 Noriko Komatsu1,3 and Hiroshi Takayanagi1,2,3*
Noriko Komatsu1,3 and Hiroshi Takayanagi1,2,3*- 1 Department of Cell Signaling, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Tokyo, Japan
- 2 Takayanagi Osteonetwork Project, Exploratory Research for Advanced Technology, Japan Science and Technology Agency, Tokyo, Japan
- 3 Global Center of Excellence Program, International Research Center for Molecular Science in Tooth and Bone Diseases, Tokyo, Japan
Rheumatoid arthritis (RA) is an immune-mediated disease of the joints that is characterized by chronic inflammation and synovial hyperplasia that eventually lead to cartilage and bone destruction. Synovial fibroblasts are mesenchymal cells recognized as a key cell population in RA due to their hyperproliferative and hypersensitive properties in the inflammatory milieu and hyperproduction of both inflammatory cytokines and matrix-degrading enzymes. On the immune cell side, a wealth of evidence has shown that CD4+T-cells, especially IL-17 producing helper T (Th17) cells, play a prominent role, particularly in the initiation of systemic immune response in RA. However, it is still unclear how the local chronic inflammation in the joint is elicited by a systemic immune response. Recent studies have shed light on the importance of the interaction between immune and mesenchymal cells in joints including synovial fibroblasts. In particular, mesenchymal cells contribute to the Th17-mediated chronic inflammation in RA by promoting the migration of Th17 cells to the inflamed site and then the homeostatic proliferation and concomitant increase in IL-17 production. In addition, recent progress in osteoimmunology has provided new insight into the pathogenesis of the bone destruction which takes place in RA. Th17-related cytokines have been shown to enhance osteoclastogenesis, mainly via synovial fibroblasts. Thus, mesenchymal cells are a determinant of the development of RA that links the systemic immune response and the local disorder in the joints. In addition, the interaction of immune and mesenchymal cells plays a key role in both the chronic inflammation and bone destruction seen in RA. Elucidation of the precise events involved in this interaction will lead to a better understanding of the mechanisms by which chronic inflammation and bone destruction in joint results from a systemic immune response, and also will help provide a molecular basis for novel therapeutic strategies to treat RA.
Introduciton
Rheumatoid arthritis (RA) afflicts up to 1% of the general population worldwide. It is a chronic inflammatory disease characterized by synovial hyperplasia and bone destruction in multiple joints (Firestein, 2003). Three of the outstanding questions in RA pathogenesis are how the systemic immune response is elicited by genetic and/or environmental factors, how this in turn results in local joint inflammation and how inflammation causes bone destruction. In the affected joints, hyperplasia of the synovial membrane is a hallmark of RA pathology, which is characterized by both hyperproliferation of synovial fibroblasts and massive infiltration of inflammatory immune cells, including CD4+T-cells and innate immune cells. Synovial fibroblasts have certain unique characteristics, such as hyperproliferative and hyperactive properties in response to an inflammatory environment, and are recognized as prominent determinants of the joint-specificity seen in RA. Therefore, it is important to establish how these pathogenetic immune cells migrate into joints and contribute to the chronic inflammation and bone destruction, especially via activation of the mesenchymal cells resident in joint, such as synovial fibroblasts. However, so far, despite the clearly evident importance and considerable effort expended, the interplay of immune and mesenchymal cells in joint is still not fully understood. Though significant roles of B-cells and antibody production are also widely appreciated in RA, here, we summarize recent findings on the RA pathogenesis by focusing on T-cells and synovial fibroblasts.
CD4+T-Cells are Indispensable for the Initiation of RA
Although the etiology of RA remains unclear, the primary role of CD4+T-cells in RA has been suggested by various findings: (1) the extensive infiltration of CD4+T-cell into the inflammatory synovium, (2) the presence of autoantibodies, (3) the association of RA susceptibility with HLA-DRB1 alleles (Perricone et al., 2011) and genes related to T-cell function such as PTPN22 (Begovich et al., 2004; Lee et al., 2005) and CCR6 (Kochi et al., 2010; Stahl et al., 2010), and (4) the efficacy of a T-cell directed therapeutic drug, CTLA-4 Ig (Linsley and Nadler, 2009). Importantly, studies in a variety of animal models have further supported the importance of CD4+T-cells in RA development.
Collagen-induced arthritis (CIA) and K/BxN mouse (Kouskoff et al., 1996) model that recapitulate the whole process of RA are the most widely used mouse models. In these models, arthritogenic antibodies contribute to the development of RA, since transfer of arthritogenic antibodies can induce arthritis. CD4+T-cells are required for the full induction of CIA (Ranges et al., 1985) and K/BxN models (Kouskoff et al., 1996), as the administration of anti-CD4 depleting antibodies suppresses both the production of autoantibodies and disease severity (Table 1). In contrast, CAIA (Kagari et al., 2002) and K/BxN serum transfer arthritis (Korganow et al., 1999) do not require T-cells or B-cells, since this form of arthritis can be induced effectively in T and B-cell-deficient mice. Thus, it is indicated that CD4+T-cells are required for the initiation phase of CIA and K/BxN arthritis partly by producing arthritogenic autoantibodies. CD4+T-cells are not strictly necessary after the production of arthritogenic autoantibodies, although this does not mean CD4+T-cells have no further role in the disease. Indeed, they are demonstrably capable of exacerbating arthritis, because adoptive transfer of a CD4+T-cell subset augments the severity in the CAIA (Nandakumar et al., 2004) and K/BxN serum transfer models (Jacobs et al., 2009).
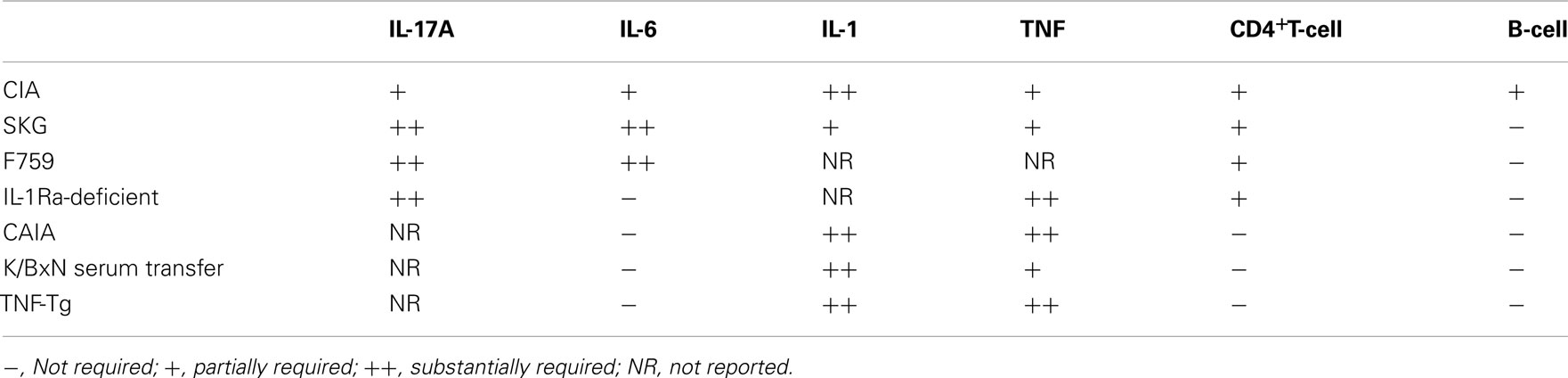
Table 1. The contribution of proinflammatory cytokines and lymphocytes to the development of mouse models of RA.
In this review, we divide a whole process of arthritis into the “initiation,” “inflammatory,” and “bone destruction” phases (Figure 1). We define the initiation phase as a phase when immune responses are triggered by an assumed antigen(s) and any apparent symptoms in joints are not yet observed. Inflammatory phase starts when any inflammatory symptoms such as swelling are recognized in joints and continues until any structural changes occur. Bone destruction phase is defined as a phase when structural damages in bone and cartilage are observed. Although the start point of initiation phase is difficult to tell in human, this phase must exist because it takes some time from the start to the point when clinical symptoms are observed. Thus, it is considered that both human RA and animal models of RA consist of all these phases.
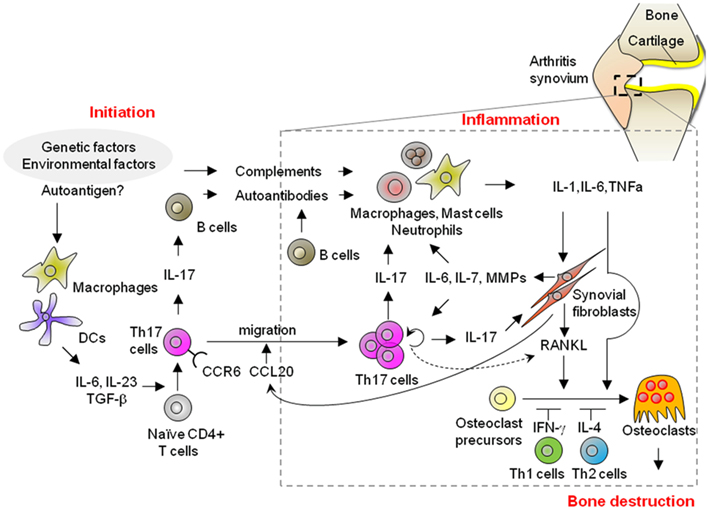
Figure 1. Possible mechanisms of the initiation, inflammatory, and bone destruction phases in RA. A variety of different cell populations, including lymphocytes, innate immune cells, synovial fibroblasts, and osteoclasts, play a role in the development of RA. Th17 cells contribute to the development of arthritis in each of the initiation, inflammatory, and bone destructive phases through the production of autoantibodies as well as the activation of innate immunity and synovial fibroblasts, and then ultimately, bone destruction. Importantly, synovial fibroblasts contribute to Th17 immunity in the inflammatory phase of arthritis by promoting the migration of Th17 cells into the inflammatory joint, and then homeostatic proliferation with an increase in IL-17 production. Thus, the interaction of CD4+T-cells and mesenchymal cells in joints plays a key role in the pathogenesis of RA in both the inflammation and bone destruction phases.
