Abstract
Genetic defects in the adenosine deaminase (ADA) gene are among the most common causes for severe combined immunodeficiency (SCID). ADA-SCID patients suffer from lymphopenia, severely impaired cellular and humoral immunity, failure to thrive, and recurrent infections. Currently available therapeutic options for this otherwise fatal disorder include bone marrow transplantation (BMT), enzyme replacement therapy with bovine ADA (PEG-ADA), or hematopoietic stem cell gene therapy (HSC-GT). Although varying degrees of immune reconstitution can be achieved by these treatments, breakdown of tolerance is a major concern in ADA-SCID. Immune dysregulation such as autoimmune hypothyroidism, diabetes mellitus, hemolytic anemia, and immune thrombocytopenia are frequently observed in milder forms of the disease. However, several reports document similar complications also in patients on long-term PEG-ADA and after BMT or GT treatment. A skewed repertoire and decreased immune functions have been implicated in autoimmunity observed in certain B-cell and/or T-cell immunodeficiencies, but it remains unclear to what extent specific mechanisms of tolerance are affected in ADA deficiency. Herein we provide an overview about ADA-SCID and the autoimmune manifestations reported in these patients before and after treatment. We also assess the value of the ADA-deficient mouse model as a useful tool to study both immune and metabolic disease mechanisms. With focus on regulatory T- and B-cells we discuss the lymphocyte subpopulations particularly prone to contribute to the loss of self-tolerance and onset of autoimmunity in ADA deficiency. Moreover we address which aspects of immune dysregulation are specifically related to alterations in purine metabolism caused by the lack of ADA and the subsequent accumulation of metabolites with immunomodulatory properties.
The ADA Metabolism
The ADA enzyme
As an enzyme of the purine salvage pathway, adenosine deaminase (ADA) catalyzes the deamination of adenosine and 2′-deoxyadenosine, as well as several naturally occurring methylated adenosine compounds (Hirschhorn and Ratech, 1980; Ratech et al., 1989). The deamination of adenosine and 2′-deoxyadenosine gives rise to inosine and deoxyinosine, respectively (Hirschhorn and Candotti, 2006). Further conversion of these deaminated nucleosides leads to hypoxanthine, which can be either transformed irreversibly into uric acid or salvaged into mononucleosides (Figure 1).
Figure 1
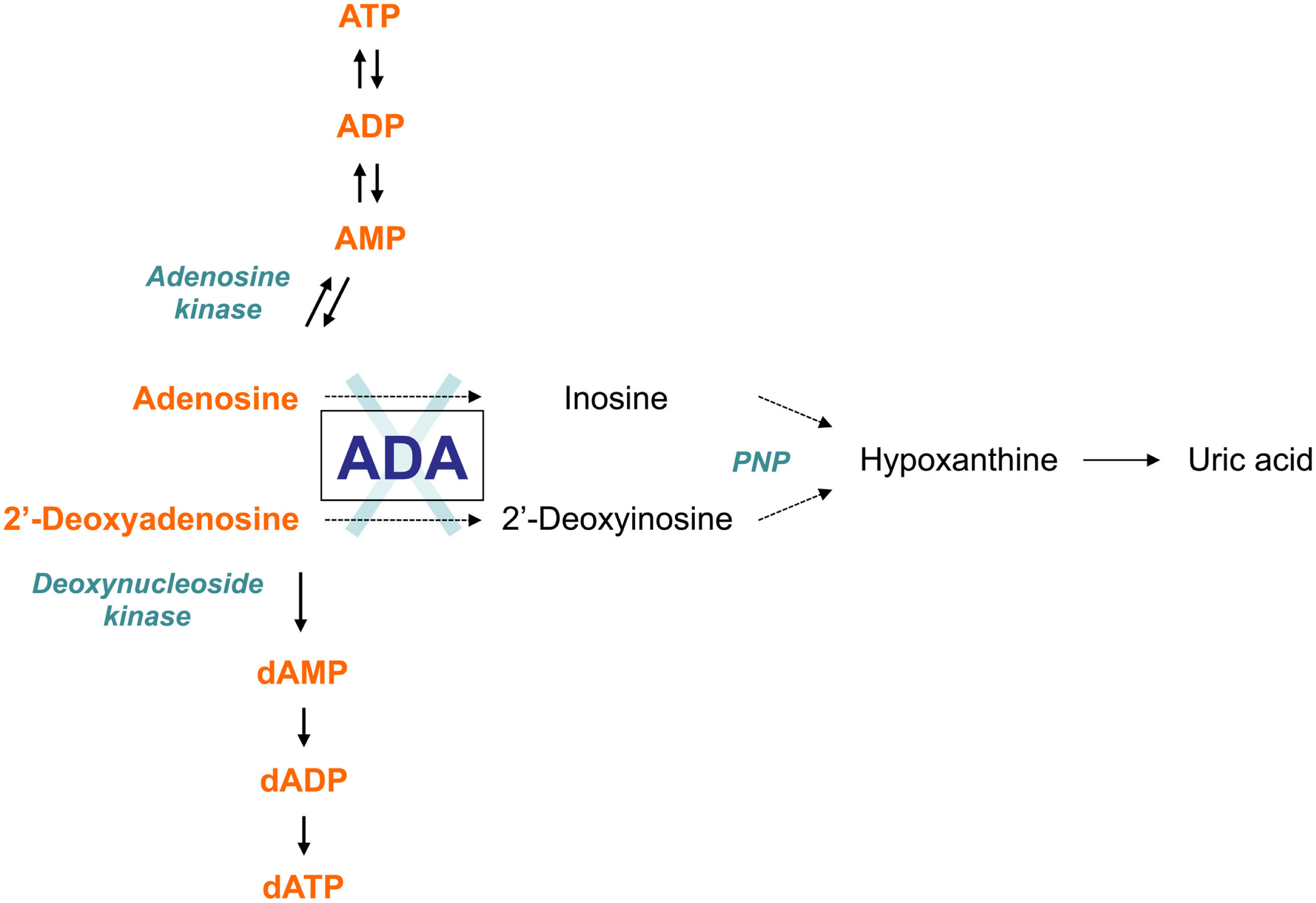
The adenosine deaminase (ADA) metabolism. ADA is an enzyme of the purine salvage pathway, which catalyzes the irreversible deamination of adenosine and 2′-deoxyadenosine into inosine and 2′-deoxyinosine, respectively. Most adenosine derives from endogenous breakdown of ATP and degradation of RNA, or is taken up exogenously by ubiquitously expressed nucleoside transporters. Unlike adenosine, 2′-deoxyadenosine is formed by DNA degradation is predominantly catabolized by ADA. Further conversion of inosine nucleoside leads to hypoxanthine, which can either enter a non-reversible pathway to uric acid or salvaged back into other mononucleosides. In the absence of ADA, the presence of these alternative “bypass” pathways results in normal concentrations of the catabolic products of the enzyme reaction in patients with ADA-SCID. Conversely, the levels of ADA substrates, adenosine and 2′-deoxyadenosine, are not only found in increased amounts in extracellular body fluids, but they also “spill over” into additional pathways normally only minimally utilized, thus contributing to the pathogenic mechanisms of the disease.
Although ADA is present in all cell types, its enzyme activity differs considerably among tissues. The highest amounts in humans are found in lymphoid tissues, particularly the thymus, the brain, and gastrointestinal tract. The ADA enzyme is ubiquitously expressed both intracellularly and on the cell surface where it complexes with two molecules of CD26 as a combined protein (Kameoka et al., 1993).
The ADA substrates adenosine and 2′-deoxyadenosine
2′-Deoxyadenosine is a component of DNA and primarily derives from its breakdown. Therefore, 2′-deoxyadenosine concentration is expected to be highest at sites of cell death, such as the bone marrow and thymus, where lymphocytes undergo apoptotic death during differentiation and selection. 2′-Deoxyadenosine behaves as a cytotoxic metabolite and is generally considered the primary cause of lymphotoxicity in ADA-severe combined immunodeficiency (SCID; Hirschhorn and Candotti, 2006). The most striking metabolic alteration in ADA deficiency is the accumulation of massive amounts of dATP in erythrocytes and lymphocytes (Hirschhorn et al., 1992). This results from uptake of increasing 2′-deoxyadenosine present in surrounding body fluids with subsequent intracellular phosphorylation and trapping.
Adenosine on the other hand is a component of adenine nucleotides including ATP and RNA (Hirschhorn and Candotti, 2006). Elevated adenosine levels, as occurring in ADA deficiency contribute to apoptosis and block in the differentiation of thymocytes, causing severe T lymphopenia in mice and humans (Apasov et al., 2001; Gaspar et al., 2009; Poliani et al., 2009). Moreover adenosine, acting through cell surface G protein-coupled receptors, functions as an extracellular signal transducer in a variety of physiological processes (Olah and Stiles, 1995). Apart from T-cell receptor signaling (Huang et al., 1997), adenosine is involved in the control of heart rate and blood pressure (Fukunaga et al., 1982; Belardinelli et al., 1989), renal function (Churchill and Bidani, 1982), inflammatory responses (Blackburn, 2003), and in neurotransmission (Fredholm and Dunwiddie, 1988).
ADA-SCID
Adenosine deaminase deficiency is the second-most prevalent form (approximately 20%) of SCID. The overall incidence in Europe is estimated to range between 1:375,000 and 1:660,000 live births. ADA-deficient patients suffer from lymphopenia, severely impaired cellular and humoral immune function, failure to thrive, and a rapidly fatal course due to infection (Hirschhorn and Candotti, 2006). Moreover, autoimmune manifestations are commonly observed in milder forms of the disease. Currently available therapeutic options include bone marrow transplantation (BMT), enzyme replacement therapy with bovine ADA (PEG-ADA), or hematopoietic stem cell gene therapy (HSC-GT).
Immune defects
Lymphopenia and attrition of immune function over time are the two findings common to all presentations of ADA deficiency. It is associated with thymic hypoplasia and a severe depletion of all three major categories of lymphocytes, T-, B-, and NK-cells (Buckley et al., 1997). Absence of cellular and humoral immunity and a rapidly fatal course due to infections with fungal, viral, and opportunistic agents are characteristic of early onset forms of ADA deficiency (Giblett et al., 1972; Buckley et al., 1997). Total immunoglobulin levels may be only slightly depressed at birth due to the maternal contribution of IgG, whereas both IgM and IgA, which ordinarily do not cross the placental barrier, are often absent. However, once IgG levels decline as maternal antibodies are cleared, a pronounced hypogammaglobulinemia signals the absence of humoral immunity (Morgan et al., 1987; Hirschhorn and Candotti, 2006). About 20% of ADA-SCID cases occur later in childhood (delayed) or beyond (late/adult onset). Delayed or late-onset patients have significant immunodeficiency, but variable clinical manifestations (Ozsahin et al., 1997). These forms show progressive immunological and clinical deterioration, often associated with autoimmune manifestations, including hemolytic anemia, and immune thrombocytopenia (Parkman et al., 1975; Aiuti et al., 2003). Serum immunoglobulin levels are altered in late-onset patients, with IgG2 levels being highly reduced or absent. IgE levels are elevated and often associated to eczema and asthma. An inability to produce antibodies against polysaccharide and pneumococcal antigens was frequently found in ADA-SCID patients with milder forms of the disease (Levy et al., 1988).
Non-immune defects
The initial and most devastating presentation of ADA-SCID is due to the immune defects (Gaspar et al., 2009). Nonetheless, several non-immune abnormalities have been described in ADA deficiency, indicating that this disease should be considered a systemic metabolic disorder (Aiuti et al., 2003; Hirschhorn and Candotti, 2006). ADA is ubiquitously expressed in all cell types; when absent, the systemic metabolic toxicity is frequently associated with organ damage (Sauer and Aiuti, 2009). These include hepatic and renal disease (Bollinger et al., 1996), skeletal alterations (Sauer et al., 2009), neurological abnormalities (Honig et al., 2007; Titman et al., 2008), and behavioral impairments (Rogers et al., 2001). Because complications from infections usually predominate in the clinical presentation of infants with ADA deficiency, the full spectrum of non-immunologic manifestations and their natural course may be obscured (Honig et al., 2007). It is important to note, that several abnormalities have been described in few patients only, and might reflect effects of infectious agents rather than primary defects due to ADA deficiency: i.e., renal and adrenal abnormalities, phyloric stenosis, and hepatic disease (Hirschhorn and Candotti, 2006).
Therapies for ADA Deficiency
Bone marrow transplantation with allogeneic HSC has long been considered the mainstay of ADA-SCID treatment. However, unlike other SCID forms, two other treatment options are available for ADA-SCID: enzyme replacement therapy with pegylated bovine ADA (PEG-ADA) and autologous HSC-GT (Hershfield et al., 1987; Aiuti et al., 2009). The availability of different treatment modalities presents an opportunity for improved patient care but also difficulties in deciding upon the specific choice of treatment for individual patients (Figure 2). Making the correct choice is further complicated by the fact that ADA deficiency is not purely an immune defect, and that the systemic manifestations, which can be of major clinical consequence, must also be managed (Gaspar et al., 2009).
Figure 2
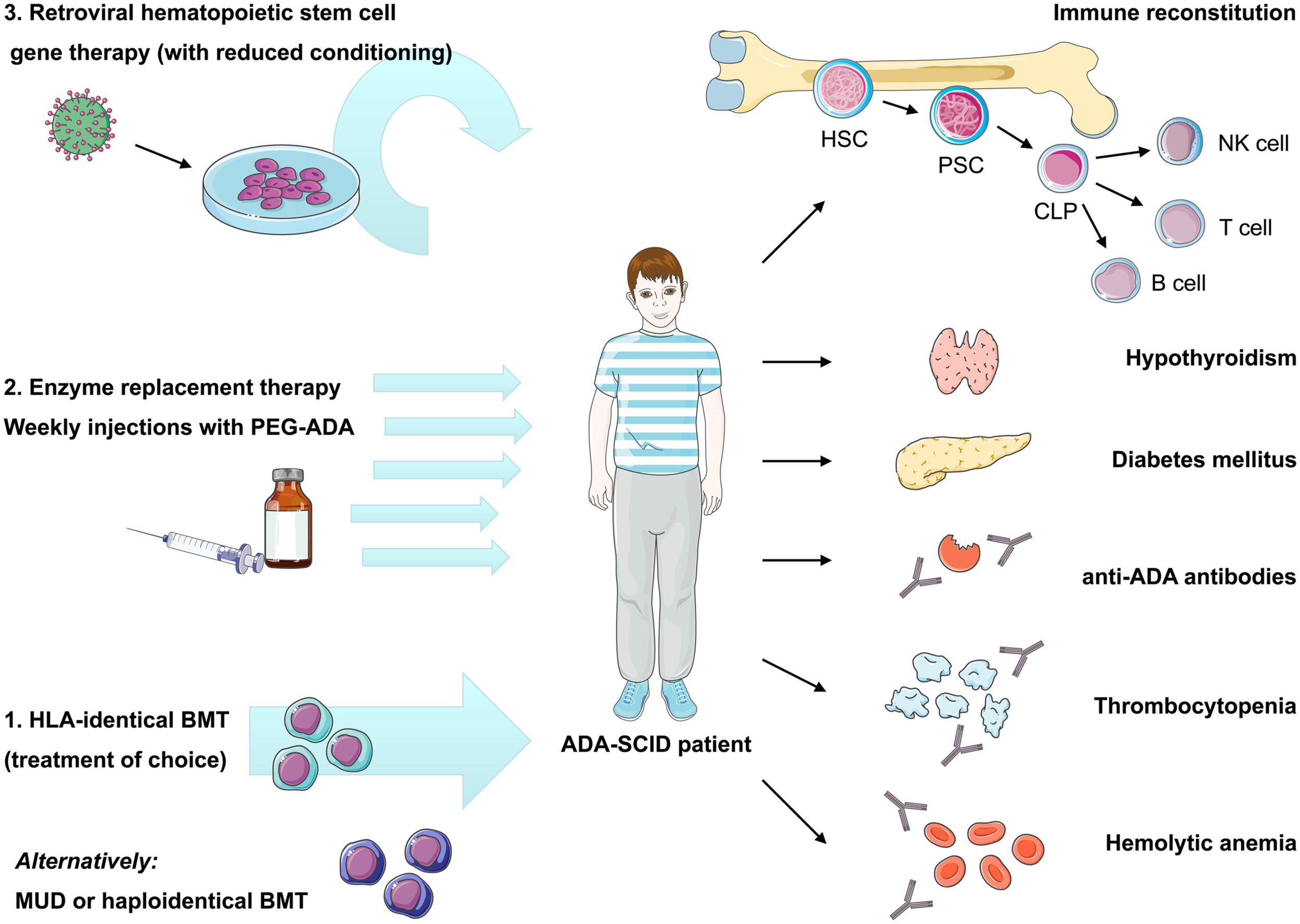
Current therapeutic options in ADA-SCID and reported autoimmune manifestations after treatment. Immune reconstitution in ADA deficiency can be achieved by bone marrow transplantation, enzyme replacement, or gene therapy, nonetheless recovery of immune functions may vary depending on the applied treatment and patient’s characteristics. Treatment of choice remains bone marrow transplantation from an HLA-identical sibling donor, while transplants from alternative donors are associated with high morbidity and mortality. Enzyme replacement therapy using pegylated bovine ADA is a non-curative treatment requiring weekly intramuscular injections with PEG-ADA. ADA-SCID has been the pioneer disease for the development of human gene therapy. It is based on the reinfusion of autologous HSC transduced with a retroviral vector containing the ADA cDNA. Variable degrees of immune reconstitution can be achieved by these treatments, but onset of autoimmunity is of concern in post-treatment ADA-SCID patients. Reported autoimmune manifestations include: autoimmune hypothyroidism, diabetes mellitus, thrombocytopenia, hemolytic anemia, and development of anti-ADA antibodies. HLA, human leukocyte antigen; BMT, bone marrow transplantation; MUD, matched unrelated donor; PEG-ADA, pegylated bovine ADA; HSC, hematopoietic stem cell; PSC, pluripotent stem cell; CLP, committed lymphocyte precursor; NK, natural killer cell.
