 Friederike Jönsson1,2
Friederike Jönsson1,2 Marc Daëron1,2*
Marc Daëron1,2*- 1 Institut Pasteur, Département d’Immunologie, Unité d’Allergologie Moléculaire et Cellulaire, Paris, France
- 2 Inserm, Unité 760, Paris, France
Classically, allergy depends on IgE antibodies and on high-affinity IgE receptors expressed by mast cells and basophils. This long accepted IgE/FcεRI/mast cell paradigm, on which the definition of immediate hypersensitivity was based in the Gell and Coomb’s classification, appears too reductionist. Recently accumulated evidence indeed requires that not only IgE but also IgG antibodies, that not only FcεRI but also FcγR of the different types, that not only mast cells and basophils but also neutrophils, monocytes, macrophages, eosinophils, and other myeloid cells be considered as important players in allergy. This view markedly changes our understanding of allergic diseases and, possibly, their treatment.
Introduction
A provocative paper was published at the very beginning of the twentieth century (Richet and Portier, 1902). Louis Pasteur had just taught the world that immunity is not only the fortune of those individuals who survived an epidemic and became protected from that disease. It can be deliberately induced and protect from all kinds of infections, by variations of an empirical practice promoted one century earlier by Edward Jenner to prevent small pox. Immunity is not a privilege. Anyone can be immunized, and thus protected. But in 1901, Charles Richet and Paul Portier make a disturbing observation. Instead of protecting, an immunization with a low dose of toxin can induce a state of hypersensitivity, which kills a dog within minutes following a second exposure to the same harmless dose of toxin. They repeat their experiments, they confirm their observation with another toxin, they convince themselves that they have unraveled a new phenomenon. They name it anaphylaxis, i.e., the opposite of Pasteur’s prophylaxis. The fame of Pasteur is immense, worldwide. Richet and Portier have the intellectual courage to publish their finding: immunity is not necessarily protective, it can be deleterious and even kill. Charles Richet was awarded the Nobel price in Physiology or Medicine in 1913 “for his work on anaphylaxis.”
Anaphylaxis was soon understood as a hyper-acute systemic allergic syndrome and, since then, unique mechanisms were searched for to explain this paradoxical response of the immune system that generates disease when reacting to substances of the environment as innocuous as pollen, cat hair, house dust, or even food. In 1910, histamine, a substance isolated from rye pin, was found to reproduce anaphylactic symptoms (Dale and Laidlaw, 1910). In the 1950s, histamine was found in cells present as a minor population in tissues (Riley, 1953) and, in the 1960s, these mast cells were shown to degranulate and to release histamine upon challenge with antibody and antigen (Prouvost-Danon et al., 1966). In 1966, a unique Erythema–wheal reaction-inducing Immunoglobulin (IgE), present in minute amounts in plasma (Ishizaka et al., 1966) but, luckily, secreted in large amounts by a rare plasmocytoma (Johansson and Bennich, 1967), was described and reported to account for allergic reactions (Bennich et al., 1969). IgE were found to be cytotropic, i.e., to bind to cells (Evans and Thomson, 1972), and in the 1970s, a receptor with a uniquely high-affinity for IgE was described on mast cells and basophils (Kulczycki and Metzger, 1974), which was named FcεRI, cloned in 1989 (Blank et al., 1989), and the 3D structure of which was solved in 1998 (Garman et al., 1998). The allergen-IgE-FcεRI-mast cell-histamine-clinical symptoms sequence became the paradigm of an allergic reaction. It accounted for Type I Hypersensitivity and was enclosed in the Gell and Coombs’ classification (Gell and Coombs, 1963). The term initially coined by Clemens von Pirquet after the Greek words ἂλλoς ἔργoν (allos ergon) to designate the altered response to a given substance induced by a previous contact with that substance (von Pirquet, 1906), became increasingly used with its etymological meaning: allergy is another reaction.
In the early 1950s, however, Zoltan Ovary described passive cutaneous anaphylaxis (PCA) and used it as a sensitive technique (Ovary, 1952) to study the IgG antibodies that are responsible for this in vivo reaction in guinea pigs (Ovary et al., 1960), rabbits (Warner and Ovary, 1970), rats, and mice (Ovary et al., 1975). The same IgG antibodies were then shown to activate rat and mouse mast cells in vitro (Vaz and Prouvost-Danon, 1969), and IgG receptors were described on these cells (Tigelaar et al., 1971). When, much later, the first knock-out mice were generated, a paper reported that active systemic anaphylaxis (ASA) could be induced in IgE-deficient mice (Oettgen et al., 1994). IgE are not alone, and much more IgG antibodies are produced together with IgE, whatever the immunization protocol used. Antibodies other than IgE contribute to allergic responses. Likewise, evidence accumulated that mast cells and basophils work in concert with eosinophils, neutrophils, monocytes, T cells and NK cells to mount allergic inflammation. Conversely, mast cells and IgE are involved in biological responses other than allergy. The Gell and Coombs boxes were not sealed off. Cells of different types and antibodies of different classes sneaked in and out. Like other antibody-dependent inflammatory diseases, allergy involves the same molecular and cellular effectors as protective immunity.
Mast Cells Beyond Allergy
We know now that we have two immune systems. The innate immune system is made of a large number of differentiated cells of several types, mostly of the myeloid lineage, equipped with pattern-recognition receptors that can induce a variety of responses to pathogens without delay. The adaptive immune system is essentially made of limited numbers of lymphoid cells equipped with antigen receptors, which need to proliferate and to differentiate into effector cells of different types before they can act on specific antigens (Figure 1).
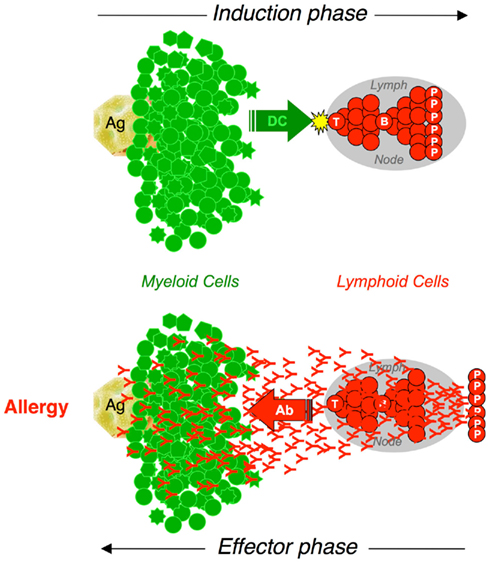
Figure 1. The interplay between myeloid and lymphoid cells in adaptive immune responses. Adaptive immune responses are initiated by the presentation of antigen by dendritic cells (DC). Cognate interactions with antigen-presenting cells activate naive T cells that proliferate and differentiate. TH cells collaborate with specific B cells that proliferate and differentiate into Plasma Cells (P) that secrete antibodies (Induction phase). Antibodies diffuse throughout the body. When interacting with Fc Receptor-expressing myeloid cells and with specific antigen, antibodies induce a wide array of biological responses that depend on the cell type (Effector phase).
Mast Cells as Effectors of Innate Immunity
Mast cells have increasingly been recognized as effector cells of innate immunity. Located everywhere in the body, but particularly at interfaces with the external world and near blood vessels, they contribute to protect against pathogens (reviewed in Abraham and St John, 2010). They are further recruited to infection sites. Mouse and human mast cells express Toll-like and NOD-like receptors through which pathogen-associated molecular patterns and proteoglycans induce them to release proteases and to secrete cytokines, chemokines, and growth factors (Supajatura et al., 2002). These, in turn, recruit neutrophils, eosinophils, NK cells and other cells that form an inflammatory infiltrate (Supajatura et al., 2001). Mouse mast cells also produce bactericidal peptides such as cathelicidin (Di Nardo et al., 2003). These mechanisms altogether account for the critical in vivo protective roles of mast cells in infection, unraveled by the cecum ligation and puncture model of acute peritonitis (Echtenacher et al., 1996) and by bacterial challenge (Supajatura et al., 2001). Rat mast cells have also been associated with helminth infection during which they proliferate in response to stem cell factor (SCF), and they contribute to worm expulsion by several mechanisms (Levy and Frondoza, 1983; Woodbury et al., 1984).
More recently, mouse mast cells were found to protect from honeybee, snake, lizard, and scorpion venoms. Venoms indeed induce mast cell degranulation and they are degraded by proteases contained in granules. Thus, carboxypeptidase A3 hydrolyzes the venom peptide sarafotoxin 6b (Metz et al., 2006) and the related mammalian vasoconstrictor peptide endothelin-1 (Maurer et al., 2004), while chymase mast cell protease 4 hydrolyzes the lizard venom helodermin and the related mammalian vaso-intestinal peptide (Akahoshi et al., 2011).
With and like dendritic cells (DC), mast cells are involved in the initiation of adaptive immunity. Mouse mast cells promote DC differentiation and, by up-regulating E-selectin expression on vascular endothelium cells, the influx of monocyte-derived DC (Shelburne et al., 2009). Mouse mast cell products modulate DC activation and antigen presentation (Amaral et al., 2007), leading to a skewed Th2 cell differentiation (Mazzoni et al., 2006). Whether mast cells themselves can present antigen has long been unclear as major histocompatibility class II (MHC-II) molecules were not found on fresh peritoneal mouse mast cells (Daëron and Voisin, 1979) or on bone marrow-derived mast cells (BMMC) cultured with IL-3 (Frandji et al., 1995). Recently, however, it was shown that MHC-II could be induced by IFN-γ and IL-4 on mouse peritoneal mast cells and on peritoneal cell-derived mast cells (PCMC; Gaudenzio et al., 2009), a cultured model of mature serosal-type mast cells that resemble peritoneal mast cells (Malbec et al., 2007), but not on BMMC. When loaded with antigen, such cytokine-primed MHC-II+ mast cells could establish cognate interactions with CD4+ T cells for antigen presentation and bidirectional cell–cell cooperation (Valitutti and Espinosa, 2010), resulting in T cell activation and enhanced responsiveness of mast cells to IgE-induced activation.
Mast Cells as Effectors of Adaptive Immunity
Mast cells are involved not only in the induction phase, but also in the effector phase of adaptive immunity. Adaptive immune responses generate effector cells and molecules. Cells include antigen receptor-expressing T cells, endowed with various effector functions, and capable of secreting numerous antigen-non-specific cytokines upon cognate interactions with specific antigen. B cell-derived plasma cells have no antigen receptor, but they secrete high amounts of antigen-specific antibodies that reach the bloodstream and diffuse in the whole body (Figure 1).
Antibodies, however, have no biological activity by themselves. Their Fab portions can bind to antigens with a variable specificity and affinity, but except in rare instances, binding to antigen has little or no biological consequences. Therapeutic antibodies raised against receptors (Li et al., 2005) or ligands (Ellis and Hicklin, 2008) can prevent ligand–receptor interactions and some anti-receptor antibodies can mimic agonists (Shan et al., 2000). It was recently shown, however, that to “neutralize” bacterial toxins (Joller et al., 2010) or viruses (Mallery et al., 2010), antibodies require that their Fc portion binds to cellular receptors that transport immune complexes to intracellular compartments where they are degraded. For antibodies to affect antigens, they indeed not only need to bind to antigens through their Fab portions, but also to interact through their Fc portion with effector systems. Among these are the many cells, mostly of hematopoietic origin, that express receptors for the Fc portion of antibodies (FcR). FcR provide these cells, which have no antigen receptors, with B Cell Receptor-like immunoreceptors and a bona fide immunological specificity (Daëron, 1997). Importantly, this specificity is an intrinsic property neither of the cell nor of the FcR, but of antibodies. When binding to FcR, specific antibodies enroll the effector cells of innate immunity in adaptive immunity (Figure 1). Mast cells are such cells.
Antibody Receptors Expressed by Mast Cells and Other Myeloid Cells
FcR are immunoreceptors of the third type. If the BCR enables B cells to interact with native antigen and if the TCR enables T cells to interact with antigen-derived peptides associated with MHC molecules, FcR enable a variety of cell types to interact with antigen under the form of immune complexes. These can thus stimulate and/or control a variety of biological responses that primarily depend on the cell type. Besides high-affinity IgE receptors, mast cells and basophils share several antibody receptors with other myeloid cells.
High- and Low-Affinity FcR
Antibodies bind to FcR with a variable affinity (Hulett and Hogarth, 1994). A proportion of high-affinity FcR, which can bind monomeric immunoglobulins in the absence of antigen, are occupied in vivo whereas low-affinity FcR, which can bind antibodies as multivalent immune complexes only, are not in spite of the high concentration of circulating immunoglobulins.
High-affinity FcR include IgA (FcαRI, in humans), IgE (FcεRI, in humans and mice), and IgG receptors (FcγRI, in humans and mice, and FcγRIV, in mice only). Low-affinity FcR include IgE (FcεRII, in humans and mice) and IgG receptors (FcγRII and III, in humans and mice). Humans have three FcγRII (FcγRIIA, B, and C), and two FcγRIII (FcγRIIIA and B) whereas mice have one receptor of each type (FcγRIIB and FcγRIIIA) only. The diversity of human FcγRII and III is further increased by polymorphisms of specific residues in their extracellular domains (Figure 2).

Figure 2. Murine and human receptors for the Fc portion (FcR) of IgE and IgG. This figure schematizes FcR for IgE and for IgG expressed by mouse and human myeloid cells.
The affinity constant (Ka) of mouse and human FcεRI for IgE is 109–10 M−1 (Kulczycki and Metzger, 1974; Metzger, 2004); those of human and mouse FcγR for human and mouse IgG subclasses, respectively, span from ±105 to 108 M−1 (Figure 3; Bruhns et al., 2009). Noticeably, low-affinity interactions were described between IgE and IgG receptors. Mouse IgE can bind to mouse FcγRIIB and FcγRIIIA with a Ka ± 2 × 104 M−1 (Takizawa et al., 1992) and to FcγRIV with a Ka ± 3 × 105 M−1 (Mancardi et al., 2008; Figure 3).

Figure 3. Affinity of mouse and human FcR for homologous IgE and IgG subclasses. Affinity constants were determined by Plasmon resonance analysis. (Dierks et al., 1993; Hibbert et al., 2005; Nimmerjahn and Ravetch, 2005; Mancardi et al., 2008; Bruhns et al., 2009).
FcR Signals
FcR trigger no signal when binding immunoglobulins. They signal when aggregated on the cell membrane by antibodies and plurivalent antigens (Maeyama et al., 1986; Metzger, 1992). Although the result is the same, the sequence of events that lead to receptor aggregation is different for high-affinity and low-affinity FcR. Monomeric antibodies bind first to high-affinity FcR that are aggregated when a plurivalent antigen binds to receptor-bound antibodies. Antibodies bind first to antigen, generating immune complexes that can bind to and, therefore, simultaneously aggregate low-affinity FcR.
