

- 1 Department of Neuropathology, University Clinic Freiburg, Freiburg, Germany
- 2 BIOSS Centre for Biological Signaling Studies, Freiburg im Breisgau, Germany
Type I interferons (IFNs) were originally identified as antiviral effector molecules that exert pleiotropic physiological processes ranging from immune modulation, control of proliferation, apoptosis to antitumor activity. However, type I IFNs were recently also shown to apply both beneficial and detrimental effects to the central nervous system (CNS) and a tightly balanced equilibrium between cellular activation and inhibition seems to be essential to maintain homeostasis within the CNS. In inflammatory pathologies affecting the CNS, type I IFNs are in the center of attention not only because interferon beta (IFN-β) is used as a standard therapeutic in the treatment of relapsing–remitting multiple sclerosis (MS), but also as type I IFN expression is associated with distinct pathologies. Despite the great efficiency of IFN-β in reducing MS relapses and attenuation of novel inflammatory lesions is well documented, underlying molecular mechanisms and cellular target specificities are just beginning to emerge. In contrast to the curative effects, aberrant activation of the type I IFN response were also recently shown to be associated with detrimental effects exemplified by the Aicardi–Goutières syndrome (AGS), a severe disabling autoimmune inflammatory encephalopathy. This review will highlight the dual role of type I interferons during chronic CNS inflammation. Recently uncovered molecular and cellular mechanisms in the etiology of AGS and experimental autoimmune encephalomyelitis (EAE), the murine model of MS will be highlighted.
Type I Interferons and Their Induction
Interferons (IFNs) represent a family of cytokines which were originally identified by their ability to mediate antiviral effects. Since their discovery more than 54 years ago (Lindenmann et al., 1957), this class of proteins now embraces around 30 members. Based on common structural, biochemical, and signaling properties as well as the source of cells producing these factors, IFNs can be classed into three distinct subfamilies namely type I, type II, and class III IFNs. While IFN-γ is the sole type II IFN and the three different IFN-λs constitute the type III IFNs, type I IFNs are a highly divergent group of cytokines encompassing 13 different IFN-α subtypes, IFN-β, IFN-κ, IFN-ε, IFN-ω, IFN-τ, IFN-δ and three different IFN-ζs (IL-28A/B and IL-29; Noppert et al., 2007).
Consistent with the functional role of type I IFNs in pathogen defense, induction of these cytokines is predominantly triggered by distinct pathogen-associated molecular patterns (PAMPs) which are recognized by specific pathogen recognition receptors (PRRs). As depicted in Figure 1, the surface toll-like receptor (TLR) 4 recognizing lipopolysaccharide from Gram-negative bacteria as well as TLRs 3, 7, 8, and 9, which recognize pathogen-derived nucleic acids, induce type I IFNs (Blasius and Beutler, 2010). TLR3 recognizes viral double-stranded RNA (dsRNA) while viral single-stranded RNA (ssRNA) is detected by TLR7 and TLR8. Viral or bacterial unmethylated DNA, commonly referred to as CpG DNA, is sensed by TLR 9 (Akira et al., 2006; Barber, 2011; Kawai and Akira, 2011).
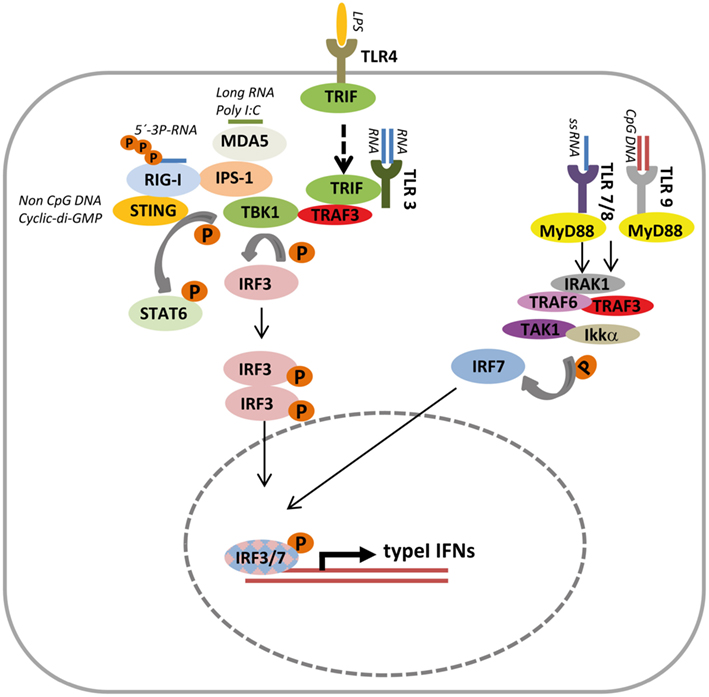
Figure 1. Overview of typical signaling cascades inducing type I Interferon expression. Upon ligand engagement, several toll-like receptors (TLRs) and RIG-I like helicases (RLHs) induce transcription of type I interferons (IFN). TLR4 located at the cell surface is typically induced extracellular while TLR3, TLR 7/8, and TLR9 sense pathogen-derived single-stranded RNA (ssRNA), double-stranded RNA (dsRNA), and unmethylated DNA (CpG DNA) within the cell sequestered from the cytoplasmic compartment. Intracellular TLRs are localized, traffic, and initiate signaling cascades in membrane surrounded compartments like the endoplasmic reticulum, endosomes, lysosomes, and phagocytic vesicles. Upon ligand binding, TLR4 is endocytosed (indicated by dashed arrows). Downstream signaling inducing type I IFN is mediated by initial binding to either MyD88 (TLR7/8/9) or TRIF (TLR3/4), followed by recruitment of multicomponent protein complexes. Typically a complex with TLR3 or TLR4 together with TRIF and TRAF3 activates the kinase TBK1 mediating phosphorylation of IRF3, which subsequently forms homodimers, translocates to the nucleus, and initiates type I IFN gene expression. MyD88 recruited to TLR7/8/9 complexes with IRAK1, TRAF6, TRAF3, and the kinases TAK1 and IKKα, which phosphorylate and thus activate IRF7 to drive type I IFN expression. The cytoplasmic RLHs MDA5 and RIG-I recognize longer RNAs like poly I:C or 5′-3-P-RNA respectively and engage IPS at the mitochondrial membrane. Recruitment of a complex containing TBK1 induces phosphorylation and thus dimerization of IRF3 followed by type I IFN gene expression. Independent from TLR and RLH intracellular, non-CpG DNA, and cyclic-di-GMP are sensed in a STING dependent manner. STING interacts with RIG-I and activates type I IFN transcription via the IRF3 axis but is also capable to recruit STAT6 to the ER followed by TBK1 mediated STAT6 phosphorylation.
