


- Center for Experimental Therapeutics and Reperfusion Injury, Department of Anesthesiology, Perioperative and Pain Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA
Endogenous mechanisms for successful resolution of an acute inflammatory response and the local return to homeostasis are of interest because excessive inflammation underlies many human diseases. In this review, we provide an update and overview of functional metabolomics that identified a new bioactive metabolome of docosahexaenoic acid (DHA). Systematic studies revealed that DHA was converted to DHEA-derived novel bioactive products as well as aspirin-triggered forms of protectins (AT-PD1). The new oxygenated DHEA-derived products blocked PMN chemotaxis, reduced P-selectin expression and platelet-leukocyte adhesion, and showed organ protection in ischemia/reperfusion injury. These products activated cannabinoid receptor (CB2 receptor) and not CB1 receptors. The AT-PD1 reduced neutrophil (PMN) recruitment in murine peritonitis. With human cells, AT-PD1 decreased transendothelial PMN migration as well as enhanced efferocytosis of apoptotic human PMN by macrophages. The recent findings reviewed here indicate that DHEA oxidative metabolism and aspirin-triggered conversion of DHA produce potent novel molecules with anti-inflammatory and organ-protective properties, opening the DHA metabolome functional roles.
The Neutrophil (PMN) in Resolution and the Programmed Outcome of Acute Inflammation
The acute inflammatory response has several potential outcomes that include progression to chronic inflammation, scarring, and fibrosis or ideally complete resolution, see Kumar et al. (2005). Acute inflammation is a response that eliminates or neutralizes foreign bodies or organisms and is protective against injury or infection (Kumar et al., 2005; Serhan et al., 2010). This process is characterized by vascular dilation, enhanced permeability of capillaries, increased blood flow, and the production of fibrin-rich exudates (Majno and Joris, 2004). Vascular permeability and increased blood flow permit leukocyte recruitment from the circulation postcapillary venules (Figure 1) to form the PMN-rich exudate (Ryan and Majno, 1977). Leukocyte recruitment is classically defined by the initial trafficking of polymorphonuclear (PMN) granulocytes followed by monocytes that differentiate locally into macrophages (Figure 1). These events in pro-resolving responses are coupled with release of factors that prevent further or excessive trafficking of leukocytes in self-limited inflammation allowing for resolution (Serhan et al., 2000, 2002; Serhan and Savill, 2005). Pro-inflammatory mediators such as prostaglandins and leukotrienes play an important role (Borgeat and Samuelsson, 1979; Samuelsson, 1983; Samuelsson et al., 1987) early in the response, stimulating vascular changes as well as PMN recruitment. These events involve LTB4 and IL-8, potent PMN chemoattractants, as well as many other endogenous and exogenous chemoattractants.
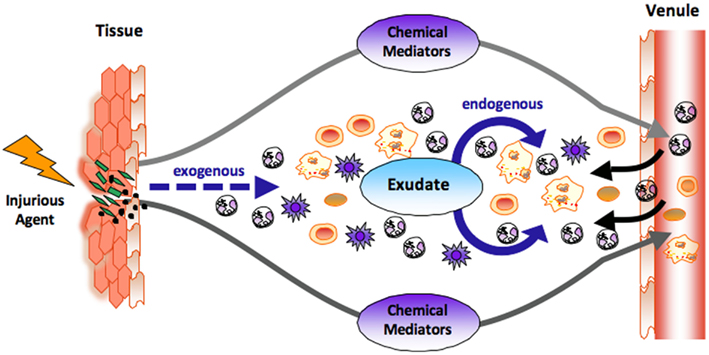
Figure 1. The role of chemical mediators in the acute inflammatory response. At the site of tissue injury or bacterial invasion, both exogenous and endogenous chemical mediators are liberated. Classic endogenous mediators such as prostaglandins and leukotrienes dilate vasculatures, enhance permeability of capillaries, increase blood flow, and stimulate the recruitment of neutrophils (PMNs) to form inflammatory exudate. Novel chemical mediators are produced in the evolution and resolution of the exudate that regulate tissue responses (see text for details). The black arrow denotes leukocyte traffic from venules and the dashed arrow denotes exogenous, i.e., bacterial components and chemoattractants.
The progression from acute to chronic inflammation as in many widely occurring human diseases such as arthritis, periodontal disease, and cardiovascular disease, is generally viewed as an excess of pro-inflammatory mediators, but may also arise via a failure in mechanisms to resolve (Serhan, 2011). Although mononuclear cells can in many settings contribute to pro-inflammatory responses, they are also critical in wound healing, tissue repair, and remodeling in a non-inflammatory, non-phlogistic manner (Stables et al., 2011; Ariel and Serhan, 2012). Most successful inflammatory processes are self-limiting (Majno and Joris, 2004), which implies the existence of endogenous anti-inflammation pathways (Serhan et al., 2004; Serhan and Savill, 2005).
