
 Ana González-Périz1,3
Ana González-Périz1,3
- 1 Department of Biochemistry and Molecular Genetics, Hospital Clínic, Centre Esther Koplowitz, Institut d’Investigacions Biomèdiques August Pi i Sunyer, Barcelona, Spain
- 2 Department of Physiological Sciences I, University of Barcelona, Barcelona, Spain
- 3 Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas, Barcelona, Spain
Obesity is causally linked to a chronic state of “low-grade” inflammation in adipose tissue. Prolonged, unremitting inflammation in this tissue has a direct impact on insulin-sensitive tissues (i.e., liver) and its timely resolution is a critical step toward reducing the prevalence of related co-morbidities such as insulin resistance and non-alcoholic fatty liver disease. This article describes the current state-of-the-art knowledge and novel insights into the role of macrophages in adipose tissue inflammation, with special emphasis on the progressive changes in macrophage polarization observed over the course of obesity. In addition, this article extends the discussion to the contribution of Kupffer cells, the liver resident macrophages, to metabolic liver disease. Special attention is given to the modulation of macrophage responses by omega-3-PUFAs, and more importantly by resolvins, which are potent anti-inflammatory and pro-resolving autacoids generated from docosahexaenoic and eicosapentaenoic acids. In fact, resolvins have been shown to work as endogenous “stop signals” in inflamed adipose tissue and to return this tissue to homeostasis by inducing a phenotypic switch in macrophage polarization toward a pro-resolving phenotype. Collectively, this article offers new views on the role of macrophages in metabolic disease and their modulation by endogenously generated omega-3-PUFA-derived lipid mediators.
Obesity and Adipose Tissue Inflammation
White adipose tissue, once considered a mere storage depot of energy in the form of fat, is today recognized as an important endocrine organ. In fact, the adipocyte or fat cell is actively involved in the balance of our body homeostasis by releasing a number of factors, collectively known as adipokines (Ouchi et al., 2011). However, the expansion of adipose tissue mass seen in obesity inadvertently interrupt the interplay among these factors and other intracellular components yielding a chronic “low-grade” inflammatory scenario in this tissue (Ferrante Jr., 2007; Ouchi et al., 2011). This “low-grade” inflammation, also known as “metabolic-triggered inflammation” or “metainflammation,” can be described as a long-term inflammatory response triggered by nutrients and metabolic surplus (Hotamisligil, 2006). It involves a similar set of molecules/signaling pathways to those involved in classical inflammation, but in this case these molecules/signaling pathways have a dual role as inflammatory mediators as well as regulators of energy metabolism. In fact, a rise in pro-inflammatory adipokines such as tumor necrosis factor (TNF) α, interleukin (IL)-6, IL-1β, monocyte chemoattractant protein (MCP)-1, leptin, and resistin, accompanied by a reduction in the anti-inflammatory and insulin-sensitizing adipokine, adiponectin has been reported to signal the onset of metabolic dysfunction (Ouchi et al., 2011).
One of the most important sequela of adipose tissue inflammation is insulin resistance (Figure 1). In fact, stress sensors activate both the c-jun-N-terminal kinase (JNK) and inhibitor of κ kinase (IKK) pathways through classical receptor-mediated mechanisms (Shoelson et al., 2006). JNK and IKK activation induce insulin resistance by disrupting serine phosphorylation of IRS-1, a protein that connects the insulin receptor to the PI(3)K signaling cascade. In parallel to the activation of these kinases and their downstream signaling cascades, there is an increased production of pro-inflammatory adipokines (i.e., TNFα, IL-6, and MCP-1) in obese subjects, whose levels directly correlate with the degree of insulin resistance (Hotamisligil et al., 1996). Adipose tissue inflammation leading to insulin resistance also has negative consequences on the liver. In fact, adipose tissue and liver have immediate access to a vast network of blood vessels that facilitate a direct connection between these two organs. The exact mechanisms linking adipose tissue dysfunction and insulin resistance with metabolic liver disease are not completely understood, but several processes have been implicated. First, increased lipolysis from visceral fat resulting in increased free fatty acid efflux to the liver (Sanyal, 2005). Second, increased secretion of pro-inflammatory and insulin-resistant adipokines (TNFα and IL-6) by adipose tissue in parallel with a reduced release of adiponectin (Sanyal, 2005; Figure 1). Finally, a combined hepatic dysregulation in free fatty acid oxidation and de novo lipogenesis secondary to altered hepatic insulin sensitivity (Tilg and Moschen, 2008).
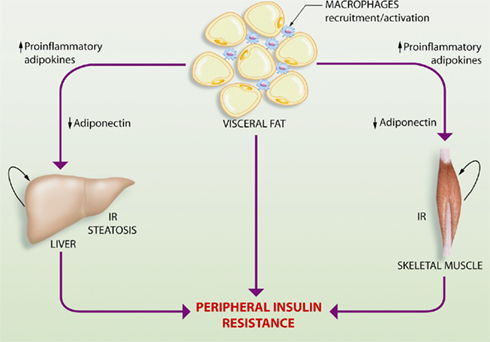
Figure 1. Schematic representation of the interplay between adipose tissue, skeletal muscle, and the liver in the obesity-related perturbation of systemic metabolic control. Obesity results in expansion of adipose tissue mass that eventually leads to a characteristic inflammatory response driven by macrophage infiltration and aberrant production and release of pro-inflammatory adipokines, accompanied by a reduction in the anti-inflammatory and insulin-sensitizing adipokine, adiponectin. This altered profile of adipokine secretion leads to insulin resistance (IR) in the liver and skeletal muscle, which are the major organs contributing to the development of peripheral insulin resistance. Hepatic insulin resistance also triggers the progression of hepatic steatosis or fatty liver.
Macrophages and Adipose Tissue Inflammation
Obesity-induced adipose tissue inflammation is a unique process characterized by an inflammatory response driven by tissue macrophages (Lumeng and Saltiel, 2011). In fact, a pathological hallmark of obesity is the presence of an increased number of adipose tissue-infiltrating macrophages, which form the characteristic “crown-like structures” that surround necrotic adipocytes and perpetuate a vicious cycle of macrophage recruitment and exacerbated production of pro-inflammatory mediators (Weisberg et al., 2003; Wellen and Hotamisligil, 2003; Cancello et al., 2005; Lesniewski et al., 2007).
