
 Tomohisa Okamura
Tomohisa Okamura
- Department of Allergy and Rheumatology, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan
Autoimmune inflammation including autoantibody-induced inflammation is responsible for the lethal organ damage. Autoantibody-induced inflammation can be separated in two components, autoantibody production, and local inflammatory responses. Accumulating evidence has suggested that regulatory T cells (Treg) control both antibody production and the numbers and functions of effector cells such as innate cells and T helper cells. Autoantibodies are produced by both the follicular and extrafollicular pathways. Recently, follicular regulatory T cells (TFR) and Qa-1 restricted CD8+ Treg were identified as populations that are capable of suppressing follicular T helper cell (TFH)-mediated antibody production. In local inflammation, CD4+CD25+Foxp3+ Treg have the capacity to control inflammation by suppressing cytokine production in T helper cells. Although complement proteins contribute to autoantibody-induced local inflammation by activating innate cells, Treg including CD4+CD25+Foxp3+ Treg are able to suppress innate cells, chiefly via IL-10 production. IL-10-secreting T cells such as T regulatory type I (Tr1) and Tr1-like cells might also play roles in the control of Th17 and innate cells. Therefore, several kinds of Tregs have the potential to control autoimmune inflammation by suppressing both autoantibody production and the local inflammatory responses induced by autoantibodies.
Introduction
Autoimmune inflammation is responsible for the lethal organ damage, and autoantibodies play a pivotal role in triggering inflammation. Immune complexes are readily detectable in the articular tissues of rheumatoid arthritis (RA) patients and the glomeruli of systemic lupus erythematosus (SLE) patients. These immune complexes are regarded as important players in the pathogeneses of these diseases as they initiate and maintain the inflammatory cascade and tissue destruction. In addition, immune complex deposition in articular tissue has been reported to have harmful effects in many experimental models of arthritis. The passive transfer of antibodies to an autoantigen that is found in the joints is an established method for inducing arthritis. For example, antibodies to glucose-6-phosphate isomerase (GPI), a ubiquitous cytoplasmic enzyme, induce spontaneous arthritis upon injection into susceptible mice (Matsumoto et al., 2002), and the administration of a cocktail of anti-type II collagen antibodies to DBA/1 mice also invokes severe arthritis (Terato et al., 1992). However, it is particularly notable that these antibody-induced arthritis models are transient and fail to achieve chronicity (Myers et al., 1997), which suggests that a continuous supply of autoantibodies is required for the development of chronic joint inflammation.
In contrast to arthritis models, there are no lupus models that are invoked by the passive transfer of autoantibodies. However, plasma cell-depletion by a proteasome inhibitor clearly demonstrated the importance of a continuous supply of autoantibodies for systemic autoimmunity; i.e., treatment with bortezomib, a proteasome inhibitor, depleted the number of plasma cells producing antibodies to double stranded DNA; eliminated autoantibody production; ameliorated glomerulonephritis; and prolonged the survival of two lupus-prone mice strains, NZB/W F1, and MRL/lpr mice. Among five bortezomib-treated mice that displayed proteinuria of >100 mg/dl before treatment, four showed proteinuria of <100 mg/dl after treatment. These findings suggest that the suppression of autoantibody production leads to reduced organ inflammation in lupus (Neubert et al., 2008).
In addition to the production of autoantibodies by B cells, antibody-induced inflammation by itself is another target of therapeutic intervention. In mouse models of arthritis, the synthesized immune complexes bind to “inflammatory” Fc-receptors on intra-articular cells and then activate complement protein (Rowley et al., 2008). Complement fragments bound to immune complexes induce tissue injury, and FcR stimulation cumulatively activates mononuclear cells in situ, causing the activated cells to release pro-inflammatory cytokines. In turn, these responses attract neutrophils and macrophages, which can damage synoviocytes and chondrocytes. As mentioned above, immune complex-induced arthritis is a prototypic inflammatory process that is characterized by the release of pro-inflammatory cytokines and the activation of degradative enzymes. In a GPI-induced arthritis model, it was found that anti-GPI autoantibodies act through FcγRIII receptors and C5a (Ji et al., 2002). Micro-positron emission tomography studies revealed that the localization of anti-GPI antibodies is dependent on mast cells, neutrophils, Fc-receptors, and immune complexes (Mandik-Nayak and Allen, 2005). In anti-collagen antibody-induced arthritis (CAIA), complement activation, and innate cells are also critical for the effector phase of arthritis (Hietala et al., 2004; Daha et al., 2011). With regard to systemic autoimmunity, MRL/lpr mice lacking factor B or factor D developed less severe nephritis than the control mice (Watanabe et al., 2000; Elliott et al., 2004). Factor B is cleaved by factor D and resulting catalytic subunit Bb forms C3 convertase. In addition, the anti-double stranded DNA antibody titer is not altered by factor D deficiency, indicating that complement activation is not required for the production of autoantibodies in MRL/lpr mice. Activated complement interacts with Fcγ receptors and complement receptors on innate effector cells (such as macrophages and monocytes) to induce local inflammation (Wasowska, 2010).
Therefore, autoantibody-induced inflammation can be separated into two components, autoantibody production and the local inflammatory response. Recently, accumulating evidence has shown that regulatory T cells (Treg) control both antibody production and the numbers and functions of effector cells such as innate cells and T helper cells. This article will discuss the Treg-mediated suppression of these two components during inflammation (Figure 1).
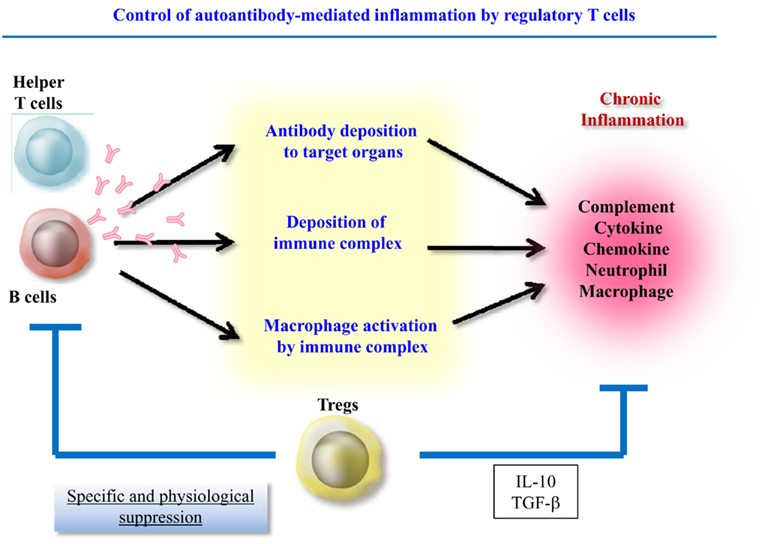
Figure 1. Control of autoantibody-induced inflammation by regulatory T cells.