Complete or successful resolution of an acute inflammatory response and the local return to homeostasis is necessary for ongoing health. The ideal outcome is the removal of leukocytes from infected or inflamed tissues without leaving remnants of the host’s combat between leukocytes, invading microbes, and/or other initiators of inflammation. The authors’ research efforts have focused on identification of mediators involved in the successful resolution process (Serhan, 2007). It was widely believed and argued that simple dilution of pro-inflammatory mediators was enough to “turn off” or “burn out” inflammation, with the subsequent responses ending naturally or passively (Kumar et al., 2005). Now, a body of evidence indicates that resolution of acute inflammation and the return to homeostasis is not simply passive, but an active and highly regulated process that can be considered programmed resolution at the tissue level (Serhan, 2007; Morris et al., 2010). In this regard, the specialized pro-resolving lipid mediators (SPMs) comprise a genus (chemically distinct families) of endogenous mediators, including lipoxins, resolvins, protectins, and maresins (Serhan et al., 2009; Figure 2; Table 1). They are actively biosynthesized during the resolution phase of acute inflammation and are potent agonists that control the magnitude and duration of inflammation. During resolution of inflammation, ω-3 fatty acids are rapidly imported (Kasuga et al., 2008) to the resolving exudate from circulation along with albumin and trafficking leukocytes that are then used as substrates to produce the local-acting E-series resolvins (Oh et al., 2011), D-series resolvins and protectins as well as maresins (Serhan et al., 2009). Each of these new families of potent local mediators carries both anti-inflammatory as well as pro-resolving actions in that they stop PMN (limit their further influx) to the site of inflammation and stimulate efferocytosis (Spite and Serhan, 2010; Serhan, 2011). Docosahexaenoic acid (DHA) is unique in this regard because it has several fates in its metabolome (Figure 2). DHA undergoes esterification to form complex lipids that circulate or reside in phospholipid membranes of cells and tissues (Bazan et al., 2010), or can undergo autooxidation of the products such as neuroisoprostanes that are potentially pro-inflammatory (Roberts et al., 1998; Roberts and Morrow, 2002), or undergo conversion in inflammatory exudates to potent pro-resolving mediators such as the SPMs. Hence, the fate of DHA is governed, in part, locally in its microenvironment and hence “when and where” determines its action and impact in vivo (Figures 2 and 3) in addition to DHA as a precursor to resolvins and their aspirin-triggered (AT) forms (Serhan et al., 2002). Systematic studies reveal that DHA is converted to DHEA-derived novel products (Yang et al., 2011) as well as AT forms of protectins (Serhan et al., 2011; see Figure 3). These are the main subjects of this review and are detailed in the next sections.
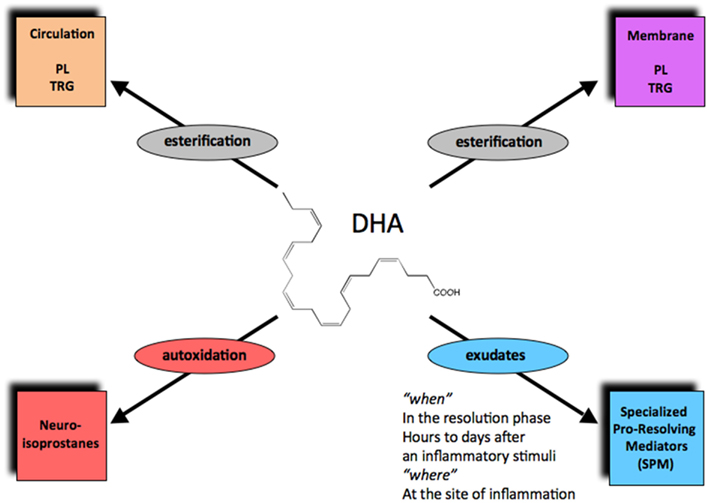
Figure 2. The fate of DHA in its new metabolome in vivo. Via esterification, DHA enters phospholipids and triglycerides that can circulate or reside in cell membranes. Autooxidation of DHA can produce neuroisoprostanes, the DHA form of isoprostanes. Enzymatic conversion in local inflammatory exudates can generate the specialized pro-resolving lipid mediators (SPMs), including D-series resolvins, protectins, and maresins.

Table 1. Key structures of the SPM: resolvins, protectins, and maresins.
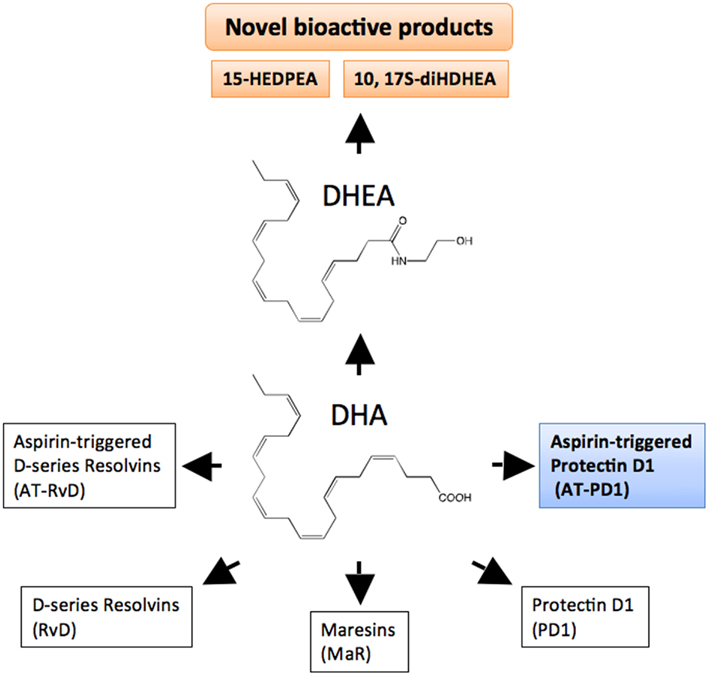
Figure 3. DHA bioactive metabolome. DHA is the precursor to D-series resolvin (RvD), aspirin-triggered resolvin D (AT-RvD), maresins (MaR), protectin D1 (PD1), and aspirin-triggered protectin D1 (AT-PD1). Another metabolic fate of DHA is conversion to docosahexaenoyl ethanolamide (DHEA). See text for details on the recently identified novel series of bioactive oxygenated DHEA-derived products including 15-HEDPEA and 10,17S-diHDHEA.
Functional Metabolomics and the New DHA Metabolome
LC–UV–MS–MS Identification of 17-HDHEA
To investigate the potential endogenous generation of DHEA-derived bioactive products in mice, brain was harvested and subject to solid phase extraction (Yang et al., 2011). Next, the methyl formate fractions were taken for LC–UV–MS–MS-based metabolomics. Tandem mass fragmentations and online UV spectrum with characteristic λmax at 237 nm were consistent with the structure as shown in Figure 4 and Yang et al. (2011). Because of the lack of suitable functional groups for direct efficient ionization and analysis of 17-hydroxy-4Z, 7Z, 10Z, 13Z, 15Z, 19Z-docosahexaenoylethanolamide (17-HDHEA) for LC–MS–MS, its acetate adduct m/z 446 = [M + CH3COOH-H] was targeted. The major tandem mass ions were assigned for: m/z 386 = [M−H], 368 = [M−H – H2O], 281 = [299 – H2O]. The m/z 288 is consistent with fragmentation at position 17 (Figure 4 and Yang et al., 2011).
