


- 1 Department of Microbiology and Immunology, Institute for Cellular Therapeutics, School of Medicine, University of Louisville, Louisville, KY, USA
- 2 Department of Integrative Health Science, Stetson University, Deland, FL, USA
The vertebrate immune system exists in equilibrium with the microbial world. The innate immune system recognizes pathogen-associated molecular patterns via a family of Toll-like receptors (TLR) that activate cells upon detection of potential pathogens. Because some microbes benefit their hosts, mobilizing the appropriate response, and then controlling that response is critical in the maintenance of health. TLR4 recognizes the various forms of lipid A produced by Gram-negative bacteria. Depending on the structural form of the eliciting lipid A molecule, TLR4 responses range from a highly inflammatory endotoxic response involving inflammasome and other pro-inflammatory mediators, to an inhibitory, protective response. Mounting the correct response against an offending microbe is key to maintaining health when exposed to various bacterial species. Further study of lipid A variants may pave the way to understanding how TLR4 responses are generally able to avoid chronic inflammatory damage.
Introduction
The innate immune response results from a highly regulated set of reactions that initiates and orchestrates the entire response to infection, injury, and tumorogenesis (Starczynowski and Karsan, 2010; Arslan et al., 2011; Osawa et al., 2011; Rock et al., 2011). However, dysregulation of the innate immune response leads to poor homeostasis that can initiate pathologic conditions including arthritis, autoimmunity, and cancer (Grivennikov et al., 2010; Bettini and Vignali, 2011; Delogu et al., 2011; Sikora and Grom, 2011; Davalos and Akassoglou, 2012).
Inflammasomes are intracellular staging areas organized to allow maturation of the powerful inflammatory cytokines interleukin (IL-) 1β, IL-18, and IL-33 from inactive precursors (Arend et al., 2008; Dunne, 2011). These cytokines are produced early upon exposure to infectious agents, often triggering receptors specific for chemistries conserved in microbial life (Janeway and Medzhitov, 2002; Kawai and Akira, 2009). The inflammatory environment produced by inflammasomes is important for the clearance of offending microbes, a process that involves the induction of other inflammatory cytokines (TNFα, IL-1β, and IL-6), chemokines (Schilling et al., 2002), and other factors that lead to the activation and differentiation of adaptive immune cells (Foell et al., 2007; Blanco et al., 2008). Dysregulation or over-activation of these cytokines can lead to massive inflammatory states so acute that they result in the death of the host after sterile clearance of infection.
Toll-like receptors (TLR) are innate immune cell receptors that recognize various conserved pathogen-associated molecular patterns (PAMP) and host-derived damage-activated molecular patterns (DAMP; Janeway and Medzhitov, 2002; Foell et al., 2007; Kawai and Akira, 2009). There are at least 13 distinct mammalian TLR that function as either homo- or hetero-dimers (Roach et al., 2005). TLR4 requires MD2 to bind its canonical ligand, lipopolysaccharide (LPS; Poltorak et al., 1998; Kennedy et al., 2004). LPS is a major cell wall component of all Gram-negative bacteria; however, its molecular structure differs depending on the bacterial species. In general, LPS molecules are made up of an O-antigen of varying lengths, the polysaccharide core, and the lipid A moiety (Raetz and Whitfield, 2002). The lipid A region is the endotoxin component of the molecule, and is the portion of the molecule that binds TLR4/MD2 (Carpenter and O’Neill, 2009).
Variations in the structure of lipid A determine inflammatory and immunostimulatory effects of TLR4 binding. The lipid A structure consists of a phosphorylated diglucosamine head group with a number of fatty acid side chains. The fatty acid (acyl) side chains are bound within the MD2 co-receptor via a large hydrophobic pocket, and the resultant complex then associates with a TLR4 monomer (Park et al., 2009). In addition, the lipid A diglucosamine head group, through phosphorylation of its 1- and 4′ carbons, associates with both TLR4 molecules in the signaling-competent dimer (Park et al., 2009). Structure-function studies of lipid A or structural mimetics demonstrate that differences in the number and the length of the acyl chain side groups are important in signaling strength of TLR4 (Coats et al., 2003; Stover et al., 2004; Reife et al., 2006). For example, Stover et al. (2004) showed that there is an optimal acyl chain length for TLR4 recognition of synthetic hexa-acyl lipid A mimetics. Analysis of the crystal structure of the ecto-domain of TLR4-associated with MD2 and lipid A suggests there is critical numbers or arrangement of the acyl chains of lipid A that are required both to bind within the MD2 pocket and associate with the monomer TLR4 molecule (Park et al., 2009).
The phosphorylation state of lipid A also affects its function through changes in the ability to engage TLR4. The prototypic lipid A structure is di-phosphorylated, with a phosphate on each of the glucosamine moieties. Unphosphorylated lipid A does not signal through TLR4, and thus acts as an inhibitor of lipid A (Coats et al., 2011). Monophosphoryl lipid A (MPLA) from Salmonella minnesota rough mutant Re 595 has been shown to be 0.1–1% as toxic (Ribi, 1984; Baldrick et al., 2002), but nearly as immunostimulatory as its parental LPS form (Thompson et al., 2005; Mata-Haro et al., 2007; Didierlaurent et al., 2009). The loss of a single phosphate from a synthetic form of lipid A also decreases the production of pro-inflammatory cytokines (Cekic et al., 2009). Recently, MPLA has been approved by the USDA as MPL® adjuvant as the first TLR agonist to be approved for use as a vaccine adjuvant based on its ability to direct adaptive immune responses with little toxicity (Didierlaurent et al., 2009).
Chronic inflammation can lead to serious conditions, including atherosclerosis, arthritis, and cancer (Coussens and Werb, 2002; Duewell et al., 2010). Various Gram-negative bacteria infect chronically and are associated with inflammatory disease and cancer. Chronic Helicobacter pylori infection causes peptic ulcers which are associated with stomach cancer (Suerbaum and Michetti, 2002). Lyme disease-associated arthritis occurs in individuals chronically infected with the Gram-negative spirochete, Borrelia burgdorferi (Murray and Shapiro, 2010). Chronic inflammation due to improper or lack of control over inflammasome function has also been linked to a variety of immune system disorders, including various forms of arthritis as well as Crohn’s disease and ulcerative colitis.