Treg-Mediated Suppression of Autoantibody Production
Two Mechanisms for Autoantibody Production
In the course of thymus-dependent responses, B cells interact with T cells in the outer T cell zones of the lymphoid organs and differentiate along either the follicular or extrafollicular pathway (Lee et al., 2011). In the follicular pathway, activated B cells form germinal centers (GC) and undergo somatic hypermutation and selection. Subsequently, they exit GC as high-affinity long-lived plasma cells or memory B cells. In the extrafollicular pathway, B cells migrate to splenic bridging channels or junction zones and the borders between T cell zones and the red pulp or extramedullary lymph node cords. These migrated B cells form clusters of short-lived plasmablasts. Thus both the follicular and extrafollicular pathways contribute to autoantibody production in murine disease models.
Extrafollicular B cell response-mediated autoantibody production
William et al. (2002) observed that the splenic autoreactive B cells of autoimmune MRL/lpr mice proliferated and undergo active somatic hypermutation at the T zone-red pulp border rather than in GC. They examined the extrafollicular generation of plasmablasts in AM14 VH transgenic (Tg) mice, which possess rheumatoid factor (RF)-producing B cells with moderate affinity for IgG2a. Intriguingly, AM14 B cells on the MRL/lpr background spontaneously differentiate into extrafollicular plasmablasts and undergo somatic hypermutation at the T zone/red pulp border. In addition, they reported that the extrafollicular plasmablast response is induced by the administration of IgG2a anti-chromatin antibodies, which presumably form immune complexes in vivo with endogenous chromatin (Herlands et al., 2007). This response was found to be T cell independent, although it was totally dependent on MyD88 signaling downstream of Toll-like receptor 7 (TLR7) and TLR9 (Herlands et al., 2008). However, another study revealed that although AM14 B cells can be activated, differentiate, and undergo isotype-switching independent of antigen-specific T helper cells, T cells dramatically enhance the AM14 B cell response via CD40L and IL-21 signaling (Sweet et al., 2011).
GC-mediated autoantibody production
Because affinity-enhancing somatic hypermutations are prevalent in autoantibodies, it has long been hypothesized that these autoantibodies are derived from GC. Mouse strains that frequently develop autoimmune diseases (NZB/W F1, BXSB, MRL/lpr, sanroque, and NOD mice) spontaneously form GC-like structures in their spleens, and the onset of autoantibody production correlates with GC formation. Recently, several pieces of evidence have suggested that dysregulated T follicular helper (TFH) cells significantly contribute to autoimmunity by inducing the aberrant selection of autoreactive B cells. The lupus-like disease that occurs in sanroque mice is caused by Roquinsan/san-induced accumulation of TFH cells that maintain spontaneously formed GC (Vinuesa et al., 2005). The glomerulonephritis and pathogenic autoantibody production displayed by sanroque mice are ameliorated by Bcl6 haploinsufficiency (Linterman et al., 2009). Moreover, SLAM-associated protein (SAP) deficiency experiments have highlighted the important roles played by TFH cells in the conditions suffered by sanroque mice. SAP interacts with a conserved tyrosine-based motif that is found in the cytoplasmic tail of SLAM family members, and Sh2d1a (the gene for SAP) deficiency abrogates TFH formation and GC responses, but not extrafollicular antibody responses. Since SAP deficiency ameliorates the lupus-like phenotype of sanroque mice, it can be assumed that aberrant TFH cell activation is responsible for the autoimmunity that they display. BXSB mice develop a severe form of lupus caused by the yaa locus, which induces the overexpression of a cluster of X-linked genes that includes Tlr7 gene. Although B6. yaa mice are not overtly autoimmune, the introduction of Sle1, which contains the autoimmune-predisposing Slam/Cd2 haplotype, into their genome causes them to develop fetal lupus (Subramanian et al., 2006). Intriguingly, CD4+ T cells from B6. Sle1.yaa mice develop the molecular signature of TFH cells and also show altered expression levels of various cytokines and chemokines.
The source of human autoantibodies revealed by B cell-depletion therapy
As discussed above, dysregulation of the follicular or extrafollicular pathway can cause systemic autoimmune disease in mice. However, the contributions of follicular and extrafollicular checkpoints to the production of disease-associated autoantibodies are more difficult to evaluate in humans than in mice. Levels of autoantibodies do not always correlate with disease activity and response to treatment. For example, the serum concentrations of some autoantibodies correlate with disease activity (i.e., anti-double stranded DNA antibodies and anti-PR3 antibodies), while the titers of other autoantibodies [i.e., anti-ribonucleoprotein (RNP) antibodies and anti-Ro and La antibodies] remain stable irrespective of disease status. The heterogeneous autoantibody effects have also been observed in patients treated with anti-CD20 monoclonal antibody, which depletes B cells and plasmablasts but not long-lived plasma cells (Cambridge et al., 2003, 2006; Lu et al., 2009). In lupus patients, the levels of anti-nucleosome and anti-double stranded DNA antibodies are significantly decreased at 6–8 months after the administration of anti-CD20 monoclonal antibody. In contrast, the same treatment does not significantly alter the levels of anti-Ro, Sm, or RNP antibodies (Cambridge et al., 2006). This suggests that anti-nucleosome and anti-double stranded DNA antibodies are produced through extrafollicular responses, which usually generate short-lived plasma cells, while antibodies to nucleic acid-associated antigens (Ro, Sm, and RNP) are derived from follicular responses, which generate long-lived plasma cells. In RA patients, the levels of IgA-RF, IgG-RF, and IgG anti-cyclic citrullinated peptide (CCP) antibodies are decreased at 6 months after the administration of anti-CD20 monoclonal antibody, and the decreases are proportionately greater than the decreases in their respective total immunoglobulin classes (Cambridge et al., 2003). Plasmablasts and short-lived plasma cells originating from the extrafollicular response might be the major source of RF and anti-CCP antibodies (Looney et al., 2008). Therefore, both extrafollicular- and follicular-mediated antibody productions should be controlled in the treatment of human autoimmune inflammation.
