

- 1 Laboratory of Experimental Immunology, WPI Immunology Frontier Research Center, Osaka University, Suita, Japan
- 2 Department of Experimental Pathology, Institute for Frontier Medical Sciences, Kyoto University, Kyoto, Japan
Foxp3+ regulatory T cells (Tregs) are a constitutively immunosuppressive cell type critical for the control of autoimmunity and inflammatory pathology. A range of mechanisms of Treg suppression have been identified and it has not always been clear how these different mechanisms interact in order to properly suppress autoimmunity and excessive inflammation. In recent years it has become clear that, while all Tregs seem to share some core suppressive mechanisms, they are also able to adapt to their surroundings in response to a variety of stimuli by homing to the sites of inflammation and exerting ancillary suppressive functions. In this review, we discuss the relevance and possible modes of Treg adaptability and put forward a modular model of Treg suppressive function. Understanding this flexibility may hold the key to understanding the full spectrum of Treg suppressive behavior.
Introduction
T-regulatory cells (Tregs) are a suppressive subset of T cells, which make up around 10% of CD4+ cells, crucial to the proper maintenance of immune self-tolerance and homeostasis. Natural Tregs are produced in the thymus but some Tregs may also be induced in the periphery (Sakaguchi et al., 2008). Since the discovery that CD25-expressing T cells in the normal immune system have suppressive function (Sakaguchi et al., 1995), the field of regulatory T cells has advanced with the discovery that the transcription factor Foxp3 is critical to the suppressive function of Tregs as illustrated by the finding that ectopic expression of Foxp3 can induce regulatory function in naïve T cells (Fontenot et al., 2003; Hori et al., 2003). A loss of Foxp3 function results in Treg deficiency or dysfunction and is responsible for the autoimmune disorders seen in both Scurfy mice (Brunkow et al., 2001; Khattri et al., 2003) and the human immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) syndrome (Wildin et al., 2001). Foxp3 regulates expression of a large number of genes including those responsible for key features of Tregs, such as an absence of IL-2 production and high expression of cytotoxic T lymphocyte antigen-4 (CTLA-4) and CD25. While it has been clear for some time that Tregs are critical to the maintenance of immune self-tolerance and homeostasis, the exact mechanisms by which they are able to do this has been a source of considerable debate (Rudensky and Campbell, 2006; Sakaguchi et al., 2009; Shevach, 2009).
Core Mechanisms of Treg Suppression
A number of different Treg suppressive mechanisms have been proposed. It seems that in part this is due to Tregs being able to adapt to environmental cues and alter their suppressive mechanisms to fit the circumstances. However, while there is some heterogeneity of function, all Foxp3-expressing Tregs may share a set of core suppressive mechanisms. As discussed below, CTLA-4 and IL-2 are most stably activated or repressed, respectively, by Foxp3 and appear to be involved in such core mechanisms.
CTLA-4-Dependent Suppression
CTLA-4 is constitutively expressed by Foxp3+ Tregs and competes with the closely related CD28 for binding with the co-stimulatory molecules, CD80 and CD86, on the surface of APCs. While CTLA-4 is also expressed on activated T-effector cells, mice with a Treg specific CTLA-4 depletion suffer from fatal lymphoproliferative disease, similar to that seen in totally CTLA-4-deficient mice, despite the development of Tregs in normal numbers (Waterhouse et al., 1995; Wing et al., 2008). CTLA-4-deficient Tregs also show impaired in vitro suppressive activity (Wing et al., 2008), while mice with mixed bone marrow chimeras of CTLA-4-deficient and sufficient donors do not develop autoimmune disease, demonstrating cell extrinsic function (Bachmann et al., 1999). For some time the mechanisms of CTLA-4 function have been obscure but recent work has elegantly demonstrated that CTLA-4-mediated trans-endocytosis and degradation of CD80/86 from the surface of APCs is a key mechanism of CTLA-4 function (Qureshi et al., 2011). However, this may not be the sole mechanism of CTLA-4 function as CTLA-4 ligation of CD80/86 also induces nuclear translocation of the transcription factor Foxo3 inhibiting dendritic cell (DC) expression of IL-6 and TNFα, while Foxo3 KO mice have enhanced CD80/86 expression (Dejean et al., 2009). In addition, CTLA-4 engagement may cause DCs to express the immunosuppressive tryptophan catabolizing enzyme IDO (Grohmann et al., 2002). These findings collectively suggest that CTLA-4 is critical to the suppressive function of Tregs and appears to act primarily via cell extrinsic effects on APCs (Wing et al., 2008) in which Treg expression of CTLA-4 reduces APC expression of CD80 and 86 thus reducing their co-stimulatory capacity (Wing et al., 2011).
