihood of observing a particular pose.
In this study, we extend our previous in silico work in studying carbohydrate–antibody recognition to carbohydrate–lectin recognition. Specifically, we investigate the application of molecular docking and site mapping to key carbohydrate–lectin systems (galectins, DC-SIGN, langerin, and SP-D), for which high quality crystal structures as complexes with carbohydrates were available. Our results demonstrate that docking algorithms can identify the correct binding modes, but cannot effectively score them. Site mapping offers substantial improvements in interaction prediction compared to the top ranked pose. The computational approach described alongside future developments should be generally applicable for investigating carbohydrate recognition by lectins involved in innate immunity.
Materials and Methods
Selection and Preparation of Test Systems
A series of 15 high resolution (≤2.0 Å) human-derived carbohydrate–lectin complexes were selected from the Protein Data Bank (Table 1). For multimeric structures, only a single monomer was used (Table 1) and all other chains were removed from the structure. Once reduced to the appropriate monomer, the structures were prepared using the Protein Preparation Wizard workflow implemented in Maestro 9.1. All water molecules were removed from the structure. Metal ions within the binding site were retained. The Prime Refinement tool was used to predict side chains for incomplete residues. Ligands were extracted from the crystal structures within Maestro.
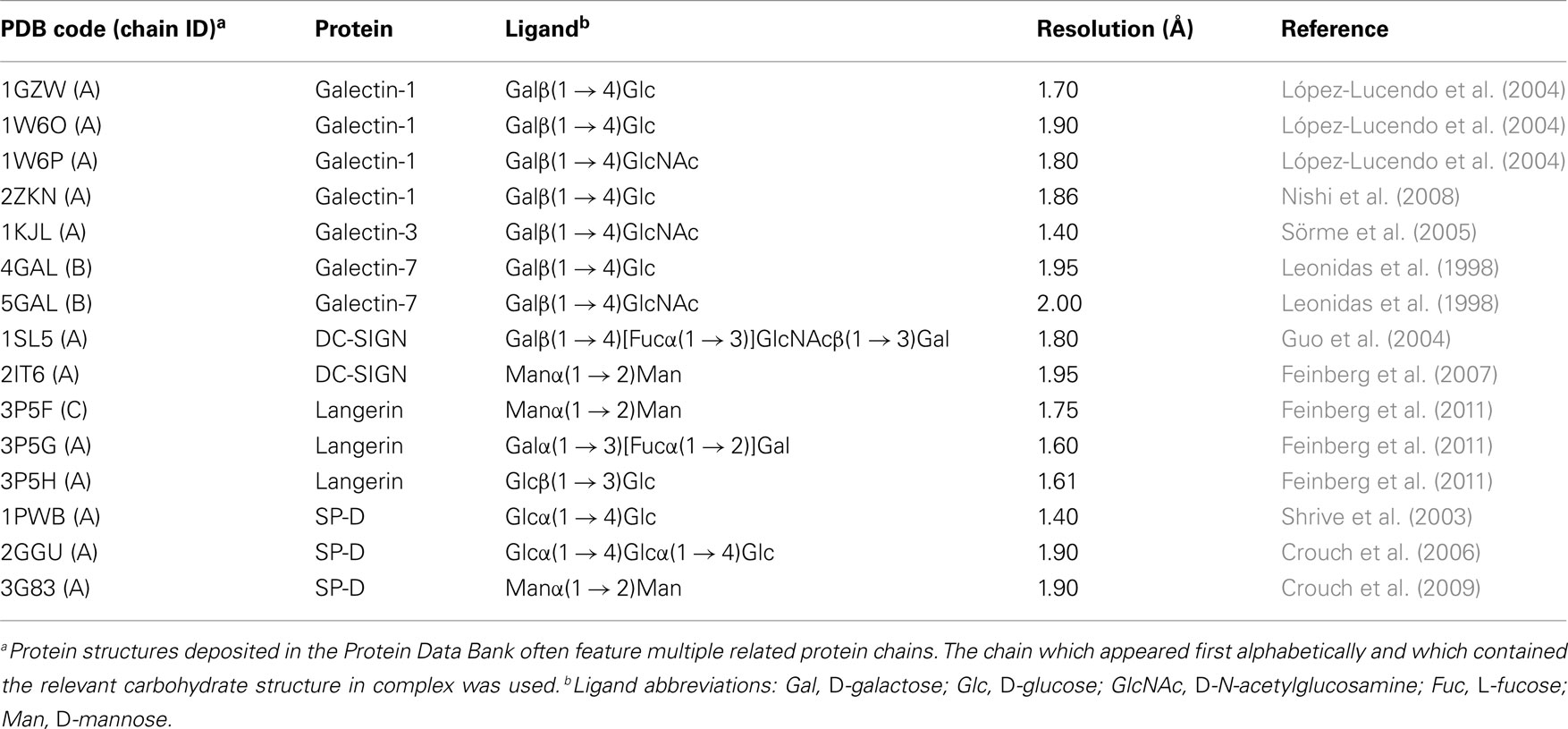
Table 1. Carbohydrate–lectin complexes selected for the test set.
Docking Programs
Glide 5.6 (with Maestro 9.1; Friesner et al., 2004), Autodock 4.2 (with Autodock Tools 1.5.4; Morris et al., 1998), DOCK 6.4 (Lang et al., 2009), and GOLD 4.1.1 (with Hermes 1.3.1; Verdonk et al., 2003) were investigated. Rigid receptor docking was used for all cases (i.e., induced-fit effects were not investigated). Default options were used unless otherwise stated. In each case, the top 100 poses per ligand were retained, clustered using a root-mean-square deviation (rmsd) threshold of 2.0 Å. The rmsd values were calculated for each docked pose relative to crystal structure ligand using the Superposition tool within Maestro. The pose for which the lowest rmsd value was obtained was designated as the best pose.
Glide
The grid box was centered at the ligand centroid and built using default options. The ligand and protein were parameterized with the OPLS force field. Docking was performed using standard precision mode. The option to sample ring conformations was disabled in order to maintain the input conformation, and to prevent the generation of unrealistic ring conformations. Poses were scored using GlideScore.
Autodock
The Autodock Tools interface was used to generate the required input files for Autodock. Gasteiger charges were added to the ligand and protein. The charge on metal ions was set to +2.0. The grid box was centered at the ligand centroid. The genetic algorithm (GA) parameters were modified to ensure thorough exploration of conformational space and energetic convergence of each run. Specifically, a modified set of parameters (Agostino et al., 2009b) compared to those used previously for carbohydrates (Rockey et al., 2000) were employed: 200 runs per ligand, population size of 200 and 106 evaluations per run. All other GA parameters were kept as defaults.
DOCK
AMBER FF99 charges were loaded for the protein using SYBYL-X 1.0. Gasteiger–Marsili charges were calculated for the ligand. Residues within 5.0 Å of the ligand were identified and surfaced using the DMS tool. Normals were calculated for surface points (-n option). The probe radius was set to 1.4 Å for surface preparation. Spheres were generated using the SPHGEN_CPP tool with default options and sphere clusters were converted to PDB format using the SHOWSPHERE tool. The PDB format sphere clusters were visualized using Maestro, and those which best represented the binding site were selected for box generation. A grid box enclosing the selected spheres was automatically generated using the SHOWBOX tool. An additional 5.0 Å margin was added in all directions, to ensure that ligand poses could be well accommodated within the box. For grid generation, the bump filter was used, with a bump overlap of 0.75. For docking, the maximum number of orientations generated per ligand was set to 4000. A minimum anchor size of five heavy atoms was used (rings are not sampled and are therefore typically selected as anchors), and clash overlap was set to 0.75. These options are adapted from our earlier work in studying carbohydrate–antibody and peptide–antibody recognition (Yuriev et al., 2001, 2008).