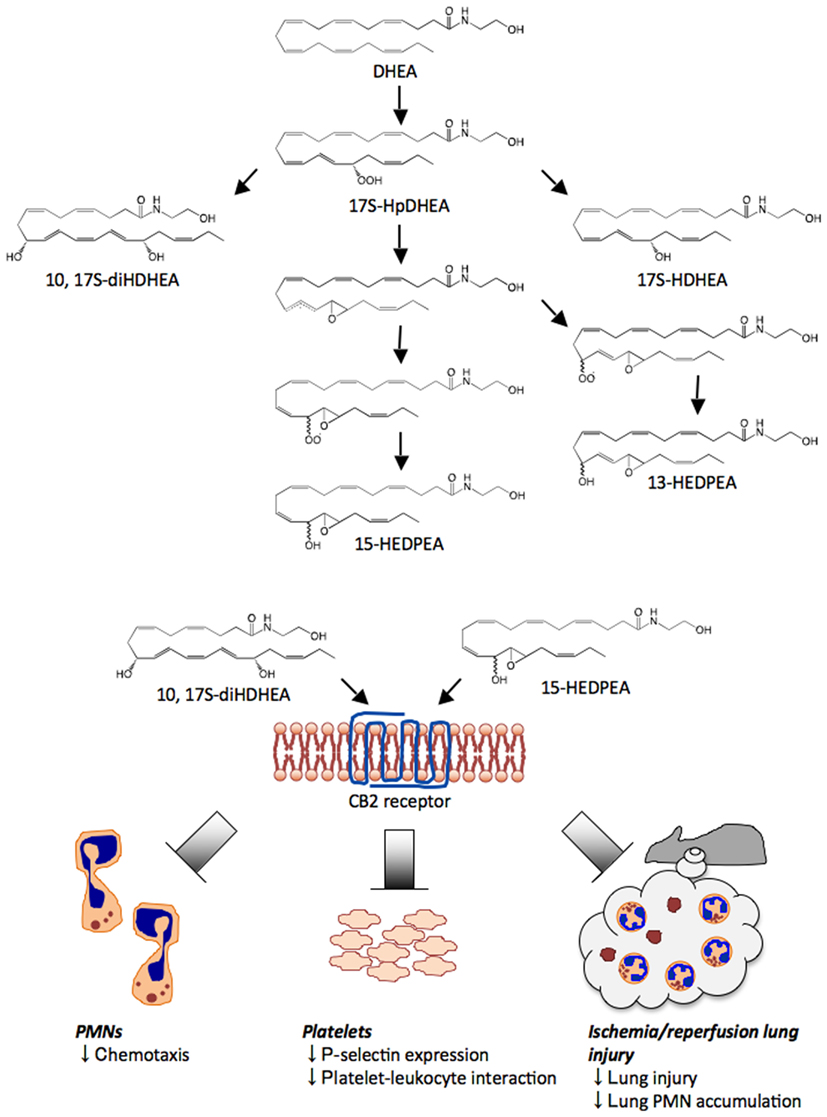
Figure 4. Novel pathways and oxygenation products from DHEA. DHEA is first converted to 17-HpDHEA by 15-LOX. Next, 17-HpDHEA is partially reduced to the oxide radical, which reacts with the vicinal double bond at the 16-position to yield the 16(17)-epoxide radical (see text and Yang et al., 2011). Non- or low stereospecific addition of oxygen to the intermediate leads to formation of two types of peroxide radical diastereomers; their further reduction produces 13-HEDPEA and 15-HEDPEA. Via a LOX-related mechanism, 17-HpDHEA is converted to 10, 17S-diHDHEA or directly reduced to 17S-HDHEA. 10, 17S-diHDHEA, and 15-HEDPEA block PMN chemotaxis, reduce P-selectin expression and aggregation of platelets and leukocytes, and show organ protection in ischemia/reperfusion injury. Although these new products directly act on recombinant CB2 receptors in vitro, the activation of CB2 in vivo and/or additional receptors in vivo by 10,17S-diHDHEA and 15-HEDPEA remains of interest.
Because of the limited quantities of endogenous 17-HDHEA produced in brain tissue, further studies and in vitro enzymatic preparation were carried out by incubating DHEA with 15-LOX followed by reduction with NaBH4. Endogenous 17-HDHEA and the enzymatically prepared compound in vitro gave essentially the same LC retention times and tandem mass fragmentations. To assess their production by human and mouse tissues, DHEA was also incubated with isolated human PMN or whole mouse brain because DHEA is enriched in this tissue. LC–MS–MS-based targeted lipidomics indicated the production of a novel series of oxygenated products (see Figure 4).
Decoding
As second approach in parallel to structure elucidation, screening of HPLC-isolated DHEA metabolites obtained from mouse brain was carried out utilizing a microfluidic chamber (Irimia et al., 2006; Kasuga et al., 2008). After a gradient of IL-8 was introduced to the main channel of the device, P-selectin tethered leukocytes rapidly migrated in the IL-8 gradient at an average rate of 2.3 μm/min. Next, a mixture of isolated metabolites was infused into the main channel. Human PMN chemotaxis is dramatically reduced upon the addition of the brain metabolite mixture. For example, the average human PMN chemotaxis velocity dropped from 2.3 to ∼ 0.7 μm/min (Yang et al., 2011). These results indicated that the brain fraction of metabolites contained products that stopped PMN chemotaxis. Results from this screening uncovered that at least one bioactive product was present among the mixture of DHEA metabolites. Hence, we next pursued the metabolic fates of DHEA and 17-HpDHEA/17-HDHEA identified in mouse brain (Yang et al., 2011) using LC/UV/MS/MS-based lipidomics. As with 17-HDHEA, acetate adducts of potential DHEA-derived novel metabolites [M + CH3COOH-H] were targeted for tandem mass analysis. From these studies we identified the presence and production of novel products in the DHEA metabolome (Figure 4).
