


- 1 Dipartimento di Medicina Interna e Specialità Mediche, Sapienza Università di Roma, Rome, Italy
- 2 Dipartimento di Scienze e Biotecnologie Medico-Chirurgiche, Sapienza Università di Roma, Rome, Italy
In the present article, we discuss the various ambiguous aspects of the immune system that render this complex biological network so highly flexible and able to defend the host from different external invaders. This ambiguity stems mainly from the property of the immune system to be both protective and harmful. Immunity cannot be fully protective without producing a certain degree of damage (immunopathology) to the host. The balance between protection and tissue damage is, therefore, critical for the establishment of immune homeostasis and protection. In this review, we will consider as ambiguous, various immunological tactics including: (a) the opposing functions driving immune responses, immune-regulation, and contra-regulation, as well as (b) the phenomenon of chronic immune activation as a result of a continuous cross-presentation of apoptotic T cells by dendritic cells. All these plans participate principally to maintain a state of chronic low-level inflammation during persisting infections, and ultimately to favor the species survival.
Introduction
To better understand the mechanisms underlying the immune response, immunologists have attempted to establish some general rules of how pathogens are fought and possibly defeated. One of the predominant views has considered the immune response as a network where different types of specialized immune cells interact with each other to ensure host survival. This “cooperative” interpretation has been recently challenged by the observation that the immune system is committed not only to attacking “invaders,” but also to suppressing the ongoing immune response, most likely to limit excessive immunopathology. The concept that the immune response might act to maintain a homeostatic balance between aggression and suppression has, therefore, emerged. It has been also hypothesized that the inhibitory activity of the immune system is the predominant one. For example, the immune system allows bacteria of the intestinal flora to survive and to produce substances (e.g., vitamin B12) that are essential for the well-being of the host (Barthlott et al., 2003; Coombes et al., 2005). However, even this hypothesis does not appear to be completely satisfying. The possible emergence of self-reactive immune responses, of useless or even harmful antibodies, of chronic inflammatory infectious diseases represents part of the “ambiguous” aspects of the immune system. We will, therefore, discuss how this ambiguity has paradoxically evolved to favor host survival, and how the immune system represents a valid biological model to highlight the evolutionistic value of the ambiguity itself.
The etymology of the term ambiguity is derived from Latin ambagere, which means the ability “to push something in different directions” or “to be understood in different ways.” Semantics identifies ambiguity with the polysemy (from the Greek word: polysemos), which is the property of a word to express more than one meaning (Jakobson, 1995). In artwork, ambiguity is a critical element of esthetical information. Aristotle, the Greek philosopher, celebrated that no human work can be defined as “art” in the absence of a certain degree of ambiguity (Hintikka, 1959). In philosophy, ambiguity has been recently considered as “a productive element that is both against and beyond any metaphysical dogma, passing through the crisis of subjectivity” or, alternatively, “the action by which any event is made possible, by either direct creation or construction from pre-existing elements” (Pasqualotto, 1997). Thus, ambiguity can represent a prerequisite of the creativity that has, psychologically speaking, “the ability to produce ideas or elements providing novel and/or alternative solutions to a wide array of problems.” From a biological point of view, all these definitions seem to be quite theoretical or even intangible. However, ambiguity and the resulting creativity could be concretely related to the capacity of a biological system to put into action different mechanisms simultaneously that are only apparently opposite but ultimately result in an evolutionary advantage for the host1. To better understand how ambiguity can be applied to the immune response, a short review of the fundamental aspects of immunity should be conducted.
Ambiguity and B or T Cell Ontogenesis
The adaptive immune response is mediated by the B and T cells, both recognizing antigens through highly specialized and clonally distributed B cell or T cell receptors (BCRs and TCRs). To allow the generation of a huge number of antigen specificity, the immune system has evolved a very effective molecular organization of gene expression for the production of BCRs and TCRs following the encounter with self antigens, which occurs at the level of central lymphoid tissues (the bone marrow for B cells and the thymus for T cells). In particular, the recombination mechanisms (including gene rearrangement, junctional diversity, and N-region addition) enable a limited number of variable (V), diversity (D), and joining (J) minigene elements to produce about 1013–1018 different genes encoding for an equal multiplicity of antigen receptors (Tonegawa, 1983; Yoshikai et al., 1984; Pullen et al., 1989; Oettinger et al., 1990; Shinkai et al., 1992; Wayne et al., 1994; Agrawal et al., 1998; West et al., 2005; Murphy et al., 2007). This enormous BCR or TCR diversity confers the potential immune identity to each individual, because the lymphocyte repertoire – and, hence, the capacity to respond to antigens – is customized for each individual, being different even amongst monozygotic twins. However, such an extraordinarily elegant mechanism, the aim of which is to protect the host from any possible invader, also produces undesired effects. The main side effect of such a broad response is the generation of “unwanted” B or T lymphocytes (“first level of ambiguity”). Indeed, a multitude of developing lymphocytes will result (a) ignorant because they will never meet their specific antigens throughout the life of a single individual; (b) apparently useless such as those able to recognize selective pathogen-associated epitopes, but not critical for the pathogen neutralization; and (c) dangerous, such as those autoreactive. To limit the generation and/or the harmful activity of these cells, the host has evolved different checkpoints to render the immune response mostly effective in fighting dangerous microorganisms without damaging host tissues. With regard to the T cell development, the first checkpoint determines that only a tiny population of thymocytes likely recognizing ubiquitous self antigens that are presented by cortical thymic epithelial cells (cTECs) can survive (positive selection mechanism; Snodgrass et al., 1985; Marrack et al., 1989; Jameson and Bevan, 1998; McCaughtry et al., 2008). This mechanism allows the deletion of a huge number of useless or harmful T cells that otherwise would flood and destroy the “vital space” at the level of central or peripheral lymphoid tissues, or may potentially induce autoimmunity (McCaughtry et al., 2008). As a consequence, only the thymocytes that have been positively selected through the recognition of ubiquitous self-epitopes could progress and reach the thymus medulla, where they will be submitted to a second checkpoint (medullar negative selection). A fundamental role is played by the autoimmune regulator (AIRE) gene, which allows the expression of a wide array of peripheral tissue-specific proteins in medullary (m)TECs (Anderson et al., 2002; Liston et al., 2003; Kuroda et al., 2005; Zhu et al., 2006; Gillard et al., 2007). Thymocytes with high affinity/avidity to tissue-specific self-proteins presented by both mTEC or thymic dendritic cells (DCs; which have phagocytosed mTEC) are deleted, a process which protects the host from the generation of autoreactive T cells with a high potential to induce organ-specific autoimmunity (Killeen et al., 1998). Similarly, self-reactive B lymphocytes are deleted in the bone marrow (Neuberger, 1997; Pillai, 1999; Hardy and Hayakawa, 2001). Nevertheless, a number of autoreactive T or B cells with high affinity antigen receptors have a second chance to survive because of a mechanism known as receptor editing (Tiegs et al., 1993; McGargill et al., 2000). As a consequence, all mature B or T lymphocytes migrating into the periphery on the one hand can recognize self-epitopes with low affinity, but they can be potentially protective by recognition of microbial antigens with high affinity, on the other hand. This process is extremely advantageous for the economy of the immune system because it allows: (a) dangerous lymphocytes recognizing self-proteins with high affinity/avidity are deleted, thus minimizing the emergence of autoimmunity; (b) a restrict lymphocyte repertoire as compared with what is potentially generated upon somatic recombination harbors the peripheral lymphoid tissues to defend the host from possible harmful aliens; (c) this repertoire of mature T or B lymphocytes is however enough to recognize an almost limitless number of microbial determinants and to control the continuous emergence of mutations of immunodominant epitopes.
In some T cells, the ambiguity of antigen recognition may be further amplified by a dual TCR expression. Indeed, the inefficiency of allelic exclusion following the rearrangement of TCR alpha chains (Casanova et al., 1991; Borgulya et al., 1992) allows the generation of a significant proportion of mature T cells harboring two functional TCRs with distinct antigen specificities (Padovan et al., 1993). Controversy still exists about the significance of dual TCR expression in the responses to foreign (He et al., 2002; Dash et al., 2010) or self antigens (Elliott and Altmann, 1995), in the development of regulatory T (Treg) cells (Tuovinen et al., 2006) or in microbial triggers of autoimmunity (Ji et al., 2010). Anyway, the availability of a second specificity by a stimulated T cell may be interpreted as a “reserve” of response, which may extend immunity to additional exogenous as well to endogenous antigens.
Therefore, the intrinsic ambiguity of this process [i.e., maturation of protective lymphocytes is dependent on (low affinity) self-antigen recognition] provides the host with a significant advantage, which outweighs the side effects (autoimmunity, chronic inflammatory diseases…) that are generally controlled by the mechanisms of peripheral tolerance.
Evolution may have selected those processes of T cell development aimed at maximizing repertoire width, even at the expenses of the single individual safeguard. A proof of this possibility comes from the hypothesis that vertebrates developed the thymus from gut-associated lymphoid tissue (GALT; Matsunaga and Rahman, 2001). Indeed, due to structural constraints, in GALT only negative but not positive selection of T cells can efficiently take place (Matsunaga and Rahman, 2001). This scenario may suggest interpreting ambiguity in antigen recognition as an advantageous feature that has been fixed by natural selection.
The Ambiguity of the Peripheral Immune Response
Innate Immune Cells and Signals
As described above, the development of the mature B- and T-cell repertoires is dependent on recognition of self antigens in the thymus and bone marrow, respectively: mature B and T cells can then fight invaders as a result of their capacity to cross-react with single pathogen-associated epitopes. This high level of ambiguity of the immune system has led to the hypothesis that the main function of the immune response is to discriminate the dangerous/infectious from the non-dangerous/non-infectious rather than the foreign from the non-foreign (Matzinger, 1994; Gallucci and Matzinger, 2001; Janeway and Medzhitov, 2002; Medzhitov and Janeway, 2002). According to these theories, innate immunity plays a key role through the presence of sensors, such as the toll-like receptors (TLRs) expressed mainly by innate immune cells (e.g., monocytes, neutrophils, DCs…) and B lymphocytes, or intracellular nuclear oligomerization domain (NOD)-like receptors (Matzinger, 2002; Inohara et al., 2005; Akira et al., 2006; Fritz et al., 2006; Meylan et al., 2006; Petrilli et al., 2007). These receptors can identify dangerous/infectious signals because they recognize molecular patterns common to different pathogens [pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS), bacterial DNA, or viral RNA], or dangerous compounds, such as endogenous (e.g., uric acid causing the gout…) or exogenous (e.g., asbestos causing mesothelioma or asbestosis, silica dust causing silicosis…) crystals (Meylan et al., 2006; Otsuki et al., 2007; Petrilli et al., 2007; Pope and Tschopp, 2007). As regards B cells, PAMPs deliver via TLRs critical costimulatory signals required for the plasma cell differentiation and the generation of memory B cells (Lanzavecchia and Sallusto, 2007). Once activated via TLRs, innate immune cells do the following: (a) induce an adequate inflammatory response in order to respond very early to pathogens and to limit the microbial invasion and (b) generate an effective adaptive immune response through cooperation (Zarember and Godowski, 2002; Viglianti et al., 2003; Hoebe et al., 2004; Schulz et al., 2005; Kawai and Akira, 2006; Matzinger, 2007). In addition, the tissues undergoing inflammation critically interact with innate immune cells to influence the type of local adaptive immune responses (Cho, 2008; Coombes and Powrie, 2008). Matzinger and Kamala (2011) have proposed that tissue-derived, rather than pathogen-derived, signals mostly dictate which classes of effector cells and molecules will be induced to achieve the maximal protection with the minimal damage in a given organ. On the one hand, this complex network has evolved to determine a high defense/offense ratio at the level of the different types of inflamed tissues; on the other hand, it can produce tissue damage and develop immunopathology under certain conditions. The critical issue is if and how this ambiguous aspect of the immune system is biologically advantageous and/or necessary.
B Cell Responses
As discussed above, the enormous BCR repertoire, which has evolved to recognize as many different pathogens as possible, has two main – and apparently contradictory – effects. The principal protective effect results in the production of neutralizing antibodies addressed to fight pathogens, and to control the continuous emergence of microbial mutants (Figure 1). In contrast, the “side effect” is the emergence of potentially harmful antibodies (i.e., the autoreactive), or of those apparently useless, such as the wide range of antimicrobial antibodies that are generated in response to a given pathogen, but do not appear to be critical for pathogen clearance (non-neutralizing or non-protective antibodies; Figure 1). In particular, autoreactive antibodies can be elicited through the cross-reactivity between self and non-self antigens (Rose and Mackay, 2000; Benoist and Mathis, 2001), or generated in response to cryptic self-epitopes that have been unveiled from injured cells, during the course of a given infection or an inflammatory process (Salemi et al., 1995; Barnaba, 1996; Di Rosa and Barnaba, 1998; Rice et al., 2005). Moreover, both autoantibodies and pathogen-specific antibodies (particularly, the non-neutralizing, which represent the majority of antibodies generated in response to a given pathogen!) can produce damage via the formation of circulating immune complexes (ICs). The resulting systemic inflammatory processes can provoke damage in various cells (e.g., blood cells) or the formation of vasculitis phenomena, which affect several tissues (e.g., joints, skin, kidney, brain; Oates and Gilkeson, 2002; Rice et al., 2005; Alard et al., 2008; Klareskog et al., 2008). However, these injuries are generally self-limited and rarely cause organ or tissue failure. Indeed, they disappear in relation to the contraction of the protective immune responses that have promptly cleared the pathogen. Even under these conditions, ICs can participate to the quick amplification of the inflammatory cascade (via the activation of the complement system, the innate immune cells and the huge variety of inflammatory molecules) that is mandatory for both the control of the pathogen and the cooperation between the innate immunity and the adaptive T and B cell mediated responses, ultimately leading to recovery (Avrameas, 1991). The antibodies, already intrinsically ambiguous per se (because of the simultaneous generation of both protective and dangerous antibodies that, as previously described, is inevitable during the development of the lymphocyte repertoire), perform their potential functions through equally ambiguous phenomena – protective effects strictly related to prompt inflammatory reactions.
