 Claudia M. Calvet
Claudia M. Calvet Tatiana G. Melo Luciana R. Garzoni Francisco O. R. Oliveira Jr. Dayse T. Silva Neto Maria N. S. L. Meirelles
Tatiana G. Melo Luciana R. Garzoni Francisco O. R. Oliveira Jr. Dayse T. Silva Neto Maria N. S. L. Meirelles Mirian C. S. Pereira*
Mirian C. S. Pereira*- Laboratório de Ultra-estrutura Celular, Fundação Oswaldo Cruz, Instituto Oswaldo Cruz, Rio de Janeiro, Rio de Janeiro, Brazil
Trypanosoma cruzi, the etiological agent of Chagas disease, exhibits multiple strategies to ensure its establishment and persistence in the host. Although this parasite has the ability to infect different organs, heart impairment is the most frequent clinical manifestation of the disease. Advances in knowledge of T. cruzi–cardiomyocyte interactions have contributed to a better understanding of the biological events involved in the pathogenesis of Chagas disease. This brief review focuses on the current understanding of molecules involved in T. cruzi–cardiomyocyte recognition, the mechanism of invasion, and on the effect of intracellular development of T. cruzi on the structural organization and molecular response of the target cell.
Introduction
Chagas disease, caused by Trypanosoma cruzi infection, has emerged as an important global public health problem due to the many Latin American T. cruzi-infected immigrants in non-endemic countries (Pérez-Molina et al., 2012). Although public health programs in the Southern Cone countries have reduced transmission by 70% (Moncayo and Silveira, 2009), blood and organ transplant transmissions in non-endemic countries (Rassi et al., 2009) and outbreaks of foodborne transmission (Pereira et al., 2009; Ríos et al., 2011) have drawn attention to Chagas disease. An estimated 8–15 million individuals in 18 endemic countries in Central and South America are infected, with approximately 30 million people at risk (WHO, 2010; Rassi et al., 2012). Chronic chagasic cardiomyopathy, the most relevant clinical manifestation, is the leading cause of death from heart failure in endemic countries, and accounts for a significant burden of ischemic and inflammatory heart disease in the USA and Europe due to “globalization” of Chagas disease (Moncayo and Silveira, 2009; Moolani et al., 2012). In this review, we summarize current knowledge of the biology of the T. cruzi–host cell interaction, highlighting molecular aspects of T. cruzi–cardiomyocyte interplay, with a focus on early infection events and the effect of intracellular parasite development on the structure and function of the target cell.
Cell Recognition and Invasion Process
T. cruzi–Cardiomyocyte Recognition
Interplay between parasite and host cell is essential for T. cruzi to successfully adjust to the different microenvironments it occupies in its vertebrate and invertebrate hosts. In the obligatory intracellular phase of its life cycle in the mammalian host, infection is driven by adhesion and internalization events involving a large variety of ligands and/or receptors on the surface of both the parasite and host cell interacting with one another to achieve recognition and invasion. Several different surface molecules in the cardiomyocyte have been implicated in adhesion and internalization by the parasite (Figure 1). Carbohydrate residues of membrane glycoconjugates in cardiomyocytes, including galactosyl, mannosyl, and sialyl residues, participate in T. cruzi cytoadherence (Barbosa and Meirelles, 1992, 1993), while mannose receptors at the surface of cardiomyocytes modulate parasite entry and are down-regulated by T. cruzi infection (Soeiro et al., 1999).
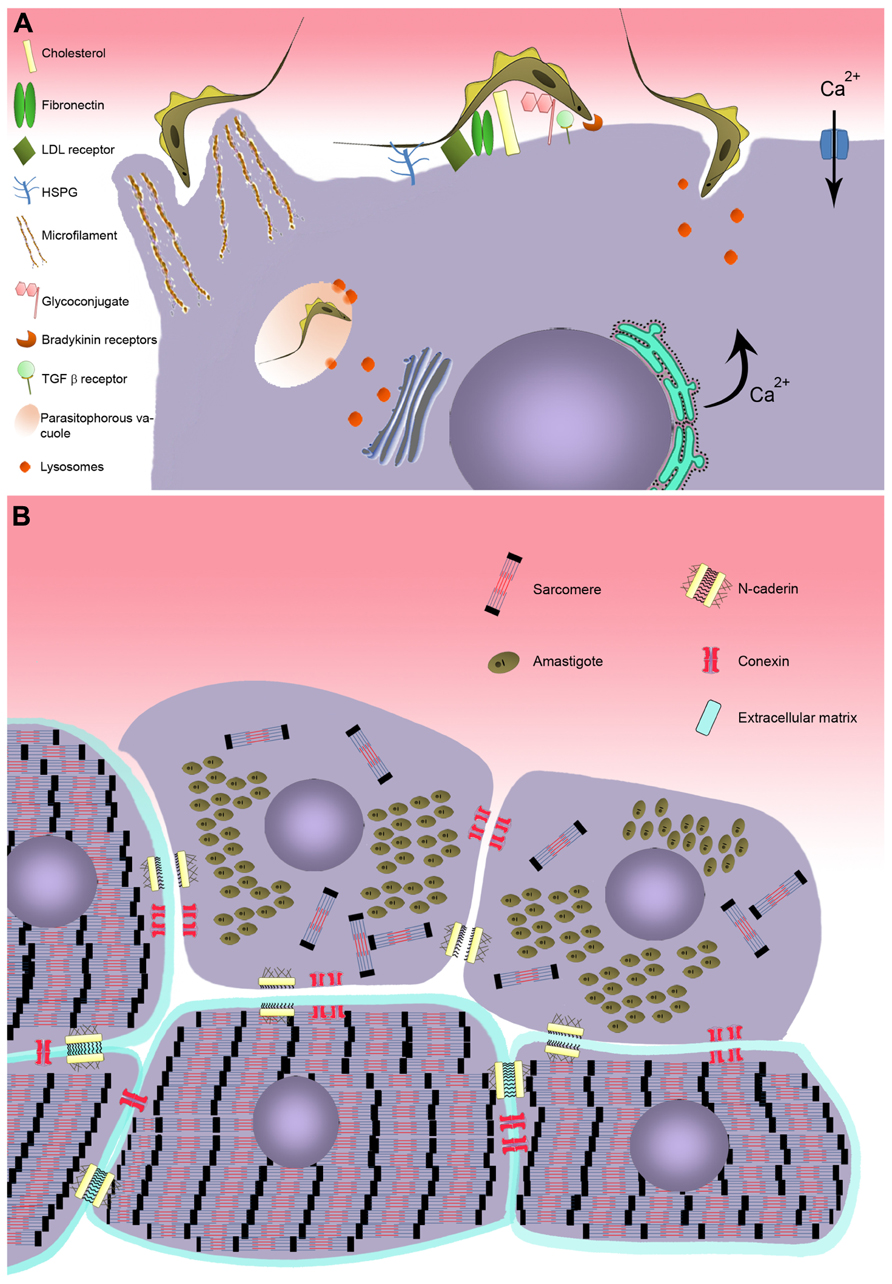
FIGURE 1. Model of T. cruzi invasion in cardiomyocyte. (A) Schematic model representing the recognition step involving cell surface molecules of cardiomyocyte during T. cruzi invasion. Two distinct mechanism of invasion are represented: actin-dependent and lysosome-dependent mechanisms. (B) Effect of T. cruzi infection on cardiomyocyte structure. Disturbance on the cardiomyocyte cytoarchitecture is evidenced after T. cruzi infection, showing breakdown of myofibrillar and disruption of adherent and gap junctions.