FcR can trigger activation signals and/or inhibition signals. The nature of signals primarily depends on molecular motifs contained in the intracytoplasmic domains of FcR or of receptor subunits with which FcR associate. Two such motifs were identified. Immunoreceptor tyrosine-based activation motifs (ITAM) consist of two YxxL motifs separated by a six to eight variable amino acid sequence (Reth, 1989). Immunoreceptor tyrosine-based inhibition motifs (ITIM) consist of a single YxxL motif preceded by a loosely conserved often hydrophobic residue at position Y-2 (Vivier and Daëron, 1997).
Activating FcR are FcαRI, FcεRI, FcγRI, FcγRIIA, FcγRIIC, FcγRIIIA, and FcγRIV. FcγRIIA and FcγRIIC are single-chain receptors the intracytoplasmic domains of which are identical and contain an ITAM. FcαRI, FcεRI, FcγRI, FcγRIIIA, and FcγRIV associate with the common FcR subunit FcRγ (Daëron, 1997). FcRγ is a homodimer that contains two ITAM (Orloff et al., 1990). FcεRI also associate with another subunit named FcRβ (Kinet et al., 1988). FcRβ is a tetraspanin that contains an ITAM. Upon receptor aggregation, ITAM are phosphorylated by src-family tyrosine kinases (Pribluda et al., 1994), which initiates the constitution of dynamic intracellular signalosomes (Kent et al., 1994) in which activation signals are dominant over inhibition signals (Malbec et al., 2004).
Inhibitory receptors are FcγRIIB. FcγRIIB are single-chain receptors the intracytoplasmic domain of which contains one ITIM (Daëron et al., 1995a). Unlike activating receptors, FcγRIIB trigger no intracellular signal upon aggregation. They trigger negative signals when they are coaggregated with activating receptors by immune complexes (Daëron et al., 1995b). Under these conditions, the ITIM of FcγRIIB is phosphorylated by the same src-family tyrosine kinase that phosphorylates ITAM in activating receptors (Malbec et al., 1998). Phosphorylated FcγRIIB recruit inhibitory molecules that are brought into signalosomes. This renders inhibition signals dominant over activation signals (Lesourne et al., 2005; Daëron and Lesourne, 2006).
Biological Responses Induced by FcR and Their Tissue Distribution
Activating FcR can trigger a variety of cellular responses, including the endocytosis of soluble immune complexes, the phagocytosis of particulate complexes, the exocytosis of preformed granular mediators, including vasoactive amines, proteolytic enzymes, and/or cytotoxic molecules, the production of newly formed lipid-derived pro-inflammatory mediators or the secretion of newly transcribed cytokines, chemokines, and growth factors. The nature of biological responses triggered by antibodies via FcR primarily depends on the cell type. They are indeed determined by the functional repertoire of the cell and, therefore, by the tissue distribution of FcR. Except NK cells that express FcγRIIIA only, B cells that express FcγRIIB only, and T cells that express none, other cells of hematopoietic origin co-express several FcR and, in most cases, activating and inhibitory FcR. FcγRIIB, however, have a more restricted tissue distribution in humans than in mice. In both mice and humans, mast cells, and basophils express constitutively not only high-affinity FcεRI, but also low-affinity FcγR of several types (Figure 4).
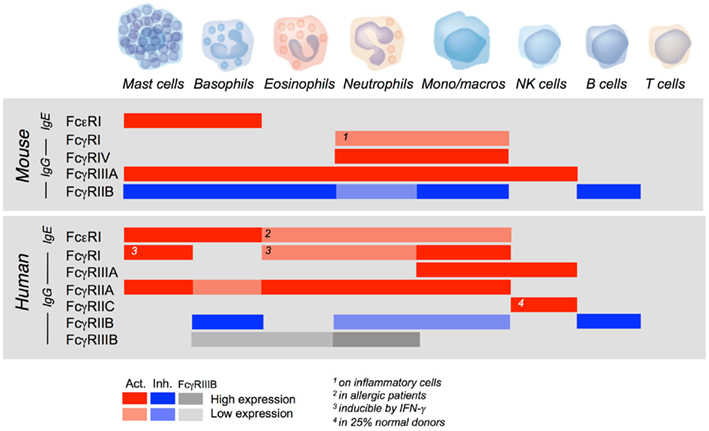
Figure 4. Tissue distribution of mouse and human FcR for IgE and IgG. The figure shows the relative expression of activating (red) and inhibitory (blue) IgE and IgG receptors of the immunoglobulin superfamily by murine and human hematopoietic cells. Gray bars show the expression of FcγRIIIB.
Multi-chain FcR need to associate with FcRγ in order to reach the plasma membrane. Their intracytoplasmic domain indeed contains a retention site that sequesters the polypeptide in the endoplasmic reticulum, unless it associates with FcRγ (Lobell et al., 1993; Letourneur et al., 1995). The expression of these receptors therefore depends on the tissue distribution of FcRγ, and FcRγ-deficient mice have no activating FcR (Takai et al., 1994). Mouse FcεRI, but not human FcεRI, also need to associate with FcRβ (Kinet, 1999). As FcRβ is expressed by mast cells and basophils only in both species, the expression of FcεRI is restricted to these cells in mice but not in humans (Gounni et al., 1994, 2001; Joseph et al., 1997). Noticeably, mouse FcγRIIIA, the expression of which requires FcRγ but not FcRβ, associate with both subunits in mast cells (Kurosaki et al., 1992).
Differential Effects of IgG Antibodies in Mast Cells and Basophils
The pattern of FcR expression enables mast cells to be activated not only by IgE, but also by IgG antibodies. IgE-induced responses of mast cells and basophils are well known. IgG-induced responses of mast cells are less known. Likewise, basophils co-express receptors for IgE and IgG. Unlike mast cells, however, basophils are not activated by IgG.
Mast Cell Responses to IgG Antibodies
Besides FcεRI, mouse mast cells express constitutively, FcγRIIB and FcγRIIIA (Benhamou et al., 1990). As FcεRI and FcγRIIIA are expressed in association with the same subunits in mouse mast cells, they can trigger the same biological responses. The aggregation of mouse FcγRIIIA was demonstrated to trigger both the release of serotonin (Daëron et al., 1992) and the secretion of TNF-α (Latour et al., 1992) when expressed in rat basophilic leukemia (RBL) 2H3 transfectants. Likewise, the engagement of FcγRIIIA on BMMC or on PCMC triggered a release of β-hexosaminidase and a secretion of TNF-α (Malbec et al., 2007). FcεRI- and FcγRIIIA-dependent responses of BMMC were qualitatively similar (they released the same granular mediators and secreted the same cytokines). IgE- and IgG-induced responses were quantitatively similar in mouse peritoneal mast cells and in PCMC.
Although mouse BMMC and PCMC express similar amounts of FcγRIIB and FcγRIIIA, PCMC can be readily activated by IgG immune complexes, but not BMMC. The reason is that FcγRIIB has a potent inhibitory effect on FcγRIIIA-dependent activation in BMMC, but a much milder one in PCMC. FcγRIIB-deficient BMMC indeed responded similarly to IgG immune complexes as FcγRIIB-deficient PCMC, and both mast cell types contained similar amounts of the SH2 domain-containing inositolphosphatidyl 5-phosphatase SHIP1 that accounts for FcγRIIB-dependent negative regulation (Ono et al., 1996; Huber et al., 1998). The difference was that, for an unknown reason, BMMC use SHIP1 more efficiently than PCMC (Malbec et al., 2007).
Besides FcεRI, human skin mast cells express constitutively FcγRIIA (Ghannadan et al., 1998; Zhao et al., 2006). All human mast cells, however, may not express FcγRIIA as lung mast cells were reported to be CD32-negative (Agis et al., 1996). Human cord blood-derived mast cells (CBMC) were reported to express FcγRIIB (Kepley et al., 2004), but not skin-derived human mast cells (Zhao et al., 2006). Because they reside in tissues, mast cells are not readily accessible in humans. Tissue samples obtained following surgery are the only available source of mature human mast cells. Although, like mouse BMMC, they are immature cells that have no known physiological equivalent, CBMC have been more often used. CBMC express FcγRIIA (our unpublished observation) and FcγRIIB (Kepley et al., 2004). The human mast cell line HMC-1 also expresses FcγRIIA, but no FcεRI. The aggregation of FcγRIIA with specific antibodies induced the release of serotonin by RBL-2H3 transfectants (Daëron et al., 1995a). The engagement of FcγRIIA by IgG immune complexes triggered peritoneal mast cells and PCMC from FcγRIIA-transgenic mice to degranulate and to release histamine (Jönsson et al., 2011b). Finally, anti-FcγRIIA antibodies triggered human skin-derived mast cells to release histamine (Jönsson et al., 2011b) and to secrete lipid-derived mediators and cytokines (Zhao et al., 2006).
In addition to low-affinity IgG receptors, high-affinity IgG receptors (FcγRI) could be induced by IFN-γ on human (Okayama et al., 2000), but not on mouse (Tan et al., 2003) mast cells, and it was found on skin mast cells from lesions of psoriasis patients (Tkaczyk et al., 2002) and intestinal mast cells from Crohn’s disease patients (Kobayashi et al., 2007). These receptors associated with the FcRγ subunit in IFN-γ-treated human mast cells. Whether they also associated with FcRβ is unknown. FcγRI aggregation induced the release of histamine, the production of prostaglandin D2 and leukotriene C4, and the secretion of IL-3, IL-13, TNF-α, and GM-CSF by IFN-γ-treated human mast cells (Okayama et al., 2001; Tkaczyk et al., 2002).
Basophil Responses to IgG Antibodies
Besides FcεRI, mouse basophils express constitutively FcγRIIIA and FcγRIIB, i.e., the same FcR as mouse mast cells. Unlike PCMC, but like BMMC, they fail to respond to IgG immune complexes. This absence of response results from an active inhibition of FcγRIIIA-dependent activation signals by FcγRIIB. Basophils from wt mice indeed secreted IL-4 in response to FcγRIIIA engagement by FcγRIIIA-specific antibodies or by IgG immune complexes when FcγRIIB were blocked by a FcγRIIB-specific antibody. Confirming these results, basophils from FcγRIIB-deficient mice secreted high amounts of IL-4 in response to IgG immune complexes. FcγRIIB therefore prevent mouse basophils to respond to IgG antibodies (our unpublished data).
Besides FcεRI, human basophils express constitutively FcγRIIA, FcγRIIB (Kepley et al., 2000), and small amounts of FcγRIIIB (Meknache et al., 2009). Basophils from normal donors could be readily activated, as assessed by CD203c up-regulation and histamine release, upon FcεRI engagement with F(ab′)2 fragments of anti-IgE antibodies. They were not detectably activated upon FcγRIIIB engagement by F(ab′)2 fragments of anti-FcγRIIIB antibodies (Meknache et al., 2009). Whether the absence of response to FcγRIIIB engagement is due to the very low expression of this receptor on basophils (10- to 100-fold lower than on neutrophils) or by the inability of this glycosylphosphatidyl-anchored receptor to trigger activation signals by itself is unclear. Normal basophils could be also activated by F(ab′)2 fragments of anti-FcγRIIA antibodies, especially when cells were primed by an overnight incubation with human IL-3, but much less than by anti-IgE. They failed to detectably respond when challenged with IgG immune complexes or heat-aggregated human IgG, even if primed with IL-3. The poor response of basophils to anti-FcγRIIA antibodies and their absence of response to IgG immune complexes can be explained by the relative expression of FcγRIIA and FcγRIIB, respectively. We indeed found that human basophils are among the blood cells that display the lowest levels of FcγRIIA, and that they express more FcγRIIB than any other blood cells (our unpublished data).
Like mouse basophils but, probably not for the same reason, human basophils therefore do not respond to IgG antibodies. It follows that FcγR do not exert the same functions in mast cells and basophils: when FcγR are engaged by IgG immune complexes, activation is dominant in mast cells whereas inhibition is dominant in basophils.
Mast Cells and Basophils in in vivo Models of Allergy and Autoimmunity
Contrasting with the in vitro properties of mast cells and basophils, their contribution to human diseases or even to murine models of human disease has been more difficult to delineate. One reason is that, until very recently, there was no satisfactory mean to investigate the contributions of mast cells and basophils in vivo.
Strengths and Weaknesses of Available Tools Used to Evaluate the Contribution of Mast Cells and Basophils in in vivo Models of Inflammatory Diseases
Chemical tools, so called mast cell stabilizers, like cromolyn, ketotifen or tranilast, have been used to treat asthmatic and other allergic patients for almost three decades (Patalano and Ruggieri, 1989; Ruggieri and Patalano, 1989). They have sometimes been used also as pharmacological inhibitors of mast cell functions (Costa-Pinto et al., 2007; Kano et al., 2008). The mechanism by which these drugs inactivate mast cells is poorly understood and, cromolyn also affects eosinophils and neutrophils. Results obtained using such tools are therefore difficult to interpret (Moqbel et al., 1986).
More importantly, mast cell-deficient mice have been widely used to investigate the involvement of mast cells in vivo, in a variety of experimental models of human diseases. There are several such mice. WCB6F1/J-KitlSl/KitlSl-d (Sl/Sl-d) mice do not express membrane SCF on fibroblasts and other cells (Zsebo et al., 1990). WBB6F1-KitW/KitW-v (W/W-v) mice are compound mutants for the SCF receptor Kit (one allele being null, the other impaired). C57BL/6-KitW-sh (Wsh) mice carry an inversion of a regulatory element, leading to Kit deficiency, but also to a disruption of the gene encoding the transmembrane protease corin that is expressed on cardiomyocytes and regulates blood pressure (Nigrovic et al., 2008). Experimental diseases that were either abrogated or milder in these mice than in wt mice were proposed to depend on mast cells. Data obtained with these mice must, however, be interpreted with caution. If they are mast cell-deficient, these mice indeed exhibit numerous additional abnormalities. The reason is that Kit is expressed not only by mast cells, but also by hematopoietic progenitor cells, germinal cells, and a few differentiated cells such as melanocytes and intestinal Cajal cells. Thus, Sl/Sl-d and W/W-v mice are sterile and anemic. W/W-v mice are neutropenic, they lack Cajal cells and intraepithelial γδ T cells, they are partially deaf and prone to dermatitis and gastritis. Wsh mice are fully fertile, but they frequently develop myeloid and megakaryocytic proliferation in the spleen and bone marrow, resulting in neutrophilia, basophilia, increased mast cell progenitor numbers in the spleen and elevated platelet numbers in the blood (Mancardi et al., 2011). Illustrating the problem, discrepant results were observed in models of contact hypersensitivity in W/W-v and in Wsh mice (van Loveren et al., 1983; Galli and Hammel, 1984; Mekori and Galli, 1985; Biedermann et al., 2000; Grimbaldeston et al., 2007).