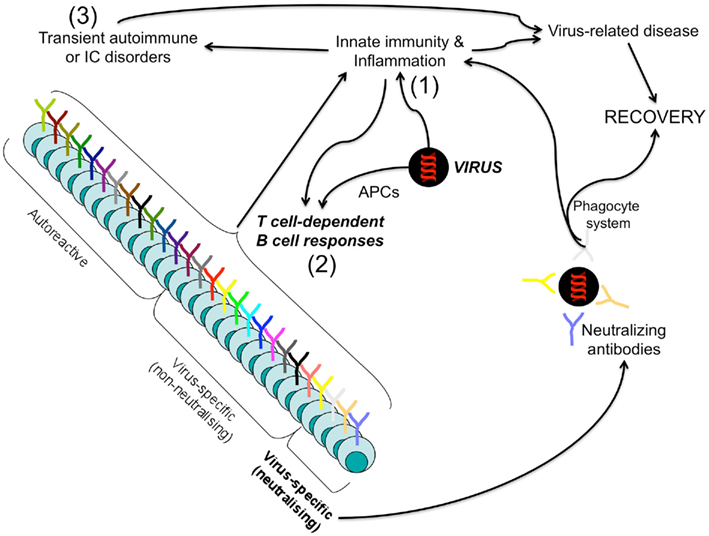
Figure 1. Protective and harmful B cell responses leading to infection resolution. Upon infection by a given pathogen (i.e., a virus), innate immunity is firstly elicited (1) and acts both to promptly control the pathogen spread and to cooperate with the virus-related antigens (presented by professional APCs) for the priming of the adaptive immunity harm (2). Both pathogen-specific antibodies (particularly, the non-neutralizing, which represent the majority of antibodies generated in response to a given pathogen!) and autoantibodies (that can be generated via cross-reactivity or bystander mechanisms) can form immune complexes (ICs) and produce damage through the cooperation with the innate immune system (macrophages, complement factors, inflammatory cytokines…) (3). However, neutralizing, non-neutralizing virus-specific B cells, as well as the autoreactive, act in concert to fight the pathogen: the neutralizing in an antigen-specific manner, the non-neutralizing and autoreactive in a bystander manner via the activation of the phagocytic system by ICs resulting in the amplification of the inflammatory responses (acute disease). This inflammatory storm is self-limited, when the pathogen is cleared.
T Cell Responses
Dendritic cells are essential to prime T cell responses, upon the processing and presentation of exogenous antigens, which are preferentially presented on major histocompatibility complex (MHC) class II molecules, or endogenous antigens, which vice-versa are preferentially presented on MHC class I molecules. However, the capacity of DCs to present exogenous antigens derived from other cells (usually necrotic or apoptotic cells) or soluble antigens on class I molecules is defined as cross-presentation (Guermonprez et al., 2003; Norbury et al., 2004; Accapezzato et al., 2005; Savina et al., 2006; Burgdorf et al., 2007; Dudziak et al., 2007).
Distinct DC subsets spread throughout the body, and although they share common features, they also have specialized functions. Humans and mice display two major DC types: myeloid DCs (myDCs, also called conventional DCs), and plasmacytoid DCs (pDCs; Palucka et al., 2010). In humans, myDCs are subdivided in two populations, on the basis of expression of BDCA-1 (CD1c) or BDCA-3 (CD141). CD1c+ DCs represent the most abundant population of myDCs, express a wider repertoire of TLRs than the CD141+ DCs, and hence they play a key role in sensing infectious/danger signals, in turn essential for their activation, migration, and T cell priming (see below). CD141+ DCs represent the human counterpart of mouse lymphoid CD8+ DCs. Indeed, both these subsets perform the cross-presentation mechanism with high efficiency, express the chemokine receptor XCR1 allowing them to migrate in response to the specific ligand (XCR1L) that is produced by NK and activated CD8+ T cells (Bachem et al., 2010; Crozat et al., 2010), and express the adhesion molecule Necl2 binding to class-I-restricted T-cell-associated molecule (CRTAM), a cell surface protein primarily expressed by NK, NK-T, and activated CD8+ T cells. Thus, mouse CD8+ DCs and human CD141+ DCs appear to be addressed for generation of CD8+ T cell immunity (Shortman and Heath, 2010). BDCA-2+ pDCs express high amounts of IL-3Rα chain (CD123) and ILT-7 and are considered the front line in antiviral immunity owing to their capacity to rapidly produce high amounts of type I interferon in response to viruses (Siegal et al., 1999; Cao et al., 2006). pDCs recognize viral components and self nucleic acids through TLR7 and TLR9, and possibly other as yet unidentified receptors (Matsui et al., 2009), allowing the secretion of two sequential cytokines: type I IFN is responsible for generation of non-Ig-secreting plasma blasts and IL-6 driving their differentiation into Ig-secreting plasma cells (Jego et al., 2003). Recently, it has been proposed that, in addition to their primary role to produce high levels of antiviral IFNs of type 1, pDCs are capable of performing efficient antigen-presenting cell (APC) functions (Guiducci et al., 2006; Di Pucchio et al., 2008). Other subsets, such as DCs in B cell follicular regions or dermal CD14+ DCs in human skin, have specialized function in their selective districts. DCs in B cell follicular regions interact with B cells, inducing humoral immunity to unprocessed soluble antigen presented by these DCs (Wykes et al., 1998). Dermal CD14+ DCs express a huge repertoire of surface C-type lectins and TLRs (van der Aar et al., 2007; Klechevsky et al., 2009), and induce naïve T cells to differentiate into cells with properties of T follicular helper (TFH) cells (Klechevsky et al., 2008), able to induce naïve B cells to produce large amounts of IgM and to induce B cells to switch isotypes toward IgG and IgA.
All tissues are “patrolled” by conventional myDCs that, like bifacial Janus (the most ambiguous Roman divinity), can perform opposing (tolerogenic or stimulatory) functions, according to the context in which they work (Lanzavecchia and Sallusto, 2001; Steinman et al., 2003). Depending on the signal myDCs will undergo activation/maturation, the quality of which will determine the type of elicited adaptive immunity. Under steady state conditions, myDCs phagocytose self antigens associated with dying (apoptotic) tissue cells (derived from the physiological cell turnover), process them, present the resulting peptides on MHC class II or class I molecules and migrate into the draining lymph nodes with very low efficiency, where they can induce tolerance or cross-tolerance of autoreactive CD4+ T cells or CD8+ T cells, respectively (peripheral tolerance; Lanzavecchia and Sallusto, 2001; Steinman et al., 2003). Peripheral tolerance is enforced by the AIRE expression in extrathymic stromally derived cells resident in peripheral lymphoid organs, which hence can present a wide array of tissue-specific antigens and are capable of interacting with and deleting naïve autoreactive T cells (Gardner et al., 2008). DCs that exist at the steady state, can also establish tolerance indirectly by inducing Treg cells (Roncarolo et al., 2001; Yamazaki et al., 2006), the key cell population involved in the regulation of immune responses and homeostasis (Sakaguchi et al., 2010; see The Control of the Immune Response). These DCs may not simply be unstimulated or immature. Activation of the Wnt and β-catenin signaling pathway in DCs has been shown to promote induced Treg cell production, at least in the mouse (Jiang et al., 2007). Similarly, in the thymus, production of thymic stroma lymphopoietin (TSLP) is essential for selection of naturally occurring CD4+CD25hi Treg cells (Watanabe et al., 2005). By contrast, in an inflammatory context (mainly induced by infectious agents or tissue-derived signals), DCs are activated (for instance, through TLR engagement by PAMPs or necrotic cell products), increase for a short time (West et al., 2004) the ability to internalize and to process both microbial and self antigens, as well as they upregulate the expression of stimulatory, costimulatory, and pro-migratory molecules [i.e., the lymph node-specific chemokine receptors (Cys–Cys chemokine receptor 7, CCR7); Figure 2; Lanzavecchia and Sallusto, 2001; Steinman et al., 2003]. Then, they can reach the lymph nodes, where they prime or cross-prime both microbial- or self-antigen-specific naïve CD4+ or CD8+ T lymphocytes (Ridge et al., 1998; Schulz et al., 2005). Therefore, the generation of autoreactive T cell responses is potentially a common event occurring in relation to the protective responses against the “invaders” (ambiguity). As a consequence, both pathogen- and self-reactive effector T lymphocytes migrate to the inflamed tissues because of the newly acquired expression of tissue-specific chemokine receptors (Sprent and Surh, 2002; Masopust and Ahmed, 2004; Sallusto et al., 2004; Lang et al., 2005). According to the microenvironmental context, lymphocytes are polarized toward different types of effector capacities that can provide opposing protective and harmful effects (ambiguity; Figure 2). If DCs are conditioned by infectious or danger signals to produce adequate amounts of IL-12, in the presence of interferon (IFN)-γ, CD4+, or CD8+ T cell priming is skewed toward the polarization of either proinflammatory Th1 cells or CD8+ T cells with high cytotoxic potential, respectively: these cells will simultaneously provide protective responses against intracellular pathogens and harmful responses via their immunopathological activities (Romagnani, 1997). In the presence of IL-4, naïve T cells preferentially differentiate into Th2 (producing IL-4, IL-5, IL-13…) with protective responses against extracellular pathogens or harmful responses in the case of allergic reactions (Nelms et al., 1999; de Jong et al., 2005). Under conditions in which DCs produce IL-23 (another member of the IL-12 family), and in the presence of IL-6, transforming growth factor (TGF)-β, and IL-1β, T cells differentiate toward Th17 cells (producing IL-17), described to be responsible for causing severe immunopathological damages (i.e., in several models of autoimmunity) and for providing protection against some extra or intracellular pathogens (Acosta-Rodriguez et al., 2007; Bettelli et al., 2007; Dong, 2008; Luger et al., 2008; McGeachy and Cua, 2008; Curtis and Way, 2009). Sustained stimulation by DCs is critical for maintaining a large pool of memory T cells. Upon infection resolution, effector cells disappear, whereas memory cells remain numerically constant because of the expression of receptors specific for the homeostatic (IL-7 and IL-15) cytokines (Surh et al., 2006; Sabbagh et al., 2007). The homeostatic proliferation of memory cells, in the absence of antigen, is critical for prompt differentiation into effector cells should they re-encounter the original infecting pathogen.
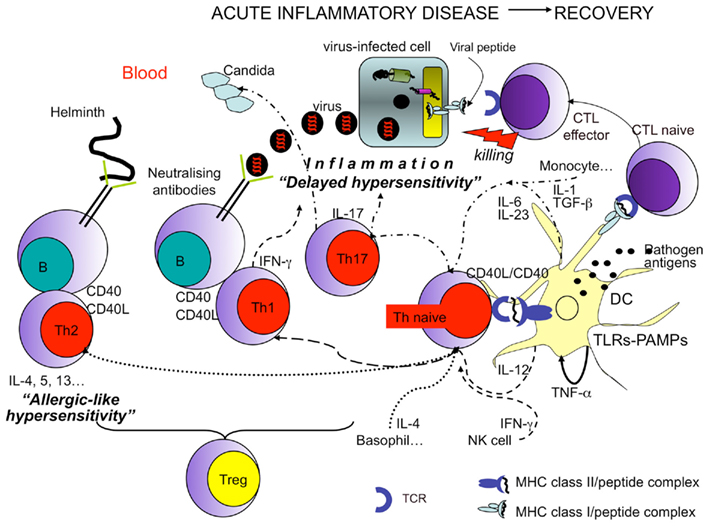
Figure 2. Protective and harmful T cell responses leading to infection resolution. Recovery from infections is dependent on the efficient priming of T cells by professional APCs (DCs) working in concert with other innate immune cells. The efficiency of DCs in migrating from inflamed tissues to lymph nodes, in presenting antigens, and in T cell priming is in turn dependent on the engagement of different types of DC sensors by pathogens (such as TLRs engaged by PAMPs). In addition (CD4+) Th1 cell polarization (dashed arrows) requires the presence of IFN-γ generally donated by NK cells, and IL-12, produced by DCs. (CD4+) Th2 cell polarization (dotted arrows) requires the presence of IL-4 particularly produced by basophils. Whereas (CD4+) Th17 cell polarization (arrows with dashes and dots) requires the presence of several cytokines, such as TGF-β, IL-6, IL-1 (particularly produced by monocytes and endothelial cells), and IL-23 (mainly produced by DCs). In all cases, the functional activity of DCs, T, or B cells is increased by the interaction between CD40, expressed by both DCs and B cells, and CD40 ligand (L), expressed by activated T cells. Then Th1 cells display protective activity against intracellular pathogens (i.e., viruses…), via both IFN-γ production (antiviral activity) and by cooperating with B cells producing IgG1 (in humans) or IgG2 (in mice) neutralizing antibodies. Th2 cells are protective against extracellular pathogens, such as helminthes, via the production of a series of cytokines (e.g., IL-4, IL-5, IL-13) addressed to provoke “allergic-like” hypersensitivity (obtained via the recruitment of various cells, including eosinophils and mast-cells) and production of protective IgE antibodies by B cells. Th17 cells seem to be important in fighting extracellular pathogens, such as fungal infections. (CD8+) Cytotoxic T lymphocytes (CTLs) directly kill infected cells via the recognition of class I molecules expressing pathogen peptides. In any case, activated T cells can eliminate the pathogen via the establishment of a severe inflammatory disease. Treg cells control exaggerated responses or the emergence of autoreactive lymphocytes.