Extracellular matrix (ECM) components are also important in parasite–host cell recognition. Fibronectin, a high molecular weight glycoprotein present at the host cell surface, is recognized by fibronectin receptors of the parasite (Ouaissi et al., 1984), which interact with the RGDS (Arg-Gly-Asp-Ser) sequence of fibronectin and mediate parasite entry (Calvet et al., 2004). Immunization with RGDS peptide induced protection in an experimental murine model of acute T. cruzi infection (Ouaissi et al., 1986). Heparan sulfate proteoglycans (HSPG), another class of ECM component widely distributed in mammalian tissues, are also involved in T. cruzi attachment and invasion (Ortega-Barria and Pereira, 1991; Calvet et al., 2003). Treatment of trypomastigotes and amastigotes, the infective forms of T. cruzi, or cardiomyocytes with soluble heparan sulfate (HS) and heparitinase II, respectively, efficiently inhibited parasite invasion (Calvet et al., 2003; Oliveira Jr. et al., 2008; Bambino-Medeiros et al., 2011). The binding of T. cruzi to HSPG involves the recognition of the N-acetylated/N-sulfated domain of the HS chain by heparin-binding proteins (HBPs) present at the surface of the parasite (Oliveira Jr. et al., 2008). Although T. cruzi HBPs are capable of binding HS and chondroitin sulfate (CS), only the HS–HBPs interaction triggers parasite invasion in cardiomyocytes (Calvet et al., 2003; Oliveira Jr. et al., 2008), while HS and CS are involved in vector–T. cruzi interactions (Oliveira Jr. et al., 2012).
Lipids also play an important role in T. cruzi–host cell interplay. Membrane rafts, enriched in cholesterol and sphingolipids, appear to participate in the invasion process (Barrias et al., 2007; Fernandes et al., 2007; Priotto et al., 2009). Recently, cholesterol has been demonstrated to modulate invasion of cardiomyocytes by T. cruzi (Hissa et al., 2012). Depletion of cholesterol from cardiac cell membrane induced an 85–90% reduction of parasite invasion by inhibiting parasites’ association with lysosomes. Additionally, the low-density lipoprotein receptor, which is up-regulated in myocardium of infected mice, also coordinates parasite entry and fusion of the parasitophorous vacuole (PV) with lysosomes (Nagajyothi et al., 2011).
Mechanisms of T.cruzi Invasion
The large number of molecules involved in recognition of target cells by T. cruzi increases the parasite’s capacity to explore multiple strategies to ensure propagation in the mammalian host. A number of different mechanisms of T. cruzi invasion have been described, involving distinct host cell type, parasite genotype, and developmental stage. At least five models of invasion have been elucidated. (i) An actin-dependent mechanism leads to the rearrangement of microfilaments, inducing the host cell membrane to enclose the parasite (Barbosa and Meirelles, 1995; Procópio et al., 1999; Rosestolato et al., 2002; Ferreira et al., 2006). (ii) Lysosome-dependent mechanisms, involving an increase of transient cytosolic Ca2+ levels induced by the parasite, generate cortical actin depolymerization and lysosome recruitment to the parasite binding site (Rodríguez et al., 1999; Hissa et al., 2012). (iii) Activated signaling pathways also participate, including tyrosine kinase receptors (TrKA and TrKC; de Melo-Jorge and PereiraPerrin, 2007; Weinkauf et al., 2011) and phosphatidylinositol 3-kinase (PI3-K; Todorov et al., 2000; Chuenkova et al., 2001; Wilkowsky et al., 2001; Vieira et al., 2002; Woolsey et al., 2003), bradykinin receptors (Scharfstein et al., 2000; Todorov et al., 2003), and transforming growth factor β (TGF-β; Ming et al., 1995; Waghabi et al., 2007). (iv) More recently, sphingomyelinase-mediated plasma membrane repair has been proposed to participate in this process (Fernandes et al., 2011; Fernandes and Andrews, 2012), as has (v) the host cell autophagy pathway (Romano et al., 2009, 2012). Finally, the combination of different mechanisms has been described as coordinating the T. cruzi invasion process (Butler and Tyler, 2012).
Elevation of transient intracellular Ca2+ levels, an invasion-related effect provoked by T. cruzi binding to the host cell membrane (Figure 1), has also been demonstrated in cardiac cells (Barr et al., 1996; Garzoni et al., 2003). The increase of cytosolic [Ca2+] has been reported to be brought about in two different ways: (i) by sarcoplasmic reticulum stores, which are sensitive to leupeptin, suggesting a cortical actin depolymerization and lysosome-dependent mechanism of invasion (Barr et al., 1996), and by (ii) extracellular Ca2+ influx through membrane Ca2+ channels, which are insensitive to leupeptin (Garzoni et al., 2003). Recently, it has been suggested that Ca2+ influx may also occur as a result of lesions on the plasma membrane, suggesting that the membrane repair pathway frequently observed in muscle cells may also be involved in cardiac cell invasion by T. cruzi (Fernandes and Andrews, 2012).
Transforming growth factor β, a multifunctional family of proteins that controls a range of biological events in most cells, including proliferation and cellular differentiation (Moustakas et al., 2002), has also been shown to participate in T. cruzi invasion of cardiomyocytes (Waghabi et al., 2005). T. cruzi directly activates latent TGF-β and modulates TGF-β signaling (Waghabi et al., 2005). Inhibition of T. cruzi infection in cardiomyocyte was achieved by blockage of the TGF-β receptor type I (TGFβRI)/Smad2 signaling pathway by SB-431542, a TGF-β signaling inhibitor (Waghabi et al., 2007). Besides impairment of parasite invasion, the inhibitor treatment also reduced T. cruzi intracellular multiplication and differentiation. Recently, the therapeutic effectiveness of GW788388, an oral inhibitor of TGF-β signaling, has been demonstrated experimentally in acute phase T. cruzi infection, leading to a reduction of parasitemia and mortality, and also preventing cardiac fibrosis (de Oliveira et al., 2012).
Bradykinin receptors (B2R/B1R) have also been reported to be involved in cardiomyocyte infection by T. cruzi (Todorov et al., 2003). This mechanism of invasion is regulated by cooperation between HSPG, kininogen, and cruzipain-1, the major cysteine protease isoform of T. cruzi, resulting in the release of kinin. Invasion through the kinin transduction pathway, activated by G protein-coupled bradykinin receptors, induces intracellular Ca2+ mobilization from stores in the endoplasmic reticulum (Scharfstein et al., 2000). The B2R agonist captopril stimulates the invasion of T. cruzi while B2R and B1R antagonists, present inhibitory effects on cardiomyocytes, suggesting that these receptors interdependently drive invasion of the parasite (Todorov et al., 2003).
