 Takashi Shichita1,2
Takashi Shichita1,2
- 1 Department of Microbiology and Immunology, School of Medicine, Keio University, Tokyo, Japan
- 2 Department of Research Promotion, Precursory Research for Embryonic Science and Technology, Tokyo, Japan
- 3 Core Research for Evolutional Science and Technology, Japan Science and Technology Agency, Tokyo, Japan
Post-ischemic inflammation is an essential step in the progression of brain ischemia-reperfusion injury. In this review, we focus on the post-ischemic inflammation triggered by infiltrating immune cells, macrophages, and T lymphocytes. Brain ischemia is a sterile organ, but injury-induced inflammation is mostly dependent on Toll-like receptor (TLR) 2 and TLR4. Some endogenous TLR ligands, high mobility group box 1 (HMGB1) and peroxiredoxin family proteins, in particular, are implicated in the activation and inflammatory cytokine expression in infiltrating macrophages. Following macrophage activation, T lymphocytes infiltrate the ischemic brain and regulate the delayed phase inflammation. IL-17-producing γδT lymphocytes induced by IL-23 from macrophages promote ischemic brain injury, whereas regulatory T lymphocytes suppress the function of inflammatory mediators. A deeper understanding of the inflammatory mechanisms of infiltrating immune cells may lead to the development of novel neuroprotective therapies.
Introduction
Stroke is a leading cause of death and disability worldwide. The most common type of stroke is ischemic stroke (e.g., approximately 70% of strokes in Japan are ischemic). However, intravenous administration of tissue plasminogen activator (tPA) is the only globally approved treatment for ischemic stroke, and it is a time-dependent therapy that must be provided within 4.5 h after stroke onset. Thus, there is a need for an efficacious therapy that can be administered beyond this time window, one that targets neuroprotection rather than clot dissolution (Lo, 2010; Moskowitz et al., 2010).
Brain infarction is tissue death caused by ischemia due to severe stenosis or occlusion of a cerebral artery. Ischemic brain tissue is deprived of oxygen, glucose, and lipids, and eventually becomes necrotic. Brain inflammation occurs in this necrotic brain tissue, following the breakdown of the blood-brain barrier (BBB) and infiltration of blood immune cells. Infiltrating immune cells promote ischemic brain inflammation by producing various inflammatory mediators, and also clear away necrotic debris. After the demolition of necrotic debris has been completed, brain inflammation subsides.
Despite intensive studies, the complexity of the brain inflammation mechanism has thus far prevented sufficient clarification (Eltzschig and Eckle, 2011; Iadecola and Anrather, 2011; Macrez et al., 2011). Macrophages and neutrophils are pivotal players in the various processes of brain inflammation, but the mechanism of their activation is still unknown. In addition, T or B lymphocytes have been also reported to participate in delayed brain inflammation. This review focuses on the mechanism of ischemic brain inflammation triggered and sustained by infiltrating immune cells.
Post-Ischemic Inflammation in the Early Phase of Brain Ischemia
Severe ischemia induces hypoxia and glucose deprivation in brain tissue. Calcium and sodium ions are stored within brain cells and glutamate is released into the extracellular compartment. The production of reactive oxygen species (ROS) activates platelets and endothelial cells, leading to microvascular occlusion. Oxidative stress reduces the beneficial effects of nitric oxide (NO), a potent vasodilator and inhibitor of platelet aggregation and leukocyte adhesion, in endothelial cells. Oxidative stress and the inflammatory cascade alter the permeability of the BBB. The activation of matrix metalloproteinases (MMPs) and the expression of various other proteases lead to BBB breakdown which exacerbates leukocyte extravasation. Intravascular leukocytes firmly adhere to activated endothelium by the interaction of endothelial expression of intercellular adhesion molecule-1 (ICAM-1) and leukocyte β2 integrins (Iadecola and Anrather, 2011). The infiltration of leukocytes is enhanced by BBB breakdown and by chemokines.
Brain cells, including astrocytes, oligodendrocytes, endothelium, and pericytes, constitute a neurovascular network, which is essential for metabolic requirement of neurons (Iadecola, 2004; Fraser, 2011). These brain cells also contribute to triggering post-ischemic inflammation by producing inflammatory mediators. TNF-α, IL-1β, NOS (nitric oxide synthetase), and MMPs which enhance cerebrovascular permeability and exaggerate brain edema (Takano et al., 2009; Morancho et al., 2010). Thus, Infiltrating leukocytes and injured brain cells produce various inflammatory mediators, leading to the beginning of post-ischemic inflammation (Barone and Feuerstein, 1999; Figure 1).
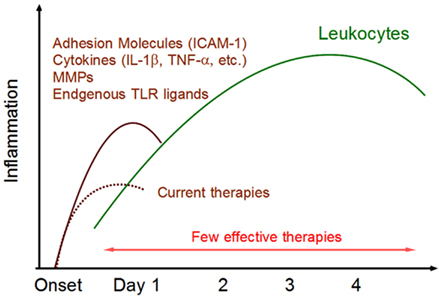
Figure 1. Post-ischemic inflammation in the brain. Within 24 h after ischemic stroke onset, various inflammatory mediators are expressed in ischemic brain tissue. ICAM-1 promotes leukocytes infiltration. Cytokines activate infiltrating leukocytes and directly induce ischemic injury in brain cells. Matrix metalloproteinases (MMPs) alter the permeability of epithelial cells and are implicated in BBB breakdown. Endogenous TLR ligands (DAMPs) are released from necrotic brain cells and activate infiltrating immune cells. These inflammatory mediators trigger post-ischemic inflammation by infiltrating leukocytes. There are currently few effective therapies for this phase of leukocyte infiltration.