Tissue macrophages display an extensive receptor repertoire and a versatile biosynthetic capacity that confer them the plasticity to adapt to different tissue microenvironments (Gordon and Taylor, 2005). Accordingly, tissue macrophages are phenotypically heterogeneous and can exhibit either pro- or anti-inflammatory properties depending on the disease stage and the signals they are exposed. Although the classification based on the Th1/Th2 nomenclature needs to be revised, macrophages are broadly characterized by their activation (polarization) state according to the M1/M2 classification system (Mantovani et al., 2007; Martínez et al., 2009). According to this classification, the M1 designation is reserved for classically activated macrophages following stimulation with interferon (IFN) γ and LPS, whereas the M2 designation is applied to the alternatively activated macrophages after in vitro stimulation with IL-4 and IL-13 (Figure 2). M1 macrophages display enhanced microbicidal capacity and secrete high levels of pro-inflammatory cytokines (TNFα, IL-1β, and IL-6) and increased concentrations of superoxide anion (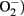 ) and oxygen and nitrogen radicals to increase their killing activity (Gordon and Taylor, 2005). Conversely, M2 macrophages dampen pro-inflammatory cytokine levels, secrete components of the extracellular matrix, and may be essential for the immune response to parasites, tissue repair, and resolution of inflammation (Gordon, 2003). In this classification system, M1 and M2 macrophages are merely regarded as two extremes of a continuum of functional stages (Mosser and Edwards, 2008). For instance, M2a designation defines those macrophages stimulated by IL-4/IL-13; M2b refers to macrophages activated by stimuli such as apoptotic cells in concert with LPS; and M2c relates to polarization in response to IL-10, transforming growth factor (TGF)-β, or glucocorticoids (Martínez et al., 2008). In mice, M1/M2 macrophage polarization can be monitored by assessing the expression of selected markers. M1-associated genes include inducible nitric oxide synthase (iNOs), the interferon responsive CXC chemokines, and classical pro-inflammatory mediators such as TNFα, IL-1β, IL-6, and MCP-1 as well as increased production of
) and oxygen and nitrogen radicals to increase their killing activity (Gordon and Taylor, 2005). Conversely, M2 macrophages dampen pro-inflammatory cytokine levels, secrete components of the extracellular matrix, and may be essential for the immune response to parasites, tissue repair, and resolution of inflammation (Gordon, 2003). In this classification system, M1 and M2 macrophages are merely regarded as two extremes of a continuum of functional stages (Mosser and Edwards, 2008). For instance, M2a designation defines those macrophages stimulated by IL-4/IL-13; M2b refers to macrophages activated by stimuli such as apoptotic cells in concert with LPS; and M2c relates to polarization in response to IL-10, transforming growth factor (TGF)-β, or glucocorticoids (Martínez et al., 2008). In mice, M1/M2 macrophage polarization can be monitored by assessing the expression of selected markers. M1-associated genes include inducible nitric oxide synthase (iNOs), the interferon responsive CXC chemokines, and classical pro-inflammatory mediators such as TNFα, IL-1β, IL-6, and MCP-1 as well as increased production of 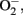 (Gordon, 2003; Martínez et al., 2008; Figure 2). M2 macrophages display up-regulation of scavenger, mannose (CD206), and galactose (Mgl-1) receptors, arginase 1, which antagonizes iNOS activity, and IL-10, in parallel with down-regulation of IL-1β and other pro-inflammatory cytokines (Gordon, 2003; Scotton et al., 2005; Martínez et al., 2008). In addition, the panel of M2 markers comprises up-regulation of other genes with unknown function such as chitinases Ym1 and Ym2, and resistin-like molecule (RELM)-α, also known as FIZZ (Figure 2).
(Gordon, 2003; Martínez et al., 2008; Figure 2). M2 macrophages display up-regulation of scavenger, mannose (CD206), and galactose (Mgl-1) receptors, arginase 1, which antagonizes iNOS activity, and IL-10, in parallel with down-regulation of IL-1β and other pro-inflammatory cytokines (Gordon, 2003; Scotton et al., 2005; Martínez et al., 2008). In addition, the panel of M2 markers comprises up-regulation of other genes with unknown function such as chitinases Ym1 and Ym2, and resistin-like molecule (RELM)-α, also known as FIZZ (Figure 2).
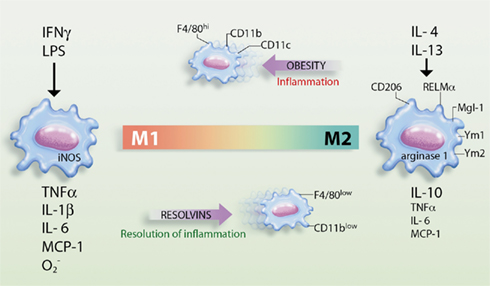
Figure 2. Schematic representation of macrophage polarization in the adipose tissue and the actions of resolvins. Obesity promotes the polarization of macrophages into the M1 phenotype, which are highly inflammatory in nature and release pro-inflammatory cytokines/chemokines [e.g., tumor necrosis factor (TNF) α, interleukin (IL)-1β, IL-6, and monocyte chemotactic peptide (MCP)-1] and superoxide anion (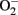 ). These macrophages express inducible nitric oxide synthase (iNOs) and cell surface markers such as F4/80, CD11b, and CD11c and act as classically activated macrophages expressing interferon (IFN) γ and lipopolysaccharide (LPS)-responsive genes. Conversely, resolvins promote the resolution of inflammation by skewing macrophages toward the M2 phenotype, which release high levels of IL-10 in parallel with reduced levels of TNFα, IL-6, and MCP-1. M2 macrophages are alternatively activated macrophages, originally identified after IL-4 and IL-13 stimulation, that up-regulate scavenger, mannose (CD206), and galactose (Mgl-1) receptors, resistin-like molecule (RELM)-α, and chitinases Ym1 and Ym2 expression and arginase 1 activity.
). These macrophages express inducible nitric oxide synthase (iNOs) and cell surface markers such as F4/80, CD11b, and CD11c and act as classically activated macrophages expressing interferon (IFN) γ and lipopolysaccharide (LPS)-responsive genes. Conversely, resolvins promote the resolution of inflammation by skewing macrophages toward the M2 phenotype, which release high levels of IL-10 in parallel with reduced levels of TNFα, IL-6, and MCP-1. M2 macrophages are alternatively activated macrophages, originally identified after IL-4 and IL-13 stimulation, that up-regulate scavenger, mannose (CD206), and galactose (Mgl-1) receptors, resistin-like molecule (RELM)-α, and chitinases Ym1 and Ym2 expression and arginase 1 activity.
