 Satoshi Ueha1,2
Satoshi Ueha1,2- 1 Department of Molecular Preventive Medicine, Graduate School of Medicine, The University of Tokyo, Bunkyo-ku, Tokyo, Japan
- 2 Japan Science and Technology Agency, Core Research for Evolutional Science and Technology, Tokyo, Japan
- 3 Department of Pharmacology, University of Melbourne, Melbourne, VIC, Australia
Organ fibrosis is a pathological condition associated with chronic inflammatory diseases. In fibrosis, excessive deposition of extracellular matrix (ECM) severely impairs tissue architecture and function, eventually resulting in organ failure. This process is mediated primarily by the induction of myofibroblasts, which produce large amounts of collagen I, the main component of the ECM. Accordingly, the origin, developmental pathways, and mechanisms of myofibroblast regulation are attracting increasing attention as potential therapeutic targets. The fibrotic cascade, from initial epithelial damage to eventual myofibroblast induction, is mediated by complex biological processes such as macrophage infiltration, a shift from Th1 to Th2 phenotype, and by inflammatory mediators such as transforming growth factor-β. Here, we review the current understanding of the cellular and molecular mechanisms underlying organ fibrosis.
Introduction
Organ fibrosis is an intractable, progressive condition that arises in multi-factorial chronic inflammatory diseases in which excessive deposition of extracellular matrix (ECM), mainly composed of collagen I (Col I), severely impairs tissue architecture and function, eventually resulting in organ failure (Kis et al., 2011). Fibrosis affects various organs following tissue injury, including the lungs, liver, and kidneys, and has become a major cause of death in the developed world.
Lung fibrosis occurs mainly in idiopathic interstitial pneumonia (IIPs), a general term describing multi-factorial conditions such as idiopathic pulmonary fibrosis (IPF), non-specific interstitial pneumonia (NSIP), and cryptogenic organizing pneumonia (COP). IPF is a chronic and progressive disease with an estimated prevalence of 20 cases per 100,000. The prognosis for patients with IPF is poor, and 50% die within 3 years of diagnosis.
Hepatic fibrosis (fibrosis of the liver) can be triggered by the hepatitis virus or alcohol. There are an estimated 350 million and 180 million carriers of the Hepatitis B (HBV) and C (HCV) viruses worldwide, respectively. In Japan, deaths from hepatic cirrhosis total around 15,000 per year (HCV, 50%; HBV, 12%; non B/non C, 4%; alcoholic hepatitis, 13%). In addition, hepatic cirrhosis is associated with hepatic cancer, which causes over 30,000 deaths annually. The prevalence of non-alcoholic steatohepatitis (NASH) ranges from 9 to 37% of the population depending on the country, and a subset of NASH patients eventually develops hepatitis and hepatic cancer.
Kidney fibrosis commonly occurs in glomerulonephritis and diabetic nephropathy. While the number of patients requiring dialysis due to chronic glomerulonephritis has decreased in recent years, the number of those with diabetic nephropathy continues to increase year by year. The cost of dialysis represents a considerable medical expense in advanced countries. In addition, organ fibrosis is associated with autoimmune diseases. About 15–30% of rheumatoid arthritis patients develop IPF, and about 30% of IIP cases are associated with autoimmune diseases.
Given the prevalence and severity of diseases involving tissue fibrosis, the prevention, and treatment of this condition remains a major medical challenge. This review focuses on the cellular and molecular bases for the accumulation of Col I producing fibroblasts and myofibroblasts, which are responsible for the excessive deposition of ECM during the fibrotic process.
The Origin of Col I Producing Fibroblasts and Myofibroblasts
Fibroblasts are non-hematopoietic, non-epithelial, non-endothelial cells that widely distribute throughout the mesenchyme where they synthesize ECM proteins that form a structural framework to support tissue architecture and function in steady-state conditions. Fibroblasts also play an important role in tissue repair following multi-factorial tissue damage by forming a provisional ECM, a process preceding re-epithelialization in successful repair. Unfortunately, dysregulated activation, proliferation, and survival of fibroblasts often results in the excessive deposition of ECM proteins and the inhibition of re-epithelialization, leading to tissue fibrosis (Gabbiani, 2003). Therefore, control of the activation, proliferation, and survival of fibroblasts is critical for the prevention and treatment of tissue fibrosis.
Fibroblasts form clusters within fibrotic tissues that are known as fibrotic foci (Visscher and Myers, 2006). These fibroblasts include α-smooth muscle actin (αSMA) expressing myofibroblasts that have the potential to produce large amounts of Col I, which has resulted in this cell population being widely considered to be the key effector cells in organ fibrosis (Gabbiani et al., 1971; Gabbiani, 2003; Sandbo and Dulin, 2011). Results in some models of organ fibrosis have suggested that there may be therapeutic benefit in targeting myofibroblasts, although the experimental approaches in these models leave-questions remaining about the selectivity of the interventions for myofibroblasts (Douglass et al., 2008). As mentioned above, fibroblasts are immunophenotypically identified as cells negative for hematopoietic, epithelial, and endothelial makers. The lack of specific markers for fibroblasts or possible subpopulations, including myofibroblasts, complicates the cellular and molecular understanding of these cells. Thus, the establishment of specific markers to identify fibroblasts and myofibroblasts remains a major challenge in this field.