The significance of CD4+T-cells in RA development is also supported by T-cell-dependent models such as the SKG mouse, which has a mutation in ZAP70 (Sakaguchi et al., 2003), the F759 mouse with a mutation in the gp130 IL-6 receptor subunit (Atsumi et al., 2002), and the IL-1 receptor antagonist (IL-1Ra)-deficient mouse (Horai et al., 2000). These mice spontaneously develop arthritis due to a defect in TCR signaling or the altered sensitivity to inflammatory cytokines. The adoptive transfer of CD4+T-cells from SKG mice into SCID mice induces arthritis, indicating that the arthritis in SKG mice is CD4+T-cell-dependent (Sakaguchi et al., 2003). In addition, the arthritis which develops in F759 mice requires the presence of CD4+T-cells, but not CD8+T-cells or B-cells, in addition to the gp130 mutation in non-hematopoietic cells (Sawa et al., 2006). Furthermore, the arthritis in IL-1Ra-deficient mice is T-cell-dependent, as the T-cells from IL-1Ra-deficient mice induce disease in nude mice (Horai et al., 2004). Taken together, those T-cell-dependent mouse models indicate that RA can be provoked by CD4+T-cells without the need of B-cell help, due to an intrinsic defect in TCR signaling or altered sensitivity to proinflammatory cytokines (Table 1).
In contrast, arthritis develops in human TNF-α transgenic (TNF-Tg) mice (Keffer et al., 1991) and mice with the myeloid-specific deletion of A20, a negative regulator of NF-κB signaling (Matmati et al., 2011). These arthritis are thought to recapitulate the inflammatory phase of RA, bypassing the initiation phase of RA. These mice develop arthritis even on the T, B-cell-deficient background (Douni et al., 1995). This suggested that hyperactivation of innate immune system is also able to induce RA (Table 1).
Considering the necessity of CD4+T-cells for the initiation phase, one of the key questions is whether arthritogenic CD4+T-cells recognize a specific antigen, and if so, a joint-specific antigen or not. In the form of arthritis in K/BxN and CIA, arthritogenic CD4+T-cells recognize antigens that are abundant in the joints, although not exclusively joint-specific. In contrast, in the arthritis of F759 mice, the recognition of joint antigens by CD4+T-cells may not be required, because F759 mice expressing a single TCR variant that recognizes a non-joint antigen do indeed develop arthritis (Murakami et al., 2011). Moreover, the antigen specificity of arthritogenic CD4+T-cells in SKG mice remains unknown. Further studies are thus needed to elucidate the antigen specificity of arthritogenic CD4+T-cells, the finding of which will provide new insight into how immunological tolerance is broken by the generation of arthritogenic CD4+T-cells.
Taken together, CD4+T-cells are necessary for at least the initiation phase of arthritis partly by producing arthritogenic antibodies. In contrast, CD4+T-cells may not be required for the inflammatory phase of the disease, especially after arthritogenic autoantibodies are generated abundantly or innate immunity is hyper-activated. Nevertheless, CD4+T-cells have been shown to at least augment the inflammatory phase of arthritis development.
Th17 Cells: An Emerging Pathogenic Subset of CD4+T-Cells
The CD4+ helper T-cells (Th cells), that are differentiated from naïve CD4+T-cells include Th1, Th2, and Th17 cell subsets. Th17 cells, via their production of IL-17, promote the development of autoimmune diseases while also protecting host against bacterial and fungal infection. IL-6 and TGF-β induce Th17 development and IL-23 promotes Th17 cell expansion (Miossec et al., 2009). In the past, Th1 cells, which predominantly produce IFN-γ, were thought to be the principal T-cell player in the pathogenesis of RA. However, accumulating evidence from animal models in fact indicates that Th17 immunity is crucially important.
In CIA, accelerated RA development is evident in IFN-γ receptor-deficient mice (Manoury-Schwartz et al., 1997; Vermeire et al., 1997). In contrast, disease development is markedly diminished in mice with IL-17A deficiency (Nakae et al., 2003a) or with antibody-mediated blockade of IL-17 (Lubberts et al., 2004). In the SKG model, RAG-deficient mice that received naïve SKG CD4+T-cells exhibited arthritis, along with concomitant Th17 generation. This arthritis is Th17-dependent, as RAG mice which received a transfer of IL-17-deficient T-cells did not exhibit any sign of arthritis (Hirota et al., 2007a). Moreover, IL-1Ra-deficient mice with IL-17A deficiency display abrogated arthritis development (Nakae et al., 2003b). Furthermore, F759 mice with IL-17A deficiency (Ogura et al., 2008) and K/BxN mice treated with a neutralizing IL-17A antibody exhibited substantially diminished arthritis (Wu et al., 2010). Taken together, as shown in Table 1, regardless of whether the dependency was on IL-6, IL-1, or TNF-α, the development of arthritis was shown to be IL-17-dependent in most T-cell-dependent models, suggesting Th17 cell is a pathogenic subset of CD4+T-cells.
As for the function of IL-17, it augments the production of proinflammatory cytokines, chemokines, and matrix-degrading enzymes of various kinds of cells such as macrophages, dendritic cells (DCs), endothelial cells, and fibroblasts (Miossec et al., 2009). Thus, Th17 cells exacerbate the inflammatory phase of arthritis through the activation of various kinds of cells in the inflamed joints. In addition, IL-17 is responsible for the production of autoantibodies in CIA (Nakae et al., 2003a) and K/BxN (Wu et al., 2010) mouse models. In particular, IL-17 has been shown to enhance germinal center (GC) formation in the K/BxN model. Thus, via IL-17 production, Th17 cells are able to exacerbate the initiation phase of arthritis through the production of autoantibodies. Moreover, IL-17 together with IL-6 amplifies the production of IL-6 by type 1 collagen+fibroblasts, which in turn enhances IL-17 production in T-cells (Ogura et al., 2008) as discussed below. Taken together, Th17 cells can exacerbate arthritis both in the initiation and inflammatory phases.
Cells other than Th17 cells are also reported to produce IL-17 in arthritis affected joints. In the synovium of CIA, γδT-cells also produce IL-17, although few IL-17+γδT-cells are in fact detected in the affected joints of SKG mice or RA patients (Ito et al., 2009). Mast cells also produce IL-17 in the inflamed joints of RA patients (Hueber et al., 2010a). Although the functional relevance of other IL-17 producing cells remains to be clarified, considering the wealth of evidence for the significance of CD4+T-cells, it can be concluded that Th17 cells play a critical role in arthritis development.
Compared with the understanding of the function of Th17 cells, it remains largely unknown how Th17 cell are generated in the context of arthritis. Recently, several studies on this issue were reported. In SKG mice, Th17 cells are generated in the presence of the IL-6 produced by tissue-resident macrophages in response to C5a, because Th17 cell development is severely impaired in SKG mice having either a C5aR deficiency or a depletion of macrophages (Hashimoto et al., 2010). In addition, a deficiency of Toll-like receptor (TLR)-4 or administration of a TLR-4 antagonist suppresses the development of arthritis in IL-1Ra-deficient mice (Abdollahi-Roodsaz et al., 2008) and CIA (Abdollahi-Roodsaz et al., 2007) by decreasing the number of Th17 cells. This suggests that TLR-4 signaling is involved in Th17 generation. Moreover, in K/BxN mice, not only arthritis, but also Th17 generation and the production of arthritogenic autoantibodies cease under germ-free conditions, whereas the administration of even a single gut-residing species, segmented filamentous bacteria, can induce Th17 generation, GC formation, and the signs of arthritis. Thus, it is suggested that the gut environment affects the generation of IL-17+ cells, presumably including Th17 cells, leading to the onset of arthritis (Wu et al., 2010).
Given the significant role of Th17 cells in arthritis in mouse models, Th17 is now recognized as a promising therapeutic target. Thus, it is important to clarify the transcriptional mechanisms regulating Th17 development. ROR nuclear receptors are essential for Th17 development (Miossec et al., 2009). IkBζ also regulates Th17 development by cooperating with RORs (Okamoto et al., 2010). Antagonizing ROR activity has been shown to be effective in suppressing Th17 differentiation and Th17-mediated autoimmunity in mice using a synthetic ligand for RORs (Solt et al., 2011) as well as digoxin and its derivatives (Huh et al., 2011). In addition, Abs against IL-17A, LY2439821 (Genovese et al., 2010), and AIN457 (Hueber et al., 2010b) have been shown to be beneficial for the treatment of RA in human, although they are unexpectedly less effective than anti-TNF Abs or anti-IL-6 Abs. This suggests that other IL-17 family members such as IL-17B or IL-17C may also contribute to RA pathogenesis (Yamaguchi et al., 2007). Alternatively, there may be a difference in the extent to which Th17 cells contribute to the pathogenesis of arthritis between mice and humans. Indeed, T-cells in the synovial fluid of patients with juvenile idiopathic arthritis (JIA) easily switch from a Th17 to Th1 phenotype via the intermediate step of a Th1/Th17 mixed phenotype (Cosmi et al., 2011), suggesting that human Th17 cells are more plastic than their mouse counterparts. In line with this, Ustekinumab, which is a human mAb against IL-12/23p40, significantly suppresses psoriatic arthritis in human (Gottlieb et al., 2009). In addition, a JAK inhibitor tofacitinib (CP690,550) which inhibits the established CIA presumably by suppressing pathogenic Th1 and Th17 cells (Ghoreschi et al., 2011), shows clinical benefit for RA (Kremer et al., 2009; Fleischmann et al., 2012). From this point of view, either an EP4 antagonist that blocks PGE2–EP4 signaling (Yao et al., 2009) or a depletion of anti-LT-α Abs (Chiang et al., 2009), which have been shown to suppress Th17-mediated autoimmune disease through the inhibition of both Th1 and Th17 immunity in mice, might be therapeutically beneficial in RA treatment.