In addition to the augmented infiltration of macrophages into the adipose tissue, obesity also induces a phenotypic switch in these cells toward the classically activated M1 phenotype (Olefsky and Glass, 2010). In fact, the majority of macrophages that accumulate in obese adipose tissue are M1-like and selectively express the cell surface markers F4/80, CD11b, and CD11c (Lumeng et al., 2007; Nguyen et al., 2007). In our laboratory, we have recently gathered data indicating the presence of a specific subset of macrophages with high expression of the surface glycoprotein F4/80 (F4/80hi) in adipose tissue from obese mice (Titos et al., 2011). This finding is consistent with that reported by Bassaganya-Riera et al. (2009) who identified two functionally distinct subsets of macrophages in adipose tissue based on their surface expression of F4/80 (F4/80lo macrophages predominate in adipose tissue of lean mice, obesity causes accumulation of both F4/80lo and F4/80hi). Importantly, lean adipose tissue macrophages are M2-like, display F4/80 and CD11b but are negative for CD11c and do not exhibit activation of the inflammatory pathways. In a series of elegant studies, Lumeng et al. (2007) and Nguyen et al. (2007) have demonstrated that adipose tissue macrophages undergo a phenotypic switch from the M2 polarization state to a more M1-like, CD11c+ polarization state upon high-fat feeding. Moreover, Patsouris et al. (2008) have reported that selective depletion of CD11c+ macrophages in adipose tissue reverses insulin resistance in high-fat diet-induced obese mice. Recently, Li et al. (2010) have reported that the M1-like, CD11c+ macrophage subset can exhibit phenotypic plasticity between inflammatory and non-inflammatory states, depending on the presence or absence of insulin resistance.
Macrophages and Liver Disease
Kupffer cells are specialized macrophages located in the liver lining the walls of the sinusoids (Ramadori et al., 2008). Kupffer cells are uniquely positioned within the liver and their location enables intimate contact with circulating blood and the clearance of pathogens and parasites by receptor-mediated phagocytosis or release of TNFα, reactive oxygen species, or proteinases. Kupffer cells are also professional antigen-presenting cells that trigger the adaptive immune system. Therefore, Kupffer cells act as true sentinels of the adaptive and immune system in the liver and protect our body from the extracorporeal environment. In cases of pathogenic infection or tissue damage, Kupffer cells act as the predominant inflammatory effector cell type to initiate the inflammatory cascade leading to liver injury (Ramadori et al., 2008). In fact, activation of Kupffer cells and the subsequent release of cytokines, reactive oxygen species, and inflammatory lipid mediators (i.e., eicosanoids) are considered an early step in the pathogenesis of liver damage and tissue remodeling, as they stimulate inflammatory and fibrogenic events in the liver (Titos et al., 2003, 2005; Ramadori et al., 2008; Table 1). Depletion of Kupffer cells by treatment with either gadolinium chloride, liposomal clodronate, or conditional ablation of the diphtheria toxin receptor appears to confer a protective role in the liver by reducing the production of inflammatory mediators and collagen content (Ramadori et al., 2008).
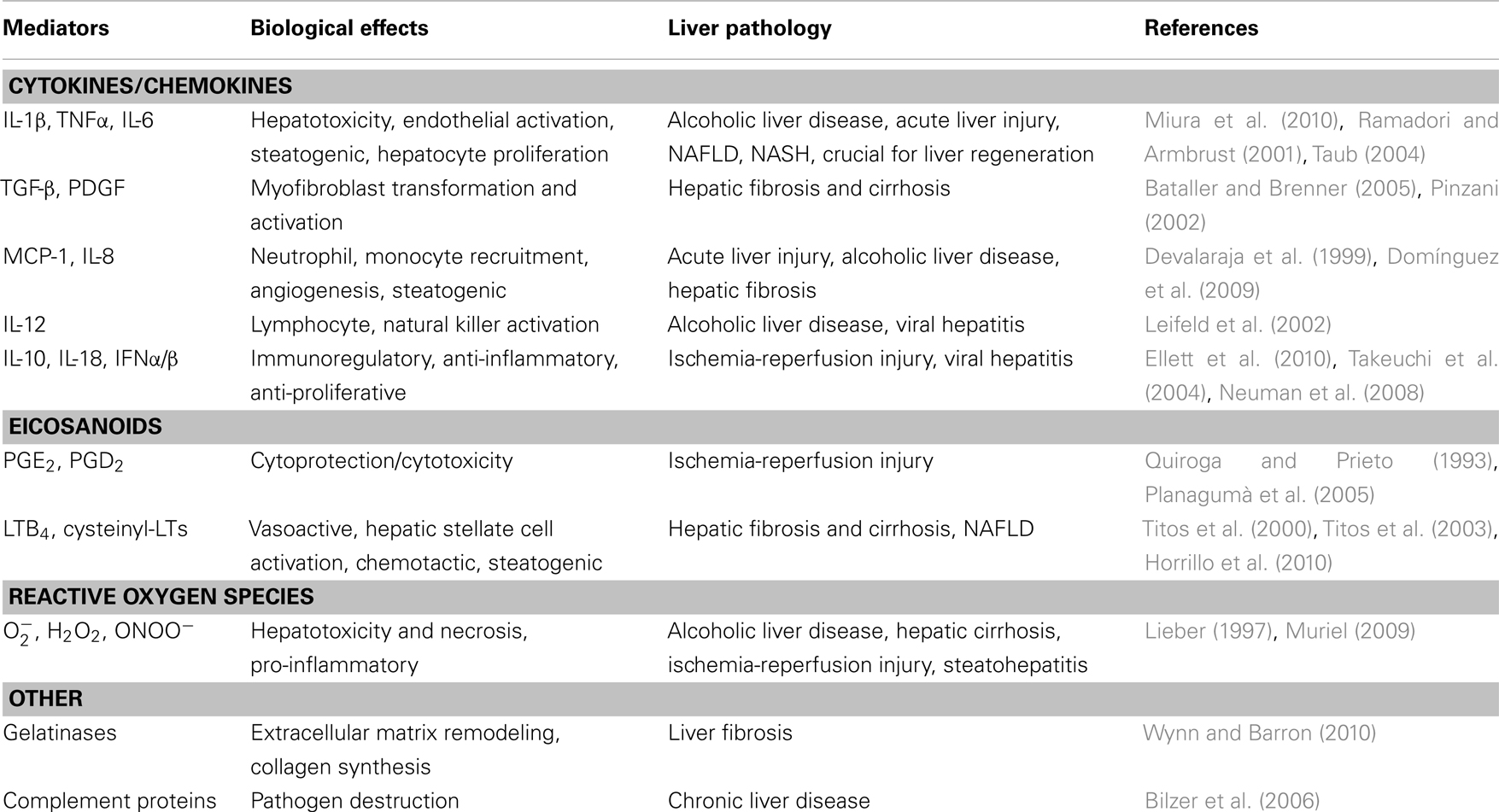
Table 1. Kupffer cell-derived mediators and associated liver pathologies.