GOLD
The Wizard within the Hermes interface was used to generate the required input files for GOLD. The binding site was defined using the centroid of the ligand. The binding site cavity was automatically detected based on the input coordinates. The goldscore_p450_csd configuration template was loaded and modified as subsequently detailed. Ligands were subject to 200 GA runs, resulting in 200 poses per ligand. Early termination of the docking run was permitted if the top two ranked poses were within an rmsd of 2.0 Å of one another. The option to generate diverse solutions was enabled. Diverse solutions were clustered to rmsd 2.0 Å. The GA search options were set to very flexible (200% search efficiency). To ensure retention of diverse solutions, the results from the docking run were clustered using the CLUSTER script from the Silico library (obtained from http://silico.sourceforge.net).
Site Mapping Studies
Site mapping was carried out for the 15 complexes according to our previously published procedures using GOLD-generated docking poses (Agostino et al., 2009b). The quality of the maps was assessed using the metrics of reproduction and correctness, which we have previously used to assess the quality of maps of carbohydrate-recognizing antibodies (Agostino et al., 2009b). The map reproduction is computed by dividing the number of crystal structure interactions reproduced by the map by the number of crystal structure interactions observed. Reproduction values close to one indicate that the map is extremely successful at identifying crystal structure interactions. The map correctness is computed by dividing the number of crystal structure interactions reproduced by the map by the total number of mapped interactions. Correctness values close to one indicate that few additional interactions are included in the map. Reproduction and correctness values were also obtained for the top ranked docked pose in each case. In these instances, the number of crystal structure interactions reproduced by the top ranked docked pose and the total number of interactions taking place in the top ranked docked pose were used to compute reproduction and correctness values. Full details of the ligand-contacting residues in the crystal structure, in the top ranked pose, and in the site maps are available for each case in the Appendix (Tables A1–A15).
Results
Evaluation of Molecular Docking for Carbohydrate–Lectin Interactions
While the four docking programs evaluated were generally found to identify reasonable carbohydrate poses in each case, the top ranked pose was rarely found to be the best pose obtained by any of the programs (Table 2). DOCK was able to correctly rank the best pose in 6 out of the 15 cases, followed closely by Glide. The failure to rank the best pose as the top pose in the majority cases is most likely due to a scoring problem. However, for Glide and, to a lesser extent, GOLD, the best poses were typically found within the top 10 ranked poses. Autodock was generally unable to accurately identify the correct pose. This kind of performance discrepancy between Glide, GOLD, and Autodock has been previously observed in both carbohydrate–antibody docking (Agostino et al., 2009a) and carbohydrate–lectin docking (Nurisso et al., 2008). In terms of pose prediction accuracy – regardless of scoring/ranking – GOLD is clearly the best program, with an average rmsd for the best poses of 1.4 Å compared to 2.2–2.7 Å for the other three programs. This docking study highlights that considering the top ranked docking pose alone is not suitable for studying carbohydrate–lectin interactions. However, there were specific cases where docking alone was successful, and also cases where it was surprisingly unsuccessful. Examining these cases in more detail allowed us to identify some of the specific successes and shortcomings of the docking approach when applied to these systems.
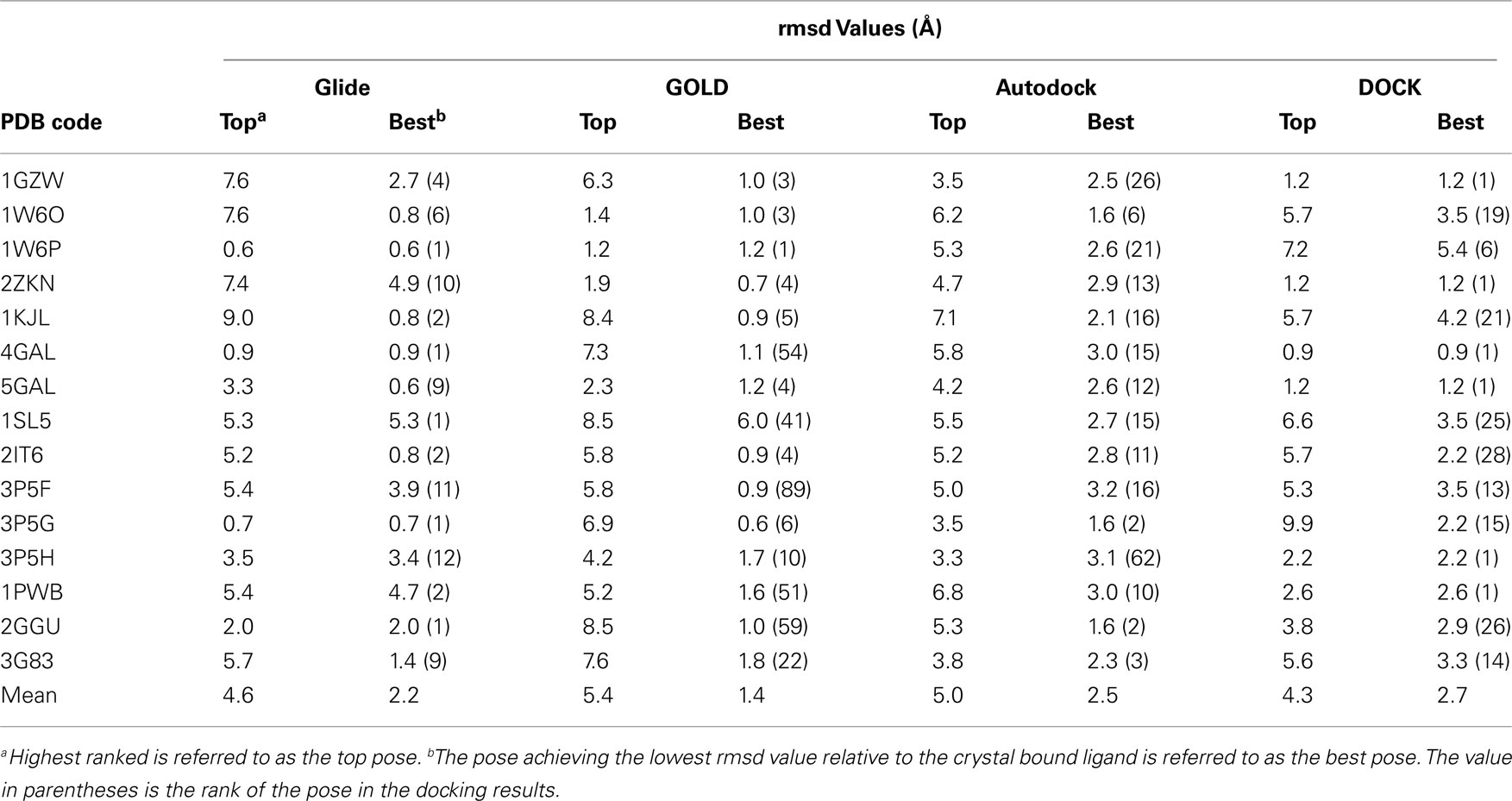
Table 2. Cognate docking performance for the test set.