Innate Inflammatory Cytokines and Inflammatory Mediators
Various cytokines and mediators are produced from infiltrating immune cells and brain cells as a result of ischemic changes of brain. IL-1β is expressed in ischemic brain tissue within 30 min after stroke onset. IL-1β directly induces apoptosis of neuronal cells and enhances the expression of chemokine (RANTES, etc.) in microglia and astrocytes. IL-1β is considered to be a neurotoxic mediator, given that the loss of IL-1β function is reported to reduce infarct size (Boutin et al., 2001). IL-1β is produced in an inactivate form, pro-IL-1β, which is cleaved by caspase-1 to become an active 17 kDa form. Recently, the mechanism of IL-1β production and caspase-1 activation mediated by inflammasome has been the subject of particular attention. Inflammasome has been shown to be present in neurons, astrocytes, microglia, and macrophages in the ischemic brain (Abulafia et al., 2009; Chakraborty et al., 2010). Several types of inflammasome have been discovered, denoted NALP1, NALP3, AIM, and so on, but the particular type most important in ischemic brain injury remains unknown. Hypoxia or ATP is reported to activate inflammasome, which then activates caspase-1 and induces IL-1β production (Martinon et al., 2002). In addition, IL-1β is mostly produced from monocytes which are activated by endogenous Toll-like receptor (TLR) ligands, given that IL-1β mRNA level in infiltrating mononuclear cells is drastically reduced in TLR2/4-double deficient mice after ischemic brain injury.
TNF-α is another important mediator implicated in the pathology of the ischemic brain. TNF-α is expressed in ischemic brain tissue within 1 h after stroke onset, and upregulation of the TNF receptors is observed thereafter. TNF-α exercises neurotoxic effects by inducing apoptotic neuronal cell death and enhancing MHC class II and ICAM-1 expression in astrocytes, leading to leukocyte infiltration and BBB breakdown. TNF-α gene knockout (KO) mice or anti-TNF-α neutralizing antibody administration has been shown to reduce infarct volume, compared with that in control mice. However, TNFR KO mice, which lack both p75 and p50 genes, exhibit enlargement of infarct volume on day 1 following ischemic brain injury, indicating that TNF-α can be considered to function as both a neurotoxic and a neuroprotective mediator (Hallenbeck, 2002). It appears that the opposing functions, toxic or protective, depends on the type of brain cell involved. TNF-α promotes post-ischemic inflammation but also participates in a negative feedback loop to suppress inflammatory signal cascades, and it controls the duration of post-ischemic inflammation by regulating these two functions.
IL-6 is also expressed in ischemic brain tissue, but ischemic brain damage is not attenuated in IL-6 KO mice or in anti-IL-6R antagonistic antibody-treated mice (Yamashita et al., 2005). However, it has been recently reported that IL-6 produced from brain cells contributes to neoangiogenesis and neuronal survival through STAT3 activation (Jung et al., 2011; Gertz et al., 2012). Consistent with this, the inhibition of JAK/STAT pathway or the enhanced role of SOCS3 (negative regulator of JAK/STAT pathway) has been reported to promote neuronal cell death (Yadav et al., 2005; Yamashita et al., 2005). Thus, it is possible that IL-6 protects neuron from cell death, although a significant role of IL-6 in brain ischemia has not yet been established.
Matrix metalloproteinases are essential neurotoxic mediators that promote BBB breakdown and post-ischemic inflammation. Functionally similar to IL-1β, MMPs induce apoptotic neuronal cell death by TNF-α and FasL processing. The neurotoxic function of MMP-9 is particularly established, given that infarct size is reduced in MMP-9-deficient mice compared to that in control mice (Asahi et al., 2000). Intercellular adhesion molecule-1 (ICAM-1) is another neurotoxic mediator. The increased expression of ICAM-1 observed in cerebrovascular endothelial cells is implicated in the promotion of leukocyte infiltration. ICAM-1-deficient mice reveal attenuated ischemic damage and the administration of anti-ICAM-1 antibody decreases the number of infiltrating immune cells (Connolly et al., 1996; Liesz et al., 2011).
Chemokines are also important enhancers of post-ischemic inflammation. Chemokines (RANTES, MCP-1, IL-8, etc.) have been reported to promote leukocyte infiltration into the ischemic brain (Terao et al., 2008, 2009; Strecker et al., 2011). Although the chemokines for lymphocyte infiltration remain unknown, CCL12, CCL20, and their receptor, CCR6, are essential for the exacerbation of experimental autoimmune encephalomyelitis (EAE; Martin et al., 2009; Reboldi et al., 2009). Whether these lymphocyte chemokines also function in acute organ injury, such as brain ischemia, should be elucidated in the future.
Sphingosine-1-phosphate (S1P) is a bioactive phospholipid. At sites of tissue injury, S1P is mainly released from platelets and mediates its effect via activation of cell-surface S1P receptors, which are ubiquitously expressed in brain cells (Dev et al., 2008). S1P receptors are also present on the surface of T lymphocytes; therefore, S1P has been thought to play an essential role in T lymphocyte infiltration of inflammatory tissue. Recently, the therapeutic effect of FTY720 (fingolimod), a functional S1P receptor antagonist, has attracted attention. The administration of FTY720 has been shown to attenuate ischemic brain damage and decrease the number of infiltrating T lymphocytes in the ischemic brain (Shichita et al., 2009; Hasegawa et al., 2010). Furthermore, S1P receptors are expressed in neurons, astrocytes, and microglial cells. S1P acts on these cells directly and exerts effects that include astrocyte proliferation and migration, oligodendrocyte differentiation and cell survival, and neurite outgrowth and neurogenesis (Dev et al., 2008). Thus, S1P is considered to be an important inflammatory mediator in the ischemic brain.
Role of TLR
Leukocyte infiltration is an essential step in the progression of post-ischemic inflammation. However, the mechanisms that activate these infiltrating immune cells, macrophages, and lymphocytes, are not yet fully clarified. TLRs are an essential type of receptor for innate and non-specific immune response to general pathogens such as bacteria, viruses, and so on. The demonstration that TLR2 and TLR4 are pivotal for sterile organ injury, including ischemic brain injury, has recently attracted attention (Chen et al., 2007; Tang et al., 2007).