Human PMN incubated with either DHEA or 17-HpDHEA led to the generation of 17-HDHEA, 4,17-diHDHEA, 10,17-diHDHEA, and 15-HEDPEA (see Figure 4 and Yang et al., 2011). Human hemoglobin, which can be liberated upon tissue damage (Kumar et al., 2005), was next incubated with 17-HpDHEA. This gave 13-HEDPEA and 15-HEDPEA as prominent products, as well as 17-HDHEA (see Figure 4). Mouse brain homogenates with DHEA also produced 17-HDHEA and 4,17-diHDHEA as major products with smaller amounts of 7,17-diHDHEA, 10,17-diHDHEA, and 15-HEDPEA. The adduct parent ion, analyte parent ion and the ions resulted from neutral loss are m/z 462 = [M + CH3COOH-H], 402 = [M−H], 384 = [M−H – H2O], 366 = [M−H – 2H2O], which are common signature ions for all dihydroxy-containing DHEA products. The ions m/z 333, 315 = [333 – H2O], 304, 286 = [304-H2O] were assigned as diagnostic ions for fragmentations at position 17. Fragmentations at position 4 can lead to m/z 144, 257 and 239 = [257-H2O]. Its UV spectrum displayed characteristic maximum absorbance at 238 nm, which was consistent with the presence of two separated conjugated diene structures in this compound. Diagnostic ions m/z 304, 286 = [304-H2O], 184, 156, corresponded to the fragmentations at positions 7 and 17 of 7,17-diHDHEA respectively (Yang et al., 2011). The UV spectrum of the compound displayed maximum absorbance, λmax, at 246 nm (Tjonahen et al., 2006), consistent with the presence of two diene structures separated by a methylene group. For 10,17-diHDHEA, m/z 333, 315 = [333-H2O], 304, 286 = [304-H2O], 196 came from fragmentations at positions 10 and 17 (Yang et al., 2011). The presence of a conjugated triene structure in 10,17-diHDHEA was confirmed by the characteristic UV spectrum with λmax at 270 nm. Tandem mass spectrum of 13-HEDPEA (Yang et al., 2011) gave signature fragmentation ions m/z 320, 304, 286 = [304-H2O], 236. GC/MS was also utilized for additional structural analysis with 13-HEDPEA and 15-HEDPEA that helped to confirm the tandem MS assignments.
To determine concentrations, as well as to further confirm structures, HPLC-isolated 13-HEDPEA and 15-HEDPEA were characterized using proton NMR (1H NMR). In the chemical shift assignments (Yang et al., 2011) for 15-HEDPEA, the proton at position 15 (H-15) displayed two distinct chemical shifts. Because of limited amounts of materials and the lack of informative UV chromophores present in these compounds, NMR spectroscopy was also used for quantitation using 17-HDHA as an internal standard with known concentrations. Each NMR quantitated compound was then used for HPLC calibration and quantitation (Yang et al., 2011).
Each of these HPLC-isolated dioxygenated DHEA products were screened for direct PMN actions using microfluidic chambers and freshly isolated PMN (Yang et al., 2011). Infusion of isolated 15-HEDPEA at 10 nM to the main channel stimulated changes in morphology and chemotaxis of human PMN in the IL-8 gradient and stopped further PMN migration (Figure 4). For direct comparison, PMN chemotaxis velocity did not change with time with the IL-8 gradient. Also, at 10 nM, 4,17-diHDHEA, 7,17-diHDHEA, or 10,17-diHDHEA did not significantly regulate chemotaxis, whereas at higher concentrations, e.g., 10 μM, 10,17-diHDHEA rapidly stopped PMN. Together, these results indicated that 15-HEDPEA is the most potent of this series in regulating human PMN shape change and motility.
Cannabinoid Receptors and Novel DHEA Products
Because AEA exerts a wide range of bioactions via activating cannabinoid receptor(s) (Devane et al., 1992; Felder et al., 1993), we therefore tested whether DHEA, 10,17-diHDHEA or 15-HEDPEA also activated CB receptors. For these experiments, we used recombinant human CB receptors over-expressed in a beta-arrestin system. HPLC purified AEA was used for direct comparison as a known receptor agonist of CB1 (Janero and Makriyannis, 2007; Vemuri et al., 2008). In these experiments, activation of CB2 by AEA gave EC50 ∼ 1.1 × 1010 M and DHEA 9.8 × 10−9 M. For comparison, EC50 for metabolically oxygenated products, 10,17-diHDHEA and 15-HEDPEA, were 3.9 × 10−10 M and 1.0 × 10−10 M (Yang et al., 2011). These results demonstrated that the novel enzymatic oxidation products from DHEA are activators of CB2 receptors, and that 10,17-diHDHEA and 15-HEDPEA also activated CB1 receptors but required much higher concentrations. In this system, 15(S)-HETE ethanolamide (oxygenated product of AEA) did not stimulate CB2 receptors in this dose range. CB2 receptor-ligand interactions were confirmed with dose response of CB2 specific antagonist AM630. When incubated with GPCR CB2 over-expressed cells, AM630 inhibited activation stimulated with 15-HEDPEA (10 nM) and AEA (10 nM). AM630 also inhibited GPCR CB2 interaction with 10,17-diHDHEA at higher concentration (Yang et al., 2011).
Novel DHEA Products Reduced Platelet-Leukocyte Aggregate
Platelet-leukocyte interactions play important roles in hemostasis, thrombosis, and inflammation (for a recent review see van Gils et al., 2009 and references within). At concentrations as low as 10 pM, 10,17S-diHDHEA or 15-HEDPEA decreased PAF- (100 nM) stimulated platelet-monocyte aggregate formation in human whole blood by ∼30% (Yang et al., 2011). The inhibitory action of 10,17-diHDHEA displayed a bell-shaped dose response and reached maximum reduction at ∼40% with 100 pM. The formation of PMN-platelet aggregates with PAF (100 nM) was also inhibited by 10,17-diHDEA at concentrations as low as 10 pM, as was the surface expression of P-selectin on platelets in human whole blood (Figure 4). By comparison, the precursor DHEA was not active in this dose range.