Hematopoietic stem cell transplantation
Hematopoietic stem cell-transplantation (BMT) from allogeneic human leukocyte antigen (HLA)-compatible sibling donors resulting in long-term survival and effective immune reconstitution is the treatment of choice for patients with ADA-SCID and other severe variants of primary immunodeficiencies. Since less than 20% of ADA-SCID patients have access to HLA-matched family donors, transplants are often performed from mismatched family or matched unrelated donors (Antoine et al., 2003; Gaspar et al., 2009; Ferrua et al., 2010). A recent retrospective analysis on the specific outcome of transplants for ADA-SCID collected data from several multicenter studies and analyzed the survival of 106 patients who received a total of 119 transplants (Hassan et al., 2012). BMT from matched sibling and family donors had a significantly better overall survival (86 and 81%) in comparison to BMT from matched unrelated (66%) and haploidentical donors (43%). Indicating that despite recent progress in transplantation, the use of alternative donors is still associated with a reduced overall survival (Gaspar et al., 2009). This is further complicated by the fact that ADA-SCID patients are more difficult to transplant especially from unrelated and haploidentical donors possibly due to their need for conditioning and the underlying metabolic nature of the disease (Gaspar et al., 2009; Sauer et al., 2009). While superior survival was seen in patients who received unconditioned transplants in comparison to myeloablative procedures (81 and 54%), non-engraftment was a major problem after unconditioned haploidentical transplants (Hassan et al., 2012).
Long-term immune recovery showed that regardless of transplant type, overall T-cell numbers were similar although a faster rate of T-cell recovery was observed following matched sibling or matched unrelated BMT. Humoral immunity and donor B cell engraftment was achieved in nearly all evaluable surviving patients and most patients were able to discontinue immunoglobulin replacement, suggesting that immune recovery is relatively complete (Hassan et al., 2012). According to the available data, the immunological and metabolic recovery after transplant is well maintained even after 10 years or longer in some patients (Gaspar et al., 2009).
Nevertheless delayed or suboptimal immune reconstitution as a result of poor early engraftment or gradual decline in immune functions is observed in a significant fraction of surviving patients (Gaspar et al., 2009). Complications such as graft-versus-host disease, autoimmune and inflammatory manifestations, persistent infections, and disease-related issues have been described (Honig et al., 2007; Titman et al., 2008; Mazzolari et al., 2009).
In summary, the results obtained with transplantation from HLA-identical siblings or family donors indicate superior donor/host compatibility and outcome both in terms of survival and sustained immune recovery. Whereas the current evidence suggests that haploidentical donor transplants (performed with or without conditioning) have a poor chance of success and are therefore only undertaken if no other treatment options are available (Gaspar et al., 2009).
Enzyme replacement therapy with PEG-ADA
Enzyme replacement therapy with PEG-ADA was developed as lifesaving, not curative treatment for patients lacking an HLA-compatible donor. Attachment of PEG through lysine residues confers several therapeutically beneficial properties to ADA (Abuchowski et al., 1977; Davis et al., 1981). This chemical modification of the bovine enzyme reduces its immunogenicity and prevents its degradation by plasmatic proteases as well as the binding of neutralizing antibodies (Abuchowski et al., 1977; Davis et al., 1981). Thereby the circulating life of the compound is prolonged from minutes to days as clearance from the circulation is inhibited (Booth and Gaspar, 2009). Cellular uptake of PEG-ADA is insignificant and its distribution is limited to the plasma. Enzymatically active ADA continuously circulates and eliminates accumulating adenosine and 2′-deoxyadenosine metabolites (Chan et al., 2005). The principle of exogenous PEG-ADA administration is based on the direct conversion of accumulating ADA substrates in the plasma and the indirect reduction of intracellular toxic metabolites by diffusion.
To date more than 150 patients worldwide have received this treatment (Booth and Gaspar, 2009; Gaspar et al., 2009). PEG-ADA is usually administered weekly or bi-weekly by intramuscular injections throughout life. In general, PEG-ADA treatment seems to be well tolerated, with clinical benefits appreciable after the first month of therapy (Figure 3). Studies have shown that upon the initiation of PEG-ADA therapy, the absolute numbers of circulating T- and B-lymphocytes and NK-cells increase and protective immune function develops (Weinberg et al., 1993). Although only limited information is available, some analysis indicated that about half of PEG-ADA treated patients discontinued IVIg (Gaspar et al., 2009), whereas long-term follow-up suggests that immune recovery is often incomplete (Booth and Gaspar, 2009). Two retrospective studies showed that despite initial improvements, the lymphocyte counts of all PEG-ADA treated patients were below the normal range at all times. A gradual decline of mitogenic proliferative responses occurred after a few years of treatment and normal antigenic responses occurred less than expected (Kohn, 2008; Serana et al., 2010). No toxic or hypersensitivity reactions have been reported with PEG-ADA administration. However, several other side effects have been reported including manifestations of immune dysregulations including autoimmunity (type I diabetes, hypothyroidism, immune thrombocytopenia, hemolytic anemia) and allergic manifestations (Notarangelo et al., 1992; Ozsahin et al., 1997). An additional concern with PEG-ADA beyond about 8–10 years is the emergence of serious complications, including lymphoid and hepatic malignancies, and progression of chronic pulmonary insufficiency (Gaspar et al., 2009).
Figure 3
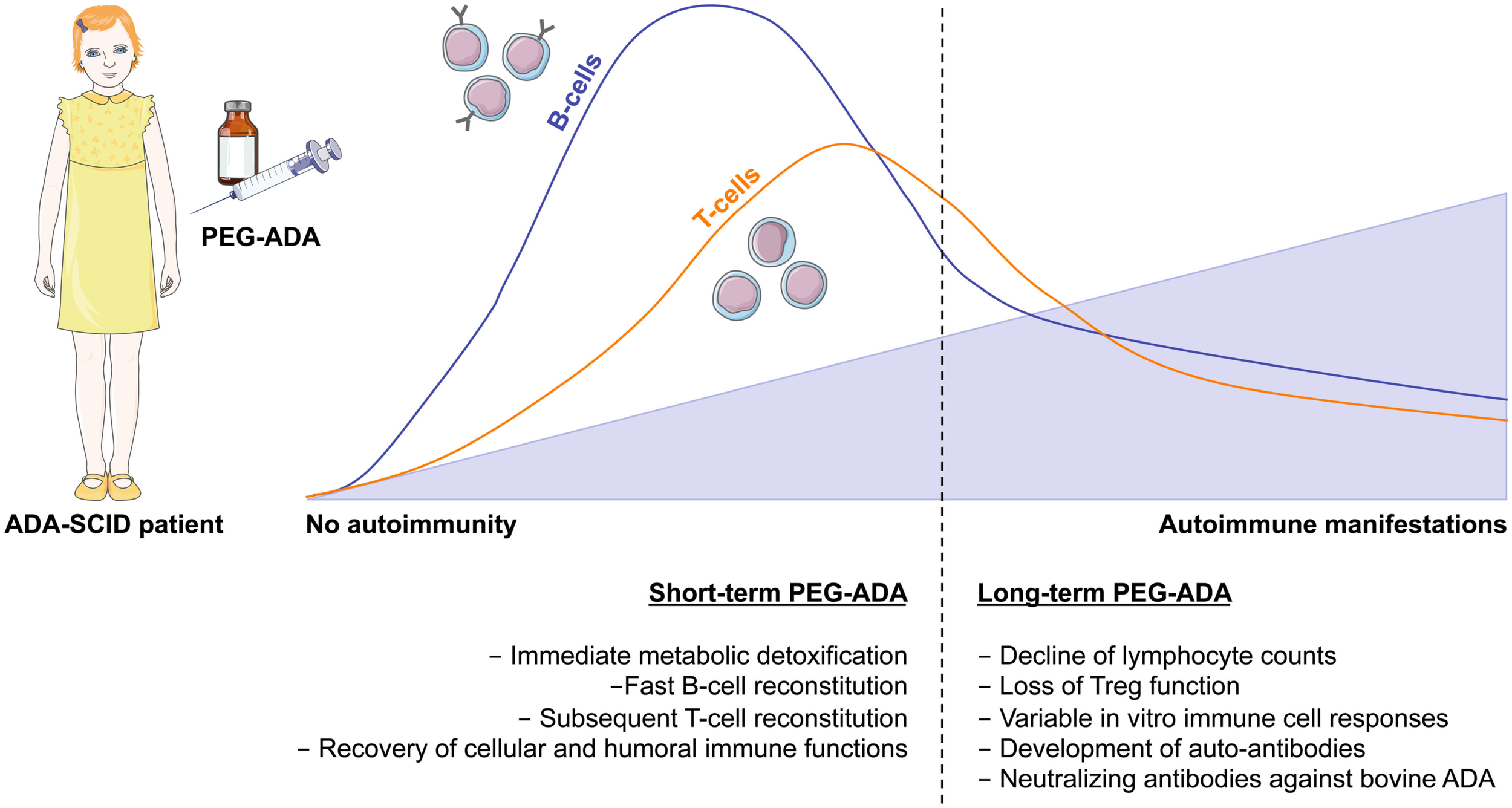
Immune reconstitution and development of autoimmunity after PEG-ADA treatment. Enzyme replacement therapy with pegylated bovine ADA is a lifesaving but non-curative treatment for ADA-SCID patients. It provides metabolic detoxification and protective immune function with patients remaining clinically well, but immune reconstitution is often suboptimal and may not be long-lived. Shortly after initiation of PEG-ADA treatment, patients show recovery of B-cell counts, followed by a gradual increase in T-cell numbers and reconstitution of immune cell functions. However, the long-term consequences of PEG-ADA treatment are unknown. Immune recovery in B and T- cells is below normal levels. Major concerns are the susceptibility to opportunistic infections and the development of autoimmunity due to lymphopenia with gradual decline of immune functions and perturbation of T- and B-cell tolerance.
The main side effect associated with the use of PEG-ADA is the development of anti-ADA antibody. The development of specific IgG antibody to bovine peptide epitopes of PEG-ADA has been reported by several groups and often coincides with an improvement in humoral immunity (Chaffee et al., 1992; Lainka et al., 2005; Booth et al., 2007). In about 10% of treated patients, inhibitory antibodies lead to the enhanced clearance of PEG-ADA with subsequent decline in metabolic parameters and immune function (Chaffee et al., 1992; Hershfield, 1995; Lainka et al., 2005).
Gene therapy
Hematopoietic stem cell gene therapy is a promising therapeutic option for genetic disorders of the immune system (Bordignon and Roncarolo, 2002; Fischer et al., 2005). ADA-deficient SCID has been under intensive preclinical and clinical investigation and nowadays represents a paradigmatic model of gene therapy for inherited disorders (Aiuti et al., 2003, 2007). The strong rationale for somatic gene therapy and the need for alternative treatments led to the design of clinical trials based on retroviral-mediated gene transfer of the normal ADA gene into autologous HSCs (Aiuti, 2004). Replication-deficient, recombinant retroviruses derived from the backbone of Moloney murine leukemia virus (MLV) were selected for these trials because of the available long-term experience and their ability to efficiently insert the therapeutic gene into the genome of dividing hematopoietic cells.
Since 2000, 37 patients have been treated in Italy, UK, and USA, achieving substantial clinical benefit in the majority of them. All patients received reduced intensity conditioning and PEG-ADA was discontinued to exploit the selective growth advantage for gene corrected over defective cells. At present, all patients are alive and in 26 patients PEG-ADA is no longer required (Aiuti et al., 2009; Gaspar et al., 2011; Montiel-Equihua et al., 2012). Gene therapy resulted in sustained engraftment of transduced cells, increased lymphocyte counts, improvement of cellular and humoral responses, and effective metabolic detoxification (Aiuti et al., 2009; Gaspar et al., 2011). Gene corrected cells were detected in myeloid and lymphoid subsets, the latter being more represented due to their survival advantage (Aiuti et al., 2009; Gaspar et al., 2011). In the HSR-TIGET study, all children maintained stable engraftment of vector ADA-transduced CD34+ cells with sustained systemic detoxification (Aiuti et al., 2009). At present, 15 of the 18 treated children do not require enzyme replacement therapy, with the longest follow-up at 11 years after treatment (Aiuti et al., 2009; Ferrua et al., 2010). These findings demonstrated the clinical efficacy of ADA gene transfer in restoring normal immune function and metabolic functions of ADA-SCID patients.
Unlike trials with gammaretroviral vectors in other diseases like X-linked SCID (Hacein-Bey-Abina et al., 2008; Howe et al., 2008), chronic granulomatous disease (Ott et al., 2006) and Wiskott–Aldrich Syndrome (Trobridge, 2011), the cumulative experience of these studies for ADA-SCID (Aiuti et al., 2009; Ferrua et al., 2010; Montiel-Equihua et al., 2012) did not reveal leukemic or oncogenic events, indicating that ADA-SCID gene therapy has a favorable risk/benefit profile. Unique risk factors may have contributed to the differential outcome of the other trials, such as vector constructs or promoters, inappropriate expression of transgenes involved in cell signaling (Kohn, 2008), cooperation between transgene and cellular oncogenes (Dave et al., 2009), or the disease background itself (Shou et al., 2006).
Autoimmunity in ADA-SCID
Immunodeficiency and autoimmune phenomena may occur concomitantly in the same individual (Etzioni, 2003). Immune dysregulation, which often manifests as multiple forms of autoimmunity, can affect both the adaptive and innate immune system, indicating that all these immune components are required for the appropriate development of tolerance in humans (Cunningham-Rundles, 2011). Since varying degrees of immune reconstitution can be achieved by the available treatment options for ADA-SCID, breakdown of tolerance and development of autoimmunity can represent a major concern. Autoimmune dysregulation are frequently observed in patients with milder forms of the disease or late-onset patients. They may manifest as autoimmune hypothyroidism, diabetes mellitus, hemolytic anemia, and immune thrombocytopenia (Notarangelo et al., 1992; Ozsahin et al., 1997; Figure 2).