In this Review, we will discuss the role of TLR4 on inflammasome activation, with a focus on the NLRP3 form of the inflammasome. In addition, the phenomenon of decreased TLR4 inflammatory signals when the lipid A agonist structure is changed in various ways will be addressed.
NLRP3 Inflammasome Activation
Inflammasomes are multi protein structures formed in the cytoplasm of activated innate immune cells that lead to the maturation of IL-1β and IL-18 from inactive pro-proteins to their active, mature forms. The NLRP3 [nucleotide-binding oligomerization domain (NOD)-like receptor family, pryin domain containing 3]-inflammasome is most often associated with TLR4 activation. The other proteins of the NLRP3 inflammasome are ASC (apoptosis-associated speck-like protein containing a CARD domain), the pro-inflammatory caspase, caspase-1, and the precursor forms of IL-1β or IL-18 or both (Martinon et al., 2002; Schroder and Tschopp, 2010).
Two distinct signals are required for the production and secretion of mature IL-1β or IL-18 via the NLRP3 inflammasome (Figure 1). First, a priming signal occurs either through TLR4 or IL-1 receptor. Priming leads to transcription and translation of the inactive pro-forms of IL-1β or IL-18. We and others have shown that an increase in NLRP3 protein abundance occurs during this priming step, and that this increase plays a key role in the ultimate maturation of IL-1β (Bauernfeind et al., 2009; Embry et al., 2011). Priming alone does not lead to secretion of mature IL-1β because primed cells simply harbor immature pro-IL-1β; an activation signal that leads to inflammasome assembly and the proteolytic activity of caspase-1 is also needed. Known activation signals include extracellular ATP, taken up through the P2X7 purinergic receptor (Pelegrin et al., 2008), and phagocytosed crystalline (Martinon et al., 2006) or particulate structures that lyse phagocytic vesicles via a cathepsin B-dependent mechanism (Niemi et al., 2011). After the activation signals, NLRP3 acts as the scaffolding protein that allows the spontaneous assembly of the accessory inflammasome components. In the presence of other inflammasome components, expression of NLRP3 leads to the production of one very large inflammasome or “speck” cluster per cell (Stutz et al., 2009); single specks are visible in primed/activated cells within 5 h (Embry et al., 2011). The assembled inflammasome includes enzymatically active caspase-1 that cleaves the precursor form of IL-1β, which can lead to secretion of both the cytokine and active caspase-1 from the cells (Figure 1a). Tight regulation of pro-IL-1β maturation and related pro-inflammatory proteins underscores the importance of avoiding potential damage to healthy tissue. This regulation is evidenced by the observation that two distinct steps are required to generate and secrete the active forms cytokine suggesting several different levels at which therapeutics may be applied to prevent or control inflammation.
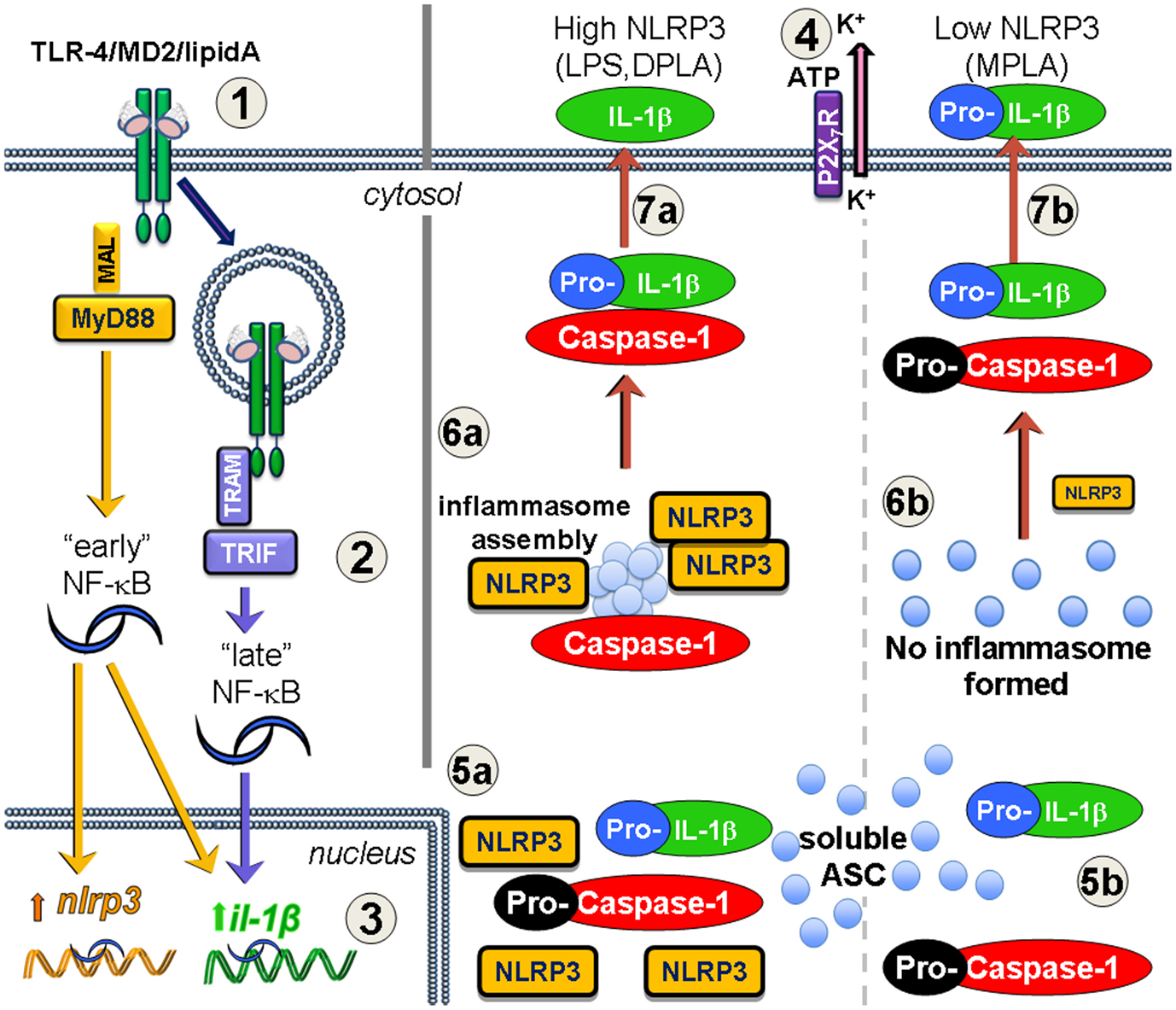
Figure 1. Increased NLRP3 expression via MyD88 leads to inflammasome formation and processing of IL-1β after lipid A treatment. Priming (1–3) and activation (4–7) are required for mature IL-1β secretion. (1) TLR4/MD2 binds lipid A forming endotoxin receptor dimers that signal through either MyD88 or TRIF, or both (2) to activate NF-κB. The MyD88-dependent “early” phase of NF-κB activity results in increased transcription of both nlrp3 and il1b genes. TRIF-dependent TLR4 signaling occurs after receptor-mediated endocytosis of TLR4 leading to the “late” phase of NF-κB activity, and increases il1b gene expression, but not nlrp3. Inflammasome activation occurs in response to such agonists as extracellular ATP which is sensed by the P2X7R and leads to K+ loss through the pannexin-1 ion channel (4). If TLR4 signaling increases NLRP3 protein levels (5a), such as with MyD88-competent LPS or DPLA, then the activation event permits the newly expressed NLRP3 to aggregate with soluble ASC protein and pro-caspase-1, leading to caspase-1 processing to its active form and inflammasome assembly (6a). Pro-IL-1β that is associated with the inflammasome is processed to mature IL-1β and secreted into the extracellular space (7a). However, when TLR4 priming does not adequately increase NLRP3 expression (5b), as is the case with MPLA treatment, then there is little to no NLRP3 protein to direct inflammasome assembly, pro-caspase-1 does not get efficiently activated (6b), and any IL-1β that is secreted is generally in the inactive pro-IL-1β form (7b).