Recent studies have revealed a novel role for Kupffer cells in metabolic liver disease. In fatty livers, similar to that occurring in obese adipose tissue, macrophages are in close proximity to fat-laden parenchymal cells (the hepatocytes) and may establish a cross-talk by secreting insulin-resistant cytokines such as TNFα and IL-6, thus regulating hepatic fat and glucose homeostasis and the progression of fatty liver (Baffy, 2009). In fact, excessive exposure of Kupffer cells to fatty acids may induce the activation of these cells via Toll-like receptors thus connecting an important mechanism by which lipids regulate inflammation and immune response in the liver (Kim, 2006). In a mouse model of steatohepatitis, Miura et al. (2010) convincingly showed that TLR9 signaling induces production of IL-1β by Kupffer cells, leading to steatosis, inflammation, and fibrosis. These authors have also shown that JNK activation in Kupffer cells contribute to the development of chronic inflammation and fibrosis in an experimental model of diet-induced steatohepatitis (Kodama et al., 2009). Lanthier et al. (2010) have elegantly demonstrated that early hepatic insulin resistance and steatosis are concurrent with Kupffer cell activation, and that selective Kupffer cell depletion through intravenous clodronate injection is sufficient to improve hepatic insulin signaling. Interestingly, as earlier described for adipose tissue macrophages, alternative M2 activation of Kupffer cells appears to ameliorate insulin resistance and to retard the progression to steatohepatitis in mice (Odegaard et al., 2008).
Clinical Impact of Omega-3-PUFAs in Diabetes and Metabolic Liver Disease
The first evidences of beneficial actions of omega-3-PUFAs in humans were provided by Endres et al. (1989). Since then, several in vivo and in vitro studies both in human and rodents have demonstrated the therapeutic potential of omega-3-PUFAs in pathologies with an important inflammatory component (Dinarello, 2010). A number of pre-clinical and clinical studies have demonstrated that regular consumption of modest amounts of omega-3-PUFAs (≤3 g/day) improves serum lipid profiles, exerts cardiovascular protective actions, and may reduce the risk of conversion from impaired glucose tolerance to type-2 diabetes (Nettleton and Katz, 2005). The use of enriched omega-3-PUFA diets in patients with non-alcoholic fatty liver disease could also represent an important nutritional strategy for their clinical management (Shapiro et al., 2011). However, there is a concern that most of studies addressing the effects of omega-3-PUFAs on glucose metabolism and insulin sensitivity did not have a control group and that dosages of fatty acids were sometimes higher than those sufficient to obtain beneficial end-points in these patients (De Caterina et al., 2007). This point out that new, more specific approaches are needed (i.e., compare potency and specificity of resolvins to their substrate precursors, see below).
Effective Resolution of Inflammation: Role of Macrophages
Since prolonged inflammation is detrimental to the host, higher organisms have evolved protective mechanisms to ensure resolution of the inflammatory response in a limited and specific time- and space-manner (Serhan et al., 2007). Once thought as a mere passive process of dilution of inflammation, resolution is today envisioned as a highly orchestrated process coordinated by a complex regulatory network of cells and mediators. Among the molecules that facilitate resolution, lipoxins generated from the omega-6-PUFA arachidonic acid, and resolvins and protectins generated from omega-3-PUFAs, are the lipid mediators that have attracted most attention. These endogenous anti-inflammatory and pro-resolving mediators counteract the effects of pro-inflammatory signaling systems and act as “braking signals” of the persistent vicious cycle leading to unremitting inflammation (Serhan et al., 2008). In fact, the same pro-inflammatory factors that initially trigger the inflammatory response also signal the termination of inflammation by stimulating the biosynthesis of pro-resolving lipid mediators (Serhan et al., 2008). For instance, both PGE2 and PGD2 transcriptionally activate the expression of 15-LO in human PMN, switching the mediator profile of these cells from the pro-inflammatory LTB4 to the anti-inflammatory lipoxin A4, which was the first identified omega-6-PUFA-derived anti-inflammatory lipid mediator (Serhan et al., 2007, 2008). Another example of this class switch is the displacement of pro-inflammatory lipid mediators derived from omega-6-PUFAs by anti-inflammatory mediators (i.e., resolvins and protectins) derived from omega-3-PUFAs (Serhan, 2011). These anti-inflammatory and pro-resolving mediators exert a strict control of the resolution process and pave the way for monocyte migration and their differentiation to phagocytosing macrophages, which remove dead cells and then terminate the inflammatory response (Tabas, 2010; Serhan, 2011).
Resolvins
Resolvins are a novel family of anti-inflammatory and pro-resolving mediators generated from the omega-3-PUFAs docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA). By using a lipidomics-based approach that combines liquid chromatography and tandem-mass spectrometry, Serhan et al. (2000, 2002) identified a library of omega-3-PUFA-derived lipid mediators present within exudates obtained from mice dorsal skin pouches during the “spontaneous resolution” phase of acute inflammation. These novel bioactive lipid autacoids were termed resolvins and were classified as either resolvin E-series, if the biosynthesis is initiated from EPA, or resolvin D-series, if they are generated from DHA. Schematically, the biosynthesis of resolvin E1 is initiated when EPA is converted to 18R-hydroperoxy-EPE by endothelial cells expressing COX-2 treated with aspirin (Serhan et al., 2000). Alternatively, 18R-hydroperoxy-EPE can be produced through cytochrome P450 activity (Haas-Stapleton et al., 2007). By transcellular biosynthesis, 18R-hydroperoxy-EPE generated by endothelial cells is transformed by 5-LO of neighboring leukocytes into resolvin E1 (5S,12R,18R-trihydroxy-EPA) via a 5(6)epoxide intermediate (Serhan et al., 2000, 2002). Resolvin D1 biosynthesis is also initiated in endothelial cells expressing COX-2 treated with aspirin, which transform DHA into 17R-hydroxy-DHA which is further transformed by leukocyte 5-LO into resolvin D1 (Serhan et al., 2000, 2002). More importantly from a physiological point of view, resolvin D1 can also be formed from endogenous sources of DHA without the requirement of aspirin. In this case, endogenous DHA is converted via 15-LO/5-LO interactions that give rise to a 17S alcohol-containing series of resolvins, including resolvin D1 and resolvin D2 (Hong et al., 2003). Finally, DHA is also transformed into a dihydroxy-containing DHA derivative, 17S-hydroxy-DHA via an intermediate epoxide that opens via hydrolysis and subsequent rearrangements to form protectin D1 (10R,17S-dihydroxy-docosa-DHA) (Serhan et al., 2000, 2002; Hong et al., 2003).