Novel DHEA Products are Organ Protective in Ischemia/Reperfusion Injury
15-HEDPEA displayed potent bioactions with human PMN at single-cell level (Figure 4 and Yang et al., 2011) and in human whole blood; hence, we questioned whether it had protective actions in vivo in murine hind limb ischemia (1 h) and second organ reperfusion (2 h) injury (Qiu et al., 2000). Indeed, following reperfusion, 15-HEDPEA significantly reduced lung PMN accumulation in mice and associated lung injury at 1 μg/mouse (Figure 4 and Yang et al., 2011; ∼50% reduction directly compared with vehicle).
Lipidomic investigation of DHEA functional metabolome uncovered a series of novel oxygenated products that (1) are potent CB2 agonists, (2) regulate single-cell PMN chemotactic responses, (3) modulate platelet-leukocyte interaction in whole blood, and (4) are organ protective. In view of the role of lipid mediators in inflammation and its resolution as well as homeostasis (Serhan et al., 2008), the present new DHEA metabolome documented herein may serve as a counter-regulatory system in neural tissues and those rich in DHEA as well as in administration of DHA (Calder, 2010) to regulate leukocyte-mediated tissue damage. The link between CB2 receptor activation in vitro and in vivo actions of these novel products remains to be established. Also, it is possible that additional receptors in vivo may be functionally regulated by these new DHEA products.
Aspirin and DHA: Uncovering the AT-(NPD1/PD1) Pathway
Aspirin is well appreciated for its ability to inhibit COX-1 and inactivate this enzyme. On the other hand, the catalytic region of COX-2 is larger than that of COX-1, and when it is acetylated by aspirin, acetylated COX-2 remains active, producing lipoxygenase-like products, demonstrated in vivo in humans (Chiang et al., 2004; Chiang and Serhan, 2004). Aspirin is unique in that it can jump-start resolution by triggering production of E-series and D-series resolvins and AT-lipoxins (Serhan et al., 2000, 2002; Schwab et al., 2007; Morris et al., 2009). The previously unrecognized mediators and mechanisms involve the biosynthesis of AT lipid mediators. With Dr. Bazan (LSU) and Dr. Petasis (USC), we established DHA conversion to the AT-NPD1 pathway (Serhan et al., 2011).
To determine the complete stereochemical assignment and bioactions of AT-(NPD1/PD1), we directly compared the physical and biological properties of DHA-derived AT-(NPD1/PD1) and related 10,17 dihydroxy-docosatriene stereoisomers produced by leukocytes to those prepared in stereochemically pure form by total organic synthesis (Serhan et al., 2011). Since AT-(NPD1/PD1) was identified in resolving murine exudates treated with aspirin (Serhan et al., 2002), biologic AT-(NPD1/PD1) was obtained from resolving murine exudates in vivo. To this end, peritonitis was initiated via intra-peritoneal (i.p.) administration of zymosan A (1 mg/mouse), and exudates were harvested 24 h post injection (i.e., within the resolution phase; Bannenberg et al., 2005). Exudates were subject to solid phase extraction and analyzed using LC–UV–MS–MS-based mediator lipidomics. AT-(NPD1/PD1) was also isolated and identified from activated human leukocytes, namely, aspirin-treated human PMN. Collectively, by matching biologic materials from both human and murine exudate AT-(NPD1/PD1) with synthetic candidates, these results established the complete stereochemistry of endogenous AT-(NPD1/PD1) as 10R,17R-dihydroxydocosa-4Z, 7Z, 11E, 13E, 15Z, 19Z-hexaenoic acid (Serhan et al., 2011).
Anti-Inflammatory Actions of AT-NPD1/PD1
Earlier results indicated that NPD1/PD1 exerted potent anti-inflammatory actions regulating leukocyte trafficking in murine systems (Serhan et al., 2002; Hong et al., 2003). We compared the bioactions of NPD1/PD1 to those of AT-(NPD1/PD1) carried out in parallel with the physical matching experiments (Serhan et al., 2011). Synthetic AT-(NPD1/PD1) limited PMN infiltration into the peritoneum in TNFα-stimulated peritonitis (Figure 5). Both NPD1/PD1 (0.1–10 ng) and AT-(NPD1/PD1; 0.01–10.0 ng) proved to be significant regulators of TNFα-stimulated leukocyte infiltration into the peritoneum. AT-(NPD1/PD1) reduced total leukocyte population of the exudate including PMN, monocyte and lymphocyte infiltrates, reaching a maximal reduction at 1 ng/mouse by as much as 50.4 ± 8.8%. The PMN population was also reduced with AT-(NPD1/PD1) reaching a maximal PMN reduction at 1 ng/mouse (62.2 ± 7.8%; Serhan et al., 2011). The flow cytometry results obtained with murine exudates indicated a reduction in the Ly6G+CD11b+ population as compared to TNFα alone. Of note, the Δ15-trans-AT-(NPD1/PD1) isomer did not reduce either the total exudate leukocyte population or PMN infiltration. Since NPD1/PD1 limited PMN infiltration in zymosan A-initiated peritonitis (Serhan et al., 2006), we determined whether AT-(NPD1/PD1) also reduced PMN infiltration in zymosan A-stimulated peritonitis. AT-(NPD1/PD1; 0.1–100.0 ng) significantly reduced total leukocytes, as well as PMN infiltration, reaching a maximal reduction of 47.8 ± 10.0% and 49.1 ± 11.9%. In comparison to AT-(NPD1/PD1; 1.0 and 10.0 ng), equal doses of either the precursor DHA or Δ15-trans-AT-NPD1/PD1 did not reduce either total leukocyte infiltration or PMN infiltration (Serhan et al., 2011). These findings indicated that AT-(NPD1/PD1) regulates inflammatory responses induced by the pro-inflammatory cytokine TNFα and the TLR ligand, zymosan A.