IL-2-Dependent Suppression
Several cardinal features of Foxp3+ Tregs are their absence of IL-2 production, constitutively high expression of the alpha chain receptor for IL-2 (CD25) and consequent ability to bind IL-2 with high affinity (Sakaguchi et al., 2007). A central role for IL-2 in Treg biology has been demonstrated by in vivo studies of mice either deficient in IL-2 production (Schorle et al., 1991) or IL-2 receptor (Suzuki et al., 1995;Willerford et al., 1995): both suffer from fatal lymphoproliferative disease in a manner similar to Foxp3-deficient scurfy mice. Tregs are able to suppress both IL-2 mRNA and protein production by responder T cells (Takahashi et al., 1998; Thornton and Shevach, 1998), most likely indirectly due to a CTLA-4-mediated reduction in CD80/86 co-stimulation from APCs and also consume IL-2, thus denying it to local T-effector cells (de la Rosa et al., 2004). This may in turn lead to arrest of proliferation and apoptosis of T-effector cells (Pandiyan et al., 2007). Since Tregs are dependent on exogenous IL-2 for their survival and function (Furtado et al., 2002; Setoguchi et al., 2005) but suppress its production by T cells, it seems that IL-2 is used as a feedback mechanism to prevent Treg overgrowth that might otherwise result in excessive immunosuppression.
Foxp3 regulated features of Treg biology such as high expression of CTLA-4, CD25 and an absence of IL-2 production are critical to the function of Tregs, efficiently controlling APC-dependent activation and proliferation of T cells. As a result, knockout of either CTLA-4 or CD25 expression replicates the phenotype of Foxp3-deficient mice. However, in addition to this “core module” of suppression, Foxp3+ Tregs are also able to adapt to their environment and take on additional suppressive functions in order to fit the immune context they find themselves in. This may be of particular importance in the more inflammatory conditions found at barrier sites such as the gut in which CTLA-4 and IL-2-based suppression may be sometimes overwhelmed due to an excess of inflammatory signals leading to a requirement for additional mechanisms such as IL-10 (Rubtsov et al., 2008; Yamaguchi et al., 2011).
Adaptability of Suppressive Mechanisms and Induction of Additional Transcription Factors
CTLA-4-dependent suppression is not the only mechanism by which Tregs are able to suppress the activity of other cells. In vitro experiments give a clear indication of this: while normal Tregs suppress T-effector proliferation in a CTLA-4-dependent manner, Tregs derived from total CTLA-4-deficient mice also retain in vitro suppressive capacity. In this case it seems likely that Tregs derived from CTLA-4-deficient mice are highly activated due to the ongoing autoimmune pathology and as such may have induced suppressive functions such as IL-10 and TGFβ secretion. A wide range of other suppressive molecules have been identified in Tregs including: IL-10, TGFβ, Galectin, Neuropilin, IL-35, cAMP, Granzyme-B, perforin, and cAMP (Sakaguchi et al., 2009; Shevach, 2009). Many of these molecules are upregulated in effector CD44high Tregs.
In addition to adopting an effector phenotype in response to stimulation, recent work has demonstrated that Tregs are able to adapt to environmental signals and further differentiate from their base state by expression of transcription factors normally associated with other T cell subsets (Campbell and Koch, 2011). It seems likely that Tregs share much of the same molecular machinery as conventional CD4+ T cells and respond to the same differentiation stimuli. This induction/co-opting of additional transcription factors seems to be critical to Treg suppression of particular T cell subsets and their associated immunopathology.
T-bet – Th1
Koch et al. (2009) have demonstrated that the transcription factor T-bet, normally required for the linage development of Th1 cells, is upregulated in Tregs in response to IFNγ, suggesting that the induction of this Treg subset occurs in reaction to a Th1 immune response (Koch et al., 2009). T-bet causes the Th1-associated chemokine receptor CXCR3 to be upregulated on the surface of Tregs and a small increase in IFNγ expression was also observed. CXCR3 homes the cell to the sites of Th1 inflammation via its IFNγ-induced ligands CXCL9, CXCL10, and CXCL11, allowing Tregs to provide in situ suppression. T-bet-deficient Tregs are unable to accumulate at the sites of Th1 inflammation and properly control the Th1 immune response (Koch et al., 2009). In addition, T-bet-deficient Tregs retain their in vitro suppressive activity (Bettelli et al., 2004) and in vivo have only a modest (approximately 50%) reduction in mRNA expression of the effector molecules, IL-10 and TGFβ (Koch et al., 2009). This suggests that the primary role of T-bet in Treg suppression of Th1 responses may be the control of CXCR3 expression and Treg localization.