NLRP3 Mutations Lead to Auto-Inflammatory Diseases
In humans as well as in mouse models, gain-of-function polymorphisms, or mutations in the gene encoding NLRP3 are associated with several similar inflammatory conditions that are episodic in nature, occurring in flares after periods of non-activity. These conditions include familial cold auto-inflammatory syndrome, Muckle–Wells syndrome, cryopyrin-associated periodic syndromes (CAPS), and neonatal onset multisystem inflammatory disease (NOMID; Akazawa et al., 2004; Koike et al., 2007; Kubota and Koike, 2010). Chronic inflammatory conditions are associated with an increased risk for development of colorectal cancer and inflammatory bowel diseases, such as ulcerative colitis, and Crohn’s disease (McDermott and Aksentijevich, 2002). The auto-inflammatory episodes typical of these conditions make sense in the framework of inflammasome formation and regulation. Even if NLRP3 is expressed at inappropriate levels, other priming signals are required for the expression of pro-inflammatory cytokines. Therefore, therapeutic potential lies in gaining control of the NLRP3 inflammasome priming factors and inhibitory signals, or both.
Although increased function of NLRP3 is associated with increased inflammation, deletion mutants of NLRP3 do not result in the opposite phenotype. In mice, single deletion mutants of NLRP3 inflammasome components NLRP3, caspase-1, or ASC, are also more susceptible to both dextran sulfate sodium (DSS)-induced colitis and inflammation-associated colon cancer. However, NLRP3 deficient mice have a less pronounced association with cancer development than caspase-1 deficient mice. Further studies are required to determine if the difference is related to a regulatory function of the NLRP3 inflammasome in controlling cancer development caused by chronic inflammation or if it is possibly due to a decrease in apoptosis via reduced caspase-1 activity (Allen et al., 2010). Another study that found increased DSS-induced colitis in NLRP3 deficient mice showed poor regulation of intestinal homeostasis, including decreases in IL-10 levels, changes in the composition of intestinal microbiota, and decreases of β-defensin production (Hirota et al., 2011). Changes in gut microbiota in NLRP3 deficient mice include the presence of potentially pathogenic Enterobacteriaceae genera. Similar changes in the Enterobacteriaceae genera in the gut microbiota, including Proteus species, occur in the TRUC (T-bet−/−Rag1−/− ulcerative colitis) mouse model of spontaneous inflammatory bowel disease (Garrett et al., 2010). These studies suggest a complex role for NLRP3 in the control of local inflammation, such as in the intestine. Therefore, the role of the NLRP3 inflammasome in determining the composition of a healthy gut microbiota needs to be determined before NLRP3 should be used as a therapeutic target to control of chronic inflammation that leads to colon cancers. A selective inhibitor of NLRP3 pro-inflammatory effects could be useful if capable of suppressing only the pro-inflammatory effects without affecting its beneficial roles, such as controlling the homeostasis of healthy microbiota.
MyD88-Dependent Signaling is Required to Prime the NLRP3 Inflammasome Downstream of TLR4 and Lipid A
Preparations of MPLA from the Re595 rough mutant of S. minnesota have been shown to possess much lower toxicity than parental, di-phosphorylated lipid A preparations (Ribi, 1984; Thompson et al., 2005). In vivo, MPLA decreases inflammatory signaling from TLR4 with minimal impairment of its immunostimulatory adjuvant effect on the initial clonal expansion of T cell (Thompson et al., 2005; Mata-Haro et al., 2007). MPLA exerted effective immunostimulatory effects at this stage of the T cell response since it retained to a significant amount of TLR4 TRIF-dependent signaling. In contrast, the TLR4 MyD88-dependent pro-inflammatory signaling was markedly reduced, but not completely absent (Mata-Haro et al., 2007). This, “TRIF-biased” signaling profile obtained with MPLA has been modeled in vitro using dendritic cell cultures and synthetic versions of the E. coli form of monophosphoryl lipid A (sMPLA) or diphosphoryl lipid A (sDPLA; Cekic et al., 2009, 2011). These molecules represent the detoxified or the toxic versions of lipid A, respectively. Following the work of Okemoto et al. (2006) showing decreased IL-1β production in response to MPLA, we hypothesized that the difference in toxicity would at least partially be caused by the reduced maturation of IL-1β using a TLR4 agonist that is “poor” in MyD88-dependent signaling. However, in our cultured dendritic cell system, both MyD88 and TRIF-signaling mutants increased the Il1b mRNA abundance to a similar extent when exposed to sDPLA. When using sDPLA or sMPLA to prime dendritic cell cultures with subsequent ATP activation, IL-1β protein levels were significantly higher after sDPLA treatment as compared to sMPLA treatment. Through Western Blot analysis of the culture supernatants, we showed that the IL-1β that was secreted after sMPLA treatment was actually the inactive, pro-IL-1β rather than mature IL-1β present in the supernatants from sDPLA-treated dendritic cells. We assessed the TLR4 signaling requirements for these priming effects and determined that although both TRIF and MyD88 adaptors contributed to the increase in IL-1β transcription, MyD88-signaling was required for the maturation of IL-1β from its inactive pro-form to its cleaved mature form (Embry et al., 2011). The MyD88-dependent component was then determined to be, at least in part, due to an increase in transcription and production of NLRP3 during priming (Figure 1a). Failure to generate sufficient MyD88-dependent signaling resulted in low expression of NLRP3, which limited the formation of inflammasomes when the activation signal, ATP, was provided (Figure 1b). Not only was decreased inflammasome formation assessed by decreased mature IL-1β secretion in response to ATP signals, we also observed a near absence of “specks” (using antibody against ASC), representing the fully formed NLRP3 inflammasome in confocal microscopy. In summary, these observations indicated a failure of inflammasome assembly in cells primed with sMPLA (decreased MyD88-signaling), as compared to sDPLA (intact MyD88-signaling; Embry et al., 2011).