Carbohydrate recognition could be well reproduced by most programs for at least a few of the galectin test cases (Table 2). Specifically, carbohydrate recognition by galectin-1 was very accurately reproduced by GOLD. In the crystallographic binding modes obtained for ligands in complex with galectin-1, the terminal galactose residue is sandwiched between His52 and Trp68 (Figure 1A). This placement of the galactose residue allows it to interact with a range of polar amino acids in proximity to that portion of the binding site. However, the placement of the ligand is fairly shallow within the binding site. In addition, there is a region adjacent to the binding site that appears able to accommodate ligands. In most of the programs, except for GOLD, the top scoring pose was docked into this adjacent site (Figure 1B). This is not unexpected, since docking algorithms generally aim to maximize the contact surface and number of favorable interactions between the ligand and receptor. Since the ligand is small enough that it can be fully accommodated into this alternative space, and can therefore form more interactions with the protein, such binding modes are favored by scoring functions. In contrast to the other docking programs, GOLD was successful in identifying the binding mode in three out of the four cases (PDB codes 1W6O, 1W6P, and 2ZKN). This may be due to its better treatment of solvation effects, or possibly due to a better consideration of enthalpic versus entropic contributions to binding (Verdonk et al., 2005). It must be noted that subtle changes in binding site organization can have a large influence on docking success, as observed in the cases of 1GZW, 1W6O, and 2ZKN, all of which feature lactose binding to galectin-1, but are reproduced with varying levels of success by each of the programs.
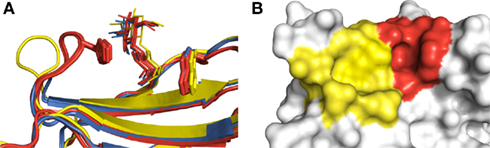
Figure 1. Structural aspects of ligand recognition by galectins. (A) Galectin-1 (PDB codes 1GZW, 1W6O, 1W6P, and 2ZKN, shown in red) features a loop with a histidine residue (His52) which associates closely with the ligand. The terminal galactose is bound between this histidine and an opposite-facing tryptophan (Trp68), effectively sandwiched between the two residues. Galectin-3 (PDB code 1KJL, shown in yellow) does not feature the histidine residue, and the loop does not closely associate with the ligand. In galectin-7 (PDB codes 4GAL and 5GAL, shown in blue), the loop is considerably shortened from that of galectin-1 and galectin-3. (B) Surface view of galectin-3, highlighting the crystallographically observed binding site (red) and the alternative site to which ligands were typically docked (yellow). Galectins-1 and 7 feature similar alternative sites. All structures rendered using PyMOL. For figure (A), structures were overlaid using Maestro.
Galectin-3 and galectin-7 do not feature the His-Trp sandwich binding mode, as the histidine residue is absent from the loop. Furthermore, as the loop has moved away from the bound carbohydrate, the binding sites are considerably more open compared to galectin-1, making accurate scoring by molecular docking quite challenging. This is reflected in almost every program being able to generate the correct binding mode for each of the three cases, but in most cases failing to correctly rank them at the top of the list (Table 2). DOCK was successful in ranking the best as the top pose for the carbohydrate ligands of the two galectin-7 structures (PDB codes 4GAL and 5GAL), suggesting that it correctly placed the anchoring groups in these cases.
The complex of Galα(1 → 3)[Fucα(1 → 2)]Galβ (B antigen) with langerin (PDB 3P5G) was well reproduced by all of the programs, but was only correctly ranked by Glide (Table 2). The binding site of langerin is very broad and shallow, although there appears to be a number of potential subsites (Figure 2A). In the crystal structure of the langerin:B antigen complex (Figure 2B), a fucose-binding site and a galactose-binding site can be observed; the fucose-binding site is formed by a cleft between Lys299 and Ala289, and the α-galactose binds in a site formed by the loop from Pro283 to Asn287. The β-galactose forms a bridge between the two terminal residues and does not directly interact with langerin. Another subsite formed by a cleft between Lys299 and Lys313 is utilized to bind sulfated ligands (Feinberg et al., 2011). Despite the shallow nature of the binding site, it should be possible to successfully dock ligands to langerin, assuming that the docking program used can correctly identify where anchoring segments of the molecule should be placed. While all of the programs can do this, only Glide successfully identified the correct pose. The Glide scoring function was able to identify that the α-galactose and fucose residues form an optimal set of interactions with their respective subsites. The other programs have likely penalized such a binding mode due to the β-galactose portion being exposed to the solvent, failing to identify the importance of the interactions made by the other two residues. While DOCK is successful at ranking the best binding mode for Glcβ(1 → 3)Glc (PDB 3P5H), the pose itself is quite distant from the crystal bound conformation, with an rmsd of 2.2 Å.
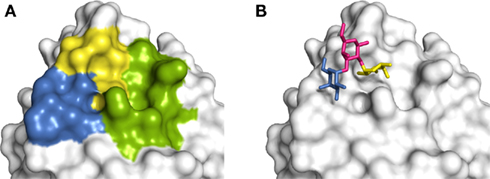
Figure 2. Carbohydrate-binding subsites of langerin. (A) Langerin has three discernable subsites within its binding site, two of which are demonstrated to be involved in carbohydrate recognition. Color guide: blue – α-galactose-binding subsite, yellow – α-fucose-binding subsite, green – potential additional binding site. (B) Langerin in complex with blood group B antigen (Galα(1 → 3)[Fucα(1 → 2)]Galβ). Color guide: blue – α-galactose, yellow – α-fucose, pink – β-galactose. Figures generated in PyMOL using the langerin-B antigen complex structure (PDB code 3P5H).
The complex of Galβ(1 → 4)[Fucα(1 → 3)]GlcNAcβ(1 → 3)Gal with DC-SIGN (PDB 1SL5) was not well reproduced by any of the docking programs. This carbohydrate contains the Lewis X trisaccharide, which, along with other Lewis antigens, is known to be structurally rigid both bound and in solution (Yuriev et al., 2005). Due to the structural rigidity of this section of the molecule, the conformational search undertaken during the docking procedure should find relatively few ligand conformations, and the problem should be largely reduced to that of ligand orientation and placement. To determine if this was indeed the case, we generated Ramachandran-like plots for the Galβ(1 → 4)GlcNAc, Fucα(1 → 3)GlcNAc, and GlcNAcβ(1 → 3)Gal linkages based on the docking results for each program (Figure 3). It was found that the Galβ(1 → 4)GlcNAc and the Fucα(1 → 3)GlcNAc linkages exhibited quite specific conformational minima, which agreed with the experimental values of Φ/ψ = −77.0/131.6° for Galβ(1 → 4)GlcNAc, and Φ/ψ = −53.4/−90.9° for Fucα(1 → 3)GlcNAc. The overall rigidity of these linkages has been found in previous computational and experimental investigations (Yuriev et al., 2005; Jackson et al., 2009; Agostino et al., 2010). The GlcNAcβ(1 → 3)Gal linkage exhibited broader minima, also in agreement with previous findings (Jackson et al., 2009). The Ramachandran-like plots highlight that Glide, GOLD, and DOCK can effectively identify appropriate conformations of the Lewis X trisaccharide in the majority of instances; the experimentally determined conformations occur within the clusters obtained via the conformational search implementation of molecular docking. Autodock performed very poorly in predicting the conformations of Lewis X. The crystallographic conformation for the GlcNAcβ(1 → 3)Gal linkage was not well reproduced by any of the docking programs. Although this part of the molecule is not directly involved in binding in the crystal structure, docking algorithms fit it into the binding site in a variety of ways by rotating this linkage (Figure 4). Such fitting results in an overall greater contacting surface area between the ligand and the receptor. Although the Lewis X portion of the molecule is generally well reproduced, the high flexibility of the GlcNAcβ(1 → 3)Gal linkage affects the ability of docking programs to correctly predict the binding mode. The effect that the shallow nature of the DC-SIGN binding site has on docking success cannot be ignored, and probably plays the more significant role in influencing the quality of docking results. Similar carbohydrates binding to more restrictive binding sites, for example, Lewis Y binding to the antibody hu3S913 (Ramsland et al., 2004; Farrugia et al., 2009), have been extremely well reproduced by docking (Agostino et al., 2009a). However, it is remarkable to note that in the variety of binding modes generated, many still feature the terminal galactose and fucose portions bound at approximately the same location as in the crystal structure. Therefore, there is some possibility that such binding modes are not merely artifacts of docking, but potentially valid in describing ligand recognition by DC-SIGN.