An additional support for the idea that DCs carry out opposing functions according to their maturational stage and the microenvironment in which they work is provided by studies investigating the complex interplay amongst cross-presentation, apoptotic cells, DCs, and stimulatory signals. The current view suggests that, under steady state conditions, apoptotic cells are consistently captured by immature DCs that in turn induce tolerance (or cross-tolerance) of apoptotic cell-derived antigen-specific T cells (Albert, 2004; Kazama et al., 2008). Otherwise, DCs mature and cross-prime these T cells in the presence of sustained infectious/inflammatory mediators, necrotic cell products, or CD4+ Th cells inducing DC maturation via the CD40/CD40L interaction (Inaba et al., 1998; Propato et al., 2001; Albert, 2004; Blachere et al., 2005; Rawson et al., 2007). The rationale behind this model is that apoptosis may be a physiological or a pathological event, depending upon the circumstances involved. Thus, apoptosis occurring physiologically during the development of a given tissue should cause tolerance, whereas apoptosis caused by a microbial infection or other inflammatory processes should result in T cell priming (Propato et al., 2001; Winau et al., 2006). In the steady state conditions, apoptotic cells have been proposed to induce T cell deletion, anergy, immune deviation from Th1 to Th2 responses, or Treg cell induction (Kurts et al., 1998; Ferguson et al., 2002; Griffith et al., 2007), via a series of not completely clear mechanisms, including the capacity of apoptotic cells to release immunosuppressive cytokines (i.e., IL-10 or TGF-β), or to engage receptors (i.e., CD36 or phosphatidyl serine receptors) subverting the stimulatory DC functions (Gao et al., 1998; Chen et al., 2001; Serhan and Savill, 2005). In addition, it has been demonstrated that apoptotic cell-dependent tolerance can be determined by the caspase-induced production of a reactive oxygen species scavenger leading to oxidization and inactivation of the high mobility group protein B1, a powerful danger signal normally involved in the full DC activation and immune response initiation (Kazama et al., 2008). However, the presence of sustained infectious/inflammatory mediators, necrotic cell products or CD4+ T cell help can bypass the tolerogenic effects of apoptotic cells and induce DCs to prime or cross-prime T cells (Inaba et al., 1998; Propato et al., 2001; Albert, 2004; Blachere et al., 2005; Rawson et al., 2007; Tesniere et al., 2008).
Our previous data proposed that an alternative mechanism of abrogation of the apoptosis-associated tolerance can take place via the expression of CD40L by apoptotic cells (Figure 3; Propato et al., 2001). CD40L+ apoptotic cells can be derived from activated CD40L+ T cells that undergo apoptosis once they have performed their effector function in a given inflamed tissue or upon infection with proapoptotic viruses, such as HIV. CD40L+ apoptotic T cells directly induce DC maturation and condition them to induce cross-priming of CD8+ T cells specific to apoptotic cell-associated self antigens, irrespective of additional exogenous signals (Figure 3). In contrast, if apoptotic T cells are CD40L− (such as those derived from resting T cells), the help of a third party activated T cell or surrogate CD40L molecule is needed for priming (Figure 3). The finding that CD40L+ apoptotic T cells induce DC maturation and cross-priming without the addition of exogenous stimuli indicates that the surface phenotype and possibly the lineage of origin of apoptotic cells may ultimately dictate the outcome of cross-presentation. This notion may help reconcile several apparently contradictory findings. For example, it explains why apoptotic cells derived from epithelial or resting T cells that are CD40L− are unable to provide DC maturation stimuli and are tolerogenic in the absence of sustained infectious/inflammatory signals (De Vita et al., 1998; Propato et al., 2001; Albert, 2004; Kazama et al., 2008). Thus, the balance between CD40L+ and CD40L− apoptotic cells during cross-presentation appears to dictate tolerance or induction of CD8 T cell responses against T-cell-associated epitopes, and to maintain or to stop the related responses in the course of an inflammatory process. These responses have a critical role in the amplification of chronic inflammation via the continuous bystander effects of inflammatory cytokines produced by T cells specific for the apoptotic T-cell-associated antigens (Propato et al., 2001; Rawson et al., 2007).
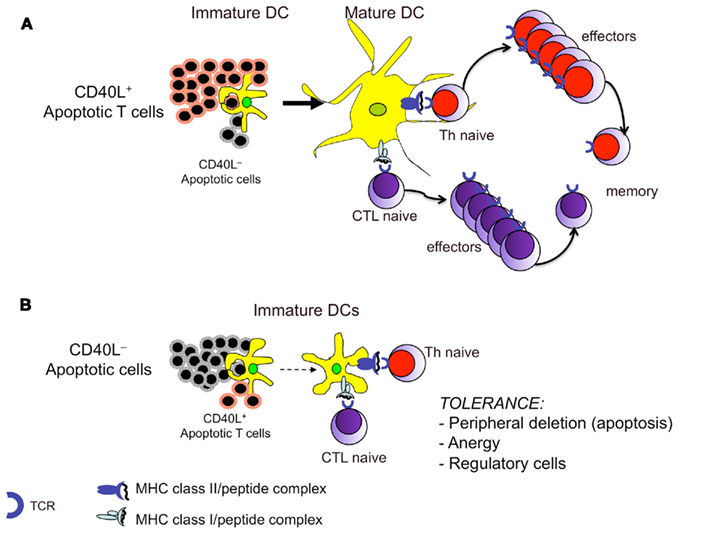
Figure 3. The balance between CD40L+ and CD40L− apoptotic cells during cross-presentation dictates induction or tolerance of T cell responses. (A) Apoptotic T cells expressing CD40L can directly provide to DCs both (apoptotic cell-derived) self antigens and the necessary maturation stimuli for T cell priming. (B) Under conditions in which the CD40L+ apoptotic T cells are overwhelmed by the CD40L− apoptotic cells (i.e., under normal conditions or when an inflammatory process is terminated), the latter will be unable to provide the appropriate signals to DCs, which will deliver tolerance signals to autoreactive T cells, even though they carry apoptotic cell-derived peptides.
For all these reasons, adaptive immune responses clearly show a deeper level of ambiguity: (a) an effective T cell response depends on an adequate level of tissue inflammation, which in turn also induce tissue injury; (b) effector lymphocyte responses are both protective and harmful at the same time.
However, the ambiguity appears to be not only advantageous but also necessary. Indeed, the control of a given infectious agent (protection) is dependent on the immunopathological process at the level of the site of infection (damage), which generally results in an acute disease undergoing recovery when the anti-pathogen responses are prompt and efficient. In this situation, the contraction of both protective effector responses and the associated immunopathological process (e.g., delayed hypersensitivity Th1 or likely Th17 cell responses, including the autoreactive, “allergic-like” hypersensitivity Th2 cell responses) occurs in relation to the clearance of intra or extracellular pathogens (Figure 2). In addition, even the autoreactive and the apparently useless T or B responses that can arise together with the protective responses via the mechanisms described previously are beneficial in supporting the immunopathology required for the recovery: also, these responses generally disappear in relation with the pathogen clearance (Di Rosa and Barnaba, 1998; Murali-Krishna et al., 1998).
Paradoxically, the possibility of recovering from a given infection is dependent on the high level of immunopathology (acute disease; Figure 2). Conversely, weak responses are generally associated with low-grade, long-lasting inflammation that, although causes minimal tissue injury, will be unable to clear the pathogen, which in turn will persist and, together with the persistent weak immune responses, will lead to irreversible chronic diseases.
The Control of the Immune Response
The ambiguity of the immune response can be found at further other levels. One important aspect is the downregulation of the immune response itself. The mechanisms evolved to control the ongoing effector response may be achieved by multiple, non-mutually exclusive mechanisms of peripheral tolerance, including T cell anergy, apoptosis, or exhaustion (Lanzavecchia and Sallusto, 2001; Walker and Abbas, 2002; Steinman et al., 2003; Barron et al., 2008).
The extinction of effector responses often involves the secretion of immunosuppressive cytokines by T helper themselves, such as IL-10 and IL-22. These two molecules, belonging to the same family, operate to rescue immune and tissue homeostasis, respectively (Sanjabi et al., 2009). Indeed, IL-10R is mainly expressed by immune cells, mediating the inhibition of proinflammatory signals; conversely, IL-22R is present predominantly on the surface of epithelial and tissue cells. This cytokine has revealed some functional ambiguity: on the one hand, during acute inflammatory reactions in gut and liver, IL-22 protects the tissue from immune-mediated injury, facilitates regeneration, promotes secretion of antimicrobial substances and maintains barrier integrity against bacterial translocation (Zenewicz et al., 2008; Zheng et al., 2008); on the other hand, IL-22 becomes pathogenic in skin diseases, fostering keratinocyte hyperplasia, and local inflammation (Duhen et al., 2009; Eyerich et al., 2009). Thus, the activities of IL-22 may result beneficial or detrimental to the host depending on tissue type, cytokine milieu, and target cells.
A special emphasis has been recently placed on the PD-1, a death receptor over-expressed by chronically stimulated lymphocytes that induces peripheral T or B cell tolerance upon the simultaneous interaction of TCR or BCR with antigens and of PD-1 with its own ligands: PD-L1, which is virtually expressed on all somatic cells (particularly from inflamed tissues; Keir et al., 2006; Sharpe et al., 2007), and PD-L2, which is mainly expressed by DCs. However, this mechanism becomes detrimental when the PD-1/PD-L1 interaction takes place in tissues infected with persistent pathogens, via its capacity to induce exhaustion of antimicrobial T cell responses (Probst et al., 2005; Barber et al., 2006; Day et al., 2006; Sharpe et al., 2007). Through this mechanism, effector lymphocytes express the death receptor PD-1 and are blocked in their functional abilities when interact with cells expressing PD-L1, the counter-receptor, which is the case with epithelial cells when chronically infected by a virus or during an inflammatory process (Probst et al., 2005; Barber et al., 2006; Day et al., 2006; Keir et al., 2006; Sharpe et al., 2007).
The extrinsic mechanisms of peripheral tolerance are put into action mainly by CD4+CD25+Foxp3+ Treg cells (Shevach, 2002; Bluestone and Abbas, 2003; von Boehmer, 2005). These cells develop either in the thymus (natural) or in the periphery from conventional CD4+ T cells (induced), and express the transcription factor Foxp3 (Shevach, 2002; Bluestone and Abbas, 2003; Fontenot et al., 2003; Hori et al., 2003; Sakaguchi, 2004; von Boehmer, 2005; Ziegler, 2006). A lack of Foxp3 expression results in the complete absence of Treg cells, which leads to the development of severe autoimmunity, as observed in immuno dysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome. Treg cells induce suppression via several mechanisms, involving membrane molecules, such as CTLA-4 or adenosine receptors, suppressive cytokine production, such as TGF-β or IL-10, dominant absorption of IL-2 by high CD25 expression (Shevach et al., 2001; Shevach, 2002; von Boehmer, 2005; Belkaid, 2007; Deaglio et al., 2007; Pandiyan et al., 2007; Vignali et al., 2008). Because of the expression of the Il2 gene-inhibitory Foxp3 transcription factor, Treg cells do not produce IL-2 and are unable to respond to antigens (anergy; Schubert et al., 2001; Coffer and Burgering, 2004; Fontenot et al., 2005). However, they promptly proliferate in response to relevant antigens in the presence of paracrine IL-2, which is mainly produced by effector T lymphocytes but is dominantly absorbed by Treg cells via the high expression of IL-2 receptors (CD25 high; de la Rosa et al., 2004; Barthlott et al., 2005; Kretschmer et al., 2005; Scheffold et al., 2005; Setoguchi et al., 2005). The main physiological functions of Treg cells are as follows: (a) to participate in the establishment of peripheral tolerance by inhibiting autoreactive lymphocytes that escaped either thymus or bone marrow checkpoints (central tolerance), (b) to suppress ongoing protective immune responses once they are no longer necessary or become harmful after the elimination of the pathogen, and (c) to limit excessive immunopathology during chronic inflammatory diseases (Shevach, 2002; Fontenot et al., 2003, 2005; Hori et al., 2003; Sakaguchi, 2004; Kretschmer et al., 2005; Setoguchi et al., 2005; von Boehmer, 2005; Ziegler, 2006; Belkaid, 2007). The immune system, by using highly specialized molecular mechanisms, on the one hand attacks the invading agents, on the other hand it suppresses the same responses. Again, this ambiguous aspect results in an advantage for the host, because it is necessary to defend, but also to limit potentially harmful proinflammatory responses.