Taken together, Th17 cells are crucial immune cells that are required for the initiation of arthritis and contribute to the augmentation of chronic inflammation in joints through the activation of both innate immunity and mesenchymal cells such as synovial fibroblasts in joints.
Innate Immune Cells: An Accelerator of Arthritis
In addition to T-cell infiltration, RA exhibits a massive infiltration into affected joints innate immune cells, including macrophages, neutrophils, mast cells, and DCs. These cells react to complement or the Fc portion of IgG isotypes via receptors expressed on their surface. They also produce proinflammatory cytokines, chemokines, and matrix-degrading enzymes that drive chronic inflammation.
The importance of innate immunity in arthritis development has been shown in both T-cell-dependent and independent mouse models. In the T-cell-dependent models, SKG mice fail to develop arthritis when they are raised under a specific pathogen free (SPF) condition, whereas SKG mice raised under a conventional environment do develop arthritis. In addition, SKG mice under an SPF condition develop severe arthritis when administrated zymosan, a crude yeast cell wall extract. Proinflammatory cytokines, presumably including TNF-α, which are produced by Dectin-1 expressing DCs or macrophages in response to zymosan, are involved in this process (Yoshitomi et al., 2005). In addition, macrophages produce IL-6 in response to C5a, leading to the generation of Th17 cells in SKG mice (Hashimoto et al., 2010). These findings indicate that activation of adaptive immunity requires innate immunity in the initiation phase of arthritis.
Among the T-cell-independent models, the K/BxN serum transfer model has helped address the mechanisms by which activation of innate immune system triggered by autoantibodies leads to the development of arthritis. In the K/BxN model, the autoantigen is the glucose-6-phosphate isomerase (GPI) that is expressed in the joint, although it is not joint-specific. GPI-anti-GPI immune complexes bind to articular surfaces, leading to the local augmentation of immune effecter responses in the joint (Matsumoto et al., 2002). K/BxN serum transfer arthritis requires complement C5 and FcγRIII (Ji et al., 2002). Neutrophils and mast cells are also required, as mice depleted of neutrophils (Wipke and Allen, 2001) and mice lacking mast cells (Lee et al., 2002) are both resistant to the arthritis.
As I mentioned above, TNF-Tg mice and mice with selective deletion of A20 (TNFAIP3) in myeloid cells do not require either T-cells or B-cells for the development of arthritis (Douni et al., 1995; Matmati et al., 2011). These studies suggest that hyperactivation of innate immunity is sufficient to induce arthritis. In addition, these findings prompt a consideration of how it is that a systemic gene mutation results in local joint inflammation. There may be certain joint-specific factors, possibly expressed by synovial fibroblasts, which drive the preferential migration of activated innate immune cells and thus the amplification of chronic inflammation in the affected joints.
Taken together, adaptive immunity requires the activity of innate immunity for the development of full blown arthritis both in the initiation and inflammatory phases. In addition, the hyperactivation of the innate immune response by itself is able to induce arthritis, presumably through an interaction with synovial fibroblasts, a unique mesenchymal cell population in joints.
Synovial Fibroblasts: Mesenchymal Cells that Determine Joint-Specificity
In general, all RA patients and RA model mice exhibit proliferative and erosive synovitis in regions adjacent to cartilage and bone, regardless of differences in the initiating mechanisms. Synoviocytes are divided into synovial fibroblasts of mesenchymal origin and macrophage-like synoviocytes, depending on their surface markers. RA synovial fibroblasts are key cells in the chronic inflammation which occurs in RA.
Synovial fibroblasts express not only receptors for proinflammatory cytokines, but also TLRs (Bartok and Firestein, 2010). In synovitis, synovial fibroblasts exhibit high proliferative activity and produce large amounts of cytokines, chemokines, and matrix-degrading enzymes in response to proinflammatory cytokines and TLR ligands, which lead to the exacerbation of synovitis and joint destruction. For instance, Tenascin-C, an extracellular matrix glycoprotein specifically expressed in inflamed joints, was shown to be an endogenous activator of the TLR-4 expressed by synovial fibroblasts and macrophages, and is also essential for maintaining synovitis in K/BxN serum transfer arthritis (Midwood et al., 2009). Interestingly, the microparticles produced by activated platelets amplify inflammatory arthritis in the K/BxN serum transfer model via a collagen-receptor expressed on synovial fibroblasts (Boilard et al., 2010).
The invasive characteristics of synovial fibroblasts from RA synovium have been reported in following studies. Cultured synovial fibroblasts from human RA synovium were shown to invade and destroy cartilage when co-transplanted with cartilage into SCID mice (Muller-Ladner et al., 1996). These transplanted RA synovial fibroblasts specifically migrate into a distal cartilage even in the absence of other immune cells (Lefèvre et al., 2009). Thus, it is suggested that synovial fibroblasts appear to have intrinsically invasive properties and to be destined to localize specifically in the joint. In addition, the invasive characteristics of synovial fibroblasts have also been reported in synoviocyte clones obtained from TNF-Tg mice (Aidinis et al., 2003). These results suggest that the intrinsically invasive properties of synovial fibroblasts from inflamed joints are stably maintained even after several passages in culture and that epigenetic modification may be involved in this process. Indeed, the DNA of RA synovial fibroblasts is hypomethylated both in synovial tissues and in vitro (Karouzakis et al., 2009). In addition, the ratio of histone acetylase/deacetylase activity is higher in RA synovial tissue than that in normal synovial tissue (Huber et al., 2007). Furthermore, synovial fibroblasts preferentially express microRNA 146a and 155, among microRNAs which function as posttranscriptional repressors of gene expression (Stanczyk et al., 2008). Further studies are needed to clarify the mechanisms of epigenetic modification and their role in the maintenance of the activated phenotype of synovial fibroblasts in arthritic joints.
Given that the infiltration of CD4+T-cells in inflamed joints is a hallmark of RA pathology, the interaction of synovial fibroblasts and CD4+T-cells is assumed to play an important role. By in vitro co-culture experiments, it has been demonstrated that RA synovial fibroblasts and CD4+T-cells activate each other through the ICAM-2 and LFA expressed on synovial fibroblasts and CD4+T-cells, respectively (Singh et al., 2008). In addition, the IL-15 expressed on RA synovial fibroblasts augments the production of effecter cytokines from CD4+CD25− cells, while also enhancing the proliferation of CD4+CD25+Treg cells (Benito-Miguel et al., 2009). Several reports suggest an antigen-presenting role for synovial fibroblasts. RA synovial fibroblasts in tissue express MHC class (Burmester et al., 1987) IFN-γ treated synovial fibroblasts in vitro stimulate T-cell activation in an MHC class II dependent manner (Tran et al., 2007). However, the capacity for MHC class II restricted antigen presentation in synovial fibroblasts and its role in RA development in vivo remain to be demonstrated.
Importantly, several recent reports have shed light on the relevance of the interaction of CD4+T-cells and mesenchymal cells in the affected joint in the development of arthritis. In the SKG model, synovial fibroblasts produce CCL20 in response to proinflammatory cytokines such as TNF-α, leading to the recruitment of CCR6+Th17 cells into the affected joint. This recruitment is essential, as the administration of a neutralizing anti-CCR6 antibody ameliorates the development of arthritis (Hirota et al., 2007b). Likewise, in F759 arthritis, type 1 collagen+fibroblasts produce CCL20 in response to local stimuli such as microbleeding and preferentially recruit CD4+T-cells into inflamed joints. The relevance of this recruitment has been demonstrated, because the inhibition of CCL20 diminished arthritis development (Murakami et al., 2011). In addition, non-hematopoietic cells, presumably synovial fibroblasts, produce elevated levels of IL-7 and IL-6, which enhances the homeostatic proliferation of CD4+T-cells and the production of IL-17 in Th17 cells, respectively (Sawa et al., 2006; Ogura et al., 2008). Moreover, IL-6 together with IL-17 amplifies IL-6 production of synovial fibroblasts (Ogura et al., 2008). In line with this, by in vitro co-culture system, a JAK inhibitor, Tofacitinib suppress the production of IL-6 by RA synovial fibroblasts through the inhibition of IL-17 and IFN-γ by RA CD4+T-cells (Maeshima et al., 2011).
Considering the important role of synovial fibroblasts, they may be a good therapeutic target for RA treatment. The induction of the cell senescence gene in synovial tissues successfully inhibits rat adjuvant-induced arthritis (Taniguchi et al., 1999). Yet only a few molecules have been identified as specific markers of synovial fibroblasts to date. Cadherin-11, a relatively specific marker, is required for the cellular connectivity of synovial fibroblasts. Cadherin-11-deficient mice exhibit a hypoplastic synovial lining of the synovium membrane and much less severe arthritis. Importantly, cadherin-11-directed therapeutics also markedly reduces synovial inflammation (Lee et al., 2007). Mechanistically, cadherin-11 contributes to the production of IL-6 in synovial fibroblasts (Chang et al., 2011). The identification of additional specific markers of synovial fibroblasts will ultimately lead to the establishment of “joint-preferential” therapeutic strategies.
These findings, taken together, indicate that synovial fibroblasts function as a unique “disease amplifier” in the inflammatory phase of RA through both the innate and acquired immunity pathways, due to their intrinsically invasive, hypersensitive, and hyperproliferative properties. Studies on animal models of RA have revealed the role of synovial fibroblasts in Th17 immunity, i.e., promoting the migration of Th17 cells to the affected joints and then homeostatic proliferation with an accompanying increase in IL-17 production, ultimately leading to the augmentation of the chronic inflammation which characterizes RA (Figure 1).