Toll-like receptors are expressed on both leukocytes and brain cells, although whether the effect of TLRs on brain cells is neurotoxic or neuroprotective remains unclear. TLR stimulation in macrophages and lymphocytes induces strong and various inflammatory responses. In ischemic brain injury, post-ischemic inflammation and subsequent ischemic damage depend on TLR2 and TLR4, but not TLR9 (Tang et al., 2007; Hyakkoku et al., 2010). TLR2- or TLR4-deficient mice demonstrate both significant reduction of infarct volume and suppression of neurotoxic inflammatory responses. Analysis of bone marrow chimeric mice indicates that TLRs in infiltrated immune cells, but not in residential microglia, have a neurotoxic effect on ischemic brain injury (Yang et al., 2011). In addition, mice lacking MyD88, the adaptor protein under almost all TLR signaling cascades other than TLR3, are reported to show no improvement of ischemic brain injury (Famakin et al., 2011). These results indicate that the function of TLRs may be dependent on kind of cells in brain.
Toll-like receptor-2 or TLR4 deficiency suppresses inflammatory cytokine expression in infiltrating immune cells on day 1 after brain ischemia (Shichita et al., 2012). Although both TLR2 and TLR4 signaling cascades are essential triggers for post-ischemic inflammation, and activators of infiltrating immune cells, the particular molecules that activate TLR2 and TLR4 in the ischemic brain remain unclear. Because the brain is a sterile organ, pathogens derived from bacteria or viruses are completely lacking in the normal and ischemic brain. This indicates that certain endogenous molecules released from necrotic brain cells become TLR stimulators, and several endogenous molecules have indeed been reported to activate TLR signaling cascades. Such endogenous molecules are called danger associated molecular patterns (DAMPs), and are considered to be danger signals or alarm molecules that warn immune cells of tissue and cellular injury.
DAMPs in Injured Brain
Several molecules in the brain have been reported as DAMPs. Heat shock proteins (HSPs), β-amyloid (Aβ), hyaluronan, heparin sulfate, DNA or RNA immune complex, oxidized low-density lipoproteins (oxLDL), and others, can stimulate TLRs (Marsh et al., 2009; Rivest, 2009; Yanai et al., 2009; Stewart et al., 2010; Zhang et al., 2010). However, it remains unclear which molecule is the most important for triggering post-ischemic inflammation and inflammatory cytokine expression.
High mobility group box 1 (HMGB1) is a well-elucidated DAMP and is also implicated in ischemic brain injury (Kim et al., 2006; Liu et al., 2007; Hayakawa et al., 2010). HMGB1 increases vascular permeability and promotes BBB breakdown (Zhang et al., 2011). HMGB1, which is localized in cell nuclei in the normal brain, translocates into the cytosolic compartment and is released into the extracellular compartment in the ischemic condition. The administration of anti-HMGB1-neutralizing antibody protects the BBB and reduces infarct volume. Thus, HMGB1 is an essential DAMP in ischemic brain injury. Extracellular release of HMGB1 is observed within 6 h after stroke onset, but is diminished by 12 h after the onset (Qiu et al., 2008; Zhang et al., 2011). Thereafter, the infiltration of immune cells and the production of inflammatory cytokines become evident. This indicates that HMGB1 may not directly activate infiltrating immune cells in the ischemic brain.
We have identified the peroxiredoxin (Prx) family proteins as strong inducers of inflammatory cytokines in infiltrating immune cells (Shichita et al., 2012). Prx family proteins are known to exert a protective effect by catalyzing ROS. In the ischemic brain, the expression of Prx within brain cells is increased by ischemic stress, and such intracellular Prx is thought to be neuroprotective (Patenaude et al., 2005; Rashidian et al., 2009; Figure 2). However, as necrosis occurs, this Prx is released into the extracellular compartment where they induce inflammatory cytokine expression in infiltrating immune cells by stimulating TLR2 and TLR4 (Figure 2). It is interesting to note that the Prx family proteins have a common active region for TLR activation and are extracellularly released over 12 h after stroke onset, which coincides with the timing of leukocyte infiltration. Thus, Prx has two opposing functions, one inside, and one outside, brain cells. Furthermore, there is a time lag as well as functional differences between HMGB1 and Prx (Figure 3).
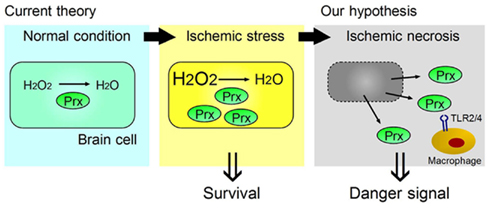
Figure 2. Two opposing functions of Prx, one inside, and one outside, brain cells. Ischemic stress increases Prx expression within brain cells, which contributes to their survival by catabolizing reactive oxygen species (ROS). When ischemic phenomena finally result in necrosis, the Prx released from necrotic brain cells into the extracellular compartment then functions as a strong TLR2 and TLR4 stimulator (DAMP) for the infiltrating macrophages in ischemic brain tissue.
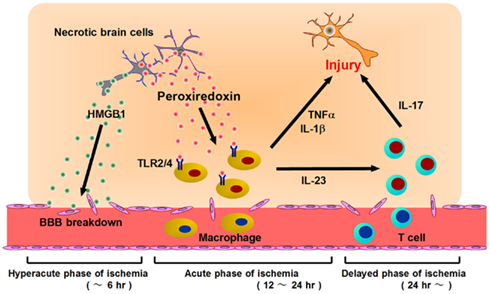
Figure 3. Post-ischemic inflammation triggered by DAMPs and infiltrating immune cells. At the hyperacute phase of brain ischemia (within 6 h after stroke onset), HMGB1 is released from brain cells and induces BBB breakdown. Following this, blood cells begin to infiltrate into ischemic brain tissue via disrupted vessels during the acute phase of ischemia (12–24 h after stroke onset). Prx is extracellularly released from necrotic brain cells and activates infiltrating macrophages via TLR2 and TLR4. Activated macrophages produce inflammatory cytokines (IL-23, IL-1β, TNF-α, etc.) which promote ischemic brain injury. At the delayed phase of brain ischemia (more than 24 h after stroke onset), IL-23 induces IL-17 production from γδT lymphocytes, which further enhances ischemic damage. Thus, HMGB1 is a hyperacute DAMP, while Prx is secondarily active in post-ischemic inflammation, during the acute phase.