In the model of hepatitis C virus (HCV) infection, CD4+CD25+Foxp3+ Treg cells have been proposed to participate in the establishment of a fine equilibrium between immunopathology and immune protection, ultimately resulting in the long-lasting survival of the host during chronic infections (Shevach, 2002; Accapezzato et al., 2004; von Boehmer, 2005; Belkaid, 2007; Ward et al., 2007; Ebinuma et al., 2008; Heeg et al., 2009; Figure 2). This would be dependent on a compromise between a status of chronic low-level hepatic inflammation and the generation of antiviral responses that, although unable to clear HCV, are enough to limit excessive viral spread. It is unclear how Treg cells control unwarranted inflammation without completely suppressing the protective immune responses. High CD25 expression by Treg cells drives a positive feedback loop, as the dominant IL-2 capture increases STAT-5 phosphorylation (pSTAT-5) that in turn drives Treg cell proliferation and function. We recently showed that PD-1 is over-expressed on Foxp3+ Treg cells and limits Treg cell proliferation and function during chronic HCV infection. The expression of PD-1, upon the contact with its own ligands, inhibits pSTAT-5 via the activation of Src homology 2-containing tyrosine phosphatases (Franceschini et al., 2009; Figure 4). As a consequence, responder T cells can escape from excessive expansion of Treg cells and render them available for responding to possible novel waves of infection. This negative feedback loop assumes a different significance during chronic infections, such as HCV. The incapacity to clear HCV by the immune system (due to the various mechanisms emphasized above) maintains a vicious spiral, whereby responder T cells are chronically stimulated to produce IL-2 that will be dominantly adsorbed by CD25hi Treg cells that in turn will continuously suppress the effector responses. The PD-1 upregulation limits the excessive expansion of Treg cells by controlling pSTAT-5 and fine-tunes the Treg function in order to minimize the immunopathology without completely switch off those intended to limit excessive viral spread (Figure 4). This may represent a critical contra-suppression mechanism that has evolved to control that Treg cells have a limited suppression. Homeostatic balance participates in establishing a status of chronic low-level liver inflammation that is in turn instrumental to ensure a long-lasting survival of the host.
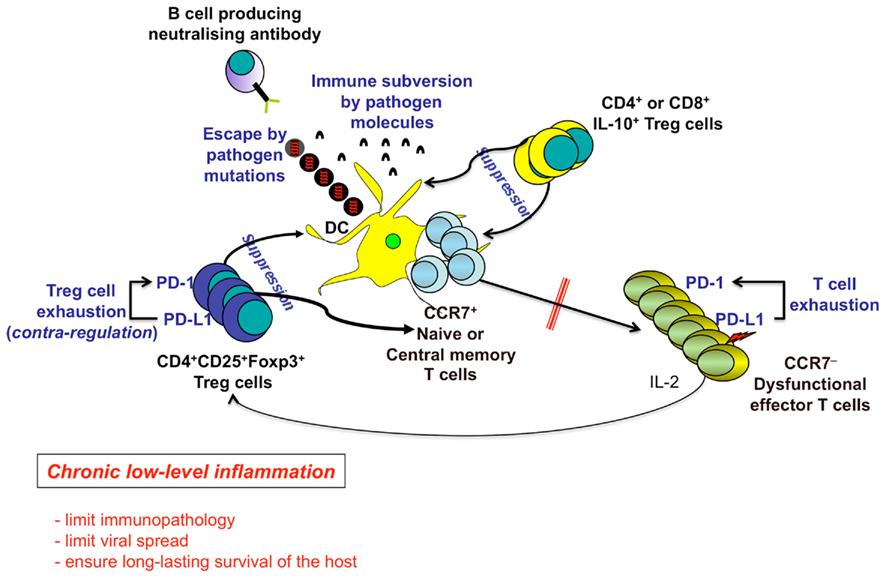
Figure 4. Chronic low-level inflammation as an advantage for the long-term human survival. Several mechanisms induced by both persistent pathogens (e.g., escape by pathogen mutations, pathogen-related immune subversion) and the host [e.g., PD-1-dependent T cell exhaustion, Regulatory T (Treg) cells] contribute to the maintenance of chronic inflammatory diseases, by down-modulating DC functions, T and B cell priming, generation of memory cells, and ultimately differentiation of terminal effector cells. Treg cells can be represented by the following: (a) CD4+CD25+Foxp3+ Treg cells, which suppress via cell–cell contact, IL-2 capture to effector cells, TGF-β production, etc.; (b) Treg cells producing immunosuppressive cytokines (e.g., IL-10, TGF-β). PD-1 is upregulated on both responder and Treg cells and upon contact with PD-L1/2 limits both T effector memory (TEM) cell responses and excessive Treg cell function, respectively. The resulting contra-regulation of Treg cells will have an important role to limit excessive suppression of immune responses allowing the control of the viral spread, at the cost of the maintenance of the chronic low-level liver immunopathology. This mechanism establishes a long-lasting survival of the host.
A further level of Treg contra-suppression may be achieved with Treg trans-differentiation into alternative fates. Indeed, many recent observations point to an inherent plasticity of Treg cells, particularly prone to acquire proinflammatory functions under adequate microenvironmental cues (Zhou et al., 2009a). For instance, an unexpected discovery is that human Treg cells have considerable plasticity that allows them to produce the proinflammatory cytokine IL-17 under certain conditions particularly related to autoimmunity (Cvetanovich and Hafler, 2010). The Treg plasticity has been related to a CpG-rich intronic enhancer region known as the Treg-specific demethylated region (TSDR), present at the level of four conserved, non-coding regions in the human Foxp3 locus, and containing the most strikingly Treg-specific pattern of CpG methylation (Huehn et al., 2009; Lal and Bromberg, 2009). The methylated state of TSDR in activated conventional T cells and TGF-β-induced Treg cells allows these cells to transiently express Foxp3. In contrast, the demethylated state of the TSDR in human Treg cells allows them to be the only cells that generally exhibit long-term stability of Foxp3 expression. The Foxp3 instability may account for the capacity of conventional T cells and TGF-β-induced Treg cells expressing Foxp3 to convert into Th17 cells, particularly when they are strongly activated in the presence of proinflammatory cytokines during different forms of autoimmune diseases (Dominguez-Villar et al., 2011). The participation of TGF-β in the differentiation of Th17 cells places the Th17 lineage in close relationship with Treg cells, as TGF-β also induces differentiation of naive T cells into Foxp3+ Treg cells (Korn et al., 2009). Controversy still exists about the stability of Foxp3 expression along Treg reprogramming (Zhou et al., 2009b; Rubtsov et al., 2010). In any case, Treg cells have been recognized as the preferential precursors for follicular helper T cells in gut Peyer’s patches (Tsuji et al., 2009) and for CD40L-expressing helper T cells in the establishment of antitumor immunity (Sharma et al., 2010). Hence, Treg cells may rapidly turn from immune-suppressive into immune-protective cells in an “innate-like” fashion (Zhou et al., 2009a). Accordingly, Treg ablation impaired, rather than promoting, early antiviral local responses in a mouse model of infection (Lund et al., 2008). The extracellular signals driving Treg plasticity are mostly unknown and may comprise not only proinflammatory cytokines but also costimulatory receptors. OX40 is a receptor belonging to the TNFR superfamily that is constitutively expressed by Treg cells, supporting their homeostasis (Piconese et al., 2010) and regulating Treg contra-suppression (Piconese et al., 2008). However, it is becoming increasingly clear that the outcome of OX40 signal is somehow modulated by the cytokine milieu and that, in turn, OX40 may affect Treg responses to cytokines. Indeed, OX40 enhances Treg susceptibility to IL-2 (Piconese et al., 2010), and different outcomes of OX40 stimulation depend on the cytokine context (Ruby et al., 2009). Therefore, we may envisage a role for OX40, and possibly for other costimulatory molecules showing comparable behaviors, in fine-tuning Treg ambiguity in response to microenvironmental soluble signals.
Immunity/Immunopathology Balance, as a Model of Ambiguity
As suggested above, we can assume that the contradictory aspect of the immune response is the result of the evolution of biological systems. This evolution proceeds through the selection of the most advantageous processes to ensure the survival of the species. Conversely, these selected processes may have a high cost for the single individual in terms of morbidity and/or mortality. Therefore, a fine line exists between immunity and immunopathology, because, as previously discussed, it is not possible to obtain an effective immune response without the onset of immunopathology. A problem arises when the immunopathologic reactions become persistent or chronic, producing irreversible damage to tissues and organs. Indeed, chronic inflammation may be interpreted as an adaptive response to challenges producing irreversible modifications of tissue homeostasis, often at the expenses of tissue functions (Medzhitov, 2010). A sustained inflammatory response can also be responsible for the development of tumors. In the next section, we will discuss the main mechanisms of chronic inflammation and the ambiguity of this process, which not only helps to control persistent infections but also may induce severe disease of the host (i.e., autoimmunity, tumors).
Chronic Inflammation as an Advantage for Species Survival
The induction of a state of chronic inflammation secondary to infections occurs for those pathogens that are able to evade the immune response and establish a status of persistence. Typical microorganisms capable of evading the immune system and of establishing chronic diseases in humans include HIV, hepatitis B virus (HBV), HCV, Mycobacterium tuberculosis, and Leishmania (Zinkernagel, 1996; Zinkernagel et al., 1999; Tortorella et al., 2000; Phillips, 2002; Klenerman and Hill, 2005).
The relationship between the host and the latent viruses, particularly the herpesviruses (i.e., herpes simplex virus, Epstein–Barr virus, cytomegalovirus), is completely different. These viruses generally establish latency in specific cell types or tissues, are continuously controlled by the immune system, and, in contrast to the persistent pathogens previously mentioned, remain lethargic with regard to their replication capacity (Nikolich-Zugich, 2008). Only occasionally, these viruses are reactivated and stimulate memory T cells, which are generally capable of returning the virus to a state of latency. In a very tiny number of healthy carriers, these viruses can degenerate in severe forms of tumors. However, the intermittent (but not continuous, as in the case of persistent pathogens) T cell stimulation does not result in the impairment or exhaustion of antigen-specific T cells and the development of chronic diseases. Latent viruses seem to play a critical role in expanding memory T cell populations and in maintaining constant their frequencies in old age. Therefore, we will not discuss latent viruses further in this review, which aims to define the role of chronic inflammation in long-term host survival during persistent infections.
Through the different (non-mutually exclusive) mechanisms illustrated above, the host could survive for a long time in parallel with both the “partially controlled” persistent agent and a low-grade inflammation. Therefore, chronic inflammatory processes are paradoxically useful, supporting the concept that the ambiguity is advantageous for the evolutionary process. If the immune responses were invariantly strong and aggressive during a persistent infection, they would be unable to eliminate the persistent pathogen, because of the acquired capacity by the persistent pathogen to escape or to subvert them. In such a situation, exuberant (but non-protective) responses would produce irreversible tissue damage in the host, leading to catastrophic epidemic infections. Considering this point of view, chronic (low-level) inflammatory diseases seem to represent a sort of safeguard for the human survival!
We can assume that chronic inflammation may be defined as the “Yin and Yang” of the immune system, because, via the sophisticated mechanisms mentioned previously, it guarantees the long-term survival of human hosts despite the pathogen persistence. However, the imbalance of the homeostatic mechanisms maintaining chronic inflammation may degenerate into severe “side effects” (i.e., the development of either autoimmune diseases or tumors) in some individuals (Figure 5).

Figure 5. Chronic inflammatory diseases as the “Yin and Yang” of the immune system. Under conditions in which the immune system has been bypassed by persistent pathogens, chronic low-level inflammation guarantees long-term survival of the majority of infected individuals in spite of pathogen persistence. The imbalance of the homeostatic mechanisms maintaining chronic inflammation may degenerate into severe “side effects” (such as autoimmune disorders or tumors) in some individuals. However, these side effects are considered irrelevant in terms of the survival of the species.