The ability to activate innate immune system cells in a manner that limits the pro-inflammatory products produced is a goal for clinical treatment of inflammatory conditions. These results suggest that MPLA may prove safe and effective to use as a single-agent therapy for certain chronic inflammatory conditions. Indeed, MPLA promotes endotoxin receptor “tolerance” in the absence of inflammatory outcomes (Madonna et al., 1986; Cekic et al., 2011). The role of IL-1β in perpetuating pro-inflammatory reactions makes the control of its production and function a promising target in controlling inflammation and diseases associated with inflammatory states. Therefore, the ability to activate the immunostimulatory arm of TLR4 signaling without increasing production of mature IL-1β could be beneficial in controlling chronic inflammation in certain situations, especially where low-level infection is a suspected cause.
Naturally-Occurring Differences in Lipid A Structure Correlate with Microbial Virulence
For some time, we have been intrigued by the bifurcation of the TLR4 signaling system: why is it able to independently activate a largely pro-inflammatory program of events (MyD88-dependent) and a separate program that can be thought of as anti-viral and co-stimulatory (TRIF-dependent). This partial separation of signaling events has lead to speculation that responses to various lipid A agonists of TLR4 may be regulated separately, possibly through unique binding characteristics of different ligands. Naturally occurring forms of lipid A possess structural variations that elicit differential effects in vivo that are known to correlate with clinical presentations and microbial pathogenesis. For example, Jarvis and colleagues have shown that the structural heterogeneity of lipid oligosaccharide (LOS) produced by various clinical isolates of Neisseria correlates with their abilities to induce TNFα production by monocytes (John et al., 2009a,b). A pathogenic isolate of N. meningitidis, strain 89I, produces a more highly phosphorylated LOS structure that elicits higher responses from both sides of the TLR4 signaling branches in human monocytic cells. The LOS from that particular isolate induces higher levels of MyD88-dependent products, such as TNFα, IL-1β, and IL-12 p70 as well as TRIF-dependent products including IFNβ and CD80 (a co-stimulatory molecule), compared to the responses elicited by the LOS from two other Neisseria isolates (N. gonorrhoeae), which have lower phosphorylated states (Liu et al., 2010). Hence, the phosphorylation status of LOS affects both MyD88-dependent as well as TRIF-dependent signals (Liu et al., 2010).
Bordetella is another bacterial genus with species-specific differences in lipid A structure and TLR4 reactivity that can be correlated with infectious profiles. The easily cleared B. bronchiseptica highly stimulates TLR4, and this stimulation is required for clearance of infection. The more pathogenic species, B. pertussis and B. parapertussis are less reactive with TLR4, and indeed TLR4 deficiency does not change susceptibility to infection (Mann et al., 2004, 2005). The structural differences between these LPS forms do not seem to be based on phosphorylation status but rather which form of saccharide group is present: B. bronchiseptica and B. parapertussis species have smooth LPS (long-chain polysaccharide), whereas B. pertussis have rough LPS (short-chain polysaccharide; Di Fabio et al., 1992) and exhibits very different effects on cultured human dendritic cells than the smooth forms (Fedele et al., 2008).
Another example of structural changes in lipid A affecting virulence of Gram-negative bacterial species is in Yersinia pestis. Y. pestis expresses a TLR4-inhibiting, tetra-acylated form of LPS when grown at 37°C, the temperature of mammalian hosts. However, a TLR4-activating, hexa-acylated LPS form is expressed when Y. pestis is grown at 26°C, the temperature of insect hosts. Recombinant Y. pestis that have been forced to express only hexa-acylated LPS regardless of temperature cannot sustain a lethal infection in mice. Lethality of infection with this recombinant Y. pestis is restored in hosts that are deficient in TLR4, CD14, or MyD88, but not TRIF or TRAM deficient mice, indicating that MyD88-associated TLR4 outcomes are required to control Y. pestis infection in vivo (Montminy et al., 2006), and that the bacteria has evolved a way to evade TLR4 signaling that allows infection in mammalian hosts.
Naturally Occurring Forms of Monophosphoryl Lipid A and their Effects
Darveau and colleagues have worked extensively with structurally defined forms of lipid A isolated from various organisms, in order to study their inherent differences in inflammatory signaling. Several of the lipid As are naturally monophosphorylated. Two such forms have been isolated from the oral bacterium Porphyromonas gingivalis grown in different hemin concentrations, which regulate the expression of bacteria-produced phosphatases. P. gingivalis grown in high hemin concentrations leads primarily to the production of antagonistic, dephosphorylated, or monophosphorylated, tetra-acylated lipid A forms within the Gram-negative cell wall (Reife et al., 2006). These modifications provide the organism with a unique evasion mechanism that prevents detection by TLR4 while also decreasing the effectiveness of cationic anti-microbial peptides due to the neutralization of the negative charge of the bacterial cell wall (Coats et al., 2009).