Approaches to Autoantibody Suppression
Antibody suppression with CD4+CD25+ Foxp3+ Treg
In general, T cells are indispensable sources of help signals, which promote B cell antibody production. Therefore, control of antibody production at the level of T cells is a rational approach to autoantibody suppression. Indeed, several T cell populations are able to suppress B cell antibody production. In humans, CD4+CD25+CD69− Treg that suppress antibody production in vitro have been found in GC. The fact that these CD4+CD25+CD69− Treg hardly express CXCR5 suggests that they mainly reside in the T cell-rich zones of secondary lymphoid tissues (Lim et al., 2004). However, T cell activation switches their chemokine receptor expression pattern from CCR7 to CXCR5 and switches their chemotactic responses from CCL19 to CXCL13. Thus, activation might change the migratory behavior of CD4+CD25+CD69− Treg so that they can migrate to GC. After migrating to GC, CD4+CD25+CD69− Treg negatively regulate T cell-dependent B cell responses through their suppressive activity toward T cells.
The preferential killing of antigen-presenting B cells by CD4+CD25+ Treg was reported in C57BL/6 mice (Zhao et al., 2006). B cell death is not mediated by the Fas–Fas ligand pathway, but instead is mediated by a granzyme-dependent, partially perforin-dependent pathway. Direct suppression of B cells by Treg was also reported in chronic systemic autoimmunity (Iikuni et al., 2009). For example, CD4+CD25+ Treg have been demonstrated to inhibit B cell antibody production in in vitro models of murine and human lupus. Treg use granule exocytosis pathways involving perforin and granzyme to induce contact-dependent apoptosis in B cells. However, in spite of the fact that CD4+CD25+ Treg from both young and old NZB/W F1 mice retain a capacity to suppress IgG production in B cells, autoantibodies continuously accumulate in these mice. Therefore, whether CD4+CD25+ Treg could be used to efficiently control autoantibody production in systemic autoimmunity needs to be examined further.
Recently, several groups simultaneously identified mice CD4+CD25+Foxp3+ Treg subpopulations that are able to suppress B cell antibody production (Chung et al., 2011; Linterman et al., 2011;Wollenberg et al., 2011). Chung et al. (2011) identified a subset of Treg cells that express CXCR5 and Bcl6 and localize to GC in mice and humans. The expression of CXCR5 on Treg depends on Bcl6, and CXCR5+Bcl6+ Treg are absent from the thymus but can be generated from CXCR5−Foxp3+ natural Treg precursors. A deficiency of CXCR5+ Treg results in enhanced GC reactions involving B cells, affinity maturation of antibodies, and plasma cell differentiation. These results demonstrated that the Bcl6–CXCR5 axis of Treg is one mechanism by which GC responses are controlled. In addition, they observed that Foxp3-mutated scurfy mice display a moderate increase in their TFH population but a markedly increased number of GL7+CD95+ B cells. Collectively, these observations suggest that Foxp3+ follicular regulatory (TFR) cells are more specialized for controlling the generation of GC B cells. Linterman et al. (2011) also described a population of Foxp3+Blimp-1+CD4+ T cells that accounted for 10–25% of the CXCR5highPD-1highCD4+ T cells found in immunized GC. In the absence of these TFR cells, they noted outgrowths of non-antigen-specific B cells in GC and a decreased number of antigen-specific B cells. Therefore, both groups revealed that TFR play a role in controlling GC reactions by inhibiting the selection of antigen-specific- and non-specific B cells. Because CXCR5-expressiong TFR localize to GC, TFR may suppress GC-mediated autoantibody production. However, whether TFR actually suppress autoantibody production and whether TFR deficiency results in autoimmunity remain to be addressed.
Antibody suppression with Qa-1 restricted CD8+ Treg and other Treg subsets
A recent study reported that Qa-1 restricted CD8+ Treg cells directly inhibit Qa-1+ TFH cells. Qa-1 is a non-classical MHC class Ib molecule presenting a peptide derived from the signal sequence of classical MHC class I proteins, named Qa-1 determinant modifier (Qdm), as well as peptides derived from proteins associated with infectious or inflammatory responses (Lu et al., 2006). Previously, a subpopulation of CD8+ T cells was reported to suppress T cell help to B cells (Noble et al., 1998), and subsequent studies have shown that Qa-1 restricted CD8+ T cells inhibit experimental autoimmune encephalomyelitis (EAE) by targeting autoreactive CD4+ cells (Hu et al., 2004). Nevertheless, although Qa-1 deficient mice showed dysregulated immune responses to immunization with self and foreign antigens, Qa-1−/− mice do not develop spontaneous autoimmunity. Since Qa-1 interacts with both the T cell receptor (TCR) on CD8+ T cells and the CD94/NKG2A receptor expressed by activated CD4+ T cells, Qa-1 knock-in mice, B6 Qa-1(D227K) mice, were generated. B6 Qa-1 (D227K) mice harbor a Qa-1 amino acid exchange mutation that disrupts the binding of Qa-1 to the TCR/CD8 complex, but has no effect on its binding to the inhibitory NKG2A receptor. Intriguingly, the B6 Qa-1 (D227K) mice exhibit lupus-like systemic autoimmune disease and a fivefold to sixfold increase in their numbers of TFH cells (Kim et al., 2010).