The localization of nucleic acid sensing TLRs at the endoplasmic reticulum and endosomal membranes limits the detection of viruses by TLRs to this specific compartment. In general, signal transduction for type I IFN induction via the TLRs mentioned above starts with the recruitment of either Toll-IL-1 receptor (Tir) domain-containing factor (TRIF; for TLR4, TLR3) and/or myeloid differentiation primary response gene 88 (MyD88; for TLR7, TLR9) to the activated receptor. Subsequent signaling events involving the molecules interleukin-1 receptor-associated kinase (IRAK) 1, IRAK 4, tumor necrosis associated factor (TRAF) 6 and TRAF3 activate the kinases TGF-β activated kinase 1 (TAK1) and TANK-binding kinase 1 (TBK1). While TAK1 acts as a common activator of nuclear factor of kappa light polypeptide gene enhancer in B cells (NF-κB) signaling via the I kappa B kinase (IKK) complex which mediates phosphorylation and degradation of the inhibitory component IκBα, TBK1 phosphorylates interferon regulatory factor (IRF) 3 leading to its dimerization (Mori et al., 2004; Tamura et al., 2008). Finally, dimeric IRF3 and NF-κB translocate into the nucleus to induce type I IFN expression. In addition, engagement of distinct TLRs also causes IRF7 phosphorylation, which preferentially activates IFN-α but also leads to IFN-β induction (Honda and Taniguchi, 2006).
Alternative to the TLRs, the cell employs the retinoic acid-inducible gene-I (RIG-I) like helicases (RLHs) RIG-I and the melanoma differentiation-associated gene 5 (MDA5) to detect viral infection (Pichlmair and Reis e Sousa, 2007). RIG-I detects 5′-triphosphate-containing RNA which for example is typical for vesicular stomatitis virus (VSV; Hornung et al., 2006; Schlee et al., 2009). Characteristic for MDA5 is the recognition of longer RNA molecules as exemplified by the synthetic dsRNA poly I:C or picornavirus genomes (Kato et al., 2006). RLHs are characterized by a helicase domain for RNA binding and a caspase recruitment domain (CARD) mediating protein interaction for downstream signaling. Binding to viral RNA induces homodimerization of these sensors resulting in the engagement of MAVS (IPS-1, VISA, CARDIF; Kawai et al., 2005; Meylan et al., 2005; Seth et al., 2005; Xu et al., 2005) located in the mitochondrial membrane followed by the recruitment of TRAF3, TRAF6, TBK1, and IKKα/β. As a result of these signaling events, IRF3, IRF7, and NF-κB are activated and together with cJUN/ATF2 and the coactivators CBP/p300 initiate the expression of type I IFNs (Kawai and Akira, 2011).
The observation that TLR9 deficient cells are still capable to efficiently mount type I IFN production when transfected with double-stranded DNA (dsDNA) led to the search of alternative DNA sensing molecules especially in non-plasmacytoid dendritic cells (pDCs; Kawai and Akira, 2009). These investigations led to the identification of the DNA sensor endoplasmic reticulum IFN stimulator (ERIS) now better known as stimulator of interferon genes (STING) which is located in the ER membrane and plays an important role in the induction of IFN by non-CpG intracellular DNA species (Ishikawa and Barber, 2008; Zhong et al., 2008). Analysis of STING−/− mice revealed that the protein, which has no homology to other DNA sensors, is essential for host defense against DNA pathogens such as herpes simplex virus 1 (HSV-1; Ishikawa et al., 2009). Recent work also provided strong evidence that STING also acts as a direct sensor for the bacterial second messenger cyclic-di-GMP, which was shown previously to elicit a type I IFN response in host cells (McWhirter et al., 2009; Woodward et al., 2010; Burdette et al., 2011). It was recently shown that STING recruits STAT6 to the endoplasmatic reticulum (ER) followed by TBK-mediated phosphorylation. Remarkably, this STAT6 activation at the ER by STING differs from cytokine induced STAT6 phosphorylation at the plasma membrane as Janus kinases (JAKs) are not involved and phosphorylation is not restricted to distinct cell types. The virus-induced STAT6 activation also occurs in IFNAR2−/− or IRF3−/− cells as well as in cells lacking individual JAKs providing strong evidence that STING-triggered STAT6 activation represents a novel signaling module. While the canonical STAT6 activation is MAVS independent this non-canonical STAT6 activation required the presence of MAVS and STAT6 phosphorylation is mediated by TBK1. Consistent with a fundamental role of STAT6 in antiviral activity, STAT6 deficient mice exhibited exacerbated pathologies when challenged with either VSV, Sendai virus, or herpes simplex virus (Chen et al., 2011).
IFN Signaling
As a common feature all interferons activate the JAK– signal transducers of activation and transcription (STAT) signaling pathway. A hallmark of type I IFNs is that they all bind and signal via the interferon alpha receptor (IFNAR), which is a heterodimer composed of interferon receptor 1 (IFNAR1) and interferon receptor 2 (IFNAR2). It is a unique feature compared to other cytokine receptors that IFNAR can bind and mediate signaling of multiple ligands. Type I IFNs bind to the IFNAR and as a consequence they activate the kinases JAKs Tyk 2 and JAK 1 which are associated with IFNAR1 and IFNAR2 respectively and phosphorylate receptor tyrosine residues on the receptor (de Weerd et al., 2007). Via SH2 domains STAT bind to the phosphotyrosines on the receptor and become themselves phosphorylated. Phosphorylated STAT1 forms heterodimers with STAT2 and together with IRF9 constitutes a transcription factor (ISGF3γ) which binds to interferon-stimulated response elements (ISREs) of interferon-stimulated target genes (ISGs) inducing their expression. Further complexity is added to the system by the formation of STAT1 and STAT3 homo- and heterodimers which activate GAS elements controlling the expression of other ISGs which are also inducible by IFN-γ. In addition, also STAT independent pathways are activated (Boxel-Dezaire et al., 2006). Ultimately, hundreds of ISGs, which might vary depending on the cell type, are induced and translated into the effector proteins responsible to mediate biological effects like antiviral activity, control of cellular proliferation, apoptosis, and immune regulation.
Inflammatory Brain Pathologies Associated with Aberrant Type I IFN Induction
Interferon beta has proinflammatory properties and contributes to the pathology of autoimmune diseases like systemic lupus erythematosus (SLE), rheumatoid arthritis, and psoriasis (Preble et al., 1982; Baechler et al., 2006). Within the central nervous system (CNS) proinflammatory detrimental effects of IFN-β were described for Aicardi–Goutières syndrome (AGS) and neuromyelitis optica (NMO).