The lipid A produced by the human gut symbiant, Bacteroides thetaiotaomicron is structurally very similar to that from P. gingivalis grown in low hemin concentrations. Although both these lipid A forms are penta-acylated and monophosphorylated, they differ in the placement of the phosphate group on the diglucosamine head group. B. thetaiotaomicron LPS, has its single phosphate on the 1-carbon, whereas the phosphate on the P. gingivalis LPS is on the 4′-carbon. This seemingly minor structural change results in differences in the ability to stimulate NF-κB in human TLR4/human MD2-expressing HEK-293 cells in vitro (Coats et al., 2011). However, NF-κB signaling was used as a read-out in these in vitro studies, which is downstream of both MyD88- and TRIF-dependent signaling pathways. Further differences may be appreciated if other signaling readouts were to be examined.
Overall, the differences in lipid A structure described above have marked effects on TLR4 signaling, resulting in important effects on bacterial pathogenesis. Changes that result in decreased inflammatory responses increase the ability of the bacteria to establish infection, thus demonstrating lipid A modifications as an under-appreciated mechanism for microbial evasion of the immune system. Both pathogenic (P. gingivalis) and commensal (B. thetaiotaomicron) bacteria appear to have taken advantage of monophosphorylation to evade sterilizing responses. More direct assessment is needed to determine the relative usage of the MyD88- and TRIF-dependent pathways after TLR4 engagement when using structurally distinct forms of lipid A, and whether or not such usage is correlated with the potential for immediate inflammatory damage (MyD88-associated innate response) or long-term protective immunity (TRIF-associated adaptive immunity).
Why has the mammalian LPS recognition system not evolved to counter this threat? One can imagine, for example, that mutations in TLR4 or MD2, or both, could permit much more efficient recognition of hypophosphorylated forms of LPS, which would lead to MyD88-dependent innate responses that conceivably could be of tremendous benefit to a mammalian host in responding to P. gingivalis or other monophosphorylated pathogens. We propose, however, that poor activation of MyD88-signaling by monophosphorylated lipid A is a critically important concession to prevent highly inflammatory responses from damaging host tissue, such as oral or gut epithelium, while also tolerating the preponderance of microbes that are beneficial in terms of enhancing nutrition and preventing colonization by truly pathogenic bacteria. Because even monophosphorylated commensal bacteria can be dangerous if they escape their niche in the gut or oral cavity (Teng et al., 2004; Goldstein et al., 2006), TRIF-dependent responses that are particularly relevant to adaptive immunity may provide “insurance” in the form of rapid responses in the event of niche escape.
Detoxification of Lipid A by Tissue Alkaline Phosphatases: Inflammatory Implications
Because lipid A-mediated inflammation can lead to septic shock and death if left unchecked, the ability to detoxify lipid A molecules is critical for host survival. In the late 1990s, Poelstra et al. showed that tissue-specific alkaline phosphatases (AP) decrease the phosphorylation state of various lipid A molecules in vitro. From this, they hypothesized that AP work in vivo to detoxify endotoxin and decrease its systemic effects (Poelstra et al., 1997a,b). It was interesting, however, that the same group also showed that MPLA is not completely dephosphorylated by AP (Bentala et al., 2002). Although that study did not discuss the possible biologic implications of failing to dephosphorylate MPLA fully, the results suggest that the monophosphorylated form of lipid A is less of a threat in locations with high levels of commensal bacteria (i.e., mucosal sites), than the di- or tri-phosphate forms. Additionally, by failing to form the non-phosphorylated lipid A versions of MPLA, non-signaling TLR4 antagonists that could have served as inhibitors of TLR4 binding, and activity are not produced.
Tissue AP belong to a family of proteins that are expressed by most tissues, but their activities are particularly high in the intestine, liver, and placenta. The intestines harbor large numbers of Gram-negative bacteria with an abundance of lipid A. Although intestinal alkaline phosphatase (IAP) is known to help in the uptake of nutrients such as pyridoxal phosphate and pyridoxamine phosphate (two forms of B6 vitamins; Waymire et al., 1995) as well as phosphate, its ability to dephosphorylate LPS in the gastrointestinal tract arguably helps in the maintenance of commensal or mutual Gram-negative bacterial species in the gut without causing either continual stimulation or endotoxin tolerance to pathologic Gram-negative bacteria.
Bates et al. demonstrated in a zebra fish model of vertebrate intestine that the presence of innocuous or beneficial Gram-negative bacterial species drives the expression of IAP in a MyD88-dependent manner. Furthermore, IAP-deficient zebrafish are hypersensitive to their own microbiota (Bates et al., 2007). Oral co-administration of IAP inhibitors and fully phosphorylated LPS to rats leads to increased levels of LPS in the serum (Koyama et al., 2002). IAP-deficient mice exhibit a local gut endotoxin tolerance, but are fully responsive to LPS systemically (Chen et al., 2011). Although IAP deletion mutants are also more sensitive to DSS-induced colitis than wild-type mice, feeding them calf IAP abrogates this effect of the deletion (Ramasamy et al., 2011). In humans, Crohn’s disease and ulcerative colitis are known to be associated with lower IAP expression (Tuin et al., 2009). Whitehead has proposed that rosacea, a chronic inflammatory condition of the face, is caused by overactive immune responses to commensal species of the skin, and has correlated rosacea with decreases in IAP expression (Whitehead, 2009). Interestingly, dietary habits that increase IAP expression can help to decrease flairs in other inflammatory diseases (Blanchard and Cousins, 2000; Tuin et al., 2006; Kaur et al., 2007). In addition, expression of the TLR4-associated co-receptor, CD14, was shown to be co-localized with AP activity in the intestine, liver, and kidneys (Tuin et al., 2006). Together, these studies suggest that detoxification of LPS by tissue AP is a natural mechanism that helps control chronic inflammation in the gut. Maintenance of monophosphorylated forms of LPS, which are detoxified but remain immunostimulatory via TRIF-biased signaling by TLR4, may be essential for the peaceful co-existence of the vertebrate host with its microbiota.
Conclusion
The signaling of TLR4 and subsequent priming and activation of the NLRP3 inflammasome provides several potential avenues to control chronic inflammation, especially in localized sites such as the intestine. Therefore, developing therapeutics that can signal through TLR4 without the danger of inflammasome activation could not only safely activate the innate immune response, but could also open new therapeutic avenues to chronic inflammatory conditions in which either IL-1β or IL-18 plays an amplifying role. The studies discussed here emphasize the importance of controlling microbiogenic inflammation to microbiota to maintain tissue homeostasis, while retaining the ability to tolerate monophosphorylated forms of lipid A. This scenario is of utmost importance in the intestine. Linking these studies with the control of NLRP3 inflammasome will increase our understanding of inflammatory disease progression and help in determining potential for therapeutic interventions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akazawa, T., Masuda, H., Saeki, Y., Matsumoto, M., Takeda, K., Tsujimura, K., Kuzushima, K., Takahashi, T., Azuma, I., Akira, S., Toyoshima, K., and Seya, T. (2004). Adjuvant-mediated tumor regression and tumor-specific cytotoxic response are impaired in MyD88-deficient mice. Cancer Res. 64, 757–764.