In view of the many abnormalities that affect Kit-mutant mice, the conclusion that an experimental process depends on mast cells because it does not occur in such mice is generally confirmed by the demonstration that it can be restored if deficient mice are reconstituted with mast cells. Conclusions drawn from these experiments must, however, also be taken with caution. Cultured mast cells, i.e., essentially BMMC, are commonly used to reconstitute mast cell-deficient mice, and reconstitution requires long periods of time (usually 8–12 weeks). Reconstitution is rarely complete and it greatly varies with the anatomical site (Grimbaldeston et al., 2005). In addition, BMMC are immature cells, and even after prolonged differentiation in IL-3 containing culture, they may contain residual pluripotent hematopoietic progenitors. What happens to these cells after they are injected is unknown, and the possibility that an in vivo differentiation into cells other than mast cells occurs cannot be excluded.
Another approach to determine the in vivo roles of mast cells and basophils consists of depleting these cells by injecting mice with specific antibodies. Thus, studies that used the anti-mouse FcεRI mAb MAR-1 or the anti-mouse CD117 (Kit) mAb ACK2, led to the conclusion that mast cells, which express both FcεRI and Kit, are required for experimental oral allergen-induced diarrhea (Brandt et al., 2003). Antibody-induced mast cell depletion has several drawbacks. Indeed, no mast cell-specific antibody is available. MAR-1 has been extensively used to delete basophils (Denzel et al., 2008; Sokol et al., 2008, 2009; Perrigoue et al., 2009; Yoshimoto et al., 2009). Since it targets FcεRI-expressing cells, it however depletes also mast cells. Furthermore, its specificity is questionable as it depletes DC as well (Hammad et al., 2010). ACK2 targets all Kit-expressing cells, i.e., mast cells, Cajal cells, and hematopoietic stem cells. Likewise, the anti-CD200R3 antibody Ba103 was reported to selectively delete basophils when injected intravenously (Obata et al., 2007). CD200R3 is however expressed not only by basophils, but also by mast cells (Obata et al., 2007). CD200R3 functions as an activating receptor whose transmembrane domain contains a lysine residue that is likely to mediate its association with an ITAM-containing subunit such as FcRγ or DAP12 and, indeed, Ba103 activates both basophils and mast cells in vitro (Kojima et al., 2007). How it depletes basophils is unknown. mAb-induced cell depletion is thought to involve activating FcγR-expressing effector cells through antibody-dependent cell-mediated cyotoxicity (ADCC). One may wonder whether, following intensive ADCC, the function of these cells, which are critical in inflammation, might not be altered for subsequent experiments. Finally one can also wonder about the in vivo consequences of a massive release of biologically active molecules stores in the granules of cells when mast cells and basophils are either systemically activated or destroyed by ADCC.
Because of the many difficulties discussed above, we chose to use another approach to investigate the in vivo contributions of mast cells and other myeloid cells in anaphylaxis. This approach consists of using mice whose mast cells and basophils are normally present, but cannot respond to a given stimulus because they lack the necessary receptors. Thus, mast cells and basophils cannot respond to IgG antibodies in FcγRIIB−/−FcγRIIIA−/− mice, but cells that, unlike mast cells, express FcγRIV, and/or FcγRI can. The differential tissue distribution of the various FcR (Figure 4) can thus be exploited to dissect the relative in vivo contributions of different cell types in experimental models of inflammatory diseases.
Recently, genetically modified mice have been generated that enable mast cells and/or basophils to be selectively targeted using promoters of proteases specifically expressed in one cell type or the other (Table 1). Mcpt5 (Scholten et al., 2008), Cpa3 (Feyerabend et al., 2009, 2011; Lilla et al., 2011), or chymase (Musch et al., 2008) were used to target mast cells, whereas Mcpt8 (Ohnmacht et al., 2010; Wada et al., 2010; Sullivan et al., 2011) was used to target basophils. Cell deletion was either inducible or constitutive. Cells expressing the diphtheria toxin receptor (DTR) were depleted following diphtheria toxin-α (DTA) injection, whereas cells expressing DTA were deleted constitutively. Another system, based on the use of the Cre recombinase, happened to lead to the selective constitutive destruction of cells that express high Cre levels, due to Cre toxicity. Mast cells and basophils are such cells when Cre is under the control of the Cpa3 (Feyerabend et al., 2011) or Mcpt8 (Ohnmacht et al., 2010) promoters, respectively. A variation of this system was very recently reported, in which a floxed anti-apoptotic factor was deleted in mast cells and basophils that expressed Cre under the control of Cpa3 (Lilla et al., 2011). Using the Mcpt5 promoter to control either DTR or DTA expression, led to the selective depletion of skin and peritoneal mast cells, i.e., of connective tissue mast cells (CTMC; Dudeck et al., 2011), whereas using the Cpa3 promoter to control Cre expression led to the depletion of both CTMC and mucosal mast cells (MMC; Feyerabend et al., 2011; Lilla et al., 2011). The latter system also led to the depletion of a significant proportion of basophils. When under the control of the chymase promoter, Cre was selectively expressed in MMC (Musch et al., 2008). These mice should enable one to deplete MMC only when crossed with mice expressing DTR or DTA. Using the Mcpt8 promoter to control the expression of DTR (Wada et al., 2010), DTA or Cre (Ohnmacht et al., 2010) led to the selective depletion of basophils (Table 1).
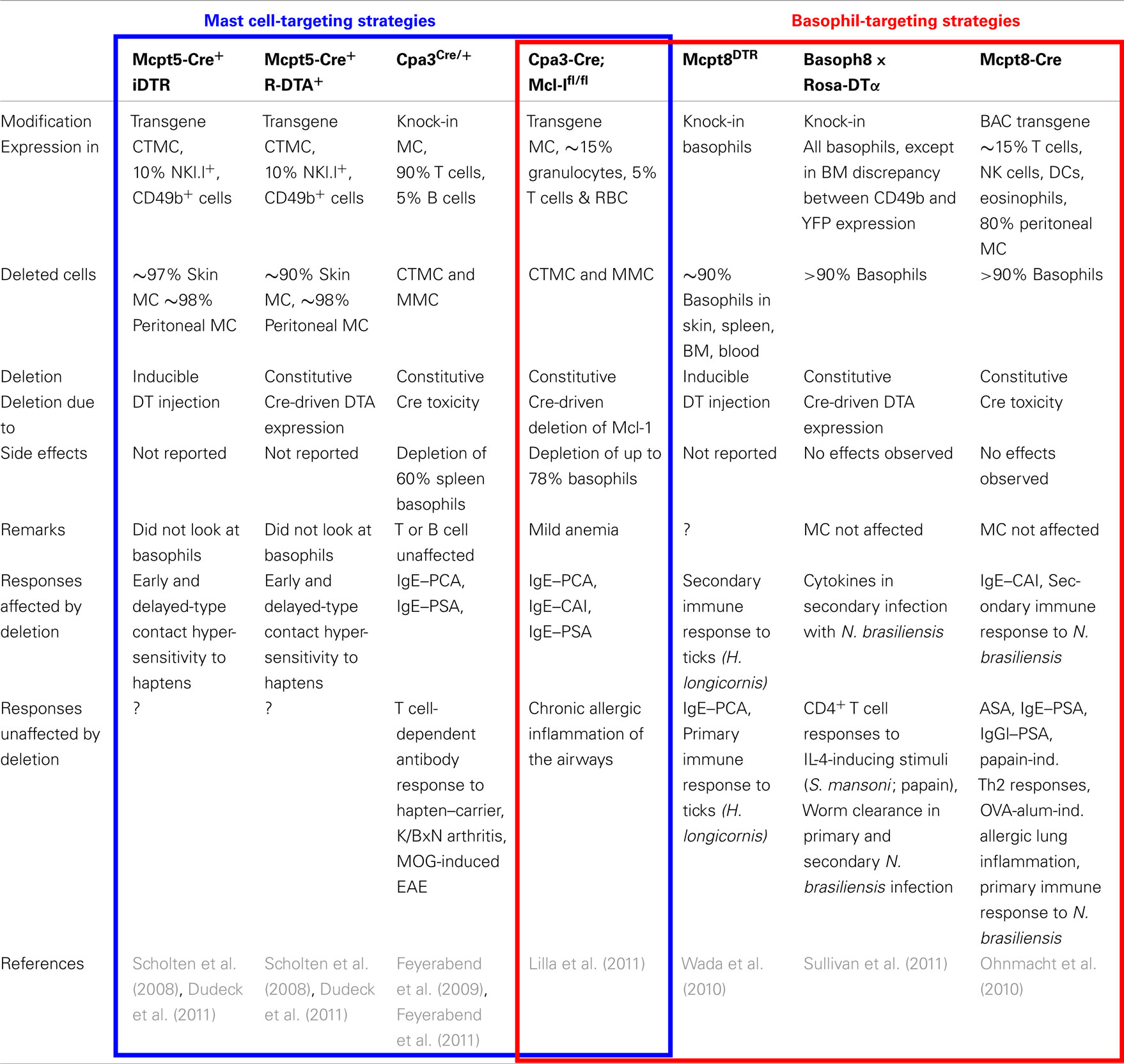
Table 1. Comparison of genetically modified mast cell- and/or basophil-deficient mice.
Due to the above-discussed limitations of the approaches previously used to study mast cell function in vivo, some conclusions on the role of mast cells in allergic and autoimmune inflammation have been challenged by other approaches, and they need to be reconsidered.
Mast Cells in Autoimmune Inflammation
Indirect evidence suggests that FcγR-dependent mast cell activation is critical in IgG-induced autoimmune diseases, both in mouse models and in patients. Numerous mast cells are found in multiple sclerosis plaques, mast cell-specific tryptase is elevated in the cerebrospinal fluid (Rozniecki et al., 1995), and FcεRI and tryptase transcripts, are upregulated in lesions (Pedotti et al., 2003). Likewise, mast cells were found to accumulate in brain lesions in experimental allergic encephalomyelitis (EAE), a model of multiple sclerosis induced by immunizing mice with myelin or myelin-derived peptides. Clinical symptoms were markedly decreased, as well as T cell infiltrates and IFN-γ production in W/W-v mice, and they could be restored by BMMC from wt mice, but not from FcRγ- or FcγRIIIA-deficient mice (Tanzola et al., 2003).
Mast cells have also been implicated in rheumatoid arthritis, Sjögren syndrome, systemic sclerosis, and thyroiditis in humans (Hebbar et al., 1995; Tetlow and Woolley, 1995; Akimoto et al., 1998; Skopouli et al., 1998; Toda et al., 2000; Hueber et al., 2010). K/BxN serum-induced arthritis, a mouse model of passive rheumatoid arthritis (Monach et al., 2007), was abrogated in FcRγ-deficient mice (Kyburz et al., 2000). It was also abrogated in W/W-v or in Sl/Sl-d mice, and it was restored by transferring BMMC from wt mice (Lee et al., 2002). Zhou et al. (2007) however reported that antibody-mediated arthritis was not impaired in Wsh mice. We could confirm this result in K/BxN arthritis using Wsh, but also in mice that lack activating FcR on mast cells (Mancardi et al., 2011). Finally, Tsuboi et al. (2011) reported that a transfer of neutrophils was sufficient to restore K/BxN arthritis in W/W-v mice.
Very recently, both K/BxN arthritis and EAE were found to develop normally in Cpa3Cre/+ mice that lack both connective-type and MMC (Feyerabend et al., 2011). This result strongly challenges the previously drawn conclusion that mast cells are mandatory for IgG-induced experimental autoimmune diseases. It does not exclude that mast cells are involved, but their role is unknown and their presence in lesions may be a consequence rather than a cause. Together with previous results obtained in Kit-mutant mice, these recent results also indicate that a Kit deficiency is more critical in experimental arthritis and encephalomyelitis than a mast cell, and probably, a basophil deficiency.
Mast Cells and Basophils in Allergy
The role of IgE-induced FcεRI-dependent mast cell activation in experimental anaphylaxis (Metcalfe et al., 2009; Kalesnikoff and Galli, 2010) and in allergy (Galli and Tsai, 2010) is well known and documented. It was recently confirmed in new mast cell-deficient mice, as IgE-induced passive systemic anaphylaxis (PSA) was abrogated in Cpa3Cre/+ mice and could be restored following reconstitution with wt BMMC (Feyerabend et al., 2011). The role of IgG-induced FcγR-dependent mast cell activation in not as clear in every experimental model.
Passive cutaneous anaphylaxis is one model that most probably depends on mast cells only, both when induced with IgE or with IgG antibodies. IgE-induced PCA (IgE–PCA) was abrogated in FcεRI-deficient mice (Dombrowicz et al., 1993), in W/W-v mice (Wershil et al., 1987; Arimura et al., 1990), in Wsh mice (Zhou et al., 2007), and in histidine decarboxylase-deficient mice that have reduced numbers of mast cells which contain no histamine (Ohtsu et al., 2002). It could be restored by a short-term reconstitution of mast cell-deficient mice with mast cells injected intradermally (Wershil et al., 1987). Recently, IgE–PCA was found to be abrogated in Cpa3Cre/+ mice (Feyerabend et al., 2011). IgG-induced PCA was abrogated in FcγRIIIA-deficient (Hazenbos et al., 1996). We found recently that mouse IgG1-induced or heat-aggregated human IgG-induced PCA was abrogated in multiple FcR-deficient mice the mast cells of which expressed no FcγR. PCA was however observed if these mice were crossed with FcγRIIA-transgenic mice the mast cells of which expressed this human receptor, but not if Wsh mice were crossed with the same FcγRIIA-transgenic mice (Jönsson et al., 2011b).
As expected, mouse IgG1-induced PSA was shown to depend on FcγRIIIA in mice (Miyajima et al., 1997). Unexpectedly, it was found not to be abrogated in mast cell-deficient mice and, therefore, not to depend on mast cells (Miyajima et al., 1997). The depletion of basophils by Ba103, however, abolished PSA as assessed by body temperature drop, leading to the conclusion that basophils, but not mast cells, account for IgG1-induced PSA (Tsujimura et al., 2008). This conclusion, however, was soon challenged by the finding that IgG1-induced PSA still occurred in basophil-deficient Mcpt8-Cre mice (Ohnmacht et al., 2010). Using FcR-deficient mice that expressed receptors for IgG2 but no receptor for IgG1, we could show that PSA can be induced by IgG2 antibodies, that IgG2-induced PSA can occur in the absence of activating FcR on mast cells and basophils, and that neutrophils largely contribute to this reaction (Jönsson et al., 2011a).
The latter observation demonstrates that anaphylaxis may not depend only on mast cells or basophils. If, indeed, these cells stand among the myeloid cells involved in inflammation, whether physiological or pathological, and in the latter case, whether allergic or autoimmune, they are not alone. One needs to assess the contribution of cells other than mast cells and basophils in both types of diseases. Using FcR-deficient mice whose mast cells and basophils express no activating FcR, we found an unexpected role of neutrophils in systemic anaphylaxis (Jönsson et al., 2011a).
Neutrophils in Anaphylaxis
Passive anaphylaxis is a highly sensitive and convenient mean to study anaphylactic antibodies. Naive animals injected with specific antibodies, however, differ markedly from actively immunized animals. We therefore investigated molecular and cellular mechanisms that account for ASA using FcR-deficient mice.