Unlike their precursors DHA and EPA, resolvins exert biological actions at the nanomolar range. Resolvin E1, decreases PMN infiltration and T cell migration, reduces TNFα and IFNγ secretion, inhibits chemokine formation and blocks IL-1-induced NF-κB activation (Gronert et al., 2005; Schwab et al., 2007; Bannenberg and Serhan, 2010). Resolvin E1 also stimulates macrophage phagocytosis of apoptotic PMN and is a potent modulator of pro-inflammatory leukocyte expression adhesion molecules (i.e., L-selectin) (Schwab et al., 2007; Dona et al., 2008). In vivo resolvin E1 exerts potent anti-inflammatory actions in experimental models of periodontitis, colitis, and peritonitis and protects mice against brain ischemia-reperfusion (Arita et al., 2005; Bannenberg and Serhan, 2010). Furthermore, Haworth et al. (2008) have identified a resolvin E1-initiated resolution program for allergic airway responses. Finally, a recent study has identified resolvin D2 as a potent endogenous regulator of excessive inflammatory responses in mice with microbial sepsis (Spite et al., 2009).
Our laboratory has recently provided evidence that adipose tissue expresses all the enzymes necessary for the formation of bioactive lipid mediators derived from both omega-6 and omega-3-PUFAs (i.e., 12/15-LO, 5-LO, FLAP, LTA4 hydrolase, and LTC4 synthase; Horrillo et al., 2010). Importantly, by means of liquid chromatography–tandem mass spectrometry (LC/MS/MS) analysis we have detected the presence of the omega-6 products PGE2, PGF2α, TXB2, 5-HETE, 12-HETE, and 15-HETE as well as the formation of the omega-3-derived mediators resolvin D1, protectin D1, and 17-hydroxy-DHA (González-Périz et al., 2009). Interestingly, the administration of a DHA-enriched diet to ob/ob mice, an experimental model of obesity-induced insulin resistance and fatty liver disease, resulted in the amplification of the formation of resolvin D1, protectin D1, and 17-hydroxy-DHA, accompanied by an inhibition of the formation of omega-6-derived inflammatory mediators (González-Périz et al., 2009). In these animals, DHA significantly increased adipose tissue levels of adiponectin which alleviated hepatic steatosis and insulin resistance (González-Périz et al., 2009). Of interest, intraperitoneal injection of resolvin E1 at the nanomolar levels elicited significant insulin-sensitizing effects by inducing adiponectin, GLUT-4, and IRS-1 expression in adipose tissue and conferred significant protection against hepatic steatosis (González-Périz et al., 2009). There is also evidence that omega-3-PUFAs may also signal independently of the 12/15-LO pathway by exerting potent anti-inflammatory and insulin-sensitizing actions through a G-protein-coupled 120 receptor (GPR120) (Oh et al., 2010).
Recent findings from our laboratory also indicate that DHA (at micromolar concentrations) and resolvin D1 (at nanomolar concentrations) consistently induce hallmarks of alternative macrophage activation in adipose tissue including stimulation of arginase 1 expression and non-phlogystic macrophage phagocytosis and attenuation of IFNγ/LPS-induced Th1 cytokine secretion (Titos et al., 2011). These results are in agreement with those reported by Schif-Zuck et al. (2011) who recently identified a novel phenotype of macrophages with pro-resolving properties emerging during the resolution of murine peritonitis. These macrophages had a low marker expression of CD11b (CD11blow), engulfed significantly higher numbers of apoptotic PMN than CD11bhigh macrophages, responded poorly to activation by different TLR ligands in terms of cytokine and chemokine secretion, lost their phagocytic potential and were prone to migrate to lymphoid organs (Schif-Zuck et al., 2011). In addition, these CD11blow macrophages expressed low or moderate levels of COX-2, metalloproteinase-9, and 12/15-LO, but not detectable levels of iNOS and arginase 1. Importantly, in vivo administration of resolvin E1, resolvin D1, and glucocorticoids to peritonitis-affected mice clearly enhanced the appearance of CD11blow macrophages by reducing the number of engulfment-related events required for macrophage deactivation and by reducing the ability of peritoneal macrophages to produce pro-inflammatory cytokines upon LPS stimulation (Schif-Zuck et al., 2011). The ability of resolvins to modify tissue macrophage plasticity has also been demonstrated by Hellmann et al. (2011). These authors were able to improve insulin resistance by administering resolvin D1 to obese-diabetic mice, which reduced macrophage F4/80+CD11c+ cell accumulation and increased the percentage of positive F4/80 cells expressing Mgl-1, a marker of alternatively activated macrophages, in adipose tissue (Hellmann et al., 2011).
Studies on experimental models of liver injury have elucidated a protective role of DHA and DHA-derived lipid mediators against hepatic inflammation. In fact, feeding of a DHA-enriched diet ameliorated hepatotoxic-induced necroinflammatory liver injury in mice (González-Périz et al., 2006). The hepatoprotective actions of DHA were associated with a decrease in the hepatic formation of PGE2 and a concomitant increase in the generation of protective DHA-derived lipid mediators (i.e., PD1 and 17S-HDHA). The beneficial role of these DHA-derived lipid signals was further supported by experiments in vitro demonstrating attenuated DNA damage and oxidative stress in hepatocytes. More important, DHA and DHA-derived autacoids reduced TNFα release in macrophages, recognized as the predominant effector cells involved in the inflammatory cascade leading to hepatocyte damage (Decker, 1990). A significant down-regulation of 5-LO protein expression was also noticed in macrophages treated with 17S-HDHA and in liver tissue from mice receiving DHA in the diet (González-Périz et al., 2006). This is relevant because the presence of an active 5-LO pathway in the liver is restricted to Kupffer cells and inhibition of the 5-LO pathway in these resident macrophages has been shown to attenuate necroinflammatory liver injury and fibrosis (Titos et al., 2000, 2003, 2005).