Similar complications, such as autoimmune hemolytic anemia and autoimmune thyroiditis, have also been reported in at least nine patients after long-term PEG-ADA treatment (Ratech et al., 1989; Notarangelo et al., 1992; Ozsahin et al., 1997; Gaspar et al., 2009; Serana et al., 2010). Refractory hemolytic anemia was fatal in three patients (Gaspar et al., 2009). Two additional studies assessed defects in the lymphoid compartments of ADA-SCID patients following PEG-ADA. Different degrees of abnormalities in the B-cell compartment and inability to respond to vaccines, despite the presence of normal serum-Ig or hypogammaglobulinemia were reported (Malacarne et al., 2005). Moreover, a retrospective longitudinal analysis in ADA-SCID patients treated with PEG-ADA showed that decreased levels of newly produced B cells underlie the progressive and significant decrease in circulating B cells in these patients (Serana et al., 2010). Since long-term PEG-ADA treatment is associated with abnormalities in B cell subsets, but often also with a decrease in T-cell functions (Malacarne et al., 2005), a limited B or T-cell repertoire combined with alterations in peripheral tolerance could further favor breakdown of tolerance (Figure 3).
No specific reports on immune dysregulation or autoimmunity in BMT-treated ADA-SCID patients are available in literature (Serana et al., 2010). Nevertheless, autoimmune manifestations have been reported in larger single-center studies on BMT-treated patients with various kinds of immunodeficiencies, including ADA deficiency (Mazzolari et al., 2009; Neven et al., 2009). The major immune dysregulations observed in both studies included thyroid autoimmunity, autoimmune hemolytic anemia, and glomerulonephritis (Mazzolari et al., 2009; Neven et al., 2009).
Most recently autoimmune manifestations have also been described in patients treated with HSC-GT (Aiuti et al., 2009). Four ADA-SCID patients, including one patient that already showed immune dysregulation while on PEG-ADA, developed signs of autoimmunity, such as hemolytic anemia, thrombocytopenia, autoimmune hepatitis, and autoimmune thyroiditis (Aiuti et al., 2009 and unpublished observation).
ADA-Deficient Mouse Model
The availability of a genetic animal model for ADA deficiency allowed a wide range of biochemical and immunological experiments that are not feasible in humans. The first attempts to generate ADA-deficient mice lead to their perinatal death due to severe liver damage (Blackburn et al., 1998). Subsequent studies suggested that ADA expression in trophoblast cells of the placenta is critical for fetal development in the mouse. Thus, ADA-deficient mice were successfully generated by specifically targeting expression of an ADA minigene to the trophoblast lineage of ADA+/− mice and by inter-crossing these mice. This gave rise to litters that contained mice expressing the ADA minigene in their placenta that were also homozygous for the ADA null allele (ADA−/−; Blackburn et al., 1998).
Untreated ADA−/− mice
The ADA−/− mouse reproduces not only the biochemical but also the immunological abnormalities of the human disease phenotype. They manifest both combined immunodeficiency as well as metabolic abnormalities and are therefore commonly used to assess the effect of ADA deficiency not only on the lymphoid organs and peripheral blood, but also its systemic organ toxicity. ADA−/− deficient mice die at approximately 3 weeks of age from severe respiratory distress (Blackburn et al., 1998).
Initial examinations of the thymus and spleen revealed a substantial decrease in organ size. The cellular proportion from the thymus of ADA−/− mice showed a significant increase in the percentage double-negative immature thymocytes, accompanied by a decrease in the percentage of CD4+ or CD8+ single-positive thymocytes. T-cell apoptosis was abundant in the ADA-deficient thymi (Blackburn et al., 1998). ADA−/− splenic B lymphocytes showed defects in proliferation and activation with high propensity to undergo B cell receptor-mediated apoptosis. As a result, profound loss of germinal center architecture was noted, which may be responsible for impaired B cell development (Aldrich et al., 2003). Lymphopenia was also seen in the peripheral circulation, confirming that this model of ADA deficiency exhibits a SCID phenotype.
At death, the severe immune deficiency and organ alterations are the most prominent features, whereas no apparent autoimmune manifestations can be observed. The almost complete absence of effector T- and B-cell populations in these mice and the high levels of anti-inflammatory adenosine might prevent their development in the first 3 weeks of life. Reconstitution of effector T- and B-cells as well as metabolic detoxification after treatment might therefore be requirements for the onset of autoimmunity (Sauer et al., 2012a).
Model for autoimmunity in ADA-deficient mice
Similarly to ADA-SCID patients, ADA−/− mice can be treated with PEG-ADA, HSC-GT with transduced BM ADA−/− cells, or BMT with wild type donor cells (Mortellaro et al., 2006; Sauer et al., 2009). A dose of 1000 U/kg/week of PEG-ADA starting from postnatal day 10 provides rescue and metabolic detoxification in ADA−/− mice (Blackburn et al., 2000). HSC-GT is performed using a SIN-lentiviral vector driving ADA expression from the phosphoglycerate kinase (PGK) promoter (Mortellaro et al., 2006), instead of the gammaretroviral vector used in the clinical trial. A long-term comparative approach between these three treatment options revealed important new information on their efficacy and established a model for autoimmunity in the context of long-term PEG-ADA treatment (Sauer et al., 2012a).
The long-term survival of PEG-ADA, HSC-GT, and BMT-treated mice was comparable between the three groups (60–70% with respect to wildtype). This outcome was the result of an early mortality in the BMT and HSC-GT treated groups, while PEG-ADA treated mice had a less stable long-term survival. As expected from the fact that PEG-ADA remains in circulation without entering in cells, ADA activity in PEG-ADA treated mice was exclusively detectable in the plasma. Reconstitution of enzymatic activity in RBC, BM, spleen, and thymus from BMT-treated mice was comparable to wildtype, while only slightly lower in HSC-GT treated mice (Sauer et al., 2012a).
Strikingly, ADA−/− mice treated with PEG-ADA developed multiple autoantibodies and hypothyroidism in contrast to mice treated with HSC-GT or BMT. Proliferation of various lymphocyte subpopulations, including B cells and highly abnormal antibody production affecting all types of antibody subclasses was observed in PEG-ADA treated mice. Moreover, autoantibodies that reacted to ADA, platelets, the thyroid, and the gastrointestinal tract were detected in the sera from PEG-ADA treated mice. Focal atresia with non-secreting follicles, an increase in apoptotic cells in affected tissue areas and significantly elevated levels of thyroid-stimulating hormone (TSH) represent signs of autoimmune hypothyroidism. The role of autoantibodies against the stomach and intestine developing in PEG-ADA treated mice, without causing gross pathological alterations, remained unclear. However, it was hypothesized that the occurrence of antibody responses to GI tissues not only interferes with nutrient uptake, but also reflect alterations in gastrointestinal immunity (Sauer et al., 2012a). The established mouse model for autoimmunity after PEG-ADA treatment represents a valuable model for future studies on the in vivo effects of PEG-ADA on immune cell function and inflammatory responses.
Interestingly, PEG-ADA treated mice produced antibodies to ADA, platelets, the thyroid, and gastrointestinal tract, but not other organs such as the pancreas or endocrine glands. The strong overlap of autoimmune manifestations observed in this model of autoimmunity in ADA−/− mice with those reported in ADA-deficient patients suggests that a component of autoimmune susceptibility may map to the target tissue. In both humans and in mouse models, single genetic loci have been linked with susceptibility to multiple autoimmune diseases. The genes underlying such loci, including AIRE, FoxP3, CTLA-4, and PTPN22, are likely to confer a general predisposition to the failure of immune tolerance and development of an auto-aggressive immune response (Hill et al., 2007). However, other loci are clearly disease specific, and presumably modify a generalized predisposition to confer organ/disease specificity. Interestingly, recent studies have implicated ADA polymorphisms in the development of type1 diabetes and rheumatoid arthritis (Sebastiani et al., 2006; Saccucci et al., 2009).
Role of ADA Metabolites in Immune Cell Development and Function
Although autoimmunity is frequently observed in certain immunodeficiencies, there is accumulating evidence that ADA deficiency predisposes to this phenomenon not only through general mechanisms of immune dysregulation but also through specific alterations caused by the accumulating ADA metabolites. Main feature of ADA deficiency is the gradual accumulation of adenosine and 2′-deoxyadenosine nucleosides. In the absence of ADA, these nucleosides are metabolized differently into AXP or dAXP, respectively, and exert distinct biochemical action (AXP: AMP, ADP, or ATP; dAXP: dAMP, dADP, or dATP). Several pathophysiological mechanisms have been proposed to describe the role of ADA substrates in cytotoxicity as well as their immunomodulatory properties in patients and in the ADA-deficient mouse model (Hirschhorn and Candotti, 2006). The major effects of adenosine, 2′-deoxyadenosine and their nucleotide byproducts are summarized in Table 1.
Table 1
| ADA metabolite | Cell type | Mouse/human | Mode of action | Reference |
|---|---|---|---|---|
| 2′-Deoxyadenosine | Lymphocytes | Human | Inhibition of SAHH activity results in accumulation of S-adenosylhomocysteine and inhibition of transmethylation reactions | Hershfield et al. (1979) and Benveniste et al. (1995) |
| Fibroblasts | Mouse | SAHH acts as a physiological modulator of Fas-mediated cell death | Ratter et al. (1996) | |
| T cells | Human | Inhibition of T-cell activation by aberrant Adora2a signaling and PKA hyperactivation | Cassani et al. (2008) | |
| dATP | Lympocytes and RBC | Human | Intracellular ATP depletion | Siaw et al. (1980), Simmonds et al. (1982), Koller et al. (1984), and Simmonds et al. (1984) |
| T cells | Human | Inhibition of ribonucleotide reductase causes an imbalance of dNTPs and an impairment of DNA synthesis | Waddell and Ullman (1983), Benveniste et al. (1995) | |
| T cells | Human | Accumulation of DNA single strand breaks | Cohen and Thompson (1986) and Gangi-Peterson et al. (1999) | |
| T cells | Mouse | Inhibition of thymocyte development past the CD4−/CD8− double-negative stage | Van de Wiele et al. (2002) and Van de Wiele et al. (2006) | |
| B cells | Human | Nucleotide pool imbalance affects TdT activity during V(D)J recombination in the bone marrow | Gangi-Peterson et al. (1999) | |
| Adenosine | T cells | Human | Compromised TCR/CD28-driven proliferation and cytokine production, defective activation of NF-κB transcriptional events | Cassani et al. (2008) |
| Resting T cells | Human | Upregulation of CD152, CTLA-4, normally involved in the termination of immune responses | Vendetti et al. (2002) | |
| T cells | Mouse | Decreased TCR-triggered activation and upregulation of activation markers | Apasov and Sitkovsky (1999), Apasov et al. (2001) | |
| Activated T cells | Mouse | Inhibition of IL-2, TNFα, and INFγ secretion | Erdmann et al. (2005) and Lappas et al. (2005) | |
| B cells | Human | Adora2a signaling interferes with BCR- and TLR-function, inhibition of B-cell activation after stimulation | Sauer et al. (2012b) | |
| B cells | Mouse | Profound loss of GC, susceptibility to apoptosis, defects in B-cell proliferation and activation, block in Ag-dependent B-cell maturation | Aldrich et al. (2003) | |
| B cells | Mouse | Increase of intracellular cAMP suppresses the activation of NF-κB after BCR and TLR-stimulation | Minguet et al. (2005) | |
| Tregs | Mouse/human | Adora signaling causes alterations in the CD39/CD73 adenosinergic machinery, upregulation of FoxP3 | Sauer et al. (2012a) | |
| Adaptive Tregs | Mouse | Adora2a signaling inhibits the generation of adaptive effector T cells and promotes the induction of adaptive Tregs, upregulation of FoxP3 | Zarek et al. (2008) | |
| ATP | T cells | Human | Purinergic stimulation through P2X receptors prolongs TCR-initiated activation and IL-2 secretion | Yip et al. (2009) |
| T cells | Mouse | Antagonism of P2X blunts TCR-mediated activation and results in unresponsiveness to subsequent stimulation | Schenk et al. (2008) | |
| Tregs | Mouse | Activation of P2X7 inhibits the suppressive potential and stability of Tregs | Schenk et al. (2011) |
Effects of ADA metabolites on lymphocyte development and function.
Adora2a, adenosine 2a receptor; ATP, adenosine-5′-triphosphate; BCR, B cell receptor; cAMP, 3′,5′-cyclic adenosine monophosphate; dATP, 2′-deoxyadenosine 5′-triphosphate; dNTPs, deoxynucleotides; GC, germinal center; INFγ, interferon gamma; NF-κB, nuclear factor kappa B; P2X/P2X7, purinergic receptors; PKA, cAMP-dependent protein kinase A; RBC, red blood cells; SAHH, S-adenosylhomocysteine hydrolase; TCR, T-cell receptor; TdT, terminal deoxynucleotidyl transferase; TNFα, tumor necrosis factor alpha; Tregs, naturally occurring regulatory T cells.
Cytotoxicity of 2′-deoxyadenosine and dATP
Based upon in vivo and in vitro findings, several mechanisms are believed to account for the block of lymphocyte development in ADA-SCID (Hirschhorn and Candotti, 2006). The biochemical hallmarks of ADA deficiency consist of the general belief, that 2′-deoxyadenosine is the primary cause of lymphotoxicity in ADA-SCID, which exerts its effects at the nucleoside level or after conversion to dATP. Although 2′-deoxyadenosine is a weak substrate for adenosine kinase and deoxycytidine kinase, in the absence of ADA these enzymes can phosphorylate 2′-deoxyadenosine. In turn, the resulting dATP pool expansion may interfere with a number of critical metabolic pathways.
These ADA substrate accumulations inhibit methyl-transfer reactions by suicide inactivation of S-adenosylhomocysteine (SAH) hydrolase (Hershfield et al., 1979). dATP is known to be a feedback inhibitor of ribonucleotide reductase. Its inhibition causes an imbalance of deoxynucleotides (dNTP), leading to an impairment of DNA synthesis, which is critical for the expansion of lymphocytes in response to antigenic challenge (Benveniste et al., 1995).
Role of adenosine as anti-inflammatory mediator
By binding to G-coupled adenosine receptors present on the surface of target cells, adenosine acts as an extracellular signal transducer to exert suppressive functions (Sitkovsky et al., 2004). Physiologically, adenosine-mediated triggering of these receptors can promote a fine-tuning of the inflammatory responses. In the context of defective ADA metabolizing enzyme, where the extracellular levels of adenosine are increased, this regulation may be exaggerated and cause immune dysfunction. This process is mostly regulated by the aberrant engagement of adenosine 2a receptor (Adora2a)-mediated signaling.
Functional studies on T cells from ADA-deficient mice and patients showed an increased susceptibility to apoptosis as well as altered intra- and extracellular signaling leading to impaired T-cell function (Apasov and Sitkovsky, 1999; Apasov et al., 2001; Cassani et al., 2008). As summarized in Figure 4, the TCR-dependent activation defect in ADA deficiency is augmented by the immunosuppression through extracellular adenosine receptor triggering. Extracellular adenosine induces increased levels of cAMP in T-lymphocytes, which inhibits both proximal signaling events after TCR triggering as well as other downstream effector functions (Huang et al., 1997; Lappas et al., 2005; Ohta et al., 2009). In accordance with previous data obtained in thymocytes from ADA−/− mice (Apasov et al., 2001), IκBα phosphorylation after TCR triggering was low or undetectable in ADA-deficient cells (Cassani et al., 2008). Reduced levels of IκBα phosphorylation and degradation leads to low levels of NF-κB translocation and transcription of target genes in the nucleus, thereby contributing to the functional impairments of ADA-deficient T cells.