Myofibroblasts have classically been considered to differentiate from tissue-resident fibroblasts. However, recent studies have suggested alternative sources of myofibroblasts (Hinz et al., 2007). Bone marrow-derived fibrocytes express both hematopoietic markers (CD45, CD11b, and HLADR) and ECM proteins (Col I and vimentin). These cells have been shown to be recruited from the circulation to inflamed tissues via chemokine receptors CXCR4 and CCR1, 2, 5, and 7, after which they differentiate into myofibroblasts (Phillips et al., 2004; Keeley et al., 2011). Epithelial cells are reported to trans-differentiate into myofibroblasts via chronic inflammation-induced epithelial–mesenchymal transition (EMT) in several fibrosis models (Kalluri and Neilson, 2003). In addition, blood vessel wall smooth muscle cells have been proposed as myofibroblast progenitors. Meanwhile, stellate cells (Ito cells), a type of hepatic pericyte, have attracted interest as a major precursor of Col I producing fibroblasts and myofibroblasts in the liver (Atzori et al., 2009). Despite these studies, overall understanding of the origin and differentiation pathways of Col I producing fibroblasts and myofibroblasts remains poor. Identification of the major developmental pathway of these cells will be an essential step toward the development of therapeutic interventions for organ fibrosis.
Challenging the EMT Hypothesis
Epithelial–mesenchymal transition is a process that was originally characterized in the context of embryonic development, in which epithelial cells lose their original phenotypic and functional features, including cell–cell adhesion and cell polarity, while acquiring migratory and invasive properties (Thiery et al., 2009). In vitro cell culture studies have shown clearly and reproducibly that transforming growth factor-β (TGFβ) treatment of epithelial cells induces expression of mesenchymal markers and morphology with a concomitant loss of epithelial markers (Qi et al., 2005; Venkov et al., 2007). Over the past 15 years, numerous studies have proposed that EMT also contributes to the activated fibroblast pool in various regenerative and pathogenic processes. For example, transition from epithelial tumor cells to mesenchymal cells occurs at the invasive front of many tumors, driving tumor progression and metastasis. In addition, inflammation-induced epithelial cell damage in parenchymal organs such as the liver, lungs, and kidneys recapitulates part of the EMT process in that epithelial cells acquire mesenchymal cell-like properties and migrate beyond the basal membrane to the interstitium, where they differentiate into Col I producing fibroblasts and myofibroblasts. However, the inflammation-associated EMT hypothesis has been challenged by an increasing number of studies, and lacks convincing evidence (Wells, 2010; Kriz et al., 2011).
For example, the EMT hypothesis for kidney fibrosis was first reported by Strutz et al. (1995), when the authors used FSP-1 (fibroblast specific protein-1/S100A4) as a marker of mesenchymal lineage. However, subsequent characterization revealed that FSP-1 is not a mesenchymal cell specific marker, and is expressed on leukocytes and endothelial cells as well. Similarly, expression of vimentin, another marker commonly used in EMT studies, is not enough on its own to identify mesenchymal cells, because a subset of epithelial cells express vimentin in both resting and inflammatory-states (Grone et al., 1987; Witzgall et al., 1994). Moreover, recent extensive and well designed cell-fate tracing studies have not provided any evidence for inflammation-associated EMT (Humphreys et al., 2010; Scholten et al., 2010). Unless the inflammation-induced conversion of epithelial cells into Col I producing fibroblasts and myofibroblasts in vivo can be demonstrated more convincingly, the role of EMT in organ fibrosis should be reconsidered.
Fibrocytes Make Only a Minimal Contribution to Organ Fibrosis
The existence of bone marrow-derived fibrocytes was originally reported by Bucala et al. (1994). Later, Strieter and colleagues reported that fibrocytes express several chemokine receptors and are recruited to inflamed tissues in a CXCR4 dependent manner, where they contribute to the Col I producing myofibroblast pool after bleomycin-induced epithelial injury in the lungs (Phillips et al., 2004). We have also demonstrated that blocking chemokine receptors CCR1, 2, 5, and 7 in mouse lung or kidney fibrosis models reduces the number of myofibroblasts detected and ameliorates organ fibrosis (Sakai et al., 2006; Ishida et al., 2007). However, it remains unclear whether the cognate chemokines regulate organ fibrosis through the recruitment of fibrocytes to the inflamed tissues, by influencing the activation or differentiation of fibroblasts, or through the recruitment of inflammatory cells such as macrophages and neutrophils that subsequently influence the tissue microenvironment. While many studies have confirmed the presence of fibrocytes in fibrotic disease, accumulating experimental evidence suggests that the contribution of bone marrow-derived cells to the Col I producing fibroblast/myofibroblast pool is limited (Higashiyama et al., 2009, 2011).