Conclusion
Obesity is not only a matter of appearance and beauty, but a serious health issue because the global obesity epidemic will result in increased incidence and risk of cardiovascular disease, type-2 diabetes, dyslipidemia, and fatty liver disease. The prevalence of obesity-related metabolic disorders is tightly associated with the appearance of a chronic “low-grade” inflammatory state in the adipose tissue, which severely disrupts the endocrine function of this organ. Indeed, a number of studies have appreciated that expansion of adipose tissue during weight gain is associated with an inflammatory phenotype characterized by the recruitment of inflammatory cells, mainly macrophages, in this tissue. A very provocative strategy to manipulate this exacerbated inflammatory response is to replace the use of drugs that inhibit the formation of pro-inflammatory mediators by the use of endogenous-generated autacoids that boost the resolution of inflammation. Therefore, adipose tissue inflammation in obesity appears to be the perfect scenario for testing the novel anti-inflammatory and pro-resolving lipid mediators, designated resolvins. Notably, these inflammation-resolving factors can induce a proper skew of macrophages toward a unique pro-resolving phenotype, thus ameliorating the incidence of obesity-related metabolic disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Our laboratory receives grant support from the Ministerio de Ciencia e Innovación (SAF 09/08767) and is a Consolidated Research Group recognized by the Generalitat de Catalunya (2009SGR1484). CIBERehd is funded by the Instituto de Salud Carlos III. This work was carried out at the Esther Koplowitz Centre. We would like to thank Pere Lluís Leon for his contribution in figure illustration. We apologize to our many colleagues whose work was not cited due to space limitations.
References
Arita, M., Yoshida, M., Hong, S., Tjonahen, E., Glickman, J. N., Petasis, N. A., Blumberg, R. S., and Serhan, C. N. (2005). Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc. Natl. Acad. Sci. U.S.A. 102, 7671–7676.
Baffy, G. (2009). Kupffer cells in non-alcoholic fatty liver disease: the emerging view. J. Hepatol. 51, 212–223.
Bannenberg, G., and Serhan, C. N. (2010). Specialized pro-resolving lipid mediators in the inflammatory response: an update. Biochim. Biophys. Acta 1801, 1260–1273.
Bassaganya-Riera, J., Misyak, S., Guri, A. J., and Hontecillas, R. (2009). PPAR gamma is highly expressed in F4/80(hi) adipose tissue macrophages and dampens adipose-tissue inflammation. Cell. Immunol. 258, 138–146.
Bilzer, M., Roggel, F., and Gerbes, A. L. (2006). Role of Kupffer cells in host defense and liver disease. Liver Int. 26, 1175–1186.
Cancello, R., Henegar, C., Viguerie, N., Taleb, S., Poitou, C., Rouault, C., Coupaye, M., Pelloux, V., Hugol, D., Bouillot, J. L., Bouloumie, A., Barbatelli, G., Cinti, S., Svensson, P. A., Barsh, G. S., Zucker, J. D., Basdevant, A., Langin, D., and Clement, K. (2005). Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes 54, 2277–2286.
De Caterina, R., Madonna, R., Bertolotto, A., and Schmidt, E. B. (2007). n-3 fatty acids in the treatment of diabetic patients: biological rationale and clinical data. Diabetes Care 30, 1012–1026.
Decker, K. (1990). Biologically active products of stimulated liver macrophages (Kupffer cells). Eur. J. Biochem. 192, 245–261.
Devalaraja, M. N., Mcclain, C. J., Barve, S., Vaddi, K., and Hill, D. B. (1999). Increased monocyte MCP-1 production in acute alcoholic hepatitis. Cytokine 11, 875–881.
Domínguez, M., Miquel, R., Colmenero, J., Moreno, M., García-Pagán, J. C., Bosch, J., Arroyo, V., Ginès, P., Caballeria, J., and Bataller, R. (2009). Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology 136, 1639–1650.
Dona, M., Fredman, G., Schwab, J. M., Chiang, N., Arita, M., Goodarzi, A., Cheng, G., von Andrian, U. H., and Serhan, C. N. (2008). Resolvin E1, an EPA-derived mediator in whole blood, selectively counterregulates leukocytes and platelets. Blood 112, 848–855.
Ellett, J. D., Atkinson, C., Evans, Z. P., Amani, Z., Balish, E., Schmidt, M. G., van Rooijen, N., Schnellmann, R. G., and Chavin, K. D. (2010). Murine Kupffer cells are protective in total hepatic ischemia/reperfusion injury with bowel congestion through IL-10. J. Immunol. 184, 5849–5858.
Endres, S., Ghorbani, R., Kelley, V. E., Georgilis, K., Lonnemann, G., van der Meer, J. W., Cannon, J. G., Rogers, T. S., Klempner, M. S., Weber, P. C., Schaefer, E. J., Wolff, S. M., and Dinarello, M. D. (1989). The effect of dietary supplementation with n-3 polyunsaturated fatty acids on the synthesis of interleukin-1 and tumor necrosis factor by mononuclear cells. N. Engl. J. Med. 320, 265–271.
Ferrante, A. W. Jr. (2007). Obesity-induced inflammation: a metabolic dialogue in the language of inflammation. J. Intern. Med. 262, 408–414.
González-Périz, A., Horrillo, R., Ferré, N., Gronert, K., Dong, B., Morán-Salvador, E., Titos, E., Martínez-Clemente, M., López-Parra, M., Arroyo, V., and Clària, J. (2009). Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: a role for resolvins and protectins. FASEB J. 23, 1946–1957.
González-Périz, A., Planagumà, A., Gronert, K., Miquel, R., López-Parra, M., Titos, E., Horrillo, R., Ferré, N., Deulofeu, R., Arroyo, V., Rodés, J., and Clària, J. (2006). Docosahexaenoic acid (DHA) blunts liver injury by conversion to protective lipid mediators: protectin D1 and 17S-hydroxy-DHA. FASEB J. 20, 2537–2539.
Gordon, S., and Taylor, P. R. (2005). Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5, 953–964.