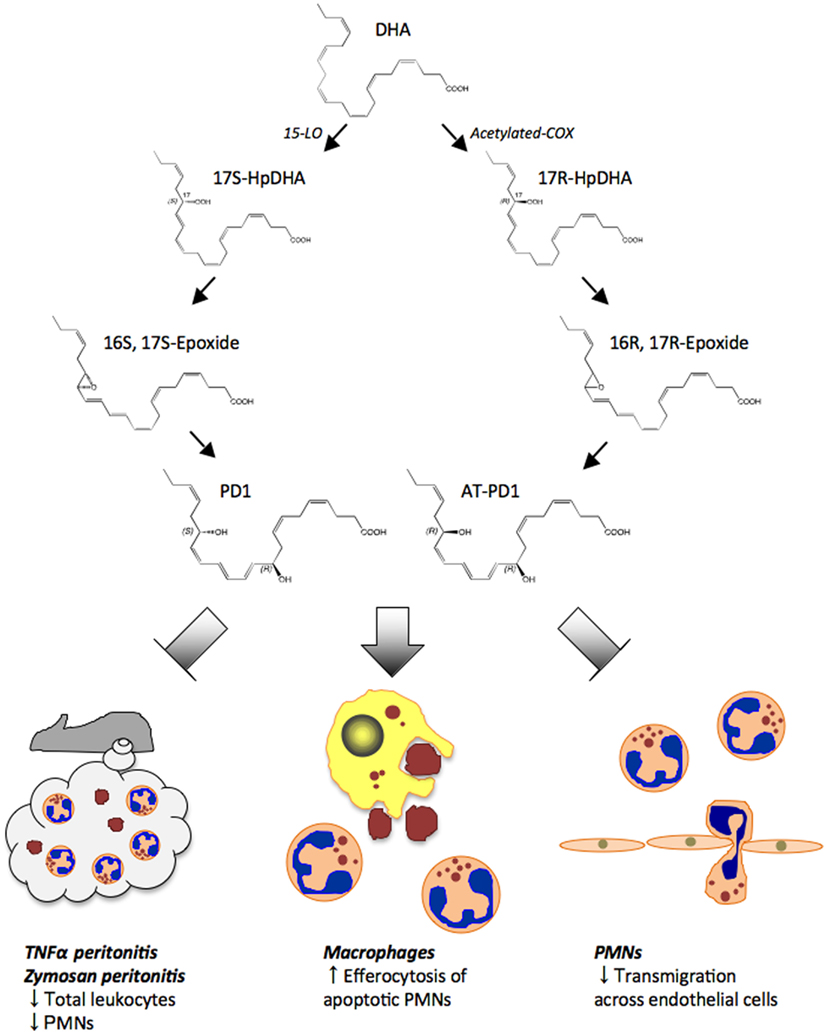
Figure 5. Proposed biosynthesis scheme for protectin D1 (PD1) and aspirin-triggered protectin D1 (AT-PD1). DHA is first converted to 17S-HpDHA intermediate by 15-LOX. Then PD1 is generated through 16S, 17S-epoxide. In the aspirin-triggered pathway, 17R-HpDHA is generated from DHA via acetylated-COX, and converted to AT-PD1 via a 16R, 17R-containing epoxide intermediate. Both PD1 and AT-PD1 reduce leukocytes and PMN infiltration in peritonitis stimulated by either TNF-α or zymosan A. PD1 and AT-PD1 each enhance human macrophage efferocytosis of apoptotic human PMNs and limit human PMN transmigration across human endothelial cells, the defining pro-resolving bioactions.
PMN-Transendothelial Migration and Enhanced Efferocytosis
PMN-transendothelial migration is the first committed step of leukocytes in acute inflammation (Kumar et al., 2005). AT-(NPD1/PD1) and NPD1/PD1 (0.1–10.0 nM) reduced (∼30 and ∼50%) PMN-transendothelial migration stimulated by LTB4. Equal concentrations of the Δ15-trans isomer of AT-(NPD1/PD1) where the conjugated triene portion of the molecule was in the trans rather than cis configuration did not reduce PMN-transendothelial migration. Again, the precursor DHA (Kasuga et al., 2008) did not reduce LTB4-stimulated PMN-transendothelial migration (Serhan et al., 2011). To corroborate these, we also used an electric cell-substrate impedance sensing system (ECIS) that sensitively quantitates cellular responses in two cell systems by real-time monitoring of barrier impedance (Tsikitis et al., 2004). Both AT-(NPD1/PD1) and NPD1/PD1 (1nM) decreased LTB4-stimulated PMN-transendothelial migration, and AT-(NPD1/PD1) also enhanced the uptake of apoptotic human PMN by human macrophages at concentrations as low as 0.1 nM, as did NPD1/PD1 when compared directly. The response was bell-shaped and consistent with the dose response relationship observed for efferocytosis and pro-resolving lipid mediators such as RvE1 (Hong et al., 2008).
Some Considerations for the Future
In summation, the novel products and pathway metabolome reviewed here might have protective and anti-inflammatory actions relevant in humans. This important level of evidence is of interest and remains to be established in human trials. For example, since RvE1 has a direct protective effect on cardiomyocytes against ischemia-reperfusion injury limiting infarct size when administered intravenously just before reperfusion (Keyes et al., 2010), the novel bioactive DHEA products discussed in this review may carry potential clinical applications. These new DHEA products (Figure 4) and/or related structures might be useful in the treatment of acute myocardial infarction, because platelet and neutrophil interactions have a key regulatory role in the site of inflammation (Marcus et al., 1982; Phillipson and Kubes, 2011). Along these lines, Pillai et al. (2012) from this laboratory recently reported on the time course and identification of inflammatory mediators (both cytokine and lipid mediators) after abdominal aortic aneurysm (AAA) surgery. Chemical mediator profiles from patients that had undergone AAA in that study assembled into two groups. The temporal profiles for local chemical mediators from these patients were either those with a pro-inflammatory profile of local mediators or a second group with a potential resolving profile of local mediators. These recent reports and results reviewed herein suggest that the temporal biosynthesis of local chemical mediators following inflammation or tissue injury can have an acute local impact as well as long-term effects following surgical intervention and tissue remodeling. Hence, knowledge of these new mediators and pathways as well as their potential roles may serve as a basis for new and more effective therapeutics that could improve the outcome of diseases, surgery and post-operative events.