Origin of Capillary Pericytes and Their Similarity with Tissue Fibroblasts
Recently, a novel role for pericytes as precursors of pro-fibrotic Col I producing cells has been described. Studies using Col 1α2–GFP transgenic mice have demonstrated that CD73+PDGFRβ+ pericytes/fibroblasts migrate from capillaries to the interstitial space and differentiate to Col 1 producing myofibroblasts in kidney and liver fibrosis models (Lin et al., 2008; Higashiyama et al., 2009). In addition, Goritz et al. (2011) recently demonstrated that a specific pericyte subtype gives rise to scar-forming stromal cells in the injured spinal cord. However, because fibroblasts in the interstitial space not only provide a scaffold for micro-tissue architecture such as nephrons and renal tubules (in the case of the kidneys), but also come into direct contact with microvessels, it is often difficult to distinguish between pericytes and tissue fibroblasts under steady-state conditions (Kriz et al., 2011). The similarities, differences, and lineage relationship between pericytes and tissue fibroblasts remain to be elucidated.
The Role of Inflammatory Cells in Fibrotic Tissue
Macrophage infiltration into inflamed tissues has been implicated in chronic inflammation-induced organ fibrosis (Wynn and Barron, 2010). Inflamed tissue-infiltrating macrophages are derived from CCR2+ inflammatory monocytes or CX3CR1hi resident monocytes (Ricardo et al., 2008). The phenotype of these macrophages is generally reported to match that of alternatively activated cells (M2) rather than classically activated cells (M1). M2 macrophages express immunosuppressive molecules such as IL-10 and arginase I, which suppress the induction of Th1 cells that produce the anti-fibrotic cytokine IFNγ. On the other hand, M1 macrophages express IL-1, IL-12, IL-23, and induce Th1 cell infiltration and activation. However, it remains to be established whether a particular macrophage subset with M2-type properties preferentially infiltrates into fibrotic tissues, or whether it is the pro-fibrotic microenvironment that drives macrophage polarization toward an M2 phenotype. In addition to their roles in immune regulation, macrophages play a pivotal role in matrix regression during the recovery phase of fibrosis (Duffield et al., 2005) and in the regulation of stellate cell proliferation (Olaso et al., 2011). In the future, conditional and lineage specific depletion or gene targeting approaches may help to reveal the specific function and overall role of each macrophage subset in tissue fibrosis.
The contribution of T lymphocytes to organ fibrosis seems to be context dependent. While a number of studies suggest an exacerbating role of T cells in fibrosis, T cells also appear to be dispensable because T cell-deficient mice develop fibrosis in some models (Luzina et al., 2008). The general concept is that prolonged inflammation induces a shift from a Th1 to Th2 phenotype, and the resulting production of Th2 cytokines induces the infiltration of pro-fibrotic eosinophils via cognate chemokine (e.g., eotaxin) production. On the other hand, a role for recently identified functional T cell subsets such as Th17 and regulatory T cells in tissue fibrosis has also begun to emerge. For example, adoptive transfer of CD4 T cells restored bacterial-induced lung inflammatory and fibrotic responses in TCRβ deficient mice with an accompanying increase in lung IL-17A protein levels, and IL-17 receptor α deficient mice develop less severe inflammation and fibrosis than wild type counterparts (Simonian et al., 2009). Recently, platelet-derived growth factor (PDGF)-producing CD4+Foxp3+Tregs have been shown to promote lung fibrosis by activating fibroblasts (Lo Re et al., 2011). A better understanding of the roles that inflammatory cells play in the fibrotic process may reveal new points of therapeutic intervention, which may be able to induce a shift from a pro-fibrotic microenvironment to an anti-fibrotic microenvironment.
Regulation of Fibrosis by Inflammatory Mediators
The fibrotic signaling cascade that occurs during chronic inflammation, which is initiated by epithelial injury and results in irreversible organ damage, is regulated by various inflammatory mediators. The pro-fibrotic roles of plasma components, platelet-derived soluble factors, and cytokines produced by activated tissue cells and infiltrating leukocytes, have been demonstrated in animal models. These mediators include factors induced as a part of an inflammatory cascade, regulatory molecules that provide feedback during the inflammatory response, and factors constitutively expressed in the body.
Transforming growth factor-β plays a central role in fibroblast activation and fibroblast-to-myofibroblast differentiation, and induces the expression of genes for ECM components including Col 1. However, despite its great potential as a therapeutic target for fibrosis, inhibition of TGFβ signaling has unacceptable side effects due to the critical role of this cytokine in the maintenance of homeostasis (Leask, 2010).
Bone morphogenic proteins (BMPs) belong to the TGFβ family and regulate proliferation and differentiation of both mesenchymal cells and epithelial cells (Rider and Mulloy, 2010). Recent studies have revealed that BMP7 prevents fibrosis by promoting epithelial regeneration, while BMP antagonists such as gremlin and ectodin drive organ fibrosis by inhibiting BMP7 signaling. Interestingly, there is a direct Smad-dependent counteraction of the TGFβ pathway by BMP7 signaling, and vice versa (Zeisberg et al., 2003).