As evidenced in other non-professional phagocytic cells (Rosestolato et al., 2002; Ferreira et al., 2006), T. cruzi entry is also mediated by an endocytic process in cardiac muscle. A protrusion of cardiomyocyte plasma membrane, orchestrated by cytoskeleton rearrangement, is observed during T. cruzi–cardiomyocyte interplay. A dense actin-based membrane skeleton meshwork projects from the sarcolemma and encloses the entering parasite (Barbosa and Meirelles, 1995). This event was drastically inhibited (75%) when cardiac cells were treated with cytochalasin D, an agent that depolymerizes actin filaments, prior to T. cruzi infection; no parasite invasion was observed in fixed cardiomyocytes (Barbosa and Meirelles, 1995). Once inside the cells, the parasite is located within a PV that lacks Ca2+–Mg2+-ATPase, adenylate cyclase, and anionic sites (Meirelles et al., 1986) but has carbohydrate residues such as N-acetylglucosamine and N-acetylgalactosamine (Barbosa and Meirelles, 1992, 1993). Ultrastructural cytochemistry for the lysosomal enzymes aryl sulfatase and acid phosphatase has revealed the fusion of the parasite-containing vacuole with lysosomes (Meirelles et al., 1987). The acidification of the PV by lysosomal fusion, leading to the activation of TC-TOX and disruption of the PV membrane (Andrews et al., 1990; Hall, 1993), is a prerequisite for the trypomastigote to exit the phagosome, also allowing the parasite to be retained intracellularly and complete its life cycle (Andrade and Andrews, 2004, 2005; Mott and Burleigh, 2008).
Effect of T. cruzi Infection in Cardiomyocyte Physiology
During the T. cruzi–cardiomyocyte interaction the parasite gains control of overall host cell gene expression, including expression of genes related to immune response, inflammation, cytoskeletal organization, cell–cell and cell–matrix interactions, apoptosis, cell cycle, and oxidative stress (Goldenberg et al., 2009; Manque et al., 2011). The intense trypanocidal immune response generated in cardiomyocytes in response to infection by T. cruzi results in the production of cytokines, chemokines, and nitric oxide that, while essential elements of the defensive reaction in cardiac tissue (Machado et al., 2000, 2008; Manque et al., 2011), can also result in cardiac hypertrophy (Petersen and Burleigh, 2003; Waghabi et al., 2009). Several studies report that T. cruzi infection stimulates production of nitric oxide synthase 2, matrix metalloproteinase-2 (MMP-2) and MMP-9 in cardiomyocytes, as well as interleukin-6 (IL-6), IL-1β, tumor necrosis factor-alpha and TGF-β (Petersen and Burleigh, 2003; Petersen et al., 2005; Gutierrez et al., 2008; Waghabi et al., 2009; Nogueira de Melo et al., 2010). Peroxisome proliferator-activated receptor γ is also implicated in regulating the inflammatory process (Hovsepian et al., 2011). Moreover, IL-1β-mediated development of cardiomyocyte hypertrophy is orchestrated by Toll-like receptor 2 (Petersen et al., 2005). Proinflammatory cytokines also modulate production of mitochondrial reactive oxygen species, impairing the efficiency of the respiratory chain (Gupta et al., 2009). Mitochondrial disturbance has been identified as an important effect of chagasic cardiomyopathy (Garg et al., 2003; Báez et al., 2011). Inflammatory mediators have also been reported to regulate Rabs expression (Stein et al., 2003) thereby interfering with host cell trafficking. Down-regulation of Rab GTPase proteins, including the effector molecule of Rab5 (EEA1), Rab7, and Rab11, has been demonstrated in T. cruzi-infected cardiomyocytes, and it has been proposed that a delayed endocytic pathway may favor microbicidal activity and increase antigen processing (Batista et al., 2006).
Changes in cytoskeletal proteins have also been shown during parasite intracellular development (Figures 1 and 2). The complex cytoskeleton organization of cardiomyocytes involved in the contraction–relaxation process of the heart is affected by T. cruzi infection (Pereira et al., 1993; Taniwaki et al., 2006). Breakdown of myofibrils has been seen in areas of amastigote nests (Pereira et al., 1993; Taniwaki et al., 2006) and disturbance of intermediate filaments (desmin) and microtubules was also induced by parasite proliferation (Pereira et al., 1993). Interestingly, the actin isoform mRNAs, α-cardiac and β-actin mRNAs, are altered during the parasite intracellular cycle (Pereira et al., 2000). Down-regulation of α-cardiac actin mRNA concomitant with up-regulation of β-actin mRNA suggested the reactivation of non-differentiated cell program. Also within the context of cytoskeletal changes, actin-binding proteins have been demonstrated to be altered in T. cruzi-infected cardiomyocytes. Alpha-actinin, an F-actin crosslinker protein that anchors actin to the Z line, and costameres, repeating adhesion structures consisting of vinculin involved in the lateral transmission of contractility force to the sarcolemma, are disrupted and down-regulated in T. cruzi-infected cells, reducing strength and force transduction (Melo et al., 2004, 2006). These cytoskeletal disorders are accompanied by deregulation of Ca2+ influx, affecting cardiac cell contractility (Taniwaki et al., 2006). One striking feature of trypanocidal drugs is their effect on the recovery of cardiomyocyte cytoskeleton (Garzoni et al., 2004; Silva et al., 2006; Adesse et al., 2011a). Posaconazole, an ergosterol biosynthesis inhibitor with potent trypanocidal activity currently in clinical trials, has been demonstrated to promote the reassembly of the contractile apparatus and microtubule network in T. cruzi-infected cardiomyocytes (Silva et al., 2006). The reorganization of myofibrils leads to recovery of cardiomyocyte functionality. Similarly, treatment of T. cruzi-infected cardiomyocyte cultures with bisphosphonate risedronate, a farnesyl pyrophosphate synthase inhibitor, and amiodarona, an anti-arrhythmic drug, also fostered the recovery of myofibrils (Garzoni et al., 2004; Adesse et al., 2011a) and may represent interesting alternatives for Chagas therapy.