Gronert, K., Maheshwari, N., Khan, N., Hassan, I. R., Dunn, M., and Laniado, S. M. (2005). A role for the mouse 12/15-lipoxygenase pathway in promoting epithelial wound healing and host defense. J. Biol. Chem. 280, 15267–15278.
Haas-Stapleton, E. J., Lu, Y., Hong, S., Arita, M., Favoreto, S., Nigam, S., Serhan, C. N., and Agabian, N. (2007). Candida albicans modulates host defense by biosynthesizing the pro-resolving mediator resolvin E1. PLoS ONE 2, e1316. doi: 10.1371/journal.pone.0001316
Haworth, O., Cernadas, M., Yang, R., Serhan, C. N., and Levy, B. D. (2008). Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat. Immunol. 9, 873–879.
Hellmann, J., Tang, Y., Kosuri, M., Bhatnagar, A., and Spite, M. (2011). Resolvin D1 decreases adipose tissue macrophage accumulation and improves insulin sensitivity in obese-diabetic mice. FASEB J. 25, 2399–2407.
Hong, S., Gronert, K., Devchand, P. R., Moussignac, R. L., and Serhan, C. N. (2003). Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J. Biol. Chem. 278, 14677–14687.
Horrillo, R., González-Périz, A., Martínez-Clemente, M., López-Parra, M., Ferré, N., Titos, E., Morán-Salvador, E., Deulofeu, R., Arroyo, V., and Clària, J. (2010). 5-lipoxygenase activating protein signals adipose tissue inflammation and lipid dysfunction in experimental obesity. J. Immunol. 184, 3978–3987.
Hotamisligil, G. S., Peraldi, P., Budavari, A., Ellis, R., White, M. F., and Spiegelman, B. M. (1996). IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 271, 665–668.
Kim, J. K. (2006). Fat uses a TOLL-road to connect inflammation and diabetes. Cell Metab. 4, 417–419.
Kodama, Y., Kisseleva, T., Iwaisako, K., Miura, K., Taura, K., De Minicis, S., Osterreicher, C. H., Schnabl, B., Seki, E., and Brenner, D. A. (2009). c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology 137, 1467–1477.
Lanthier, N., Molendi-Coste, O., Horsmans, Y., van Rooijen, N., Cani, P. D., and Leclercq, I. A. (2010). Kupffer cell activation is a causal factor for hepatic insulin resistance. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G107–G116.
Leifeld, L., Cheng, S., Ramakers, J., Dumoulin, F. L., Trautwein, C., Sauerbruch, T., and Spengler, U. (2002). Imbalanced intrahepatic expression of interleukin 12, interferon gamma, and interleukin 10 in fulminant hepatitis B. Hepatology 36, 1001–1008.
Lesniewski, L. A., Hosch, S. E., Neels, J. G., de Luca, C., Pashmforoush, M., Lumeng, C. N., Chiang, S. H., Scadeng, M., Saltiel, A. R., and Olefsky, J. M. (2007). Bone marrow-specific Cap gene deletion protects against high-fat diet-induced insulin resistance. Nat. Med. 13, 455–462.
Li, P., Lu, M., Nguyen, M. T., Bae, E. J., Chapman, J., Feng, D., Hawkins, M., Pessin, J. E., Sears, D. D., Nguyen, A. K., Amidi, A., Watkins, S. M., Nguyen, U., and Olefsky, J. M. (2010). Functional heterogeneity of CD11c-positive adipose tissue macrophages in diet-induced obese mice. J. Biol. Chem. 285, 15333–15345.
Lieber, C. S. (1997). Role of oxidative stress and antioxidant therapy in alcoholic and nonalcoholic liver diseases. Adv. Pharmacol. 38, 601–628.
Lumeng, C. N., Bodzin, J. L., and Saltiel, A. R. (2007). Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 117, 175–184.
Lumeng, C. N., and Saltiel, A. R. (2011). Inflammatory links between obesity and metabolic disease. J. Clin. Invest. 121, 2111–2117.
Mantovani, A., Sica, A., and Locati, M. (2007). New vistas on macrophage differentiation and activation. Eur. J. Immunol. 37, 14–16.
Martínez, F. O., Helming, L., and Gordon, S. (2009). Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 27, 451–483.
Martínez, F. O., Sica, A., Mantovani, A., and Locati, M. (2008). Macrophage activation and polarization. Front. Biosci. 13, 453–461.
Miura, K., Kodama, Y., Inokuchi, S., Schnabl, B., Aoyama, T., Ohnishi, H., Olefsky, J. M., Brenner, D. A., and Seki, E. (2010). Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 139, 323–324.
Mosser, D. M., and Edwards, J. P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969.
Nettleton, J. A., and Katz, R. (2005). n-3 Long-chain polyunsaturated fatty acids in type 2 diabetes: a review. J. Am. Diet. Assoc. 105, 428–440.
Neuman, M. G., Sha, K., Esguerra, R., Zakhari, S., Winkler, R. E., Hilzenrat, N., Wyse, J., Cooper, C. L., Seth, D., Gorrell, M. D., Haber, P. S., McCaughan, G. W., Leo, M. A., Lieber, C. S., Voiculescu, M., Buzatu, E., Ionescu, C., Dudas, J., Saile, B., and Ramadori, G. (2008). Inflammation and repair in viral hepatitis C. Dig. Dis. Sci. 53, 1468–1487.
Nguyen, M. T., Favelyukis, S., Nguyen, A. K., Reichart, D., Scott, P. A., Jenn, A., Liu-Bryan, R., Glass, C. K., Neels, J. G., and Olefsky, J. M. (2007). A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 282, 35279–35292.
Odegaard, J. I., Ricardo-Gonzalez, R. R., Red, E. A., Vats, D., Morel, C. R., Goforth, M. H., Subramanian, V., Mukundan, L., Ferrante, A. W., and Chawla, A. (2008). Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 7, 496–507.
Oh, D. Y., Talukdar, S., Bae, E. J., Imamura, T., Morinaga, H., Fan, W., Li, P., Lu, W. J., Watkins, S. M., and Olefsky, J. M. (2010). GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142, 687–698.
Olefsky, J. M., and Glass, C. K. (2010). Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 72, 219–246.
Ouchi, N., Parker, J. L., Lugus, J. J., and Walsh, K. (2011). Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 11, 85–97.
Patsouris, D., Li, P. P., Thapar, D., Chapman, J., Olefsky, J. M., and Neels, J. G. (2008). Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 8, 301–309.