IRF4 and GATA-3 – Th2
The transcription factor interferon regulatory factor 4 (IRF4) is critical for the differentiation of Th2 cells and may also play a role in the differentiation of Th17 cells (Lohoff et al., 2002; Brustle et al., 2007). Foxp3 is able to bind to the promoter region of IRF4 and enhance its expression (Zheng et al., 2009). Additionally, Treg-specific deletion of Irf4 prevents Tregs from efficiently suppressing Th2 immune responses leading to an uncontrolled Th2 immune response characterized by significantly enhanced IL-4 and IL-5 production, but not IFNγ, IL-2, or IL-17 (Zheng et al., 2009). Additionally, IRF4-deficient Tregs have decreased expression of the genes encoding the suppressive molecules IL-10 and Granzyme B, while also reducing CCR8 expression, the chemokine receptor important for migration to the sites of Th2 inflammation (Zheng et al., 2009).
Cretney et al. (2011) demonstrated a wider role for IRF4 expression with the finding that IRF4 may also be required for T-bet and Blimp-1 expression. Blimp-1 is a transcriptional repressor normally associated with plasma cell differentiation, but in Tregs it appears to be crucial to the expression of IL-10 (Cretney et al., 2011). This would suggest that IRF4 expression by Tregs may also be critical to IL-10-dependent effector mechanisms. As a result it seems likely that part of the reason for the failure of IRF4-deficient Tregs to regulate Th2 responses may be due to a lack of IL-10 production.
It has also become clear that the canonical Th2 transcription factor, GATA-3, may also expressed by Tregs (Campbell, 2011; Wang et al., 2011; Wohlfert et al., 2011). In GATA-3-deficient Tregs, expression of Foxp3 and constitutively expressed Treg markers/effector molecules such as CLTA-4, GITR, and CD25 were all reduced both at the mRNA and protein levels. GATA-3-deficient Tregs are unable to prevent the induction of systemic lymphoproliferative disease and increased production of Th1, Th2, and Th17 cytokines (Wang et al., 2011). Thus, unlike STAT3 and T-bet, whose expression in Tregs appears to be largely linked to the control of the T cell subtypes with which they are associated, it seems that GATA-3 has a wider function in the maintenance of immunity. This is perhaps not surprising as while GATA-3 is often primarily associated with Th2 cells, it is also expressed in a range of other cell types and is required for thymocyte development (Pai et al., 2003).
BCL-6 – T-Follicular
During a T-dependent immune response CD4+ T cells are able to express the transcription factor BCL-6 that converts them to a PD-1+ CXCR5+ T-follicular helper subtype (Tfh; Crotty, 2011). These cells then home to the follicle, due to the action of the chemokine receptor CXCR5 guided by CXCL13 produced by follicular stromal cells, and specialize in assisting B cell affinity maturation and memory cell formation within the germinal center (Crotty, 2011). Recent work has demonstrated that thymically derived Tregs are also able to effectively mimic this phenotype by induction of BCL-6 giving a CXCR5+ PD-1+ phenotype that home to the follicular region. These cells appear to differentiate from thymically produced Tregs in response to the same SLAM-associated protein (SAP), CD28, and B cell-dependent signals that induce Tfh differentiation (Chung et al., 2011; Linterman et al., 2011). This follicular Treg subset then travels to the germinal center and controls the germinal center reaction (Chung et al., 2011; Linterman et al., 2011; Wollenberg et al., 2011). At this time the primary suppression mechanisms used by follicular Tregs are not clear, although CTLA-4 protein levels and IL-10 mRNA expression is increased, while Granzyme B is reduced in comparison to normal Tregs (Linterman et al., 2011). Surprisingly, given their expression of BCL-6, normally a mutual repressor of Blimp-1, these cells also express high levels of prdm1 mRNA encoding the transcriptional repressor Blimp-1 (Chung et al., 2011; Linterman et al., 2011) crucial to the expression of IL-10.
STAT3 and RORγt – Th17
The transcription factor STAT3 is essential for the proper development of Th17 cells (Mathur et al., 2007). More recently a role for STAT3 in Treg biology has been demonstrated by the finding that STAT3-deficient Tregs are unable to prevent fatal Th17-mediated colitis (Chaudhry et al., 2009). Interestingly, these Tregs retain in vitro suppressive capacity, presumably due to intact CTLA-4 function, but have reduced expression of IL-10, IL-35, and CCR6 (Chaudhry et al., 2009). STAT3 phosphorylation by Tregs is induced in an IL-10-dependent manner with IL-10 receptor-deficient Tregs failing to phosphorylate STAT3 and restrain Th17 responses and the development of colitis (Chaudhry et al., 2011). Additionally, IL-10 receptor (IL10R) expression by Th17 cells is critical for prevention of their inflammatory pathology (Huber et al., 2011).