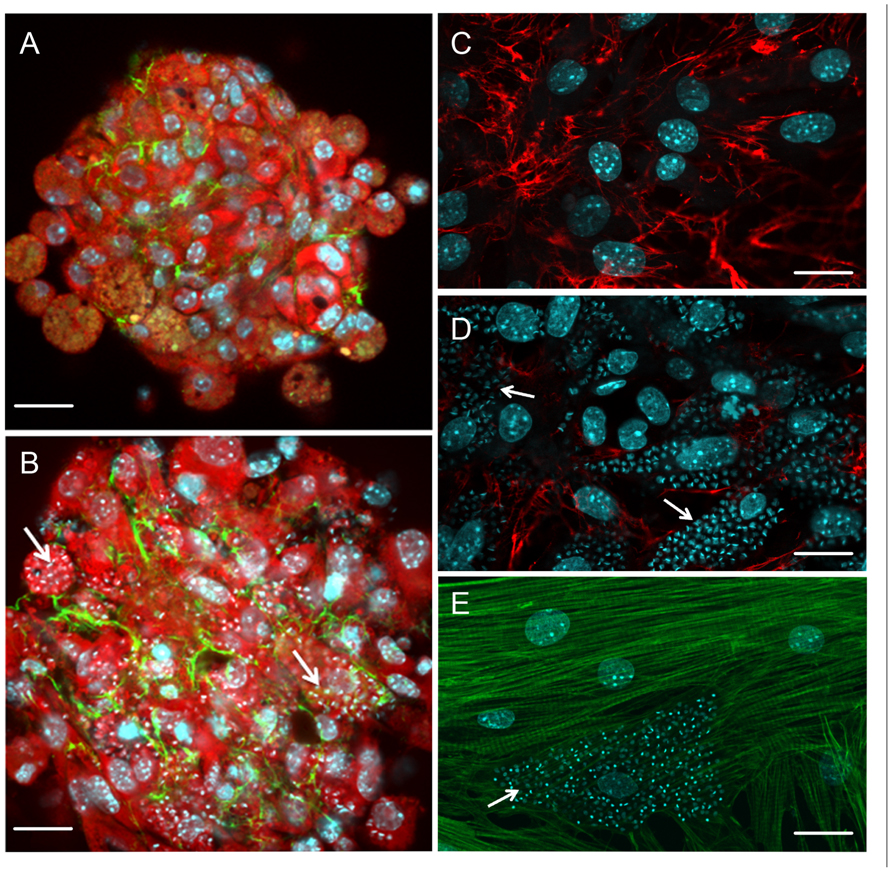
FIGURE 2. Effect of T. cruzi infection on cardiomyocyte cytoarchitecture and extracellular matrix remodeling (A–E). T. cruzi-infected cardiomyocyte 3D-culture revealed fibrosis and hypertrophy. Note an increase in spheroid size and the spatial distribution of fibronectin (FN; green) in T. cruzi-infected (B) compared with the uninfected (A) 3D-culture system. However, the FN labeling fainted in highly T. cruzi-infected cells. In contrast, the analysis of FN (red) distribution in cardiomyocyte 2D-culture (C) demonstrates a reduction of this extracellular matrix component in T. cruzi-infected cardiomyocyte culture (D). The low expression of FN in the cells harboring the parasites and the factors involved in the enhancement of FN in 3D-culture are unclear and focus of future investigation. (E) Cytoskeletal changes were also evidenced in T. cruzi-infected cardiomyocytes, showing complete disorganization of myofibrils. Cardiomyocytes were stained with Evans-blue (red; A and B) and DAPI (blue; A–E), a DNA dye. Arrows indicate intracellular parasites. Bar = 20 µm.
In addition to disruption of the cytoskeletal architecture by the parasite, cell–cell adhesion (adherens junctions) and intercellular communication (gap junctions), which play important physiological roles in cardiac tissue, are also been disrupted by T. cruzi infection (Adesse et al., 2008, 2011b; Melo et al., 2008). Alteration in spatial distribution and down-regulation of the adherence junction proteins N-cadherin and β-catenin in T. cruzi-infected cardiomyocytes (Melo et al., 2008) may interfere with tissue integrity and perturb the function of the cardiac conduction system, as has been proposed to be the case in arrhythmogenic cardiomyopathies (Mezzano and Sheikh, 2012). Additionally, electrical conduction disturbance, frequently seen in both acute and chronic phases of Chagas diseases, seems to be related to altered gap junction (connexin-43) coupling of cardiomyocytes induced by T. cruzi (de Carvalho et al., 1992, 1994; Adesse et al., 2008, 2011b). Connexin-43 dysregulation has also been attributed to increased levels of TGF-β (Waghabi et al., 2009). Following treatment of T. cruzi-infected cardiomyocyte cultures with amiodarone and SB-431542 causes reversal of the disorganization of gap junctions and return to their normal distribution (Waghabi et al., 2009; Adesse et al., 2011a), making these compounds potential therapeutic candidates for treatment of Chagas disease.
Besides their involvement in the early steps of T. cruzi–cardiomyocyte recognition, ECM components also present a striking role in chagasic cardiomyopathy pathogenesis since their accumulation leads to fibrosis, disposing patients to heart failure and ventricular arrhythmias (Rassi et al., 2010, 2012). In experimental systems, ECM accumulation begins during the late acute phase of infection (Andrade et al., 1989; Calvet et al., 2004), concomitantly with the onset of inflammatory infiltrates, indicating that the process of fibrogenesis is triggered in the early stages of T. cruzi infection. A general increase in ECM transcripts and expression was detected by microarray analysis in acute infection (Garg et al., 2003). Cardiac hypertrophy and ECM remodeling were also seen in a T. cruzi-infected 3D cardiomyocyte model (Garzoni et al., 2008; Figure 2). Surprisingly, reduction of ECM in T. cruzi-infected cardiomyocytes was detected by silver staining in acute infection in mice (Factor et al., 1993). Additionally, T. cruzi-mediated down-regulated ECM gene expression in cardiomyocyte cultures (Goldenberg et al., 2009; Manque et al., 2011) and a reduction in the synthesis and spatial distribution of fibronectin were detected in heavily infected cardiomyocytes (Calvet et al., 2004; Figure 2) even after TGF-β stimulation (Calvet et al., 2009), suggesting that despite the general enhancement of ECM in the heart, the cells harboring the parasites display low ECM expression. The anti-fibrogenic effect of T. cruzi has also been seen in human dermal fibroblasts, with repression of transcription factors that regulate expression of fibroblast genes involved in wound repair and tissue remodeling, including ctgf/ccn2 connective tissue growth factor gene, followed by down-regulation of ECM proteins such as fibronectin and collagen I, suggesting another route of parasite dissemination and infection (Unnikrishnan and Burleigh, 2004; Mott et al., 2011).
Another point worth discussing relates to the ability of T. cruzi to modulate host cell apoptosis, or programed cell death, a physiological process of cell replacement to maintain tissue homeostasis (Mondello and Scovassi, 2010). Pathogens can hijack the host cell apoptotic machinery as an offensive strategy to eliminate the host’s immune response (Lamkanfi and Dixit, 2010). Both anti- and pro-apoptotic gene expression are differentially modulated during T. cruzi–cardiomyocyte infection, leading to a balance between cell death and survival at different stages of infection (Manque et al., 2011). Induction of apoptosis by T. cruzi infection is controversial and seems to be dependent on host cell and parasite genotype (de Souza et al., 2003;
Aoki et al., 2006; Petersen et al., 2006). While fibroblasts are refractory to apoptosis, cardiomyocytes and macrophages differentially undergo apoptosis after T. cruzi infection, the latter cell type being highly susceptible. Still, cardiomyocytes infected with T. cruzi clone Dm28c have higher levels of apoptosis compared to infection with strains Y and CL (de Souza et al., 2003). Furthermore, the intracellular parasites themselves also undergo apoptosis, hinting at a host attempt to control parasite burden (de Souza et al., 2003, 2010). Interestingly, it has been shown that α2-macroglobulin, a plasma proteinase inhibitor, regulates apoptosis in T. cruzi-infected cardiomyocytes and macrophages, impairing the cell death process (de Souza et al., 2008). In contrast, an anti-apoptotic effect has also been demonstrated in cardiac cells (Petersen et al., 2006). The prevention of apoptosis appears to be related to NF-κB activation by inhibiting the signaling of caspases, thus avoiding cell death. Thus, avoidance of apoptosis reduces cardiac damage and may be responsible for the persistence of T. cruzi infection.