Aicardi–Goutières syndrome represents a fatal genetic disease that manifests as an encephalopathy. The disease is characterized by increased lymphocyte numbers in the cerebrospinal fluid, demyelination of the white matter and calcification of basal ganglia thus mimicking pathological consequences of congenital infection which often leads to misdiagnoses (Rice et al., 2007). One hallmark of AGS are increased levels of IFN-α in serum and cerebrospinal fluid (Dussaix et al., 1985). Increased levels of IFN-α are also a key feature of SLE and indeed, molecular mechanisms underlying AGS and SLE exhibit striking parallels allocating AGS into the group of autoimmune diseases. Furthermore, some children with AGS also exhibit an early onset form of SLE (De Laet et al., 2005). AGS is inherited in an autosomal recessive trait and mutations in different genetic loci were identified. It was shown before that recessive mutations in the genes encoding either human 3′repair exonuclease 1 (TREX1), different components of the RNASEH2 complex or the SAM-domain HD-domain-containing protein 1 (SAMHD1) can cause this severe inflammatory disease (Crow et al., 2006a,b; Rice et al., 2009). TREX1 was shown to be a 3′–5′ DNA exonuclease preferentially binding and cleaving single-stranded DNA (ssDNA; Mazur and Perrino, 1999, 2001). Initially appearing paradox for a DNAse, TREX1 is predominantly localized in the cytoplasm where the protein is associated with the endoplasmic reticulum. TREX1 is part of the SET complex and in response to oxidative stress the protein can translocate to the nucleus and is involved in Granzyme A-mediated apoptosis (Martinvalet et al., 2005; Chowdhury et al., 2006). In concordance with an extranuclear function, absence of TREX in mice and humans results in the accumulation of cytoplasmic ssDNA (Yang et al., 2007; Stetson et al., 2008). It was reported that excessive and mislocalized ssDNA originates from excision mediated DNA repair (Yang et al., 2007). Work by Medzhitov’s group identified TREX1 as an important negative regulator of the interferon stimulatory DNA (ISD) response. They provided compelling evidence that the ssDNA accumulating in the absence of TREX arises from endogenous retroelements which are no longer degraded properly by the TREX nuclease. As a consequence, the elevated levels of ssDNA which might mimic viral infection are sensed by an unidentified DNA sensor, initiate the ISD pathway and activate IRF3 resulting in the production of high levels of type I IFN and lymphocyte mobilization (Stetson et al., 2008). In line with an aberrant activation of the innate immune system TREX1−/− mice develop an inflammatory myocarditis (Morita et al., 2004). The observed cardiomyopathy in TREX1 deficient mice might be analogous to the encephalopathy in AGS patients associated with an autoinflammatory response which arises in the absence of an infection. The observation that the phenotype of TREX1−/− mice can be rescued in the absence of either IRF3, IFNAR, or the recombination activating gene (RAG) further underlines that the autoimmune mechanism is triggered by activation of IRF3 mediated type I IFN induction (Stetson et al., 2008) but also shows that secondary mechanisms like leukocyte recruitment are essential for disease manifestation. In line with the central role of TREX in the maintenance of immune homeostasis is the observation that mutations in this gene are also associated with SLE (Lee-Kirsch et al., 2007).
The central role of TREX1 as a negative regulator of type I IFN induction is exploited by the human immunodeficiency virus (HIV) type 1. Recent work showed that TREX1 not only degrades DNA from endogenous retroelements to prevent autoimmune activation. The nuclease is also active against HIV DNA which arises during HIV infection after reverse transcription (RT; Geijtenbeek, 2010; Yan et al., 2010). Apparently, the virus hijacks TREX1’s specificity for retrovirus-derived DNA to avoid activation of cytoplasmic nucleic acid sensors, which usually trigger the antiviral effector system. TREX1-mediated degradation of HIV DNA thus represents one mechanism how HIV escapes immune recognition and helps to explain why infection of T cells and macrophages do not elicit an effective antiviral interferon response (Geijtenbeek, 2010).
Interestingly, also another AGS susceptibility gene, SAMHD1, which is highly expressed in macrophages and dendritic cells and upregulated upon viral infection was proposed to act as a negative regulator of the innate immune response (Lafuse et al., 1995; Li et al., 2000; Rice et al., 2009). However, the molecular function until recently remained elusive. SAMHD1 function was now shown to be tightly linked to the control of retroviral infection (Hrecka et al., 2011; Laguette et al., 2011). HIV is incapable to transduce dendritic cells and impaired in the transduction of macrophages because efficient viral cDNA synthesis is inhibited in these cells. In contrast, other retroviruses encoding Vpx accessory proteins circumvent this inhibition (Ayinde et al., 2010). Two recent publications identified SAMHD1 as the factor restricting HIV infection in myeloid and dendritic cells and showed that Vpx proteins counteract this restriction by mediating proteasomal degradation of SAMHD1 (Hrecka et al., 2011; Laguette et al., 2011).
Recently, also the molecular function of SAMHD1 was identified. It was shown that the protein is an effective dGTP-stimulated deoxyguanosine triphosphate triphosphohydrolase, which catalyzes the conversion of dGTP to guanosine and inorganic triphosphate (Goldstone et al., 2011). SAMHD1 rapidly hydrolyzes dGTP but not dATP, dCTP, and dTTP. However, SAMHD1 is allosterically activated by dGTP which binds to SAMHD1 leading to a more promiscuous substrate specificity which is extended to dATP, dCTP, and dTTP. dGTP is thus both a substrate and an activator of the enzyme against the other dNTPs. The catalytic activity should strongly limit the dNTP pool within the cytoplasm and thus exert a role in nucleic acid metabolism. It is reasonable to hypothesize that SAMHD1 limits the dNTP pool and thus interferes with HIV replication by inhibiting endogenous RT. With respect to the etiology of AGS, loss of SAMHD1 activity would no longer counteract RT of endogenous retroviral elements as the dNTP pool is no longer restricted. As a consequence, the loss of RT restriction might promote the accumulation of cytoplasmic DNA similar to what is observed in TREX1 mutants. Although further experimental evidence needs to be added, this model would be a straight forward explanation for the similar phenotypic manifestations arising from human mutations in SAMHD1 and TREX genes. In summary, TREX1 and SAMHD1 provide excellent examples for the intricate relationship between the control of retroviral infection and the control of cell intrinsic nucleic acids harboring the risk to trigger autoimmune inflammation induced by type I IFNs.
In addition, also mutations in subunits of the RNase H2 complex were shown to cause AGS (Crow et al., 2006b). The RNase H2 is the dominant cytoplasmic RNase in human cells and composed of a heterotrimeric complex in which all three components are needed to constitute the active enzyme. RNase H2 enzymes recognize RNA:DNA hybrids, degrade the polyribonucleotide strand and thus also play a central role in nucleic acid metabolism (Eder and Walder, 1991; Rydberg and Game, 2002). Although direct experimental evidence is still missing, it is likely that RNase H2 mutations which impair degradation of RNA:DNA hybrids alter cytoplasmic nucleic acid homeostasis resulting in the activation of the ISD response.
As depicted in Figure 2, analysis of the molecular mechanisms underlying AGS revealed a role of all the susceptibility genes mentioned above in the control of cytoplasmic nucleic acid homeostasis which needs to be tightly controlled in order to prevent autoimmunity induced by type I IFNs.