G-protein coupled receptor ligands also regulate chronic inflammation and the fibrotic cascade. Angiotensin II (Ang II) induces the expression of pro-fibrotic factors such as connective tissue growth factor (CTGF; Ruperez et al., 2003; Esteban et al., 2004), and recent studies have revealed that there is an intracellular cross-talk between Ang II signaling and TGFβ signaling that cooperatively promotes fibrosis (Campbell and Katwa, 1997; Schultz Jel et al., 2002; Gao et al., 2009). Leukotrienes (LTs) not only induce fibroblast migration, proliferation, and matrix protein synthesis, but also promote fibrosis through the stimulation and activation of TGFβ (Shim et al., 2006). On the contrary, prostaglandin E2 (PGE2), which has well established anti-inflammatory activities, may suppress fibrosis by inhibiting the proliferation, migration, and differentiation of myofibroblasts (Kohyama et al., 2001; Lama et al., 2002; Thomas et al., 2007). Recent studies have demonstrated that PGF2a receptor deficient mice are resistant against bleomycin-induced lung fibrosis (Oga et al., 2009), and that LTB4 receptor inhibitors and LPA1 inhibitors suppress bleomycin-induced lung fibrosis (Tager et al., 2008). Lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P) are liberated from stored lipid precursors through enzymatic activation and provide migration, proliferation, and differentiation signals to a variety of cells through the LPA receptors (LPA1–8) and S1P receptors (S1P1–5), respectively (Pattanaik and Postlethwaite, 2010). LPA1 deficient mice are protected from bleomycin-induced lung fibrosis and unilateral ureteral ligation induced-renal fibrosis (Tager et al., 2008). The pro-fibrotic role of LPA is reportedly mediated in part by the induction of fibroblast-to-myofibroblast differentiation (Yin et al., 2008). S1P plays a critical role in the circulation of lymphocytes, and accordingly, inhibition of the S1P–S1P1 axis results in strong immunosuppressive effects. In addition, S1P also regulates the migration and activation of fibroblasts, and recent studies have revealed cross-talk between the S1P3 and TGFβ – Smad signaling pathways that promote cardiac fibrosis (Takuwa et al., 2010).
Plasma coagulation cascade proteases are also involved in fibrosis (Chambers and Laurent, 2002); thrombin, factor VII, and factor Xa activate protease-activated receptor-1 (PAR-1) on fibroblasts and induce their proliferation. In addition, these proteases promote fibrosis through the induction of pro-fibrotic molecules such as platelet-derived growth factors and CTGF. CTGF mediates mesenchymal stem cell (MSC)-to-fibroblast differentiation as well as fibroblast activation (Ponticos et al., 2009; Lee et al., 2010), while PDGFs induce the proliferation and activation of fibroblasts leading to vascular diseases and fibrosis. Ijichi et al. (2011) have demonstrated that CXC chemokines induce CTGF expression in fibroblasts, and that the inhibition of CXCR2 in tumor-bearing mice impairs tumor progression.
Matrix metalloproteinases (MMPs) and their inhibitors, tissue inhibitors of MMPs (TIMPs), play an important role in the regulation of ECM turnover in fibrotic tissues. While the degradation of pathological fibrillar collagen by MMPs is a key event in the resolution of fibrosis, the degradation of normal ECM components in the early stages of fibrosis promotes deposition of newly synthesized collagen (Hemmann et al., 2007).
ATP released from damaged epithelial cells serves as a danger signal to alert the immune system of tissue damage, and may also trigger a fibrotic cascade (Mortaz et al., 2010). Activation of the Wnt/β-catenin signaling pathway, which regulates epithelial and mesenchymal proliferation and activation, has been demonstrated in lung epithelial cells of IPF patients. Overall, this activation drives fibrosis rather than epithelial repair, possibly due to cross-talk with other pro-fibrotic factors such as TGFβ and CTGF (Konigshoff and Eickelberg, 2010). Furthermore, inhibition of Wnt signaling (Henderson et al., 2010) and the BMP binding protein ectodin (Tanaka et al., 2010) ameliorates renal fibrosis. A better understanding of the role of each inflammatory mediator in the fibrotic cascade is likely to reveal novel molecular targets for the early diagnosis, prevention, and treatment of fibrotic disease.
Conclusion and Future Perspectives
In recent years, confusion has surrounded the major source of myofibroblasts in fibrosis, with attention centering on tissue-resident fibroblasts and pericytes (Figure 1). However, the relative importance of the various developmental pathways of Col I producing fibroblasts and myofibroblasts needs to be re-examined by lineage tracing approaches, utilizing cell-type specific promoters, and inducible systems in a range of fibrosis models. It will also be important to further elucidate the mechanisms underlying the maintenance of myofibroblasts during chronic inflammation. It is possible that precursor cells provide a continuous supply of myofibroblasts, that myofibroblasts have proliferative potential, or that the myofibroblast lifespan is relatively long. A deeper understanding of the population dynamics of myofibroblasts and their precursors may reveal new points of therapeutic intervention with the potential to halt myofibroblast accumulation in fibrotic tissue.