Figure 4
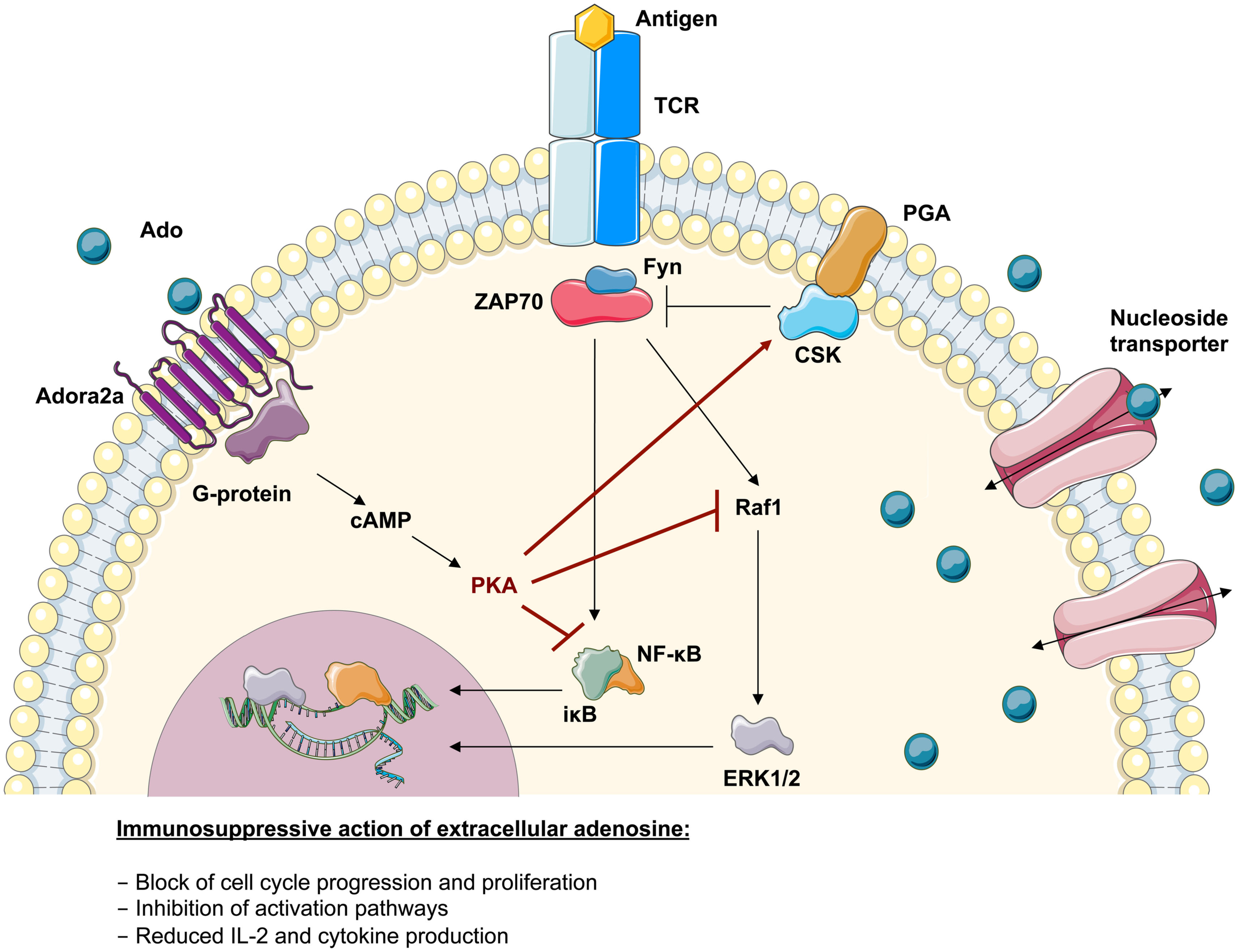
Immunosuppressive action of adenosine in lymphoid cells. Upon TCR-antigen-binding and T-cell activation, the translocation of activated transcription factors and the expression of anti-apoptotic factors supports their survival, proliferation, and differentiation. The adenosine 2a receptor (Adora2a) present on T-lymphocytes interacts with coupled G-proteins to stimulate cAMP formation. The effects of cAMP in T cells are almost entirely mediated by the cAMP-dependent protein kinase A (PKA). It has been shown that the inhibitory properties of PKA are mediated via activation of Csk or inhibition of Raf1. Consistently it has been shown that the severely compromised effector functions in ADA-deficient T cells associates with an intrinsically reduced ERK1/2 signaling, a decreased phosphorylation of IκB and an altered nuclear recruitment of NF-κB. This suggests that the accumulating adenosine levels in an ADA-deficient environment can directly antagonize biochemical events arising from TCR engagement.
Less information is available about the effects exerted by adenosine on B-cell function. Similarly to the alterations in T cells described above, adenosine receptor ligation in B cells inhibits downstream responses to antigen receptor engagement like BCR-induced IκB phosphorylation and the NF-κB pathway after BCR or TLR4 stimulation (Minguet et al., 2005). Adenosine may thereby drive BCR-stimulated B cells toward an anergic rather than an immunogenic response. Recent findings showing defects in BCR and TLR signaling as well as in tolerance checkpoint control in human B cells are discussed and illustrated separately in section “Defects in B-cell tolerance in ADA-SCID.”
Overall, these evidences strongly suggest that rather than controlling a single pathway downstream of the TCR or BCR, the immune defects in ADA-deficient lymphocytes may involve multiple pathways converging toward the defective induction of lymphocyte activation. They also illustrate how extracellular adenosine levels can interfere with the downstream signaling transduction upon activation, thereby exerting its immunosuppressive activity on the transcriptional machinery. Because of cell-specific expression and regulation, aberrant adenosine receptor-mediated signaling might also contribute to the occurrence of autoimmune manifestations observed in some ADA-SCID patients (Kohn, 1996; Ozsahin et al., 1997).
Role of ATP and other purinergic receptors
Stimulation of almost all mammalian cell types leads to the release of cellular ATP and autocrine feedback through a diverse array of purinergic receptors (Junger, 2011). ATP binds to two classes of purinergic P2 receptors in the plasma membrane of eukaryotic cells: P2X receptors, which are ligand-gated ion channels, and heterotrimeric G protein-coupled P2Y receptors (Schenk et al., 2011). Depending on the types of purinergic receptors that are involved, autocrine signaling can promote or inhibit immune cell activation and fine-tune adaptive immune responses (Junger, 2011; Schenk et al., 2011).
In addition to the autocrine feedback mechanisms that regulate the function of healthy immune cells, purinergic receptors allow immune cells to recognize ATP that is released from damaged or stressed host cells. Thus, the purinergic signaling systems of immune cells serve an important function in the recognition of danger signals. ATP that is released by stressed cells guides phagocytes to inflammatory sites and promotes clearance of damaged and apoptotic cells (Elliott et al., 2009; Junger, 2011).
To date, little information is available on alterations in the ATP-induced regulation of immune cells in ADA deficiency. It is reported that dATP accumulation in the absence of ADA leads to a cellular depletion of ATP (Siaw et al., 1980; Simmonds et al., 1982, 1984; Koller et al., 1984). The pool of extracellular ATP on the other hand might well be augmented in ADA-deficient lymphoid organs, due to the increased percentage of cells undergoing apoptosis. It can therefore be hypothesized that alterations in ATP concentrations in ADA deficiency also influence T-cell responses on the level of TCR induced activation and in response to stimuli from an inflammatory microenvironment.
Break of Tolerance and Contribution of Lymphocytes to Autoimmunity in ADA Deficiency
Adaptive immunity requires sophisticated regulatory mechanisms to ensure protection to a variety of pathogenic microbes while maintaining immune self-tolerance and preventing autoimmunity (Sakaguchi et al., 2008). The main mechanisms for the induction and maintenance of a self-tolerant repertoire, which is diverse in antigen recognition, are central and peripheral tolerance. Central tolerance is the mechanism able to eliminate newly developing T cells and B cells that have high affinity to self (Mathis and Benoist, 2004). Central tolerance is distinct from peripheral tolerance in that it occurs while cells are still present in the primary lymphoid organs, whereas emigrant cells are controlled through peripheral tolerance mechanisms, after they reach the periphery (Wardemann and Nussenzweig, 2007; Klein et al., 2009). These include suppression of autoreactive cells by regulatory T cells and the generation of hyporesponsiveness (anergy) in lymphocytes, which encounter antigen in the absence of the co-stimulatory signals that accompany inflammation (Meffre and Wardemann, 2008).
Numerous mechanisms have been proposed to explain the break of tolerance and development of autoimmune manifestations, such as defective negative selection of autoreactive T-lymphocytes in the thymus, alterations in the number and/or function of regulatory T cells, defects of the central and peripheral B-cell tolerance checkpoints, impaired apoptosis of autoreactive lymphocytes, break of tolerance due to increased or decreased clearance of apoptotic cells and pathogens, or increased homeostatic lymphoid proliferation and cytokine secretion associated with lymphopenia (Carneiro-Sampaio and Coutinho, 2007; Westerberg et al., 2008; Notarangelo, 2009; Meffre, 2012).
T-cell tolerance
Central T-cell tolerance mechanisms are based on the elimination or negative selection of the majority of T cells recognizing self with high affinity for negative selection in the thymus. Nonetheless thymic selection is not a tight process and T cells expressing low-avidity TCR on their cell surface are frequently released in the periphery, where they are potentially dangerous to the host as they can be effectively recruited into an autoimmune response (Parish and Heath, 2008).
A major cause of tolerance breakdown is associated with lymphopenia (Daikeler and Tyndall, 2007). This typical state of primary immunodeficiencies may contribute to the induction of spontaneous homeostatic proliferation of residual T cells allowing peripheral expansion of autoreactive cells with a skewed repertoire. Particularly, after conditioning or transplantation these cells may persist, since insufficient thymic reconstitution may affect the control of self-reactivity due to defective negative selection in the thymus and/or reduced regulatory T-cell development and function (Hauri-Hohl et al., 2007). In the case of ADA deficiency, it has been hypothesized that the structure and functions of the thymic microenvironment might be altered, either directly, by toxicity of purine metabolites, or indirectly, by failure of T cells arrested in their development to deliver supportive signals to the thymic stroma (Apasov et al., 2001).
Peripheral tolerance depends on the balance between immune responses to invading pathogens and immune tolerance to self-antigens. In the context of tissue damage and frequently occurring infections in primary immunodeficient patients, apoptotic cells represent a major source of autoantigen. Since apoptosis plays a major role in the deletion of autoreactive lymphocytes and the removal of virus-infected cells, defects in cell death have been implicated in the development of autoimmune diseases and persistent viral infection (Utz et al., 2000). The release of self-antigen into the intracellular space and their presentation mediated by dendritic cells or other antigen-presenting cells may prime naive autoreactive T cells, which were not eliminated by depletion or anergy (Waldner et al., 2004). Several mechanisms exist, including a spectrum of CD4+ regulatory T cells (Tregs), to suppress self-reactive T cells that escape thymic clonal deletion and attenuate anti-pathogen effector mechanisms from inducing immune pathology (Piccirillo and Thornton, 2004). There is ample evidence that Tregs actively mediate suppression to control immune responses to self- and non-self-antigens and the onset of autoimmunity (Bach, 2003; Sakaguchi, 2005). Lessons from other primary immunodeficiencies have provided unequivocal evidence for the essential role of Tregs in suppressing autoreactive T cells in the periphery (Westerberg et al., 2008). Rising of autoimmunity may not only be linked to a reduction in Treg numbers but also to attenuation of their suppressive activity (Sakaguchi et al., 2008). While this is principally mediated by cell–cell contact, recent findings revealed additional mechanisms of Treg-mediated suppression, including secretion of immunosuppressive cytokines, functional modification or killing of APC, and metabolic disruption (Vignali et al., 2008). Moreover, extracellular adenosine produced by Tregs, has been identified as one of the mechanisms mediating their suppressive activity (Sitkovsky et al., 2008; Mandapathil et al., 2010). Treg cells possess a unique biochemical signature amongst T cells in that they generate and sustain high adenosine concentrations. Since Tregs primarily mediate peripheral control of autoreactive T cells, it is conceivable that this compartment might be specifically affected in ADA-SCID (see also Defective Regulatory T Cell Function in ADA Deficiency). Consequently the autoimmune manifestations associated with ADA deficiency might be the result of an altered purine metabolism interfering with normal regulatory T-cell function (Sauer et al., 2012a).
B-cell tolerance
A variety of mechanisms ranging from clonal deletion to functional inactivation by anergy of autoreactive B cells serve to shape the peripheral B-cell repertoire. Nevertheless, dysregulation of B cell development and autoantibody production is a characteristic of most autoimmune diseases including rheumatoid arthritis, systemic lupus erythematosus, and type 1 diabetes, but also immunodeficiencies such as CVID, Wiskott–Aldrich Syndrome, and X-linked agammaglobulinemia (Park et al., 2005; Cuss et al., 2006; Westerberg et al., 2008). In ADA deficiency, some of the observed immune dysregulation were hypothesized to be associated with a more restricted B-cell repertoire due to abnormalities in central B-cell generation or to a dysregulated expansion of these cells in the periphery.
Autoantibodies appear in the serum many years before the onset of clinical disease suggesting an early break in B-cell tolerance (Wardemann et al., 2003). Some of B-cell mediated autoimmune diseases, such as myasthenia gravis (MG), idiopathic autoimmune thrombocytopenic purpura (AITP), and Graves’ disease are characterized by auto-Abs production that destroy target tissues (Barsalou et al., 2011; Cunningham-Rundles, 2011). A remarkably high proportion of autoantibodies associated with systemic autoimmune diseases binds DNA, RNA, or macromolecular complexes that contain DNA or RNA. It has been hypothesized, that under certain circumstances these intracellular autoantigens become visible to the immune system when they accumulate during apoptosis. In fact the impaired clearance of apoptotic cell debris and dsDNA by macrophages might induce TLR signaling and differentiation of autoreactive B cells (Gaipl et al., 2006). Response to nucleic acid-containing immunecomplexes relies on the coengagement of endosomal members of the TLR family, TLR9 and TLR7 (Marshak-Rothstein, 2006). Therefore, self-antigens that can effectively engage both the BCR and either TLR7 or TLR9 might stimulate autoreactive B cells that are normally quiescent, through inherent adjuvant activity and trigger the development of systemic autoimmune disease (Marshak-Rothstein, 2006). In ADA deficiency, the metabolic basis underlying immune cell deficiency is the cytotoxic effect impact of the ADA substrates deoxyadenosine and dATP, leading to apoptosis of lymphocytes. It is therefore conceivable that developing B lymphocytes in affected lymphoid organs encounter massive amounts of nucleic acid. Nucleic acid-sensing TLRs might therefore represent Achilles’ heel in susceptible ADA-deficient patients by which relative tolerance for nucleic acid-containing antigens is breached and autoimmunity occurs (Kono et al., 2009).
New Insights into Immune Cell Dysfunction and Onset of Autoimmunity in ADA Deficiency
Recent in-depth studies have revealed specific defects in ADA deficiency that may contribute to the onset of autoimmunity in these patients. Herein we discuss alterations in the adenosinergic machinery of ADA-deficient regulatory T cells and in B-cell tolerance in the absence of functional ADA.
Defective regulatory T-cell function in ADA deficiency
Although autoimmune manifestations are frequent findings in ADA-deficient patients with milder forms or in patients under PEG-ADA, mechanisms causing the loss of peripheral tolerance and onset of autoimmunity have remained elusive. CD4+CD25+FoxP3+ Tregs actively suppress pathological and physiological immune responses in order to maintain peripheral immune self-tolerance and prevent autoimmunity (Sakaguchi et al., 2008; Sitkovsky et al., 2008). Extracellular adenosine produced by Tregs has been described as one of the mechanisms mediating their suppressive activity (Figure 5A). Concordant expression of the ectoenzymes CD39 and CD73 has been reported both for murine and human Tregs (Borsellino et al., 2007; Deaglio et al., 2007; Mandapathil et al., 2010). The CD39 ectoenzyme produces AMP from ATP or ADP, which is subsequently converted into extracellular adenosine by the CD73 ectoenzyme (Hasko et al., 2008). Treg function requires the coordinated expression of the Adora2a on activated T effector cells to enable adenosine-mediated immunosuppression (Sitkovsky et al., 2008). Moreover, Tregs have been shown to express low levels of ADA, whereas T effector cells are enriched in ADA but express low levels of CD39 and CD73 (Mandapathil et al., 2010; Sauer et al., 2012a). This molecular profile of Tregs (CD39+CD73+ADAlow) has functional importance, as it not only confers Tregs the capability to produce extracellular adenosine but also to sustain relatively high concentrations due to low ADA expression (Mandapathil et al., 2010).