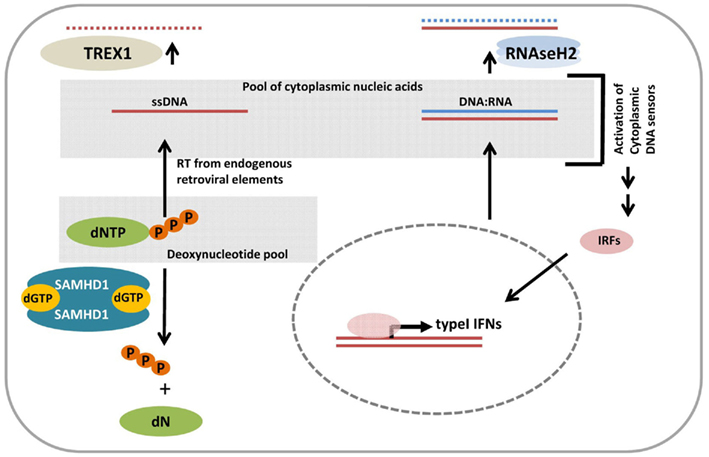
Figure 2. Model for the role of cytoplasmic nucleic acid homeostasis in the etiology of Aicardi–Goutieres syndrome (AGS). TREX1 is an 5′–3′ Exonuclease which degrades cytoplasmic single-stranded DNA (ssDNA) originating from reverse transcription of endogenous retrotransposons. Lack of TREX1 activity results in the congestion of ssDNA thereby augmenting the pool of nucleic acids in the cytoplasm. SAMHD1 cleaves inorganic triphosphate from deoxynucleotides and thus restricts the cytoplasmic dNTP concentrations hence constraining reverse transcription by limiting the amount of “building blocks” for DNA synthesis. SAMHD1 mutations would eliminate this dNTP control mechanism, enhance the dNTP pool, and could thus fuel reverse transcription from endogenous retroelements resulting in increased levels of cytoplasmic ssDNA. RNASEH2 is an endonuclease composed of three subunits and required to cleave ribonucleotides from RNA:DNA duplexes which arise from intracellular processes. Lack of function mutations in RNASeH2 would thus also impair the control of nucleic acid homeostasis. Thus mutations in any kind of the genes mentioned above which were found in AGS patients would lead to the accumulation of nucleic acids in the cytoplasm. These aberrant levels are sensed by cytoplasmic DNA sensors originally designated to detect viral infections. Consequently the initiated signaling cascade activates interferon regulatory transcription factors (IRFs) for type I IFN induction and drive autoimmune disease onset.
Neuromyelitis Optica (Devic’s Disease) and Type I IFNs
Neuromyelitis optica represents a neuroinflammatory disease previously considered to be a variant of relapsing–remitting multiple sclerosis (RRMS). However, in contrast to RRMS, NMO is characterized by demyelination in the spinal cord and optic nerve (Lucchinetti et al., 2002). Autoantibodies directed against the aquaporin 4 water channel are found in these patients and occurrence of anti-aquaporin antibodies also is the major parameter in the diagnosis of NMO (Paul et al., 2007). Driven by the supposed analogy to RRMS, IFN-β was tried as a therapeutic but it was quickly clear that IFN-β treatment even worsens the disease and induces severe relapses (Warabi et al., 2007; Palace et al., 2010; Shimizu et al., 2010; Uzawa et al., 2010). Notably, the dominant infiltrating cells are granulocytes which is in sharp contrast to classical multiple sclerosis (MS). This clearly different infiltratory make up might explain the different response upon IFN-β treatment. Interestingly, levels of IL-17 were found to be elevated in the cerebrospinal fluid of people suffering from NMO and it has been proposed that IL-17 mediated induction of IL-8, granulocyte stimulating factor (G-CSF) and Gro-alpha causes granulocyte recruitment (Axtell et al., 2011).
Anti-Inflammatory Effects of Type I IFNs in Relapsing–Remitting Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis
As exemplified for SLE and AGS, type I IFNs were frequently causally associated with the development of autoimmune pathologies. In contrast, type I IFNs were also shown to exert anti-inflammatory effects and IFN-β is used as a routine therapeutic for the treatment of RRMS in patients (Jacobs et al., 2000; Filippi et al., 2004). MS is an autoimmune inflammatory disease of the CNS of unclear etiology characterized by demyelination of axons in brain and spinal cord. Characteristic for the disease is the infiltration of the CNS with inflammatory cells like monocytes, TH1 and TH17 cells. In RRMS, phases with no or only minor disease progression are followed by unpredictable acute relapses causing deficits which might at least partially resolve in times of remission.
According to results from clinical trials, IFN-β treatment reduces relapse rates by about 30%, decreases the formation of inflammatory lesions in the CNS and extends remission periods (Schwid and Panitch, 2007). However, a major problem is that a high proportion of about 20% of the patients do not or only poorly respond to IFN-β treatment. Despite excessive attempts to define biomarkers for responders and non-responders it is still impossible to predict whether an individual patient will respond to IFN-β therapy. Mechanism underlying this variety include development of antibodies against IFN-β, polymorphisms in components of the IFN signaling pathway and IFN effector genes as well as variable MS pathomechanisms (Killestein and Polman, 2011). By further dissecting type I IFN signatures in MS patients, IFN-β non-responders were found to exhibit increased monocyte-specific type I IFN secretion upon innate immune stimuli via TLR 4, by increased endogenous production of type I interferon, and by an elevated activation status of myeloid dendritic cells (Comabella et al., 2009). These findings indicate that perturbations of the type I IFN signaling pathway in monocytes are related to a lack of response to IFN-β and type I IFN-regulated genes may be used as response markers in IFN-β treatment in MS. Consistent with the concept that genetic variants define the individual response is that induction of interferon response genes among different patients varies significantly but is remarkably stable within a long time period.
A frequently used system to gain insight into the molecular and cellular events underlying MS is the model of experimental autoimmune encephalomyelitis (EAE). In this well-established mouse model active immunization with myelin components together with a strong adjuvant induces T-cell mediated neuroinflammation and demyelination resembling multiple aspects of MS (Owens et al., 2001; Gold et al., 2006).
Consistent with the beneficial effect of IFN-β in human MS, EAE in rodents can be suppressed by injection of recombinant IFN-β. In addition, mice lacking IFN-β exhibit strongly exacerbated disease parameters in the EAE model (Teige et al., 2003). Furthermore, IFNβ is stronger induced in the CNS than in the periphery. To trace down the cell type mediating the “protective” immune regulation of type I IFNs via the type I receptor (IFNAR), mice lacking this receptor only on distinct cell types were used for EAE experiments and IFN-β treatment (Prinz et al., 2008). It could be shown that negative regulation of autoimmunity in the EAE model relies on the presence of IFNAR on cells of the myeloid lineage while absence of IFNAR on T cells, B cells, and cells of neuroectodermal origin does not enhance disease severity. These experiments suggest a scenario where locally produced IFN-β within the CNS acts on invading myeloid cells to attenuate autoimmune damage especially in the effector phase of EAE. Another study which also employed EAE in IFNAR deficient mice provided evidence for a functional role of type I interferon in negatively regulating the development of TH17 cells. Numbers of encephalitogenic TH17 cells were found by this group to be elevated after EAE induction in IFNAR1 and TRIF deficient mice (Guo et al., 2008). TH17 cells develop from naive T cells which induced by TGFβ and IL-6 secrete IL-21. This cytokine subsequently induces the transcription factor RORγt in an autocrine manner which triggers the TH17 differentiation program (Veldhoen et al., 2006; Stockinger and Veldhoen, 2007). IFN-β induces expression of IL-27 which acts on naive CD4+ T cells as a negative regulator of TH17 development (Prinz and Kalinke, 2010). IL-27 also promotes IL-10 secretion by T cells which was shown to suppress autoimmune inflammation of the CNS (Fitzgerald et al., 2007). IFN-β was furthermore shown to inhibit IL-23 induced proliferation of TH17 cells (Harrington et al., 2005). Increased levels of TH17 cells in IFNAR−/− mice would thus be explained by the lack of these inhibitory effects of type I IFNs.