Analysis of the surface phenotype of Qa-1 restricted CD8+ Treg indicated that they express CD44, ICOSL, and CXCR5 and the CD44+ICOSL+CD8+ T cells inhibit the generation of high-affinity antibodies and Qa-1+ TFH cells. This observation provides a clue that might greatly increase our understanding of autoantibody production. However, the antigen-specificity of Qa-1 restricted CD8+ Treg during TFH cell suppression remains unclear because the repertoire of peptides presented by Qa-1 is substantially smaller than the repertoire of classical MHC molecules (Lu et al., 2006). Only a small number of peptides have been identified that bind to Qa-1 and stimulate CD8+ T cells, including dominant Qdm as well as peptides from HSP60, insulin, Salmonella GroEL, and TCR Vβ chains. Thus, Qa-1 restricted CD8+ Treg might suppress TFH cells irrespective of the antigen-specificity of the TCR on TFH cells. Because Qa-1 restricted CD8+ Treg express CXCR5 and migrate to lymphoid follicles (Kim et al., 2010), Qa-1 restricted CD8+ Treg may suppress GC-mediated autoantibody production.
TFR and Qa-1 restricted CD8+ Treg appear to be important checking mechanisms for antibody production. However, no Treg populations that control autoantibody production and autoimmunity in an antigen-specific manner have yet been identified. Although the importance of T regulatory type I (Tr1) cells for controlling immune responses has been described in a number of reports, anti-CD46-induced IL-10-secreting T cells even enhance antibody production by B cells (Fuchs et al., 2009). Recently, several CD4+ T cell populations that possess regulatory activity have been identified (Fujio et al., 2010). CD4+CD25−LAP+ T cells and CD4+NKG2D+ T cells produce both IL-10 and TGF-β (Oida et al., 2003; Dai et al., 2009), and CD4+CD25−IL-7R− T cells and CD4+CD25−LAG3+ T cells produce large amounts of IL-10 (Haringer et al., 2009; Okamura et al., 2009). The association between these recently identified Treg and antigen-specific autoantibody suppression should be investigated. In particular, CD4+CD25−LAG3+ T cells, which characteristically express the anergy-linked transcription factor Egr2, might be associated with autoantibody suppression, because T cell-specific Egr2-deficient mice exhibit lupus-like disease (Zhu et al., 2008) and polymorphisms in the EGR2 gene are associated with human SLE susceptibility (Myouzen et al., 2010). Although both TFR cells and Qa-1 restricted CD8+ Treg express CXCR5 and may suppress GC-mediated autoantibody production, Treg populations which suppress extrafollicular response are yet to be identified.
Treg-Mediated Suppression of Local Inflammation
IL-10-Mediated Suppression of Inflammation
Nguyen et al. (2007) reported a role of CD4+CD25+Foxp3+ Treg in antibody-induced arthritis at several levels. They examined the effect of the scurfy loss of function mutation of the Foxp3 gene in K/BxN mouse model. These mice carry the KRN transgene, which encodes a TCR reactive against a peptide from GPI and the autoreactive T cells promote the production of vast quantities of anti-GPI antibodies, which are sufficient to induce arthritis after transfer into normal recipients (Korganow et al., 1999). The absence of CD4+CD25+Foxp3+ Treg led to more accelerated aggressive arthritis with significantly earlier autoantibody production. However, the broadened spectrum of affected joints in Foxp3-mutated K/BxN mice was not due to the earlier appearance of autoantibodies and could not be reproduced by increasing anti-GPI antibody load. Therefore, CD4+CD25+Foxp3+ Treg are supposed to play a role in effector phase manifestations. Their another observation that Foxp3+ Treg accumulated in inflamed joint of K/BxN serum-transferred B6 mice suggested that Foxp3+ Treg actively migrate to the site of antibody-induced inflammation and control the local inflammatory process. Although the mechanism of this Foxp3+ Treg-mediated suppression was not clarified, IL-10 was mentioned as a candidate mediator.
Furthermore, a single transfer of CD4+CD25+ Treg markedly slowed the progression of collagen-induced arthritis (CIA), which could not be attributed to the loss of systemic type II collagen-specific T and B cell responses (Morgan et al., 2005). The transferred CD4+CD25+ Treg were found in the inflamed synovium soon after the transfer, indicating that regulation occurs locally in the joints. It is unlikely that the transferred CD4+CD25+ Treg acted against CIA solely via the suppression of T cell immunity involving the Th17 cell response since the effector phase of CIA depends on T cell-independent immune responses (Ehinger et al., 2001). Thus, the transferred CD4+CD25+ Treg might have interacted with local innate cells as well as effector T cells.
CD4+CD25+ Treg-mediated control of innate cells was found to be IL-10 and TGF-β dependent in a colitis model (Maloy et al., 2003). Indeed, IL-10 production might be a key factor controlling local inflammation. Human IL-10 suppresses the expression of MHC class II, co-stimulatory, and adhesion molecules (De Waal Malefyt et al., 1991; Willems et al., 1994). IL-10 also inhibits the production of inflammatory cytokines and the T cell stimulating capacity of antigen-presenting cells (APC; Fiorentino et al., 1991; Allavena et al., 1998), and local IL-10 production has been shown to suppress TNF-α and IL-1α production (Lubberts et al., 2000). CD4+CD25+ Treg downregulate the expression of co-stimulatory molecules on APC (Cederbom et al., 2000) and restrain the maturation and antigen-presenting function of dendritic cells in an IL-10-dependent manner (Misra et al., 2004; Houot et al., 2006). Furthermore, IL-10 was recently reported to suppress Th17 cells (Huber et al., 2011). Interestingly, both CD4+Foxp3+ Treg and CD4+Foxp3−IL-10-producing cells (Tr1) are able to control Th17 cell numbers in an IL-10-dependent manner. Therefore, it was suggested that Tr1 cells can compensate for a paucity of Foxp3+ Treg and vice versa during the suppression of innate and Th17 cells.
Tr1 cells are considered to be different from Th1, Th2, and Th17 cells based on their cytokine production profile; i.e., they secrete high levels of IL-10. Tr1 cells are inducible in vitro and in vivo, and they can also be isolated from humans and mice in steady state conditions (Roncarolo et al., 2011). Tr1 cells are able to suppress Th1-mediated colitis induced by the transfer of naïve CD4+CD45RBhi cells into SCID mice as well as EAE (Roncarolo et al., 2001). Although few reports have directly compared IL-10 production between CD4+CD25+ Treg and Tr1-like cells, CD4+CD25−LAG3+ Treg secrete significantly higher amounts of IL-10 than CD4+CD25+ Treg (Okamura et al., 2009). Thus, Tr1 cells and Tr1-like cells might have the ability to control innate immune cells.