Conflict of Interest Statement
The authors declare that this review was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Mary H. Small for skillful manuscript preparation.
References
Ariel, A., and Serhan, C. N. (2012). New lives given by cell death: macrophage differentiation following their encounter with apoptotic leukocytes during the resolution of inflammation. Front. Immun. 3, 4. doi: 10.3389/fimmu.2012.00004
Bannenberg, G. L., Chiang, N., Ariel, A., Arita, M., Tjonahen, E., Gotlinger, K. H., Hong, S., and Serhan, C. N. (2005). Molecular circuits of resolution: formation and actions of resolvins and protectins. J. Immunol. 174, 4345–4355.
Bazan, N. G., Calandria, J. M., and Serhan, C. N. (2010). Rescue and repair during photoreceptor cell renewal mediated by docosahexaenoic acid-derived neuroprotectin D1. J. Lipid Res. 51, 2018–2031.
Borgeat, P., and Samuelsson, B. (1979). Arachidonic acid metabolism in polymorphonuclear leukocytes: effects of ionophore A23187. Proc. Natl. Acad. Sci. U.S.A. 76, 2148–2152.
Calder, P. C. (2010). Rationale and uses of n-3 fatty acids in artificial nutrition. Proc. Nutr. Soc. 69, 565–573.
Chiang, N., Bermudez, E. A., Ridker, P. M., Hurwitz, S., and Serhan, C. N. (2004). Aspirin triggers anti-inflammatory 15-epi-lipoxin A4 and inhibits thromboxane in a randomized human trial. Proc. Natl. Acad. Sci. U.S.A. 101, 15178–15183.
Chiang, N., and Serhan, C. N. (2004). Aspirin triggers formation of anti-inflammatory mediators: new mechanism for an old drug. Discov. Med. 4, 470–475.
Devane, W. A., Hanus, L., Breuer, A., Pertwee, R. G., Stevenson, L. A., Griffin, G., Gibson, D., Mandelbaum, A., Etinger, A., and Mechoulam, R. (1992). Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258, 1946–1949.
Felder, C. C., Briley, E. M., Axelrod, J., Simpson, J. T., Mackie, K., and Devane, W. A. (1993). Anandamide, an endogenous cannabimimetic eicosanoid, binds to the cloned human cannabinoid receptor and stimulates receptor-mediated signal transduction. Proc. Natl. Acad. Sci. U.S.A. 90, 7656–7660.
Hong, S., Gronert, K., Devchand, P., Moussignac, R. -L., and Serhan, C. N. (2003). Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood and glial cells: autacoids in anti-inflammation. J. Biol. Chem. 278, 14677–14687.
Hong, S., Porter, T. F., Lu, Y., Oh, S. F., Pillai, P. S., and Serhan, C. N. (2008). Resolvin E1 metabolome in local inactivation during inflammation-resolution. J. Immunol. 180, 3512–3519.
Irimia, D., Liu, S. -Y., Tharp, W. G., Samadani, A., Toner, M., and Poznansky, M. C. (2006). Microfluidic system for measuring neutrophil migratory responses to fast switches of chemical gradients. Lab. Chip 6, 191–198.
Janero, D. R., and Makriyannis, A. (2007). Targeted modulators of the endogenous cannabinoid system: future medications to treat addiction disorders and obesity. Curr. Psychiatry Rep. 9, 365–373.
Kasuga, K., Yang, R., Porter, T. F., Agrawal, N., Petasis, N. A., Irimia, D., Toner, M., and Serhan, C. N. (2008). Rapid appearance of resolvin precursors in inflammatory exudates: novel mechanisms in resolution. J. Immunol. 181, 8677–8687.
Keyes, K. T., Ye, Y., Lin, Y., Zhang, C., Perez-Polo, J. R., Gjorstrup, P., and Birnbaum, Y. (2010). Resolvin E1 protects the rat heart against reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 299, H153–H164.
Kumar, V., Abbas, A. K., Fausto, N., Robbins, S. L., and Cotran, R. S. (2005). Robbins and Cotran Pathologic Basis of Disease. Philadelphia: Elsevier/Saunders.
Majno, G., and Joris, I. (2004). Cells, Tissues, and Disease: Principles of General Pathology. New York: Oxford University Press.
Marcus, A. J., Broekman, M. J., Safier, L. B., Ullman, H. L., Islam, N., Serhan, C. N., Rutherford, L. E., Korchak, H. M., and Weissmann, G. (1982). Formation of leukotrienes and other hydroxy acids during platelet-neutrophil interactions in vitro. Biochem. Biophys. Res. Commun. 109, 130–137.
Morris, T., Stables, M., Colville-Nash, P., Newson, J., Bellingan, G., De Souza, P. M., and Gilroy, D. W. (2010). Dichotomy in duration and severity of acute inflammatory responses in humans arising from differentially expressed proresolution pathways. Proc. Natl. Acad. Sci. U.S.A. 107, 8842–8847.
Morris, T., Stables, M., Hobbs, A., De Souza, P., Colville-Nash, P., Warner, T., Newson, J., Bellingan, G., and Gilroy, D. W. (2009). Effects of low-dose aspirin on acute inflammatory responses in humans. J. Immunol. 183, 2089–2096.
Oh, S. F., Pillai, P. S., Recchiuti, A., Yang, R., and Serhan, C. N. (2011). Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J. Clin. Invest. 121, 569–581.
Phillipson, M., and Kubes, P. (2011). The neutrophil in vascular inflammation. Nat. Med. 17, 1381–1390.