ASA Requires Neither FcεRI nor FcγRIIIA
We found first that, when immunized with antigen in Freund’s adjuvant, mice produced not only high amounts of specific IgG1 and (much) lower amounts of IgG2 but also some IgE, whereas mice immunized with antigen in alum produced high amounts of IgG1 and some IgE, but no detectable IgG2. Surprisingly, mice lacking either IgE receptors (both FcεRI and FcεRII) or FcγRIIIA underwent similar shocks as wt mice when immunized with antigen in Freund’s adjuvant and challenged with antigen intravenously. Even more surprisingly, when immunized and challenged under the same conditions, quintuple knock-out (5KO) mice lacking FcεRI, FcεRII, FcγRI, FcγRIIB, and FcγRIIIA, underwent also anaphylactic shocks of similar intensity and leading to similar mortality as wt mice.
ASA may be Induced by IgG2 Antibodies via FcγRIV
The only known FcR expressed by 5KO mice are FcRn and FcγRIV. Evidence that FcγRIV were responsible for the shocks was obtained using FcγRIV-specific blocking antibodies that abrogated both clinical symptoms and mortality. FcγRIV are high-affinity receptors that bind IgG2, but not IgG1 (Nimmerjahn et al., 2005; Mancardi et al., 2008). This suggested that IgG2, but not IgG1, antibodies were responsible for ASA in 5KO mice immunized with antigen in Freund’s adjuvant. Indeed, 5KO mice underwent no ASA if immunized with antigen in alum.
Neutrophils are Necessary and Sufficient for FcγRIV-Dependent ASA
FcγRIV are expressed by monocyte/macrophages and neutrophils, but not by mast cells, basophils, or eosinophils. We therefore investigated the possible contribution of these two cell types. Monocyte/macrophage depletion by clodronate-containing liposomes or inactivation by gadolinium had no effect on ASA in 5KO mice immunized in Freund’s adjuvant. However, neutrophil depletion with anti-Gr1 antibodies abrogated ASA in these mice. Noticeably, neutrophil depletion was transient, and ASA was restored spontaneously as neutrophil numbers returned to normal. Supporting this correlation, a rapid systemic release of myeloperoxidase could be observed in 5KO mice injected with luminol intraperitoneally before being challenged for ASA. Finally, demonstrating the role of neutrophils, ASA, which was abrogated in FcRγ-deficient mice, was restored by an intravenous injection of purified neutrophils from 5KO mice, but not from FcRγ-deficient mice.
Neutrophils Play a Dominant Role in ASA in Wt Mice
If neutrophils account for ASA in 5KO mice, they may not be critical in wt mice. We therefore examined the effect of depleting neutrophils, basophils, or both on ASA in wt mice. Basophil depletion had a mild effect on body temperature drop and on mortality observed following antigen challenge. Neutrophil depletion had a more marked effect on temperature drop and mortality. The depletion of both neutrophils and basophils had a more marked effect on temperature drop and the same effect on mortality. Both cell types therefore contribute to ASA in wt mice, but neutrophils play a dominant role.
Neutrophils Can Induce IgG–PSA
If as discussed above, mast cells are the effector cells of IgE-induced PSA, (Metcalfe et al., 2009; Kalesnikoff and Galli, 2010), cells responsible for IgG1–PSA have not been formally identified. We found that neutrophils can play a major role in IgG–PSA. Immune complexes made of GPI and polyclonal IgG anti-GPI antibodies or of DNP-HSA and monoclonal IgG2b anti-DNP antibodies could indeed induce anaphylaxis when injected i.v. into 5KO mice. This FcγRIV-dependent IgG2–PSA was abrogated following neutrophil but not monocyte/macrophage depletion. Neutrophils are therefore not only capable of inducing ASA, they can also induce IgG2–PSA. Whether they can contribute to IgG1–PSA was not investigated. It is a likely possibility since FcγRIIIA, whose deletion sufficed to abrogate this reaction (our unpublished data) and which bind efficiently IgG1 immune complexes (Figure 3), are expressed in mouse neutrophils (Figure 4).
Human Neutrophils Can Induce ASA
Noticeably, ASA could be restored by injecting actively immunized FcRγ-deficient mice not only with mouse neutrophils, but also with human neutrophils from normal donors shortly before antigen challenge. No shock was induced in non-immunized mice, excluding that human neutrophils were non-specifically activated in mouse blood, and the magnitude of temperature drop was proportional to the amount of human neutrophils injected in immunized mice. Supporting these in vivo findings, human neutrophils could be activated in vitro by preformed immune complexes made with mouse IgG1 or IgG2, as assessed by the down regulation of CD62L expression.
FcγRIIA Can Trigger Anaphylaxis
Human neutrophils do not express FcγRIV that do not exist in humans. They however express two IgG receptors: the ITAM-containing single-chain FcγRIIA and the GPI-anchored FcγRIIIB that do not exist in mice. Because FcγRIIA, but not FcγRIIIB, have cell-activating capabilities and because FcγRIIA, but not FcγRIIIB, bind immune complexes made with mouse IgG antibodies, we investigated the role of FcγRIIA in ASA.
To this end, we crossed human FcγRIIA-transgenic (FcγRIIATg) mice with FcRγ-deficient mice. Importantly, because it was placed under the control of its own promoter, FcγRIIA had the same tissue distribution in transgenic mice as in humans. When actively immunized and challenged with antigen, FcRγ−/− FcγRIIATg mice underwent ASA of comparable magnitude as wt mice (Figure 5), and both temperature drop and mortality were abrogated by treating mice with the blocking anti-FcγRIIA antibody IV.3. ASA can therefore be triggered by FcγRIIA. Cells responsible for FcγRIIA-dependent ASA were not identified. Similar results were observed in FcRγ−/− FcγRIIATg mice immunized with antigen in Freund’s adjuvant, which induces both IgG1 and IgG2 antibodies, or with antigen in Alum, which induces IgG1 antibodies only. FcγRIIA has indeed an affinity for both mouse IgG1 and IgG2 (our unpublished data).
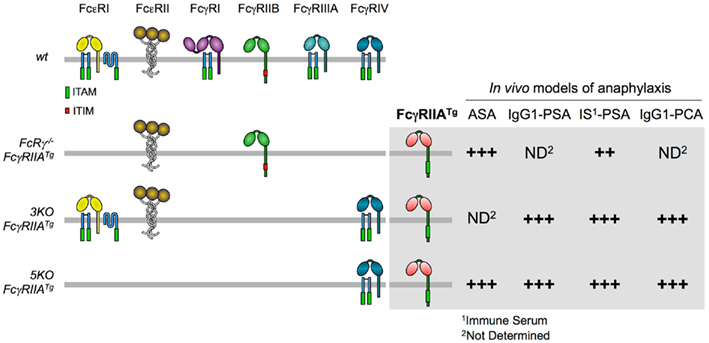
Figure 5. Anaphylactic properties of FcγRIIATg in FcR-KO mice. The figure shows the expression of FcR in FcgRIIATg mice used in our studies (left) and the magnitude of their responses in four models of anaphylaxis (right).
To further investigate the role of FcγRIIA in anaphylaxis, we crossed FcγRIIATg mice with 5KO or 3KO (lacking FcγRI, FcγRIIB, and FcγRIIIA) mice. One advantage of the latter two mutant mice is that they express FcγRIV whereas FcRγ−/− FcγRIIATg mice do not. This activating receptor can be used for depletion experiments with IgG2 antibodies, but it will not be engaged if IgG1 antibodies are used for anaphylaxis. Conversely, FcRγ−/− FcγRIIATg mice express FcγRIIB whereas 3KO-FcγRIIATg and 5KO-FcγRIIATg mice do not. IgG-induced reactions are therefore not submitted to FcγRIIB-dependent inhibition in these mice (Figure 5).
IgG–PSA could be induced in both 3KO-FcγRIIATg and 5KO-FcγRIIATg mice and, as expected, shocks observed in these mice were more severe than shocks observed in FcRγ−/− FcγRIIATg mice. Likewise, IgG–PCA could be induced in 5KO-FcγRIIATg mice injected with heat-aggregated polyclonal human IgG or with ova-mouse IgG1 anti-ova immune complexes i.d. (Figure 5). In accordance with these findings, PCMC from 3KO-FcγRIIATg mice degranulated, released granular mediators and secreted lipid mediators upon challenge with ova-IgG1 anti-ova or with GPI–IgG anti-GPI immune complexes. To confirm that, as suggested by these results, mast cells account for FcγRIIA-dependent PCA, 3KO, and 3KO-FcγRIIATg mice were crossed with Wsh mice. PCA was observed neither in 3KO-Wsh nor in 3KO-FcγRIIATg Wsh mice whereas it occurred in 3KO-FcγRIIATg mice.
Other Myeloid Cells in Allergy
Macrophage/Monocytes in PSA
Contrasting with these results that altogether indicate that mast cells are the cell type that accounts for FcγRIIA-dependent PCA, FcγRIIA-dependent PSA developed normally in 3KO-FcγRIIATg Wsh mice. Basophil depletion with Ba103 did not affect either FcγRIIA-dependent PSA in 3KO-FcγRIIATg mice. Both mast cells and basophils are therefore dispensable for FcγRIIA-dependent PSA. GPI–IgG anti-GPI immune complex-induced PSA was however reduced following neutrophil depletion with anti-Gr1 or anti-Ly6G antibodies in the same mice. It was also reduced following monocyte/macrophage depletion with clodronate-containing liposomes or inactivation with gadolinium, and abrogated following depletion of both neutrophils and monocyte/macrophages. Neutrophils, but also monocyte/macrophages therefore contribute to FcγRIIA-dependent PSA. Both cell types express high levels of FcγRIIA in humans (Figure 4).
A contribution of macrophages in a model of active anaphylaxis was previously reported. In this model, mice immunized with goat anti-mouse IgD antibodies, which induced robust anti-goat IgG responses, as well as mastocytosis, were challenged with goat IgG used as an antigen. Anaphylaxis induced under these conditions was of similar magnitude in wt mice, in IgE- or FcεRI-deficient mice, and in W/W-v mice. It was reduced in mice treated with gadolinium, but not or in mice injected with neuraminidase, which depletes platelets, or in mice injected with a rat IgG2b anti-granulocyte mAb. The conclusion was that macrophages, but not mast cells, platelets, or granulocytes accounted for this anaphylactic reaction (Strait et al., 2002).
Eosinophils in Asthma
Eosinophils reside in mucosal tissues. They are well characterized as effector cells in helminth infections (Klion and Nutman, 2004), during which they release a panoply of different cytokines, such as TGF-β, lipid mediators such as platelet-activating factor (PAF), leukotriene C4 (LTC4) and other effector molecules such as major basic protein (MBP), and eosinophil peroxidase (EPO; Kita, 2011). These functions make the eosinophil a powerful weapon against parasites, however their activation is often associated with detrimental effects on the host environment. Two allergic conditions have been associated with eosinophil activation: allergic bronchial asthma and eosinophilic esophagitis (EOE).
Allergic bronchial asthma is characterized by airway hyperresponsiveness (AHR), bronchoobstruction, and increased mucus production, resulting in a chronic inflammation and, ultimately, in airway remodeling. Since Ehrlich (1879) described eosinophils in tissue and sputum of patients with asthma, these cells have been identified as the prime target of therapeutic intervention in bronchial asthma. Indeed, eosinophils have been implicated in all aspects of asthma pathogenesis. Whereas MBP and EPO can cause tissue destruction and AHR, PAF and LTC4 are potent bronchoconstrictors and increase mucus production, and TGF-β is thought to contribute to airway remodeling. Various strategies that interfere with eosinophil recruitment, activation, and survival have been proven successful in animal models of experimental asthma. These strategies included inhibition of eosinophil recruitment and activation by targeting IL-5 (Foster et al., 1996; Yoshida et al., 1996; Lee et al., 1997), CD131 (Nicola et al., 1996; Sun et al., 1999), CD300a (Munitz et al., 2006), CD23 (Dasic et al., 1999; Haczku et al., 2000), CD48 (Munitz et al., 2007), CCR-3 (Pope et al., 2005; Wegmann et al., 2007), or PGD2 signaling (Sugimoto et al., 2003; Uller et al., 2007; Shichijo et al., 2009; Stearns et al., 2009). Other strategies aimed at reducing eosinophil numbers by using mAbs anti-CCR-3 (Justice et al., 2003) or mAbs anti-Siglec F (Song et al., 2009). Although all of these approaches ameliorated the symptoms of experimental asthma in mice, the first clinical trials targeting IL-5 (Leckie et al., 2000; Flood-Page et al., 2003, 2007), CD23 (Rosenwasser and Meng, 2005), CCR-3 (Pease and Horuk, 2009), and/or CD131 (Gauvreau et al., 2008) did not meet the therapeutic expectations. This might have been due to the fact that experimental asthma models in mice often strictly depend on airway hypereosinophilia and, therefore, may not reflect human asthma. Recently however, two studies reported a significant improvement of severe corticosteroid-refractory highly eosinophilic asthma, using an anti-IL-5 mAb (Haldar et al., 2009; Nair et al., 2009).
Eosinophilic esophagitis is an atopic disease linked to ingested and inhaled allergens. It is characterized by an eosinophilic infiltrate in the mucosa of the esophagus that can result in a narrowed esophageal lumen (Liacouras et al., 2011). Patients suffer from dysphagia, heartburn, or chest pain. In most cases EoE is associated with other forms of allergy like, asthma, hay fever, or eczema. Common treatments consist of dietary elimination and topical administration of corticosteroids or fluticasone propionate, which mobilizes the eosinophilic infiltrate, and improve symptoms (Remedios et al., 2011). Anti-IL-5 mAb (Assa’Ad et al., 2011) and leukotriene receptor antagonists (Lucendo et al., 2011) have also been used with variable successes.
Mast Cells and Company
Considering the variety of antibodies, receptors, and cells that were found to be involved in experimental models of anaphylaxis (Table 2), the long accepted IgE/FcεRI/mast cell paradigm, on which the definition of immediate hypersensitivity is based in the Gell and Coomb’s classification, and on which most therapeutic approaches of allergy depend, appears much too reductionist. One must consider not only IgE but also IgG antibodies, not only FcεRI, but also FcγR of the different types, not only mast cells and basophils but also neutrophils, monocytes, macrophages, eosinophils, and possibly other myeloid cells.