Autoimmunity and Cancer
From an evolutionary point of view, the onset of autoimmune diseases or of some tumors might be the price to pay following the establishment of chronic inflammation. It has been suggested that the intensity and nature of the inflammation might explain the apparent contradiction between the inducing and inhibiting effects on tumor survival. Indeed, vigorous acute inflammatory processes can favor an immune effector response potentially able to induce tumor regression (Mantovani et al., 2008a,b). Conversely, a status of pre-existing chronic inflammation can participate to the development of cancer, by the production of growth and angiogenic factors eventually promoting cancer-cell survival, implantation, and growth. In addition, chronic inflammation can affect the immune-surveillance directly via its own intrinsic mechanisms (i.e., expansion of Treg cells, T cell exhaustion, etc.), and indirectly by the incapacity to limit the immunosuppressive effects by tumors. In these conditions, the suppression of the immune response, that is certainly useful to terminate the effector responses or to limit excessive immunopathology, can result detrimental. In fact, the chronic activity of Treg cells is believed to play a crucial role in suppressing the immune-surveillance against tumors by CD8+, Th1, and probably Th17 cells (Curiel et al., 2004; Zou, 2006; Colombo and Piconese, 2007; Curiel, 2007; Nair et al., 2007; Zhou and Levitsky, 2007; Piconese et al., 2008).
The production of soluble factors (i.e., proinflammatory or cell growth cytokines) that favor cell proliferation, generally needed to the immune system to defend the host efficaciously, can facilitate the mitotic cycle also of non-lymphoid cells. In the long run, this prolonged stimulation, which is characteristic of chronic inflammatory processes, can induce tissue damage, as in the case of liver cirrhosis by both HBV and HCV, where the phenomena of necrosis, cell renewal, and even neoplastic transformation might simultaneously occur (Tan et al., 2008). The sustained recognition of antigens that cannot be eliminated by the immune system can induce errors in VDJ rearrangement by B cells or cause chromosomal translocations. These conditions can generate the onset of lymphomas. A typical case is the mucosa-associated lymphoid tissue (MALT) lymphoma associated with the presence of several infectious agents including Helicobacter pylori, Campylobacter jejuni, Borrelia burgdorferi, Chlamydia psittaci, and HCV, inducing gastric lymphomas, immunoproliferative small intestinal disease, cutaneous lymphoma, ocular lymphoma, and spleen lymphoma, respectively, following the chronic stimulation of local B cells (Suarez et al., 2006; Sagaert et al., 2007). In addition, the chronic stimulation of B lymphocytes by HCV infection can induce the monoclonal expansion of anti-IgG antibodies, which are responsible for the formation of cryoglobulins or even the establishment of follicular B cell lymphomas (Landau et al., 2007).
Another complex issue deserving discussion is how autoreactive T lymphocytes become pathogenetic and develop chronic autoimmune diseases. Under conditions in which the stimulatory capacities gain the upper hand over the tolerogenic capacities of DCs (i.e., during inflammatory or infectious processes), they phagocytose both microbial and self antigens and migrate with high efficiency into the draining lymph nodes, where they efficiently prime both pathogen-specific and autoreactive T cells (Lang et al., 2005; Marshak-Rothstein, 2006). Generally (as previously discussed in the B Cell Responses and T Cell Responses), these responses are switched off when the pathogen has been cleared, including the autoreactive responses that have participated in the amplification of the protective responses. Under certain conditions, several genetic and environmental factors (many of which are still unknown) can, however, allow the expansion of autoreactive clones, which escape the mechanisms of peripheral tolerance, and can induce autoimmune diseases.
In this context, it is of interest our recent observation indicating that the phenomenon of chronic immune activation, commonly observed during different viral or autoimmune diseases (Propato et al., 2001; Chernysheva et al., 2002; Bangs et al., 2006; Grossman et al., 2006), is in part caused by a vicious cycle, which is maintained by a continuous cross-presentation of apoptotic lymphoid cells that are derived from chronically activated lymphocytes (Figure 6; Rawson et al., 2007). The phagocytosis of these apoptotic cells by DCs leads to the generation of a huge number of apoptotic cell-derived self-epitopes. In a chronic inflammatory context and in virtue of the activatory signals provided by apoptotic T cells expressing CD40L, this activity also leads to the cross-priming of a large repertoire of apoptotic epitope-specific T cells, which in turn expand the inflammation and undergo apoptosis after they have performed their effector functions, and so on (Propato et al., 2001; Rawson et al., 2007). Thus, chronic immune activation can establish a milieu favoring the emergence and the maintenance of autoimmune responses, and ultimately contribute to the irreversible impairment of the immune system, such as in the case of HIV infection or in the final stages of several systemic autoimmune diseases (Propato et al., 2001; Rawson et al., 2007).
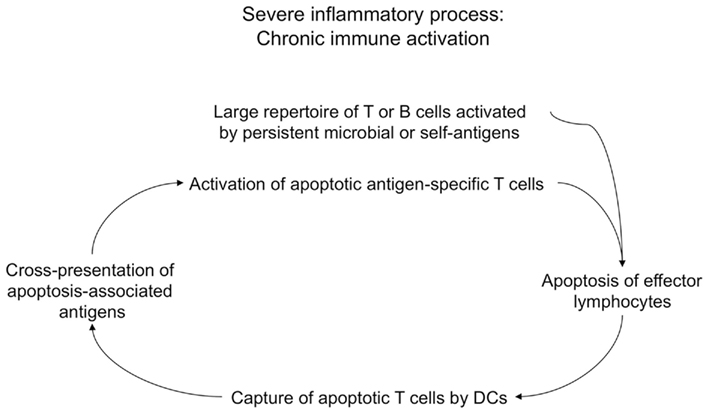
Figure 6. Chronic immune activation as a result of cross-presentation of apoptotic T cells. During severe inflammatory processes, chronic immune activation may be in part maintained by a vicious spiral, which may be initiated by activated lymphocytes (e.g., specific for persistent microbial antigens or self antigens) undergoing apoptosis upon they performed their effector functions. These apoptotic cells are cross-presented by DCs, which cross-prime a huge repertoire of autoreactive T cells specific for a multitude of apoptotic cell-associated epitopes. Apoptotic antigen-specific T cells participate to the maintenance of chronic immune activation via their effector performances, then they will undergo apoptosis, and the resulting apoptotic cells will be cross-presented by DCs that will cross-prime new waves of apoptotic antigen-specific T cells, and so on.
Finally, functional defects of Treg cells seem to play a key role in the induction of autoimmune processes. Defects of these cells have been clearly documented in several autoimmune diseases (e.g., rheumatoid arthritis, type I diabetes, multiple sclerosis; Chatenoud et al., 2001; Shevach et al., 2001; Bluestone and Abbas, 2003; von Herrath and Harrison, 2003; Ehrenstein et al., 2004; Hsieh et al., 2004; Lerman et al., 2004; Tang et al., 2004; Viglietta et al., 2004; Lindley et al., 2005; Sakaguchi, 2005) as well as in allergic diseases (Medoff et al., 2008). Similar mechanisms play a crucial role in the uncontrolled generation of autoantibodies recognizing apoptotic cell-associated self-epitopes, as in the case of several systemic autoimmune diseases (i.e., systemic lupus erythematosus, progressive systemic sclerosis, Sjogren’s diseases; Chatenoud et al., 2001; Shevach et al., 2001; Bluestone and Abbas, 2003; von Herrath and Harrison, 2003; Ehrenstein et al., 2004; Hsieh et al., 2004; Lerman et al., 2004; Tang et al., 2004; Viglietta et al., 2004; Lindley et al., 2005; Sakaguchi, 2005).
Conclusion
Based on the definition of ambiguity as “a symptom of the crisis of the subject, a productive function against and beyond the metaphysic (creativity),” we can consider this concept as a hallmark of biological systems. When the definition of ambiguity is applied to the immune system, the “crisis” should derive by its capacity to recognize everything and thus to risk to offend and not only to defend the host. Therefore, the “creativity” of the immune system consists of its capacity to express simultaneously different strategies that are only apparently opposite but eventually result in an evolutionary advantage. On the one hand, the immune response contributes to the species survival; on the other hand, it can lead to the sacrifice of single individuals. During the evolutionary process, the selective pressure led to the generation of multiple ambiguous mechanisms to better counteract the aggression of infectious agents. Although this is obtained at the cost of severe side effects (tumor development, autoimmune diseases) in several individuals, these side effects are considered irrelevant in terms of the survival of the species.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by: European Union grants (IMECS no. 201169, FP7-Health-2007-A and SPHYNX no. 261365, FP7-Health-2010); MinisterodellaSanità [Ricercafinalizzata (RFPS-2006-3-337923 and RFPS-2007-1-636647); IstitutoSuperiore di Sanità (Progetti AIDS-2006 and -2008)]; Ministerodell’Istruzione, dell’Università e dellaRicerca [MIUR; Programmi di ricerca di interessenazionale (PRIN)-2008/10 no. 7245/1; Fondo per gliinvestimenti di ricerca di base (FIRB)-2011/13 no. RBAP10TPXK]; FondazioneCariplo (progetti no. 13535 and 3603 2010/12); AssociazioneItaliana per la RicercasulCancro [AIRC; Investigator Grant (IG)-2010/13 no. 10756]. Silvia Piconese is supported by My First AIRC grant (no. 8726).
Footnote
- ^Here, we will not consider the definition of ambiguity generally attributed by the psychoanalysis, as the incapacity to discriminate between ego and non-ego potentially leading to severe social and psychic disorders Bleger (1967). Simbiosis y ambiguedad, estudio psiconalitico. Editorial Paidos, Buenos Aires. If ever, according to the etymologic, semantic, or philosophic definitions reported above, we will consider the biological ambiguities (such as those cellular and molecular largely described for the immunology in this review) as critical events participating to the general homeostasis of the individual that ultimately favors the species survival.
References
Accapezzato, D., Francavilla, V., Paroli, M., Casciaro, M., Chircu, L. V., Cividini, A., Abrignani, S., Mondelli, M. U., and Barnaba, V. (2004). Hepatic expansion of a virus-specific regulatory CD8(+) T cell population in chronic hepatitis C virus infection. J. Clin. Invest. 113, 963–972.
Accapezzato, D., Visco, V., Francavilla, V., Molette, C., Donato, T., Paroli, M., Mondelli, M. U., Doria, M., Torrisi, M. R., and Barnaba, V. (2005). Chloroquine enhances human CD8+ T cell responses against soluble antigens in vivo. J. Exp. Med. 202, 817–828.
Acosta-Rodriguez, E. V., Napolitani, G., Lanzavecchia, A., and Sallusto, F. (2007). Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat. Immunol. 8, 942–949.
Agrawal, A., Eastman, Q. M., and Schatz, D. G. (1998). Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature 394, 744–751.
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801.
Alard, J. E., Dueymes, M., Youinou, P., and Jamin, C. (2008). HSP60 and anti-HSP60 antibodies in vasculitis: they are two of a kind. Clin. Rev. Allergy Immunol. 35, 66–71.
Albert, M. L. (2004). Death-defying immunity: do apoptotic cells influence antigen processing and presentation? Nat. Rev. Immunol. 4, 223–231.
Anderson, M. S., Venanzi, E. S., Klein, L., Chen, Z., Berzins, S. P., Turley, S. J., von Boehmer, H., Bronson, R., Dierich, A., Benoist, C., and Mathis, D. (2002). Projection of an immunological self shadow within the thymus by the aire protein. Science 298, 1395–1401.
Avrameas, S. (1991). Natural autoantibodies: from ‘horror autotoxicus’ to ‘gnothi seauton.’ Immunol. Today 12, 154–159.
Bachem, A., Guttler, S., Hartung, E., Ebstein, F., Schaefer, M., Tannert, A., Salama, A., Movassaghi, K., Opitz, C., Mages, H. W., Henn, V., Kloetzel, P. M., Gurka, S., and Kroczek, R. A. (2010). Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J. Exp. Med. 207, 1273–1281.
Bangs, S. C., McMichael, A. J., and Xu, X. N. (2006). Bystander T cell activation – implications for HIV infection and other diseases. Trends Immunol. 27, 518–524.
Barber, D. L., Wherry, E. J., Masopust, D., Zhu, B., Allison, J. P., Sharpe, A. H., Freeman, G. J., and Ahmed, R. (2006). Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687.
Barron, L., Knoechel, B., Lohr, J., and Abbas, A. K. (2008). Cutting edge: contributions of apoptosis and anergy to systemic T cell tolerance. J. Immunol. 180, 2762–2766.
Barthlott, T., Kassiotis, G., and Stockinger, B. (2003). T cell regulation as a side effect of homeostasis and competition. J. Exp. Med. 197, 451–460.
Barthlott, T., Moncrieffe, H., Veldhoen, M., Atkins, C. J., Christensen, J., O’Garra, A., and Stockinger, B. (2005). CD25+ CD4+ T cells compete with naive CD4+ T cells for IL-2 and exploit it for the induction of IL-10 production. Int. Immunol. 17, 279–288.
Belkaid, Y. (2007). Regulatory T cells and infection: a dangerous necessity. Nat. Rev. Immunol. 7, 875–888.
Benoist, C., and Mathis, D. (2001). Autoimmunity provoked by infection: how good is the case for T cell epitope mimicry? Nat. Immunol. 2, 797–801.
Bettelli, E., Korn, T., and Kuchroo, V. K. (2007). Th17: the third member of the effector T cell trilogy. Curr. Opin. Immunol. 19, 652–657.