Figure 5
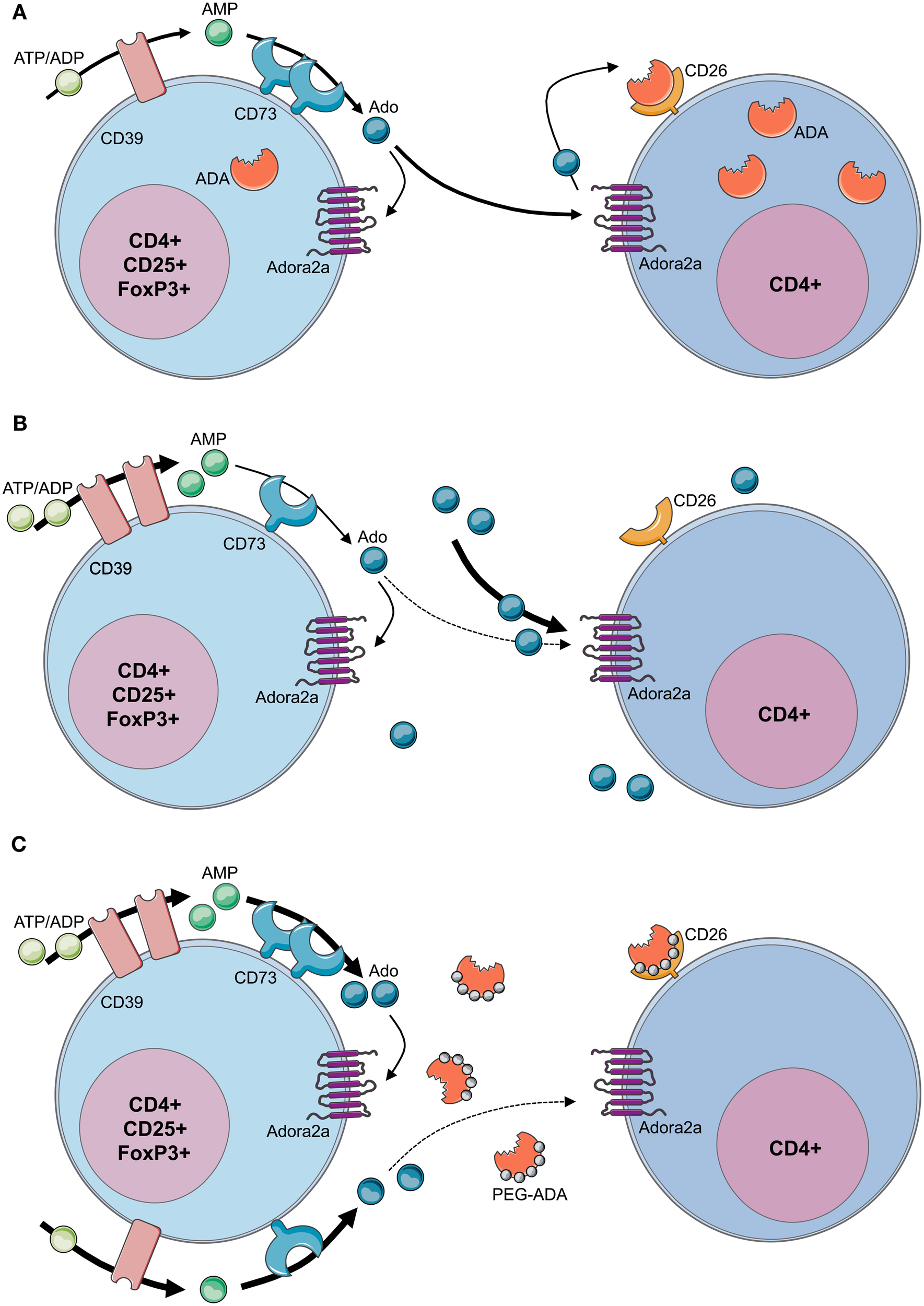
Loss of regulatory T-cell function in ADA-SCID. (A) By concomitant expression of CD39 and CD73, Tregs have the enzymatic machinery to generate and maintain high levels of extracellular adenosine. Contrarily to T effector cells, Tregs express low levels of ADA and CD26. Extracellular ATP or ADP is converted by the ectonucleotidase CD39 into AMP, which is further converted into adenosine by the CD73 ectoenzyme. The produced adenosine binds to the Adora2A receptor expressed on activated effector T cells, which are enriched in ADA and the surface-bound glycoprotein CD26. The coordinated expression of CD39 and CD73 on Tregs and Adora2a on T effector cells enables adenosine-mediated immunosuppression. (B) In the absence of ADA, deficient Tregs express high levels of CD39, increasing their capacity for ATP hydrolysis, but reduced levels of CD73 on their surface. Although both CD39 and CD73 are rate limiting for extracellular adenosine generation, CD73 is the last component of the ectoenzymatic chain. With accumulating adenosine levels, possibly to avoid a further increase of extracellular concentrations, CD73 is reduced and ADA−/− Tregs display a decreased suppressive activity toward T effector cells. Nevertheless no apparent autoimmune manifestations can be observed at onset, likely due to the severely reduced effector T- and B-cell populations. Moreover, the accumulating extracellular adenosine is likely to maintain an anti-inflammatory environment in these mice. (C) After PEG-ADA treatment, the adenosinergic machinery of CD39 and CD73 are upregulated, indicating an increased requirement for ATP hydrolysis and enhanced adenosine production. Despite the initial rescue of suppressive activity by upregulation of CD73 for elevated adenosine production, long-term PEG-ADA treatment interferes with Treg function by augmenting adenosine turnover. PEG-ADA present in the extracellular space eliminates adenosine produced by the ectoenzymatic chain and hinders adenosine-mediated suppression by interfering between adenosine and Adora2a expressed on T effector cells.
Figure 5B summarizes recently described defects and functional alterations of the adenosinergic pathway in Tregs from ADA-deficient mice and patients (Sauer et al., 2012a). ADA−/− Tregs showed significantly higher expression of CD39, while expressing significantly less CD73. ADA−/− Tregs are sensitive to extracellular adenosine concentrations and the expression of CD73 is regulated by this metabolite. With adenosine accumulating in ADA−/− mice, possibly to avoid a further increase of extracellular concentrations, CD73 is reduced and ADA−/− Tregs display a decreased suppressive activity toward T effector cells. The underlying mechanism accounting for increased CD39 expression in ADA−/− Tregs remains to be elucidated. However, intracellular cAMP levels, which are elevated in the absence of ADA, have been reported to increase CD39 expression (Liao et al., 2010).
In order to dissect the cellular mechanisms leading to loss of peripheral tolerance, ADA−/− mice were studied after treatment with PEG-ADA, HSC-GT, and BMT. Although short-term PEG-ADA treatment initially rescued Treg-mediated suppression in comparison to untreated ADA−/− mice, their functionality became exhausted by long-term PEG-ADA treatment. Tregs from PEG-ADA treated animals maintained increased expression of CD39 and upregulated CD73 expression in comparison to age-matched wildtype controls. Consistently, CD39 activity measured by ATP consumption and AMP formation, as well as adenosine production by CD73 were significantly increased in comparison with wildtype Tregs. These results were confirmed in a cohort of patients including 7 PEG-ADA treated and 11 retroviral HSC-GT treated patients. The percentage of CD4+CD25+FOXP3+CD127−/low Tregs was significantly reduced in PEG-ADA treated patients and their expression of CD39 and CD73 ectonucleotidase were significantly increased. Unlike Tregs from HSC-GT treated patients and HD, Tregs isolated from PEG-ADA treated patients were unable to suppress the proliferation of effector cells (Sauer et al., 2012a).
The obtained results revealed an elevated adenosine catabolism in the presence of PEG-ADA, characterized by alterations in the adenosinergic machinery producing high levels of adenosine and a significantly increased turnover by the enzymatic activity of PEG-ADA. Upregulation of CD73 in treated ADA−/− mice and patients can therefore be interpreted as a compensatory mechanism representing a higher requirement for ATP/ADP to adenosine conversion in the presence of extracellular PEG-ADA. Despite the initial rescue of suppressive activity by upregulation of CD73 for elevated adenosine production, long-term PEG-ADA treatment interfered with Treg function by augmenting adenosine turnover. These findings fit the hypothesis that PEG-ADA present in the extracellular space eliminates adenosine produced by this ectoenzymatic chain and hinders adenosine-mediated suppression by interfering between adenosine and Adora2a expressed on T effector cells (Sauer et al., 2012a; Figure 5C).
Defects in B-cell tolerance in ADA-SCID
Although PEG-ADA induces metabolic detoxification, BMT and HSC-GT provide superior restoration of purine metabolism and immune functions. However, it had remained unclear how patient’s B cells contribute to autoimmune complications and if B-cell tolerance is established properly in ADA-deficient patients before and after treatment.
Random V(D)J recombination produces large numbers of antibodies displaying self-reactive specificities and during normal B-cell development the majority of these antibodies are removed at two distinct checkpoints in the bone marrow and periphery (Wardemann et al., 2003). Large numbers of self-reactive antibodies are removed from the B cell repertoire during the immature B cell stage in the bone marrow, where BCR-mediated selection plays a crucial role in controlling B-cell survival based on excessively strong or weak BCR signals that identify autoreactive or functionally unfit B cells (Goodnow, 1996; Nemazee et al., 2000; Cancro, 2009; Figure 6). Alterations of BCR signaling thresholds result in a defective central B-cell tolerance checkpoint and interfere with the removal of developing autoreactive B cells in humans (Ng et al., 2004; Menard et al., 2011). In addition to their BCRs, B cells also express TLRs that were originally described to bind microbial components but that are also able to recognize self-antigens (Marshak-Rothstein, 2006) and are involved in the removal of developing anti-nuclear antibody (ANA)-expressing B cells (Isnardi et al., 2008). Both BCR- and TLR-mediated B-cell responses have been reported to be modulated by adenosine receptor signaling and intracellular cyclic adenosine monophospate (cAMP), which are increased in ADA deficiency (Apasov et al., 2001; Hershfield, 2005; Minguet et al., 2005; Power Coombs et al., 2011).
Figure 6
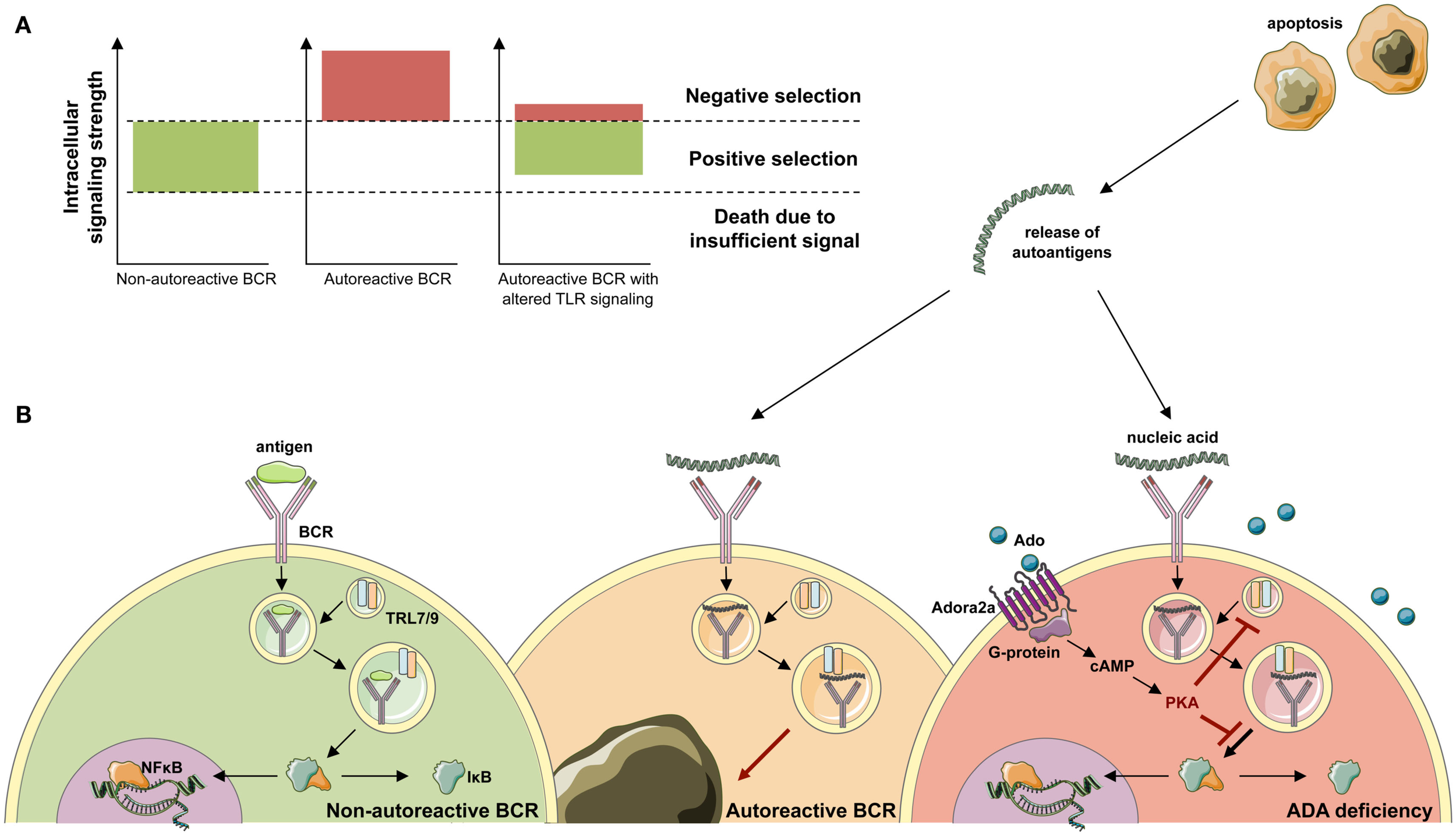
Development of ANA-expressing B-cell clones in ADA deficiency. (A) During B-cell development in the bone marrow, BCR signals provide a cell-intrinsic measure for negative or positive selection. By excessively weak or strong BCR signals, two thresholds identify functionally unfit or autoreactive B-cell clones. B cells that fail to rearrange and signal through their BCR die of neglect. Negative selection against autoreactive BCR specificities occurs following high avidity BCR interactions with antigen. While positive selection of B-cells requires persistent intermediate BCR signaling in both developing and mature B-cells. (B) Upon antigen-binding the BCR is internalized and transported to the cytoplasmic compartments containing Toll-like receptor 9 (TLR9) or TLR7. Physiological engagement of immunoreceptors leads to the phosphorylation and proteasomal degradation of IκB, thereby releasing NF-κB into the cytoplasm. Subsequently, NF-κB translocates to the nucleus and initiates the transcription of NF-κB target genes required for immune cell development and function. During the bone marrow differentiation of B cells, a major source of autoantigen is developing lymphocytes, which undergo apoptosis. Autoantigens are released in the extracellular space and form immune complexes. Binding of nucleic acid-containing immune complexes to an autoreactive BCR produces a strong activation signal through simultaneous activation of TLRs and leads to the depletion or receptor editing of the B cell. It can be hypothesized that in ADA deficiency this negative selection is dampened by adenosine present in the extracellular place and engagement of the Adora2a. Activation of Adora2a elevates intracellular cAMP through activation of adenylyl cyclase. In turn, cAMP activates PKA that blocks the BCR-induced phosphorylation of IκB to inhibit immunoreceptor-mediated NF-κB activation in the cytoplasm. The strong depletion signal coming from BCR and TLR coengagement is thereby lowered and ADA-deficient B cells carrying an autoreactive BCR egress into the periphery.
B-cell tolerance checkpoints in ADA-SCID patients were assessed by cloning antibodies expressed by single B cells before and after successful HSC-GT (Sauer et al., 2012b). New emigrant/transitional and mature naïve B cells from ADA-deficient patients before HSC-GT contained high frequencies of autoreactive and ANA-expressing clones compared to healthy donors, revealing defective central and peripheral B-cell tolerance checkpoints in the absence of functional ADA.
The receptor candidates for the removal of ANA-expressing clones are nucleic acid-sensing endosomal members of the TLR family, TLR7 and TLR9 (Marshak-Rothstein, 2006), thereby suggesting that ADA impinges not only on BCR but more importantly TLR signaling. A similar mechanism has also been hypothesized to contribute to B-cell dysfunctions, defective B-cell proliferation, and activation observed in ADA-deficient mice (Aldrich et al., 2003; Hershfield, 2005). The accumulating adenosine blocks NF-κB activation in murine B cells stimulated through BCRs or TLR4 by LPS (Minguet et al., 2005; Power Coombs et al., 2011). In line with this hypothesis, we found that stimulation through TLR7 and TLR9 were significantly dependent on proper ADA enzymatic activity and adenosine receptor engagement, further underlining the inability of these receptors to function in the absence of functional ADA (Figure 6).