While our knowledge of T. cruzi–host cell interactions has greatly improved, many questions remain open. There are still gaps in our understanding of the molecular interactions involved in cellular recognition and/or signaling pathway in most of the mechanisms of invasion. What are the critical links between these processes? And little is still known about the cooperative role played by the host cell in parasite intracellular growth and differentiation. These questions demand deeper investigation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
Acknowledgments
The authors thank Mr Potter Wickware for a critical reading of the manuscript. This work was supported by grants from Fundação Oswaldo Cruz (FIOCRUZ), Conselho Nacional de Desenvolvimento Científico e Tecnológico (Mirian C. S. Pereira, grant number 477292/2010-0; 400120/2011-9), Programa Estratégico de Apoio à Pesquisa em Saúde and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro – FAPERJ (Mirian C. S. Pereira, grant number E-26/110.322/2012)
References
Adesse, D., Azzam, E. M., Meirelles, M. N., Urbina, J. A., and Garzoni, L. R. (2011a). Amiodarone inhibits Trypanosoma cruzi infection and promotes cardiac cell recovery with gap junction and cytoskeleton reassembly in vitro. Antimicrob. Agents Chemother. 55, 203–210.
Adesse, D., Goldenberg, R. C., Fortes, F. S., Jasmin Iacobas, D. A., Iacobas, S., Campos de Carvalho, A. C., et al. (2011b). Gap junctions and Chagas disease. Adv. Parasitol. 76, 63–81.
Adesse, D., Garzoni, L. R., Huang, H., Tanowitz, H. B., Meirelles, M. N., and Spray, D. C. (2008). Trypanosoma cruzi induces changes in cardiac connexin43 expression. Microbes Infect. 10, 21–28.
Andrade, L. O., and Andrews, N. W. (2004). Lysosomal fusion is essential for the retention of Trypanosoma cruzi inside host cells. J. Exp. Med. 200, 1135–1143.
Andrade, L. O., and Andrews, N. W. (2005). The Trypanosoma cruzi–host-cell interplay: location, invasion, retention. Nat. Rev. Microbiol. 3, 819–823.
Andrade, S. G., Grimaud, J. A., and Stocker-Guerret, S. (1989). Sequential changes of the connective matrix components of the myocardium (fibronectin and laminin) and evolution of cardiac fibrosis in mice infected with Trypanosoma cruzi. Am. J. Trop. Med. Hyg. 40, 252–260.
Andrews, N. W., Abrams, C. K., Slatin, S. L., and Griffiths, G. (1990). A T. cruzi-secreted protein immunologically related to the complement component C9: evidence for membrane pore-forming activity at low pH. Cell 61, 1277–1287.
Aoki, M. P., Cano, R. C., Pellegrini, A. V., Tanos, T., Guiñazú, N. L., Coso, O. A., et al. (2006). Different signaling pathways are involved in cardiomyocyte survival induced by a Trypanosoma cruzi glycoprotein. Microbes Infect. 8, 1723–1731.
Báez, A., Lo Presti, M. S., Rivarola, H. W., Mentesana, G. G., Pons, P., Fretes, R., et al. (2011). Mitochondrial involvement in chronic chagasic cardiomyopathy. Trans. R. Soc. Trop. Med. Hyg. 105, 239–246.
Bambino-Medeiros, R., Oliveira, F. O., Calvet, C. M., Vicente, D., Toma, L., Krieger, M. A., et al. (2011). Involvement of host cell heparan sulfate proteoglycan in Trypanosoma cruzi amastigote attachment and invasion. Parasitology 138, 593–601.
Barbosa, H. S., and Meirelles, M. N. (1992). Ultrastructural detection in vitro of WGA-, RCA I-, and Con A-binding sites involved in the invasion of heart muscle cells by Trypanosoma cruzi. Parasitol. Res. 78, 404–409.
Barbosa, H. S., and Meirelles, M. N. (1993). The role of RCA-binding sites in the adhesion of Trypanosoma cruzi to heart muscle cells, as revealed by electron spectroscopic imaging. J. Submicrosc. Cytol. Pathol. 25, 47–51.
Barbosa, H. S., and Meirelles, M. N. (1995). Evidence of participation of cytoskeleton of heart muscle cells during the invasion of Trypanosoma cruzi. Cell Struct. Funct. 20, 275–284.
Barr, S. C., Han, W., Andrews, N. W., Lopez, J. W., Ball, B. A., Pannabecker, T. L., et al. (1996). A factor from Trypanosoma cruzi induces repetitive cytosolic free Ca2+ transients in isolated primary canine cardiac myocytes. Infect. Immun. 64, 1770–1777.
Barrias, E. S., Dutra, J. M., De Souza, W., and Carvalho, T. M. (2007). Participation of macrophage membrane rafts in Trypanosoma cruzi invasion process. Biochem. Biophys. Res. Commun. 363, 828–834.
Batista, D. G., Silva, C. F., Mota, R. A., Costa, L. C., Meirelles, M. N., Meuser-Batista, M., et al. (2006). Trypanosoma cruzi modulates the expression of Rabs and alters the endocytosis in mouse cardiomyocytes in vitro. J. Histochem. Cytochem. 54, 605–614.
Butler, C. E., and Tyler, K. M. (2012). Membrane traffic and synaptic cross-talk during host cell entry by Trypanosoma cruzi. Cell. Microbiol. 14, 1345–1353.
Calvet, C. M., Meuser, M., Almeida, D., Meirelles, M. N., and Pereira, M. C. (2004). Trypanosoma cruzi–cardiomyocyte interaction: role of fibronectin in the recognition process and extracellular matrix expression in vitro and in vivo. Exp. Parasitol. 107, 20–30.
Calvet, C. M., Oliveira, F. O. Jr., Araújo-Jorge, T. C., and Pereira, M. C. (2009). Regulation of extracellular matrix expression and distribution in Trypanosoma cruzi-infected cardiomyocytes. Int. J. Med. Microbiol. 299, 301–312.
Calvet, C. M., Toma, L., De Souza, F. R., Meirelles, M. N., and Pereira, M. C. (2003). Heparan sulfate proteoglycans mediate the invasion of cardiomyocytes by Trypanosoma cruzi. J. Eukaryot. Microbiol. 50, 97–103.