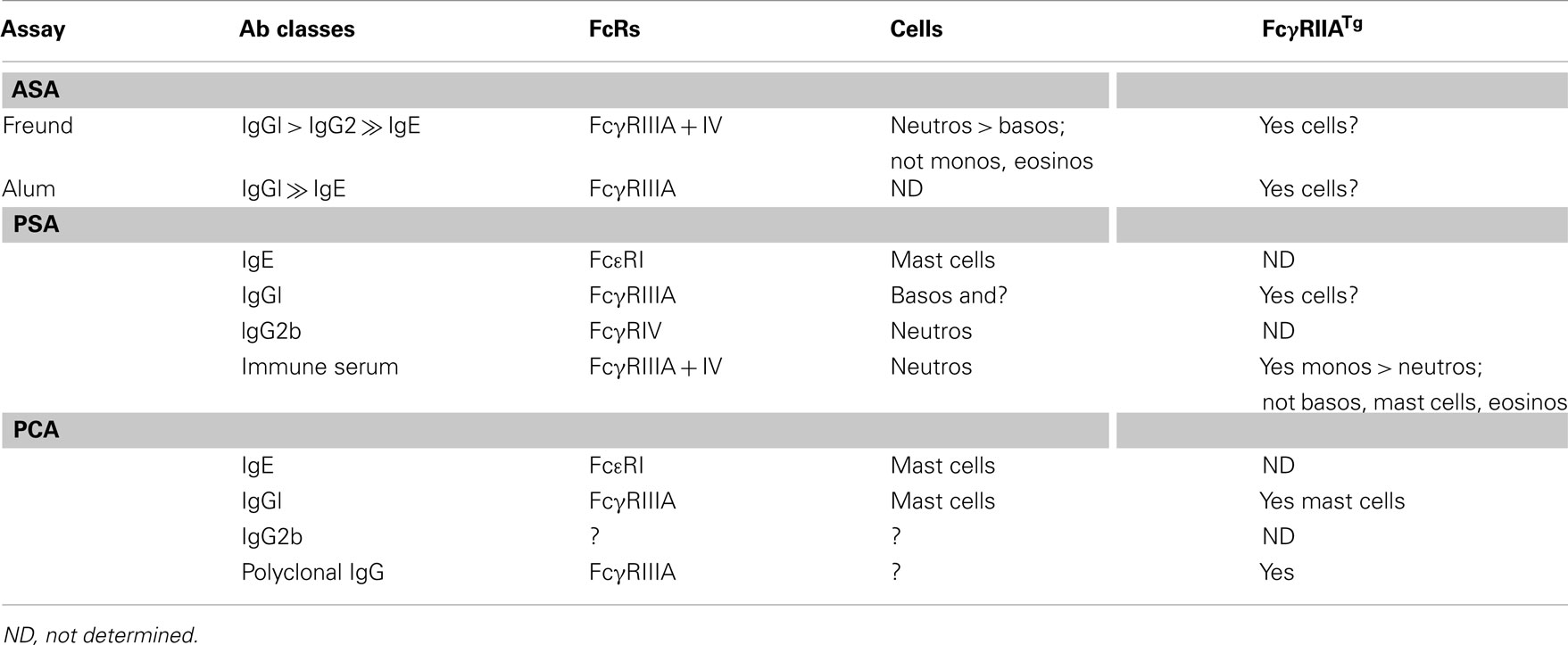
Table 2. Antibodies, receptors, and cells involved in experimental anaphylaxis.
IgE/Mast Cells and IgG/Myeloid Cells in Anaphylaxis
The most surprising conclusion of the works discussed in this review is that the IgE/FcεRI/mast cell paradigm has probably a minor contribution, if any, in ASA. Indeed, ASA developed normally in the absence of IgE, in FcεRI-deficient mice, and in mice lacking mast cells. IgG antibodies (not only IgG1, but also IgG2), FcγR (not only FcγRIIIA whose deletion did not prevent ASA, but also FcγRIV), and granulocytes (not only basophils whose depletion hardly diminished the magnitude of ASA, but essentially neutrophils) appeared as the major players that were involved in and accounted for ASA. One can argue that this conclusion lies on the results of experiments done in murine models that may not faithfully mimic anaphylactic shocks observed in patients. Mice were immunized by repeated injections of relatively high amounts of antigen in Freund’s adjuvant or in alum, which favors IgG responses rather than IgE responses. The role of IgG2 antibodies could be unraveled using mice that expressed IgG2 receptors only, and this antibody subclass is quantitatively minor in mouse immune responses. The blocking of FcγRIV, however, reduced ASA, albeit less than the deletion of FcγRIIIA that bind both IgG1 and IgG2, in wt mice immunized with antigen in Freund’s adjuvant. IgG1 are several-fold more abundant than IgG2 in serum from normal and immunized mice, even following immunization in Freund’s adjuvant. They are exceedingly more abundant than IgE in mice, whatever the immunization protocol used, but also in humans, including in allergic patients.
ASA and PSA
Another unexpected finding is that PSA does not appear as an accurate model of ASA. Passive anaphylaxis (PSA in experimental animals and, later, the Prausnitz–Küstner reaction, its equivalent in humans) was developed soon after Richet (1907) demonstrated that anaphylactic sensitization could be passively transferred by the serum of actively immunized animals. A major difference is that the immune system of animals injected with antibodies has not “seen” antigen before challenge, whereas that of immunized animals has previously mounted a specific adaptive immune response, with all its effectors, and it reacts for the second time to antigen, with these effectors, when challenged with antigen. Another difference is that a whole array of antibodies, with relatively well defined respective concentrations, have been generated in immunized animals, whereas antibodies of a single class are usually injected into recipients, especially since monoclonal antibodies became available. Under these conditions, specific FcR are targeted by antibodies of one class or another and, in most cases, specific cell types, depending on the FcR they express. Thus, passively administered IgE target primarily FcεRI that are exclusively expressed by mast cells and basophils in mice. Why mast cells, but not basophils, account for IgE–PSA is unclear. IgG1 antibodies target FcγRIIIA, which are the only activating FcR that bind this antibody subclass in mice. FcγRIIIA are widely expressed by mouse cells, and not only by mast cells and basophils. Why neither mast cells nor, possibly, basophils seem to account for IgG1–PSA is unknown, as well as the responsible cell type. IgG2 antibodies target FcγRIIIA as well, but also FcγRIV that are expressed by monocyte/macrophages and neutrophils only. Why IgG2–PSA involves neutrophils, but neither monocyte/macrophages, which also express FcγRIIIA and FcγRIV, nor mast cells or basophils, which express FcγRIIIA, may possibly be explained by the crosstalk between activating and inhibitory receptors.
Activating FcR and Inhibitory FcR
IgG antibodies engage both activating and inhibitory receptors, depending on the cell type, which can modulate not only IgG- but also IgE-induced responses. This regulation does not occur when monomeric purified IgE are injected to prepare mice for PSA. An example of FcγRIIB-dependent inhibition is as follows. Monocytes and neutrophils from wt mice express the same FcR: FcγRIIB, FcγRIIIA, FcγRIV, and possibly FcγRI. Mouse monocytes and neutrophils can both induce anaphylaxis. Yet, neutrophils, but not monocytes accounted for anti-GPI serum-induced PSA in wt mice, whereas monocytes, more than neutrophils, accounted for the same reaction in FcγRIIATg mice. The dominant contribution of neutrophils in wt mice can be explained by the lower expression of FcγRIIB on neutrophils than on monocytes (Figure 4). Thus, IgG antibody-induced activation signals of a similar magnitude could be inhibited by FcγRIIB more efficiently in monocytes than in neutrophils. Differing from wt mice, 3KO-, and 5KO-FcγRIIATg mice do not express FcγRIIB. Their neutrophils and their monocytes expressed similar levels of transgenic FcγRIIA the activation signals of which were therefore not inhibited by FcγRIIB and could induce anaphylactic reactions of comparable magnitudes.
The Tree and the Forest
Finally, the findings and conclusions discussed in this review are less provocative than they may seem. Serum-induced PSA is a better model of ASA than monoclonal antibody-induced PSA. The reason is that both assays involve the same polyclonal antibodies. Indeed, anti-GPI serum, which contains a whole array of polyclonal antibodies of the various classes and subclasses, induced PSA that involved both the same receptors (FcγRIIIA and FcγRIV) and the same cells (primarily neutrophils) as ASA in wt type mice (Table 2). Serum-induced PSA, was described initially, and later used to demonstrate that ASA depends on antibodies, before purified antibodies, first and, later, monoclonal antibodies were increasingly used for PSA. If, however, it does not recapitulate ASA, monoclonal antibody-induced PSA remains a powerful analytical tool that targets cells and receptors.
Our findings make sense if one simply keeps in mind that neutrophils are 100-fold more numerous than basophils in blood and that IgG antibodies are 105- to 106-fold more abundant than IgE antibodies in plasma. The IgE/mast cell/basophil tree was somehow hiding the forest of IgG/myeloid cells. This change in perspective may have important consequences.
Concluding Remarks
Indeed, allergy is not another reaction. It is not due to unique effectors, whether molecular or cellular, that are exclusively pathogenic, but to the same cells and molecules that concur to protective immune responses. Allergy is an acute adaptive inflammatory reaction initiated by antibodies. Inflammation is a physiological defense response. It associates destruction and repair mechanisms that are instrumental in wound healing and protection against infection. Inflammation is also potentially harmful, but in normal conditions, it is kept under the control of efficient regulatory mechanisms.
Antibody-induced inflammation depends on a multitude of parameters. (1) It depends first on the presence of antigen. No effect can be induced in the absence of antigen, even if antibodies are present. (2) It depends on the availability of antibodies. Antibodies are present in relatively high concentrations in blood and in hematopoietic organs, but in lower concentrations in peripheral tissues, such as skin for instance. Local concentrations of antibodies (e.g., IgE) can, however, be high. An example is nasal polyps that are associated with allergic rhinitis and that contain IgE-secreting plasma cells (Takhar et al., 2005). (3) It depends on the isotypic composition of immune complexes at a given time and at a given place. This composition is itself shaped by the nature and the amount of immunizing antigen, by adjuvants, by the route and mode of immunization. (4) It depends on the availability of FcR on a given cell. High-affinity FcR may be occupied, at least in part, by antibodies of a different specificity. Some FcR are inducible only, others can be up- and down-regulated by cytokines. (5) It depends on the relative expression of FcRs, especially of activating vs. inhibitory FcR, on myeloid cells that are present at the same site. (6) It depends on the functional repertoire of each of the cell types involved and on the cocktail of pro-inflammatory mediators they can release or secrete. (7) It depends on the availability and on the proportion of cells of the different types that are present initially and that are recruited in the inflammatory infiltrate. (8) It finally depends on the mediators that are released by effector cells upon activation and on the target cells that express receptors for these mediators present in the environment.
Allergies are remarkably polymorphic in their clinical expression. This polymorphism is usually explained by the location where the acute reaction occurs and on the target cells that respond to released and secreted mediators. It may also depend on the nature of effector cells that are activated by antibodies and, therefore, on the antibody classes and subclasses. If this view is correct, one may consider that several nosological entities are possibly hidden behind that of allergy. One may therefore envision that the therapeutic approaches of allergies be diversified, whether symptomatic or etiologic, and adapted to the effector cells and molecules involved. An antihistamine treatment, for instance, may be efficient in a mast cell-dependent reaction, but much less in a neutrophil-dependent reaction.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Works of our laboratory discussed in this review were supported by the Institut Pasteur, the Institut National de la Santé et de la Recherche Médicale (INSERM), the Agence Nationale pour la Recherche (ANR), the Fondation Recherche Médicale (FRM), the Société Française d’Allergologie (SFA), and the company Balsan. Friederike Jönsson was a recipient of a fellowship from FRM; Marc Daëron is a Directeur de Recherche de classe exceptionnelle (DRE) at INSERM and a Chef de Laboratoire at Institut Pasteur.
References
Abraham, S. N., and St John, A. L. (2010). Mast cell-orchestrated immunity to pathogens. Nat. Rev. Immunol. 10, 440–452.
Agis, H., Fureder, W., Bankl, H. C., Kundi, M., Sperr, W. R., Willheim, M., Boltz-Nitulescu, G., Butterfield, J. H., Kishi, K., Lechner, K., and Valent, P. (1996). Comparative immunophenotypic analysis of human mast cells, blood basophils and monocytes. Immunology 87, 535–543.
Akahoshi, M., Song, C. H., Piliponsky, A. M., Metz, M., Guzzetta, A., Abrink, M., Schlenner, S. M., Feyerabend, T. B., Rodewald, H. R., Pejler, G., Tsai, M., and Galli, S. J. (2011). Mast cell chymase reduces the toxicity of Gila monster venom, scorpion venom, and vasoactive intestinal polypeptide in mice. J. Clin. Invest. 121, 4180–4191.
Akimoto, S., Ishikawa, O., Igarashi, Y., Kurosawa, M., and Miyachi, Y. (1998). Dermal mast cells in scleroderma: their skin density, tryptase/chymase phenotypes and degranulation. Br. J. Dermatol. 138, 399–406.
Amaral, M. M., Davio, C., Ceballos, A., Salamone, G., Canones, C., Geffner, J., and Vermeulen, M. (2007). Histamine improves antigen uptake and cross-presentation by dendritic cells. J. Immunol. 179, 3425–3433.
Arimura, A., Nagata, M., Takeuchi, M., Watanabe, A., Nakamura, K., and Harada, M. (1990). Active and passive cutaneous anaphylaxis in WBB6F1 mouse, a mast cell-deficient strain. Immunol. Invest. 19, 227–233.
Assa’Ad, A. H., Gupta, S. K., Collins, M. H., Thomson, M., Heath, A. T., Smith, D. A., Perschy, T. L., Jurgensen, C. H., Ortega, H. G., and Aceves, S. S. (2011). An antibody against IL-5 reduces numbers of esophageal intraepithelial eosinophils in children with eosinophilic esophagitis. Gastroenterology 141, 1593–1604.
Benhamou, M., Bonnerot, C., Fridman, W. H., and Daëron, M. (1990). Molecular heterogeneity of murine mast cell Fc gamma receptors. J. Immunol. 144, 3071–3077.
Bennich, H., Ishizaka, K., Ishizaka, T., and Johansson, S. G. (1969). A comparative antigenic study of gamma E-globulin and myeloma-IgND. J. Immunol. 102, 826–831.
Biedermann, T., Kneilling, M., Mailhammer, R., Maier, K., Sander, C. A., Kollias, G., Kunkel, S. L., Hultner, L., and Rocken, M. (2000). Mast cells control neutrophil recruitment during T cell-mediated delayed-type hypersensitivity reactions through tumor necrosis factor and macrophage inflammatory protein 2. J. Exp. Med. 192, 1441–1452.
Blank, U., Ra, C., Miller, L., White, K., Metzger, H., and Kinet, J. P. (1989). Complete structure and expression in transfected cells of high affinity IgE receptor. Nature 337, 187–189.
Brandt, E. B., Strait, R. T., Hershko, D., Wang, Q., Muntel, E. E., Scribner, T. A., Zimmermann, N., Finkelman, F. D., and Rothenberg, M. E. (2003). Mast cells are required for experimental oral allergen-induced diarrhea. J. Clin. Invest. 112, 1666–1677.
Bruhns, P., Iannascoli, B., England, P., Mancardi, D. A., Fernandez, N., Jorieux, S., and Daëron, M. (2009). Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 113, 3716–3725.
Costa-Pinto, F. A., Basso, A. S., and Russo, M. (2007). Role of mast cell degranulation in the neural correlates of the immediate allergic reaction in a murine model of asthma. Brain Behav. Immun. 21, 783–790.
Daëron, M., Bonnerot, C., Latour, S., and Fridman, W. H. (1992). Murine recombinant Fc gamma RIII, but not Fc gamma RII, trigger serotonin release in rat basophilic leukemia cells. J. Immunol. 149, 1365–1373.