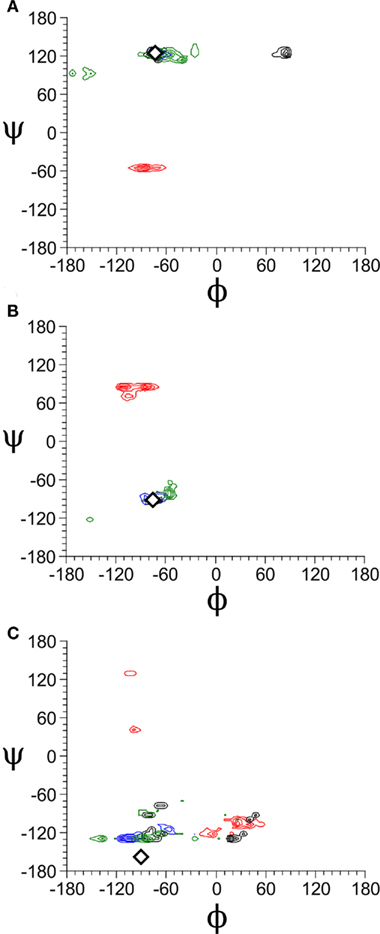
Figure 3. Ramachandran-like plots obtained for cognate docking to PDB 1SL5. (A) Plot obtained for Galβ(1 → 4)GlcNAc linkage. (B) Plot obtained for Fucα(1 → 3)GlcNAc linkage. (C) Plot obtained for GlcNAcβ(1 → 3)Gal linkage. The Φ/ψ angles were calculated for each linkage in each pose given by each of the four programs. The angles were calculated according to the IUPAC convention (Liebecq, 1992); for a given 1 → x linkage, Φ is the dihedral of O5-C1-O1-Cx, and ψ is the dihedral of C1-O1-Cx-Cx−1. Contour plots were generated using QTIPLOT. Color guide: black – Glide, green – GOLD, red – Autodock, blue – DOCK. The crystal structure angle is highlighted by a white diamond in each plot.
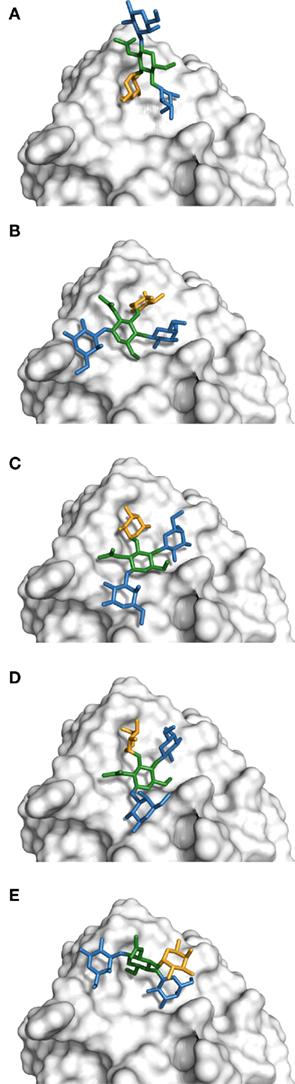
Figure 4. Rotation of the GlcNAcβ(1 → 3)Gal linkage allows many different tetrasaccharide binding modes with DC-SIGN. (A) Crystal structure binding mode (Φ = −84.2, ψ = −157.4; PDB code 1SL5). (B) Top ranked binding mode (Φ = −76.6, ψ = −126.8). (C) Third ranked binding mode (Φ = −81.8, ψ = −130.6). (D) Twelfth ranked binding mode (Φ = −132.4, ψ = −131.5). (E) Twenty-seventh ranked binding mode (Φ = −89.6, ψ = −88.6). Figures (B–E) generated based on representative examples of each conformational cluster of GlcNAcβ(1 → 3)Gal linkage (see Figure 3C) obtained by GOLD docking results. Color guide: blue – galactose, green – N-acetylglucosamine, yellow – fucose.
Although the carbohydrate-binding mode could be generated for each of the human lung SP-D examples, these were generally ranked poorly by all of the programs, often outside of the top 10 poses (Table 2). The binding site of SP-D cannot be easily discerned when examining a surface representation of the protein as it is extremely broad and shallow. Poses docked into such binding sites are generally not effectively scored by docking programs, as noted for our other test systems. The most successful case is the prediction of the complex of SP-D with Glcα(1 → 4)Glcα(1 → 4)Glc (PDB code 2GGU). Glide was able to correctly identify and rank the binding mode, while Autodock ranked the correct binding mode in second place. This suggests that an optimal balance between the size of the binding site and the size of the carbohydrate has been reached, with a trisaccharide motif being able to participate in all of the likely interactions, and for those interactions to be appropriately scored above others (at least by Glide and Autodock). By comparison, reproducing disaccharide binding modes is far more difficult. Since the binding site is so broad and shallow, disaccharides could be more easily made to fit anywhere in the binding site. In the case of reproducing the Glcα(1 → 4)Glc binding mode (PDW 1PWB), DOCK was able to correctly rank the best binding mode, however, it is moderately distant from the crystal bound conformation, with an rmsd of 2.6 Å. In general, the potential for accurate discrimination between binding modes by the scoring functions for a wide range of cases is limited and, consequently, the results of docking are generally not very informative.
Evaluation of Site Mapping for Carbohydrate–Lectin Interactions
It is clear from our molecular docking test cases that some additional interpretation and post-processing of the carbohydrate poses is needed in order to derive useful information regarding carbohydrate recognition by lectins. We previously developed a site mapping technique which we applied to investigate carbohydrate–antibody recognition (Agostino et al., 2009b) and peptide–antibody recognition (Agostino et al., 2011). We wanted to investigate whether this technique could be applied to carbohydrate–lectin systems, to augment the output of molecular docking. The docked poses from GOLD were used as input for site mapping, since this program performed the best at molecular docking in the carbohydrate–lectin systems, at least in terms of identifying the best poses in a given docking run.