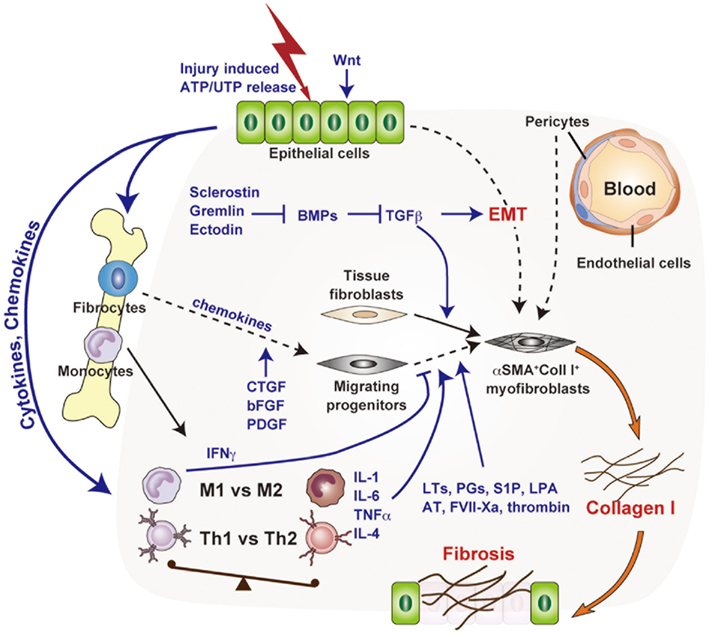
Figure 1. Molecular and cellular mechanisms of chronic inflammation-associated organ fibrosis. Organ fibrosis is mediated primarily by the induction of myofibroblasts, which produce large amounts of collagen I. Tissue fibroblasts, transdifferentiated epithelial cells (EMT), bone marrow-derived fibrocytes, and pericytes have attracted interest as potential myofibroblast precursors. The fibrotic cascade, from initial epithelial damage to eventual myofibroblast induction, is mediated by complex biological processes such as macrophage infiltration, a shift from Th1 to Th2 phenotype, and by inflammatory mediators such as transforming growth factor-β.
Although removal of the cause of chronic inflammation is essential and effective for the prevention and treatment of tissue fibrosis (for example, virus clearance by interferon effectively prevents viral hepatitis-associated fibrosis), this can be challenging as the precise cause of the inflammation is often unclear. Given that in most cases steroids are largely ineffective against fibrosis, currently there is no effective drug available for patients with clinically significant organ fibrosis. Further elucidation of the molecular and cellular bases for chronic inflammation-associated organ fibrosis is imperative for the development of effective anti-fibrotic therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was in part supported by The Ministry of Education, Culture, Sports, Science and Technology (MEXT) KAKENHI Grants-in-Aid for Young Scientists (B) 23790432 (S. Ueha) and Grants-in-Aid for Scientific Research (B) 22390095, and Grants-in-Aid for Scientific Research on Priority Areas 19059004 (K. Matsushima).
Abbreviations
smooth muscle actin; Ang II, angiotensin II; BMP, bone morphogenic protein; Col I, collagen I; CTGF, connective tissue growth factor; EMT, epithelial–mesenchymal transition; FSP-1, fibroblast specific protein-1; HBV/HCV, hepatitis B/C virus; IIP, idiopathic interstitial pneumonia; IPF, idiopathic pulmonary fibrosis; LPA, lysophosphatidic acid; LT, leukotriene; MMP, matrix metalloproteinase; MSC, mesenchymal stem cell; PDGF, platelet-derived growth factor; PGE2, prostaglandin E2; S1P, sphingosine-1-phosphate; TGFβ, transforming growth factor-β; TIMP, tissue inhibitor of matrix metalloproteinases.
References
Atzori, L., Poli, G., and Perra, A. (2009). Hepatic stellate cell: a star cell in the liver. Int. J. Biochem. Cell Biol. 41, 1639–1642.
Bucala, R., Spiegel, L. A., Chesney, J., Hogan, M., and Cerami, A. (1994). Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med. 1, 71–81.
Campbell, S. E., and Katwa, L. C. (1997). Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts. J. Mol. Cell. Cardiol. 29, 1947–1958.
Chambers, R. C., and Laurent, G. J. (2002). Coagulation cascade proteases and tissue fibrosis. Biochem. Soc. Trans. 30, 194–200.
Douglass, A., Wallace, K., Parr, R., Park, J., Durward, E., Broadbent, I., Barelle, C., Porter, A. J., and Wright, M. C. (2008). Antibody-targeted myofibroblast apoptosis reduces fibrosis during sustained liver injury. J. Hepatol. 49, 88–98.
Duffield, J. S., Forbes, S. J., Constandinou, C. M., Clay, S., Partolina, M., Vuthoori, S., Wu, S., Lang, R., and Iredale, J. P. (2005). Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest. 115, 56–65.