Allen, I. C., TeKippe, E. M., Woodford, R. M., Uronis, J. M., Holl, E. K., Rogers, A. B., Herfarth, H. H., Jobin, C., and Ting, J. P. (2010). The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 207, 1045–1056.
Arend, W. P., Palmer, G., and Gabay, C. (2008). IL-1, IL-18, and IL-33 families of cytokines. Immunol. Rev. 223, 20–38.
Arslan, F., de Kleijn, D. P., and Pasterkamp, G. (2011). Innate immune signaling in cardiac ischemia. Nat. Rev. Cardiol. 8, 292–300.
Baldrick, P., Richardson, D., Elliott, G., and Wheeler, A. W. (2002). Safety evaluation of monophosphoryl lipid A (MPL): an immunostimulatory adjuvant. Regul. Toxicol. Pharmacol. 35, 398–413.
Bates, J. M., Akerlund, J., Mittge, E., and Guillemin, K. (2007). Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe 2, 371–382.
Bauernfeind, F. G., Horvath, G., Stutz, A., Alnemri, E. S., MacDonald, K., Speert, D., Fernandes-Alnemri, T., Wu, J., Monks, B. G., Fitzgerald, K. A., Hornung, V., and Latz, E. (2009). Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791.
Bentala, H., Verweij, W. R., Huizinga-van der Vlag, A., van Loenen-Weemaes, A. M., Meijer, D. K., and Poelstra, K. (2002). Removal of phosphate from lipid A as a strategy to detoxify lipopolysaccharide. Shock 18, 561–566.
Bettini, M., and Vignali, D. A. (2011). T cell-driven initiation and propagation of autoimmune diabetes. Curr. Opin. Immunol. 23, 754–760.
Blanchard, R. K., and Cousins, R. J. (2000). Regulation of intestinal gene expression by dietary zinc: induction of uroguanylin mRNA by zinc deficiency. J. Nutr. 130, 1393S–1398S.
Blanco, P., Palucka, A. K., Pascual, V., and Banchereau, J. (2008). Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev. 19, 41–52.
Carpenter, S., and O’Neill, L. A. (2009). Recent insights into the structure of Toll-like receptors and post-translational modifications of their associated signalling proteins. Biochem. J. 422, 1–10.
Cekic, C., Casella, C. R., Eaves, C. A., Matsuzawa, A., Ichijo, H., and Mitchell, T. C. (2009). Selective activation of the p38 MAPK pathway by synthetic monophosphoryl lipid A. J. Biol. Chem. 284, 31982–31991.
Cekic, C., Casella, C. R., Sag, D., Antignano, F., Kolb, J., Suttles, J., Hughes, M. R., Krystal, G., and Mitchell, T. C. (2011). MyD88-dependent SHIP1 regulates proinflammatory signaling pathways in dendritic cells after monophosphoryl lipid A stimulation of TLR4. J. Immunol. 186, 3858–3865.
Chen, K. T., Malo, M. S., Beasley-Topliffe, L. K., Poelstra, K., Millan, J. L., Mostafa, G., Alam, S. N., Ramasamy, S., Warren, H. S., Hohmann, E. L., and Hodin, R. A. (2011). A role for intestinal alkaline phosphatase in the maintenance of local gut immunity. Dig. Dis. Sci. 56, 1020–1027.
Coats, S. R., Berezow, A. B., To, T. T., Jain, S., Bainbridge, B. W., Banani, K. P., and Darveau, R. P. (2011). The lipid A phosphate position determines differential host Toll-like receptor 4 responses to phylogenetically related symbiotic and pathogenic bacteria. Infect. Immun. 79, 203–210.
Coats, S. R., Reife, R. A., Bainbridge, B. W., Pham, T. T., and Darveau, R. P. (2003). Porphyromonas gingivalis lipopolysaccharide antagonizes Escherichia coli lipopolysaccharide at toll-like receptor 4 in human endothelial cells. Infect. Immun. 71, 6799–6807.
Coats, S. R., To, T. T., Jain, S., Braham, P. H., and Darveau, R. P. (2009). Porphyromonas gingivalis resistance to polymyxin B is determined by the lipid A 4’-phosphatase, PGN_0524. Int. J. Oral Sci. 1, 126–135.
Davalos, D., and Akassoglou, K. (2012). Fibrinogen as a key regulator of inflammation in disease. Semin. Immunopathol. 34, 43–62.
Delogu, L. G., Deidda, S., Delitala, G., and Manetti, R. (2011). Infectious diseases and autoimmunity. J. Infect. Dev. Ctries. 5, 679–687.
Di Fabio, J. L., Caroff, M., Karibian, D., Richards, J. C., and Perry, M. B. (1992). Characterization of the common antigenic lipopolysaccharide O-chains produced by Bordetella bronchiseptica and Bordetella parapertussis. FEMS Microbiol. Lett. 76, 275–281.
Didierlaurent, A. M., Morel, S., Lockman, L., Giannini, S. L., Bisteau, M., Carlsen, H., Kielland, A., Vosters, O., Vanderheyde, N., Schiavetti, F., Larocque, D., Van, M. M., and Garcon, N. (2009). AS04, an aluminum salt- and TLR4 agonist-based adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J. Immunol. 183, 6186–6197.
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., Abela, G. S., Franchi, L., Nunez, G., Schnurr, M., Espevik, T., Lien, E., Fitzgerald, K. A., Rock, K. L., Moore, K. J., Wright, S. D., Hornung, V., and Latz, E. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361.
Dunne, A. (2011). Inflammasome activation: from inflammatory disease to infection. Biochem. Soc. Trans. 39, 669–673.
Embry, C. A., Franchi, L., Nunez, G., and Mitchell, T. C. (2011). Mechanism of impaired NLRP3 inflammasome priming by monophosphoryl lipid A. Sci. Signal. 4, ra28.