Pillai, P. S., Leeson, S., Porter, T. F., Owens, C. D., Kim, J. M., Conte, M. S., Serhan, C. N., and Gelman, S. (2012). Chemical mediators of inflammation and resolution in post-operative abdominal aortic aneurysm patients. Inflammation 35, 98–113.
Qiu, F.-H., Wada, K., Stahl, G. L., and Serhan, C. N. (2000). IMP and AMP deaminase in reperfusion injury down-regulates neutrophil recruitment. Proc. Natl. Acad. Sci. U.S.A. 97, 4267–4272.
Roberts, L. J. II, Montine, T. J., Markesbery, W. R., Tapper, A. R., Hardy, P., Chemtob, S., Dettbarn, W. D., and Morrow, J. D. (1998). Formation of isoprostane-like compounds (neuroprostanes) in vivo from docosahexaenoic acid. J. Biol. Chem. 273, 13605–13612.
Roberts, L. J. II, and Morrow, J. D. (2002). Products of the isoprostane pathway: unique bioactive compounds and markers of lipid peroxidation. Cell. Mol. Life Sci. 59, 808–820.
Samuelsson, B. (1983). Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation. Science 220, 568–575.
Samuelsson, B., Dahlen, S. E., Lindgren, J. A., Rouzer, C. A., and Serhan, C. N. (1987). Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science 237, 1171–1176.
Schwab, J. M., Chiang, N., Arita, M., and Serhan, C. N. (2007). Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447, 869–874.
Serhan, C. N. (2007). Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu. Rev. Immunol. 25, 101–137.
Serhan, C. N. (2011). The resolution of inflammation: the devil in the flask and in the details. FASEB J. 25, 1441–1448.
Serhan, C. N., Chiang, N., and Van Dyke, T. E. (2008). Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361.
Serhan, C. N., Clish, C. B., Brannon, J., Colgan, S. P., Chiang, N., and Gronert, K. (2000). Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 192, 1197–1204.
Serhan, C. N., Fredman, G., Yang, R., Karamnov, S., Belayev, L. S., Bazan, N. G., Zhu, M., Winkler, J. W., and Petasis, N. A. (2011). Novel proresolving aspirin-triggered DHA pathway. Chem. Biol. 18, 976–987.
Serhan, C. N., Gotlinger, K., Hong, S., and Arita, M. (2004). Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their aspirin-triggered endogenous epimers: an overview of their protective roles in catabasis. Prostaglandins Other Lipid Mediat. 73, 155–172.
Serhan, C. N., Gotlinger, K., Hong, S., Lu, Y., Siegelman, J., Baer, T., Yang, R., Colgan, S. P., and Petasis, N. A. (2006). Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. J. Immunol. 176, 1848–1859.
Serhan, C. N., Hong, S., Gronert, K., Colgan, S. P., Devchand, P. R., Mirick, G., and Moussignac, R.-L. (2002). Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter pro-inflammation signals. J. Exp. Med. 196, 1025–1037.
Serhan, C. N., and Petasis, N. A. (2011). Resolvins and protectins in inflammation-resolution. Chem. Rev. 111, 5922–5943.
Serhan, C. N., and Savill, J. (2005). Resolution of inflammation: the beginning programs the end. Nat. Immunol. 6, 1191–1197.
Serhan, C. N., Ward, P. A., and Gilroy, D. W. (eds). (2010). Fundamentals of Inflammation. New York: Cambridge University Press.
Serhan, C. N., Yang, R., Martinod, K., Kasuga, K., Pillai, P. S., Porter, T. F., Oh, S. F., and Spite, M. (2009). Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 206, 15–23.
Spite, M., and Serhan, C. N. (2010). Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ. Res. 107, 1170–1184.
Stables, M. J., Shah, S., Camon, E. B., Lovering, R. C., Newson, J., Bystrom, J., Farrow, S., and Gilroy, D. W. (2011). Transcriptomic analyses of murine resolution-phase macrophages. Blood 118, e192–e208.
Tjonahen, E., Oh, S. F., Siegelman, J., Elangovan, S., Percarpio, K. B., Hong, S., Arita, M., and Serhan, C. N. (2006). Resolvin E2: identification and anti-inflammatory actions: pivotal role of human 5-lipoxygenase in resolvin E series biosynthesis. Chem. Biol. 13, 1193–1202.
Tsikitis, V. L., Morin, N. A., Harrington, E. O., Albina, J. E., and Reichner, J. S. (2004). The lectin-like domain of complement receptor 3 protects endothelial barrier function from activated neutrophils. J. Immunol. 173, 1284–1291.
van Gils, J. M., Zwaginga, J. J., and Hordijk, P. L. (2009). Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J. Leukoc. Biol. 85, 195–204.
Vemuri, V. K., Janero, D. R., and Makriyannis, A. (2008). Pharmacotherapeutic targeting of the endocannabinoid signaling system: drugs for obesity and the metabolic syndrome. Physiol. Behav. 93, 671–686.
Keywords: resolvins, protectins, specialized pro-resolving mediator, DHEA, neutrophil, aspirin
Citation: Shinohara M, Mirakaj V and Serhan CN (2012) Functional metabolomics reveals novel active products in the DHA metabolome. Front. Immun. 3:81. doi: 10.3389/fimmu.2012.00081
Received: 07 February 2012; Paper pending published: 21 February 2012;
Accepted: 29 March 2012; Published online: 17 April 2012.
Edited by:
Masaaki Murakami, Osaka University, JapanReviewed by:
Takayuki Yoshimoto, Tokyo Medical University, JapanHiroki Yoshida, Saga University Faculty of Medicine, Japan
Daisuke Kamimura, Osaka University, Japan
Copyright: © 2012 Shinohara, Mirakaj and Serhan. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Charles N. Serhan, Center for Experimental Therapeutics, Harvard Institutes of Medicine, 77 Avenue Louis Pasteur, HIM 829, Boston, MA 02115-5727, USA. e-mail:Y25zZXJoYW5AemV1cy5id2guaGFydmFyZC5lZHU=