Blachere, N. E., Darnell, R. B., and Albert, M. L. (2005). Apoptotic cells deliver processed antigen to dendritic cells for cross-presentation. PLoS Biol. 3, e185. doi:10.1371/journal.pbio.0030185
Bluestone, J. A., and Abbas, A. K. (2003). Natural versus adaptive regulatory T cells. Nat. Rev. Immunol. 3, 253–257.
Borgulya, P., Kishi, H., Uematsu, Y., and von Boehmer, H. (1992). Exclusion and inclusion of alpha and beta T cell receptor alleles. Cell 69, 529–537.
Burgdorf, S., Kautz, A., Bohnert, V., Knolle, P. A., and Kurts, C. (2007). Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 316, 612–616.
Cao, W., Rosen, D. B., Ito, T., Bover, L., Bao, M., Watanabe, G., Yao, Z., Zhang, L., Lanier, L. L., and Liu, Y. J. (2006). Plasmacytoid dendritic cell-specific receptor ILT7-Fc epsilonRI gamma inhibits toll-like receptor-induced interferon production. J. Exp. Med. 203, 1399–1405.
Casanova, J. L., Romero, P., Widmann, C., Kourilsky, P., and Maryanski, J. L. (1991). T cell receptor genes in a series of class I major histocompatibility complex-restricted cytotoxic T lymphocyte clones specific for a Plasmodium berghei nonapeptide: implications for T cell allelic exclusion and antigen-specific repertoire. J. Exp. Med. 174, 1371–1383.
Chatenoud, L., Salomon, B., and Bluestone, J. A. (2001). Suppressor T cells – they’re back and critical for regulation of autoimmunity! Immunol. Rev. 182, 149–163.
Chen, W., Frank, M. E., Jin, W., and Wahl, S. M. (2001). TGF-beta released by apoptotic T cells contributes to an immunosuppressive milieu. Immunity 14, 715–725.
Chernysheva, A. D., Kirou, K. A., and Crow, M. K. (2002). T cell proliferation induced by autologous non-T cells is a response to apoptotic cells processed by dendritic cells. J. Immunol. 169, 1241–1250.
Cho, J. H. (2008). The genetics and immunopathogenesis of inflammatory bowel disease. Nat. Rev. Immunol. 8, 458–466.
Coffer, P. J., and Burgering, B. M. (2004). Forkhead-box transcription factors and their role in the immune system. Nat. Rev. Immunol. 4, 889–899.
Colombo, M. P., and Piconese, S. (2007). Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat. Rev. Cancer 7, 880–887.
Coombes, J. L., and Powrie, F. (2008). Dendritic cells in intestinal immune regulation. Nat. Rev. Immunol. 8, 435–446.
Coombes, J. L., Robinson, N. J., Maloy, K. J., Uhlig, H. H., and Powrie, F. (2005). Regulatory T cells and intestinal homeostasis. Immunol. Rev. 204, 184–194.
Crozat, K., Guiton, R., Contreras, V., Feuillet, V., Dutertre, C. A., Ventre, E., Vu Manh, T. P., Baranek, T., Storset, A. K., Marvel, J., Boudinot, P., Hosmalin, A., Schwartz-Cornil, I., and Dalod, M. (2010). The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J. Exp. Med. 207, 1283–1292.
Curiel, T. J., Coukos, G., Zou, L., Alvarez, X., Cheng, P., Mottram, P., Evdemon-Hogan, M., Conejo-Garcia, J. R., Zhang, L., Burow, M., Zhu, Y., Wei, S., Kryczek, I., Daniel, B., Gordon, A., Myers, L., Lackner, A., Disis, M. L., Knutson, K. L., Chen, L., and Zou, W. (2004). Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 10, 942–949.
Curtis, M. M., and Way, S. S. (2009). Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology 126, 177–185.
Cvetanovich, G. L., and Hafler, D. A. (2010). Human regulatory T cells in autoimmune diseases. Curr. Opin. Immunol. 22, 753–760.
Dash, P., McClaren, J. L., Oguin, T. H. III, Rothwell, W., Todd, B., Morris, M. Y., Becksfort, J., Reynolds, C., Brown, S. A., Doherty, P. C., and Thomas, P. G. (2010). Paired analysis of TCRalpha and TCRbeta chains at the single-cell level in mice. J. Clin. Invest. 121, 288–295.
Day, C. L., Kaufmann, D. E., Kiepiela, P., Brown, J. A., Moodley, E. S., Reddy, S., Mackey, E. W., Miller, J. D., Leslie, A. J., DePierres, C., Mncube, Z., Duraiswamy, J., Zhu, B., Eichbaum, Q., Altfeld, M., Wherry, E. J., Coovadia, H. M., Goulder, P. J., Klenerman, P., Ahmed, R., Freeman, G. J., and Walker, B. D. (2006). PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443, 350–354.
de Jong, E. C., Smits, H. H., and Kapsenberg, M. L. (2005). Dendritic cell-mediated T cell polarization. Springer Semin. Immunopathol. 26, 289–307.
de la Rosa, M., Rutz, S., Dorninger, H., and Scheffold, A. (2004). Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur. J. Immunol. 34, 2480–2488.
De Vita, L., Accapezzato, D., Mangino, G., Morrone, S., Santilio, I., Casciaro, M. A., Fava, D., Bruno, G., Del Prete, G., Santoni, A., and Barnaba, V. (1998). Defective Th1 and Th2 cytokine synthesis in the T-T cell presentation model for lack of CD40/CD40 ligand interaction. Eur. J. Immunol. 28, 3552–3563.
Deaglio, S., Dwyer, K. M., Gao, W., Friedman, D., Usheva, A., Erat, A., Chen, J. F., Enjyoji, K., Linden, J., Oukka, M., Kuchroo, V. K., Strom, T. B., and Robson, S. C. (2007). Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 204, 1257–1265.
Di Pucchio, T., Chatterjee, B., Smed-Sorensen, A., Clayton, S., Palazzo, A., Montes, M., Xue, Y., Mellman, I., Banchereau, J., and Connolly, J. E. (2008). Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat. Immunol. 9, 551–557.
Di Rosa, F., and Barnaba, V. (1998). Persisting viruses and chronic inflammation: understanding their relation to autoimmunity. Immunol. Rev. 164, 17–27.
Dominguez-Villar, M., Baecher-Allan, C. M., and Hafler, D. A. (2011). Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat. Med. 17, 673–675.
Dong, C. (2008). TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat. Rev. Immunol. 8, 337–348.
Dudziak, D., Kamphorst, A. O., Heidkamp, G. F., Buchholz, V. R., Trumpfheller, C., Yamazaki, S., Cheong, C., Liu, K., Lee, H. W., Park, C. G., Steinman, R. M., and Nussenzweig, M. C. (2007). Differential antigen processing by dendritic cell subsets in vivo. Science 315, 107–111.
Duhen, T., Geiger, R., Jarrossay, D., Lanzavecchia, A., and Sallusto, F. (2009). Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat. Immunol. 10, 857–863.
Ebinuma, H., Nakamoto, N., Li, Y., Price, D. A., Gostick, E., Levine, B. L., Tobias, J., Kwok, W. W., and Chang, K. M. (2008). Identification and in vitro expansion of functional antigen-specific CD25+ FoxP3+ regulatory T cells in hepatitis C virus infection. J. Virol. 82, 5043–5053.
Ehrenstein, M. R., Evans, J. G., Singh, A., Moore, S., Warnes, G., Isenberg, D. A., and Mauri, C. (2004). Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J. Exp. Med. 200, 277–285.
Elliott, J. I., and Altmann, D. M. (1995). Dual T cell receptor alpha chain T cells in autoimmunity. J. Exp. Med. 182, 953–959.
Eyerich, S., Eyerich, K., Pennino, D., Carbone, T., Nasorri, F., Pallotta, S., Cianfarani, F., Odorisio, T., Traidl-Hoffmann, C., Behrendt, H., Durham, S. R., Schmidt-Weber, C. B., and Cavani, A. (2009). Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Invest. 119, 3573–3585.
Ferguson, T. A., Green, D. R., and Griffith, T. S. (2002). Cell death and immune privilege. Int. Rev. Immunol. 21, 153–172.
Fontenot, J. D., Gavin, M. A., and Rudensky, A. Y. (2003). Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4, 330–336.
Fontenot, J. D., Rasmussen, J. P., Gavin, M. A., and Rudensky, A. Y. (2005). A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol. 6, 1142–1151.
Franceschini, D., Paroli, M., Francavilla, V., Videtta, M., Morrone, S., Labbadia, G., Cerino, A., Mondelli, M. U., and Barnaba, V. (2009). PD-L1 negatively regulates CD4+CD25+Foxp3+ Tregs by limiting STAT-5 phosphorylation in patients chronically infected with HCV. J. Clin. Invest. 119, 551–564.
Fritz, J. H., Ferrero, R. L., Philpott, D. J., and Girardin, S. E. (2006). Nod-like proteins in immunity, inflammation and disease. Nat. Immunol. 7, 1250–1257.
Gallucci, S., and Matzinger, P. (2001). Danger signals: SOS to the immune system. Curr. Opin. Immunol. 13, 114–119.
Gao, Y., Herndon, J. M., Zhang, H., Griffith, T. S., and Ferguson, T. A. (1998). Antiinflammatory effects of CD95 ligand (FasL)-induced apoptosis. J. Exp. Med. 188, 887–896.
Gardner, J. M., Devoss, J. J., Friedman, R. S., Wong, D. J., Tan, Y. X., Zhou, X., Johannes, K. P., Su, M. A., Chang, H. Y., Krummel, M. F., and Anderson, M. S. (2008). Deletional tolerance mediated by extrathymic Aire-expressing cells. Science 321, 843–847.
Gillard, G. O., Dooley, J., Erickson, M., Peltonen, L., and Farr, A. G. (2007). Aire-dependent alterations in medullary thymic epithelium indicate a role for Aire in thymic epithelial differentiation. J. Immunol. 178, 3007–3015.
Griffith, T. S., Kazama, H., VanOosten, R. L., Earle, J. K. Jr., Herndon, J. M., Green, D. R., and Ferguson, T. A. (2007). Apoptotic cells induce tolerance by generating helpless CD8+ T cells that produce TRAIL. J. Immunol. 178, 2679–2687.
Grossman, Z., Meier-Schellersheim, M., Paul, W. E., and Picker, L. J. (2006). Pathogenesis of HIV infection: what the virus spares is as important as what it destroys. Nat. Med. 12, 289–295.
Guermonprez, P., Saveanu, L., Kleijmeer, M., Davoust, J., Van Endert, P., and Amigorena, S. (2003). ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 425, 397–402.
Guiducci, C., Ott, G., Chan, J. H., Damon, E., Calacsan, C., Matray, T., Lee, K. D., Coffman, R. L., and Barrat, F. J. (2006). Properties regulating the nature of the plasmacytoid dendritic cell response to toll-like receptor 9 activation. J. Exp. Med. 203, 1999–2008.
Hardy, R. R., and Hayakawa, K. (2001). B cell development pathways. Annu. Rev. Immunol. 19, 595–621.
He, X., Janeway, C. A. Jr., Levine, M., Robinson, E., Preston-Hurlburt, P., Viret, C., and Bottomly, K. (2002). Dual receptor T cells extend the immune repertoire for foreign antigens. Nat. Immunol. 3, 127–134.
Heeg, M. H., Ulsenheimer, A., Gruner, N. H., Zachoval, R., Jung, M. C., Gerlach, J. T., Raziorrouh, B., Schraut, W., Horster, S., Kauke, T., Spannagl, M., and Diepolder, H. M. (2009). FOXP3 expression in hepatitis C virus-specific CD4+ T cells during acute hepatitis C. Gastroenterology 137, 1280–1288. e1281–e1286.
Hoebe, K., Janssen, E., and Beutler, B. (2004). The interface between innate and adaptive immunity. Nat. Immunol. 5, 971–974.
Hori, S., Nomura, T., and Sakaguchi, S. (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061.
Hsieh, C. S., Liang, Y., Tyznik, A. J., Self, S. G., Liggitt, D., and Rudensky, A. Y. (2004). Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity 21, 267–277.
Huehn, J., Polansky, J. K., and Hamann, A. (2009). Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat. Rev. Immunol. 9, 83–89.
Inaba, K., Turley, S., Yamaide, F., Iyoda, T., Mahnke, K., Inaba, M., Pack, M., Subklewe, M., Sauter, B., Sheff, D., Albert, M., Bhardwaj, N., Mellman, I., and Steinman, R. M. (1998). Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J. Exp. Med. 188, 2163–2173.
Inohara, N., Chamaillard, M., McDonald, C., and Nunez, G. (2005). NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu. Rev. Biochem. 74, 355–383.
Jakobson, R. (1995). “On realism in art,” in On Language, Chap. I, eds. L. Waugh and M. Monville-Burston (Cambridge: Harvard University Press), 21–23.
Janeway, C. A. Jr., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216.
Jego, G., Palucka, A. K., Blanck, J. P., Chalouni, C., Pascual, V., and Banchereau, J. (2003). Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 19, 225–234.