Chuenkova, M. V., Furnari, F. B., Cavenee, W. K., and Pereira, M. A. (2001). Trypanosoma cruzi trans-sialidase: a potent and specific survival factor for human Schwann cells by means of phosphatidylinositol 3-kinase/Akt signaling. Proc. Natl. Acad. Sci. U.S.A. 98, 9936–9941.
de Carvalho, A. C., Masuda, M. O., Tanowitz, H. B., Wittner, M., Goldenberg, R. C., and Spray, D. C. (1994). Conduction defects and arrhythmias in Chagas’ disease: possible role of gap junctions and humoral mechanisms. J. Cardiovasc. Electrophysiol. 5, 686–698.
de Carvalho, A. C., Tanowitz, H. B., Wittner, M., Dermietzel, R., Roy, C., Hertzberg, E. L., et al. (1992). Gap junction distribution is altered between cardiac myocytes infected with Trypanosoma cruzi. Circ. Res. 70, 733–742.
de Melo-Jorge, M., and PereiraPerrin, M. (2007). The Chagas’ disease parasite Trypanosoma cruzi exploits nerve growth factor receptor TrkA to infect mammalian hosts. Cell Host Microbe 1, 251–261.
de Oliveira, F. L., Araújo-Jorge, T. C., de Souza, E. M., de Oliveira, G. M., Degrave, W. M., Feige, J. J., et al. (2012). Oral administration of GW788388, an inhibitor of transforming growth factor beta signaling, prevents heart fibrosis in Chagas disease. PLoS Negl. Trop. Dis. 6, e1696. doi: 10.1371/journal.pntd.0001696
de Souza, E. M., Araújo-Jorge, T. C., Bailly, C., Lansiaux, A., Batista, M. M., Oliveira, G. M., et al. (2003). Host and parasite apoptosis following Trypanosoma cruzi infection in in vitro and in vivo models. Cell Tissue Res. 314, 223–235.
de Souza, E. M., Meuser-Batista, M., Batista, D. G., Duarte, B. B., Araújo-Jorge, T. C., and Soeiro, M. N. (2008). Trypanosoma cruzi: alpha-2-macroglobulin regulates host cell apoptosis induced by the parasite infection in vitro. Exp. Parasitol. 118, 331–337.
de Souza, E. M., Nefertiti, A. S., Bailly, C., Lansiaux, A., and Soeiro, M. N. (2010). Differential apoptosis-like cell death in amastigote and trypomastigote forms from Trypanosoma cruzi-infected heart cells in vitro. Cell Tissue Res. 341, 173–180.
Factor, S. M., Tanowitz, H., Wittner, M., and Ventura, M. C. (1993). Interstitial connective tissue matrix alterations in acute murine Chagas’ disease. Clin. Immunol. Immunopathol. 68, 147
Fernandes, M. C., and Andrews, N. W. (2012). Host cell invasion by Trypanosoma cruzi: a unique strategy that promotes persistence. FEMS Microbiol. Rev. 36, 734–747.
Fernandes, M. C., Cortez, M., Geraldo Yoneyama, K. A., Straus, A. H., Yoshida, N., and Mortara, R. A. (2007). Novel strategy in Trypanosoma cruzi cell invasion: implication of cholesterol and host cell microdomains. Int. J. Parasitol. 37, 1431–1441.
Fernandes, M. C., de Andrade, L. R., Andrews, N. W., and Mortara, R. A. (2011). Trypanosoma cruzi trypomastigotes induce cytoskeleton modifications during HeLa cell invasion. Mem. Inst. Oswaldo Cruz 106, 1014–1016.
Ferreira, D., Cortez, M., Atayde, V. D., and Yoshida, N. (2006). Actin cytoskeleton-dependent and -independent host cell invasion by Trypanosoma cruzi is mediated by distinct parasite surface molecules. Infect. Immun. 74, 5522–5528.
Garg, N., Popov, V. L., and Papaconstantinou, J. (2003). Profiling gene transcription reveals a deficiency of mitochondrial oxidative phosphorylation in Trypanosoma cruzi-infected murine hearts: implications in chagasic myocarditis development. Biochim. Biophys. Acta 1638, 106–120.
Garzoni, L. R., Adesse, D., Soares, M. J., Rossi, M. I., Borojevic, R., and Meirelles, M. N. (2008). Fibrosis and hypertrophy induced by Trypanosoma cruzi in a three-dimensional cardiomyocyte-culture system. J. Infect. Dis. 197, 906–915.
Garzoni, L. R., Masuda, M. O., Capella, M. M., Lopes, A. G., and Meirelles, M. N. (2003). Characterization of [Ca2+]i responses in primary cultures of mouse cardiomyocytes induced by Trypanosoma cruzi trypomastigotes. Mem. Inst. Oswaldo Cruz 98, 487–493.
Garzoni, L. R., Waghabi, M. C., Baptista, M. M., de Castro, S. L., Meirelles, M. N., Britto, C. C., et al. (2004). Antiparasitic activity of risedronate in a murine model of acute Chagas’ disease. Int. J. Antimicrob. Agents 23, 286–290.
Goldenberg, R. C., Iacobas, D. A., Iacobas, S., Rocha, L. L., da Silva de Azevedo Fortes, F., et al. (2009). Transcriptomic alterations in Trypanosoma cruzi-infected cardiac myocytes. Microbes Infect. 11, 1140–1149.
Gupta, S., Bhatia, V., Wen, J. J., Wu, Y., Huang, M. H., and Garg, N. J. (2009). Trypanosoma cruzi infection disturbs mitochondrial membrane potential and ROS production rate in cardiomyocytes. Free Radic. Biol. Med. 47, 1414–1421.
Gutierrez, F. R., Lalu, M. M., Mariano, F. S., Milanezi, C. M., Cena, J., Gerlach, R. F., et al. (2008). Increased activities of cardiac matrix metalloproteinases matrix metalloproteinase (MMP)-2 and MMP-9 are associated with mortality during the acute phase of experimental Trypanosoma cruzi infection. J. Infect. Dis. 197, 1468–1476.
Hall, B. F. (1993). Trypanosoma cruzi: mechanisms for entry into host cells. Semin. Cell Biol. 4, 323–333.
Hissa, B., Duarte, J. G., Kelles, L. F., Santos, F. P., del Puerto, H. L., Gazzinelli-Guimarães, P. H., et al. (2012). Membrane cholesterol regulates lysosome-plasma membrane fusion events and modulates Trypanosoma cruzi invasion of host cells. PLoS Negl. Trop. Dis. 6, e1583. doi: 10.1371/journal.pntd.0001583
Hovsepian, E., Mirkin, G. A., Penas, F., Manzano, A., Bartrons, R., and Goren, N. B. (2011). Modulation of inflammatory response and parasitism by 15-Deoxy-Δ(12,14) prostaglandin J(2) in Trypanosoma cruzi-infected cardiomyocytes. Int. J. Parasitol. 41, 553.
Lamkanfi, M., and Dixit, V. M. (2010). Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 8, 44–54.
Machado, F. S., Martins, G. A., Aliberti, J. C., Mestriner, F. L., Cunha, F. Q., and Silva, J. S. (2000). Trypanosoma cruzi-infected cardiomyocytes produce chemokines and cytokines that trigger potent nitric oxide-dependent trypanocidal activity. Circulation 102, 3003–3008.
Machado, F. S., Souto, J. T., Rossi, M. A., Esper, L., Tanowitz, H. B., Aliberti, J., et al. (2008). Nitric oxide synthase-2 modulates chemokine production by Trypanosoma cruzi-infected cardiac myocytes. Microbes Infect. 10, 1558–1566.
Manque, P. A., Probst, C. M., Pereira, M. C., Rampazzo, R. C., Ozaki, L. S., Pavoni, D. P., et al. (2011). Trypanosoma cruzi infection induces a global host cell response in cardiomyocytes. Infect. Immun. 79, 1855–1862.