Fedele, G., Nasso, M., Spensieri, F., Palazzo, R., Frasca, L., Watanabe, M., and Ausiello, C. M. (2008). Lipopolysaccharides from Bordetella pertussis and Bordetella parapertussis differently modulate human dendritic cell functions resulting in divergent prevalence of Th17-polarized responses. J. Immunol. 181, 208–216.
Foell, D., Wittkowski, H., and Roth, J. (2007). Mechanisms of disease: a “DAMP” view of inflammatory arthritis. Nat. Clin. Pract. Rheumatol. 3, 382–390.
Garrett, W. S., Gallini, C. A., Yatsunenko, T., Michaud, M., DuBois, A., Delaney, M. L., Punit, S., Karlsson, M., Bry, L., Glickman, J. N., Gordon, J. I., Onderdonk, A. B., and Glimcher, L. H. (2010). Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8, 292–300.
Goldstein, E. J., Citron, D. M., Vaidya, S. A., Warren, Y. A., Tyrrell, K. L., Vreni, M. C., and Fernandez, H. (2006). In vitro activity of 11 antibiotics against 74 anaerobes isolated from pediatric intra-abdominal infections. Anaerobe 12, 63–66.
Grivennikov, S. I., Greten, F. R., and Karin, M. (2010). Immunity, inflammation, and cancer. Cell 140, 883–899.
Hirota, S. A., Ng, J., Lueng, A., Khajah, M., Parhar, K., Li, Y., Lam, V., Potentier, M. S., Ng, K., Bawa, M., McCafferty, D. M., Rioux, K. P., Ghosh, S., Xavier, R. J., Colgan, S. P., Tschopp, J., Muruve, D., Macdonald, J. A., and Beck, P. L. (2011). NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 17, 1359–1372.
Janeway, C. A. Jr., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216.
John, C. M., Liu, M., and Jarvis, G. A. (2009a). Natural phosphoryl and acyl variants of lipid A from Neisseria meningitidis strain 89I differentially induce tumor necrosis factor-alpha in human monocytes. J. Biol. Chem. 284, 21515–21525.
John, C. M., Liu, M., and Jarvis, G. A. (2009b). Profiles of structural heterogeneity in native lipooligosaccharides of Neisseria and cytokine induction. J. Lipid Res. 50, 424–438.
Kaur, J., Madan, S., Hamid, A., Singla, A., and Mahmood, A. (2007). Intestinal alkaline phosphatase secretion in oil-fed rats. Dig. Dis. Sci. 52, 665–670.
Kawai, T., and Akira, S. (2009). The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 21, 317–337.
Kennedy, M. N., Mullen, G. E., Leifer, C. A., Lee, C., Mazzoni, A., Dileepan, K. N., and Segal, D. M. (2004). A complex of soluble MD-2 and lipopolysaccharide serves as an activating ligand for Toll-like receptor 4. J. Biol. Chem. 279, 34698–34704.
Koike, R., Kubota, T., Hara, Y., Ito, S., Suzuki, K., Yanagisawa, K., Uchibori, K., and Miyasaka, N. (2007). A case of Muckle-Wells syndrome caused by a novel H312P mutation in NALP3 (cryopyrin). Mod. Rheumatol. 17, 496–499.
Koyama, I., Matsunaga, T., Harada, T., Hokari, S., and Komoda, T. (2002). Alkaline phosphatases reduce toxicity of lipopolysaccharides in vivo and in vitro through dephosphorylation. Clin. Biochem. 35, 455–461.
Kubota, T., and Koike, R. (2010). Cryopyrin-associated periodic syndromes: background and therapeutics. Mod. Rheumatol. 20, 213–221.
Liu, M., John, C. M., and Jarvis, G. A. (2010). Phosphoryl moieties of lipid A from Neisseria meningitidis and N. gonorrhoeae lipooligosaccharides play an important role in activation of both MyD88- and TRIF-dependent TLR4-MD-2 signaling pathways. J. Immunol. 185, 6974–6984.
Madonna, G. S., Peterson, J. E., Ribi, E. E., and Vogel, S. N. (1986). Early-phase endotoxin tolerance: induction by a detoxified lipid A derivative, monophosphoryl lipid A. Infect. Immun. 52, 6–11.
Mann, P. B., Elder, K. D., Kennett, M. J., and Harvill, E. T. (2004). Toll-like receptor 4-dependent early elicited tumor necrosis factor alpha expression is critical for innate host defense against Bordetella bronchiseptica. Infect. Immun. 72, 6650–6658.
Mann, P. B., Wolfe, D., Latz, E., Golenbock, D., Preston, A., and Harvill, E. T. (2005). Comparative toll-like receptor 4-mediated innate host defense to Bordetella infection. Infect. Immun. 73, 8144–8152.
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426.
Martinon, F., Petrilli, V., Mayor, A., Tardivel, A., and Tschopp, J. (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241.
Mata-Haro, V., Cekic, C., Martin, M., Chilton, P. M., Casella, C. R., and Mitchell, T. C. (2007). The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 316, 1628–1632.
McDermott, M. F., and Aksentijevich, I. (2002). The autoinflammatory syndromes. Curr. Opin. Allergy Clin. Immunol. 2, 511–516.
Montminy, S. W., Khan, N., McGrath, S., Walkowicz, M. J., Sharp, F., Conlon, J. E., Fukase, K., Kusumoto, S., Sweet, C., Miyake, K., Akira, S., Cotter, R. J., Goguen, J. D., and Lien, E. (2006). Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat. Immunol. 7, 1066–1073.
Niemi, K., Teirila, L., Lappalainen, J., Rajamaki, K., Baumann, M. H., Oorni, K., Wolff, H., Kovanen, P. T., Matikainen, S., and Eklund, K. K. (2011). Serum amyloid A activates the NLRP3 inflammasome via P2×7 receptor and a cathepsin B-sensitive pathway. J. Immunol. 186, 6119–6128.
Okemoto, K., Kawasaki, K., Hanada, K., Miura, M., and Nishijima, M. (2006). A potent adjuvant monophosphoryl lipid A triggers various immune responses, but not secretion of IL-1beta or activation of caspase-1. J. Immunol. 176, 1203–1208.
Osawa, R., Williams, K. L., and Singh, N. (2011). The inflammasome regulatory pathway and infections: role in pathophysiology and clinical implications. J. Infect. 62, 119–129.
Park, B. S., Song, D. H., Kim, H. M., Choi, B. S., Lee, H., and Lee, J. O. (2009). The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195.