Daëron, M., Latour, S., Malbec, O., Espinosa, E., Pina, P., Pasmans, S., and Fridman, W. H. (1995a). The same tyrosine-based inhibition motif, in the intracytoplasmic domain of Fc gamma RIIB, regulates negatively BCR-, TCR-, and FcR-dependent cell activation. Immunity 3, 635–646.
Daëron, M., Malbec, O., Latour, S., Arock, M., and Fridman, W. H. (1995b). Regulation of high-affinity IgE receptor-mediated mast cell activation by murine low-affinity IgG receptors. J. Clin. Invest. 95, 577–585.
Daëron, M., and Lesourne, R. (2006). Negative signaling in Fc receptor complexes. Adv. Immunol. 89, 39–86.
Daëron, M., and Voisin, G. A. (1979). Mast cell membrane antigens and Fc receptors in anaphylaxis. I. Products of the major histocompatibility complex involved in alloantibody-induced mast cell activation. Immunology 38, 447–458.
Dale, H. H., and Laidlaw, P. P. (1910). The physiological action of beta-iminazolylethylamine. J. Physiol. (Lond.) 41, 318–344.
Dasic, G., Juillard, P., Graber, P., Herren, S., Angell, T., Knowles, R., Bonnefoy, J. Y., Kosco-Vilbois, M. H., and Chvatchko, Y. (1999). Critical role of CD23 in allergen-induced bronchoconstriction in a murine model of allergic asthma. Eur. J. Immunol. 29, 2957–2967.
Denzel, A., Maus, U. A., Rodriguez Gomez, M., Moll, C., Niedermeier, M., Winter, C., Maus, R., Hollingshead, S., Briles, D. E., Kunz-Schughart, L. A., Talke, Y., and Mack, M. (2008). Basophils enhance immunological memory responses. Nat. Immunol. 9, 733–742.
Di Nardo, A., Vitiello, A., and Gallo, R. L. (2003). Cutting edge: mast cell antimicrobial activity is mediated by expression of cathelicidin antimicrobial peptide. J. Immunol. 170, 2274–2278.
Dierks, S. E., Bartlett, W. C., Edmeades, R. L., Gould, H. J., Rao, M., and Conrad, D. H. (1993). The oligomeric nature of the murine Fc epsilon RII/CD23. Implications for function. J. Immunol. 150, 2372–2382.
Dombrowicz, D., Flamand, V., Brigman, K. K., Koller, B. H., and Kinet, J. P. (1993). Abolition of anaphylaxis by targeted disruption of the high affinity immunoglobulin E receptor alpha chain gene. Cell 75, 969–976.
Dudeck, A., Dudeck, J., Scholten, J., Petzold, A., Surianarayanan, S., Kohler, A., Peschke, K., Vohringer, D., Waskow, C., Krieg, T., Muller, W., Waisman, A., Hartmann, K., Gunzer, M., and Roers, A. (2011). Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity 34, 973–984.
Echtenacher, B., Mannel, D. N., and Hultner, L. (1996). Critical protective role of mast cells in a model of acute septic peritonitis. Nature 381, 75–77.
Ehrlich, P. (1879). Beiträge zur Kenntnis der granulierten Bindegewebszellen und der eosinophilen Leukozyten. Arch. Anat. Physiol. 166–169.
Ellis, L. M., and Hicklin, D. J. (2008). VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat. Rev. Cancer 8, 579–591.
Evans, D. P., and Thomson, D. S. (1972). Histamine release from rat mast cells passively sensitised with homocytotropic (IgE) antibody. Int. Arch. Allergy Appl. Immunol. 43, 217–231.
Feyerabend, T. B., Terszowski, G., Tietz, A., Blum, C., Luche, H., Gossler, A., Gale, N. W., Radtke, F., Fehling, H. J., and Rodewald, H. R. (2009). Deletion of Notch1 converts pro-T cells to dendritic cells and promotes thymic B cells by cell-extrinsic and cell-intrinsic mechanisms. Immunity 30, 67–79.
Feyerabend, T. B., Weiser, A., Tietz, A., Stassen, M., Harris, N., Kopf, M., Radermacher, P., Moller, P., Benoist, C., Mathis, D., Fehling, H. J., and Rodewald, H. R. (2011). Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity 35, 832–844.
Flood-Page, P., Swenson, C., Faiferman, I., Matthews, J., Williams, M., Brannick, L., Robinson, D., Wenzel, S., Busse, W., Hansel, T. T., and Barnes, N. C. (2007). A study to evaluate safety and efficacy of mepolizumab in patients with moderate persistent asthma. Am. J. Respir. Crit. Care Med. 176, 1062–1071.
Flood-Page, P. T., Menzies-Gow, A. N., Kay, A. B., and Robinson, D. S. (2003). Eosinophil’s role remains uncertain as anti-interleukin-5 only partially depletes numbers in asthmatic airway. Am. J. Respir. Crit. Care Med. 167, 199–204.
Foster, P. S., Hogan, S. P., Ramsay, A. J., Matthaei, K. I., and Young, I. G. (1996). Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J. Exp. Med. 183, 195–201.
Frandji, P., Tkaczyk, C., Oskeritzian, C., Lapeyre, J., Peronet, R., David, B., Guillet, J. G., and Mecheri, S. (1995). Presentation of soluble antigens by mast cells: upregulation by interleukin-4 and granulocyte/macrophage colony-stimulating factor and downregulation by interferon-gamma. Cell. Immunol. 163, 37–46.
Galli, S. J., and Hammel, I. (1984). Unequivocal delayed hypersensitivity in mast cell-deficient and beige mice. Science 226, 710–713.
Galli, S. J., and Tsai, M. (2010). Mast cells in allergy and infection: versatile effector and regulatory cells in innate and adaptive immunity. Eur. J. Immunol. 40, 1843–1851.
Garman, S. C., Kinet, J. P., and Jardetzky, T. S. (1998). Crystal structure of the human high-affinity IgE receptor. Cell 95, 951–961.
Gaudenzio, N., Espagnolle, N., Mars, L. T., Liblau, R., Valitutti, S., and Espinosa, E. (2009). Cell-cell cooperation at the T helper cell/mast cell immunological synapse. Blood 114, 4979–4988.
Gauvreau, G. M., Boulet, L. P., Cockcroft, D. W., Baatjes, A., Cote, J., Deschesnes, F., Davis, B., Strinich, T., Howie, K., Duong, M., Watson, R. M., Renzi, P. M., and O’Byrne, P. M. (2008). Antisense therapy against CCR3 and the common beta chain attenuates allergen-induced eosinophilic responses. Am. J. Respir. Crit. Care Med. 177, 952–958.
Gell, P. G. H., and Coombs, R. R. A. (1963). The Classification of Allergic Reactions Underlying Disease. Oxford: Blackwell.
Ghannadan, M., Baghestanian, M., Wimazal, F., Eisenmenger, M., Latal, D., Kargul, G., Walchshofer, S., Sillaber, C., Lechner, K., and Valent, P. (1998). Phenotypic characterization of human skin mast cells by combined staining with toluidine blue and CD antibodies. J. Invest. Dermatol. 111, 689–695.
Gounni, A. S., Lamkhioued, B., Delaporte, E., Dubost, A., Kinet, J. P., Capron, A., and Capron, M. (1994). The high-affinity IgE receptor on eosinophils: from allergy to parasites or from parasites to allergy? J. Allergy Clin. Immunol. 94, 1214–1216.
Gounni, A. S., Lamkhioued, B., Koussih, L., Ra, C., Renzi, P. M., and Hamid, Q. (2001). Human neutrophils express the high-affinity receptor for immunoglobulin E (Fc epsilon RI): role in asthma. FASEB J. 15, 940–949.
Grimbaldeston, M. A., Chen, C. C., Piliponsky, A. M., Tsai, M., Tam, S. Y., and Galli, S. J. (2005). Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am. J. Pathol. 167, 835–848.
Grimbaldeston, M. A., Nakae, S., Kalesnikoff, J., Tsai, M., and Galli, S. J. (2007). Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat. Immunol. 8, 1095–1104.
Haczku, A., Takeda, K., Hamelmann, E., Loader, J., Joetham, A., Redai, I., Irvin, C. G., Lee, J. J., Kikutani, H., Conrad, D., and Gelfand, E. W. (2000). CD23 exhibits negative regulatory effects on allergic sensitization and airway hyperresponsiveness. Am. J. Respir. Crit. Care Med. 161, 952–960.
Haldar, P., Brightling, C. E., Hargadon, B., Gupta, S., Monteiro, W., Sousa, A., Marshall, R. P., Bradding, P., Green, R. H., Wardlaw, A. J., and Pavord, I. D. (2009). Mepolizumab and exacerbations of refractory eosinophilic asthma. N. Engl. J. Med. 360, 973–984.
Hammad, H., Plantinga, M., Deswarte, K., Pouliot, P., Willart, M. A., Kool, M., Muskens, F., and Lambrecht, B. N. (2010). Inflammatory dendritic cells – not basophils – are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J. Exp. Med. 207, 2097–2111.
Hazenbos, W. L., Gessner, J. E., Hofhuis, F. M., Kuipers, H., Meyer, D., Heijnen, I. A., Schmidt, R. E., Sandor, M., Capel, P. J., Daëron, M., Van De Winkel, J. G., and Verbeek, J. S. (1996). Impaired IgG-dependent anaphylaxis and Arthus reaction in Fc gamma RIII (CD16) deficient mice. Immunity 5, 181–188.
Hebbar, M., Lassalle, P., Janin, A., Vanhee, D., Bisiau, S., Hatron, P. Y., Tonnel, A. B., and Gosselin, B. (1995). E-selectin expression in salivary endothelial cells and sera from patients with systemic sclerosis. Role of resident mast cell-derived tumor necrosis factor alpha. Arthritis Rheum. 38, 406–412.
Hibbert, R. G., Teriete, P., Grundy, G. J., Beavil, R. L., Reljic, R., Holers, V. M., Hannan, J. P., Sutton, B. J., Gould, H. J., and Mcdonnell, J. M. (2005). The structure of human CD23 and its interactions with IgE and CD21. J. Exp. Med. 202, 751–760.
Huber, M., Helgason, C. D., Damen, J. E., Liu, L., Humphries, R. K., and Krystal, G. (1998). The src homology 2-containing inositol phosphatase (SHIP) is the gatekeeper of mast cell degranulation. Proc. Natl. Acad. Sci. U.S.A. 95, 11330–11335.
Hueber, A. J., Asquith, D. L., Miller, A. M., Reilly, J., Kerr, S., Leipe, J., Melendez, A. J., and Mcinnes, I. B. (2010). Mast cells express IL-17A in rheumatoid arthritis synovium. J. Immunol. 184, 3336–3340.
Hulett, M. D., and Hogarth, P. M. (1994). Molecular basis of Fc receptor function. Adv. Immunol. 57, 1–127.
Ishizaka, K., Ishizaka, T., and Hornbrook, M. M. (1966). Physico-chemical properties of human reaginic antibody. IV. Presence of a unique immunoglobulin as a carrier of reaginic activity. J. Immunol. 97, 75–85.
Johansson, S. G., and Bennich, H. (1967). Immunological studies of an atypical (myeloma) immunoglobulin. Immunology 13, 381–394.
Joller, N., Weber, S. S., Muller, A. J., Sporri, R., Selchow, P., Sander, P., Hilbi, H., and Oxenius, A. (2010). Antibodies protect against intracellular bacteria by Fc receptor-mediated lysosomal targeting. Proc. Natl. Acad. Sci. U.S.A. 107, 20441–20446.
Jönsson, F., Mancardi, D. A., Kita, Y., Karasuyama, H., Iannascoli, B., Van Rooijen, N., Shimizu, T., Daëron, M., and Bruhns, P. (2011a). Mouse and human neutrophils induce anaphylaxis. J. Clin. Invest. 121, 1484–1496.
Jönsson, F., Mancardi, D. A., Zhao, W., Kita, Y., Iannascoli, B., Khun, H., van Rooijen, N., Shimizu, T., Schwartz, L. B., Daëron, M., and Bruhns, P. (2011b). Human FcγRIIA induces anaphylactic and allergic reactions. Blood. PMID: 22138510. [Epub ahead of print].
Joseph, M., Gounni, A. S., Kusnierz, J. P., Vorng, H., Sarfati, M., Kinet, J. P., Tonnel, A. B., Capron, A., and Capron, M. (1997). Expression and functions of the high-affinity IgE receptor on human platelets and megakaryocyte precursors. Eur. J. Immunol. 27, 2212–2218.
Justice, J. P., Borchers, M. T., Crosby, J. R., Hines, E. M., Shen, H. H., Ochkur, S. I., Mcgarry, M. P., Lee, N. A., and Lee, J. J. (2003). Ablation of eosinophils leads to a reduction of allergen-induced pulmonary pathology. Am. J. Physiol. Lung Cell Mol. Physiol. 284, L169–L178.
Kalesnikoff, J., and Galli, S. J. (2010). Anaphylaxis: mechanisms of mast cell activation. Chem. Immunol. Allergy 95, 45–66.
Kano, S., Tyler, E., Salazar-Rodriguez, M., Estephan, R., Mackins, C. J., Veerappan, A., Reid, A. C., Silver, R. B., and Levi, R. (2008). Immediate hypersensitivity elicits renin release from cardiac mast cells. Int. Arch. Allergy Immunol. 146, 71–75.
Kent, U. M., Mao, S. Y., Wofsy, C., Goldstein, B., Ross, S., and Metzger, H. (1994). Dynamics of signal transduction after aggregation of cell-surface receptors: studies on the type I receptor for IgE. Proc. Natl. Acad. Sci. U.S.A. 91, 3087–3091.
Kepley, C. L., Cambier, J. C., Morel, P. A., Lujan, D., Ortega, E., Wilson, B. S., and Oliver, J. M. (2000). Negative regulation of FcepsilonRI signaling by FcgammaRII costimulation in human blood basophils. J. Allergy Clin. Immunol. 106, 337–348.
Kepley, C. L., Taghavi, S., Mackay, G., Zhu, D., Morel, P. A., Zhang, K., Ryan, J. J., Satin, L. S., Zhang, M., Pandolfi, P. P., and Saxon, A. (2004). Co-aggregation of FcgammaRII with FcepsilonRI on human mast cells inhibits antigen-induced secretion and involves SHIP-Grb2-Dok complexes. J. Biol. Chem. 279, 35139–35149.
Kinet, J. P. (1999). The high-affinity IgE receptor (Fc epsilon RI): from physiology to pathology. Annu. Rev. Immunol. 17, 931–972.
Kinet, J. P., Blank, U., Ra, C., White, K., Metzger, H., and Kochan, J. (1988). Isolation and characterization of cDNAs coding for the beta subunit of the high-affinity receptor for immunoglobulin E. Proc. Natl. Acad. Sci. U.S.A. 85, 6483–6487.