Meirelles, M. N., Araujo Jorge, T. C., de Souza, W., Moreira, A. L., and Barbosa, H. S. (1987). Trypanosoma cruzi: phagolysosomal fusion after invasion into non professional phagocytic cells. Cell Struct. Funct. 12, 387–393.
Meirelles, M. N., Araujo-Jorge, T. C., Miranda, C. F., de Souza, W., and Barbosa, H. S. (1986). Interaction of Trypanosoma cruzi with heart muscle cells: ultrastructural and cytochemical analysis of endocytic vacuole formation and effect upon myogenesis in vitro. Eur. J. Cell Biol. 41, 198–206.
Melo, T. G., Almeida, D. S., Meirelles, M. N., and Pereira, M. C. (2004). Trypanosoma cruzi infection disrupts vinculin costameres in cardiomyocytes. Eur. J. Cell Biol. 83, 531–540.
Melo, T. G., Almeida, D. S., Meirelles, M. N., and Pereira, M. C. (2006). Disarray of sarcomeric alpha-actinin in cardiomyocytes infected by Trypanosoma cruzi. Parasitology 133, 171–178.
Melo, T. G., Meirelles, M. N., and Pereira, M. C. (2008). Trypanosoma cruzi alters adherens junctions in cardiomyocytes. Microbes Infect. 10, 1405–1410.
Mezzano, V., and Sheikh, F. (2012). Cell–cell junction remodeling in the heart: possible role in cardiac conduction system function and arrhythmias? Life Sci. 90, 313–321.
Ming, M., Ewen, M. E., and Pereira, M. E. (1995). Trypanosome invasion of mammalian cells requires activation of the TGF beta signaling pathway. Cell 82, 287–296.
Moncayo, A., and Silveira, A. C. (2009). Current epidemiological trends for Chagas disease in Latin America and future challenges in epidemiology, surveillance and health policy. Mem. Inst. Oswaldo Cruz 104, 17–30.
Mondello, C., and Scovassi, A. I. (2010). Apoptosis: a way to maintain healthy individuals. Subcell. Biochem. 50, 307–323.
Moolani, Y., Bukhman, G., and Hotez, P. J. (2012). Neglected tropical diseases as hidden causes of cardiovascular disease. PLoS Negl. Trop. Dis. 6, e1499. doi: 10.1371/journal.pntd.0001499
Mott, G. A., and Burleigh, B. A. (2008). The role of host cell lysosomes in Trypanosoma cruzi invasion. Subcell. Biochem. 47, 165–173.
Mott, G. A., Costales, J. A., and Burleigh, B. A. (2011). A soluble factor from Trypanosoma cruzi inhibits transforming growth factor-β-induced MAP kinase activation and gene expression in dermal fibroblasts. PLoS ONE 6, e23482. doi: 10.1371/journal.pone.0023482
Moustakas, A., Pardali, K., Gaal, A., and Heldin, C. H. (2002). Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation. Immunol. Lett. 82, 85–91.
Nagajyothi, F., Weiss, L. M., Silver, D. L., Desruisseaux, M. S., Scherer, P. E., Herz, J., et al. (2011). Trypanosoma cruzi utilizes the host low density lipoprotein receptor in invasion. PLoS Negl. Trop. Dis. 5, e953. doi: 10.1371/journal.pntd.0000953
Nogueira de Melo, A. C., de Souza, E. P., Elias, C. G., dos Santos, A. L., Branquinha, M. H., d’Avila-Levy, C. M., et al. (2010). Detection of matrix metallopeptidase-9-like proteins in Trypanosoma cruzi. Exp. Parasitol. 125, 256–263.
Oliveira, F. O. Jr., Alves, C. R., Calvet, C. M., Toma, L., Bouças, R. I., Nader, H. B., et al. (2008). Trypanosoma cruzi heparin-binding proteins and the nature of the host cell heparan sulfate-binding domain. Microb. Pathog. 44, 329–338.
Oliveira, F. O. Jr., Alves, C. R., Souza-Silva, F., Calvet, C. M., Côrtes, L. M., Gonzalez, M. S., et al. (2012). Trypanosoma cruzi heparin-binding proteins mediate the adherence of epimastigotes to the midgut epithelial cells of Rhodnius prolixus. Parasitology 139, 735–743.
Ortega-Barria, E., and Pereira, M. E. (1991). A novel T. cruzi heparin-binding protein promotes fibroblast adhesion and penetration of engineered bacteria and trypanosomes into mammalian cells. Cell 67, 411–421.
Ouaissi, M. A., Afchain, D., Capron, A., and Grimaud, J. A. (1984). Fibronectin receptors on Trypanosoma cruzi trypomastigotes and their biological function. Nature 308, 380–382.
Ouaissi, M. A., Cornette, J., Afchain, D., Capron, A., Gras-Masse, H., and Tartar, A. (1986). Trypanosoma cruzi infection inhibited by peptides modeled from a fibronectin cell attachment domain. Science 234, 603–607.
Pereira, K. S., Schmidt, F. L., Guaraldo, A. M., Franco, R. M., Dias, V. L., and Passos, L. A. (2009). Chagas’ disease as a foodborne illness. J. Food Prot. 72, 441–446.
Pereira, M. C., Costa, M., Chagas Filho, C., and Meirelles, M. N. (1993). Myofibrillar breakdown and cytoskeletal alterations in heart muscle cells during invasion by Trypanosoma cruzi: immunological and ultrastructural study. J. Submicrosc. Cytol. Pathol. 25, 559–569.
Pereira, M. C., Singer, R. H., and Meirelles, M. N. (2000). Trypanosoma cruzi infection affects actin mRNA regulation in heart muscle cells. J. Eukaryot. Microbiol. 47, 271–279.
Pérez-Molina, J. A., Norman, F., and López-Vélez, R. (2012). Chagas disease in non-endemic countries: epidemiology, clinical presentation and treatment. Curr. Infect. Dis. Rep. 14, 263–274.
Petersen, C. A., and Burleigh, B. A. (2003). Role for interleukin-1 beta in Trypanosoma cruzi-induced cardiomyocyte hypertrophy. Infect. Immun. 71, 4441–4447.
Petersen, C. A., Krumholz, K. A., and Burleigh, B. A. (2005). Toll-like receptor 2 regulates interleukin-1beta-dependent cardiomyocyte hypertrophy triggered by Trypanosoma cruzi. Infect. Immun. 73, 6974–6980.
Petersen, C. A., Krumholz, K. A., Carmen, J., Sinai, A. P., and Burleigh, B. A. (2006). Trypanosoma cruzi infection and nuclear factor kappa B activation prevent apoptosis in cardiac cells. Infect. Immun. 74, 1580–1587.
Priotto, S., Sartori, M. J., Repossi, G., and Valentich, M. A. (2009). Trypanosoma cruzi: participation of cholesterol and placental alkaline phosphatase in the host cell invasion. Exp. Parasitol. 122, 70–73.
Procópio, D. O., Barros, H. C., and Mortara, R. A. (1999). Actin-rich structures formed during the invasion of cultured cells by infective forms of Trypanosoma cruzi. Eur. J. Cell Biol. 78, 911–924.