Suppression of T Cell Cytokine Production
In several antibody-induced autoimmune inflammations such as RA-synovitis and lupus nephritis, co-existence of antibody deposition and T cell infiltration is frequently observed. RA is a prototypic autoimmune disease characterized by chronic joint inflammation and the production of cytokines, including TNF-α, IL-6, IL-15, IL-17, and IL-1β. These cytokines are thought to be derived from both innate cells and effector T cells. In the K/BxN arthritis model, T cells can augment antibody-induced arthritis independently of their influence on antibody production (Jacobs et al., 2009). This enhancement was mediated by IL-17 producing CD4+ T cells preferentially recruited to the environment of the arthritic joint. Therefore, Treg-mediated suppression of effector T cells may be also beneficial in controlling autoantibody-induced inflammation accompanied with T cell infiltration. In the past, IFN-γ producing Th1 cells were thought to be the principal mediators of autoimmune inflammation such as that observed in RA. However, IL-17 has emerged as a key driver of inflammation and is detectable in the RA synovium. IL-17 and IL-17F promote inflammation on several levels, as their receptors IL-17RA and IL-17RC are expressed on both hematopoietic and non-hematopoietic cells. IL-17 and IL-17F induce the production of pro-inflammatory cytokines like IL-6, IL-1β, and TNF-α, and pro-inflammatory chemokines such as CXCL1, GCP-2, and IL-8 and thus promote tissue inflammation and neutrophil recruitment at sites of inflammation (Bettelli et al., 2008).
CD4+CD25+ Treg not only suppress the proliferation of conventional T cells, but also their production of inflammatory cytokines, such as TNF-α and IFN-γ. In contrast, IL-17 production is not suppressed when human CD4+CD25+ Treg are added to responder T cells in vitro (Annunziato et al., 2008; Flores-Borja et al., 2008), and murine CD4+CD25+ Treg promote Th17 cell development both in vitro and in vivo (Chen et al., 2011; Pandiyan et al., 2011). As IL-17 is important for infection control, the resistance of Th17 cells to suppression by CD4+CD25+ Treg cells makes sense. However, in a previous study CD4+CD25+Foxp3+ Treg-specific ablation of STAT3 resulted in the development of fetal intestinal inflammation due to the loss of Th17 cell suppression in mice (Chaudhry et al., 2009). Moreover, other studies have suggested that some subpopulations of CD4+CD25+ Treg cells are capable of regulating Th17 cell responses. For example, CD4+CD25+ Treg expressing CD39 (an ectonucleotidase that hydrolyzes ATP) were reported to be able to suppress Th17 cell responses (Fletcher et al., 2009). In addition, CD4+CD25+CD39+ Treg numbers are reduced in patients with multiple sclerosis (MS), suggesting that an association exists between this Treg population and the suppression of pathogenic Th17 cells. Therefore, at least some CD4+CD25+ Treg are suspected to suppress the production of inflammatory cytokines in inflamed organs.
EAE is an animal model of MS that is induced by the injection of myelin components. Until recently, the pathogeneses of MS and EAE were thought to be initiated by myelin-specific Th1 cells. However, a number of lines of evidence have indicated that Th17 cells induce central nervous system (CNS) inflammation (Oukka, 2007). For example, it was reported that the Th17:Th1 ratio of infiltrating T cells in EAE determines where inflammation occurs in the CNS (Stromnes et al., 2008), and T cell infiltration and inflammation in the brain parenchyma only occur when Th17 cells outnumber Th1 cells and trigger a disproportionate increase in IL-17 expression in the brain. In contrast, T cells showing a wide range of Th17:Th1 ratios induce spinal cord parenchymal inflammation. Tg mice bearing a TCR against the myelin basic protein (MBP) that had been crossed with recombination-activating gene 1 (Rag1)-deficient mice (Tg MBP/Rag−/−) developed spontaneous EAE, whereas Tg MBP/Rag+/+ mice did not (Lafaille et al., 1994). This discrepancy can be explained by the existence of Treg in the Rag+/+ mice but not the Rag−/− mice because the adoptive transfer of CD4+CD25+ Treg from wild-type mice to Tg MBP/Rag−/− mice prevented the development of spontaneous EAE (Hori et al., 2002). Moreover, adoptive transfer experiments have revealed that transferring large numbers of CD4+CD25+ Treg purified from the peripheral lymph nodes of naive mice reduced the incidence and severity of EAE (Kohm et al., 2003). In a study conducted by Matsumoto et al. (2007), peripheral CD4+CD25+ Treg from mice with EAE suppressed the development of chronic EAE in the recipient rats. Therefore, CD4+CD25+Foxp3+ Treg apparently have the capability to suppress T helper cell-mediated organ inflammation, and this effect may be beneficial in the control of the antibody-induced inflammation accompanied with effector T cell infiltration.
Conclusion
The current standard treatment for autoimmune disease is non-specific immunosuppression with steroids and immunosup-pressants, which inevitably leads to opportunistic infections. As autoantibodies are key components in the development of autoimmune inflammation, targeting autoantibody-induced immunity is a rational approach to the treatment of autoimmune diseases. The modulation of Treg function is a promising physiological approach to suppressing both autoantibody production and autoantibody-induced local inflammation. Further examinations of CD4+CD25+ Treg and other Treg subsets are necessary in future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the Japan Society for the Promotion of Science, Ministry of Health, Labor and Welfare, and the Ministry of Education, Culture, Sports, Science, and Technology (MEXT; in part by Global COE Program Chemical Biology of the Diseases, by the MEXT), Japan.
References
Allavena, P., Piemonti, L., Longoni, D., Bernasconi, S., Stoppacciaro, A., Ruco, L., and Mantovani, A. (1998). IL-10 prevents the differentiation of monocytes to dendritic cells but promotes their maturation to macrophages. Eur. J. Immunol. 28, 359–369.
Annunziato, F., Cosmi, L., Liotta, F., Maggi, E., and Romagnani, S. (2008). The phenotype of human Th17 cells and their precursors, the cytokines that mediate their differentiation and the role of Th17 cells in inflammation. Int. Immunol. 20, 1361–1368.