Strikingly, ADA-deficient patients treated with HSC-GT displayed quasi-normal early B-cell tolerance checkpoints as evidenced by restored efficient removal of developing autoreactive and anti-nuclear B cells. Hence, ADA plays an essential role in the establishment of early B-cell tolerance and the removal of developing autoreactive B cells in humans (Luning Prak, 2012; Sauer et al., 2012b).
Concluding Remarks on the Occurrence of Autoimmunity after ADA-SCID Treatment
In summary, the available literature provides supporting evidence for a predisposition to autoimmunity in ADA deficiency. Alterations in both central and peripheral tolerance in T- and B-cells have been described to contribute to the pathogenesis of autoimmunity. Moreover, it is becoming increasingly clear that tolerance mechanisms and immune responses are specifically altered by the lack of ADA and the accumulation of its substrates.
Particularly, the impact of accumulating adenosine as anti-inflammatory mediator has to be underlined in ADA deficiency. The ligation of Adora2a receptors leads to an increase in cAMP levels, which in cooperation with PKA induces immunosuppression, attenuation of proximal signaling events after TCR and BCR triggering, and inhibition of downstream effector functions (Skalhegg et al., 1992; Huang et al., 1997; Lappas et al., 2005; Cassani et al., 2008; Sauer et al., 2012b). It can further be hypothesized that dampening of TCR- and BCR-downstream signaling interferes with the depletion signals during negative selection in central tolerance, thereby allowing the survival of autoreactive cells in ADA deficiency. In addition to its effects on T- and B-cells, adenosine is an important regulator, physiologically involved in inhibiting a variety of activated immune cells and in protecting tissues from acute inflammatory damage (Panther et al., 2003; Sitkovsky et al., 2004). Indeed we showed that Tregs require a balanced adenosine metabolism to exert their suppressive activity, since excessively high adenosine concentrations, or excessive conversion of extracellularly produced adenosine by PEG-ADA interferes with their suppressive function (Luning Prak, 2012; Sauer et al., 2012a).
The precise role of PEG-ADA alongside other treatment options is still undetermined, but it certainly allows rapid detoxification and stabilization of patients awaiting more definitive treatment (Booth and Gaspar, 2009). With a progressive loss of lymphocyte functions, the occurrence of neutralizing anti-ADA antibodies and autoimmune manifestations, long-term immunological reconstitution in PEG-ADA patients is often incomplete. It has been hypothesized that partial ADA correction resulting in low enzymatic activity may mimic late-onset patients, which typically display a higher prevalence of autoimmune manifestations (Ochs et al., 1992; Ozsahin et al., 1997; Luning Prak, 2012). Indeed, recent data underlined the importance of intracellular ADA expression and superior efficacy of gene therapy over PEG-ADA treatment for the development of functional T- and B-cell tolerance, including Tregs (Sauer et al., 2012a,b).
Both BMT and HSC-GT provide efficient reconstitution of the immune system through endogenous enzymatic ADA activity. BMT from an HLA-identical sibling donor remains the treatment of choice, while transplants from alternative donors are associated with high morbidity and mortality. The occurrence of mixed chimerism in transplanted patients with other primary immunodeficiencies is associated with a higher incidence of autoimmune manifestations (Moratto et al., 2011) and might well play a role also in ADA deficiency (Cancrini et al., 2010). Moreover, transplantation-induced lymphopenia is a possible cause for the homeostatic expansion of autoreactive T- and/or B-cells with subsequent loss of self-tolerance (Daikeler and Tyndall, 2007). Phenomena of immune dysregulation as occurring in the context of pre-transplant conditioning and BMT might further trigger the onset of autoimmunity (Etzioni, 2003).
In accordance with the current guidelines of the European group for Blood and Marrow Transplantation (EBMT) and given the long-term experience in gene therapy (Aiuti et al., 2009), this treatment can now be considered for all ADA-SCID patients lacking an HLA-identical sibling donor (Gaspar et al., 2009). After HSC-GT, high levels (50–90% on average) of gene correction were detected in T- and B- and NK-cells (Aiuti et al., 2009), leading to an efficient systemic detoxification and recovery of immune cell functions. However, as suggested by the cloning of single B-cell receptors, non-gene corrected cells may carry autoreactive specificities (Sauer et al., 2012b). It can be hypothesized that the co-existence of non-corrected autoreactive T- or B-cells and the restored functional T cell help could allow the development of autoimmune manifestations in ADA-SCID patients after HSC-GT (Aiuti et al., 2009). Modification of preparatory regimen or increased gene transfer efficiency by more robust approaches such as lentiviral vectors may further improve HSC-GT outcome for ADA deficiency (Mortellaro et al., 2006).
Adenosine deaminase-SCID remains a challenging condition to treat (Gaspar et al., 2009). With large-scale outcome studies still lacking, the choice between lifelong PEG-ADA, unrelated BMT and HSC-GT is currently based on the risk/benefit ratio, the availability, and costs of the three different treatment options (Gaspar et al., 2009). Due to the rarity of the disease and the small cohort numbers, accurate survey and long-term follow-up will be essential to determine the outcome following different treatments and their efficacy in restoring immune tolerance.
Statements
Acknowledgments
Supported by grants from the Italian Telethon Foundation: TIGET, core grant A1; the European Commission: Advanced Cell-based Therapies for the treatment of Primary ImmunoDeficiency (Cell-PID), HEALTH F5-2010-261387; and the Ministero della Salute: Ricerca Finalizzata, 005/RF-2009-1485896. Figures 2–6 were produced using Servier Medical Art.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ADA, adenosine deaminase; Adora, adenosine receptor(s); ANA, anti-nuclear antibody; BCR, B cell receptor; BMT, bone marrow transplantation; HLA, human leukocyte antigen; HSC, hematopoietic stem cell; HSC-GT, hematopoietic stem cell gene therapy; PEG-ADA, pegylated bovine ADA; SCID, severe combined immunodeficiency; TCR, T-cell receptor; TLR, Toll-like receptor; Tregs, naturally occurring regulatory T cells.
References
1
Abuchowski A. van Es T. Palczuk N. C. Davis F. F. (1977). Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J. Biol. Chem.252, 3578–3581.
2
Aiuti A. (2004). Gene therapy for adenosine-deaminase-deficient severe combined immunodeficiency. Best Pract. Res. Clin. Haematol.17, 505–516.10.1016/j.beha.2004.05.012
3
Aiuti A. Cassani B. Andolfi G. Mirolo M. Biasco L. Recchia A. Urbinati F. Valacca C. Scaramuzza S. Aker M. Slavin S. Cazzola M. Sartori D. Ambrosi A. Di Serio C. Roncarolo M. G. Mavilio F. Bordignon C. (2007). Multilineage hematopoietic reconstitution without clonal selection in ADA-SCID patients treated with stem cell gene therapy. J. Clin. Invest.117, 2233–2240.10.1172/JCI31666
4
Aiuti A. Cattaneo F. Galimberti F. Benninghoff U. Cassani B. Callegaro L. Scaramuzza S. Andolfi G. Mirolo M. Brigida I. Tabucchi A. Carlucci F. Eibl M. Aker M. Slavin S. Al-Mousa H. Al Ghonaium A. Ferster A. Duppenthaler A. Notarangelo L. Wintergerst U. Buckley R. Bregni M. Valsecchi M. Rossi P. Ciceri F. Miniero R. Bordignon C. Roncarolo M. (2009). Long-term safety and efficacy of gene therapy for adenosine deaminase (ADA)-deficient severe combined immunodeficiency. N. Engl. J. Med.360, 447–458.10.1056/NEJMoa0805817
5
Aiuti A. Ficara F. Cattaneo F. Bordignon C. Roncarolo M. G. (2003). Gene therapy for adenosine deaminase deficiency. Curr. Opin. Allergy Clin. Immunol.3, 461–466.10.1097/00130832-200312000-00007
6
Aldrich M. B. Chen W. Blackburn M. R. Martinez-Valdez H. Datta S. K. Kellems R. E. (2003). Impaired germinal center maturation in adenosine deaminase deficiency. J. Immunol.171, 5562–5570.
7
Antoine C. Muller S. Cant A. Cavazzana-Calvo M. Veys P. Vossen J. Fasth A. Heilmann C. Wulffraat N. Seger R. Blanche S. Friedrich W. Abinun M. Davies G. Bredius R. Schulz A. Landais P. Fischer A. (2003). Long-term survival and transplantation of haemopoietic stem cells for immunodeficiencies: report of the European experience 1968–1999. Lancet361, 553–560.10.1016/S0140-6736(03)12513-5
8
Apasov S. G. Blackburn M. R. Kellems R. E. Smith P. T. Sitkovsky M. V. (2001). Adenosine deaminase deficiency increases thymic apoptosis and causes defective T cell receptor signaling. J. Clin. Invest.108, 131–141.10.1172/JCI10360
9
Apasov S. G. Sitkovsky M. V. (1999). The extracellular versus intracellular mechanisms of inhibition of TCR-triggered activation in thymocytes by adenosine under conditions of inhibited adenosine deaminase. Int. Immunol.11, 179–189.10.1093/intimm/11.2.179
10
Bach J. F. (2003). Regulatory T cells under scrutiny. Nat. Rev. Immunol.3, 189–198.10.1038/nri1026
11
Barsalou J. Saint-Cyr C. Drouin E. Le Deist F. Haddad E. (2011). High prevalence of primary immune deficiencies in children with autoimmune disorders. Clin. Exp. Rheumatol.29, 125–130.
12
Belardinelli L. Linden J. Berne R. M. (1989). The cardiac effects of adenosine. Prog. Cardiovasc. Dis.32, 73–97.10.1016/0033-0620(89)90015-7
13
Benveniste P. Zhu W. Cohen A. (1995). Interference with thymocyte differentiation by an inhibitor of S-adenosylhomocysteine hydrolase. J. Immunol.155, 536–544.
14
Blackburn M. R. (2003). Too much of a good thing: adenosine overload in adenosine-deaminase-deficient mice. Trends Pharmacol. Sci.24, 66–70.10.1016/S0165-6147(02)00045-7
15
Blackburn M. R. Aldrich M. Volmer J. B. Chen W. Zhong H. Kelly S. Hershfield M. S. Datta S. K. Kellems R. E. (2000). The use of enzyme therapy to regulate the metabolic and phenotypic consequences of adenosine deaminase deficiency in mice. Differential impact on pulmonary and immunologic abnormalities. J. Biol. Chem.275, 32114–32121.10.1074/jbc.M005153200
16
Blackburn M. R. Datta S. K. Kellems R. E. (1998). Adenosine deaminase-deficient mice generated using a two-stage genetic engineering strategy exhibit a combined immunodeficiency. J. Biol. Chem.273, 5093–5100.10.1074/jbc.273.9.5093
17
Bollinger M. E. Arredondo-Vega F. X. Santisteban I. Schwarz K. Hershfield M. S. Lederman H. M. (1996). Brief report: hepatic dysfunction as a complication of adenosine deaminase deficiency. N. Engl. J. Med.334, 1367–1371.10.1056/NEJM199605233342104
18
Booth C. Gaspar H. B. (2009). Pegademase bovine (PEG-ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID). Biologics3, 349–358.
19
Booth C. Hershfield M. Notarangelo L. Buckley R. Hoenig M. Mahlaoui N. Cavazzana-Calvo M. Aiuti A. Gaspar H. B. (2007). Management options for adenosine deaminase deficiency; proceedings of the EBMT satellite workshop (Hamburg, March 2006). Clin. Immunol.123, 139–147.10.1016/j.clim.2006.12.009
20
Bordignon C. Roncarolo M. G. (2002). Therapeutic applications for hematopoietic stem cell gene transfer. Nat. Immunol.3, 318–321.10.1038/nrg784
21
Borsellino G. Kleinewietfeld M. Di Mitri D. Sternjak A. Diamantini A. Giometto R. Hopner S. Centonze D. Bernardi G. Dell’acqua M. L. Rossini P. M. Battistini L. Rotzschke O. Falk K. (2007). Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood110, 1225–1232.10.1182/blood-2006-12-064527
22
Buckley R. H. Schiff R. I. Schiff S. E. Markert M. L. Williams L. W. Harville T. O. Roberts J. L. Puck J. M. (1997). Human severe combined immunodeficiency: genetic, phenotypic, and functional diversity in one hundred eight infants. J. Pediatr.130, 378–387.10.1016/S0022-3476(97)70199-9
23
Cancrini C. Ferrua F. Scarselli A. Brigida I. Romiti M. L. Barera G. Finocchi A. Roncarolo M. G. Caniglia M. Aiuti A. (2010). Role of reduced intensity conditioning in T-cell and B-cell immune reconstitution after HLA-identical bone marrow transplantation in ADA-SCID. Haematologica95, 1778–1782.10.3324/haematol.2010.025098
24
Cancro M. P. (2009). Signalling crosstalk in B cells: managing worth and need. Nat. Rev. Immunol.9, 657–661.10.1038/nri2621
25
Carneiro-Sampaio M. Coutinho A. (2007). Tolerance and autoimmunity: lessons at the bedside of primary immunodeficiencies. Adv. Immunol.95, 51–82.10.1016/S0065-2776(07)95002-6
26
Cassani B. Mirolo M. Cattaneo F. Benninghoff U. Hershfield M. Carlucci F. Tabucchi A. Bordignon C. Roncarolo M. G. Aiuti A. (2008). Altered intracellular and extracellular signaling leads to impaired T-cell functions in ADA-SCID patients. Blood111, 4209–4219.10.1182/blood-2007-05-092429
27
Chaffee S. Mary A. Stiehm E. R. Girault D. Fischer A. Hershfield M. S. (1992). IgG antibody response to polyethylene glycol-modified adenosine deaminase in patients with adenosine deaminase deficiency. J. Clin. Invest.89, 1643–1651.10.1172/JCI115761
28
Chan B. Wara D. Bastian J. Hershfield M. S. Bohnsack J. Azen C. G. Parkman R. Weinberg K. Kohn D. B. (2005). Long-term efficacy of enzyme replacement therapy for adenosine deaminase (ADA)-deficient severe combined immunodeficiency (SCID). Clin. Immunol.117, 133–143.10.1016/j.clim.2005.07.006
29
Churchill P. C. Bidani A. K. (1982). Hypothesis: adenosine mediates hemodynamic changes in renal failure. Med. Hypotheses8, 275–285.10.1016/0306-9877(82)90124-4
30
Cohen A. Thompson E. (1986). DNA repair in nondividing human lymphocytes: inhibition by deoxyadenosine. Cancer Res.46, 1585–1588.
31
Cunningham-Rundles C. (2011). Autoimmunity in primary immune deficiency: taking lessons from our patients. Clin. Exp. Immunol.164(Suppl. 2), 6–11.10.1111/j.1365-2249.2011.04388.x
32
Cuss A. K. Avery D. T. Cannons J. L. Yu L. J. Nichols K. E. Shaw P. J. Tangye S. G. (2006). Expansion of functionally immature transitional B cells is associated with human-immunodeficient states characterized by impaired humoral immunity. J. Immunol.176, 1506–1516.
33
Daikeler T. Tyndall A. (2007). Autoimmunity following haematopoietic stem-cell transplantation. Best Pract. Res. Clin. Haematol.20, 349–360.10.1016/j.beha.2006.09.008
34
Dave U. P. Akagi K. Tripathi R. Cleveland S. M. Thompson M. A. Yi M. Stephens R. Downing J. R. Jenkins N. A. Copeland N. G. (2009). Murine leukemias with retroviral insertions at Lmo2 are predictive of the leukemias induced in SCID-X1 patients following retroviral gene therapy. PLoS Genet.5, e1000491.10.1371/journal.pgen.1000491
35
Davis S. Abuchowski A. Park Y. K. Davis F. F. (1981). Alteration of the circulating life and antigenic properties of bovine adenosine deaminase in mice by attachment of polyethylene glycol. Clin. Exp. Immunol.46, 649–652.