Proinflammatory Cytokines Mediate the Interplay between Immune Cells and Joints
In RA synovium, elevated levels of the proinflammatory cytokines IL-1, IL-6, and TNF-α are produced by macrophages and synovial fibroblasts. These proinflammatory cytokines both directly and indirectly exert their effects through the production of additional proinflammatory cytokines and chemokines as well as matrix-degrading enzymes, resulting in a cytokine “storm” in the inflamed synovium. The relative contribution of IL-1, IL-6, and TNF-α to the development and progression of arthritis is different in the various mouse models (Table 1).
In CIA, the blockade of IL-1 prevents arthritis (Joosten et al., 1996). IL-6 deficiency suppresses disease development (Alonzi et al., 1998). However, administration of a neutralizing anti-IL-6 mAb suppresses arthritis development when given early, but the suppressive effect is not observed when given in the later phases (Fujimoto et al., 2008). Likewise, the blockade of TNF-α markedly decreases inflammation and joint destruction when given early (Williams et al., 1992; Joosten et al., 1996). Recently, the growth factor progranulin was shown to bind to TNF receptors and block TNF-α/TNFR signaling. Progranulin reverses inflammatory arthritis in TNF-Tg mice and prevents the development of both CIA and CAIA (Tang et al., 2011). In addition, inhibition of migration of pathogenic T-cells into the joints and the prevention of emigration out of draining lymph nodes are observed in CIA mice in which TNF/TNFR signaling has been blocked (Notley et al., 2008). In line with this, impaired migration of T-cells into the joints is also observed in human RA patients treated with an anti-TNFα mAb (Taylor et al., 2000).
Overall, it is clear that there is a substantial difference in the relative contribution of these inflammatory cytokines to the development of arthritis (Table 1). Dependency on IL-1 and TNFα in both the T-cell-dependent and independent arthritis models suggests that IL-1 and TNFα may be involved in the inflammatory phase of arthritis in mice. As for human RA, anti-TNF therapies achieved clinical remission while the IL-1 inhibitor IL-1Ra was less effective than would be expected from mouse studies, suggesting that IL-1 in RA may not be as important as it is in mouse arthritis (Buch et al., 2004). In contrast, the different pattern of dependency on IL-6 in the T-cell-dependent and T-cell-independent arthritis models suggests that IL-6 may be critically involved in T-cell mediated arthritis and affect pathogenesis of T-cells. Indeed, the protective effect of IL-6 blockade in CIA correlates with the inhibition of Th17 differentiation. In this model, IL-6 blockade was shown to be effective when administered at an early initiation phase (Fujimoto et al., 2008). However, a significant number of RA patients with the blockade of IL-6 signaling achieved clinical remission suggesting that IL-6 plays an important role even in the inflammatory phase in human.
Taken together, proinflammatory cytokines mediate the interplay between immune cells and joints, leading to the initiation and augmentation of chronic inflammation in RA. The substantial differences in cytokine-dependency in animal models may reflect the different effect of each cytokine in each phase of arthritis progression, in association with the triggering arthritogenic stimuli and type of the cells that constitute the inflammatory synovium.
Osteoclasts; A Key Player in the Bone Destruction Which Occurs in RA
In the pathology of RA, chronic inflammation leads to bone destruction. The synovium is a site where the immune system interferes with normal bone homeostasis. Bone homeostasis is maintained by a balance between the continuous resorption activity of osteoclasts and formation by osteoblasts. In RA, the bone destruction which takes place is mainly due to the excessive bone resorption activity of osteoclasts.
Osteoimmunology is a cross-disciplinary research field that investigates the interplay of the bone and immune system at the molecular level (Takayanagi, 2009). The interaction of osteoclasts and immune cells is a major topic of interest in this field. Studies of the relationship of osteoclasts and macrophages have led to important mechanistic insights into osteoclast differentiation. In addition, studies of the interaction of osteoclasts and T-cells have contributed to an improved understanding of the mechanism of bone destruction in RA.
Historically, increased numbers of osteoclast-like giant cells had been identified in the synovium of RA joints by the early 1980s (Bromley and Woolley, 1984). Based on these pathological findings, it was therefore suggested that osteoclasts have an important role in bone resorption in arthritis. Importantly, osteoclast formation from cultured synovial cells was successfully performed without the need of any other cells, demonstrating that rheumatoid synovial cells contain both osteoclast precursor cells and osteoclastogenesis-supporting cells (Takayanagi et al., 1997). However, the molecular mechanism still remained unclear until the identification of RANKL as an osteoclast differentiation factor expressed on synovial cells (Gravallese et al., 2000; Takayanagi et al., 2000a).
Osteoclasts are formed when bone marrow cells are cultured in the presence of M-CSF and RANKL in vitro. Osteoclasts also are differentiated from bone marrow cells when co-cultured with mesenchymal cells, such as osteoblasts, in the presence of osteoclastogenic factors, including 1,25-dihydroxylvitamin D3, which induce RANKL expression on mesenchymal cells. Recent studies indicate that osteocytes, which are embedded in bone, express a higher amount of RANKL than osteoblasts and are thus the major source of RANKL in bone remodeling in vivo (Nakashima et al., 2011; Xiong et al., 2011).
RANKL is essential for osteoclast differentiation, as RANKL-deficient mice exhibit an osteopetrotic phenotype (Theill et al., 2002). Of note, a critical role for both RANKL and osteoclasts in arthritic bone destruction was demonstrated in mouse models of RA (Pettit et al., 2001; Redlich et al., 2002). Bone destruction did not occur in the absence of osteoclasts in either of these models, but a level of inflammation similar to that in their wild-type counterparts was observed, indicating that RANKL and osteoclasts are indispensable for bone destruction, but not for inflammation. There is a long-standing debate whether cells other than synovial fibroblasts express RANKL and thus contribute to osteoclastogenesis in arthritis. RANKL was originally identified as being expressed on activated T-cells (Wong et al., 1997). Histologically, in the RA synovium, RANKL is expressed by both synovial cells and T-cells (Kong et al., 1999; Gravallese et al., 2000; Takayanagi et al., 2000a). In addition, RANKL expression on B-cells in the arthritic joints of RA patients was reported (Yeo et al., 2012). However, it still remains unclear the extent to which lymphocytes, as a source of RANKL, contribute to the bone destruction in arthritis. Mice bearing a cell type-specific deletion of RANKL will be required to decide this issue. Given the important role of RANKL in osteoclastogenesis, RANKL is a promising pharmacological target for the prevention of joint destruction. Indeed, an anti-RANKL antibody was recently shown to inhibit joint destruction in human RA patients (Dore et al., 2010).
The discovery of RANKL shed light on the importance of understanding the molecular mechanisms that underlie osteoclast differentiation and function, which has led to the identification of NFATc1 as a master transcription regulator of osteoclastogenesis (Takayanagi et al., 2002) and other related signaling molecules. Notably, tyrosine kinases Btk and Tec regulate osteoclastogenesis and the inhibition of Tec kinase reduce inflammation-induced bone destruction (Shinohara et al., 2008). Further studies regarding precise mechanisms of osteoclast differentiation and function are required for a precise molecular basis for novel therapeutic strategies.
Synovial Fibroblasts Promote Osteoclastogenesis via Interaction with Immune Cells
Activated T-cells express not only RANKL but also effector cytokines, including cytokines with either stimulatory or inhibitory effects on osteoclastogenesis, as shown in Table 2 (Takayanagi et al., 2000b; Takayanagi, 2009). Thus, the osteoclastogenic capacity of T-cells is determined by both RANKL and cytokine expression.
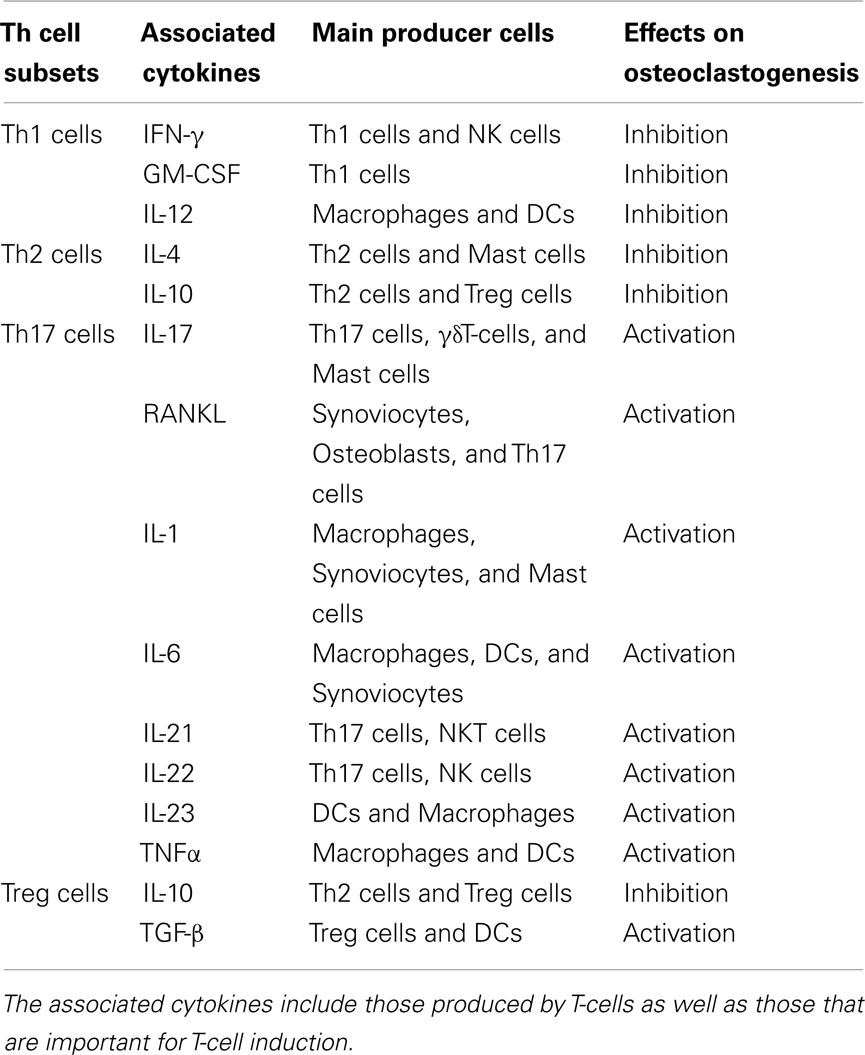
Table 2. T-cell-related cytokines and osteoclastogenesis.