Disease symptoms and generation of antigen specific TH17 cells were also enhanced in TRIF deficient mice suggesting that type I IFN production counteracting autoimmunity in EAE is triggered by a TRIF dependent mechanism (Guo et al., 2008). Interestingly, type I IFN-related genes were found to be strongly induced in toxic mouse models of demyelination whereas the presence of IFNAR could neither modulate demyelination or myelin repair (Schmidt et al., 2009).
Just recently, also cytosolic RLHs were shown to act as negative regulators of sterile inflammation within the CNS (Dann et al., 2012). Mice lacking the RIG-I like helicase adaptor IPS-1 developed exacerbated disease symptoms. In a reciprocal experiment the same study showed that on the other hand activation of RLHs via IFN-inducing 5′-triphosphate RNA oligonucleotides ameliorated disease outcome providing compelling evidence that RLHs mediate signals counteracting encephalitogenic immune responses. While RLH stimulation did not affect T-cell differentiation, the maintenance and expansion of TH1 and TH17 cells was strongly suppressed upon RLH engagement (Figure 3). Repression of proinflammatory TH1 and TH17 cells could only be observed when the type I interferon receptor was present on dendritic cells by using CD11cCre mice crossed with conditional IFNAR animals. Remarkably, absence of IFNAR1 on macrophages or microglia did not interfere with RLH mediated TH1 or TH17 repression (Dann et al., 2012). These results imply that engagement of RLHs might be a passable way to stimulate endogenous type I IFNs for therapeutic intervention. Replacing systemic administration with the endogenous induction of type I IFNs should prevent the development of neutralizing antibodies. It might also be more effective than systemic administration as type I IFNs are biologically designed to exert their effects in a spatially restricted area affecting cells in their direct neighborhood.
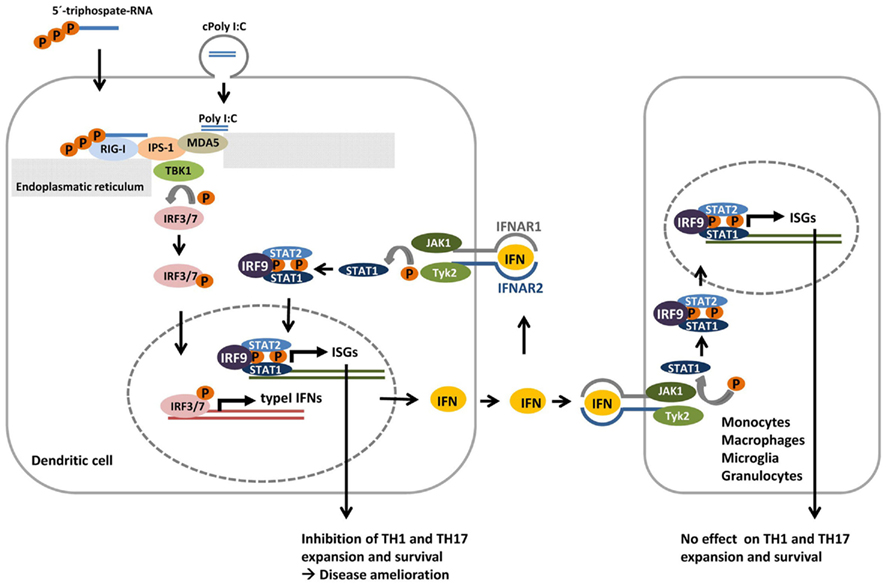
Figure 3. Model for negative regulation of sterile CNS inflammation by RIG-I like helicases (RLHs). In dendritic cells activation of RLHs RIG-I or MDA5 by 5′triphosphate RNA or polyI:C complexed to liposomes (cPolyI:C) respectively, recruits IPS-1 to the RLH. This initiates a signaling cascade which involves TBK1 mediated phosphorylation of IRF3 and/or IRF7 which upon dimerization enter the nucleus and lead to the expression of type I IFN. Secreted type IFNs (IFN) can either bind to type I interferon receptor (IFNAR) in an autocrine or paracrine manner and stimulate the expression of interferon-stimulated target genes (ISGs). While lack of IPS-1 leads to exacerbated disease pathology in the EAE model of sterile inflammation, RLH stimulation improved clinical signs of disease and inhibits TH1 and TH17 expansion and survival. This regulation is type I IFN mediated as the “therapeutic effect” of RLH stimulation is abrogated in IFNAR deficient mice. However, mice specifically lacking IFNAR on monocytes, macrophages, microglia, or granulocytes like wildtype mice showed reduced symptoms upon RLH induction. IFNAR signaling on these cells is thus dispensable for the suppressive effect of RLH engagement. In contrast, when mice specifically lacking IFNAR on dendritic cells were used, RLH treatment did not ameliorate autoimmunity clearly showing that interferon stimulation in this particular celltype is essential for IFN mediated TH1 and TH17 inhibition and disease improvement.
Recognition of triphosphate RNA by RIG-I like helicases followed by IFN induction is largely independent from the sequence used for the dsRNA (Hornung et al., 2006). Thus, a specific short interfering RNA (RNAi) suitable to suppress expression of a specific target protein can be used. This, at least in principle, offers therapeutic strategies where activation of the type I IFN response can be combined with the suppression of harmful gene products. As a proof of principle for the feasibility of such an approach it was shown in a melanoma model that BCL2 specific siRNA carrying 5′ triphosphate ends efficiently activated type I IFN via RIG-I and in parallel silenced Bcl2 expression (Poeck et al., 2008). As a consequence, the synergistic activation of the innate immune response combined with suppression of a pro survival signal caused massive apoptosis of tumor cells, prolonged survival of the animals, and reduced tumor size.
It might thus also be suitable to combine type I IFN induction with RNAi mediated inhibition of proinflammatory factors or proteins triggering the differentiation of deleterious cells. Such a strategy where for example type I IFN induction could be combined with RNAi mediated inhibition of RORγt or T-bet to simultaneously repress TH17 and TH1 differentiation might potentially open new ways to modulate the immune response and counteract autoimmunity within the CNS.
Conclusion and Future Perspectives
Despite extensively analyzed, the functions of type I IFNs within the CNS are still far from being well understood. One principal problem to generalize type I IFN effects is surely based on the fact that hundreds of genes, which even vary among different cell types, are controlled by this particular group of cytokines. In addition, the levels and kind of IFN target genes induced are differentially regulated depending on the situation, interaction and environment of the cell. Further complexity is added to the system as multiple negative regulators of the IFN signaling system modulate the outcome and refractory mechanisms blunt restimulation. A major task for the future will be to identify the particular interferon effector genes responsible for the anti-inflammatory and therapeutic properties which even might considerably differ between various cell types. With respect to the etiology of type I IFN associated autoimmune disease like AGS it will be interesting to see which particular molecules are involved in the pathways sensing enhanced levels of endogenous nucleic acids in the cytoplasm.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801.
Axtell, R. C., Raman, C., and Steinman, L. (2011). Interferon-beta exacerbates Th17-mediated inflammatory disease. Trends Immunol. 32, 272–277.