The ability of the site maps to identify lectin residues involved in ligand recognition was compared to that of the top pose obtained for each system (Table 3). Using the metrics of reproduction and correctness (see Materials and Methods), it was found that site mapping performed better (reproduction) or comparably (correctness) to the top pose. However, there are several cases where site mapping clearly outperformed the top pose. The most notable examples were the site maps produced for DC-SIGN. The docking programs generally failed to accurately predict the carbohydrate-binding modes of both Galβ(1 → 4)[Fucα(1 → 3)]GlcNAcβ(1 → 3)Gal (PDB code 1SL5) and Manα(1 → 2)Man (PDB code 2IT6) in complex with DC-SIGN, most likely due to the shallow nature of the binding site. Since GOLD failed to correctly identify the ligand binding mode in either case, the accuracy is low for the interactions taking place between DC-SIGN and the top carbohydrate poses (reproduction of 0.25 and 0.55, respectively). In both cases, site mapping was able to identify almost all of the interactions taking place (reproduction of 0.88 and 0.73, respectively). In the case of 1SL5 (Figure 5), several additional contacts appear in the map, as indicated by the relatively low correctness. However, these contacts, not observed in the crystal structure, may yet be important in the dynamics of carbohydrate recognition by DC-SIGN. Regardless, the relative success of site mapping compared to the top pose prediction indicates that this is the preferred technique for studying the interaction of large carbohydrates with shallow binding sites.
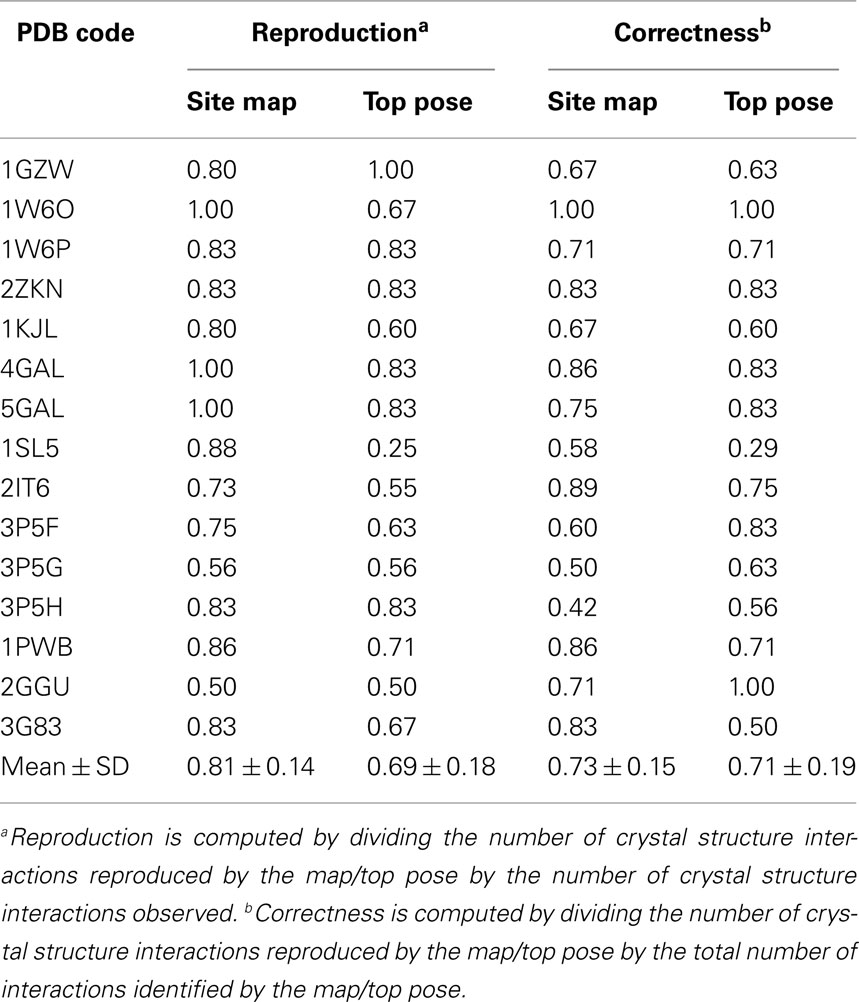
Table 3. Site mapping results for carbohydrate–lectin interactions.
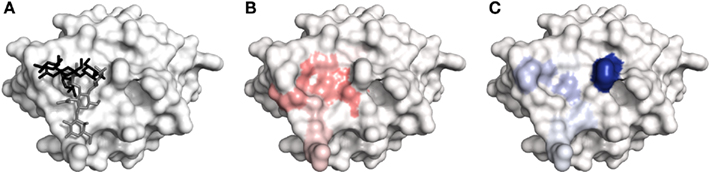
Figure 5. The recognition of Galβ(1 → 4)[Fucα(1 → 3)]GlcNAcβ(1 → 3)Gal by DC-SIGN. (A) Comparison of crystal structure binding mode (PDB code 1SL5; black) with top ranked docked pose (gray). (B) Hydrogen bonding map. (C) van der Waals interaction map. Color depth corresponds to importance of the mapped residue for recognition. All figures generated in PyMOL.
Site mapping was overall found to be quite successful for investigating carbohydrate recognition by the various galectins (Table 3). Notable improvements in interaction prediction made by site mapping, compared to considering the top ranked docked pose alone, were obtained for galectin-3 complexed to N-acetyllactosamine (PDB code 1KJL) and galectin-7 complexed to lactose (PDB code 4GAL). The reason for the observed improvement is the same in each case. In each of the galectins (including galectin-1), the galactose-binding site is centered at Trp68. The terminal galactose moiety interacts with Trp68 via CH-π interactions, and can also form various other interactions. Adjacent to the galactose-binding site is another potential binding site, which appears to be “joined” to the galactose-binding site (Figure 1B). Due to the binding topography, this additional site is unavoidably selected as a potential binding site for docking during protein preparation. Without the aid of a bound ligand, or further experimentally derived knowledge, it would be virtually impossible to discern between the true binding site and this additional site. However, in these examples, we have achieved this using the site mapping technique.
Site mapping generally performed well compared to the top pose in interaction prediction for each of the SP-D cases. The interactions taking place in the two disaccharide complexes were better predicted by the respective site maps rather than by the top docked poses (Figures 6A–C). As noted earlier, it was particularly difficult for molecular docking to identify and score the correct disaccharide poses. Therefore, these cases highlight a situation where site mapping clearly outperforms molecular docking: disaccharide recognition by a lectin with a shallow binding site. However, trisaccharide binding by SP-D was not as well reproduced by site mapping (Figures 6D–F). The reason for these observations is likely to be associated with the size and shape of the ligands. A disaccharide is more likely to be able to discriminate between potential subsites in a broad, shallow binding site. However, in such a binding site, a linear trisaccharide is able to spread out across multiple subsites without discriminating between any of them. It is important to note that such an effect is likely to be observed only when studying recognition of linear, as opposed to branched, oligosaccharides, as they are somewhat easier to fit to the contours of a binding site. The effect is possibly exacerbated in this case by the removal of water molecules from the binding site (carried out as part of the protein preparation), which creates access to protein subsites that were previously occupied (Van Dijk and Bonvin, 2006; Englebienne and Moitessier, 2009). The removal of crystallographic water has a limited impact on the quality of molecular docking in reproducing carbohydrate recognition by some antibodies, particularly hu3S913 (PDB code 1S3K), owing to the carbohydrate being largely tightly bound by the antibody (Agostino et al., 2009a).