T Lymphocytes in Ischemic Brain Injury
It has been recently suggested that T lymphocytes play a role as mediators in the delayed phase of brain ischemia (Yilmaz et al., 2006). The number of infiltrating T lymphocytes in ischemic brain tissue increases over 24 h after stroke onset and reaches its peak in the delayed phase (around day 3; Schroeter et al., 1994; Jander et al., 1995). Infiltrating T lymphocytes appear to be localized to the infarct boundary zones, typically close to blood vessels. By Percoll gradient centrifugation, infiltrating immune cells have been analyzed (Shichita et al., 2009). The number of infiltrating immune cells reaches the peak at day 3 after stroke onset and most of these are macrophages. Approximately 1 ∼ 1.5% of immune cells are T lymphocytes which are consisted of 30 ∼ 40% CD4+ helper T lymphocytes, 20 ∼ 30% γδT lymphocytes, and 20 ∼ 30% CD8+ cytotoxic T lymphocytes. Inflammatory cytokines, IL-1β, TNF-α, IL-23, and IL-12 have been shown to be produced from macrophage and play important roles in promoting brain injury. However, role of various cytokines from T lymphocytes, such as IFNγ and IL-17 in ischemic brain injury has not been clarified.
Although the function of these infiltrating T lymphocytes in the ischemic brain is not yet clear, T lymphocytes, on the whole, are considered to act as a neurotoxic effector. This is indicated by the fact that severe combined immunodeficient (SCID) mice, and recombination activating gene (RAG)-deficient mice, both of which lack T and B lymphocytes, reveal significant reduction of infarct volume (Yilmaz et al., 2006; Hurn et al., 2007). In addition, the depletion of CD4+ (helper) or CD8+ (cytotoxic) T lymphocytes, but not one of the B lymphocytes, is reported to attenuate ischemic brain damage. There is a report that regulatory B lymphocytes protect brain from ischemic damages; however, the neurotoxic effect of lymphocytes is considered to be majorly made by T lymphocytes (Ren et al., 2011). It will be important to elucidate which subtype of lymphocytes is implicated in ischemic brain injury (see T Lymphocyte Cytokines in Ischemic Brain Injury).
It is not yet clear whether a specific antigen in the brain is involved in the activation of these infiltrating T lymphocytes. Up to now, it has been thought likely that these T lymphocytes mediate antigen-independent, innate inflammatory responses, because post-ischemic inflammatory responses have been shown to be driven by the innate immune system. However, some reports suggest the importance of antigen recognition by T lymphocytes in ischemic brain injury. Treatment with T cell receptor (TCR) ligands, which are major histocompatibility complex (MHC) class II molecules bound to myelin peptides, is protective against ischemic brain injury (Subramanian et al., 2009). Infarct size in myelin basic protein (MBP) tolerized animals has been shown to be reduced compared to that in control mice. Thus, there is a possibility that some T lymphocyte subsets specifically tolerized to brain proteins could be protective to ischemic brain injury (Becker et al., 2003; Becker, 2009). This idea is supported by the recent finding that regulatory T lymphocytes are protective to ischemic brain injury (Liesz et al., 2009).
T Lymphocyte Cytokines in Ischemic Brain Injury
T lymphocytes are considered to mediate ischemic brain injury by producing various cytokines, and IFN-γ and IL-4 are well-known classical examples. In ischemic injury, IFN-γ is thought to be neurotoxic, as it acts on neurons directly and induces apoptotic neuronal cell death in vitro (Lambertsen et al., 2004). However, a protective effect by the IFN-γ deficiency has not been observed, and the role of IFN-γ in ischemic brain injury is controversial (Lambertsen et al., 2004; Yilmaz et al., 2006). IL-12 is produced from myeloid cells such as macrophages, dendritic cells, neutrophils, and so on, and is important for the differentiation of IFN-γ-producing helper T lymphocytes (Th1). IL-12 is expressed in ischemic brain tissue by infiltrating immune cells, but its function has not been fully elucidated.
IL-4 may have the potential to attenuate ischemic damage and promote tissue repair, since IL-4-deficient mice demonstrate exacerbated ischemic damage and neurological deficit (Xiong et al., 2011). Although the emerging recognition of the function of IL-4 for tissue repair has recently attracted attention, whether or not IL-4 is directly implicated in tissue repair in ischemic brain injury is still unclear (Chen et al., 2012).
IL-10 is an immunosuppressive cytokine and is thought to have a neuroprotective effect in ischemic brain injury. IL-10 is produced from regulatory T lymphocytes (Treg) and suppresses the neurotoxic function of TNF-α and IFN-γ (Liesz et al., 2009). The overexpression of IL-10 by an adenovirus vector protects hippocampal neurons against apoptotic cell death (Ooboshi et al., 2006).