Rassi, A. Jr., Rassi, A., and Marcondes de Rezende, J. (2012). American trypanosomiasis (Chagas disease). Infect. Dis. Clin. North Am. 26, 275–291.
Rassi, A. Jr., Rassi, A., and Marin-Neto, J. á. (2009). Chagas heart disease: pathophysiologic mechanisms, prognostic factors and risk stratification. Mem. Inst. Oswaldo Cruz 104, 152–158.
Ríos, J. F., Arboleda, M., Montoya, A. N., and Alarcón, E. P. (2011). Probable outbreak of oral transmission of Chagas disease in Turbo, Antioquia. Biomedica 31, 185–195.
Rodríguez, A., Martinez, I., Chung, A., Berlot, C. H., and Andrews, N. W. (1999). cAMP regulates Ca2+-dependent exocytosis of lysosomes and lysosome-mediated cell invasion by trypanosomes. J. Biol. Chem. 274, 16754–16759.
Romano, P. S., Arboit, M. A., Vázquez, C. L., and Colombo, M. I. (2009). The autophagic pathway is a key component in the lysosomal dependent entry of Trypanosoma cruzi into the host cell. Autophagy 5, 6–18.
Romano, P. S., Cueto, J. A., Casassa, A. F., Vanrell, M. C., Gottlieb, R. A., and Colombo, M. I. (2012). Molecular and cellular mechanisms involved in the Trypanosoma cruzi/host cell interplay. IUBMB Life 64, 387–396.
Rosestolato, C. T., Dutra, J. M., De Souza, W., and Carvalho, T. M. (2002). Participation of host cell actin filaments during interaction of trypomastigote forms of Trypanosoma cruzi with host cells. Cell Struct. Funct. 27, 91–98.
Scharfstein, J., Schmitz, V., Morandi, V., Capella, M. M., Lima, A. P., Morrot, A., et al. (2000). Host cell invasion by Trypanosoma cruzi is potentiated by activation of bradykinin B(2) receptors. J. Exp. Med. 192, 1289–1300.
Silva, D. T., Meirelles, M. N., Almeida, D., Urbina, J. A., and Pereira, M. C. (2006). Cytoskeleton reassembly in cardiomyocytes infected by Trypanosoma cruzi is triggered by treatment with ergosterol biosynthesis inhibitors. Int. J. Antimicrob. Agents 27, 530–537.
Soeiro, M. N., Paiva, M. M., Barbosa, H. S., Meirelles, M. N., and Araújo-Jorge, T. C. (1999). A cardiomyocyte mannose receptor system is involved in Trypanosoma cruzi invasion and is down-modulated after infection. Cell Struct. Funct. 24, 139–149.
Stein, M. P., Dong, J., and Wandinger-Ness, A. (2003). Rab proteins and endocytic trafficking: potential targets for therapeutic intervention. Adv. Drug Deliv. Rev. 55, 1421–1437.
Taniwaki, N. N., Machado, F. S., Massensini, A. R., and Mortara, R. A. (2006). Trypanosoma cruzi disrupts myofibrillar organization and intracellular calcium levels in mouse neonatal cardiomyocytes. Cell Tissue Res. 324, 489–496.
Todorov, A. G., Andrade, D., Pesquero, J. B., Araujo, R. C., Bader, M., Stewart, J., et al. (2003). Trypanosoma cruzi induces edematogenic responses in mice and invades cardiomyocytes and endothelial cells in vitro by activating distinct kinin receptor (B1/B2) subtypes. FASEB J. 17, 73–75.
Todorov, A. G., Einicker-Lamas, M., de Castro, S. L., Oliveira, M. M., and Guilherme, A. (2000). Activation of host cell phosphatidylinositol 3-kinases by Trypanosoma cruzi infection. J. Biol. Chem. 275, 32182–32186.
Unnikrishnan, M., and Burleigh, B. A. (2004). Inhibition of host connective tissue growth factor expression: a novel Trypanosoma cruzi-mediated response. FASEB J. 18, 1625–1635.
Vieira, M., Dutra, J. M., Carvalho, T. M., Cunha-e-Silva, N. L., Souto-Padrón, T., and De Souza, W. (2002). Cellular signaling during the macrophage invasion by Trypanosoma cruzi. Histochem. Cell Biol. 118, 491–500.
Waghabi, M. C., Coutinho-Silva, R., Feige, J. J., Higuchi, M. L., Becker, D., Burnstock, G., et al. (2009). Gap junction reduction in cardiomyocytes following transforming growth factor-beta treatment and Trypanosoma cruzi infection. Mem. Inst. Oswaldo Cruz 104, 1083–1090.
Waghabi, M. C., Keramidas, M., Bailly, S., Degrave, W., Mendonça-Lima, L., Soeiro, M. N., et al. (2005). Uptake of host cell transforming growth factor-beta by Trypanosoma cruzi amastigotes in cardiomyocytes: potential role in parasite cycle completion. Am. J. Pathol. 167, 993–1003.
Waghabi, M. C., Keramidas, M., Calvet, C. M., Meuser, M., Soeiro, M. N., Mendonça-Lima, L., et al. (2007). SB-431542, a transforming growth factor beta inhibitor, impairs Trypanosoma cruzi infection in cardiomyocytes and parasite cycle completion. Antimicrob. Agents Chemother. 51, 2905–2910.
Weinkauf, C., Salvador, R., and Pereiraperrin, M. (2011). Neurotrophin receptor TrkC is an entry receptor for Trypanosoma cruzi in neural, glial, and epithelial cells. Infect. Immun. 79, 4081–4087.
WHO (2010). Available at: http://www.who.int/mediacentre/factsheets/fs340/en/index.html
Wilkowsky, S. E, Barbieri, M. A, Stahl, P. and Isola, E. L. (2001). Trypanosoma cruzi: phosphatidylinositol 3-kinase and protein kinase B activation is associated with parasite invasion. Exp. Cell Res. 264, 211–218.
Keywords: Trypanosoma cruzi, cardiomyocyte, cell recognition, endocytosis, cytoskeleton, cell junction, extracellular matrix, apoptosis
Citation: Calvet CM, Melo TG, Garzoni LR, Oliveira FOR Jr., Silva Neto DT, Meirelles MNSL and Pereira MCS (2012) Current understanding of the Trypanosoma cruzi–cardiomyocyte interaction. Front. Immun. 3:327. doi: 10.3389/fimmu.2012.00327
Received: 31 August 2012; Paper pending published: 04 October 2012;
Accepted: 16 October 2012; Published online: 30 October 2012.
Edited by:
Wanderley De Souza, Universidade Federal do Rio de Janeiro, BrazilReviewed by:
Emilio Luis Malchiodi, University of Buenos Aires, ArgentinaWanderley De Souza, Universidade Federal do Rio de Janeiro, Brazil
Copyright: © 2012 Calvet, Melo, Garzoni, Oliveira Jr., Silva Neto, Meirelles and Pereira. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Mirian C. S. Pereira, Laboratório de Ultra-estrutura Celular, Fundação Oswaldo Cruz, Instituto Oswaldo Cruz, Av. Brasil 4365, Manguinhos, Rio de Janeiro, Rio de Janeiro 21045-900, Brazil. e-mail:bWlyaWFuQGlvYy5maW9jcnV6LmJy