Planagumà, A., Clària, J., Miquel, R., López-Parra, M., Titos, E., Masferrer, J. L., Arroyo, V., and Rodés, J. (2005). The selective cyclooxygenase-2 inhibitor SC-236 reduces liver fibrosis by mechanisms involving non-parenchymal cell apoptosis and PPARgamma activation. FASEB J. 19, 1120–1122.
Quiroga, J., and Prieto, J. (1993). Liver cytoprotection by prostaglandins. Pharmacol. Ther. 58, 67–91.
Ramadori, G., and Armbrust, T. (2001). Cytokines in the liver. Eur. J. Gastroenterol. Hepatol. 13, 777–784.
Ramadori, G., Moriconi, F., Malik, I., and Dudas, J. (2008). Physiology and pathophysiology of liver inflammation, damage and repair. J. Physiol. Pharmacol. 59(Suppl. 1), 107–117.
Sanyal, A. J. (2005). Mechanisms of disease: pathogenesis of nonalcoholic fatty liver disease. Nat. Clin. Pract. Gastroenterol. Hepatol. 2, 46–53.
Schif-Zuck, S., Gross, N., Assi, S., Rostoker, R., Serhan, C. N., and Ariel, A. (2011). Saturated-efferocytosis generates pro-resolving CD11b low macrophages: modulation by resolvins and glucocorticoids. Eur. J. Immunol. 41, 366–379.
Schwab, J. M., Chiang, N., Arita, M., and Serhan, C. N. (2007). Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447, 869–874.
Scotton, C. J., Martinez, F. O., Smelt, M. J., Sironi, M., Locati, M., Mantovani, A., and Sozzani, S. (2005). Transcriptional profiling reveals complex regulation of the monocyte IL-1 beta system by IL-13. J. Immunol. 174, 834–845.
Serhan, C. N. (2011). The resolution of inflammation: the devil in the flask and in the details. FASEB J. 25, 1441–1448.
Serhan, C. N., Brain, S. D., Buckley, C. D., Gilroy, D. W., Haslett, C., O’Neill, L. A., Perretti, M., Rossi, A. G., and Wallace, J. L. (2007). Resolution of inflammation: state of the art, definitions and terms. FASEB J. 21, 325–332.
Serhan, C. N., Chiang, N., and Van Dyke, T. E. (2008). Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361.
Serhan, C. N., Clish, C. B., Brannon, J., Colgan, S. P., Chiang, N., and Gronert, K. (2000). Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 192, 1197–1204.
Serhan, C. N., Hong, S., Gronert, K., Colgan, S. P., Devchand, P. R., Mirick, G., and Moussignac, R. L. (2002). Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 196, 1025–1037.
Shapiro, H., Tehilla, M., Attal-Singer, J., Bruck, R., Luzzatti, R., and Singer, P. (2011). The therapeutic potential of long-chain omega-3 fatty acids in nonalcoholic fatty liver disease. Clin. Nutr. 30, 6–19.
Shoelson, S. E., Lee, J., and Goldfine, A. B. (2006). Inflammation and insulin resistance. J. Clin. Invest. 116, 1793–1801.
Spite, M., Norling, L. V., Summers, L., Yang, R., Cooper, D., Petasis, N. A., Flower, R. J., Perretti, M., and Serhan, C. N. (2009). Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461, 1287–1291.
Tabas, I. (2010). Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 10, 36–46.
Takeuchi, D., Yoshidome, H., Kato, A., Ito, H., Kimura, F., Shimizu, H., Ohtsuka, M., Morita, Y., and Miyazaki, M. (2004). Interleukin 18 causes hepatic ischemia/reperfusion injury by suppressing anti-inflammatory cytokine expression in mice. Hepatology 39, 699–710.
Tilg, H., and Moschen, A. R. (2008). Insulin resistance, inflammation, and non-alcoholic fatty liver disease. Trends Endocrinol. Metab. 19, 371–379.
Titos, E., Clària, J., Bataller, R., Bosch-Marcé, M., Ginès, P., Jiménez, W., Arroyo, V., Rivera, F., and Rodés, J. (2000). Hepatocyte-derived cysteinyl leukotrienes modulate vascular tone in experimental cirrhosis. Gastroenterology 119, 794–805.
Titos, E., Clària, J., Planagumà, A., López-Parra, M., González-Périz, A., Gaya, J., Miquel, R., Arroyo, V., and Rodés, J. (2005). Inhibition of 5-lipoxygenase-activating protein abrogates experimental liver injury: role of Kupffer cells. J. Leukoc. Biol. 78, 871–878.
Titos, E., Clària, J., Planagumà, A., López-Parra, M., Villamor, N., Parrizas, M., Carrió, A., Miquel, R., Jiménez, W., Arroyo, V., Rivera, F., and Rodés, J. (2003). Inhibition of 5-lipoxygenase induces cell growth arrest and apoptosis in rat Kupffer cells: implications for liver fibrosis. FASEB J. 17, 1745–1747.
Titos, E., Rius, B., González-Périz, A., López-Vicario, C., Morán-Salvador, E., Martínez-Clemente, M., Arroyo, V., and Clària, J. (2011). Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J. Immunol. (in press).
Weisberg, S. P., McCann, D., Desai, M., Rosenbaum, M., Leibel, R. L., and Ferrante, A. W. Jr. (2003). Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112, 1796–1808.
Wellen, K. E., and Hotamisligil, G. S. (2003). Obesity-induced inflammatory changes in adipose tissue. J. Clin. Invest. 112, 1785–1788.
Keywords: obesity, adipocytes, M2 macrophages, resolvins, docosahexaenoic acid, Kupffer cells
Citation: Clària J, González-Périz A, López-Vicario C, Rius B and Titos E (2011) New insights into the role of macrophages in adipose tissue inflammation and fatty liver disease: modulation by endogenous omega-3 fatty acid-derived lipid mediators. Front. Immun. 2:49. doi: 10.3389/fimmu.2011.00049
Received: 17 August 2011;
Paper pending published: 26 August 2011;
Accepted: 15 September 2011;
Published online: 05 October 2011.
Edited by:
Amiram Ariel, University of Haifa, IsraelCopyright: © 2011 Clària, González-Périz, López-Vicario, Rius and Titos. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Joan Clària and Esther Titos, Department of Biochemistry and Molecular Genetics, Hospital Clínic, Villarroel 170, Barcelona 08036, Spain. e-mail:amNsYXJpYUBjbGluaWMudWIuZXM=;ZXN0aGVyLnRpdG9zQGNpYmVyZWhkLm9yZw==