IL-17 is an emerging therapeutic target for various organ injuries. IL-23 has been reported to be essential for IL-17 induction from T lymphocytes, and to play a critical role in EAE (Cua et al., 2003). In ischemic brain injury, IL-23 is produced by infiltrating macrophages on day 1, and it induces IL-17 production from γδT lymphocytes in the delayed phase (Figure 3). Both IL-23 KO mice and IL-17 KO mice show significantly attenuated ischemic brain damage on day 4. The IL-17 receptor is ubiquitously expressed in brain cells and modifies various inflammatory responses in the central nervous system. IL-17 has been reported to promote the expression of inflammatory cytokines and chemokines from macrophages (Fossiez et al., 1996). IL-17 also modulates the epithelial barrier function by promoting MMPs and ICAM-1 expression (Kebir et al., 2007; Ifergan et al., 2008). Although it remains unknown whether IL-17 directly affects neurons, IL-17 is thought to be a promising therapeutic target for suppressing post-ischemic inflammation. Thus, IL-23-induced IL-17-producing γδT lymphocytes play a pivotal role in the delayed phase of brain ischemia (Figure 4). IL-17 production from γδT lymphocytes requires only IL-1β and IL-23 stimulation, but not specific TCR stimulation, and IL-6 and TGFβ stimulation are indispensable for IL-17-producing helper T lymphocyte (Th17) differentiation (Sutton et al., 2009). Thus, it is reasonable that γδT lymphocytes mediate ischemic brain injury, given that γδT lymphocytes produce IL-17 more rapidly than does Th17.
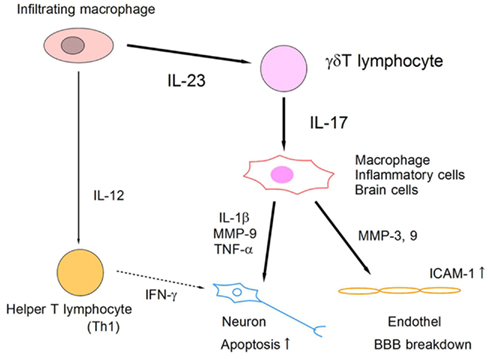
Figure 4. Schematic model of IL-23/IL-17 inflammatory pathway in ischemic brain tissue. Infiltrating macrophages produce IL-23 and IL-12, which induce IL-17-producing γδT lymphocytes and IFN-γ-producing helper T lymphocytes (Th1), respectively. IL-17 from γδT lymphocytes acts on macrophages and brain cells directly, and promotes the expression of inflammatory mediators that enhance apoptotic neuronal cell death and BBB breakdown.
The Possibility of Medical Intervention in Post-Ischemic Inflammation
Post-ischemic inflammation in the brain is considered to have two opposing functions in stroke patients; one beneficial, the other harmful. Post-ischemic inflammation promotes brain swelling (brain edema) which leads to the compression of normal brain tissue surrounding the ischemic core and the exacerbation of neurological deficits. This undesirable effect of post-ischemic brain inflammation should be suppressed by medical intervention when possible. However, post-ischemic inflammation is also thought to promote tissue repair in the recovery phase of ischemic stroke. Thus, suppression of all inflammatory responses in ischemic brain tissue is not always effective, due to their involvement in both ischemic injury and tissue repair processes. To create novel neuroprotective strategy, it may be possible to control the balance between the neurotoxic and neuroprotective effects of post-ischemic inflammation by targeting specific inflammatory mediators. For example, therapy which both suppresses the inflammatory subset of T lymphocytes (e.g., IL-17-producing γδT lymphocytes) and promotes Treg function may be desirable.
It is possible that administration of γδTCR-depleting antibody or anti-IL-12/IL-23p40 antibody [p40 is a common subunit of the IL-12 heterodimer (p35/p40) and IL-23 heterodimer (p19/p40)] will become a therapeutic tool for ischemic stroke (Shichita et al., 2009; Konoeda et al., 2010). Thus, neuroprotective therapies for ischemic brain injury may be developed by targeting the IL-23/IL-17 inflammatory pathway. Furthermore, one of the most advantageous points of therapy targeting IL-17-producing T lymphocytes is its long therapeutic time window. Since current globally approved therapy is limited to intravenous administration of tPA, which should be performed within 4.5 h after stroke onset, further elucidation of the inflammatory mechanisms of the T lymphocytes that infiltrate the ischemic brain during the 24-h-period after stroke onset is needed.
FTY720 is one of the most promising therapeutic tools for ischemic stroke at this time (Wei et al., 2011). FTY720 decreases the number of infiltrating T lymphocytes, including γδT lymphocytes. The most troublesome side effect of FTY720 administration is the increased incidence of bacterial pneumonia after stroke (Meisel and Meisel, 2011). It is possible that FTY720 interferes with peripheral lymphocyte distribution in the body after stroke and thus inhibits the protective function of peripheral T lymphocytes against bacterial infection. Although it has been reported that FTY720 does not promote spontaneous bacterial infection after experimental stroke in mice, future studies aimed at developing effective clinical interventions for stroke patients should seek to minimize this kind of detrimental effect of FTY720 (Pfeilschifter et al., 2011).
In conclusion, research has gradually shed light on the mechanisms of post-ischemic inflammation. A deeper understanding of these intricacies in the context of medical intervention should enable the development of novel neuroprotective strategies that are more effective and have a longer therapeutic time window (Figure 5).
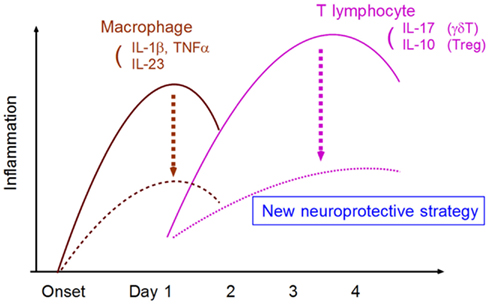
Figure 5. Strategy for developing neuroprotective therapy by suppressing neurotoxic inflammatory response. The targeting of specific inflammatory mediators from macrophages and T lymphocytes can attenuate neurotoxic inflammatory reactions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abulafia, D. P., de Rivero Vaccari, J. P., Lozano, J. D., Lotocki, G., Keane, R. W., and Dietrich, W. D. (2009). Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J. Cereb. Blood Flow Metab. 29, 534–544.