Ayinde, D., Maudet, C., Transy, C., and Margottin-Goguet, F. (2010). Limelight on two HIV/SIV accessory proteins in macrophage infection: is Vpx overshadowing Vpr? Retrovirology 7, 35.
Baechler, E. C., Batliwalla, F. M., Reed, A. M., Peterson, E. J., Gaffney, P. M., Moser, K. L., Gregersen, P. K., and Behrens, T. W. (2006). Gene expression profiling in human autoimmunity. Immunol. Rev. 210, 120–137.
Boxel-Dezaire, A. H., Rani, M. R., and Stark, G. R. (2006). Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 25, 361–372.
Burdette, D. L., Monroe, K. M., Sotelo-Troha, K., Iwig, J. S., Eckert, B., Hyodo, M., Hayakawa, Y., and Vance, R. E. (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518.
Chen, H., Sun, H., You, F., Sun, W., Zhou, X., Chen, L., Yang, J., Wang, Y., Tang, H., Guan, Y., Xia, W., Gu, J., Ishikawa, H., Gutman, D., Barber, G., Qin, Z., and Jiang, Z. (2011). Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 147, 436–446.
Chowdhury, D., Beresford, P. J., Zhu, P., Zhang, D., Sung, J. S., Demple, B., Perrino, F. W., and Lieberman, J. (2006). The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol. Cell 23, 133–142.
Comabella, M., Lunemann, J. D., Rio, J., Sanchez, A., Lopez, C., Julia, E., Fernandez, M., Nonell, L., Camina-Tato, M., Deisenhammer, F., Caballero, E., Tortola, M. T., Prinz, M., Montalban, X., and Martin, R. (2009). A type I interferon signature in monocytes is associated with poor response to interferon-beta in multiple sclerosis. Brain 132, 3353–3365.
Crow, Y. J., Hayward, B. E., Parmar, R., Robins, P., Leitch, A., Ali, M., Black, D. N., van Bokhoven, H., Brunner, H. G., Hamel, B. C., Corry, P. C., Cowan, F. M., Frints, S. G., Klepper, J., Livingston, J. H., Lynch, S. A., Massey, R. F., Meritet, J. F., Michaud, J. L., Ponsot, G., Voit, T., Lebon, P., Bonthron, D. T., Jackson, A. P., Barnes, D. E., and Lindahl, T. (2006a). Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat. Genet. 38, 917–920.
Crow, Y. J., Leitch, A., Hayward, B. E., Garner, A., Parmar, R., Griffith, E., Ali, M., Semple, C., Aicardi, J., Babul-Hirji, R., Baumann, C., Baxter, P., Bertini, E., Chandler, K. E., Chitayat, D., Cau, D., Dery, C., Fazzi, E., Goizet, C., King, M. D., Klepper, J., Lacombe, D., Lanzi, G., Lyall, H., Martinez-Frias, M. L., Mathieu, M., McKeown, C., Monier, A., Oade, Y., Quarrell, O. W., Rittey, C. D., Rogers, R. C., Sanchis, A., Stephenson, J. B., Tacke, U., Till, M., Tolmie, J. L., Tomlin, P., Voit, T., Weschke, B., Woods, C. G., Lebon, P., Bonthron, D. T., Ponting, C. P., and Jackson, A. P. (2006b). Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat. Genet. 38, 910–916.
Dann, A., Poeck, H., Croxford, A. L., Gaupp, S., Kierdorf, K., Knust, M., Pfeifer, D., Maihoefer, C., Endres, S., Kalinke, U., Meuth, S. G., Wiendl, H., Knobeloch, K. P., Akira, S., Waisman, A., Hartmann, G., and Prinz, M. (2012). Cytosolic RIG-I-like helicases act as negative regulators of sterile inflammation in the CNS. Nat. Neurosci. 15, 98–106.
De Laet, C., Goyens, P., Christophe, C., Ferster, A., Mascart, F., and Dan, B. (2005). Phenotypic overlap between infantile systemic lupus erythematosus and Aicardi-Goutieres syndrome. Neuropediatrics 36, 399–402.
de Weerd, N. A., Samarajiwa, S. A., and Hertzog, P. J. (2007). Type I interferon receptors: biochemistry and biological functions. J. Biol. Chem. 282, 20053–20057.
Dussaix, E., Lebon, P., Ponsot, G., Huault, G., and Tardieu, M. (1985). Intrathecal synthesis of different alpha-interferons in patients with various neurological diseases. Acta Neurol. Scand. 71, 504–509.
Eder, P. S., and Walder, J. A. (1991). Ribonuclease H from K562 human erythroleukemia cells. Purification, characterization, and substrate specificity. J. Biol. Chem. 266, 6472–6479.
Filippi, M., Rovaris, M., Inglese, M., Barkhof, F., De Stefano, N., Smith, S., and Comi, G. (2004). Interferon beta-1a for brain tissue loss in patients at presentation with syndromes suggestive of multiple sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet 364, 1489–1496.
Fitzgerald, D. C., Zhang, G. X., El Behi, M., Fonseca-Kelly, Z., Li, H., Yu, S., Saris, C. J., Gran, B., Ciric, B., and Rostami, A. (2007). Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T cells. Nat. Immunol. 8, 1372–1379.
Gold, R., Linington, C., and Lassmann, H. (2006). Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 129, 1953–1971.
Goldstone, D. C., Ennis-Adeniran, V., Hedden, J. J., Groom, H. C., Rice, G. I., Christodoulou, E., Walker, P. A., Kelly, G., Haire, L. F., Yap, M. W., de Carvalho, L. P., Stoye, J. P., Crow, Y. J., Taylor, I. A., and Webb, M. (2011). HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480, 379–382.
Guo, B., Chang, E. Y., and Cheng, G. (2008). The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J. Clin. Invest. 118, 1680–1690.
Harrington, L. E., Hatton, R. D., Mangan, P. R., Turner, H., Murphy, T. L., Murphy, K. M., and Weaver, C. T. (2005). Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6, 1123–1132.
Honda, K., and Taniguchi, T. (2006). Toll-like receptor signaling and IRF transcription factors. IUBMB Life 58, 290–295.
Hornung, V., Ellegast, J., Kim, S., Brzozka, K., Jung, A., Kato, H., Poeck, H., Akira, S., Conzelmann, K. K., Schlee, M., Endres, S., and Hartmann, G. (2006). 5′-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997.
Hrecka, K., Hao, C., Gierszewska, M., Swanson, S. K., Kesik-Brodacka, M., Srivastava, S., Florens, L., Washburn, M. P., and Skowronski, J. (2011). Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474, 658–661.
Ishikawa, H., and Barber, G. N. (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signaling. Nature 455, 674–678.
Ishikawa, H., Ma, Z., and Barber, G. N. (2009). STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792.
Jacobs, L. D., Beck, R. W., Simon, J. H., Kinkel, R. P., Brownscheidle, C. M., Murray, T. J., Simonian, N. A., Slasor, P. J., and Sandrock, A. W. (2000). Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS Study Group. N. Engl. J. Med. 343, 898–904.