Bettelli, E., Korn, T., Oukka, M., and Kuchroo, V. K. (2008). Induction and effector functions of T(H)17 cells. Nature 453, 1051–1057.
Cambridge, G., Leandro, M. J., Edwards, J. C., Ehrenstein, M. R., Salden, M., Bodman-Smith, M., and Webster, A. D. (2003). Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis. Arthritis Rheum. 48, 2146–2154.
Cambridge, G., Leandro, M. J., Teodorescu, M., Manson, J., Rahman, A., Isenberg, D. A., and Edwards, J. C. (2006). B cell depletion therapy in systemic lupus erythematosus: effect on autoantibody and antimicrobial antibody profiles. Arthritis Rheum. 54, 3612–3622.
Cederbom, L., Hall, H., and Ivars, F. (2000). CD4+CD25+ regulatory T cells down-regulate co-stimulatory molecules on antigen-presenting cells. Eur. J. Immunol. 30, 1538–1543.
Chaudhry, A., Rudra, D., Treuting, P., Samstein, R. M., Liang, Y., Kas, A., and Rudensky, A. Y. (2009). CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 326, 986–991.
Chen, Y., Haines, C. J., Gutcher, I., Hochweller, K., Blumenschein, W. M., Mcclanahan, T., Hammerling, G., Li, M. O., Cua, D. J., and Mcgeachy, M. J. (2011). Foxp3(+) regulatory T cells promote T helper 17 cell development in vivo through regulation of interleukin-2. Immunity 34, 409–421.
Chung, Y., Tanaka, S., Chu, F., Nurieva, R. I., Martinez, G. J., Rawal, S., Wang, Y. H., Lim, H., Reynolds, J. M., Zhou, X. H., Fan, H. M., Liu, Z. M., Neelapu, S. S., and Dong, C. (2011). Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med. 17, 983–988.
Daha, N. A., Banda, N. K., Roos, A., Beurskens, F. J., Bakker, J. M., Daha, M. R., and Trouw, L. A. (2011). Complement activation by (auto-) antibodies. Mol. Immunol. 48, 1656–1665.
Dai, Z., Turtle, C. J., Booth, G. C., Riddell, S. R., Gooley, T. A., Stevens, A. M., Spies, T., and Groh, V. (2009). Normally occurring NKG2D+CD4+ T cells are immunosuppressive and inversely correlated with disease activity in juvenile-onset lupus. J. Exp. Med. 206, 793–805.
De Waal Malefyt, R., Haanen, J., Spits, H., Roncarolo, M. G., Te Velde, A., Figdor, C., Johnson, K., Kastelein, R., Yssel, H., and De Vries, J. E. (1991). Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J. Exp. Med. 174, 915–924.
Ehinger, M., Vestberg, M., Johansson, A. C., Johannesson, M., Svensson, A., and Holmdahl, R. (2001). Influence of CD4 or CD8 deficiency on collagen-induced arthritis. Immunology 103, 291–300.
Elliott, M. K., Jarmi, T., Ruiz, P., Xu, Y., Holers, V. M., and Gilkeson, G. S. (2004). Effects of complement factor D deficiency on the renal disease of MRL/lpr mice. Kidney Int. 65, 129–138.
Fiorentino, D. F., Zlotnik, A., Mosmann, T. R., Howard, M., and O’Garra, A. (1991). IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 147, 3815–3822.
Fletcher, J. M., Lonergan, R., Costelloe, L., Kinsella, K., Moran, B., O’Farrelly, C., Tubridy, N., and Mills, K. H. (2009). CD39+Foxp3+ regulatory T cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J. Immunol. 183, 7602–7610.
Flores-Borja, F., Jury, E. C., Mauri, C., and Ehrenstein, M. R. (2008). Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc. Natl. Acad. Sci. U.S.A. 105, 19396–19401.
Fuchs, A., Atkinson, J. P., Fremeaux-Bacchi, V., and Kemper, C. (2009). CD46-induced human Treg enhance B-cell responses. Eur. J. Immunol. 39, 3097–3109.
Fujio, K., Okamura, T., and Yamamoto, K. (2010). The family of IL-10-secreting CD4+ T cells. Adv. Immunol. 105, 99–130.
Haringer, B., Lozza, L., Steckel, B., and Geginat, J. (2009). Identification and characterization of IL-10/IFN-gamma-producing effector-like T cells with regulatory function in human blood. J. Exp. Med. 206, 1009–1017.
Herlands, R. A., Christensen, S. R., Sweet, R. A., Hershberg, U., and Shlomchik, M. J. (2008). T cell-independent and toll-like receptor-dependent antigen-driven activation of autoreactive B cells. Immunity 29, 249–260.
Herlands, R. A., William, J., Hershberg, U., and Shlomchik, M. J. (2007). Anti-chromatin antibodies drive in vivo antigen-specific activation and somatic hypermutation of rheumatoid factor B cells at extrafollicular sites. Eur. J. Immunol. 37, 3339–3351.
Hietala, M. A., Nandakumar, K. S., Persson, L., Fahlen, S., Holmdahl, R., and Pekna, M. (2004). Complement activation by both classical and alternative pathways is critical for the effector phase of arthritis. Eur. J. Immunol. 34, 1208–1216.
Hori, S., Haury, M., Coutinho, A., and Demengeot, J. (2002). Specificity requirements for selection and effector functions of CD25+4+ regulatory T cells in anti-myelin basic protein T cell receptor transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 99, 8213–8218.
Houot, R., Perrot, I., Garcia, E., Durand, I., and Lebecque, S. (2006). Human CD4+CD25high regulatory T cells modulate myeloid but not plasmacytoid dendritic cells activation. J. Immunol. 176, 5293–5298.
Hu, D., Ikizawa, K., Lu, L., Sanchirico, M. E., Shinohara, M. L., and Cantor, H. (2004). Analysis of regulatory CD8 T cells in Qa-1-deficient mice. Nat. Immunol. 5, 516–523.
Huber, S., Gagliani, N., Esplugues, E., O’Connor, W. Jr., Huber, F. J., Chaudhry, A., Kamanaka, M., Kobayashi, Y., Booth, C. J., Rudensky, A. Y., Roncarolo, M. G., Battaglia, M., and Flavell, R. A. (2011). Th17 cells express interleukin-10 receptor and are controlled by Foxp3 and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 34, 554–565.