A number of groups have reported that Tregs may be able to simultaneously express Foxp3 and the Th17 defining transcription factor RORγt, and may produce IL-17 (Zhou et al., 2008; Voo et al., 2009; Hovhannisyan et al., 2011). In humans, Tregs have been observed to lose suppressive function while producing IL-17 but then regain suppressive function, suggesting that this could be a temporary loss of suppressive capability in order to allow the immune response sufficient time to effectively fight pathogens before suppressive function is re-established (Beriou et al., 2009). Interestingly, only CCR6-expressing Tregs expressed IL-17 (Beriou et al., 2009). This is of relevance since CCR6 controls Treg accumulation at the sites of Th17-mediated inflammation due to the IL-17-mediated release of its ligand CCL20 by epithelial cells (Hirota et al., 2007; Yamazaki et al., 2008).
Synthesis
While all Tregs can be characterized as Foxp3+ CTLA-4+ and lacking IL-2 production, it seems that from this base state a range of transcription factors may be induced/co-opted in order to allow the Treg to adapt to inflammatory cues. It seems likely that Tregs share much of the same molecular machinery as conventional CD4+ T cells and thus respond to the same stimuli to differentiate into Thχ-like Tregs.
In this sense there is a core module of Treg suppression driven by Foxp3 expression that acts to prevent the activation, proliferation, and as a result the differentiation, of T-effector cells. This core module may be augmented by induction of additional modules, driven by transcription factors normally associated with other T cell subsets, allowing Tregs to adapt to become “Thχ-like” Tregs and deliver suppression to diverse sites within the body (Figure 1).
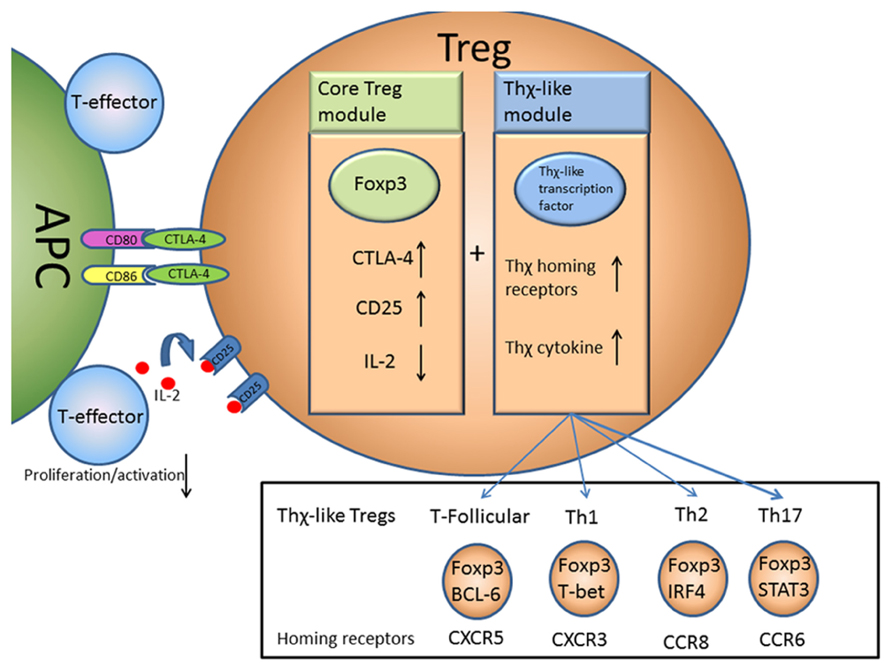
FIGURE 1. A two module model of Treg function. Foxp3 acts to control the core module of Treg suppressive function by regulating expression of a number of key molecules such as CTLA-4 and CD25. This allows Tregs to suppress T-effector activation and proliferation by suppressing APC function via CTLA-4 and possibly depriving IL-2 from other T cells. An additional Thχ-like module of suppressive function may also be induced that causes Tregs to express Thχ-like transcription factors allowing the Tregs to take on some of the properties of Th1, Th2, Th17, or follicular T cells, travel to the same sites and deliver in situ suppression.