Kato, H., Takeuchi, O., Sato, S., Yoneyama, M., Yamamoto, M., Matsui, K., Uematsu, S., Jung, A., Kawai, T., Ishii, K. J., Yamaguchi, O., Otsu, K., Tsujimura, T., Koh, C. S., Reis e Sousa, C., Matsuura, Y., Fujita, T., and Akira, S. (2006). Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105.
Kawai, T., and Akira, S. (2009). The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 21, 317–337.
Kawai, T., and Akira, S. (2011). Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650.
Kawai, T., Takahashi, K., Sato, S., Coban, C., Kumar, H., Kato, H., Ishii, K. J., Takeuchi, O., and Akira, S. (2005). IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6, 981–988.
Killestein, J., and Polman, C. H. (2011). Determinants of interferon beta efficacy in patients with multiple sclerosis. Nat. Rev. Neurol. 7, 221–228.
Lafuse, W. P., Brown, D., Castle, L., and Zwilling, B. S. (1995). Cloning and characterization of a novel cDNA that is IFN-gamma-induced in mouse peritoneal macrophages and encodes a putative GTP-binding protein. J. Leukoc. Biol. 57, 477–483.
Laguette, N., Sobhian, B., Casartelli, N., Ringeard, M., Chable-Bessia, C., Segeral, E., Yatim, A., Emiliani, S., Schwartz, O., and Benkirane, M. (2011). SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474, 654–657.
Lee-Kirsch, M. A., Chowdhury, D., Harvey, S., Gong, M., Senenko, L., Engel, K., Pfeiffer, C., Hollis, T., Gahr, M., Perrino, F. W., Lieberman, J., and Hubner, N. (2007). A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J. Mol. Med. (Berl.) 85, 531–537.
Li, N., Zhang, W., and Cao, X. (2000). Identification of human homologue of mouse IFN-gamma induced protein from human dendritic cells. Immunol. Lett. 74, 221–224.
Lindenmann, J., Burke, D. C., and Isaacs, A. (1957). Studies on the production, mode of action and properties of interferon. Br. J. Exp. Pathol. 38, 551–562.
Lucchinetti, C. F., Mandler, R. N., McGavern, D., Bruck, W., Gleich, G., Ransohoff, R. M., Trebst, C., Weinshenker, B., Wingerchuk, D., Parisi, J. E., and Lassmann, H. (2002). A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 125, 1450–1461.
Martinvalet, D., Zhu, P., and Lieberman, J. (2005). Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 22, 355–370.
Mazur, D. J., and Perrino, F. W. (1999). Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3′ → 5′ exonucleases. J. Biol. Chem. 274, 19655–19660.
Mazur, D. J., and Perrino, F. W. (2001). Structure and expression of the TREX1 and TREX2 3′ → 5′ exonuclease genes. J. Biol. Chem. 276, 14718–14727.
McWhirter, S. M., Barbalat, R., Monroe, K. M., Fontana, M. F., Hyodo, M., Joncker, N. T., Ishii, K. J., Akira, S., Colonna, M., Chen, Z. J., Fitzgerald, K. A., Hayakawa, Y., and Vance, R. E. (2009). A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J. Exp. Med. 206, 1899–1911.
Meylan, E., Curran, J., Hofmann, K., Moradpour, D., Binder, M., Bartenschlager, R., and Tschopp, J. (2005). Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172.
Mori, M., Yoneyama, M., Ito, T., Takahashi, K., Inagaki, F., and Fujita, T. (2004). Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J. Biol. Chem. 279, 9698–9702.
Morita, M., Stamp, G., Robins, P., Dulic, A., Rosewell, I., Hrivnak, G., Daly, G., Lindahl, T., and Barnes, D. E. (2004). Gene-targeted mice lacking the Trex1 (DNase III) 3′ → 5′ DNA exonuclease develop inflammatory myocarditis. Mol. Cell Biol 24, 6719–6727.
Noppert, S. J., Fitzgerald, K. A., and Hertzog, P. J. (2007). The role of type I interferons in TLR responses. Immunol. Cell Biol. 85, 446–457.
Owens, T., Wekerle, H., and Antel, J. (2001). Genetic models for CNS inflammation. Nat. Med. 7, 161–166.
Palace, J., Leite, M. I., Nairne, A., and Vincent, A. (2010). Interferon beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch. Neurol. 67, 1016–1017.
Paul, F., Jarius, S., Aktas, O., Bluthner, M., Bauer, O., Appelhans, H., Franciotta, D., Bergamaschi, R., Littleton, E., Palace, J., Seelig, H. P., Hohlfeld, R., Vincent, A., and Zipp, F. (2007). Antibody to aquaporin 4 in the diagnosis of neuromyelitis optica. PLoS. Med. 4, e133. doi:10.1371/journal.pmed.0040133
Poeck, H., Besch, R., Maihoefer, C., Renn, M., Tormo, D., Morskaya, S. S., Kirschnek, S., Gaffal, E., Landsberg, J., Hellmuth, J., Schmidt, A., Anz, D., Bscheider, M., Schwerd, T., Berking, C., Bourquin, C., Kalinke, U., Kremmer, E., Kato, H., Akira, S., Meyers, R., Hacker, G., Neuenhahn, M., Busch, D., Ruland, J., Rothenfusser, S., Prinz, M., Hornung, V., Endres, S., Tuting, T., and Hartmann, G. (2008). 5′-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat. Med. 14, 1256–1263.
Preble, O. T., Black, R. J., Friedman, R. M., Klippel, J. H., and Vilcek, J. (1982). Systemic lupus erythematosus: presence in human serum of an unusual acid-labile leukocyte interferon. Science 216, 429–431.
Prinz, M., and Kalinke, U. (2010). New lessons about old molecules: how type I interferons shape Th1/Th17-mediated autoimmunity in the CNS. Trends Mol. Med. 16, 379–386.
Prinz, M., Schmidt, H., Mildner, A., Knobeloch, K. P., Hanisch, U. K., Raasch, J., Merkler, D., Detje, C., Gutcher, I., Mages, J., Lang, R., Martin, R., Gold, R., Becher, B., Bruck, W., and Kalinke, U. (2008). Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity 28, 675–686.