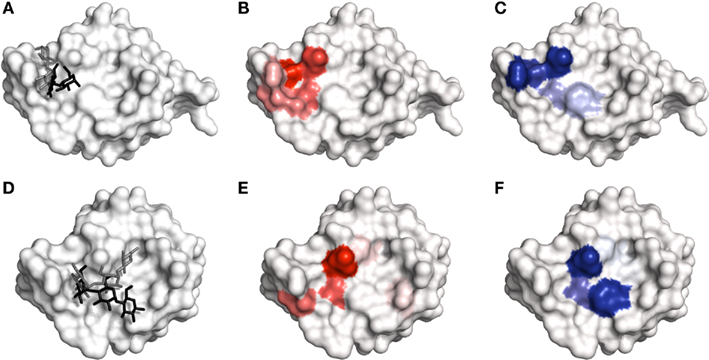
Figure 6. The recognition of Glcα(1 → 4)Glc and Glcα(1 → 4)Glcα(1 → 4)Glc by SP-D. (A) Comparison of crystal structure binding mode of Glcα(1 → 4)Glc (PDB code 1PWB; black) with top ranked docked pose (gray). (B) Hydrogen bonding map obtained for the recognition of Glcα(1 → 4)Glc. (C) van der Waals map obtained for the recognition of Glcα(1 → 4)Glc. (D) Comparison of crystal structure binding mode of Glcα(1 → 4)Glcα(1 → 4)Glc (PDB code 2GGU; black) with top ranked docked pose (gray). (E) Hydrogen bonding map obtained for the recognition of Glcα(1 → 4)Glcα(1 → 4)Glc. (F) van der Waals map obtained for the recognition of Glcα(1 → 4)Glcα(1 → 4)Glc. All figures generated in PyMOL.
Low map correctness was observed when site mapping was applied to langerin. Although most of the crystal structure interactions could be identified, many additional interactions not observed in the crystal structure were seen in the site maps (Figure 7). These interactions occur with residues located in and around the sulfate binding subsite. Although this subsite has only been demonstrated to bind sulfated glycans, it may be important in the dynamics of recognition of non-sulfated glycans by langerin. Despite this finding, it is clear from the relative intensities of the langerin site maps that residues involved in binding the carbohydrate in the crystal structure are more prominent in the site maps than the additional residues forming the sulfate binding subsite that is clearly occupied by the top ranked disaccharide pose (Figures 7A–C). Thus, even when site maps have relatively low map correctness, they may still be informative about the relative contributions that residues make to carbohydrate recognition. This feature is reflected by the clearly increased reproduction values obtained for site maps (0.81 ± 0.14) when compared to the top ranked carbohydrate poses (0.69 ± 0.18) across the various lectin systems (Table 3).
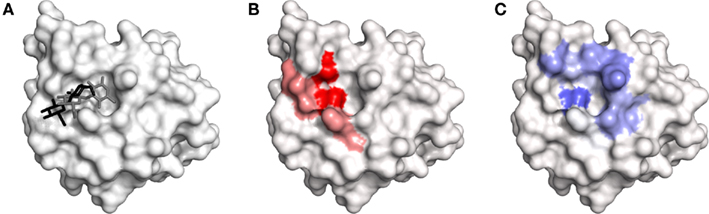
Figure 7. The recognition of Glcβ(1 → 3)Glc by langerin. (A) Comparison of crystal structure binding mode of Glcβ(1 → 3)Glc (PDB code 3P5H; black) with top ranked docked pose (gray). (B) Hydrogen bonding map. (C) van der Waals map. All figures generated in PyMOL.
Several of the lectins studied utilize calcium ions to bind their ligands. An interesting effect observed in the mapping data was that interactions to the calcium ions were fairly similar across the range of the structures (Table 4), generally accounting for ∼10% of the hydrogen bonding/ionic interactions observed. This suggests that the contribution to binding made by metal ions can effectively be ignored during mapping if the location of the metal ion is not known, i.e., in a homology model or low resolution crystal structure. In these cases, the hydrogen bonding map may be adjusted by a fixed amount to account for the potential contribution to binding made by the metal ion.
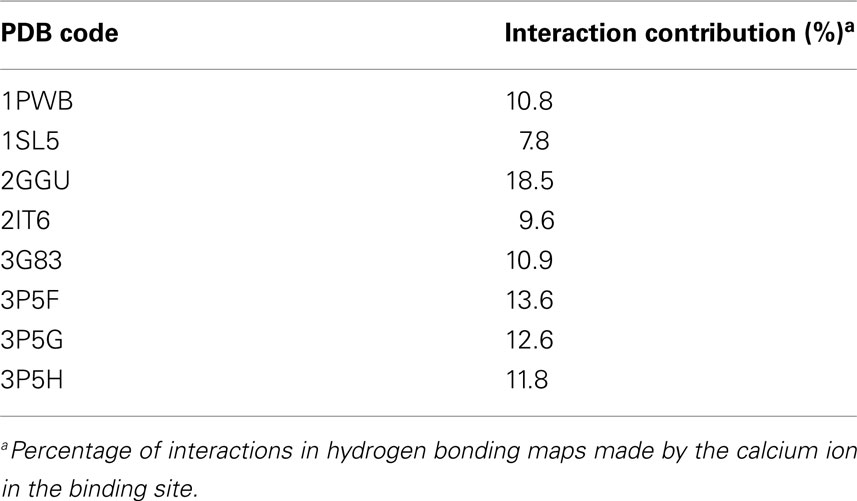
Table 4. Contributions to ligand recognition made by metal ions in lectins.
Discussion
We have shown that considering a top pose from molecular docking alone is not a suitable tool for investigating carbohydrate recognition by human lectins. The flexible nature of carbohydrates, coupled with the often shallow and broad binding sites of lectins, makes scoring very difficult for molecular docking programs. In particular, while Glide and GOLD are able to identify the correct binding mode, it is not typically scored as the best result. Alternative scoring strategies are clearly needed to address this issue in the majority of cases.
While structurally conserved water molecules may be important in carbohydrate recognition by the lectins studied (Lemieux, 1996), the contribution to binding made by such water molecules has not been considered in our investigation. A variety of solvation models which can better take into account the role of water in ligand binding have been developed such as the Generalized Born (MM/GBSA) and the Poisson–Boltzmann (MM/PBSA) models (reviewed in Yuriev et al., 2011). These scoring techniques allow for the consideration of solvation effects in binding, but do not explicitly derive the locations of structurally conserved water molecules. Aside from molecular dynamics simulation in explicit water, a variety of methods have been developed which can be used to identify where such water molecules should be placed (reviewed in De Beer et al., 2010). Of these, the GRID method has been applied to investigate the role of water in carbohydrate recognition by a bacterial enterotoxin (Minke et al., 1999). Regardless of the effect of water on carbohydrate-binding, molecular docking in isolation cannot provide information about the dynamics of binding. Molecular dynamics simulation is routinely used for such investigation, but is computationally expensive and time-consuming. The site mapping technique, which considers information from multiple binding modes, allows for some consideration of dynamic behavior.
As our results demonstrate, mapping techniques offer promising strategies for investigating carbohydrate–lectin recognition. The site mapping technique has successfully identified interacting residues in the majority of lectin cases. As in our validation of site mapping of carbohydrate-recognizing antibodies (Agostino et al., 2009b), the low map correctness in some cases is a point of concern. Low map correctness is generally indicative of the inclusion of residues, which do not appear to be involved in recognition in the crystal structures, into the maps. However, such residues, which are typically deemed as “erroneous,” may be important for the dynamics of binding. This is highly relevant for lectins, which generally feature broad and shallow binding sites, and may either recognize multiple carbohydrate-binding modes, or multiple types of carbohydrates. Therefore, site mapping provides a rational approach to identifying residues that are potentially involved in carbohydrate recognition in a dynamic setting. Furthermore, the speed with which site mapping can be employed is unparalleled by molecular dynamics simulations.