While the possibility that some Tregs may lose Foxp3 expression and become proinflammatory “ex-Tregs” is highly controversial (Zhou et al., 2009; Rubtsov et al., 2010; Hori, 2011; Miyao et al., 2012), we should perhaps not be surprised to find some evidence of proinflammatory cytokine expression by Thχ-like Tregs that broadly retain suppressive function. While Foxp3 is a master regulator it does not totally repress the loci associated with Th1, Th2, and Th17 cytokines (Wei et al., 2009; Duhen et al., 2012). As a result it is not necessarily a contradiction to find Tregs-expressing proinflammatory cytokines and it will be interesting to see if in some settings there are benefits to Treg expression of proinflammatory cytokines. For example, Treg expression of IFNγ has been reported in several settings; during infection with Toxoplasma gondii IFNγ-producing Tregs retained their in vitro suppressive capacity (Oldenhove et al., 2009) while IL-12-induced IFNγ-producing Tregs retain both in vitro suppressive capacity and the ability to prevent colitis in an IFNγ-dependent manner (Feng et al., 2011). Treg produced IFNγ may also have a protective role in transplant tolerance. Alloreactive Tregs are able to prevent rejection of a BALB/c skin graft in C57/B6 mice, while Tregs from IFNγ-deficient mice were significantly less protective (Sawitzki et al., 2005).
Thχ-like Tregs appear to have an effector Treg phenotype, with clear roles for IL-10 in STAT-3 and IRF4 driven suppression, while T-bet and BCL-6-expressing Tregs also seem to have upregulated expression of IL-10. While IL-10 is not critical to the in vitro function of Tregs (Takahashi et al., 1998; Rubtsov et al., 2008), in vivo experiments have demonstrated IL-10 to be important in the maintenance of immune homeostasis in the gut, with IL-10-deficient mice spontaneously developing intestinal inflammation (Kuhn et al., 1993), while Treg-specific deletion of il10 leads to colitis and inflammation at the lung surface (Rubtsov et al., 2008), suggesting that its role may be to control excessive inflammation from microbial stimulation at mucosal surfaces rather than maintenance of self-tolerance. At this time it is not clear if the suppressive mechanisms employed by Thχ-like Tregs are different from previously defined effector Tregs, or if the differences are primarily due to cell localization. Additionally, it seems likely that, on close examination of their transcription factor expression, there may be substantial overlap between previously identified effector Tregs and Thχ-like Tregs. As it stands, no clear mechanisms of suppression that are truly specific to a particular Thχ-like Treg have been identified. As a result, it seems likely that these are specialized subsets of effector Tregs and the primary role of the induction of these transcription factors is to allow Tregs to respond to the same stimuli as conventional T cells, travel to the same sites, and enhance or upregulate existing effector Treg suppressive mechanisms such as CTLA-4 and IL-10.
Conclusion
While Tregs have a core module of suppression driven by Foxp3 expression, they are also able to adapt to changes in their environment and harness additional modules by the expression of transcription factors normally associated with other T cell subtypes in order to better control immunopathology. Particularly in the light of ongoing work examining either transfer or depletion of Tregs in clinical settings it is critical that we understand both the core functions of Tregs and their range of flexibility that allows them to modulate their mechanisms of suppression and cell localization in response to specific stimuli.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank associate professors T. Yamaguchi and N. Ohkura for helpful discussion and critical reading of the manuscript. James B. Wing is supported by Japan Society for Promotion of Science (JSPS) postdoctoral fellowship for foreign researcher (P10426); Shimon Sakaguchi is supported by Grant-in-Aid for Specially Promoted Research (No. 20002007).
References
Bachmann, M. F., Kohler, G., Ecabert, B., Mak, T. W., and Kopf, M. (1999). Cutting edge: lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J. Immunol. 163, 1128–1131.
Beriou, G., Costantino, C. M., Ashley, C. W., Yang, L., Kuchroo, V. K., Baecher-Allan, C., and Hafler, D. A. (2009). IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood 113, 4240–4249.
Bettelli, E., Sullivan, B., Szabo, S. J., Sobel, R. A., Glimcher, L. H., and Kuchroo, V. K. (2004). Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J. Exp. Med. 200, 79–87.
Brunkow, M. E., Jeffery, E. W., Hjerrild, K. A., Paeper, B., Clark, L. B., Yasayko, S. A., Wilkinson, J. E., Galas, D., Ziegler, S. F., and Ramsdell, F. (2001). Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 27, 68–73.
Brustle, A., Heink, S., Huber, M., Rosenplanter, C., Stadelmann, C., Yu, P., Arpaia, E., Mak, T. W., Kamradt, T., and Lohoff, M. (2007). The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat. Immunol. 8, 958–966.
Campbell, D. J., and Koch, M. A. (2011). Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat. Rev. Immunol. 11, 119–130.