IL-17 is known to enhance osteoclastogenesis in vitro by acting on osteoclastogenesis-supporting cells (Kotake et al., 1999). Of note, Th17 cells, but neither Th1 cell nor Th2 cells, comprise the osteoclastogenic helper T subset. Th17 cells do not produce either IFN-γ or IL-4, each of which inhibits osteoclastogenesis, but do produce IL-17, which stimulates osteoclastogenesis by its effect on osteoblasts that act as osteoclastogenesis-supporting mesenchymal cells (Sato et al., 2006).
Therefore, the presumable roles of IL-17 in the bone destruction which occurs in RA are as follows. First, IL-17 exerts its osteoclastogenic effect by stimulating RANKL expression by synovial fibroblasts. Furthermore, IL-17 up-regulates the expression of proinflammatory cytokines such as IL-1, IL-6, and TNF-α, which promote osteoclastogenesis through their effects on osteoclast precursor cells by enhancing RANK mediated signaling, or indirectly through upregulation of RANKL expression by synovial fibroblasts. These events synergistically promote osteoclastic bone resorption in the inflamed synovium.
Besides IL-17, IL-21(Kwok et al., 2012), and IL-22 (Kim et al., 2012), which are also produced by Th17 cells, stimulate osteoclastogenesis mainly by upregulating RANKL expression in synovial fibroblasts. Thus, it is plausible that synovial fibroblasts augment their capacity to induce osteoclastogenesis in the presence of Th17 cells.
An important role for Th17 in bone destruction is supported by studies in mouse models. In CIA, the neutralization of IL-17 after the onset of arthritis reduces the severity of joint destruction (Lubberts et al., 2004). Although both Th17 cells and γδT-cells produce IL-17 in the affected joints of CIA, Th17 cells, but not γδT-cells, have been shown by antibody-mediated depletion and adoptive transfer studies to reside adjacent to osteoclasts and to play a prominent role in bone destruction in vivo (Pollinger et al., 2011).
Osteoclast precursor cells express receptors for proinflammatory cytokines. Most of the proinflammatory cytokines which augment inflammation also promote osteoclastogenesis by augmenting RANK–RANKL signaling, with the exception that TNF-α and TGF-β together induce osteoclastogenesis even in the absence of RANK (Kim et al., 2005) This suggests that the inhibition of proinflammatory cytokines by neutralizing Abs would play a dual role in the suppression of inflammation and bone destruction in RA. Interestingly, the inhibition of cathepsin K, which was thought to be expressed exclusively by osteoclasts and to play an essential role in bone degradation, has been shown to play dual role in suppression of osteoclastic bone resorption and TLR-9 mediated-activation of DCs (Asagiri et al., 2008).
Taken together, synovial fibroblasts contribute not only to chronic inflammation but also to the bone destruction which occurs in RA by promoting RANKL-mediated osteoclastogenesis through the interaction of immune cells, mainly Th17 cells.
Concluding Remarks
Rheumatoid arthritis is an immune-mediated disease, characterized by local inflammation and bone destruction in joint as a result of alteration of systemic immune response. Recent studies have revealed that Th17 cells and synovial fibroblasts are the critical regulators. As shown in Figure 1, Th17 cells, differentiated in the presence of innate immunity, help B-cells produce arthritogenic autoantibodies in the initiation phase. In inflamed joints, Th17 cells activate innate immune cells and synovial fibroblasts by upregulating proinflammatory cytokines and matrix-degrading enzymes, thereby leading to an amplification of chronic inflammation. Moreover, Th17-related cytokines stimulate the differentiation of osteoclasts, mainly via the synovial fibroblasts in the joints, which eventually leads to bone destruction. Thus, Th17 cells are not only required for the initiation of the systemic immune response, they contribute to chronic inflammation and bone destruction. Importantly, synovial fibroblasts contribute to Th17 immunity in both the inflammatory and bone destruction phases of arthritis by promoting the migration of Th17 cells into the joint, inducing homeostatic proliferation with a concomitant increase in IL-17 production and promoting osteoclastogenesis by upregulation of RANKL expression. It is thus suggested that synovial fibroblasts connect the systemic immune response to local joint disorders by their intrinsic characteristics, including their “hyper-reactivity” and “hyper-chemoattractivity” in response to inflammatory stimuli.
Collectively, the interaction of immune cells and non-hematopoietic mesenchymal cells in the joints plays a key role in the pathogenesis of RA in both the inflammatory and bone destruction phases. Elucidation of the precise mechanisms involved in this interaction will lead to a better understanding of RA and provide a molecular basis for effective therapeutic strategies against this disease. Furthermore, the findings obtained from such investigation of RA will undoubtedly prove applicable to other diseases evoked through the interaction of immune and mesenchymal cells.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to all the members of our laboratory in the Graduate School of Medical and Dental Sciences. The work in our laboratory was supported in part by a grant for the ERATO, Takayanagi Osteonetwork Project from JST; Grant-in-Aid for Postdocs for from the Japan Society for the Promotion of Science (JSPS); a grant for the Global Center of Excellence Program from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT).
References
Abdollahi-Roodsaz, S., Joosten, L. A., Koenders, M. I., Devesa, I., Roelofs, M. F., Radstake, T. R., Heuvelmans-Jacobs, M., Akira, S., Nicklin, M. J., Ribeiro-Dias, F., and van Den Berg, W. B. (2008). Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J. Clin. Invest. 118, 205–216.
Abdollahi-Roodsaz, S., Joosten, L. A., Roelofs, M. F., Radstake, T. R., Matera, G., Popa, C., Van Der Meer, J. W., Netea, M. G., and Van Den Berg, W. B. (2007). Inhibition of Toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum. 56, 2957–2967.
Aidinis, V., Plows, D., Haralambous, S., Armaka, M., Papadopoulos, P., Kanaki, M. Z., Koczan, D., Thiesen, H. J., and Kollias, G. (2003). Functional analysis of an arthritogenic synovial fibroblast. Arthritis Res. Ther. 5, R140–R157.
Alonzi, T., Fattori, E., Lazzaro, D., Costa, P., Probert, L., Kollias, G., De Benedetti, F., Poli, V., and Ciliberto, G. (1998). Interleukin 6 is required for the development of collagen-induced arthritis. J. Exp. Med. 187, 461–468.
Asagiri, M., Hirai, T., Kunigami, T., Kamano, S., Gober, H. J., Okamoto, K., Nishikawa, K., Latz, E., Golenbock, D. T., Aoki, K., Ohya, K., Imai, Y., Morishita, Y., Miyazono, K., Kato, S., Saftig, P., and Takayanagi, H. (2008). Cathepsin K-dependent toll-like receptor 9 signaling revealed in experimental arthritis. Science 319, 624–627.
Atsumi, T., Ishihara, K., Kamimura, D., Ikushima, H., Ohtani, T., Hirota, S., Kobayashi, H., Park, S. J., Saeki, Y., Kitamura, Y., and Hirano, T. (2002). A point mutation of Tyr-759 in interleukin 6 family cytokine receptor subunit gp130 causes autoimmune arthritis. J. Exp. Med. 196, 979–990.
Bartok, B., and Firestein, G. S. (2010). Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol. Rev. 233, 233–255.
Begovich, A. B., Carlton, V. E., Honigberg, L. A., Schrodi, S. J., Chokkalingam, A. P., Alexander, H. C., Ardlie, K. G., Huang, Q., Smith, A. M., Spoerke, J. M., Conn, M. T., Chang, M., Chang, S. Y., Saiki, R. K., Catanese, J. J., Leong, D. U., Garcia, V. E., Mcallister, L. B., Jeffery, D. A., Lee, A. T., Batliwalla, F., Remmers, E., Criswell, L. A., Seldin, M. F., Kastner, D. L., Amos, C. I., Sninsky, J. J., and Gregersen, P. K. (2004). A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am. J. Hum. Genet. 75, 330–337.
Benito-Miguel, M., Garcia-Carmona, Y., Balsa, A., Perez De Ayala, C., Cobo-Ibanez, T., Martin-Mola, E., and Miranda-Carus, M. E. (2009). A dual action of rheumatoid arthritis synovial fibroblast IL-15 expression on the equilibrium between CD4+CD25+ regulatory T cells and CD4+CD25- responder T cells. J. Immunol. 183, 8268–8279.
Boilard, E., Nigrovic, P. A., Larabee, K., Watts, G. F., Coblyn, J. S., Weinblatt, M. E., Massarotti, E. M., Remold-O’Donnell, E., Farndale, R. W., Ware, J., and Lee, D. M. (2010). Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 327, 580–583.
Bromley, M., and Woolley, D. E. (1984). Chondroclasts and osteoclasts at subchondral sites of erosion in the rheumatoid joint. Arthritis Rheum. 27, 968–975.
Buch, M. H., Bingham, S. J., Seto, Y., Mcgonagle, D., Bejarano, V., White, J., and Emery, P. (2004). Lack of response to anakinra in rheumatoid arthritis following failure of tumor necrosis factor alpha blockade. Arthritis Rheum. 50, 725–728.
Burmester, G. R., Jahn, B., Rohwer, P., Zacher, J., Winchester, R. J., and Kalden, J. R. (1987). Differential expression of Ia antigens by rheumatoid synovial lining cells. J. Clin. Invest. 80, 595–604.
Chang, S. K., Noss, E. H., Chen, M., Gu, Z., Townsend, K., Grenha, R., Leon, L., Lee, S. Y., Lee, D. M., and Brenner, M. B. (2011). Cadherin-11 regulates fibroblast inflammation. Proc. Natl. Acad. Sci. U.S.A. 108, 8402–8407.