Ji, Q., Perchellet, A., and Goverman, J. M. (2010). Viral infection triggers central nervous system autoimmunity via activation of CD8+ T cells expressing dual TCRs. Nat. Immunol. 11, 628–634.
Jiang, A., Bloom, O., Ono, S., Cui, W., Unternaehrer, J., Jiang, S., Whitney, J. A., Connolly, J., Banchereau, J., and Mellman, I. (2007). Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity 27, 610–624.
Kawai, T., and Akira, S. (2006). Innate immune recognition of viral infection. Nat. Immunol. 7, 131–137.
Kazama, H., Ricci, J. E., Herndon, J. M., Hoppe, G., Green, D. R., and Ferguson, T. A. (2008). Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 29, 21–32.
Keir, M. E., Liang, S. C., Guleria, I., Latchman, Y. E., Qipo, A., Albacker, L. A., Koulmanda, M., Freeman, G. J., Sayegh, M. H., and Sharpe, A. H. (2006). Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 203, 883–895.
Killeen, N., Irving, B. A., Pippig, S., and Zingler, K. (1998). Signaling checkpoints during the development of T lymphocytes. Curr. Opin. Immunol. 10, 360–367.
Klareskog, L., Widhe, M., Hermansson, M., and Ronnelid, J. (2008). Antibodies to citrullinated proteins in arthritis: pathology and promise. Curr. Opin. Rheumatol. 20, 300–305.
Klechevsky, E., Liu, M., Morita, R., Banchereau, R., Thompson-Snipes, L., Palucka, A. K., Ueno, H., and Banchereau, J. (2009). Understanding human myeloid dendritic cell subsets for the rational design of novel vaccines. Hum. Immunol. 70, 281–288.
Klechevsky, E., Morita, R., Liu, M., Cao, Y., Coquery, S., Thompson-Snipes, L., Briere, F., Chaussabel, D., Zurawski, G., Palucka, A. K., Reiter, Y., Banchereau, J., and Ueno, H. (2008). Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity 29, 497–510.
Klenerman, P., and Hill, A. (2005). T cells and viral persistence: lessons from diverse infections. Nat. Immunol. 6, 873–879.
Korn, T., Bettelli, E., Oukka, M., and Kuchroo, V. K. (2009). IL-17 and Th17 Cells. Annu. Rev. Immunol. 27, 485–517.
Kretschmer, K., Apostolou, I., Hawiger, D., Khazaie, K., Nussenzweig, M. C., and von Boehmer, H. (2005). Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 6, 1219–1227.
Kuroda, N., Mitani, T., Takeda, N., Ishimaru, N., Arakaki, R., Hayashi, Y., Bando, Y., Izumi, K., Takahashi, T., Nomura, T., Sakaguchi, S., Ueno, T., Takahama, Y., Uchida, D., Sun, S., Kajiura, F., Mouri, Y., Han, H., Matsushima, A., Yamada, G., and Matsumoto, M. (2005). Development of autoimmunity against transcriptionally unrepressed target antigen in the thymus of Aire-deficient mice. J. Immunol. 174, 1862–1870.
Kurts, C., Heath, W. R., Kosaka, H., Miller, J. F., and Carbone, F. R. (1998). The peripheral deletion of autoreactive CD8+ T cells induced by cross-presentation of self-antigens involves signaling through CD95 (Fas, Apo-1). J. Exp. Med. 188, 415–420.
Lal, G., and Bromberg, J. S. (2009). Epigenetic mechanisms of regulation of Foxp3 expression. Blood 114, 3727–3735.
Landau, D. A., Saadoun, D., Calabrese, L. H., and Cacoub, P. (2007). The pathophysiology of HCV induced B-cell clonal disorders. Autoimmun. Rev. 6, 581–587.
Lang, K. S., Recher, M., Junt, T., Navarini, A. A., Harris, N. L., Freigang, S., Odermatt, B., Conrad, C., Ittner, L. M., Bauer, S., Luther, S. A., Uematsu, S., Akira, S., Hengartner, H., and Zinkernagel, R. M. (2005). Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat. Med. 11, 138–145.
Lanzavecchia, A., and Sallusto, F. (2001). Regulation of T cell immunity by dendritic cells. Cell 106, 263–266.
Lanzavecchia, A., and Sallusto, F. (2007). Toll-like receptors and innate immunity in B-cell activation and antibody responses. Curr. Opin. Immunol. 19, 268–274.
Lerman, M. A., Larkin, J. III, Cozzo, C., Jordan, M. S., and Caton, A. J. (2004). CD4+ CD25+ regulatory T cell repertoire formation in response to varying expression of a neo-self-antigen. J. Immunol. 173, 236–244.
Lindley, S., Dayan, C. M., Bishop, A., Roep, B. O., Peakman, M., and Tree, T. I. (2005). Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes 54, 92–99.
Liston, A., Lesage, S., Wilson, J., Peltonen, L., and Goodnow, C. C. (2003). Aire regulates negative selection of organ-specific T cells. Nat. Immunol. 4, 350–354.
Luger, D., Silver, P. B., Tang, J., Cua, D., Chen, Z., Iwakura, Y., Bowman, E. P., Sgambellone, N. M., Chan, C. C., and Caspi, R. R. (2008). Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J. Exp. Med. 205, 799–810.
Lund, J. M., Hsing, L., Pham, T. T., and Rudensky, A. Y. (2008). Coordination of early protective immunity to viral infection by regulatory T cells. Science 320, 1220–1224.
Mantovani, A., Allavena, P., Sica, A., and Balkwill, F. (2008a). Cancer-related inflammation. Nature 454, 436–444.
Mantovani, A., Romero, P., Palucka, A. K., and Marincola, F. M. (2008b). Tumour immunity: effector response to tumour and role of the microenvironment. Lancet 371, 771–783.
Marrack, P., McCormack, J., and Kappler, J. (1989). Presentation of antigen, foreign major histocompat-ibility complex proteins and self by thymus cortical epithelium. Nature 338, 503–505.
Marshak-Rothstein, A. (2006). Toll-like receptors in systemic autoimmune disease. Nat. Rev. Immunol. 6, 823–835.
Masopust, D., and Ahmed, R. (2004). Reflections on CD8 T-cell activation and memory. Immunol. Res. 29, 151–160.
Matsui, T., Connolly, J. E., Michnevitz, M., Chaussabel, D., Yu, C. I., Glaser, C., Tindle, S., Pypaert, M., Freitas, H., Piqueras, B., Banchereau, J., and Palucka, A. K. (2009). CD2 distinguishes two subsets of human plasmacytoid dendritic cells with distinct phenotype and functions. J. Immunol. 182, 6815–6823.
Matsunaga, T., and Rahman, A. (2001). In search of the origin of the thymus: the thymus and GALT may be evolutionarily related. Scand. J. Immunol. 53, 1–6.
Matzinger, P. (2007). Friendly and dangerous signals: is the tissue in control? Nat. Immunol. 8, 11–13.
Matzinger, P., and Kamala, T. (2011). Tissue-based class control: the other side of tolerance. Nat. Rev. Immunol. 11, 221–230.
McCaughtry, T. M., Baldwin, T. A., Wilken, M. S., and Hogquist, K. A. (2008). Clonal deletion of thymocytes can occur in the cortex with no involvement of the medulla. J. Exp. Med. 205, 2575–2584.
McGargill, M. A., Derbinski, J. M., and Hogquist, K. A. (2000). Receptor editing in developing T cells. Nat. Immunol. 1, 336–341.
McGeachy, M. J., and Cua, D. J. (2008). Th17 cell differentiation: the long and winding road. Immunity 28, 445–453.
Medoff, B. D., Thomas, S. Y., and Luster, A. D. (2008). T cell trafficking in allergic asthma: the ins and outs. Annu. Rev. Immunol. 26, 205–232.
Medzhitov, R., and Janeway, C. A. Jr. (2002). Decoding the patterns of self and nonself by the innate immune system. Science 296, 298–300.
Meylan, E., Tschopp, J., and Karin, M. (2006). Intracellular pattern recognition receptors in the host response. Nature 442, 39–44.
Murali-Krishna, K., Altman, J. D., Suresh, M., Sourdive, D. J., Zajac, A. J., Miller, J. D., Slansky, J., and Ahmed, R. (1998). Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity 8, 177–187.
Murphy, K., Travers, P., and Walport, M. (2007). Janeway’s Immunobiology, 7th Edn. New York: Garland Science.
Nair, S., Boczkowski, D., Fassnacht, M., Pisetsky, D., and Gilboa, E. (2007). Vaccination against the forkhead family transcription factor Foxp3 enhances tumor immunity. Cancer Res. 67, 371–380.
Nelms, K., Keegan, A. D., Zamorano, J., Ryan, J. J., and Paul, W. E. (1999). The IL-4 receptor: signaling mechanisms and biologic functions. Annu. Rev. Immunol. 17, 701–738.
Neuberger, M. S. (1997). Antigen receptor signaling gives lymphocytes a long life. Cell 90, 971–973.
Nikolich-Zugich, J. (2008). Ageing and life-long maintenance of T-cell subsets in the face of latent persistent infections. Nat. Rev. Immunol. 8, 512–522.
Norbury, C. C., Basta, S., Donohue, K. B., Tscharke, D. C., Princiotta, M. F., Berglund, P., Gibbs, J., Bennink, J. R., and Yewdell, J. W. (2004). CD8+ T cell cross-priming via transfer of proteasome substrates. Science 304, 1318–1321.
Oates, J. C., and Gilkeson, G. S. (2002). Mediators of injury in lupus nephritis. Curr. Opin. Rheumatol. 14, 498–503.
Oettinger, M. A., Schatz, D. G., Gorka, C., and Baltimore, D. (1990). RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science 248, 1517–1523.
Otsuki, T., Maeda, M., Murakami, S., Hayashi, H., Miura, Y., Kusaka, M., Nakano, T., Fukuoka, K., Kishimoto, T., Hyodoh, F., Ueki, A., and Nishimura, Y. (2007). Immunological effects of silica and asbestos. Cell. Mol. Immunol. 4, 261–268.
Padovan, E., Casorati, G., Dellabona, P., Meyer, S., Brockhaus, M., and Lanzavecchia, A. (1993). Expression of two T cell receptor alpha chains: dual receptor T cells. Science 262, 422–424.
Palucka, K., Banchereau, J., and Mellman, I. (2010). Designing vaccines based on biology of human dendritic cell subsets. Immunity 33, 464–478.
Pandiyan, P., Zheng, L., Ishihara, S., Reed, J., and Lenardo, M. J. (2007). CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 8, 1353–1362.
Petrilli, V., Dostert, C., Muruve, D. A., and Tschopp, J. (2007). The inflammasome: a danger sensing complex triggering innate immunity. Curr. Opin. Immunol. 19, 615–622.
Piconese, S., Pittoni, P., Burocchi, A., Gorzanelli, A., Care, A., Tripodo, C., and Colombo, M. P. (2010). A non-redundant role for OX40 in the competitive fitness of Treg in response to IL-2. Eur. J. Immunol. 40, 2902–2913.
Piconese, S., Valzasina, B., and Colombo, M. P. (2008). OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J. Exp. Med. 205, 825–839.
Pillai, S. (1999). The chosen few? Positive selection and the generation of naive B lymphocytes. Immunity 10, 493–502.
Pope, R. M., and Tschopp, J. (2007). The role of interleukin-1 and the inflammasome in gout: implications for therapy. Arthritis Rheum. 56, 3183–3188.
Probst, H. C., McCoy, K., Okazaki, T., Honjo, T., and van den Broek, M. (2005). Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat. Immunol. 6, 280–286.
Propato, A., Cutrona, G., Francavilla, V., Ulivi, M., Schiaffella, E., Landt, O., Dunbar, R., Cerundolo, V., Ferrarini, M., and Barnaba, V. (2001). Apoptotic cells overexpress vinculin and induce vinculin-specific cytotoxic T-cell cross-priming. Nat. Med. 7, 807–813.
Pullen, A. M., Kappler, J. W., and Marrack, P. (1989). Tolerance to self antigens shapes the T-cell repertoire. Immunol. Rev. 107, 125–139.
Rawson, P. M., Molette, C., Videtta, M., Altieri, L., Franceschini, D., Donato, T., Finocchi, L., Propato, A., Paroli, M., Meloni, F., Mastroianni, C. M., d’Ettorre, G., Sidney, J., Sette, A., and Barnaba, V. (2007). Cross-presentation of caspase-cleaved apoptotic self antigens in HIV infection. Nat. Med. 13, 1431–1439.
Rice, J. S., Kowal, C., Volpe, B. T., DeGiorgio, L. A., and Diamond, B. (2005). Molecular mimicry: anti-DNA antibodies bind microbial and nonnucleic acid self-antigens. Curr. Top. Microbiol. Immunol. 296, 137–151.
Ridge, J. P., Di Rosa, F., and Matzinger, P. (1998). A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 393, 474–478.
Roncarolo, M. G., Levings, M. K., and Traversari, C. (2001). Differentiation of T regulatory cells by immature dendritic cells. J. Exp. Med. 193, F5–F9.