Chaudhry, A., Rudra, D., Treuting, P., Samstein, R. M., Liang, Y., Kas, A., and Rudensky, A. Y. (2009). CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 326, 986–991.
Chaudhry, A., Samstein, R. M., Treuting, P., Liang, Y., Pils, M. C., Heinrich, J. M., Jack, R. S., Wunderlich, F. T., Bruning, J. C., Muller, W., and Rudensky, A. Y. (2011). Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34, 566–578.
Chung, Y., Tanaka, S., Chu, F., Nurieva, R. I., Martinez, G. J., Rawal, S., Wang, Y. H., Lim, H., Reynolds, J. M., Zhou, X. H., Fan, H. M., Liu, Z. M., Neelapu, S. S., and Dong, C. (2011). Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med. 17, 983–988.
Cretney, E., Xin, A., Shi, W., Minnich, M., Masson, F., Miasari, M., Belz, G. T., Smyth, G. K., Busslinger, M., Nutt, S. L., and Kallies, A. (2011). The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat. Immunol. 12, 304–311.
de la Rosa, M., Rutz, S., Dorninger, H., and Scheffold, A. (2004). Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur. J. Immunol. 34, 2480–2488.
Dejean, A. S., Beisner, D. R., Ch'en, I. L., Kerdiles, Y. M., Babour, A., Arden, K. C., Castrillon, D. H., Depinho, R. A., and Hedrick, S. M. (2009). Transcription factor Foxo3 controls the magnitude of T cell immune responses by modulating the function of dendritic cells. Nat. Immunol. 10, 504–513.
Duhen, T., Duhen, R., Lanzavecchia, A., Sallusto, F., and Campbell, D. J. (2012). Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood 119, 4430–4440.
Feng, T., Cao, A. T., Weaver, C. T., Elson, C. O., and Cong, Y. (2011). Interleukin-12 converts Foxp3+ regulatory T cells to interferon-gamma-producing Foxp3+ T cells that inhibit colitis. Gastroenterology 140, 2031–2043.
Fontenot, J. D., Gavin, M. A., and Rudensky, A. Y. (2003). Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4, 330–336.
Furtado, G. C., Curotto De Lafaille, M. A., Kutchukhidze, N., and Lafaille, J. J. (2002). Interleukin 2 signaling is required for CD4(+) regulatory T cell function. J. Exp. Med. 196, 851–857.
Grohmann, U., Orabona, C., Fallarino, F., Vacca, C., Calcinaro, F., Falorni, A., Candeloro, P., Belladonna, M. L., Bianchi, R., Fioretti, M. C., and Puccetti, P. (2002). CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 3, 1097–1101.
Hirota, K., Yoshitomi, H., Hashimoto, M., Maeda, S., Teradaira, S., Sugimoto, N., Yamaguchi, T., Nomura, T., Ito, H., Nakamura, T., Sakaguchi, N., and Sakaguchi, S. (2007). Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J. Exp. Med. 204, 2803–2812.
Hori, S. (2011). Regulatory T cell plasticity: beyond the controversies. Trends Immunol. 32, 295–300.
Hori, S., Nomura, T., and Sakaguchi, S. (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061.
Hovhannisyan, Z., Treatman, J., Littman, D. R., and Mayer, L. (2011). Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology 140, 957–965.
Huber, S., Gagliani, N., Esplugues, E., O'Connor, W. Jr., Huber, F. J., Chaudhry, A., Kamanaka, M., Kobayashi, Y., Booth, C. J., Rudensky, A. Y., Roncarolo, M. G., Battaglia, M., and Flavell, R. A. (2011). Th17 cells express interleukin-10 receptor and are controlled by Foxp3 and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 34, 554–565.
Khattri, R., Cox, T., Yasayko, S. A., and Ramsdell, F. (2003). An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4, 337–342.
Koch, M. A., Tucker-Heard, G., Perdue, N. R., Killebrew, J. R., Urdahl, K. B., and Campbell, D. J. (2009). The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 10, 595–602.
Kuhn, R., Lohler, J., Rennick, D., Rajewsky, K., and Muller, W. (1993). Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263–274.
Linterman, M. A., Pierson, W., Lee, S. K., Kallies, A., Kawamoto, S., Rayner, T. F., Srivastava, M., Divekar, D. P., Beaton, L., Hogan, J. J., Fagarasan, S., Liston, A., Smith, K. G., and Vinuesa, C. G. (2011). Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 17, 975–982.