Esteban, V., Lorenzo, O., Ruperez, M., Suzuki, Y., Mezzano, S., Blanco, J., Kretzler, M., Sugaya, T., Egido, J., and Ruiz-Ortega, M. (2004). Angiotensin II, via AT1 and AT2 receptors and NF-kappaB pathway, regulates the inflammatory response in unilateral ureteral obstruction. J. Am. Soc. Nephrol. 15, 1514–1529.
Gabbiani, G. (2003). The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 200, 500–503.
Gabbiani, G., Ryan, G. B., and Majne, G. (1971). Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 27, 549–550.
Gao, X., He, X., Luo, B., Peng, L., Lin, J., and Zuo, Z. (2009). Angiotensin II increases collagen I expression via transforming growth factor-beta1 and extracellular signal-regulated kinase in cardiac fibroblasts. Eur. J. Pharmacol. 606, 115–120.
Goritz, C., Dias, D. O., Tomilin, N., Barbacid, M., Shupliakov, O., and Frisen, J. (2011). A pericyte origin of spinal cord scar tissue. Science 333, 238–242.
Grone, H. J., Weber, K., Grone, E., Helmchen, U., and Osborn, M. (1987). Coexpression of keratin and vimentin in damaged and regenerating tubular epithelia of the kidney. Am. J. Pathol. 129, 1–8.
Hemmann, S., Graf, J., Roderfeld, M., and Roeb, E. (2007). Expression of MMPs and TIMPs in liver fibrosis – a systematic review with special emphasis on anti-fibrotic strategies. J. Hepatol. 46, 955–975.
Henderson, W. R. Jr., Chi, E. Y., Ye, X., Nguyen, C., Tien, Y. T., Zhou, B., Borok, Z., Knight, D. A., and Kahn, M. (2010). Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc. Natl. Acad. Sci. U.S.A. 107, 14309–14314.
Higashiyama, R., Moro, T., Nakao, S., Mikami, K., Fukumitsu, H., Ueda, Y., Ikeda, K., Adachi, E., Bou-Gharios, G., Okazaki, I., and Inagaki, Y. (2009). Negligible contribution of bone marrow-derived cells to collagen production during hepatic fibrogenesis in mice. Gastroenterology 137, 1459–1466 e1451.
Higashiyama, R., Nakao, S., Shibusawa, Y., Ishikawa, O., Moro, T., Mikami, K., Fukumitsu, H., Ueda, Y., Minakawa, K., Tabata, Y., Bou-Gharios, G., and Inagaki, Y. (2011). Differential contribution of dermal resident and bone marrow-derived cells to collagen production during wound healing and fibrogenesis in mice. J. Invest. Dermatol. 131, 529–536.
Hinz, B., Phan, S. H., Thannickal, V. J., Galli, A., Bochaton-Piallat, M. L., and Gabbiani, G. (2007). The myofibroblast: one function, multiple origins. Am. J. Pathol. 170, 1807–1816.
Humphreys, B. D., Lin, S. L., Kobayashi, A., Hudson, T. E., Nowlin, B. T., Bonventre, J. V., Valerius, M. T., McMahon, A. P., and Duffield, J. S. (2010). Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 176, 85–97.
Ijichi, H., Chytil, A., Gorska, A. E., Aakre, M. E., Bierie, B., Tada, M., Mohri, D., Miyabayashi, K., Asaoka, Y., Maeda, S., Ikenoue, T., Tateishi, K., Wright, C. V., Koike, K., Omata, M., and Moses, H. L. (2011). Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J. Clin. Invest. 121, 4106–4117.
Ishida, Y., Kimura, A., Kondo, T., Hayashi, T., Ueno, M., Takakura, N., Matsushima, K., and Mukaida, N. (2007). Essential roles of the CC chemokine ligand 3-CC chemokine receptor 5 axis in bleomycin-induced pulmonary fibrosis through regulation of macrophage and fibrocyte infiltration. Am. J. Pathol. 170, 843–854.
Kalluri, R., and Neilson, E. G. (2003). Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 112, 1776–1784.
Keeley, E. C., Mehrad, B., and Strieter, R. M. (2011). The role of fibrocytes in fibrotic diseases of the lungs and heart. Fibrogenesis Tissue Repair 4, 2.
Kis, K., Liu, X., and Hagood, J. S. (2011). Myofibroblast differentiation and survival in fibrotic disease. Expert Rev. Mol. Med. 13, e27.
Kohyama, T., Ertl, R. F., Valenti, V., Spurzem, J., Kawamoto, M., Nakamura, Y., Veys, T., Allegra, L., Romberger, D., and Rennard, S. I. (2001). Prostaglandin E(2) inhibits fibroblast chemotaxis. Am. J. Physiol. Lung Cell Mol. Physiol. 281, L1257–L1263.
Konigshoff, M., and Eickelberg, O. (2010). WNT signaling in lung disease: a failure or a regeneration signal? Am. J. Respir. Cell Mol. Biol. 42, 21–31.