Asahi, M., Asahi, K., Jung, J. C., del Zoppo, G. J., Fini, M. E., and Lo, E. H. (2000). Role for matrix metalloproteinase 9 after cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J. Cereb. Blood Flow Metab. 20, 1681–1689.
Barone, F. C., and Feuerstein, G. Z. (1999). Inflammatory mediators and stroke: new opportunities for novel therapeutics. J. Cereb. Blood Flow Metab. 19, 819–834.
Becker, K., Kindrick, D., McCarron, R., Hallenbeck, J., and Winn, R. (2003). Adoptive transfer of myelin basic protein-tolerized splenocytes to naive animals reduces infarct size: a role for lymphocytes in ischemic brain injury? Stroke 34, 1809–1815.
Becker, K. J. (2009). Sensitization and tolerization to brain antigens in stroke. Neuroscience 158, 1090–1097.
Boutin, H., LeFeuvre, R. A., Horai, R., Asano, M., Iwakura, Y., and Rothwell, N. J. (2001). Role of IL-1alpha and IL-1beta in ischemic brain damage. J. Neurosci. 21, 5528–5534.
Chakraborty, S., Kaushik, D. K., Gupta, M., and Basu, A. (2010). Inflammasome signaling at the heart of central nervous system pathology. J. Neurosci. Res. 88, 1615–1631.
Chen, C. J., Kono, H., Golenbock, D., Reed, G., Akira, S., and Rock, K. L. (2007). Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 13, 851–856.
Chen, F., Liu, Z., Wu, W., Rozo, C., Bowdridge, S., Millman, A., Van Rooijen, N., Urban, J. F. Jr., Wynn, T. A., and Gause, W. C. (2012). An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat. Med. 18, 260–266.
Connolly, E. S. Jr., Winfree, C. J., Springer, T. A., Naka, Y., Liao, H., Yan, S. D., Stern, D. M., Solomon, R. A., Gutierrez-Ramos, J. C., and Pinsky, D. J. (1996). Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J. Clin. Invest. 97, 209–216.
Cua, D. J., Sherlock, J., Chen, Y., Murphy, C. A., Joyce, B., Seymour, B., Lucian, L., To, W., Kwan, S., Churakova, T., Zurawski, S., Wiekowski, M., Lira, S. A., Gorman, D., Kastelein, R. A., and Sedgwick, J. D. (2003). Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421, 744–748.
Dev, K. K., Mullershausen, F., Mattes, H., Kuhn, R. R., Bilbe, G., Hoyer, D., and Mir, A. (2008). Brain sphingosine-1-phosphate receptors: implication for FTY720 in the treatment of multiple sclerosis. Pharmacol. Ther. 117, 77–93.
Eltzschig, H. K., and Eckle, T. (2011). Ischemia and reperfusion – from mechanism to translation. Nat. Med. 17, 1391–1401.
Famakin, B. M., Mou, Y., Ruetzler, C. A., Bembry, J., Maric, D., and Hallenbeck, J. M. (2011). Disruption of downstream MyD88 or TRIF Toll-like receptor signaling does not protect against cerebral ischemia. Brain Res. 1388, 148–156.
Fossiez, F., Djossou, O., Chomarat, P., Flores-Romo, L., Ait-Yahia, S., Maat, C., Pin, J. J., Garrone, P., Garcia, E., Saeland, S., Blanchard, D., Gaillard, C., Das Mahapatra, B., Rouvier, E., Golstein, P., Banchereau, J., and Lebecque, S. (1996). T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J. Exp. Med. 183, 2593–2603.
Fraser, P. A. (2011). The role of radical generation in increasing cerebrovascular permeability. Free Radic. Biol. Med. 51, 967–977.
Gertz, K., Kronenberg, G., Kälin, R. E., Baldinger, T., Werner, C., Balkaya, M., Eom, G. D., Hellmann-Regen, J., Kröber, J., Miller, K. R., Lindauer, U., Laufs, U., Dirnagl, U., Heppner, F. L., and Endres, M. (2012). Essential role of interleukin-6 in post-stroke angiogenesis. Brain. doi: 10.1093/brain/aws075
Hallenbeck, J. M. (2002). The many faces of tumor necrosis factor in stroke. Nat. Med. 8, 1363–1368.
Hasegawa, Y., Suzuki, H., Sozen, T., Rolland, W., and Zhang, J. H. (2010). Activation of sphingosine 1-phosphate receptor-1 by FTY720 is neuroprotective after ischemic stroke in rats. Stroke 41, 368–374.
Hayakawa, K., Qiu, J., and Lo, E. H. (2010). Biphasic actions of HMGB1 signaling in inflammation and recovery after stroke. Ann. N. Y. Acad. Sci. 1207, 50–57.
Hurn, P. D., Subramanian, S., Parker, S. M., Afentoulis, M. E., Kaler, L. J., Vandenbark, A. A., and Offner, H. (2007). T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J. Cereb. Blood Flow Metab. 27, 1798–1805.
Hyakkoku, K., Hamanaka, J., Tsuruma, K., Shimazawa, M., Tanaka, H., Uematsu, S., Akira, S., Inagaki, N., Nagai, H., and Hara, H. (2010). Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience 171, 258–267.
Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 5, 347–360.
Iadecola, C., and Anrather, J. (2011). The immunology of stroke: from mechanisms to translation. Nat. Med. 17, 796–808.
Ifergan, I., Kébir, H., Bernard, M., Wosik, K., Dodelet-Devillers, A., Cayrol, R., Arbour, N., and Prat, A. (2008). The blood-brain barrier induces differentiation of migrating monocytes into Th17-polarizing dendritic cells. Brain 131, 785–799.
Jander, S., Karemer, M., Schroeter, M., Witte, O. W., and Stoll, G. (1995). Lymphocytic infiltration and expression of intercellular adhesion molecule-1 in photochemically induced ischemia of the rat cortex. J. Cereb. Blood Flow Metab. 15, 42–51.