Rice, G., Patrick, T., Parmar, R., Taylor, C. F., Aeby, A., Aicardi, J., Artuch, R., Montalto, S. A., Bacino, C. A., Barroso, B., Baxter, P., Benko, W. S., Bergmann, C., Bertini, E., Biancheri, R., Blair, E. M., Blau, N., Bonthron, D. T., Briggs, T., Brueton, L. A., Brunner, H. G., Burke, C. J., Carr, I. M., Carvalho, D. R., Chandler, K. E., Christen, H. J., Corry, P. C., Cowan, F. M., Cox, H., D’Arrigo, S., Dean, J., De Laet, C., De Praeter, C., Dery, C., Ferrie, C. D., Flintoff, K., Frints, S. G., Garcia-Cazorla, A., Gener, B., Goizet, C., Goutieres, F., Green, A. J., Guet, A., Hamel, B. C., Hayward, B. E., Heiberg, A., Hennekam, R. C., Husson, M., Jackson, A. P., Jayatunga, R., Jiang, Y. H., Kant, S. G., Kao, A., King, M. D., Kingston, H. M., Klepper, J., van der Knaap, M. S., Kornberg, A. J., Kotzot, D., Kratzer, W., Lacombe, D., Lagae, L., Landrieu, P. G., Lanzi, G., Leitch, A., Lim, M. J., Livingston, J. H., Lourenco, C. M., Lyall, E. G., Lynch, S. A., Lyons, M. J., Marom, D., McClure, J. P., McWilliam, R., Melancon, S. B., Mewasingh, L. D., Moutard, M. L., Nischal, K. K., Ostergaard, J. R., Prendiville, J., Rasmussen, M., Rogers, R. C., Roland, D., Rosser, E. M., Rostasy, K., Roubertie, A., Sanchis, A., Schiffmann, R., Scholl-Burgi, S., Seal, S., Shalev, S. A., Corcoles, C. S., Sinha, G. P., Soler, D., Spiegel, R., Stephenson, J. B., Tacke, U., Tan, T. Y., Till, M., Tolmie, J. L., Tomlin, P., Vagnarelli, F., Valente, E. M., Van Coster, R. N., Van der, A. N., Vanderver, A., Vles, J. S., Voit, T., Wassmer, E., Weschke, B., Whiteford, M. L., Willemsen, M. A., Zankl, A., Zuberi, S. M., Orcesi, S., Fazzi, E., Lebon, P., and Crow, Y. J. (2007). Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am. J. Hum. Genet. 81, 713–725.
Rice, G. I., Bond, J., Asipu, A., Brunette, R. L., Manfield, I. W., Carr, I. M., Fuller, J. C., Jackson, R. M., Lamb, T., Briggs, T. A., Ali, M., Gornall, H., Couthard, L. R., Aeby, A., Attard-Montalto, S. P., Bertini, E., Bodemer, C., Brockmann, K., Brueton, L. A., Corry, P. C., Desguerre, I., Fazzi, E., Cazorla, A. G., Gener, B., Hamel, B. C., Heiberg, A., Hunter, M., van der Knaap, M. S., Kumar, R., Lagae, L., Landrieu, P. G., Lourenco, C. M., Marom, D., McDermott, M. F., van der, M. W., Orcesi, S., Prendiville, J. S., Rasmussen, M., Shalev, S. A., Soler, D. M., Shinawi, M., Spiegel, R., Tan, T. Y., Vanderver, A., Wakeling, E. L., Wassmer, E., Whittaker, E., Lebon, P., Stetson, D. B., Bonthron, D. T., and Crow, Y. J. (2009). Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat. Genet. 41, 829–832.
Rydberg, B., and Game, J. (2002). Excision of misincorporated ribonucleotides in DNA by RNase H (type 2) and FEN-1 in cell-free extracts. Proc. Natl. Acad. Sci. U.S.A. 99, 16654–16659.
Schlee, M., Roth, A., Hornung, V., Hagmann, C. A., Wimmenauer, V., Barchet, W., Coch, C., Janke, M., Mihailovic, A., Wardle, G., Juranek, S., Kato, H., Kawai, T., Poeck, H., Fitzgerald, K. A., Takeuchi, O., Akira, S., Tuschl, T., Latz, E., Ludwig, J., and Hartmann, G. (2009). Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31, 25–34.
Schmidt, H., Raasch, J., Merkler, D., Klinker, F., Krauss, S., Bruck, W., and Prinz, M. (2009). Type I interferon receptor signaling is induced during demyelination while its function for myelin damage and repair is redundant. Exp. Neurol. 216, 306–311.
Schwid, S. R., and Panitch, H. S. (2007). Full results of the evidence of interferon dose-response-European North American comparative efficacy (EVIDENCE) study: a multicenter, randomized, assessor-blinded comparison of low-dose weekly versus high-dose, high-frequency interferon beta-1a for relapsing multiple sclerosis. Clin. Ther. 29, 2031–2048.
Seth, R. B., Sun, L., Ea, C. K., and Chen, Z. J. (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682.
Shimizu, J., Hatanaka, Y., Hasegawa, M., Iwata, A., Sugimoto, I., Date, H., Goto, J., Shimizu, T., Takatsu, M., Sakurai, Y., Nakase, H., Uesaka, Y., Hashida, H., Hashimoto, K., Komiya, T., and Tsuji, S. (2010). IFNbeta-1b may severely exacerbate Japanese optic-spinal MS in neuromyelitis optica spectrum. Neurology 75, 1423–1427.
Stetson, D. B., Ko, J. S., Heidmann, T., and Medzhitov, R. (2008). Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598.
Stockinger, B., and Veldhoen, M. (2007). Differentiation and function of Th17 T cells. Curr. Opin. Immunol. 19, 281–286.
Tamura, T., Yanai, H., Savitsky, D., and Taniguchi, T. (2008). The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 26, 535–584.
Teige, I., Treschow, A., Teige, A., Mattsson, R., Navikas, V., Leanderson, T., Holmdahl, R., and Issazadeh-Navikas, S. (2003). IFN-beta gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J. Immunol. 170, 4776–4784.
Uzawa, A., Mori, M., Hayakawa, S., Masuda, S., and Kuwabara, S. (2010). Different responses to interferon beta-1b treatment in patients with neuromyelitis optica and multiple sclerosis. Eur. J. Neurol. 17, 672–676.
Veldhoen, M., Hocking, R. J., Atkins, C. J., Locksley, R. M., and Stockinger, B. (2006). TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189.
Warabi, Y., Matsumoto, Y., and Hayashi, H. (2007). Interferon beta-1b exacerbates multiple sclerosis with severe optic nerve and spinal cord demyelination. J. Neurol. Sci. 252, 57–61.
Woodward, J. J., Iavarone, A. T., and Portnoy, D. A. (2010). c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328, 1703–1705.
Xu, L. G., Wang, Y. Y., Han, K. J., Li, L. Y., Zhai, Z., and Shu, H. B. (2005). VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19, 727–740.
Yan, N., Regalado-Magdos, A. D., Stiggelbout, B., Lee-Kirsch, M. A., and Lieberman, J. (2010). The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 11, 1005–1013.
Yang, Y. G., Lindahl, T., and Barnes, D. E. (2007). Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131, 873–886.
Keywords: interferon, experimental autoimmune encephalomyelitis, RIG-I, MDA5, TREX1, AGS, SAMHD1, RNASEH2
Citation: Prinz M and Knobeloch K-P (2012) Type I interferons as ambiguous modulators of chronic inflammation in the central nervous system. Front. Immun. 3:67. doi: 10.3389/fimmu.2012.00067
Received: 16 December 2011; Paper pending published: 07 February 2012;
Accepted: 19 March 2012; Published online: 09 April 2012.
Edited by:
Masaaki Murakami, Osaka University, JapanReviewed by:
Barbara Viviani, University of Milan, ItalyEric Huseby, University of Massachusetts Medical School, USA
Copyright: © 2012 Prinz and Knobeloch. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Marco Prinz, Department of Neuropathology, University Clinic Freiburg, Breisacher Str. 64, 79106 Freiburg im Breisgau, Germany. e-mail:bWFyY28ucHJpbnpAdW5pa2xpbmlrLWZyZWlidXJnLmRl