Chiang, E. Y., Kolumam, G. A., Yu, X., Francesco, M., Ivelja, S., Peng, I., Gribling, P., Shu, J., Lee, W. P., Refino, C. J., Balazs, M., Paler-Martinez, A., Nguyen, A., Young, J., Barck, K. H., Carano, R. A., Ferrando, R., Diehl, L., Chatterjea, D., and Grogan, J. L. (2009). Targeted depletion of lymphotoxin-alpha-expressing TH1 and TH17 cells inhibits autoimmune disease. Nat. Med. 15, 766–773.
Cosmi, L., Cimaz, R., Maggi, L., Santarlasci, V., Capone, M., Borriello, F., Frosali, F., Querci, V., Simonini, G., Barra, G., Piccinni, M. P., Liotta, F., De Palma, R., Maggi, E., Romagnani, S., and Annunziato, F. (2011). Evidence of the transient nature of the Th17 phenotype of CD4+CD161+ T cells in the synovial fluid of patients with juvenile idiopathic arthritis. Arthritis Rheum. 63, 2504–2515.
Dore, R. K., Cohen, S. B., Lane, N. E., Palmer, W., Shergy, W., Zhou, L., Wang, H., Tsuji, W., and Newmark, R. (2010). Effects of denosumab on bone mineral density and bone turnover in patients with rheumatoid arthritis receiving concurrent glucocorticoids or bisphosphonates. Ann. Rheum. Dis. 69, 872–875.
Douni, E., Akassoglou, K., Alexopoulou, L., Georgopoulos, S., Haralambous, S., Hill, S., Kassiotis, G., Kontoyiannis, D., Pasparakis, M., Plows, D., Probert, L., and Kollias, G. (1995). Transgenic and knockout analyses of the role of TNF in immune regulation and disease pathogenesis. J. Inflamm. 47, 27–38.
Fleischmann, R., Cutolo, M., Genovese, M. C., Lee, E. B., Kanik, K. S., Sadis, S., Connell, C. A., Gruben, D., Krishnaswami, S., Wallenstein, G., Wilkinson, B. E., and Zwillich, S. H. (2012). Phase 2B dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to DMARDs. Arthritis Rheum. 64, 617–629.
Fujimoto, M., Serada, S., Mihara, M., Uchiyama, Y., Yoshida, H., Koike, N., Ohsugi, Y., Nishikawa, T., Ripley, B., Kimura, A., Kishimoto, T., and Naka, T. (2008). Interleukin-6 blockade suppresses autoimmune arthritis in mice by the inhibition of inflammatory Th17 responses. Arthritis Rheum. 58, 3710–3719.
Genovese, M. C., Van Den Bosch, F., Roberson, S. A., Bojin, S., Biagini, I. M., Ryan, P., and Sloan-Lancaster, J. (2010). LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: a phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum. 62, 929–939.
Ghoreschi, K., Jesson, M. I., Li, X., Lee, J. L., Ghosh, S., Alsup, J. W., Warner, J. D., Tanaka, M., Steward-Tharp, S. M., Gadina, M., Thomas, C. J., Minnerly, J. C., Storer, C. E., Labranche, T. P., Radi, Z. A., Dowty, M. E., Head, R. D., Meyer, D. M., Kishore, N., and O’Shea, J. J. (2011). Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 186, 4234–4243.
Gottlieb, A., Menter, A., Mendelsohn, A., Shen, Y. K., Li, S., Guzzo, C., Fretzin, S., Kunynetz, R., and Kavanaugh, A. (2009). Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomised, double-blind, placebo-controlled, crossover trial. Lancet 373, 633–640.
Gravallese, E. M., Manning, C., Tsay, A., Naito, A., Pan, C., Amento, E., and Goldring, S. R. (2000). Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 43, 250–258.
Hashimoto, M., Hirota, K., Yoshitomi, H., Maeda, S., Teradaira, S., Akizuki, S., Prieto-Martin, P., Nomura, T., Sakaguchi, N., Kohl, J., Heyman, B., Takahashi, M., Fujita, T., Mimori, T., and Sakaguchi, S. (2010). Complement drives Th17 cell differentiation and triggers autoimmune arthritis. J. Exp. Med. 207, 1135–1143.
Hirota, K., Hashimoto, M., Yoshitomi, H., Tanaka, S., Nomura, T., Yamaguchi, T., Iwakura, Y., Sakaguchi, N., and Sakaguchi, S. (2007a). T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J. Exp. Med. 204, 41–47.
Hirota, K., Yoshitomi, H., Hashimoto, M., Maeda, S., Teradaira, S., Sugimoto, N., Yamaguchi, T., Nomura, T., Ito, H., Nakamura, T., Sakaguchi, N., and Sakaguchi, S. (2007b). Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J. Exp. Med. 204, 2803–2812.
Horai, R., Nakajima, A., Habiro, K., Kotani, M., Nakae, S., Matsuki, T., Nambu, A., Saijo, S., Kotaki, H., Sudo, K., Okahara, A., Tanioka, H., Ikuse, T., Ishii, N., Schwartzberg, P. L., Abe, R., and Iwakura, Y. (2004). TNF-alpha is crucial for the development of autoimmune arthritis in IL-1 receptor antagonist-deficient mice. J. Clin. Invest. 114, 1603–1611.
Horai, R., Saijo, S., Tanioka, H., Nakae, S., Sudo, K., Okahara, A., Ikuse, T., Asano, M., and Iwakura, Y. (2000). Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J. Exp. Med. 191, 313–320.
Huber, L. C., Brock, M., Hemmatazad, H., Giger, O. T., Moritz, F., Trenkmann, M., Distler, J. H., Gay, R. E., Kolling, C., Moch, H., Michel, B. A., Gay, S., Distler, O., and Jungel, A. (2007). Histone deacetylase/acetylase activity in total synovial tissue derived from rheumatoid arthritis and osteoarthritis patients. Arthritis Rheum. 56, 1087–1093.
Hueber, A. J., Asquith, D. L., Miller, A. M., Reilly, J., Kerr, S., Leipe, J., Melendez, A. J., and Mcinnes, I. B. (2010a). Mast cells express IL-17A in rheumatoid arthritis synovium. J. Immunol. 184, 3336–3340.
Hueber, W., Patel, D. D., Dryja, T., Wright, A. M., Koroleva, I., Bruin, G., Antoni, C., Draelos, Z., Gold, M. H., Durez, P., Tak, P. P., Gomez-Reino, J. J., Foster, C. S., Kim, R. Y., Samson, C. M., Falk, N. S., Chu, D. S., Callanan, D., Nguyen, Q. D., Rose, K., Haider, A., and Di Padova, F. (2010b). Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci. Transl. Med. 2, 52ra72.
Huh, J. R., Leung, M. W., Huang, P., Ryan, D. A., Krout, M. R., Malapaka, R. R., Chow, J., Manel, N., Ciofani, M., Kim, S. V., Cuesta, A., Santori, F. R., Lafaille, J. J., Xu, H. E., Gin, D. Y., Rastinejad, F., and Littman, D. R. (2011). Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORgammat activity. Nature 472, 486–490.
Ito, Y., Usui, T., Kobayashi, S., Iguchi-Hashimoto, M., Ito, H., Yoshitomi, H., Nakamura, T., Shimizu, M., Kawabata, D., Yukawa, N., Hashimoto, M., Sakaguchi, N., Sakaguchi, S., Yoshifuji, H., Nojima, T., Ohmura, K., Fujii, T., and Mimori, T. (2009). Gamma/delta T cells are the predominant source of interleukin-17 in affected joints in collagen-induced arthritis, but not in rheumatoid arthritis. Arthritis Rheum. 60, 2294–2303.
Jacobs, J. P., Wu, H. J., Benoist, C., and Mathis, D. (2009). IL-17-producing T cells can augment autoantibody-induced arthritis. Proc. Natl. Acad. Sci. U.S.A. 106, 21789–21794.
Ji, H., Ohmura, K., Mahmood, U., Lee, D. M., Hofhuis, F. M., Boackle, S. A., Takahashi, K., Holers, V. M., Walport, M., Gerard, C., Ezekowitz, A., Carroll, M. C., Brenner, M., Weissleder, R., Verbeek, J. S., Duchatelle, V., Degott, C., Benoist, C., and Mathis, D. (2002). Arthritis critically dependent on innate immune system players. Immunity 16, 157–168.
Joosten, L. A., Helsen, M. M., Van De Loo, F. A., and Van Den Berg, W. B. (1996). Anticytokine treatment of established type II collagen-induced arthritis in DBA/1 mice. A comparative study using anti-TNF alpha, anti-IL-1 alpha/beta, and IL-1Ra. Arthritis Rheum. 39, 797–809.
Kagari, T., Doi, H., and Shimozato, T. (2002). The importance of IL-1 beta and TNF-alpha, and the noninvolvement of IL-6, in the development of monoclonal antibody-induced arthritis. J. Immunol. 169, 1459–1466.
Karouzakis, E., Gay, R. E., Michel, B. A., Gay, S., and Neidhart, M. (2009). DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 60, 3613–3622.
Keffer, J., Probert, L., Cazlaris, H., Georgopoulos, S., Kaslaris, E., Kioussis, D., and Kollias, G. (1991). Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 10, 4025–4031.
Kim, K. W., Kim, H. R., Park, J. Y., Park, J. S., Oh, H. J., Woo, Y. J., Park, M. K., Cho, M. L., and Lee, S. H. (2012). IL-22 promotes osteoclastogenesis in rheumatoid arthritis through induction of RANKL in human synovial fibroblasts. Arthritis Rheum. 64, 1015–1023.
Kim, N., Kadono, Y., Takami, M., Lee, J., Lee, S. H., Okada, F., Kim, J. H., Kobayashi, T., Odgren, P. R., Nakano, H., Yeh, W. C., Lee, S. K., Lorenzo, J. A., and Choi, Y. (2005). Osteoclast differentiation independent of the TRANCE-RANK-TRAF6 axis. J. Exp. Med. 202, 589–595.