Jung, J. E., Kim, G. S., and Chan, P. H. (2011). Neuroprotection by interleukin-6 is mediated by signal transducer and activator of transcription 3 and antioxidative signaling in ischemic stroke. Stroke 42, 3574–3579.
Kebir, H., Kreymborg, K., Ifergan, I., Dodelet-Devillers, A., Cayrol, R., Bernard, M., Giuliani, F., Arbour, N., Becher, B., and Prat, A. (2007). Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 13, 1173–1175.
Kim, J. B., Sig Choi, J., Yu, Y. M., Nam, K., Piao, C. S., Kim, S. W., Lee, M. H., Han, P. L., Park, J. S., and Lee, J. K. (2006). HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J. Neurosci. 26, 6413–6421.
Konoeda, F., Shichita, T., Yoshida, H., Sugiyama, Y., Muto, G., Hasegawa, E., Morita, R., Suzuki, N., and Yoshimura, A. (2010). Therapeutic effect of IL-12/23 and their signaling pathway blockade on brain ischemia model. Biochem. Biophys. Res. Commun. 402, 500–506.
Lambertsen, K. L., Gregersen, R., Meldgaard, M., Clausen, B. H., Heibøl, E. K., Ladeby, R., Knudsen, J., Frandsen, A., Owens, T., and Finsen, B. (2004). A role for interferon-gamma in focal cerebral ischemia in mice. J. Neuropathol. Exp. Neurol. 63, 942–955.
Liesz, A., Suri-Payer, E., Veltkamp, C., Doerr, H., Sommer, C., Rivest, S., Giese, T., and Veltkamp, R. (2009). Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 15, 192–199.
Liesz, A., Zhou, W., Mracskó, É., Karcher, S., Bauer, H., Schwarting, S., Sun, L., Bruder, D., Stegemann, S., Cerwenka, A., Sommer, C., Dalpke, A. H., and Veltkamp, R. (2011). Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain 134, 704–720.
Liu, K., Mori, S., Takahashi, H. K., Tomono, Y., Wake, H., Kanke, T., Sato, Y., Hiraga, N., Adachi, N., Yoshino, T., and Nishibori, M. (2007). Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 21, 3904–3916.
Lo, E. H. (2010). Degeneration and repair in central nervous system disease. Nat. Med. 16, 1205–1209.
Macrez, R., Ali, C., Toutirais, O., Le Mauff, B., Defer, G., Dirnagl, U., and Vivien, D. (2011). Stroke and the immune system: from pathophysiology to new therapeutic strategies. Lancet Neurol. 10, 471–480.
Marsh, B. J., Williams-Karnesky, R. L., and Stenzel-Poore, M. P. (2009). Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience 158, 1007–1020.
Martin, B., Hirota, K., Cua, D. J., Stockinger, B., and Veldhoen, M. (2009). Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity 31, 321–330.
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-1beta. Mol. Cell 10, 417–426.
Meisel, C., and Meisel, A. (2011). Suppressing immunosuppression after stroke. N. Engl. J. Med. 365, 2134–2136.
Morancho, A., Rosell, A., Garcia-Bonilla, L., and Montaner, J. (2010). Metalloproteinase and stroke infarct size: role for anti-inflammatory treatment? Ann. N. Y. Acad. Sci. 1207, 123–133.
Moskowitz, M. A., Lo, E. H., and Ladecola, C. (2010). The science of stroke: mechanisms in search of treatments. Neuron 67, 181–198.
Ooboshi, H., Ibayashi, S., Shichita, T., Kumai, Y., Takada, J., Ago, T., Arakawa, S., Sugimori, H., Kamouchi, M., Kitazono, T., and Iida, M. (2006). Postischemic gene transfer of interleukin-10 protects against both focal and global brain ischemia. Circulation 111, 913–919.
Patenaude, A., Murthy, M. R., and Mirault, M. E. (2005). Emerging roles of thioredoxin cycle enzymes in the central nervous system. Cell. Mol. Life Sci. 62, 1063–1080.
Pfeilschifter, W., Czech-Zechmeister, B., Sujak, M., Foerch, C., Wichelhaus, T. A., and Pfeilschifter, J. (2011). Treatment with the immunomodulator FTY720 does not promote spontaneous bacterial infections after experimental stroke in mice. Exp. Transl. Stroke Med. 3, 2.
Qiu, J., Nishimura, M., Wang, Y., Sims, J. R., Qiu, S., Savitz, S. I., Salomone, S., and Moskowitz, M. A. (2008). Early release of HMGB-1 from neurons after the onset of brain ischemia. J. Cereb. Blood Flow Metab. 28, 927–938.
Rashidian, J., Rousseaux, M. W., Venderova, K., Qu, D., Callaghan, S. M., Phillips, M., Bland, R. J., During, M. J., Mao, Z., Slack, R. S., and Park, D. S. (2009). Essential role of cytoplasmic cdk5 and Prx2 in multiple ischemic injury models, in vivo. J. Neurosci. 29, 12497–12505.
Reboldi, A., Coisne, C., Baumjohann, D., Benvenuto, F., Bottinelli, D., Lira, S., Uccelli, A., Lanzavecchia, A., Engelhardt, B., and Sallusto, F. (2009). C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 10, 514–523.
Ren, X., Akiyoshi, K., Dziennis, S., Vandenbark, A. A., Herson, P. S., Hurn, P. D., and Offner, H. (2011). Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J. Neurosci. 31, 8556–8563.
Rivest, S. (2009). Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 9, 429–439.
Schroeter, M., Jander, S., Witte, O. W., and Stoll, G. (1994). Local immune responses in the rat middle cerebral artery occlusion. J. Neuroimmunol. 55, 195–203.