Rose, N. R., and Mackay, I. R. (2000). Molecular mimicry: a critical look at exemplary instances in human diseases. Cell. Mol. Life Sci. 57, 542–551.
Rubtsov, Y. P., Niec, R. E., Josefowicz, S., Li, L., Darce, J., Mathis, D., Benoist, C., and Rudensky, A. Y. (2010). Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671.
Ruby, C. E., Yates, M. A., Hirschhorn-Cymerman, D., Chlebeck, P., Wolchok, J. D., Houghton, A. N., Offner, H., and Weinberg, A. D. (2009). Cutting edge: OX40 agonists can drive regulatory T cell expansion if the cytokine milieu is right. J. Immunol. 183, 4853–4857.
Sabbagh, L., Snell, L. M., and Watts, T. H. (2007). TNF family ligands define niches for T cell memory. Trends Immunol. 28, 333–339.
Sagaert, X., De Wolf-Peeters, C., Noels, H., and Baens, M. (2007). The pathogenesis of MALT lymphomas: where do we stand? Leukemia 21, 389–396.
Sakaguchi, S. (2004). Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 22, 531–562.
Sakaguchi, S. (2005). Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat. Immunol. 6, 345–352.
Sakaguchi, S., Miyara, M., Costantino, C. M., and Hafler, D. A. (2010). FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 10, 490–500.
Salemi, S., Caporossi, A. P., Boffa, L., Longobardi, M. G., and Barnaba, V. (1995). HIVgp120 activates autoreactive CD4-specific T cell responses by unveiling of hidden CD4 peptides during processing. J. Exp. Med. 181, 2253–2257.
Sallusto, F., Geginat, J., and Lanzavecchia, A. (2004). Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu. Rev. Immunol. 22, 745–763.
Sanjabi, S., Zenewicz, L. A., Kamanaka, M., and Flavell, R. A. (2009). Anti-inflammatory and pro-inflammatory roles of TGF-beta, IL-10, and IL-22 in immunity and autoimmunity. Curr. Opin. Pharmacol. 9, 447–453.
Savina, A., Jancic, C., Hugues, S., Guermonprez, P., Vargas, P., Moura, I. C., Lennon-Dumenil, A. M., Seabra, M. C., Raposo, G., and Amigorena, S. (2006). NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126, 205–218.
Scheffold, A., Huhn, J., and Hofer, T. (2005). Regulation of CD4+CD25+ regulatory T cell activity: it takes (IL-) two to tango. Eur. J. Immunol. 35, 1336–1341.
Schubert, L. A., Jeffery, E., Zhang, Y., Ramsdell, F., and Ziegler, S. F. (2001). Scurfin (FOXP3) acts as a repressor of transcription and regulates T cell activation. J. Biol. Chem. 276, 37672–37679.
Schulz, O., Diebold, S. S., Chen, M., Naslund, T. I., Nolte, M. A., Alexopoulou, L., Azuma, Y. T., Flavell, R. A., Liljestrom, P., and Reis e Sousa, C. (2005). Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 433, 887–892.
Serhan, C. N., and Savill, J. (2005). Resolution of inflammation: the beginning programs the end. Nat. Immunol. 6, 1191–1197.
Setoguchi, R., Hori, S., Takahashi, T., and Sakaguchi, S. (2005). Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 201, 723–735.
Sharma, M. D., Hou, D. Y., Baban, B., Koni, P. A., He, Y., Chandler, P. R., Blazar, B. R., Mellor, A. L., and Munn, D. H. (2010). Reprogrammed foxp3(+) regulatory T cells provide essential help to support cross-presentation and CD8(+) T cell priming in naive mice. Immunity 33, 942–954.
Sharpe, A. H., Wherry, E. J., Ahmed, R., and Freeman, G. J. (2007). The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 8, 239–245.
Shevach, E. M. (2002). CD4+ CD25+ suppressor T cells: more questions than answers. Nat. Rev. Immunol. 2, 389–400.
Shevach, E. M., McHugh, R. S., Piccirillo, C. A., and Thornton, A. M. (2001). Control of T-cell activation by CD4+ CD25+ suppressor T cells. Immunol. Rev. 182, 58–67.
Shinkai, Y., Rathbun, G., Lam, K. P., Oltz, E. M., Stewart, V., Mendelsohn, M., Charron, J., Datta, M., Young, F., Stall, A. M., and Alt, F. W. (1992). RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 68, 855–867.
Siegal, F. P., Kadowaki, N., Shodell, M., Fitzgerald-Bocarsly, P. A., Shah, K., Ho, S., Antonenko, S., and Liu, Y. J. (1999). The nature of the principal type 1 interferon-producing cells in human blood. Science 284, 1835–1837.
Snodgrass, H. R., Kisielow, P., Kiefer, M., Steinmetz, M., and von Boehmer, H. (1985). Ontogeny of the T-cell antigen receptor within the thymus. Nature 313, 592–595.
Steinman, R. M., Hawiger, D., and Nussenzweig, M. C. (2003). Tolerogenic dendritic cells. Annu. Rev. Immunol. 21, 685–711.
Suarez, F., Lortholary, O., Hermine, O., and Lecuit, M. (2006). Infection-associated lymphomas derived from marginal zone B cells: a model of antigen-driven lymphoproliferation. Blood 107, 3034–3044.
Surh, C. D., Boyman, O., Purton, J. F., and Sprent, J. (2006). Homeostasis of memory T cells. Immunol. Rev. 211, 154–163.
Tan, A., Yeh, S. H., Liu, C. J., Cheung, C., and Chen, P. J. (2008). Viral hepatocarcinogenesis: from infection to cancer. Liver Int. 28, 175–188.
Tang, Q., Henriksen, K. J., Bi, M., Finger, E. B., Szot, G., Ye, J., Masteller, E. L., McDevitt, H., Bonyhadi, M., and Bluestone, J. A. (2004). In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J. Exp. Med. 199, 1455–1465.
Tesniere, A., Apetoh, L., Ghiringhelli, F., Joza, N., Panaretakis, T., Kepp, O., Schlemmer, F., Zitvogel, L., and Kroemer, G. (2008). Immunogenic cancer cell death: a key-lock paradigm. Curr. Opin. Immunol. 20, 504–511.
Tiegs, S. L., Russell, D. M., and Nemazee, D. (1993). Receptor editing in self-reactive bone marrow B cells. J. Exp. Med. 177, 1009–1020.
Tortorella, D., Gewurz, B. E., Furman, M. H., Schust, D. J., and Ploegh, H. L. (2000). Viral subversion of the immune system. Annu. Rev. Immunol. 18, 861–926.
Tsuji, M., Komatsu, N., Kawamoto, S., Suzuki, K., Kanagawa, O., Honjo, T., Hori, S., and Fagarasan, S. (2009). Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science 323, 1488–1492.
Tuovinen, H., Salminen, J. T., and Arstila, T. P. (2006). Most human thymic and peripheral-blood CD4+ CD25+ regulatory T cells express 2 T-cell receptors. Blood 108, 4063–4070.
van der Aar, A. M., Sylva-Steenland, R. M., Bos, J. D., Kapsenberg, M. L., de Jong, E. C., and Teunissen, M. B. (2007). Loss of TLR2, TLR4, and TLR5 on Langerhans cells abolishes bacterial recognition. J. Immunol. 178, 1986–1990.
Viglianti, G. A., Lau, C. M., Hanley, T. M., Miko, B. A., Shlomchik, M. J., and Marshak-Rothstein, A. (2003). Activation of autoreactive B cells by CpG dsDNA. Immunity 19, 837–847.
Viglietta, V., Baecher-Allan, C., Weiner, H. L., and Hafler, D. A. (2004). Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J. Exp. Med. 199, 971–979.
Vignali, D. A., Collison, L. W., and Workman, C. J. (2008). How regulatory T cells work. Nat. Rev. Immunol. 8, 523–532.
von Herrath, M. G., and Harrison, L. C. (2003). Antigen-induced regulatory T cells in autoimmunity. Nat. Rev. Immunol. 3, 223–232.
Walker, L. S., and Abbas, A. K. (2002). The enemy within: keeping self-reactive T cells at bay in the periphery. Nat. Rev. Immunol. 2, 11–19.
Ward, S. M., Fox, B. C., Brown, P. J., Worthington, J., Fox, S. B., Chapman, R. W., Fleming, K. A., Banham, A. H., and Klenerman, P. (2007). Quantification and localisation of FOXP3+ T lymphocytes and relation to hepatic inflammation during chronic HCV infection. J. Hepatol. 47, 316–324.
Watanabe, N., Wang, Y. H., Lee, H. K., Ito, T., Cao, W., and Liu, Y. J. (2005). Hassall’s corpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature 436, 1181–1185.
Wayne, J., Suh, H., Misulovin, Z., Sokol, K. A., Inaba, K., and Nussenzweig, M. C. (1994). A regulatory role for recombinase activating genes, RAG-1 and RAG-2, in T cell development. Immunity 1, 95–107.
West, K. L., Singha, N. C., De Ioannes, P., Lacomis, L., Erdjument-Bromage, H., Tempst, P., and Cortes, P. (2005). A direct interaction between the RAG2 C terminus and the core histones is required for efficient V(D)J recombination. Immunity 23, 203–212.
West, M. A., Wallin, R. P., Matthews, S. P., Svensson, H. G., Zaru, R., Ljunggren, H. G., Prescott, A. R., and Watts, C. (2004). Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science 305, 1153–1157.
Winau, F., Weber, S., Sad, S., de Diego, J., Hoops, S. L., Breiden, B., Sandhoff, K., Brinkmann, V., Kaufmann, S. H., and Schaible, U. E. (2006). Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity 24, 105–117.
Wykes, M., Pombo, A., Jenkins, C., and MacPherson, G. G. (1998). Dendritic cells interact directly with naive B lymphocytes to transfer antigen and initiate class switching in a primary T-dependent response. J. Immunol. 161, 1313–1319.
Yamazaki, S., Inaba, K., Tarbell, K. V., and Steinman, R. M. (2006). Dendritic cells expand antigen-specific Foxp3+ CD25+ CD4+ regulatory T cells including suppressors of alloreactivity. Immunol. Rev. 212, 314–329.
Yoshikai, Y., Anatoniou, D., Clark, S. P., Yanagi, Y., Sangster, R., Van den Elsen, P., Terhorst, C., and Mak, T. W. (1984). Sequence and expression of transcripts of the human T-cell receptor beta-chain genes. Nature 312, 521–524.
Zarember, K. A., and Godowski, P. J. (2002). Tissue expression of human toll-like receptors and differential regulation of toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 168, 554–561.
Zenewicz, L. A., Yancopoulos, G. D., Valenzuela, D. M., Murphy, A. J., Stevens, S., and Flavell, R. A. (2008). Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 29, 947–957.
Zheng, Y., Valdez, P. A., Danilenko, D. M., Hu, Y., Sa, S. M., Gong, Q., Abbas, A. R., Modrusan, Z., Ghilardi, N., de Sauvage, F. J., and Ouyang, W. (2008). Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14, 282–289.
Zhou, G., and Levitsky, H. I. (2007). Natural regulatory T cells and de novo-induced regulatory T cells contribute independently to tumor-specific tolerance. J. Immunol. 178, 2155–2162.
Zhou, X., Bailey-Bucktrout, S., Jeker, L. T., and Bluestone, J. A. (2009a). Plasticity of CD4(+) FoxP3(+) T cells. Curr. Opin. Immunol. 21, 281–285.
Zhou, X., Bailey-Bucktrout, S. L., Jeker, L. T., Penaranda, C., Martinez-Llordella, M., Ashby, M., Nakayama, M., Rosenthal, W., and Bluestone, J. A. (2009b). Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 10, 1000–1007.
Zhu, M., Chin, R. K., Christiansen, P. A., Lo, J. C., Liu, X., Ware, C., Siebenlist, U., and Fu, Y. X. (2006). NF-kappaB2 is required for the establishment of central tolerance through an Aire-dependent pathway. J. Clin. Invest. 116, 2964–2971.
Zinkernagel, R. M., Planz, O., Ehl, S., Battegay, M., Odermatt, B., Klenerman, P., and Hengartner, H. (1999). General and specific immunosuppression caused by antiviral T-cell responses. Immunol. Rev. 168, 305–315.
Keywords: ambiguity, homeostasis, inflammation, autoimmunity, cancer
Citation: Barnaba V, Paroli M and Piconese S (2012) The ambiguity in immunology. Front. Immun. 3:18. doi: 10.3389/fimmu.2012.00018
Received: 30 October 2011;
Accepted: 02 February 2012;
Published online: 22 February 2012.
Edited by:
Masaaki Murakami, Osaka University, JapanReviewed by:
Silvano Sozzani, University of Brescia, ItalyArnaud Hot, Hopital Edouard Herriot, France
Copyright: © 2012 Barnaba, Paroli and Piconese. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Vincenzo Barnaba, Dipartimento di Medicina Interna e Specialità Mediche, Sapienza Università di Roma, Viale del Policlinico 155, Rome 00161, Italy. e-mail:dmluY2Vuem8uYmFybmFiYUB1bmlyb21hMS5pdA==