Pelegrin, P., Barroso-Gutierrez, C., and Surprenant, A. (2008). P2×7 receptor differentially couples to distinct release pathways for IL-1beta in mouse macrophage. J. Immunol. 180, 7147–7157.
Poelstra, K., Bakker, W. W., Klok, P. A., Hardonk, M. J., and Meijer, D. K. (1997a). A physiologic function for alkaline phosphatase: endotoxin detoxification. Lab. Invest. 76, 319–327.
Poelstra, K., Bakker, W. W., Klok, P. A., Kamps, J. A., Hardonk, M. J., and Meijer, D. K. (1997b). Dephosphorylation of endotoxin by alkaline phosphatase in vivo. Am. J. Pathol. 151, 1163–1169.
Poltorak, A., Smirnova, I., He, X., Liu, M. Y., Van, H. C., McNally, O., Birdwell, D., Alejos, E., Silva, M., Du, X., Thompson, P., Chan, E. K., Ledesma, J., Roe, B., Clifton, S., Vogel, S. N., and Beutler, B. (1998). Genetic and physical mapping of the Lps locus: identification of the toll-4 receptor as a candidate gene in the critical region. Blood Cells Mol. Dis. 24, 340–355.
Raetz, C. R., and Whitfield, C. (2002). Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700.
Ramasamy, S., Nguyen, D. D., Eston, M. A., Alam, S. N., Moss, A. K., Ebrahimi, F., Biswas, B., Mostafa, G., Chen, K. T., Kaliannan, K., Yammine, H., Narisawa, S., Millan, J. L., Warren, H. S., Hohmann, E. L., Mizoguchi, E., Reinecker, H. C., Bhan, A. K., Snapper, S. B., Malo, M. S., and Hodin, R. A. (2011). Intestinal alkaline phosphatase has beneficial effects in mouse models of chronic colitis. Inflamm. Bowel. Dis. 17, 532–542.
Reife, R. A., Coats, S. R., Al-Qutub, M., Dixon, D. M., Braham, P. A., Billharz, R. J., Howald, W. N., and Darveau, R. P. (2006). Porphyromonas gingivalis lipopolysaccharide lipid A heterogeneity: differential activities of tetra- and penta-acylated lipid A structures on E-selectin expression and TLR4 recognition. Cell. Microbiol. 8, 857–868.
Roach, J. C., Glusman, G., Rowen, L., Kaur, A., Purcell, M. K., Smith, K. D., Hood, L. E., and Aderem, A. (2005). The evolution of vertebrate Toll-like receptors. Proc. Natl. Acad. Sci. U.S.A. 102, 9577–9582.
Rock, K. L., Lai, J. J., and Kono, H. (2011). Innate and adaptive immune responses to cell death. Immunol. Rev. 243, 191–205.
Schilling, D., Thomas, K., Nixdorff, K., Vogel, S. N., and Fenton, M. J. (2002). Toll-like receptor 4 and Toll-IL-1 receptor domain-containing adapter protein (TIRAP)/myeloid differentiation protein 88 adapter-like (Mal) contribute to maximal IL-6 expression in macrophages. J. Immunol. 169, 5874–5880.
Sikora, K. A., and Grom, A. A. (2011). Update on the pathogenesis and treatment of systemic idiopathic arthritis. Curr. Opin. Pediatr. 23, 640–646.
Starczynowski, D. T., and Karsan, A. (2010). Innate immune signaling in the myelodysplastic syndromes. Hematol. Oncol. Clin. North Am. 24, 343–359.
Stover, A. G., Da Silva, C. J., Evans, J. T., Cluff, C. W., Elliott, M. W., Jeffery, E. W., Johnson, D. A., Lacy, M. J., Baldridge, J. R., Probst, P., Ulevitch, R. J., Persing, D. H., and Hershberg, R. M. (2004). Structure-activity relationship of synthetic toll-like receptor 4 agonists. J. Biol. Chem. 279, 4440–4449.
Stutz, A., Golenbock, D. T., and Latz, E. (2009). Inflammasomes: too big to miss. J. Clin. Invest. 119, 3502–3511.
Suerbaum, S., and Michetti, P. (2002). Helicobacter pylori infection. N. Engl. J. Med. 347, 1175–1186.
Teng, L. J., Hsueh, P. R., Huang, Y. H., and Tsai, J. C. (2004). Identification of bacteroides thetaiotaomicron on the basis of an unexpected specific amplicon of universal 16S ribosomal DNA PCR. J. Clin. Microbiol. 42, 1727–1730.
Thompson, B. S., Chilton, P. M., Ward, J. R., Evans, J. T., and Mitchell, T. C. (2005). The low-toxicity versions of LPS, MPL adjuvant and RC529, are efficient adjuvants for CD4+ T cells. J. Leukoc. Biol. 78, 1273–1280.
Tuin, A., Huizinga-van der Vlag, A., van Loenen-Weemaes, A. M., Meijer, D. K., and Poelstra, K. (2006). On the role and fate of LPS-dephosphorylating activity in the rat liver. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G377–G385.
Tuin, A., Poelstra, K., de Jager-Krikken, A., Bok, L., Raaben, W., Velders, M. P., and Dijkstra, G. (2009). Role of alkaline phosphatase in colitis in man and rats. Gut 58, 379–387.
Waymire, K. G., Mahuren, J. D., Jaje, J. M., Guilarte, T. R., Coburn, S. P., and MacGregor, G. R. (1995). Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat. Genet. 11, 45–51.
Keywords: NLRP3, inflammasome, monophosphoryl lipid A, LPS, TLR4
Citation: Chilton PM, Embry CA and Mitchell TC (2012) Effects of differences in lipid A structure on TLR4 pro-inflammatory signaling and inflammasome activation. Front. Immun. 3:154. doi: 10.3389/fimmu.2012.00154
Received: 19 March 2012; Paper pending published: 03 April 2012;
Accepted: 25 May 2012; Published online: 13 June 2012.
Edited by:
Masaaki Murakami, Osaka University, JapanCopyright: © 2012 Chilton, Embry and Mitchell. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Thomas C. Mitchell, Department of Microbiology and Immunology, Institute for Cellular Therapeutics, School of Medicine, University of Louisville, 570 South Preston Street, Donald Baxter Building, Room 404, Louisville, KY 40202, USA. e-mail:dG9tLm1pdGNoZWxsQGxvdWlzdmlsbGUuZWR1