Underpinning our findings is the importance of the interplay between computational and experimental techniques in studying carbohydrate–lectin interactions, and in particular, the importance of techniques which do not rely on the availability of an experimentally determined protein structure. In order to accurately identify carbohydrate-binding modes, knowledge of lectin residues functionally involved in carbohydrate recognition is extremely useful. Such knowledge can be determined via mutagenesis studies. In vitro mutagenesis studies may be directly complemented by in silico mutagenesis studies, in particular, computational alanine scanning (Kortemme et al., 2004; Bradshaw et al., 2011). In silico mutagenesis has been used to investigate the role of specific residues in carbohydrate recognition by the Pseudomonas aeruginosa PA-IIL lectin (Adam et al., 2008). Furthermore, knowledge about the important interacting groups on the carbohydrate can be obtained using chemical mapping (Audette et al., 2003). Although this technique involves time-consuming chemical modifications to be made to the carbohydrate, it is considerably powerful for identifying the key carbohydrate positions involved in recognition.
While we have demonstrated that site mapping is useful for investigating carbohydrate–lectin recognition, the technique represents only one step in the full determination of likely binding modes. We have developed additional mapping techniques which analyze binding from the point-of-view of the ligand and, when combined with site mapping and conformational filtering, were used to suggest likely carbohydrate-binding modes in their complexes with antibodies (Agostino et al., 2010). Additional work is needed to evaluate whether ligand-based mapping and conformational filtering can be used to predict preferred carbohydrate-binding modes in lectins. It will also be important to establish, with cross-docking and cross-mapping studies, how suitable mapping approaches are for examining carbohydrate recognition in the absence of a crystal structure of the cognate carbohydrate–lectin complex. Native structures and homology models present a special challenge for structural studies of ligand binding, as the generated protein structure may not be appropriately “induced” for the ligand of interest (Agostino et al., 2009a). Induced-fit models of ligand binding can be used to provide information about likely changes in the conformation of the protein, while site mapping can be used to identify key contacts made by a range of ligand poses. In this setting, induced-fit modeling and site mapping provide complementary considerations of the dynamic nature of the binding event, with induced-fit considering protein dynamics, and site mapping considering ligand dynamics.
Conclusion
The success of site mapping in identifying lectin residues involved in interactions is an encouraging first step in the application of in silico techniques to fully characterize carbohydrate–lectin interactions. The present study demonstrates that mapping techniques are generally applicable to examining carbohydrate recognition by proteins other than antibodies, including the wide range of carbohydrate-binding proteins involved in the innate arm of the immune system.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded in part by grants from the National Health and Medical Research Council of Australia (NHMRC), and a grant from Monash Institute of Pharmaceutical Sciences (to Elizabeth Yuriev). Mark Agostino is a recipient of an Australian Postgraduate Award (APA). The authors gratefully acknowledge the contribution to this work of the Victorian Operational Infrastructure Support Program received by the Burnet Institute.
References
Adam, J., Kříž, Z., Prokop, M., Wimmerová, M., and Koca, J. (2008). In silico mutagenesis and docking studies of Pseudomonas aeruginosa PA-IIL lectin – predicting binding modes and energies. J. Chem. Inf. Model. 48, 2234–2242.
Agostino, M., Jene, C., Boyle, T., Ramsland, P. A., and Yuriev, E. (2009a). Molecular docking of carbohydrate ligands to antibodies: structural validation against crystal structures. J. Chem. Inf. Model. 49, 2749–2760.
Agostino, M., Sandrin, M. S., Thompson, P. E., Yuriev, E., and Ramsland, P. A. (2009b). In silico analysis of antibody-carbohydrate interactions and its application to xenoreactive antibodies. Mol. Immunol. 47, 233–246.
Agostino, M., Sandrin, M. S., Thompson, P. E., Ramsland, P. A., and Yuriev, E. (2011). Peptide inhibitors of xenoreactive antibodies mimic the interaction profile of the native carbohydrate antigens. Biopolymers 96, 193–206.
Agostino, M., Sandrin, M. S., Thompson, P. E., Yuriev, E., and Ramsland, P. A. (2010). Identification of preferred carbohydrate binding modes in xenoreactive antibodies by combining conformational filters and binding site maps. Glycobiology 20, 724–735.
Audette, G. F., Delbaere, L. T. J., and Xiang, J. (2003). Mapping protein: carbohydrate interactions. Curr. Protein Pept. Sci. 4, 11–20.
Bradshaw, R. T., Patel, B. H., Tate, E. W., Leatherbarrow, R. J., and Gould, I. R. (2011). Comparing experimental and computational alanine scanning techniques for probing a prototypical protein-protein interaction. Protein Eng. Des. Sel. 24, 197–207.
Crocker, P. R., Paulson, J. C., and Varki, A. (2007). Siglecs and their role in the immune system. Nat. Rev. Immunol. 7, 255–266.
Crouch, E., Hartshorn, K., Horlacher, T., Mcdonald, B., Smith, K., Cafarella, T., Seaton, B., Seeberger, P. H., and Head, J. (2009). Recognition of mannosylated ligands and influenza A virus by human surfactant protein D: contributions of an extended site and residue 343. Biochemistry 48, 3335–3345.
Crouch, E., Mcdonald, B., Smith, K., Cafarella, T., Seaton, B., and Head, J. (2006). Contributions of phenylalanine 335 to ligand recognition by human surfactant protein D: ring interactions with SP-D ligands. J. Biol. Chem. 281, 18008–18014.
Dam, T. K., and Brewer, C. F. (2010). Lectins as pattern recognition molecules: the effects of epitope density in innate immunity. Glycobiology 20, 270–279.
De Beer, S. B. A., Vermeulen, N. P. E., and Oostenbrink, C. (2010). The role of water molecules in computational drug design. Curr. Top. Med. Chem. 10, 55–66.
Englebienne, P., and Moitessier, N. (2009). Docking ligands into flexible and solvated macromolecules. 4. Are popular scoring functions accurate for this class of proteins? J. Chem. Inf. Model. 49, 1568–1580.
Farrugia, W., Scott, A. M., and Ramsland, P. A. (2009). A possible role for metallic ions in the carbohydrate cluster recognition displayed by a Lewis Y specific antibody. PLoS ONE 4, e7777. doi: 10.1371/journal.pone.0007777
Feinberg, H., Castelli, R., Drickamer, K., Seeberger, P. H., and Weis, W. I. (2007). Multiple modes of binding enhance the affinity of DC-SIGN for high mannose N-linked glycans found on viral glycoproteins. J. Biol. Chem. 282, 4202–4209.
Feinberg, H., Taylor, M. E., Razi, H., Mcbride, R., Knirel, Y. A., Graham, S. A., Drickamer, K., and Weis, W. I. (2011). Structural basis for langerin recognition of diverse pathogen and mammalian glycans through a single binding site. J. Mol. Biol. 405, 1027–1039.
Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Kilcic, J. J., Mainz, D. T., Repasky, M. P., Knoll, E. H., Shelley, M., Perry, J. K., Shaw, D. E., Francis, P., and Shenkin, P. S. (2004). Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 1739–1749.
Gabius, H.-J. (2008). Glycans: bioactive signals decoded by lectins. Biochem. Soc. Trans. 36, 1491–1496.
Guo, Y., Feinberg, H., Conroy, E., Mitchell, D. A., Alvarez, R., Blixt, O., Taylor, M. E., Weis, W. I., and Drickamer, K. (2004). Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR. Nat. Struct. Mol. Biol. 11, 591–598.
Hertje, M., Zhou, M., and Dietrich, U. (2010). Inhibition of HIV-1 entry: multiple keys to close the door. ChemMedChem 5, 1825–1835.