Kochi, Y., Okada, Y., Suzuki, A., Ikari, K., Terao, C., Takahashi, A., Yamazaki, K., Hosono, N., Myouzen, K., Tsunoda, T., Kamatani, N., Furuichi, T., Ikegawa, S., Ohmura, K., Mimori, T., Matsuda, F., Iwamoto, T., Momohara, S., Yamanaka, H., Yamada, R., Kubo, M., Nakamura, Y., and Yamamoto, K. (2010). A regulatory variant in CCR6 is associated with rheumatoid arthritis susceptibility. Nat. Genet. 42, 515–519.
Kong, Y. Y., Feige, U., Sarosi, I., Bolon, B., Tafuri, A., Morony, S., Capparelli, C., Li, J., Elliott, R., Mccabe, S., Wong, T., Campagnuolo, G., Moran, E., Bogoch, E. R., Van, G., Nguyen, L. T., Ohashi, P. S., Lacey, D. L., Fish, E., Boyle, W. J., and Penninger, J. M. (1999). Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 402, 304–309.
Korganow, A. S., Ji, H., Mangialaio, S., Duchatelle, V., Pelanda, R., Martin, T., Degott, C., Kikutani, H., Rajewsky, K., Pasquali, J. L., Benoist, C., and Mathis, D. (1999). From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity 10, 451–461.
Kotake, S., Udagawa, N., Takahashi, N., Matsuzaki, K., Itoh, K., Ishiyama, S., Saito, S., Inoue, K., Kamatani, N., Gillespie, M. T., Martin, T. J., and Suda, T. (1999). IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Invest. 103, 1345–1352.
Kouskoff, V., Korganow, A. S., Duchatelle, V., Degott, C., Benoist, C., and Mathis, D. (1996). Organ-specific disease provoked by systemic autoimmunity. Cell 87, 811–822.
Kremer, J. M., Bloom, B. J., Breedveld, F. C., Coombs, J. H., Fletcher, M. P., Gruben, D., Krishnaswami, S., Burgos-Vargas, R., Wilkinson, B., Zerbini, C. A., and Zwillich, S. H. (2009). The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 60, 1895–1905.
Kwok, S. K., Cho, M. L., Park, M. K., Oh, H. J., Park, J. S., Her, Y. M., Lee, S. Y., Youn, J., Ju, J. H., Park, K. S., Kim, S. I., Kim, H. Y., and Park, S. H. (2012). Interleukin-21 promotes osteoclastogenesis in rheumatoid arthritis in humans and mice. Arthritis Rheum. 64, 740–751.
Lee, A. T., Li, W., Liew, A., Bombardier, C., Weisman, M., Massarotti, E. M., Kent, J., Wolfe, F., Begovich, A. B., and Gregersen, P. K. (2005). The PTPN22 R620W polymorphism associates with RF positive rheumatoid arthritis in a dose-dependent manner but not with HLA-SE status. Genes Immun. 6, 129–133.
Lee, D. M., Friend, D. S., Gurish, M. F., Benoist, C., Mathis, D., and Brenner, M. B. (2002). Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science 297, 1689–1692.
Lee, D. M., Kiener, H. P., Agarwal, S. K., Noss, E. H., Watts, G. F., Chisaka, O., Takeichi, M., and Brenner, M. B. (2007). Cadherin-11 in synovial lining formation and pathology in arthritis. Science 315, 1006–1010.
Lefèvre, S., Knedla, A., Tennie, C., Kampmann, A., Wunrau, C., Dinser, R., Korb, A., Schnaker, E. M., Tarner, I. H., Robbins, P. D., Evans, C. H., Sturz, H., Steinmeyer, J., Gay, S., Scholmerich, J., Pap, T., Muller-Ladner, U., and Neumann, E. (2009). Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat. Med. 15, 1414–1420.
Linsley, P. S., and Nadler, S. G. (2009). The clinical utility of inhibiting CD28-mediated costimulation. Immunol. Rev. 229, 307–321.
Lubberts, E., Koenders, M. I., Oppers-Walgreen, B., Van Den Bersselaar, L., Coenen-De Roo, C. J., Joosten, L. A., and Van Den Berg, W. B. (2004). Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 50, 650–659.
Maeshima, K., Yamaoka, K., Kubo, S., Nakano, K., Iwata, S., Saito, K., Ohishi, M., Miyahara, H., Tanaka, S., Ishii, K., Yoshimatsu, H., and Tanaka, Y. (2011). A JAK inhibitor tofacitinib regulates synovitis through inhibition of IFN-gamma and IL-17 production by human CD4(+) T cells. Arthritis Rheum. doi: 10.1002/art.34329
Manoury-Schwartz, B., Chiocchia, G., Bessis, N., Abehsira-Amar, O., Batteux, F., Muller, S., Huang, S., Boissier, M. C., and Fournier, C. (1997). High susceptibility to collagen-induced arthritis in mice lacking IFN-gamma receptors. J. Immunol. 158, 5501–5506.
Matmati, M., Jacques, P., Maelfait, J., Verheugen, E., Kool, M., Sze, M., Geboes, L., Louagie, E., Guire, C. M., Vereecke, L., Chu, Y., Boon, L., Staelens, S., Matthys, P., Lambrecht, B. N., Schmidt-Supprian, M., Pasparakis, M., Elewaut, D., Beyaert, R., and Van Loo, G. (2011). A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat. Genet. 43, 908–912.
Matsumoto, I., Maccioni, M., Lee, D. M., Maurice, M., Simmons, B., Brenner, M., Mathis, D., and Benoist, C. (2002). How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat. Immunol. 3, 360–365.
Midwood, K., Sacre, S., Piccinini, A. M., Inglis, J., Trebaul, A., Chan, E., Drexler, S., Sofat, N., Kashiwagi, M., Orend, G., Brennan, F., and Foxwell, B. (2009). Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat. Med. 15, 774–780.
Miossec, P., Korn, T., and Kuchroo, V. K. (2009). Interleukin-17 and type 17 helper T cells. N. Engl. J. Med. 361, 888–898.
Muller-Ladner, U., Kriegsmann, J., Franklin, B. N., Matsumoto, S., Geiler, T., Gay, R. E., and Gay, S. (1996). Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am. J. Pathol. 149, 1607–1615.
Murakami, M., Okuyama, Y., Ogura, H., Asano, S., Arima, Y., Tsuruoka, M., Harada, M., Kanamoto, M., Sawa, Y., Iwakura, Y., Takatsu, K., Kamimura, D., and Hirano, T. (2011). Local microbleeding facilitates IL-6- and IL-17-dependent arthritis in the absence of tissue antigen recognition by activated T cells. J. Exp. Med. 208, 103–114.
Nakae, S., Nambu, A., Sudo, K., and Iwakura, Y. (2003a). Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J. Immunol. 171, 6173–6177.
Nakae, S., Saijo, S., Horai, R., Sudo, K., Mori, S., and Iwakura, Y. (2003b). IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc. Natl. Acad. Sci. U.S.A. 100, 5986–5990.
Nakashima, T., Hayashi, M., Fukunaga, T., Kurata, K., Oh-Hora, M., Feng, J. Q., Bonewald, L. F., Kodama, T., Wutz, A., Wagner, E. F., Penninger, J. M., and Takayanagi, H. (2011). Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 17, 1231–1234.
Nandakumar, K. S., Backlund, J., Vestberg, M., and Holmdahl, R. (2004). Collagen type II (CII)-specific antibodies induce arthritis in the absence of T or B cells but the arthritis progression is enhanced by CII-reactive T cells. Arthritis Res. Ther. 6, R544–R550.
Notley, C. A., Inglis, J. J., Alzabin, S., Mccann, F. E., Mcnamee, K. E., and Williams, R. O. (2008). Blockade of tumor necrosis factor in collagen-induced arthritis reveals a novel immunoregulatory pathway for Th1 and Th17 cells. J. Exp. Med. 205, 2491–2497.
Ogura, H., Murakami, M., Okuyama, Y., Tsuruoka, M., Kitabayashi, C., Kanamoto, M., Nishihara, M., Iwakura, Y., and Hirano, T. (2008). Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 29, 628–636.
Okamoto, K., Iwai, Y., Oh-Hora, M., Yamamoto, M., Morio, T., Aoki, K., Ohya, K., Jetten, A. M., Akira, S., Muta, T., and Takayanagi, H. (2010). IkappaBzeta regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature 464, 1381–1385.
Perricone, C., Ceccarelli, F., and Valesini, G. (2011). An overview on the genetic of rheumatoid arthritis: a never-ending story. Autoimmun. Rev. 10, 599–608.
Pettit, A. R., Ji, H., Von Stechow, D., Muller, R., Goldring, S. R., Choi, Y., Benoist, C., and Gravallese, E. M. (2001). TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am. J. Pathol. 159, 1689–1699.
Pollinger, B., Junt, T., Metzler, B., Walker, U. A., Tyndall, A., Allard, C., Bay, S., Keller, R., Raulf, F., Di Padova, F., O’Reilly, T., Horwood, N. J., Patel, D. D., and Littlewood-Evans, A. (2011). Th17 cells, not IL-17+ gammadelta T cells, drive arthritic bone destruction in mice and humans. J. Immunol. 186, 2602–2612.
Ranges, G. E., Sriram, S., and Cooper, S. M. (1985). Prevention of type II collagen-induced arthritis by in vivo treatment with anti-L3T4. J. Exp. Med. 162, 1105–1110.
Redlich, K., Hayer, S., Ricci, R., David, J. P., Tohidast-Akrad, M., Kollias, G., Steiner, G., Smolen, J. S., Wagner, E. F., and Schett, G. (2002). Osteoclasts are essential for TNF-alpha-mediated joint destruction. J. Clin. Invest. 110, 1419–1427.