36
Deaglio S. Dwyer K. M. Gao W. Friedman D. Usheva A. Erat A. Chen J. F. Enjyoji K. Linden J. Oukka M. Kuchroo V. K. Strom T. B. Robson S. C. (2007). Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med.204, 1257–1265.10.1084/jem.20062512
37
Elliott M. R. Chekeni F. B. Trampont P. C. Lazarowski E. R. Kadl A. Walk S. F. Park D. Woodson R. I. Ostankovich M. Sharma P. Lysiak J. J. Harden T. K. Leitinger N. Ravichandran K. S. (2009). Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature461, 282–286.10.1038/nature08296
38
Erdmann A. A. Gao Z. G. Jung U. Foley J. Borenstein T. Jacobson K. A. Fowler D. H. (2005). Activation of Th1 and Tc1 cell adenosine A2A receptors directly inhibits IL-2 secretion in vitro and IL-2-driven expansion in vivo. Blood105, 4707–4714.10.1182/blood-2004-04-1407
39
Etzioni A. (2003). Immune deficiency and autoimmunity. Autoimmun. Rev.2, 364–369.10.1016/S1568-9972(03)00052-1
40
Ferrua F. Brigida I. Aiuti A. (2010). Update on gene therapy for adenosine deaminase-deficient severe combined immunodeficiency. Curr. Opin. Allergy Clin. Immunol.10, 551–556.10.1097/ACI.0b013e32833fea85
41
Fischer A. Le Deist F. Hacein-Bey-Abina S. Andre-Schmutz I. Basile Gde S. De Villartay J. P. Cavazzana-Calvo M. (2005). Severe combined immunodeficiency. A model disease for molecular immunology and therapy. Immunol. Rev.203, 98–109.10.1111/j.0105-2896.2005.00223.x
42
Fredholm B. B. Dunwiddie T. V. (1988). How does adenosine inhibit transmitter release?Trends Pharmacol. Sci.9, 130–134.10.1016/0165-6147(88)90194-0
43
Fukunaga A. F. Flacke W. E. Bloor B. C. (1982). Hypotensive effects of adenosine and adenosine triphosphate compared with sodium nitroprusside. Anesth. Analg.61, 273–278.10.1213/00000539-198203000-00010
44
Gaipl U. S. Sheriff A. Franz S. Munoz L. E. Voll R. E. Kalden J. R. Herrmann M. (2006). Inefficient clearance of dying cells and autoreactivity. Curr. Top. Microbiol. Immunol.305, 161–176.10.1007/3-540-29714-6_8
45
Gangi-Peterson L. Sorscher D. H. Reynolds J. W. Kepler T. B. Mitchell B. S. (1999). Nucleotide pool imbalance and adenosine deaminase deficiency induce alterations of N-region insertions during V(D)J recombination. J. Clin. Invest.103, 833–841.10.1172/JCI4320
46
Gaspar H. B. Aiuti A. Porta F. Candotti F. Hershfield M. S. Notarangelo L. D. (2009). How I treat ADA deficiency. Blood114, 3524–3532.10.1182/blood-2009-06-189209
47
Gaspar H. B. Cooray S. Gilmour K. C. Parsley K. L. Zhang F. Adams S. Bjorkegren E. Bayford J. Brown L. Davies E. G. Veys P. Fairbanks L. Bordon V. Petropolou T. Kinnon C. Thrasher A. J. (2011). Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci. Transl. Med.3, 97ra80.10.1126/scitranslmed.3002715
48
Giblett E. R. Anderson J. E. Cohen F. Pollara B. Meuwissen H. J. (1972). Adenosine-deaminase deficiency in two patients with severely impaired cellular immunity. Lancet2, 1067–1069.10.1016/S0140-6736(72)92345-8
49
Goodnow C. C. (1996). Balancing immunity and tolerance: deleting and tuning lymphocyte repertoires. Proc. Natl. Acad. Sci. U.S.A.93, 2264–2271.10.1073/pnas.93.6.2264
50
Hacein-Bey-Abina S. Garrigue A. Wang G. P. Soulier J. Lim A. Morillon E. Clappier E. Caccavelli L. Delabesse E. Beldjord K. Asnafi V. Macintyre E. Dal Cortivo L. Radford I. Brousse N. Sigaux F. Moshous D. Hauer J. Borkhardt A. Belohradsky B. H. Wintergerst U. Velez M. C. Leiva L. Sorensen R. Wulffraat N. Blanche S. Bushman F. D. Fischer A. Cavazzana-Calvo M. (2008). Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest.118, 3132–3142.10.1172/JCI35700
51
Hasko G. Linden J. Cronstein B. Pacher P. (2008). Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov.7, 759–770.10.1038/nrd2638
52
Hassan A. Booth C. Brightwell A. Allwood Z. Veys P. Rao K. Honig M. Friedrich W. Gennery A. Slatter M. Bredius R. Finocchi A. Cancrini C. Aiuti A. Porta F. Lanfranchi A. Ridella M. Steward C. Filipovich A. Marsh R. Bordon V. Al-Muhsen S. Al-Mousa H. Alsum Z. Al-Dhekri H. Al Ghonaium A. Speckmann C. Fischer A. Mahlaoui N. Nichols K. E. Grunebaum E. Al Zahrani D. Roifman C. M. Boelens J. Davies E. G. Cavazzana-Calvo M. Notarangelo L. Gaspar H. B. (2012). Outcome of hematopoietic stem cell transplantation for adenosine deaminase deficient severe combined immunodeficiency. Blood. [Epub ahead of print].10.1182/blood-2011-12-396879
53
Hauri-Hohl M. M. Keller M. P. Gill J. Hafen K. Pachlatko E. Boulay T. Peter A. Hollander G. A. Krenger W. (2007). Donor T-cell alloreactivity against host thymic epithelium limits T-cell development after bone marrow transplantation. Blood109, 4080–4088.10.1182/blood-2006-07-034157
54
Hershfield M. (2005). New insights into adenosine-receptor-mediated immunosuppression and the role of adenosine in causing the immunodeficiency associated with adenosine deaminase deficiency. Eur. J. Immunol.35, 25–30.10.1002/eji.200425738
55
Hershfield M. S. (1995). PEG-ADA replacement therapy for adenosine deaminase deficiency: an update after 8.5 years. Clin. Immunol. Immunopathol.76, S228–S232.10.1016/S0090-1229(95)90306-2
56
Hershfield M. S. Buckley R. H. Greenberg M. L. Melton A. L. Schiff R. Hatem C. Kurtzberg J. Markert M. L. Kobayashi R. H. Kobayashi A. L. Abuchowski A. (1987). Treatment of adenosine deaminase deficiency with polyethylene glycol-modified adenosine deaminase. N. Engl. J. Med.316, 589–596.10.1056/NEJM198703053161005
57
Hershfield M. S. Kredich N. M. Ownby D. R. Ownby H. Buckley R. (1979). In vivo inactivation of erythrocyte S-adenosylhomocysteine hydrolase by 2′-deoxyadenosine in adenosine deaminase-deficient patients. J. Clin. Invest.63, 807–811.10.1172/JCI109367
58
Hill N. J. Hultcrantz M. Sarvetnick N. Flodstrom-Tullberg M. (2007). The target tissue in autoimmunity – an influential niche. Eur. J. Immunol.37, 589–597.10.1002/eji.200636368
59
Hirschhorn R. Candotti F. (2006). “Immunodeficiency due to defects of purine metabolism,” in Primary Immunodeficiency Diseases, eds OchsH. D.SmithC. I. E.PuckJ. M. (Oxford: Oxford University Press), 169–196.
60
Hirschhorn R. Nicknam M. N. Eng F. Yang D. R. Borkowsky W. (1992). Novel deletion and a new missense mutation (Glu 217 Lys) at the catalytic site in two adenosine deaminase alleles of a patient with neonatal onset adenosine deaminase – severe combined immunodeficiency. J. Immunol.149, 3107–3112.
61
Hirschhorn R. Ratech H. (1980). Isozymes of adenosine deaminase. Isozymes Curr. Top. Biol. Med. Res.4, 131–157.
62
Honig M. Albert M. H. Schulz A. Sparber-Sauer M. Schutz C. Belohradsky B. Gungor T. Rojewski M. T. Bode H. Pannicke U. Lippold D. Schwarz K. Debatin K. M. Hershfield M. S. Friedrich W. (2007). Patients with adenosine deaminase deficiency surviving after hematopoietic stem cell transplantation are at high risk of CNS complications. Blood109, 3595–3602.10.1182/blood-2006-07-034678
63
Howe S. J. Mansour M. R. Schwarzwaelder K. Bartholomae C. Hubank M. Kempski H. Brugman M. H. Pike-Overzet K. Chatters S. J. de Ridder D. Gilmour K. C. Adams S. Thornhill S. I. Parsley K. L. Staal F. J. Gale R. E. Linch D. C. Bayford J. Brown L. Quaye M. Kinnon C. Ancliff P. Webb D. K. Schmidt M. Von Kalle C. Gaspar H. B. Thrasher A. J. (2008). Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest.118, 3143–3150.10.1172/JCI35798
64
Huang S. Apasov S. Koshiba M. Sitkovsky M. (1997). Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood90, 1600–1610.
65
Isnardi I. Ng Y. S. Srdanovic I. Motaghedi R. Rudchenko S. Von Bernuth H. Zhang S. Y. Puel A. Jouanguy E. Picard C. Garty B. Z. Camcioglu Y. Doffinger R. Kumararatne D. Davies G. Gallin J. I. Haraguchi S. Day N. K. Casanova J. L. Meffre E. (2008). IRAK-4- and MyD88-dependent pathways are essential for the removal of developing autoreactive B cells in humans. Immunity29, 746–757.10.1016/j.immuni.2008.09.015
66
Junger W. G. (2011). Immune cell regulation by autocrine purinergic signalling. Nat. Rev. Immunol.11, 201–212.10.1038/nri2938
67
Kameoka J. Tanaka T. Nojima Y. Schlossman S. F. Morimoto C. (1993). Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science261, 466–469.10.1126/science.8101391
68
Klein L. Hinterberger M. Wirnsberger G. Kyewski B. (2009). Antigen presentation in the thymus for positive selection and central tolerance induction. Nat. Rev. Immunol.9, 833–844.10.1038/nri2669
69
Kohn D. B. (1996). Gene therapy for hematopoietic and immune disorders. Bone Marrow Transplant.18(Suppl. 3), S55–S58.
70
Kohn D. B. (2008). Gene therapy for childhood immunological diseases. Bone Marrow Transplant.41, 199–205.10.1038/sj.bmt.1705895
71
Koller C. A. Orringer E. P. Berkowitz L. R. Mulhern A. T. (1984). Role of glycolysis in deoxyadenosine induced ATP depletion and dATP accumulation in red cells. Prog. Clin. Biol. Res.165, 227–239.
72
Kono D. H. Haraldsson M. K. Lawson B. R. Pollard K. M. Koh Y. T. Du X. Arnold C. N. Baccala R. Silverman G. J. Beutler B. A. Theofilopoulos A. N. (2009). Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc. Natl. Acad. Sci. U.S.A.106, 12061–12066.10.1073/pnas.0905441106
73
Lainka E. Hershfield M. S. Santisteban I. Bali P. Seibt A. Neubert J. Friedrich W. Niehues T. (2005). polyethylene glycol-conjugated adenosine deaminase (ADA) therapy provides temporary immune reconstitution to a child with delayed-onset ADA deficiency. Clin. Diagn. Lab. Immunol.12, 861–866.
74
Lappas C. M. Rieger J. M. Linden J. (2005). A2A adenosine receptor induction inhibits IFN-gamma production in murine CD4+ T cells. J. Immunol.174, 1073–1080.
75
Levy Y. Hershfield M. S. Fernandez-Mejia C. Polmar S. H. Scudiery D. Berger M. Sorensen R. U. (1988). Adenosine deaminase deficiency with late onset of recurrent infections: response to treatment with polyethylene glycol-modified adenosine deaminase. J. Pediatr.113, 312–317.10.1016/S0022-3476(88)80271-3
76
Liao H. Hyman M. C. Baek A. E. Fukase K. Pinsky D. J. (2010). cAMP/CREB-mediated transcriptional regulation of ectonucleoside triphosphate diphosphohydrolase 1 (CD39) expression. J. Biol. Chem.285, 14791–14805.10.1074/jbc.M110.169599
77
Luning Prak E. T. (2012). Restoring balance to B cells in ADA deficiency. J. Clin. Invest.122, 1960–1962.10.1172/JCI63782
78
Malacarne F. Benicchi T. Notarangelo L. D. Mori L. Parolini S. Caimi L. Hershfield M. Imberti L. (2005). Reduced thymic output, increased spontaneous apoptosis and oligoclonal B cells in polyethylene glycol-adenosine deaminase-treated patients. Eur. J. Immunol.35, 3376–3386.10.1002/eji.200526248
79
Mandapathil M. Hilldorfer B. Szczepanski M. J. Czystowska M. Szajnik M. Ren J. Lang S. Jackson E. K. Gorelik E. Whiteside T. L. (2010). Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells(TREG). J. Biol. Chem.285, 7176–7186.10.1074/jbc.M109.047423
80
Marshak-Rothstein A. (2006). Toll-like receptors in systemic autoimmune disease. Nat. Rev. Immunol.6, 823–835.10.1038/nri1957
81
Mathis D. Benoist C. (2004). Back to central tolerance. Immunity20, 509–516.10.1016/S1074-7613(04)00111-6
82
Mazzolari E. de Martiis D. Forino C. Lanfranchi A. Giliani S. Marzollo R. Airo P. Imberti L. Porta F. Notarangelo L. D. (2009). Single-center analysis of long-term outcome after hematopoietic cell transplantation in children with congenital severe T cell immunodeficiency. Immunol. Res.44, 4–17.10.1007/s12026-008-8022-4
83
Meffre E. (2012). The establishment of early B cell tolerance in humans: lessons from primary immunodeficiency diseases. Ann. N. Y. Acad. Sci.1246, 1–10.10.1111/j.1749-6632.2011.06347.x
84
Meffre E. Wardemann H. (2008). B-cell tolerance checkpoints in health and autoimmunity. Curr. Opin. Immunol.20, 632–638.10.1016/j.coi.2008.09.001
85
Menard L. Saadoun D. Isnardi I. Ng Y. S. Meyers G. Massad C. Price C. Abraham C. Motaghedi R. Buckner J. H. Gregersen P. K. Meffre E. (2011). The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J. Clin. Invest.121, 3635–3644.10.1172/JCI45790
86
Minguet S. Huber M. Rosenkranz L. Schamel W. W. Reth M. Brummer T. (2005). Adenosine and cAMP are potent inhibitors of the NF-kappa B pathway downstream of immunoreceptors. Eur. J. Immunol.35, 31–41.10.1002/eji.200425524
87
Montiel-Equihua C. A. Thrasher A. J. Gaspar H. B. (2012). Gene therapy for severe combined immunodeficiency due to adenosine deaminase deficiency. Curr. Gene Ther.12, 57–65.10.2174/156652312799789253
88
Moratto D. Giliani S. Bonfim C. Mazzolari E. Fischer A. Ochs H. D. Cant A. J. Thrasher A. J. Cowan M. J. Albert M. H. Small T. Pai S. Y. Haddad E. Lisa A. Hambleton S. Slatter M. Cavazzana-Calvo M. Mahlaoui N. Picard C. Torgerson T. R. Burroughs L. Koliski A. Neto J. Z. Porta F. Qasim W. Veys P. Kavanau K. Honig M. Schulz A. Friedrich W. Notarangelo L. D. (2011). Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott–Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980–2009: an international collaborative study. Blood118, 1675–1684.10.1182/blood-2010-11-319376
89
Morgan G. Levinsky R. J. Hugh-Jones K. Fairbanks L. D. Morris G. S. Simmonds H. A. (1987). Heterogeneity of biochemical, clinical and immunological parameters in severe combined immunodeficiency due to adenosine deaminase deficiency. Clin. Exp. Immunol.70, 491–499.