Kriz, W., Kaissling, B., and Le Hir, M. (2011). Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J. Clin. Invest. 121, 468–474.
Lama, V., Moore, B. B., Christensen, P., Toews, G. B., and Peters-Golden, M. (2002). Prostaglandin E2 synthesis and suppression of fibroblast proliferation by alveolar epithelial cells is cyclooxygenase-2-dependent. Am. J. Respir. Cell Mol. Biol. 27, 752–758.
Leask, A. (2010). Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ. Res. 106, 1675–1680.
Lee, C. H., Shah, B., Moioli, E. K., and Mao, J. J. (2010). CTGF directs fibroblast differentiation from human mesenchymal stem/stromal cells and defines connective tissue healing in a rodent injury model. J. Clin. Invest. 120, 3340–3349.
Lin, S. L., Kisseleva, T., Brenner, D. A., and Duffield, J. S. (2008). Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 173, 1617–1627.
Lo Re, S., Lecocq, M., Uwambayinema, F., Yakoub, Y., Delos, M., Demoulin, J. B., Lucas, S., Sparwasser, T., Renauld, J. C., Lison, D., and Huaux, F. (2011). PDGF-producing CD4+ Foxp3+ regulatory T lymphocytes promote lung fibrosis. Am. J. Respir. Crit. Care Med. 184, 1270–1281.
Luzina, I. G., Todd, N. W., Iacono, A. T., and Atamas, S. P. (2008). Roles of T lymphocytes in pulmonary fibrosis. J. Leukoc. Biol. 83, 237–244.
Mortaz, E., Folkerts, G., Nijkamp, F. P., and Henricks, P. A. (2010). ATP and the pathogenesis of COPD. Eur. J. Pharmacol. 638, 1–4.
Oga, T., Matsuoka, T., Yao, C., Nonomura, K., Kitaoka, S., Sakata, D., Kita, Y., Tanizawa, K., Taguchi, Y., Chin, K., Mishima, M., Shimizu, T., and Narumiya, S. (2009). Prostaglandin F(2alpha) receptor signaling facilitates bleomycin-induced pulmonary fibrosis independently of transforming growth factor-beta. Nat. Med. 15, 1426–1430.
Olaso, E., Arteta, B., Benedicto, A., Crende, O., and Friedman, S. L. (2011). Loss of discoidin domain receptor 2 promotes hepatic fibrosis after chronic carbon tetrachloride through altered paracrine interactions between hepatic stellate cells and liver-associated macrophages. Am. J. Pathol. 179, 2894–2904.
Pattanaik, D., and Postlethwaite, A. E. (2010). A role for lysophosphatidic acid and sphingosine 1-phosphate in the pathogenesis of systemic sclerosis. Discov. Med. 10, 161–167.
Phillips, R. J., Burdick, M. D., Hong, K., Lutz, M. A., Murray, L. A., Xue, Y. Y., Belperio, J. A., Keane, M. P., and Strieter, R. M. (2004). Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Invest. 114, 438–446.
Ponticos, M., Holmes, A. M., Shi-wen, X., Leoni, P., Khan, K., Rajkumar, V. S., Hoyles, R. K., Bou-Gharios, G., Black, C. M., Denton, C. P., Abraham, D. J., Leask, A., and Lindahl, G. E. (2009). Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum. 60, 2142–2155.
Qi, W., Twigg, S., Chen, X., Polhill, T. S., Poronnik, P., Gilbert, R. E., and Pollock, C. A. (2005). Integrated actions of transforming growth factor-beta1 and connective tissue growth factor in renal fibrosis. Am. J. Physiol. Renal Physiol. 288, F800–F809.
Ricardo, S. D., van Goor, H., and Eddy, A. A. (2008). Macrophage diversity in renal injury and repair. J. Clin. Invest. 118, 3522–3530.
Rider, C. C., and Mulloy, B. (2010). Bone morphogenetic protein and growth differentiation factor cytokine families and their protein antagonists. Biochem. J. 429, 1–12.
Ruperez, M., Lorenzo, O., Blanco-Colio, L. M., Esteban, V., Egido, J., and Ruiz-Ortega, M. (2003). Connective tissue growth factor is a mediator of angiotensin II-induced fibrosis. Circulation 108, 1499–1505.
Sakai, N., Wada, T., Yokoyama, H., Lipp, M., Ueha, S., Matsushima, K., and Kaneko, S. (2006). Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling regulates fibrocytes in renal fibrosis. Proc. Natl. Acad. Sci. U.S.A. 103, 14098–14103.
Sandbo, N., and Dulin, N. (2011). Actin cytoskeleton in myofibroblast differentiation: ultrastructure defining form and driving function. Transl. Res. 158, 181–196.
Scholten, D., Osterreicher, C. H., Scholten, A., Iwaisako, K., Gu, G., Brenner, D. A., and Kisseleva, T. (2010). Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology 139, 987–998.