Lohoff, M., Mittrucker, H. W., Prechtl, S., Bischof, S., Sommer, F., Kock, S., Ferrick, D. A., Duncan, G. S., Gessner, A., and Mak, T. W. (2002). Dysregulated T helper cell differentiation in the absence of interferon regulatory factor 4. Proc. Natl. Acad. Sci. U.S.A. 99, 11808–11812.
Mathur, A. N., Chang, H. C., Zisoulis, D. G., Stritesky, G. L., Yu, Q., O'Malley, J. T., Kapur, R., Levy, D. E., Kansas, G. S., and Kaplan, M. H. (2007). Stat3 and Stat4 direct development of IL-17-secreting Th cells. J. Immunol. 178, 4901–4907.
Miyao, T., Floess, S., Setoguchi, R., Luche, H., Fehling, H. J., Waldmann, H., Huehn, J., and Hori, S. (2012). Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity 36, 262–275.
Oldenhove, G., Bouladoux, N., Wohlfert, E. A., Hall, J. A., Chou, D., Dos Santos, L., O'brien, S., Blank, R., Lamb, E., Natarajan, S., Kastenmayer, R., Hunter, C., Grigg, M. E., and Belkaid, Y. (2009). Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity 31, 772–786.
Pai, S. Y., Truitt, M. L., Ting, C. N., Leiden, J. M., Glimcher, L. H., and Ho, I. C. (2003). Critical roles for transcription factor GATA-3 in thymocyte development. Immunity 19, 863–875.
Pandiyan, P., Zheng, L., Ishihara, S., Reed, J., and Lenardo, M. J. (2007). CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 8, 1353–1362.
Qureshi, O. S., Zheng, Y., Nakamura, K., Attridge, K., Manzotti, C., Schmidt, E. M., Baker, J., Jeffery, L. E., Kaur, S., Briggs, Z., Hou, T. Z., Futter, C. E., Anderson, G., Walker, L. S., and Sansom, D. M. (2011). Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600–603.
Rubtsov, Y. P., Niec, R. E., Josefowicz, S., Li, L., Darce, J., Mathis, D., Benoist, C., and Rudensky, A. Y. (2010). Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671.
Rubtsov, Y. P., Rasmussen, J. P., Chi, E. Y., Fontenot, J., Castelli, L., Ye, X., Treuting, P., Siewe, L., Roers, A., Henderson, W. R. Jr., Muller, W., and Rudensky, A. Y. (2008). Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558.
Rudensky, A. Y., and Campbell, D. J. (2006). In vivo sites and cellular mechanisms of T reg cell-mediated suppression. J. Exp. Med. 203, 489–492.
Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M., and Toda, M. (1995). Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155, 1151–1164.
Sakaguchi, S., Wing, K., and Miyara, M. (2007). Regulatory T cells – a brief history and perspective. Eur. J. Immunol. 37, S116–S123.
Sakaguchi, S., Wing, K., Onishi, Y., Prieto-Martin, P., and Yamaguchi, T. (2009). Regulatory T cells: how do they suppress immune responses? Int. Immunol. 21, 1105–1111.
Sakaguchi, S., Yamaguchi, T., Nomura, T., and Ono, M. (2008). Regulatory T cells and immune tolerance. Cell 133, 775–787.
Sawitzki, B., Kingsley, C. I., Oliveira, V., Karim, M., Herber, M., and Wood, K. J. (2005). IFN-gamma production by alloantigen-reactive regulatory T cells is important for their regulatory function in vivo. J. Exp. Med. 201, 1925–1935.
Schorle, H., Holtschke, T., Hunig, T., Schimpl, A., and Horak, I. (1991). Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature 352, 621–624.
Setoguchi, R., Hori, S., Takahashi, T., and Sakaguchi, S. (2005). Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 201, 723–735.
Shevach, E. M. (2009). Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 30, 636–645.
Suzuki, H., Kundig, T. M., Furlonger, C., Wakeham, A., Timms, E., Matsuyama, T., Schmits, R., Simard, J. J., Ohashi, P. S., Griesser, H., Taniguchi, T., Paige, C. J., and Mak, T. W. (1995). Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science 268, 1472–1476.
Takahashi, T., Kuniyasu, Y., Toda, M., Sakaguchi, N., Itoh, M., Iwata, M., Shimizu, J., and Sakaguchi, S. (1998). Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 10, 1969–1980.
Thornton, A. M., and Shevach, E. M. (1998). CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med.188, 287–296.
Voo, K. S., Wang, Y. H., Santori, F. R., Boggiano, C., Arima, K., Bover, L., Hanabuchi, S., Khalili, J., Marinova, E., Zheng, B., Littman, D. R., and Liu, Y. J. (2009). Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc. Natl. Acad. Sci. U.S.A. 106, 4793–4798.