Shichita, T., Hasegawa, E., Kimura, A., Morita, R., Sakaguchi, R., Takada, I., Sekiya, T., Ooboshi, H., Kitazono, T., Yanagawa, T., Ishii, T., Takahashi, H., Mori, S., Nishibori, M., Kuroda, K., Miyake, K., Akira, S., and Yoshimura, A. (2012). Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat. Med. doi: 10.1038/nm.2749
Shichita, T., Sugiyama, Y., Ooboshi, H., Sugimori, H., Nakagawa, R., Takada, I., Iwaki, T., Okada, Y., Iida, M., Cua, D. J., Iwakura, Y., and Yoshimura, A. (2009). Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat. Med. 15, 946–950.
Stewart, C. R., Stuart, L. M., Wilkinson, K., van Gils, J. M., Deng, J., Halle, A., Rayner, K. J., Boyer, L., Zhong, R., Frazier, W. A., Lacy-Hulbert, A., El Khoury, J., Golenbock, D. T., and Moore, K. J. (2010). CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, 155–161.
Strecker, J. K., Minnerup, J., Gess, B., Ringelstein, E. B., Schäbitz, W. R., and Schilling, M. (2011). Monocyte chemoattractant protein-1-deficiency impairs the expression of IL-6, IL-1b and G-CSF after transient focal ischemia in mice. PLoS ONE 6, e25863. doi:10.1371/journal.pone.0025863
Subramanian, S., Zhang, B., Kosaka, Y., Burrows, G. G., Grafe, M. R., Vandenbark, A. A., Hurn, P. D., and Offner, H. (2009). Recombinant T cell receptor ligand treats experimental stroke. Stroke 40, 2539–2545.
Sutton, C. E., Lalor, S. J., Sweeney, C. M., Brereton, C. F., Lavelle, E. C., and Mills, K. H. (2009). Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31, 331–341.
Takano, T., Oberheim, N., Cotrina, M. L., and Nedergaard, M. (2009). Astrocytes and ischemic injury. Stroke 40, S8–S12.
Tang, S. C., Arumugam, T. V., Xu, X., Cheng, A., Mughal, M. R., Jo, D. G., Lathia, J. D., Siler, D. A., Chigurupati, S., Ouyang, X., Magnus, T., Camandola, S., and Mattson, M. P. (2007). Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl. Acad. Sci. U.S.A. 104, 13798–13803.
Terao, S., Yilmaz, G., Stokes, K. Y., Russell, J., Ishikawa, M., Kawase, T., and Granger, D. N. (2008). Blood cell-derived RANTES mediates cerebral microvascular dysfunction, inflammation, and tissue injury after focal ischemia-reperfusion. Stroke 39, 2560–2570.
Terao, Y., Ohta, H., Oda, A., Nakagaito, Y., Kiyota, Y., and Shintani, Y. (2009). Macrophage inflammatory protein-3alpha plays a key role in the inflammatory cascade in rat focal cerebral ischemia. Neurosci. Res. 64, 75–82.
Wei, Y., Yemisci, M., Kim, H. H., Yung, L. M., Shin, H. K., Hwang, S. K., Guo, S., Qin, T., Alsharif, N., Brinkmann, V., Liao, J. K., Lo, E. H., and Waeber, C. (2011). Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann. Neurol. 69, 119–129.
Xiong, X., Barreto, G. E., Xu, L., Ouyang, Y. B., Xie, X., and Giffard, R. G. (2011). Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke 42, 2026–2032.
Yadav, A., Kalita, A., Dhillon, S., and Banerjee, K. (2005). JAK/STAT3 pathway is involved in survival of neurons in response to insulin like growth factor and negatively regulated by suppressor of cytokine signal-3. J. Biol. Chem. 280, 31830–31840.
Yamashita, T., Sawamoto, K., Suzuki, S., Suzuki, N., Adachi, K., Kawase, T., Mihara, M., Ohsugi, Y., Abe, K., and Okano, H. (2005). Blockade of interleukin-6 signaling aggravates ischemic cerebral damage in mice: possible involvement of Stat3 activation in the protection of neurons. J. Neurochem. 94, 459–468.
Yanai, H., Ban, T., Wang, Z., Choi, M. K., Kawamura, T., Negishi, H., Nakasato, M., Lu, Y., Hangai, S., Koshiba, R., Savitsky, D., Ronfani, L., Akira, S., Bianchi, M. E., Honda, K., Tamura, T., Kodama, T., and Taniguchi, T. (2009). HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 462, 99–103.
Yang, Q. W., Lu, F. L., Zhou, Y., Wang, L., Zhong, Q., Lin, S., Xiang, J., Li, J. C., Fang, C. Q., and Wang, J. Z. (2011). HMBG1 mediates ischemia-reperfusion injury by TRIF-adaptor independent Toll-like receptor 4 signaling. J. Cereb. Blood Flow Metab. 31, 593–605.
Yilmaz, G., Arumugam, T. V., Stokes, K. Y., and Granger, D. N. (2006). Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation 113, 2105–2112.
Zhang, J., Takahashi, H. K., Liu, K., Wake, H., Liu, R., Maruo, T., Date, I., Yoshino, T., Ohtsuka, A., Mori, S., and Nishibori, M. (2011). Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke 42, 1420–1428.
Keywords: cytokine, inflammation, ischemia, brain, stroke, T cells, macrophages, DAMPs
Citation: Shichita T, Sakaguchi R, Suzuki M and Yoshimura A (2012) Post-ischemic inflammation in the brain. Front. Immun. 3:132. doi: 10.3389/fimmu.2012.00132
Received: 14 April 2012; Paper pending published: 30 April 2012;
Accepted: 08 May 2012; Published online: 31 May 2012.
Edited by:
Masaaki Murakami, Osaka University, JapanCopyright: © 2012 Shichita, Sakaguchi, Suzuki and Yoshimura. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Akihiko Yoshimura, Department of Microbiology and Immunology, School of Medicine, Keio University, 35 Shinanomachi, Shinjuku-ku, Tokyo 160-8582, Japan. e-mail:eW9zaGltdXJhQGE2LmtlaW8uanA=