Iikuni, N., Lourenco, E. V., Hahn, B. H., and La Cava, A. (2009). Cutting edge: regulatory T cells directly suppress B cells in systemic lupus erythematosus. J. Immunol. 183, 1518–1522.
Jacobs, J. P., Wu, H. J., Benoist, C., and Mathis, D. (2009). IL-17-producing T cells can augment autoantibody-induced arthritis. Proc. Natl. Acad. Sci. U.S.A. 106, 21789–21794.
Ji, H., Ohmura, K., Mahmood, U., Lee, D. M., Hofhuis, F. M., Boackle, S. A., Takahashi, K., Holers, V. M., Walport, M., Gerard, C., Ezekowitz, A., Carroll, M. C., Brenner, M., Weissleder, R., Verbeek, J. S., Duchatelle, V., Degott, C., Benoist, C., and Mathis, D. (2002). Arthritis critically dependent on innate immune system players. Immunity 16, 157–168.
Kim, H. J., Verbinnen, B., Tang, X., Lu, L., and Cantor, H. (2010). Inhibition of follicular T-helper cells by CD8(+) regulatory T cells is essential for self tolerance. Nature 467, 328–332.
Kohm, A. P., Carpentier, P. A., and Miller, S. D. (2003). Regulation of experimental autoimmune encephalomyelitis (EAE) by CD4+CD25+ regulatory T cells. Novartis Found. Symp. 252, 45–52; discussion 52–54, 106–114.
Korganow, A. S., Ji, H., Mangialaio, S., Duchatelle, V., Pelanda, R., Martin, T., Degott, C., Kikutani, H., Rajewsky, K., Pasquali, J. L., Benoist, C., and Mathis, D. (1999). From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity 10, 451–461.
Lafaille, J. J., Nagashima, K., Katsuki, M., and Tonegawa, S. (1994). High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell 78, 399–408.
Lee, S. K., Rigby, R. J., Zotos, D., Tsai, L. M., Kawamoto, S., Marshall, J. L., Ramiscal, R. R., Chan, T. D., Gatto, D., Brink, R., Yu, D., Fagarasan, S., Tarlinton, D. M., Cunningham, A. F., and Vinuesa, C. G. (2011). B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J. Exp. Med. 208, 1377–1388.
Lim, H. W., Hillsamer, P., and Kim, C. H. (2004). Regulatory T cells can migrate to follicles upon T cell activation and suppress GC-Th cells and GC-Th cell-driven B cell responses. J. Clin. Invest. 114, 1640–1649.
Linterman, M. A., Pierson, W., Lee, S. K., Kallies, A., Kawamoto, S., Rayner, T. F., Srivastava, M., Divekar, D. P., Beaton, L., Hogan, J. J., Fagarasan, S., Liston, A., Smith, K. G., and Vinuesa, C. G. (2011). Foxp3(+) follicular regulatory T cells control the germinal center response. Nat. Med. 17, 975–982.
Linterman, M. A., Rigby, R. J., Wong, R. K., Yu, D., Brink, R., Cannons, J. L., Schwartzberg, P. L., Cook, M. C., Walters, G. D., and Vinuesa, C. G. (2009). Follicular helper T cells are required for systemic autoimmunity. J. Exp. Med. 206, 561–576.
Looney, R. J., Srinivasan, R., and Calabrese, L. H. (2008). The effects of rituximab on immunocompetency in patients with autoimmune disease. Arthritis Rheum. 58, 5–14.
Lu, L., Werneck, M. B., and Cantor, H. (2006). The immunoregulatory effects of Qa-1. Immunol. Rev. 212, 51–59.
Lu, T. Y., Ng, K. P., Cambridge, G., Leandro, M. J., Edwards, J. C., Ehrenstein, M., and Isenberg, D. A. (2009). A retrospective seven-year analysis of the use of B cell depletion therapy in systemic lupus erythematosus at University College London Hospital: the first fifty patients. Arthritis Rheum. 61, 482–487.
Lubberts, E., Joosten, L. A., Van Den Bersselaar, L., Helsen, M. M., Bakker, A. C., Xing, Z., Richards, C. D., and Van Den Berg, W. B. (2000). Intra-articular IL-10 gene transfer regulates the expression of collagen-induced arthritis (CIA) in the knee and ipsilateral paw. Clin. Exp. Immunol. 120, 375–383.
Maloy, K. J., Salaun, L., Cahill, R., Dougan, G., Saunders, N. J., and Powrie, F. (2003). CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J. Exp. Med. 197, 111–119.
Mandik-Nayak, L., and Allen, P. M. (2005). Initiation of an autoimmune response: insights from a transgenic model of rheumatoid arthritis. Immunol. Res. 32, 5–13.
Matsumoto, I., Maccioni, M., Lee, D. M., Maurice, M., Simmons, B., Brenner, M., Mathis, D., and Benoist, C. (2002). How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat. Immunol. 3, 360–365.
Matsumoto, Y., Sakuma, H., Kohyama, K., and Park, I. K. (2007). Paralysis of CD4(+)CD25(+) regulatory T cell response in chronic autoimmune encephalomyelitis. J. Neuroimmunol. 187, 44–54.
Misra, N., Bayry, J., Lacroix-Desmazes, S., Kazatchkine, M. D., and Kaveri, S. V. (2004). Cutting edge: human CD4+CD25+ T cells restrain the maturation and antigen-presenting function of dendritic cells. J. Immunol. 172, 4676–4680.
Morgan, M. E., Flierman, R., Van Duivenvoorde, L. M., Witteveen, H. J., Van Ewijk, W., Van Laar, J. M., De Vries, R. R., and Toes, R. E. (2005). Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum. 52, 2212–2221.
Myers, L. K., Rosloniec, E. F., Cremer, M. A., and Kang, A. H. (1997). Collagen-induced arthritis, an animal model of autoimmunity. Life Sci. 61, 1861–1878.
Myouzen, K., Kochi, Y., Shimane, K., Fujio, K., Okamura, T., Okada, Y., Suzuki, A., Atsumi, T., Ito, S., Takada, K., Mimori, A., Ikegawa, S., Yamada, R., Nakamura, Y., and Yamamoto, K. (2010). Regulatory polymorphisms in EGR2 are associated with susceptibility to systemic lupus erythematosus. Hum. Mol. Genet. 19, 2313–2320.