Wang, Y., Su, M. A., and Wan, Y. Y. (2011). An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity 35, 337–348.
Waterhouse, P., Penninger, J. M., Timms, E., Wakeham, A., Shahinian, A., Lee, K. P., Thompson, C. B., Griesser, H., and Mak, T. W. (1995). Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270, 985–988.
Wei, G., Wei, L., Zhu, J., Zang, C., Hu-Li, J., Yao, Z., Cui, K., Kanno, Y., Roh, T. Y., Watford, W. T., Schones, D. E., Peng, W., Sun, H. W., Paul, W. E., O'Shea, J. J., and Zhao, K. (2009). Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30, 155–167.
Wildin, R. S., Ramsdell, F., Peake, J., Faravelli, F., Casanova, J. L., Buist, N., Levy-Lahad, E., Mazzella, M., Goulet, O., Perroni, L., Bricarelli, F. D., Byrne, G., Mceuen, M., Proll, S., Appleby, M., and Brunkow, M. E. (2001). X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 27, 18–20.
Willerford, D. M., Chen, J., Ferry, J. A., Davidson, L., Ma, A., and Alt, F. W. (1995). Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity 3, 521–530.
Wing, K., Onishi, Y., Prieto-Martin, P., Yamaguchi, T., Miyara, M., Fehervari, Z., Nomura, T., and Sakaguchi, S. (2008). CTLA-4 control over Foxp3(+) regulatory T cell function. Science 322, 271–275.
Wing, K., Yamaguchi, T., and Sakaguchi, S. (2011). Cell-autonomous and -non-autonomous roles of CTLA-4 in immune regulation. Trends Immunol. 32, 428–433.
Wohlfert, E. A., Grainger, J. R., Bouladoux, N., Konkel, J. E., Oldenhove, G., Ribeiro, C. H., Hall, J. A., Yagi, R., Naik, S., Bhairavabhotla, R., Paul, W. E., Bosselut, R., Wei, G., Zhao, K., Oukka, M., Zhu, J., and Belkaid, Y. (2011). GATA3 controls Foxp3 regulatory T cell fate during inflammation in mice. J. Clin. Invest. 121, 4503–4515.
Wollenberg, I., Agua-Doce, A., Hernandez, A., Almeida, C., Oliveira, V. G., Faro, J., and Graca, L. (2011). Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J. Immunol. 187, 4553–4560.
Yamaguchi, T., Wing, J. B., and Sakaguchi, S. (2011). Two modes of immune suppression by Foxp3(+) regulatory T cells under inflammatory or non-inflammatory conditions. Semin. Immunol. 23, 424–430.
Yamazaki, T., Yang, X. O., Chung, Y., Fukunaga, A., Nurieva, R., Pappu, B., Martin-Orozco, N., Kang, H. S., Ma, L., Panopoulos, A. D., Craig, S., Watowich, S. S., Jetten, A. M., Tian, Q., and Dong, C. (2008). CCR6 regulates the migration of inflammatory and regulatory T cells. J. Immunol. 181, 8391–8401.
Zheng, Y., Chaudhry, A., Kas, A., Deroos, P., Kim, J. M., Chu, T. T., Corcoran, L., Treuting, P., Klein, U., and Rudensky, A. Y. (2009). Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 458, 351–356.
Zhou, L., Lopes, J. E., Chong, M. M., Ivanov, Ii, Min, R., Victora, G. D., Shen, Y., Du, J., Rubtsov, Y. P., Rudensky, A. Y., Ziegler, S. F., and Littman, D. R. (2008). TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 453, 236–240.
Keywords: Tregs, suppression, transcription factors, adaptability, CTLA-4
Citation: Wing JB and Sakaguchi S (2012) Multiple Treg suppressive modules and their adaptability. Front. Immun. 3:178. doi: 10.3389/fimmu.2012.00178
Received: 13 April 2012; Paper pending published: 29 April 2012;
Accepted: 11 June 2012; Published online: 29 June 2012.
Edited by:
Kendall A. Smith, Weill Medical College of Cornell University, USAReviewed by:
Kendall A. Smith, Weill Medical College of Cornell University, USAWolfgang Kastenmuller, National Institutes of Health, USA
Georg Gasteiger, Memorial Sloan-Kettering Cancer Center, USA
Copyright: © 2012 Wing and Sakaguchi. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Shimon Sakaguchi, Department of Experimental Immunology, WPI Immunology Frontier Research Center, Osaka University, 3-1 Yamadaoka, Suita, Osaka 565-0871, Japan. e-mail:c2hpbW9uQGlmcmVjLm9zYWthLXUuYWMuanA=