Schultz Jel, J., Witt, S. A., Glascock, B. J., Nieman, M. L., Reiser, P. J., Nix, S. L., Kimball, T. R., and Doetschman, T. (2002). TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J. Clin. Invest. 109, 787–796.
Shim, Y. M., Zhu, Z., Zheng, T., Lee, C. G., Homer, R. J., Ma, B., and Elias, J. A. (2006). Role of 5-lipoxygenase in IL-13-induced pulmonary inflammation and remodeling. J. Immunol. 177, 1918–1924.
Simonian, P. L., Roark, C. L., Wehrmann, F., Lanham, A. K., Diaz del Valle, F., Born, W. K., O’Brien, R. L., and Fontenot, A. P. (2009). Th17-polarized immune response in a murine model of hypersensitivity pneumonitis and lung fibrosis. J. Immunol. 182, 657–665.
Strutz, F., Okada, H., Lo, C. W., Danoff, T., Carone, R. L., Tomaszewski, J. E., and Neilson, E. G. (1995). Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 130, 393–405.
Tager, A. M., LaCamera, P., Shea, B. S., Campanella, G. S., Selman, M., Zhao, Z., Polosukhin, V., Wain, J., Karimi-Shah, B. A., Kim, N. D., Hart, W. K., Pardo, A., Blackwell, T. S., Xu, Y., Chun, J., and Luster, A. D. (2008). The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 14, 45–54.
Takuwa, N., Ohkura, S., Takashima, S., Ohtani, K., Okamoto, Y., Tanaka, T., Hirano, K., Usui, S., Wang, F., Du, W., Yoshioka, K., Banno, Y., Sasaki, M., Ichi, I., Okamura, M., Sugimoto, N., Mizugishi, K., Nakanuma, Y., Ishii, I., Takamura, M., Kaneko, S., Kojo, S., Satouchi, K., Mitumori, K., Chun, J., and Takuwa, Y. (2010). S1P3-mediated cardiac fibrosis in sphingosine kinase 1 transgenic mice involves reactive oxygen species. Cardiovasc. Res. 85, 484–493.
Tanaka, M., Asada, M., Higashi, A. Y., Nakamura, J., Oguchi, A., Tomita, M., Yamada, S., Asada, N., Takase, M., Okuda, T., Kawachi, H., Economides, A. N., Robertson, E., Takahashi, S., Sakurai, T., Goldschmeding, R., Muso, E., Fukatsu, A., Kita, T., and Yanagita, M. (2010). Loss of the BMP antagonist USAG-1 ameliorates disease in a mouse model of the progressive hereditary kidney disease Alport syndrome. J. Clin. Invest. 120, 768–777.
Thiery, J. P., Acloque, H., Huang, R. Y., and Nieto, M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890.
Thomas, P. E., Peters-Golden, M., White, E. S., Thannickal, V. J., and Moore, B. B. (2007). PGE(2) inhibition of TGF-beta1-induced myofibroblast differentiation is Smad-independent but involves cell shape and adhesion-dependent signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 293, L417–L428.
Venkov, C. D., Link, A. J., Jennings, J. L., Plieth, D., Inoue, T., Nagai, K., Xu, C., Dimitrova, Y. N., Rauscher, F. J., and Neilson, E. G. (2007). A proximal activator of transcription in epithelial-mesenchymal transition. J. Clin. Invest. 117, 482–491.
Visscher, D. W., and Myers, J. L. (2006). Histologic spectrum of idiopathic interstitial pneumonias. Proc. Am. Thorac. Soc. 3, 322–329.
Wells, R. G. (2010). The epithelial-to-mesenchymal transition in liver fibrosis: here today, gone tomorrow? Hepatology 51, 737–740.
Witzgall, R., Brown, D., Schwarz, C., and Bonventre, J. V. (1994). Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J. Clin. Invest. 93, 2175–2188.
Wynn, T. A., and Barron, L. (2010). Macrophages: master regulators of inflammation and fibrosis. Semin. Liver Dis. 30, 245–257.
Yin, Z., Tong, Y., Zhu, H., and Watsky, M. A. (2008). ClC-3 is required for LPA-activated Cl- current activity and fibroblast-to-myofibroblast differentiation. Am. J. Physiol. Cell Physiol. 294, C535–C542.
Keywords: fibrosis, myofibroblast, fibroblast, chemokine, TGFb, mesenchymal stem cell, collagen I, pericyte
Citation: Ueha S, Shand FHW and Matsushima K (2012) Cellular and molecular mechanisms of chronic inflammation-associated organ fibrosis. Front. Immun. 3:71. doi: 10.3389/fimmu.2012.00071
Received: 16 December 2011; Accepted: 22 March 2012;
Published online: 10 April 2012.
Edited by:
Masaaki Murakami, Osaka University, JapanCopyright: © 2012 Ueha, Shand and Matsushima. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Kouji Matsushima, Department of Molecular Preventive Medicine, Graduate School of Medicine, The University of Tokyo, 7-3-1, Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. e-mail:a291amltQG0udS10b2t5by5hYy5qcA==