Neubert, K., Meister, S., Moser, K., Weisel, F., Maseda, D., Amann, K., Wiethe, C., Winkler, T. H., Kalden, J. R., Manz, R. A., and Voll, R. E. (2008). The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat. Med. 14, 748–755.
Nguyen, L. T., Jacobs, J., Mathis, D., and Benoist, C. (2007). Where FoxP3-dependent regulatory T cells impinge on the development of inflammatory arthritis. Arthritis Rheum. 56, 509–520.
Noble, A., Zhao, Z. S., and Cantor, H. (1998). Suppression of immune responses by CD8 cells. II. Qa-1 on activated B cells stimulates CD8 cell suppression of T helper 2 responses. J. Immunol. 160, 566–571.
Oida, T., Zhang, X., Goto, M., Hachimura, S., Totsuka, M., Kaminogawa, S., and Weiner, H. L. (2003). CD4+CD25- T cells that express latency-associated peptide on the surface suppress CD4+CD45RBhigh-induced colitis by a TGF-beta-dependent mechanism. J. Immunol. 170, 2516–2522.
Okamura, T., Fujio, K., Shibuya, M., Sumitomo, S., Shoda, H., Sakaguchi, S., and Yamamoto, K. (2009). CD4+CD25-LAG3+ regulatory T cells controlled by the transcription factor Egr-2. Proc. Natl. Acad. Sci. U.S.A. 106, 13974–13979.
Oukka, M. (2007). Interplay between pathogenic Th17 and regulatory T cells. Ann. Rheum. Dis. 66(Suppl. 3), iii87–iii90.
Pandiyan, P., Conti, H. R., Zheng, L., Peterson, A. C., Mathern, D. R., Hernandez-Santos, N., Edgerton, M., Gaffen, S. L., and Lenardo, M. J. (2011). CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 cells in vitro and enhance host resistance in mouse Candida albicans Th17 cell infection model. Immunity 34, 422–434.
Roncarolo, M. G., Bacchetta, R., Bordignon, C., Narula, S., and Levings, M. K. (2001). Type 1 T regulatory cells. Immunol. Rev. 182, 68–79.
Roncarolo, M. G., Gregori, S., Lucarelli, B., Ciceri, F., and Bacchetta, R. (2011). Clinical tolerance in allogeneic hematopoietic stem cell transplantation. Immunol. Rev. 241, 145–163.
Rowley, M. J., Nandakumar, K. S., and Holmdahl, R. (2008). The role of collagen antibodies in mediating arthritis. Mod. Rheumatol. 18, 429–441.
Stromnes, I. M., Cerretti, L. M., Liggitt, D., Harris, R. A., and Goverman, J. M. (2008). Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat. Med. 14, 337–342.
Subramanian, S., Tus, K., Li, Q. Z., Wang, A., Tian, X. H., Zhou, J., Liang, C., Bartov, G., Mcdaniel, L. D., Zhou, X. J., Schultz, R. A., and Wakeland, E. K. (2006). A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc. Natl. Acad. Sci. U.S.A. 103, 9970–9975.
Sweet, R. A., Ols, M. L., Cullen, J. L., Milam, A. V., Yagita, H., and Shlomchik, M. J. (2011). Facultative role for T cells in extrafollicular Toll-like receptor-dependent autoreactive B-cell responses in vivo. Proc. Natl. Acad. Sci. U.S.A. 108, 7932–7937.
Terato, K., Hasty, K. A., Reife, R. A., Cremer, M. A., Kang, A. H., and Stuart, J. M. (1992). Induction of arthritis with monoclonal antibodies to collagen. J. Immunol. 148, 2103–2108.
Vinuesa, C. G., Cook, M. C., Angelucci, C., Athanasopoulos, V., Rui, L., Hill, K. M., Yu, D., Domaschenz, H., Whittle, B., Lambe, T., Roberts, I. S., Copley, R. R., Bell, J. I., Cornall, R. J., and Goodnow, C. C. (2005). A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 435, 452–458.
Wasowska, B. A. (2010). Mechanisms involved in antibody- and complement-mediated allograft rejection. Immunol. Res. 47, 25–44.
Watanabe, H., Garnier, G., Circolo, A., Wetsel, R. A., Ruiz, P., Holers, V. M., Boackle, S. A., Colten, H. R., and Gilkeson, G. S. (2000). Modulation of renal disease in MRL/lpr mice genetically deficient in the alternative complement pathway factor B. J. Immunol. 164, 786–794.
Willems, F., Marchant, A., Delville, J. P., Gerard, C., Delvaux, A., Velu, T., De Boer, M., and Goldman, M. (1994). Interleukin-10 inhibits B7 and intercellular adhesion molecule-1 expression on human monocytes. Eur. J. Immunol. 24, 1007–1009.
William, J., Euler, C., Christensen, S., and Shlomchik, M. J. (2002). Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science 297, 2066–2070.
Wollenberg, I., Agua-Doce, A., Hernandez, A., Almeida, C., Oliveira, V. G., Faro, J., and Graca, L. (2011). Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J. Immunol. 187, 4553–4560.
Zhao, D. M., Thornton, A. M., Dipaolo, R. J., and Shevach, E. M. (2006). Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood 107, 3925–3932.
Keywords: chronic inflammation, autoantibody, regulatory T cells, IL-10, Tr1 cells
Citation: Fujio K, Okamura T, Sumitomo S and Yamamoto K (2012) Regulatory T cell-mediated control of autoantibody-induced inflammation. Front. Immun. 3:28. doi: 10.3389/fimmu.2012.00028
Received: 10 December 2011; Paper pending published: 04 January 2012;
Accepted: 10 February 2012; Published online: 27 February 2012.
Edited by:
Masaaki Murakami, Osaka University, JapanCopyright: © 2012 Fujio, Okamura, Sumitomo and Yamamoto. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Keishi Fujio, Department of Allergy and Rheumatology, Graduate School of Medicine, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, Japan. e-mail:a2Z1amlvLXRreUB1bWluLmFjLmpw