90
Mortellaro A. Jofra Hernandez R. Guerrini M. M. Carlucci F. Tabucchi A. Ponzoni M. Sanvito F. Doglioni C. Di Serio C. Biasco L. Follenzi A. Naldini L. Bordignon C. Roncarolo M. G. Aiuti A. (2006). Ex vivo gene therapy with lentiviral vectors rescues adenosine deaminase (ADA)-deficient mice and corrects their immune and metabolic defects. Blood108, 2979–2988.10.1182/blood-2006-05-023507
91
Nemazee D. Kouskoff V. Hertz M. Lang J. Melamed D. Pape K. Retter M. (2000). B-cell-receptor-dependent positive and negative selection in immature B cells. Curr. Top. Microbiol. Immunol.245, 57–71.10.1007/978-3-642-59641-4_3
92
Neven B. Leroy S. Decaluwe H. Le Deist F. Picard C. Moshous D. Mahlaoui N. Debre M. Casanova J. L. Dal Cortivo L. Madec Y. Hacein-Bey-Abina S. De Saint Basile G. De Villartay J. P. Blanche S. Cavazzana-Calvo M. Fischer A. (2009). Long-term outcome after haematopoietic stem cell transplantation of a single-centre cohort of 90 patients with severe combined immunodeficiency: long-term outcome of HSCT in SCID. Blood113, 4114–4124.10.1182/blood-2008-09-177923
93
Ng Y. S. Wardemann H. Chelnis J. Cunningham-Rundles C. Meffre E. (2004). Bruton’s tyrosine kinase is essential for human B cell tolerance. J. Exp. Med.200, 927–934.10.1084/jem.20040920
94
Notarangelo L. D. (2009). Primary immunodeficiencies (PIDs) presenting with cytopenias. Hematology Am. Soc. Hematol. Educ. Program2009, 139–143.10.1182/asheducation-2009.1.139
95
Notarangelo L. D. Stoppoloni G. Toraldo R. Mazzolari E. Coletta A. Airo P. Bordignon C. Ugazio A. G. (1992). Insulin-dependent diabetes mellitus and severe atopic dermatitis in a child with adenosine deaminase deficiency. Eur. J. Pediatr.151, 811–814.10.1007/BF01959086
96
Ochs H. D. Buckley R. H. Kobayashi R. H. Kobayashi A. L. Sorensen R. U. Douglas S. D. Hamilton B. L. Hershfield M. S. (1992). Antibody responses to bacteriophage phi X174 in patients with adenosine deaminase deficiency. Blood80, 1163–1171.
97
Ohta A. Madasu M. Kini R. Subramanian M. Goel N. Sitkovsky M. (2009). A2A adenosine receptor may allow expansion of T cells lacking effector functions in extracellular adenosine-rich microenvironments. J. Immunol.183, 5487–5493.10.4049/jimmunol.0901247
98
Olah M. E. Stiles G. L. (1995). Adenosine receptor subtypes: characterization and therapeutic regulation. Annu. Rev. Pharmacol. Toxicol.35, 581–606.10.1146/annurev.pa.35.040195.003053
99
Ott M. G. Schmidt M. Schwarzwaelder K. Stein S. Siler U. Koehl U. Glimm H. Kuhlcke K. Schilz A. Kunkel H. Naundorf S. Brinkmann A. Deichmann A. Fischer M. Ball C. Pilz I. Dunbar C. Du Y. Jenkins N. A. Copeland N. G. Luthi U. Hassan M. Thrasher A. J. Hoelzer D. Von Kalle C. Seger R. Grez M. (2006). Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med.12, 401–409.10.1038/nm1393
100
Ozsahin H. Arredondo-Vega F. X. Santisteban I. Fuhrer H. Tuchschmid P. Jochum W. Aguzzi A. Lederman H. M. Fleischman A. Winkelstein J. A. Seger R. A. Hershfield M. S. (1997). Adenosine deaminase deficiency in adults. Blood89, 2849–2855.
101
Panther E. Corinti S. Idzko M. Herouy Y. Napp M. La Sala A. Girolomoni G. Norgauer J. (2003). Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T-cell stimulatory capacity of human dendritic cells. Blood101, 3985–3990.10.1182/blood-2002-07-2113
102
Parish I. A. Heath W. R. (2008). Too dangerous to ignore: self-tolerance and the control of ignorant autoreactive T cells. Immunol. Cell Biol.86, 146–152.10.1038/sj.icb.7100161
103
Park J. Y. Shcherbina A. Rosen F. S. Prodeus A. P. Remold-O’Donnell E. (2005). Phenotypic perturbation of B cells in the Wiskott–Aldrich syndrome. Clin. Exp. Immunol.139, 297–305.10.1111/j.1365-2249.2005.02693.x
104
Parkman R. Gelfand E. W. Rosen F. S. Sanderson A. Hirschhorn R. (1975). Severe combined immunodeficiency and adenosine deaminase deficiency. N. Engl. J. Med.292, 714–719.10.1056/NEJM197504032921402
105
Piccirillo C. A. Thornton A. M. (2004). Cornerstone of peripheral tolerance: naturally occurring CD4+CD25+ regulatory T cells. Trends Immunol.25, 374–380.10.1016/j.it.2004.04.009
106
Poliani P. L. Vermi W. Facchetti F. (2009). Thymus microenvironment in human primary immunodeficiency diseases. Curr. Opin. Allergy Clin. Immunol.9, 489–495.10.1097/ACI.0b013e3283327e5c
107
Power Coombs M. R. Belderbos M. E. Gallington L. C. Bont L. Levy O. (2011). Adenosine modulates Toll-like receptor function: basic mechanisms and translational opportunities. Expert Rev. Anti. Infect. Ther.9, 261–269.10.1586/eri.10.158
108
Ratech H. Hirschhorn R. Greco M. A. (1989). Pathologic findings in adenosine deaminase deficient-severe combined immunodeficiency. II. Thymus, spleen, lymph node, and gastrointestinal tract lymphoid tissue alterations. Am. J. Pathol.135, 1145–1156.
109
Ratter F. Germer M. Fischbach T. Schulze-Osthoff K. Peter M. E. Droge W. Krammer P. H. Lehmann V. (1996). S-adenosylhomocysteine as a physiological modulator of Apo-1-mediated apoptosis. Int. Immunol.8, 1139–1147.10.1093/intimm/8.7.1139
110
Rogers M. H. Lwin R. Fairbanks L. Gerritsen B. Gaspar H. B. (2001). Cognitive and behavioral abnormalities in adenosine deaminase deficient severe combined immunodeficiency. J. Pediatr.139, 44–50.10.1067/mpd.2001.115023
111
Saccucci P. Manca Bitti M. L. Bottini N. Rapini N. Piccinini S. D’Annibale F. Chiarelli F. Verrotti A. Bottini E. Gloria-Bottini F. (2009). Type 1 diabetes: evidence of interaction between ACP1 and ADA1 gene polymorphisms. Med. Sci. Monit.15, CR511–C517.
112
Sakaguchi S. (2005). Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat. Immunol.6, 345–352.10.1038/nrm1620
113
Sakaguchi S. Yamaguchi T. Nomura T. Ono M. (2008). Regulatory T cells and immune tolerance. Cell133, 775–787.10.1016/j.cell.2008.05.009
114
Sauer A. V. Aiuti A. (2009). New insights into the pathogenesis of adenosine deaminase-severe combined immunodeficiency and progress in gene therapy. Curr. Opin. Allergy Clin. Immunol.9, 496–502.10.1097/ACI.0b013e3283327da5
115
Sauer A. V. Brigida I. Carriglio N. Jofra Hernandez R. Scaramuzza S. Clavenna D. Sanvito F. Poliani P. L. Gagliani N. Carlucci F. Tabucchi A. Roncarolo M. G. Traggiai E. Villa A. Aiuti A. (2012a). Alterations in the adenosine metabolism and CD39/CD73 adenosinergic machinery cause loss of Treg cell function and autoimmunity in ADA-deficient SCID. Blood119, 1428–1439.10.1182/blood-2011-07-366781
116
Sauer A. V. Morbach H. Brigida I. Ng Y. S. Aiuti A. Meffre E. (2012b). Defective B cell tolerance in adenosine deaminase deficiency is corrected by gene therapy. J. Clin. Invest.122, 2141–2152.10.1172/JCI61788
117
Sauer A. V. Mrak E. Jofra Hernandez R. Zacchi E. Cavani F. Casiraghi M. Grunebaum E. Roifman C. M. Cervi M. C. Ambrosi A. Carlucci F. Roncarolo M. G. Villa A. Rubinacci A. Aiuti A. (2009). ADA-deficient SCID is associated with a specific microenvironment and bone phenotype characterized by RANKL/OPG imbalance and osteoblast insufficiency. Blood114, 3216–3226.10.1182/blood-2009-03-209221
118
Schenk U. Frascoli M. Proietti M. Geffers R. Traggiai E. Buer J. Ricordi C. Westendorf A. M. Grassi F. (2011). ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci. Signal.4, ra12.10.1126/scisignal.2001270
119
Schenk U. Westendorf A. M. Radaelli E. Casati A. Ferro M. Fumagalli M. Verderio C. Buer J. Scanziani E. Grassi F. (2008). Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci. Signal.1, ra6.10.1126/scisignal.1160583
120
Sebastiani G. D. Bottini N. Greco E. Saccucci P. Canu G. Lucarelli P. Gloria-Bottini F. Fontana L. (2006). A study of Adenosine-Deaminase genetic polymorphism in rheumatoid arthritis. Int. J. Immunopathol. Pharmacol.23, 791–795.
121
Serana F. Sottini A. Chiarini M. Zanotti C. Ghidini C. Lanfranchi A. Notarangelo L. D. Caimi L. Imberti L. (2010). The different extent of B and T cell immune reconstitution after hematopoietic stem cell transplantation and enzyme replacement therapies in SCID patients with adenosine deaminase deficiency. J. Immunol.185, 7713–7722.10.4049/jimmunol.1001770
122
Shou Y. Ma Z. Lu T. Sorrentino B. P. (2006). Unique risk factors for insertional mutagenesis in a mouse model of XSCID gene therapy. Proc. Natl. Acad. Sci. U.S.A.103, 11730–11735.10.1073/pnas.0603635103
123
Siaw M. F. Mitchell B. S. Koller C. A. Coleman M. S. Hutton J. J. (1980). ATP depletion as a consequence of adenosine deaminase inhibition in man. Proc. Natl. Acad. Sci. U.S.A.77, 6157–6161.10.1073/pnas.77.10.6157
124
Simmonds H. A. Goday A. Morris G. S. Fairbanks L. D. Levinsky R. J. (1984). dATP accumulation and ATP depletion in platelets in adenosine deaminase deficiency: significance for the immune response?Biosci. Rep.4, 809–818.10.1007/BF01138162
125
Simmonds H. A. Levinsky R. J. Perrett D. Webster D. R. (1982). Reciprocal relationship between erythrocyte ATP and deoxy-ATP levels in inherited ADA deficiency. Biochem. Pharmacol.31, 947–951.10.1016/0006-2952(82)90325-2
126
Sitkovsky M. Lukashev D. Deaglio S. Dwyer K. Robson S. C. Ohta A. (2008). Adenosine A2A receptor antagonists: blockade of adenosinergic effects and T regulatory cells. Br. J. Pharmacol.153(Suppl. 1), S457–S464.
127
Sitkovsky M. V. Lukashev D. Apasov S. Kojima H. Koshiba M. Caldwell C. Ohta A. Thiel M. (2004). Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu. Rev. Immunol.22, 657–682.10.1146/annurev.immunol.22.012703.104731
128
Skalhegg B. S. Landmark B. F. Doskeland S. O. Hansson V. Lea T. Jahnsen T. (1992). Cyclic AMP-dependent protein kinase type I mediates the inhibitory effects of 3′,5′-cyclic adenosine monophosphate on cell replication in human T lymphocytes. J. Biol. Chem.267, 15707–15714.
129
Titman P. Pink E. Skucek E. O’Hanlon K. Cole T. J. Gaspar J. Xu-Bayford J. Jones A. Thrasher A. J. Davies E. G. Veys P. A. Gaspar H. B. (2008). Cognitive and behavioral abnormalities in children after hematopoietic stem cell transplantation for severe congenital immunodeficiencies. Blood112, 3907–3913.10.1182/blood-2008-04-151332
130
Trobridge G. D. (2011). Genotoxicity of retroviral hematopoietic stem cell gene therapy. Expert Opin. Biol. Ther.11, 581–593.10.1517/14712598.2011.562496
131
Utz P. J. Gensler T. J. Anderson P. (2000). Death, autoantigen modifications, and tolerance. Arthritis Res.2, 101–114.10.1186/ar75
132
Van de Wiele C. J. Joachims M. L. Fesler A. M. Vaughn J. G. Blackburn M. R. Mcgee S. T. Thompson L. F. (2006). Further differentiation of murine double-positive thymocytes is inhibited in adenosine deaminase-deficient murine fetal thymic organ culture. J. Immunol.176, 5925–5933.
133
Van de Wiele C. J. Vaughn J. G. Blackburn M. R. Ledent C. A. Jacobson M. Jiang H. Thompson L. F. (2002). Adenosine kinase inhibition promotes survival of fetal adenosine deaminase-deficient thymocytes by blocking dATP accumulation. J. Clin. Invest.110, 395–402.10.1172/JCI200215683
134
Vendetti S. Riccomi A. Sacchi A. Gatta L. Pioli C. De Magistris M. T. (2002). Cyclic adenosine 5′-monophosphate and calcium induce CD152 (CTLA-4) up-regulation in resting CD4+ T lymphocytes. J. Immunol.169, 6231–6235.
135
Vignali D. A. A. Collison L. W. Workman C. J. (2008). How regulatory T cells work. Nat. Rev. Immunol.8, 523–532.10.1038/nri2343
136
Waddell D. Ullman B. (1983). Characterization of a cultured human T-cell line with genetically altered ribonucleotide reductase activity. Model for immunodeficiency. J. Biol. Chem.258, 4226–4231.
137
Waldner H. Collins M. Kuchroo V. K. (2004). Activation of antigen-presenting cells by microbial products breaks self tolerance and induces autoimmune disease. J. Clin. Invest.113, 990–997.10.1172/JCI19388
138
Wardemann H. Nussenzweig M. C. (2007). B-cell self-tolerance in humans. Adv. Immunol.95, 83–110.10.1016/S0065-2776(07)95003-8
139
Wardemann H. Yurasov S. Schaefer A. Young J. W. Meffre E. Nussenzweig M. C. (2003). Predominant autoantibody production by early human B cell precursors. Science301, 1374–1377.10.1126/science.1086907
140
Weinberg K. Hershfield M. S. Bastian J. Kohn D. Sender L. Parkman R. Lenarsky C. (1993). T lymphocyte ontogeny in adenosine deaminase-deficient severe combined immune deficiency after treatment with polyethylene glycol-modified adenosine deaminase. J. Clin. Invest.92, 596–602.10.1172/JCI116626
141
Westerberg L. S. Klein C. Snapper S. B. (2008). Breakdown of T cell tolerance and autoimmunity in primary immunodeficiency – lessons learned from monogenic disorders in mice and men. Curr. Opin. Immunol.20, 646–654.10.1016/j.coi.2008.10.004
142
Yip L. Woehrle T. Corriden R. Hirsh M. Chen Y. Inoue Y. Ferrari V. Insel P. A. Junger W. G. (2009). Autocrine regulation of T-cell activation by ATP release and P2×7 receptors. FASEB J.23, 1685–1693.10.1096/fj.08-126458
143
Zarek P. E. Huang C. T. Lutz E. R. Kowalski J. Horton M. R. Linden J. Drake C. G. Powell J. D. (2008). A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood111, 251–259.10.1182/blood-2007-03-081646
Summary
Keywords
adenosine deaminase, severe combined immunodeficiency, ADA-SCID, autoimmunity, gene therapy
Citation
Sauer AV, Brigida I, Carriglio N and Aiuti A (2012) Autoimmune Dysregulation and Purine Metabolism in Adenosine Deaminase Deficiency. Front. Immun. 3:265. doi: 10.3389/fimmu.2012.00265
Received
08 May 2012
Accepted
02 August 2012
Published
27 August 2012
Volume
3 - 2012
Edited by
Rosa Bacchetta, San Raffaele Scientific Institute, Italy
Reviewed by
Rosa Bacchetta, San Raffaele Scientific Institute, Italy; Mirjam van Der Burg, Erasmus MC, Netherlands
Copyright
© 2012 Sauer, Brigida, Carriglio and Aiuti.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Alessandro Aiuti, San Raffaele Telethon Institute for Gene Therapy, Via Olgettina 58, Dibit 2A2, Milan 20132, Italy. e-mail: aiuti.alessandro@hsr.it
This article was submitted to Frontiers in Primary Immunodeficiencies, a specialty of Frontiers in Immunology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.