Kita, H. (2011). Eosinophils: multifaceted biological properties and roles in health and disease. Immunol. Rev. 242, 161–177.
Klion, A. D., and Nutman, T. B. (2004). The role of eosinophils in host defense against helminth parasites. J. Allergy Clin. Immunol. 113, 30–37.
Kobayashi, R., Okamura, S., Ohno, T., Saito, H., Mori, M., Ra, C., and Okayama, Y. (2007). Hyperexpression of FcgammaRI and Toll-like receptor 4 in the intestinal mast cells of Crohn’s disease patients. Clin. Immunol. 125, 149–158.
Kojima, T., Obata, K., Mukai, K., Sato, S., Takai, T., Minegishi, Y., and Karasuyama, H. (2007). Mast cells and basophils are selectively activated in vitro and in vivo through CD200R3 in an IgE-independent manner. J. Immunol. 179, 7093–7100.
Kulczycki, A. Jr., and Metzger, H. (1974). The interaction of IgE with rat basophilic leukemia cells. II. Quantitative aspects of the binding reaction. J. Exp. Med. 140, 1676–1695.
Kurosaki, T., Gander, I., Wirthmueller, U., and Ravetch, J. V. (1992). The beta subunit of the Fc epsilon RI is associated with the Fc gamma RIII on mast cells. J. Exp. Med. 175, 447–451.
Kyburz, D., Carson, D. A., and Corr, M. (2000). The role of CD40 ligand and tumor necrosis factor alpha signaling in the transgenic K/BxN mouse model of rheumatoid arthritis. Arthritis Rheum. 43, 2571–2577.
Latour, S., Bonnerot, C., Fridman, W. H., and Daëron, M. (1992). Induction of tumor necrosis factor-alpha production by mast cells via Fc gamma R. Role of the Fc gamma RIII gamma subunit. J. Immunol. 149, 2155–2162.
Leckie, M. J., Ten Brinke, A., Khan, J., Diamant, Z., O’Connor, B. J., Walls, C. M., Mathur, A. K., Cowley, H. C., Chung, K. F., Djukanovic, R., Hansel, T. T., Holgate, S. T., Sterk, P. J., and Barnes, P. J. (2000). Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet 356, 2144–2148.
Lee, D. M., Friend, D. S., Gurish, M. F., Benoist, C., Mathis, D., and Brenner, M. B. (2002). Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science 297, 1689–1692.
Lee, J. J., Mcgarry, M. P., Farmer, S. C., Denzler, K. L., Larson, K. A., Carrigan, P. E., Brenneise, I. E., Horton, M. A., Haczku, A., Gelfand, E. W., Leikauf, G. D., and Lee, N. A. (1997). Interleukin-5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathognomonic of asthma. J. Exp. Med. 185, 2143–2156.
Lesourne, R., Fridman, W. H., and Daëron, M. (2005). Dynamic interactions of Fc gamma receptor IIB with filamin-bound SHIP1 amplify filamentous actin-dependent negative regulation of Fc epsilon receptor I signaling. J. Immunol. 174, 1365–1373.
Letourneur, F., Hennecke, S., Demolliere, C., and Cosson, P. (1995). Steric masking of a dilysine endoplasmic reticulum retention motif during assembly of the human high affinity receptor for immunoglobulin E. J. Cell Biol. 129, 971–978.
Levy, D. A., and Frondoza, C. (1983). Immunity to intestinal parasites: role of mast cells and goblet cells. Fed. Proc. 42, 1750–1755.
Li, S., Schmitz, K. R., Jeffrey, P. D., Wiltzius, J. J., Kussie, P., and Ferguson, K. M. (2005). Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 7, 301–311.
Liacouras, C. A., Furuta, G. T., Hirano, I., Atkins, D., Attwood, S. E., Bonis, P. A., Burks, A. W., Chehade, M., Collins, M. H., Dellon, E. S., Dohil, R., Falk, G. W., Gonsalves, N., Gupta, S. K., Katzka, D. A., Lucendo, A. J., Markowitz, J. E., Noel, R. J., Odze, R. D., Putnam, P. E., Richter, J. E., Romero, Y., Ruchelli, E., Sampson, H. A., Schoepfer, A., Shaheen, N. J., Sicherer, S. H., Spechler, S., Spergel, J. M., Straumann, A., Wershil, B. K., Rothenberg, M. E., and Aceves, S. S. (2011). Eosinophilic esophagitis: updated consensus recommendations for children and adults. J. Allergy Clin. Immunol. 128, 3–20.
Lilla, J. N., Chen, C. C., Mukai, K., Benbarak, M. J., Franco, C. B., Kalesnikoff, J., Yu, M., Tsai, M., Piliponsky, A. M., and Galli, S. J. (2011). Reduced mast cell and basophil numbers and function in Cpa3-Cre; Mcl-1fl/fl mice. Blood 118, 6930–6938.
Lobell, R. B., Arm, J. P., Raizman, M. B., Austen, K. F., and Katz, H. R. (1993). Intracellular degradation of Fc gamma RIII in mouse bone marrow culture-derived progenitor mast cells prevents its surface expression and associated function. J. Biol. Chem. 268, 1207–1212.
Lucendo, A. J., De Rezende, L. C., Jimenez-Contreras, S., Yague-Compadre, J. L., Gonzalez-Cervera, J., Mota-Huertas, T., Guagnozzi, D., Angueira, T., Gonzalez-Castillo, S., and Arias, A. (2011). Montelukast was inefficient in maintaining steroid-induced remission in adult eosinophilic esophagitis. Dig. Dis. Sci. 56, 3551–3558.
Maeyama, K., Hohman, R. J., Metzger, H., and Beaven, M. A. (1986). Quantitative relationships between aggregation of IgE receptors, generation of intracellular signals, and histamine secretion in rat basophilic leukemia (2H3) cells. Enhanced responses with heavy water. J. Biol. Chem. 261, 2583–2592.
Malbec, O., Fong, D. C., Turner, M., Tybulewicz, V. L., Cambier, J. C., Fridman, W. H., and Daëron, M. (1998). Fc epsilon receptor I-associated lyn-dependent phosphorylation of Fc gamma receptor IIB during negative regulation of mast cell activation. J. Immunol. 160, 1647–1658.
Malbec, O., Malissen, M., Isnardi, I., Lesourne, R., Mura, A. M., Fridman, W. H., Malissen, B., and Daëron, M. (2004). Linker for activation of T cells integrates positive and negative signaling in mast cells. J. Immunol. 173, 5086–5094.
Malbec, O., Roget, K., Schiffer, C., Iannascoli, B., Dumas, A. R., Arock, M., and Daëron, M. (2007). Peritoneal cell-derived mast cells: an in vitro model of mature serosal-type mouse mast cells. J. Immunol. 178, 6465–6475.
Mallery, D. L., Mcewan, W. A., Bidgood, S. R., Towers, G. J., Johnson, C. M., and James, L. C. (2010). Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl. Acad. Sci. U.S.A. 107, 19985–19990.
Mancardi, D. A., Iannascoli, B., Hoos, S., England, P., Daëron, M., and Bruhns, P. (2008). FcgammaRIV is a mouse IgE receptor that resembles macrophage FcepsilonRI in humans and promotes IgE-induced lung inflammation. J. Clin. Invest. 118, 3738–3750.
Mancardi, D. A., Jönsson, F., Iannascoli, B., Khun, H., Van Rooijen, N., Huerre, M., Daëron, M., and Bruhns, P. (2011). Cutting edge: the murine high-affinity IgG receptor FcgammaRIV is sufficient for autoantibody-induced arthritis. J. Immunol. 186, 1899–1903.
Maurer, M., Wedemeyer, J., Metz, M., Piliponsky, A. M., Weller, K., Chatterjea, D., Clouthier, D. E., Yanagisawa, M. M., Tsai, M., and Galli, S. J. (2004). Mast cells promote homeostasis by limiting endothelin-1-induced toxicity. Nature 432, 512–516.
Mazzoni, A., Siraganian, R. P., Leifer, C. A., and Segal, D. M. (2006). Dendritic cell modulation by mast cells controls the Th1/Th2 balance in responding T cells. J. Immunol. 177, 3577–3581.
Meknache, N., Jönsson, F., Laurent, J., Guinnepain, M. T., and Daëron, M. (2009). Human basophils express the glycosylphosphatidylinositol-anchored low-affinity IgG receptor FcgammaRIIIB (CD16B). J. Immunol. 182, 2542–2550.
Mekori, Y. A., and Galli, S. J. (1985). Undiminished immunologic tolerance to contact sensitivity in mast cell-deficient W/Wv and Sl/Sld mice. J. Immunol. 135, 879–885.
Metcalfe, D. D., Peavy, R. D., and Gilfillan, A. M. (2009). Mechanisms of mast cell signaling in anaphylaxis. J. Allergy Clin. Immunol. 124, 639–646.
Metz, M., Piliponsky, A. M., Chen, C. C., Lammel, V., Abrink, M., Pejler, G., Tsai, M., and Galli, S. J. (2006). Mast cells can enhance resistance to snake and honeybee venoms. Science 313, 526–530.
Metzger, H. (2004). The high affinity receptor for IgE, FcepsilonRI. Novartis Found. Symp. 257, 51–59; discussion 59–64, 98–100, 276–185.
Miyajima, I., Dombrowicz, D., Martin, T. R., Ravetch, J. V., Kinet, J. P., and Galli, S. J. (1997). Systemic anaphylaxis in the mouse can be mediated largely through IgG1 and Fc gammaRIII. Assessment of the cardiopulmonary changes, mast cell degranulation, and death associated with active or IgE- or IgG1-dependent passive anaphylaxis. J. Clin. Invest. 99, 901–914.
Monach, P., Hattori, K., Huang, H., Hyatt, E., Morse, J., Nguyen, L., Ortiz-Lopez, A., Wu, H. J., Mathis, D., and Benoist, C. (2007). The K/BxN mouse model of inflammatory arthritis: theory and practice. Methods Mol. Med. 136, 269–282.
Moqbel, R., Walsh, G. M., Macdonald, A. J., and Kay, B. (1986). Effect of disodium cromoglycate on activation of human eosinophils and neutrophils following reversed (anti-IgE) anaphylaxis. Clin. Allergy 16, 73–83.
Munitz, A., Bachelet, I., and Levi-Schaffer, F. (2006). Reversal of airway inflammation and remodeling in asthma by a bispecific antibody fragment linking CCR3 to CD300a. J. Allergy Clin. Immunol. 118, 1082–1089.
Munitz, A., Bachelet, I., and Levi-Schaffer, F. (2007). CD48 as a novel target in asthma therapy. Recent Pat. Inflamm. Allergy. Drug. Discov. 1, 9–12.
Musch, W., Wege, A. K., Mannel, D. N., and Hehlgans, T. (2008). Generation and characterization of alpha-chymase-Cre transgenic mice. Genesis 46, 163–166.
Nair, P., Pizzichini, M. M., Kjarsgaard, M., Inman, M. D., Efthimiadis, A., Pizzichini, E., Hargreave, F. E., and O’Byrne, P. M. (2009). Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N. Engl. J. Med. 360, 985–993.
Nicola, N. A., Robb, L., Metcalf, D., Cary, D., Drinkwater, C. C., and Begley, C. G. (1996). Functional inactivation in mice of the gene for the interleukin-3 (IL-3)-specific receptor beta-chain: implications for IL-3 function and the mechanism of receptor transmodulation in hematopoietic cells. Blood 87, 2665–2674.
Nigrovic, P. A., Gray, D. H., Jones, T., Hallgren, J., Kuo, F. C., Chaletzky, B., Gurish, M., Mathis, D., Benoist, C., and Lee, D. M. (2008). Genetic inversion in mast cell-deficient (Wsh) mice interrupts corin and manifests as hematopoietic and cardiac aberrancy. Am. J. Pathol. 173, 1693–1701.
Nimmerjahn, F., Bruhns, P., Horiuchi, K., and Ravetch, J. V. (2005). FcgammaRIV: a novel FcR with distinct IgG subclass specificity. Immunity 23, 41–51.
Nimmerjahn, F., and Ravetch, J. V. (2005). Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science 310, 1510–1512.
Obata, K., Mukai, K., Tsujimura, Y., Ishiwata, K., Kawano, Y., Minegishi, Y., Watanabe, N., and Karasuyama, H. (2007). Basophils are essential initiators of a novel type of chronic allergic inflammation. Blood 110, 913–920.
Oettgen, H. C., Martin, T. R., Wynshaw-Boris, A., Deng, C., Drazen, J. M., and Leder, P. (1994). Active anaphylaxis in IgE-deficient mice. Nature 370, 367–370.
Ohnmacht, C., Schwartz, C., Panzer, M., Schiedewitz, I., Naumann, R., and Voehringer, D. (2010). Basophils orchestrate chronic allergic dermatitis and protective immunity against helminths. Immunity 33, 364–374.
Ohtsu, H., Kuramasu, A., Tanaka, S., Terui, T., Hirasawa, N., Hara, M., Makabe-Kobayashi, Y., Yamada, N., Yanai, K., Sakurai, E., Okada, M., Ohuchi, K., Ichikawa, A., Nagy, A., and Watanabe, T. (2002). Plasma extravasation induced by dietary supplemented histamine in histamine-free mice. Eur. J. Immunol. 32, 1698–1708.
Okayama, Y., Hagaman, D. D., and Metcalfe, D. D. (2001). A comparison of mediators released or generated by IFN-gamma-treated human mast cells following aggregation of Fc gamma RI or Fc epsilon RI. J. Immunol. 166, 4705–4712.
Okayama, Y., Kirshenbaum, A. S., and Metcalfe, D. D. (2000). Expression of a functional high-affinity IgG receptor, Fc gamma RI, on human mast cells: up-regulation by IFN-gamma. J. Immunol. 164, 4332–4339.
Ono, M., Bolland, S., Tempst, P., and Ravetch, J. V. (1996). Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature 383, 263–266.
Orloff, D. G., Ra, C. S., Frank, S. J., Klausner, R. D., and Kinet, J. P. (1990). Family of disulphide-linked dimers containing the zeta and eta chains of the T-cell receptor and the gamma chain of Fc receptors. Nature 347, 189–191.
Ovary, Z. (1952). Quantitative studies in passive cutaneous anaphylaxis of the guinea pig. Int. Arch. Allergy Appl. Immunol. 3, 162–174.
Ovary, Z., Caiazza, S. S., and Kojima, S. (1975). PCA reactions with mouse antibodies in mice and rats. Int. Arch. Allergy Appl. Immunol. 48, 16–21.
Ovary, Z., Fudenberg, H., and Kunkel, H. G. (1960). Anaphylactic reactions in the skin of the guinea pig with high and low molecular weight antibodies and gamma globulins. J. Exp. Med. 112, 953–961.
Patalano, F., and Ruggieri, F. (1989). Sodium cromoglycate: a review. Eur. Respir. J. 6(Suppl.), 556s–560s.
Pease, J. E., and Horuk, R. (2009). Chemokine receptor antagonists: part 1. Expert Opin. Ther. Pat. 19, 39–58.