Jackson, T. A., Robertson, V., Imberty, A., and Auzanneau, F.-I. (2009). The flexibility of the LeaLex tumor associated antigen central fragment studied by systematic and stochastic searches as well as dynamic simulations. Bioorg. Med. Chem. 17, 1514–1526.
Kerrigan, A. M., and Brown, G. D. (2009). C-type lectins and phagocytosis. Immunobiology 214, 562–575.
Kortemme, T., Kim, D. E., and Baker, D. (2004). Computational alanine scanning of protein-protein interfaces. Sci. STKE 2004, pl2.
Lang, P. T., Brozell, S. R., Mukherjee, S., Pettersen, E. F., Meng, E. C., Thomas, V., Rizzo, R. C., Case, D. A., James, T. L., and Kuntz, I. D. (2009). DOCK 6: combining techniques to model RNA-small molecule complexes. RNA 15, 1219–1230.
Lemieux, R. U. (1996). How water provides the impetus for molecular recognition in aqueous solution. Acc. Chem. Res. 29, 373–380.
Leonidas, D. D., Vatzaki, E. H., Vorum, H., Celis, J. E., Madsen, P., and Acharya, K. R. (1998). Structural basis for the recognition of carbohydrates by human galectin-7. Biochemistry 37, 13930–13940.
Liebecq, C. (ed.). (1992). Biochemical Nomenclature and Related Documents: A Compendium 1992 (International Union of Biochemistry and Molecular Biology). London: Portland Press.
López-Lucendo, M. F., Solís, D., André, S., Hirabayashi, J., Kasai, K., Kaltner, H., Gabius, H.-J., and Romero, A. (2004). Growth-regulatory human galectin-1: crystallographic characterisation of the structural changes induced by single-site mutations and their impact on the thermodynamics of ligand binding. J. Mol. Biol. 343, 957–970.
Minke, W. E., Diller, D. J., Hol, W. G. J., and Verlinde, C. L. M. J. (1999). The role of waters in docking strategies with incremental flexibility for carbohydrate derivatives: heat-labile enterotoxin, a multivalent test case. J. Med. Chem. 42, 1778–1788.
Morris, G. M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart, W. E., Belew, R. K., and Olson, A. J. (1998). Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19, 1639–1662.
Nishi, N., Abe, A., Iwaki, J., Yoshida, H., Itoh, A., Shoji, H., Kamitori, S., Hirabayashi, J., and Nakamura, T. (2008). Functional and structural bases of a cysteine-less mutant as a long-lasting substitute for galectin-1. Glycobiology 18, 1065–1073.
Nurisso, A., Kozmon, S., and Imberty, A. (2008). Comparison of docking methods for carbohydrate binding in calcium-dependent lectins and prediction of the carbohydrate binding mode to sea cucumber lectin CEL-III. Mol. Simul. 34, 469–479.
Rabinovich, G. A., and Toscano, M. A. (2009). Turning “sweet” on immunity: galectin-glycan interactions in immune tolerance and inflammation. Nat. Rev. Immunol. 9, 338–352.
Ramsland, P. A., Farrugia, W., Bradford, T. M., Hogarth, P. M., and Scott, A. M. (2004). Structural convergence of antibody binding of carbohydrate determinants in Lewis Y tumor antigens. J. Mol. Biol. 340, 809–818.
Rockey, W. M., Laederach, A., and Reilly, P. J. (2000). Automated docking of alpha-(1 → 4)- and alpha-(1 → 6)-linked glucosyl trisaccharides and maltopentaose into the soybean beta-amylase active site. Proteins 40, 299–309.
Sato, S., St-Pierre, C., Bhaumik, P., and Nieminen, J. (2009). Galectins in innate immunity: dual functions of host soluble β-galactoside-binding lectins as damage-associated molecular patterns (DAMPs) and as receptors for pathogen-associated molecular patterns (PAMPs). Immunol. Rev. 230, 172–187.
Shrive, A. K., Tharia, H. A., Strong, P., Kishore, U., Burns, I., Rizkallah, P. J., Reid, K. B. M., and Greenhough, T. J. (2003). High-resolution structural insights into ligand binding and immune cell recognition by human lung surfactant protein D. J. Mol. Biol. 331, 509–523.
Sörme, P., Arnoux, P., Kahl-Knutsson, B., Leffler, H., Rini, J. M., and Nilsson, U. J. (2005). Structural and thermodynamic studies on cation-π interactions in lectin-ligand complexes: high-affinity galectin-3 inhibitors through fine-tuning of an arginine-arene interaction. J. Am. Chem. Soc. 127, 1737–1743.
Van Der Vlist, M., and Geijtenbeek, T. B. H. (2010). Langerin functions as an antiviral receptor on Langerhans cells. Immunol. Cell Biol. 88, 410–415.
Van Dijk, A. D. J., and Bonvin, A. M. J. J. (2006). Solvated docking: introducing water into the modelling of biomolecular complexes. Bioinformatics 22, 2340–2347.
Verdonk, M. L., Chessari, G., Cole, J. C., Hartshorn, M. J., Murray, C. W., Nissink, J. W. M., Taylor, R. D., and Taylor, R. (2005). Modeling water molecules in protein-ligand docking using GOLD. J. Med. Chem. 48, 6504–6515.
Verdonk, M. L., Cole, J. C., Hartshorn, M. J., Murray, C. W., and Taylor, R. D. (2003). Improved protein-ligand docking using GOLD. Proteins 52, 609–623.
Wallis, R., Mitchell, D. A., Schmid, R., Schwaeble, W. J., and Keeble, A. H. (2010). Paths reunited: initiation of the classical and lectin pathways of complement activation. Immunobiology 215, 1–11.
Yuriev, E., Agostino, M., Farrugia, W., Christiansen, D., Sandrin, M. S., and Ramsland, P. A. (2009). Structural biology of carbohydrate xenoantigens. Expert Opin. Biol. Ther. 9, 1017–1029.
Yuriev, E., Agostino, M., and Ramsland, P. A. (2011). Challenges and advances in computational docking: 2009 in review. J. Mol. Recognit. 24, 149–164.
Yuriev, E., Farrugia, W., Scott, A. M., and Ramsland, P. A. (2005). Three-dimensional structures of carbohydrate determinants of Lewis system antigens: implications for effective antibody targeting of cancer. Immunol. Cell Biol. 83, 709–717.
Yuriev, E., Ramsland, P. A., and Edmundson, A. B. (2001). Docking of combinatorial peptide libraries into a broadly cross-reactive human IgM. J. Mol. Recognit. 14, 172–184.
Keywords: lectins, carbohydrates, molecular modeling, molecular docking
Citation: Agostino M, Yuriev E and Ramsland PA (2011) A computational approach for exploring carbohydrate recognition by lectins in innate immunity. Front. Immun. 2:23. doi: 10.3389/fimmu.2011.00023
Received: 13 April 2011; Paper pending published: 08 June 2011;
Accepted: 14 June 2011; Published online: 28 June 2011.
Edited by:
Uday Kishore, Brunel University, UKCopyright: © 2011 Agostino, Yuriev and Ramsland. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Elizabeth Yuriev, Medicinal Chemistry and Drug Action, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia. e-mail:ZWxpemFiZXRoLnl1cmlldkBwaGFybS5tb25hc2guZWR1LmF1; Paul A. Ramsland, Centre for Immunology, Burnet Institute, 85 Commercial Road, Melbourne, VIC 3004, Australia. e-mail:cHJhbXNsYW5kQGJ1cm5ldC5lZHUuYXU=