Sakaguchi, N., Takahashi, T., Hata, H., Nomura, T., Tagami, T., Yamazaki, S., Sakihama, T., Matsutani, T., Negishi, I., Nakatsuru, S., and Sakaguchi, S. (2003). Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature 426, 454–460.
Sato, K., Suematsu, A., Okamoto, K., Yamaguchi, A., Morishita, Y., Kadono, Y., Tanaka, S., Kodama, T., Akira, S., Iwakura, Y., Cua, D. J., and Takayanagi, H. (2006). Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 203, 2673–2682.
Sawa, S., Kamimura, D., Jin, G. H., Morikawa, H., Kamon, H., Nishihara, M., Ishihara, K., Murakami, M., and Hirano, T. (2006). Autoimmune arthritis associated with mutated interleukin (IL)-6 receptor gp130 is driven by STAT3/IL-7-dependent homeostatic proliferation of CD4+ T cells. J. Exp. Med. 203, 1459–1470.
Shinohara, M., Koga, T., Okamoto, K., Sakaguchi, S., Arai, K., Yasuda, H., Takai, T., Kodama, T., Morio, T., Geha, R. S., Kitamura, D., Kurosaki, T., Ellmeier, W., and Takayanagi, H. (2008). Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell 132, 794–806.
Singh, K., Colmegna, I., He, X., Weyand, C. M., and Goronzy, J. J. (2008). Synoviocyte stimulation by the LFA-1-intercellular adhesion molecule-2-Ezrin-Akt pathway in rheumatoid arthritis. J. Immunol. 180, 1971–1978.
Solt, L. A., Kumar, N., Nuhant, P., Wang, Y., Lauer, J. L., Liu, J., Istrate, M. A., Kamenecka, T. M., Roush, W. R., Vidovic, D., Schurer, S. C., Xu, J., Wagoner, G., Drew, P. D., Griffin, P. R., and Burris, T. P. (2011). Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 472, 491–494.
Stahl, E. A., Raychaudhuri, S., Remmers, E. F., Xie, G., Eyre, S., Thomson, B. P., Li, Y., Kurreeman, F. A., Zhernakova, A., Hinks, A., Guiducci, C., Chen, R., Alfredsson, L., Amos, C. I., Ardlie, K. G., Barton, A., Bowes, J., Brouwer, E., Burtt, N. P., Catanese, J. J., Coblyn, J., Coenen, M. J., Costenbader, K. H., Criswell, L. A., Crusius, J. B., Cui, J., De Bakker, P. I., De Jager, P. L., Ding, B., Emery, P., Flynn, E., Harrison, P., Hocking, L. J., Huizinga, T. W., Kastner, D. L., Ke, X., Lee, A. T., Liu, X., Martin, P., Morgan, A. W., Padyukov, L., Posthumus, M. D., Radstake, T. R., Reid, D. M., Seielstad, M., Seldin, M. F., Shadick, N. A., Steer, S., Tak, P. P., Thomson, W., Van Der Helm-Van Mil, A. H., Van Der Horst-Bruinsma, I. E., Van Der Schoot, C. E., Van Riel, P. L., Weinblatt, M. E., Wilson, A. G., Wolbink, G. J., Wordsworth, B. P., Wijmenga, C., Karlson, E. W., Toes, R. E., De Vries, N., Begovich, A. B., Worthington, J., Siminovitch, K. A., Gregersen, P. K., Klareskog, L., and Plenge, R. M. (2010). Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 42, 508–514.
Stanczyk, J., Pedrioli, D. M., Brentano, F., Sanchez-Pernaute, O., Kolling, C., Gay, R. E., Detmar, M., Gay, S., and Kyburz, D. (2008). Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 58, 1001–1009.
Takayanagi, H. (2009). Osteoimmunology and the effects of the immune system on bone. Nat. Rev. Rheumatol. 5, 667–676.
Takayanagi, H., Iizuka, H., Juji, T., Nakagawa, T., Yamamoto, A., Miyazaki, T., Koshihara, Y., Oda, H., Nakamura, K., and Tanaka, S. (2000a). Involvement of receptor activator of nuclear factor kappaB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis Rheum. 43, 259–269.
Takayanagi, H., Ogasawara, K., Hida, S., Chiba, T., Murata, S., Sato, K., Takaoka, A., Yokochi, T., Oda, H., Tanaka, K., Nakamura, K., and Taniguchi, T. (2000b). T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 408, 600–605.
Takayanagi, H., Kim, S., Koga, T., Nishina, H., Isshiki, M., Yoshida, H., Saiura, A., Isobe, M., Yokochi, T., Inoue, J., Wagner, E. F., Mak, T. W., Kodama, T., and Taniguchi, T. (2002). Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 3, 889–901.
Takayanagi, H., Oda, H., Yamamoto, S., Kawaguchi, H., Tanaka, S., Nishikawa, T., and Koshihara, Y. (1997). A new mechanism of bone destruction in rheumatoid arthritis: synovial fibroblasts induce osteoclastogenesis. Biochem. Biophys. Res. Commun. 240, 279–286.
Tang, W., Lu, Y., Tian, Q. Y., Zhang, Y., Guo, F. J., Liu, G. Y., Syed, N. M., Lai, Y., Lin, E. A., Kong, L., Su, J., Yin, F., Ding, A. H., Zanin-Zhorov, A., Dustin, M. L., Tao, J., Craft, J., Yin, Z., Feng, J. Q., Abramson, S. B., Yu, X. P., and Liu, C. J. (2011). The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science 332, 478–484.
Taniguchi, K., Kohsaka, H., Inoue, N., Terada, Y., Ito, H., Hirokawa, K., and Miyasaka, N. (1999). Induction of the p16INK4a senescence gene as a new therapeutic strategy for the treatment of rheumatoid arthritis. Nat. Med. 5, 760–767.
Taylor, P. C., Peters, A. M., Paleolog, E., Chapman, P. T., Elliott, M. J., Mccloskey, R., Feldmann, M., and Maini, R. N. (2000). Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor alpha blockade in patients with rheumatoid arthritis. Arthritis Rheum. 43, 38–47.
Theill, L. E., Boyle, W. J., and Penninger, J. M. (2002). RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu. Rev. Immunol. 20, 795–823.
Tran, C. N., Davis, M. J., Tesmer, L. A., Endres, J. L., Motyl, C. D., Smuda, C., Somers, E. C., Chung, K. C., Urquhart, A. G., Lundy, S. K., Kovats, S., and Fox, D. A. (2007). Presentation of arthritogenic peptide to antigen-specific T cells by fibroblast-like synoviocytes. Arthritis Rheum. 56, 1497–1506.
Vermeire, K., Heremans, H., Vandeputte, M., Huang, S., Billiau, A., and Matthys, P. (1997). Accelerated collagen-induced arthritis in IFN-gamma receptor-deficient mice. J. Immunol. 158, 5507–5513.
Williams, R. O., Feldmann, M., and Maini, R. N. (1992). Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc. Natl. Acad. Sci. U.S.A. 89, 9784–9788.
Wipke, B. T., and Allen, P. M. (2001). Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J. Immunol. 167, 1601–1608.
Wong, B. R., Josien, R., Lee, S. Y., Sauter, B., Li, H. L., Steinman, R. M., and Choi, Y. (1997). TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J. Exp. Med. 186, 2075–2080.
Wu, H. J., Ivanov, I. I., Darce, J., Hattori, K., Shima, T., Umesaki, Y., Littman, D. R., Benoist, C., and Mathis, D. (2010). Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32, 815–827.
Xiong, J., Onal, M., Jilka, R. L., Weinstein, R. S., Manolagas, S. C., and O’Brien, C. A. (2011). Matrix-embedded cells control osteoclast formation. Nat. Med. 17, 1235–1241.
Yamaguchi, Y., Fujio, K., Shoda, H., Okamoto, A., Tsuno, N. H., Takahashi, K., and Yamamoto, K. (2007). IL-17B and IL-17C are associated with TNF-alpha production and contribute to the exacerbation of inflammatory arthritis. J. Immunol. 179, 7128–7136.
Yao, C., Sakata, D., Esaki, Y., Li, Y., Matsuoka, T., Kuroiwa, K., Sugimoto, Y., and Narumiya, S. (2009). Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat. Med. 15, 633–640.
Yeo, L., Toellner, K. M., Salmon, M., Filer, A., Buckley, C. D., Raza, K., and Scheel-Toellner, D. (2012). Cytokine mRNA profiling identifies B cells as a major source of RANKL in rheumatoid arthritis. Ann. Rheum. Dis. 70, 2022–2028.
Yoshitomi, H., Sakaguchi, N., Kobayashi, K., Brown, G. D., Tagami, T., Sakihama, T., Hirota, K., Tanaka, S., Nomura, T., Miki, I., Gordon, S., Akira, S., Nakamura, T., and Sakaguchi, S. (2005). A role for fungal {beta}-glucans and their receptor Dectin-1 in the induction of autoimmune arthritis in genetically susceptible mice. J. Exp. Med. 201, 949–960.
Keywords: CD4+T-cell, Th17 cell, synovial fibroblast, osteoclast, inflammation, bone destruction, rheumatoid arthritis
Citation: Komatsu N and Takayanagi H (2012) Inflammation and bone destruction in arthritis: synergistic activity of immune and mesenchymal cells in joints. Front. Immun. 3:77. doi: 10.3389/fimmu.2012.00077
Received: 15 January 2012; Paper pending published: 03 February 2012;
Accepted: 27 March 2012; Published online: 13 April 2012.
Edited by:
Masaaki Murakami, Osaka University, JapanReviewed by:
Rik Lories, Katholieke Universiteit Leuven, BelgiumHideki Ogura, Osaka University, Japan
Copyright: © 2012 Komatsu and Takayanagi. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Hiroshi Takayanagi, Department of Cell Signaling, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Yushima 1-5-45, Bunkyo-ku, Tokyo 113-8549, Japan. e-mail:dGFrYS5jc2lAdG1kLmFjLmpw