Pedotti, R., Devoss, J. J., Youssef, S., Mitchell, D., Wedemeyer, J., Madanat, R., Garren, H., Fontoura, P., Tsai, M., Galli, S. J., Sobel, R. A., and Steinman, L. (2003). Multiple elements of the allergic arm of the immune response modulate autoimmune demyelination. Proc. Natl. Acad. Sci. U.S.A. 100, 1867–1872.
Perrigoue, J. G., Saenz, S. A., Siracusa, M. C., Allenspach, E. J., Taylor, B. C., Giacomin, P. R., Nair, M. G., Du, Y., Zaph, C., Van Rooijen, N., Comeau, M. R., Pearce, E. J., Laufer, T. M., and Artis, D. (2009). MHC class II-dependent basophil-CD4+ T cell interactions promote T(H)2 cytokine-dependent immunity. Nat. Immunol. 10, 697–705.
Pope, S. M., Zimmermann, N., Stringer, K. F., Karow, M. L., and Rothenberg, M. E. (2005). The eotaxin chemokines and CCR3 are fundamental regulators of allergen-induced pulmonary eosinophilia. J. Immunol. 175, 5341–5350.
Pribluda, V. S., Pribluda, C., and Metzger, H. (1994). Transphosphorylation as the mechanism by which the high-affinity receptor for IgE is phosphorylated upon aggregation. Proc. Natl. Acad. Sci. U.S.A. 91, 11246–11250.
Prouvost-Danon, A., Javierre, M. Q., and Lima, M. S. (1966). Passive anaphylactic reaction in mouse peritoneal mast cells in vitro. Life Sci. 5, 1751–1760.
Remedios, M., Jones, D., and Kerlin, P. (2011). Eosinophilic oesophagitis: epidemiology, pathogenesis and management. Drugs 71, 527–540.
Richet, C. (1907). De l’anaphylaxie en général et de l’anaphylaxie par la mytilocongestive en particulier. Ann. Inst. Pasteur 21, 497–524.
Richet, P., and Portier, P. (1902). De l’action anaphylactique de certains venins. C. R. Soc. Biol. (Paris) 54.
Rozniecki, J. J., Hauser, S. L., Stein, M., Lincoln, R., and Theoharides, T. C. (1995). Elevated mast cell tryptase in cerebrospinal fluid of multiple sclerosis patients. Ann. Neurol. 37, 63–66.
Ruggieri, F., and Patalano, F. (1989). Nedocromil sodium: a review of clinical studies. Eur. Respir. J. Suppl. 6, 568s–571s.
Scholten, J., Hartmann, K., Gerbaulet, A., Krieg, T., Muller, W., Testa, G., and Roers, A. (2008). Mast cell-specific Cre/loxP-mediated recombination in vivo. Transgenic Res. 17, 307–315.
Shan, D., Ledbetter, J. A., and Press, O. W. (2000). Signaling events involved in anti-CD20-induced apoptosis of malignant human B cells. Cancer Immunol. Immunother. 48, 673–683.
Shelburne, C. P., Nakano, H., St John, A. L., Chan, C., Mclachlan, J. B., Gunn, M. D., Staats, H. F., and Abraham, S. N. (2009). Mast cells augment adaptive immunity by orchestrating dendritic cell trafficking through infected tissues. Cell Host Microbe 6, 331–342.
Shichijo, M., Arimura, A., Hirano, Y., Yasui, K., Suzuki, N., Deguchi, M., and Abraham, W. M. (2009). A prostaglandin D2 receptor antagonist modifies experimental asthma in sheep. Clin. Exp. Allergy 39, 1404–1414.
Skopouli, F. N., Li, L., Boumba, D., Stefanaki, S., Hanel, K., Moutsopoulos, H. M., and Krilis, S. A. (1998). Association of mast cells with fibrosis and fatty infiltration in the minor salivary glands of patients with Sjogren’s syndrome. Clin. Exp. Rheumatol. 16, 63–65.
Sokol, C. L., Barton, G. M., Farr, A. G., and Medzhitov, R. (2008). A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat. Immunol. 9, 310–318.
Sokol, C. L., Chu, N. Q., Yu, S., Nish, S. A., Laufer, T. M., and Medzhitov, R. (2009). Basophils function as antigen-presenting cells for an allergen-induced T helper type 2 response. Nat. Immunol. 10, 713–720.
Song, D. J., Cho, J. Y., Lee, S. Y., Miller, M., Rosenthal, P., Soroosh, P., Croft, M., Zhang, M., Varki, A., and Broide, D. H. (2009). Anti-Siglec-F antibody reduces allergen-induced eosinophilic inflammation and airway remodeling. J. Immunol. 183, 5333–5341.
Stearns, B. A., Baccei, C., Bain, G., Broadhead, A., Clark, R. C., Coate, H., Evans, J. F., Fagan, P., Hutchinson, J. H., King, C., Lee, C., Lorrain, D. S., Prasit, P., Prodanovich, P., Santini, A., Scott, J. M., Stock, N. S., and Truong, Y. P. (2009). Novel tricyclic antagonists of the prostaglandin D2 receptor DP2 with efficacy in a murine model of allergic rhinitis. Bioorg. Med. Chem. Lett. 19, 4647–4651.
Strait, R. T., Morris, S. C., Yang, M., Qu, X. W., and Finkelman, F. D. (2002). Pathways of anaphylaxis in the mouse. J. Allergy Clin. Immunol. 109, 658–668.
Sugimoto, H., Shichijo, M., Iino, T., Manabe, Y., Watanabe, A., Shimazaki, M., Gantner, F., and Bacon, K. B. (2003). An orally bioavailable small molecule antagonist of CRTH2, ramatroban (BAY u3405), inhibits prostaglandin D2-induced eosinophil migration in vitro. J. Pharmacol. Exp. Ther. 305, 347–352.
Sullivan, B. M., Liang, H. E., Bando, J. K., Wu, D., Cheng, L. E., Mckerrow, J. K., Allen, C. D., and Locksley, R. M. (2011). Genetic analysis of basophil function in vivo. Nat. Immunol. 12, 527–535.
Sun, Q., Jones, K., Mcclure, B., Cambareri, B., Zacharakis, B., Iversen, P. O., Stomski, F., Woodcock, J. M., Bagley, C. J., D’Andrea, R., and Lopez, A. F. (1999). Simultaneous antagonism of interleukin-5, granulocyte-macrophage colony-stimulating factor, and interleukin-3 stimulation of human eosinophils by targetting the common cytokine binding site of their receptors. Blood 94, 1943–1951.
Supajatura, V., Ushio, H., Nakao, A., Akira, S., Okumura, K., Ra, C., and Ogawa, H. (2002). Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. J. Clin. Invest. 109, 1351–1359.
Supajatura, V., Ushio, H., Nakao, A., Okumura, K., Ra, C., and Ogawa, H. (2001). Protective roles of mast cells against enterobacterial infection are mediated by Toll-like receptor 4. J. Immunol. 167, 2250–2256.
Takai, T., Li, M., Sylvestre, D., Clynes, R., and Ravetch, J. V. (1994). FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell 76, 519–529.
Takhar, P., Smurthwaite, L., Coker, H. A., Fear, D. J., Banfield, G. K., Carr, V. A., Durham, S. R., and Gould, H. J. (2005). Allergen drives class switching to IgE in the nasal mucosa in allergic rhinitis. J. Immunol. 174, 5024–5032.
Takizawa, F., Adamczewski, M., and Kinet, J. P. (1992). Identification of the low affinity receptor for immunoglobulin E on mouse mast cells and macrophages as Fc gamma RII and Fc gamma RIII. J. Exp. Med. 176, 469–475.
Tan, P. S., Gavin, A. L., Barnes, N., Sears, D. W., Vremec, D., Shortman, K., Amigorena, S., Mottram, P. L., and Hogarth, P. M. (2003). Unique monoclonal antibodies define expression of Fc gamma RI on macrophages and mast cell lines and demonstrate heterogeneity among subcutaneous and other dendritic cells. J. Immunol. 170, 2549–2556.
Tanzola, M. B., Robbie-Ryan, M., Gutekunst, C. A., and Brown, M. A. (2003). Mast cells exert effects outside the central nervous system to influence experimental allergic encephalomyelitis disease course. J. Immunol. 171, 4385–4391.
Tetlow, L. C., and Woolley, D. E. (1995). Mast cells, cytokines, and metalloproteinases at the rheumatoid lesion: dual immunolocalisation studies. Ann. Rheum. Dis. 54, 896–903.
Tigelaar, R. E., Vaz, N. M., and Ovary, Z. (1971). Immunoglobulin receptors on mouse mast cells. J. Immunol. 106, 661–672.
Tkaczyk, C., Okayama, Y., Woolhiser, M. R., Hagaman, D. D., Gilfillan, A. M., and Metcalfe, D. D. (2002). Activation of human mast cells through the high affinity IgG receptor. Mol. Immunol. 38, 1289–1293.
Toda, S., Tokuda, Y., Koike, N., Yonemitsu, N., Watanabe, K., Koike, K., Fujitani, N., Hiromatsu, Y., and Sugihara, H. (2000). Growth factor-expressing mast cells accumulate at the thyroid tissue-regenerative site of subacute thyroiditis. Thyroid 10, 381–386.
Tsuboi, N., Ernandez, T., Li, X., Nishi, H., Cullere, X., Mekala, D., Hazen, M., Kohl, J., Lee, D. M., and Mayadas, T. N. (2011). Regulation of human neutrophil Fcgamma receptor IIa by C5a receptor promotes inflammatory arthritis in mice. Arthritis Rheum. 63, 467–478.
Tsujimura, Y., Obata, K., Mukai, K., Shindou, H., Yoshida, M., Nishikado, H., Kawano, Y., Minegishi, Y., Shimizu, T., and Karasuyama, H. (2008). Basophils play a pivotal role in immunoglobulin-G-mediated but not immunoglobulin-E-mediated systemic anaphylaxis. Immunity 28, 581–589.
Uller, L., Mathiesen, J. M., Alenmyr, L., Korsgren, M., Ulven, T., Hogberg, T., Andersson, G., Persson, C. G., and Kostenis, E. (2007). Antagonism of the prostaglandin D2 receptor CRTH2 attenuates asthma pathology in mouse eosinophilic airway inflammation. Respir. Res. 8, 16.
Valitutti, S., and Espinosa, E. (2010). Cognate interactions between mast cells and helper T lymphocytes. Self Nonself 1, 114–122.
van Loveren, H., Meade, R., and Askenase, P. W. (1983). An early component of delayed-type hypersensitivity mediated by T cells and mast cells. J. Exp. Med. 157, 1604–1617.
Vaz, N. M., and Prouvost-Danon, A. (1969). Behaviour of mouse mast cells during anaphylaxis in vitro. Prog. Allergy 13, 111–173.
Vivier, E., and Daëron, M. (1997). Immunoreceptor tyrosine-based inhibition motifs. Immunol. Today 18, 286–291.
Wada, T., Ishiwata, K., Koseki, H., Ishikura, T., Ugajin, T., Ohnuma, N., Obata, K., Ishikawa, R., Yoshikawa, S., Mukai, K., Kawano, Y., Minegishi, Y., Yokozeki, H., Watanabe, N., and Karasuyama, H. (2010). Selective ablation of basophils in mice reveals their nonredundant role in acquired immunity against ticks. J. Clin. Invest. 120, 2867–2875.
Warner, N. L., and Ovary, Z. (1970). A comparison of PCA reactive and hemolytic rabbit antibodies to sheep red blood cells. J. Immunol. 104, 1429–1434.
Wegmann, M., Goggel, R., Sel, S., Erb, K. J., Kalkbrenner, F., Renz, H., and Garn, H. (2007). Effects of a low-molecular-weight CCR-3 antagonist on chronic experimental asthma. Am. J. Respir. Cell Mol. Biol. 36, 61–67.
Wershil, B. K., Mekori, Y. A., Murakami, T., and Galli, S. J. (1987). 125I-fibrin deposition in IgE-dependent immediate hypersensitivity reactions in mouse skin. Demonstration of the role of mast cells using genetically mast cell-deficient mice locally reconstituted with cultured mast cells. J. Immunol. 139, 2605–2614.
Woodbury, R. G., Miller, H. R., Huntley, J. F., Newlands, G. F., Palliser, A. C., and Wakelin, D. (1984). Mucosal mast cells are functionally active during spontaneous expulsion of intestinal nematode infections in rat. Nature 312, 450–452.
Yoshida, T., Ikuta, K., Sugaya, H., Maki, K., Takagi, M., Kanazawa, H., Sunaga, S., Kinashi, T., Yoshimura, K., Miyazaki, J., Takaki, S., and Takatsu, K. (1996). Defective B-1 cell development and impaired immunity against Angiostrongylus cantonensis in IL-5R alpha-deficient mice. Immunity 4, 483–494.
Yoshimoto, T., Yasuda, K., Tanaka, H., Nakahira, M., Imai, Y., Fujimori, Y., and Nakanishi, K. (2009). Basophils contribute to T(H)2-IgE responses in vivo via IL-4 production and presentation of peptide-MHC class II complexes to CD4+ T cells. Nat. Immunol. 10, 706–712.
Zhao, W., Kepley, C. L., Morel, P. A., Okumoto, L. M., Fukuoka, Y., and Schwartz, L. B. (2006). Fc gamma RIIa, not Fc gamma RIIb, is constitutively and functionally expressed on skin-derived human mast cells. J. Immunol. 177, 694–701.
Zhou, J. S., Xing, W., Friend, D. S., Austen, K. F., and Katz, H. R. (2007). Mast cell deficiency in Kit(W-sh) mice does not impair antibody-mediated arthritis. J. Exp. Med. 204, 2797–2802.
Zsebo, K. M., Williams, D. A., Geissler, E. N., Broudy, V. C., Martin, F. H., Atkins, H. L., Hsu, R. Y., Birkett, N. C., Okino, K. H., Murdock, D. C., Jacobsen, F. W., Langley, K. E., Smith, K. A., Takeish, T., Cattanach, B. M., Galli, S. J., and Suggs, S. V. (1990). Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell 63, 213–224.
Keywords: mast cells, basophils, neutrophils, Fc receptors, antibodies, anaphylaxis, allergy
Citation: Jönsson F and Daëron M (2012) Mast cells and company. Front. Immun. 3:16. doi: 10.3389/fimmu.2012.00016
Received: 12 December 2011; Accepted: 01 February 2012;
Published online: 20 February 2012.
Edited by:
Toshiaki Kawakami, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Francesca Levi-Schaffer, The Hebrew University of Jerusalem, IsraelYoshimichi Okayama, Nihon University School of Medicine, Japan
Copyright: © 2012 Jönsson and Daëron. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Marc Daëron, Unité d’Allergologie Moléculaire et Cellulaire, Département d’Immunologie, Institut Pasteur, Bâtiment Metchnikoff, 25 rue du Docteur Roux, 75015 Paris, France. e-mail:ZGFlcm9uQHBhc3RldXIuZnI=