


- 1Laboratório de Ultraestrutura Celular Hertha Meyer, Instituto de Biofísica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
- 2Laboratório de Biologia, Instituto Nacional de Metrologia, Qualidade e Tecnologia – Inmetro Duque de Caxias, Rio de Janeiro, Brazil
Trypanosoma cruzi, the causative agent of Chagas disease, is transmitted to vertebrate hosts by blood-sucking insects. This protozoan is an obligate intracellular parasite. The infective forms of the parasite are the metacyclic trypomastigotes, amastigotes, and bloodstream trypomastigotes. The recognition between the parasite and mammalian host cell, involves numerous molecules present in both cell types, and similar to several intracellular pathogens, T. cruzi is internalized by host cells via multiple endocytic pathways. Morphological studies demonstrated that after the interaction of the infective forms of T. cruzi with phagocytic or non-phagocytic cell types, plasma membrane (PM) protrusions can form, showing similarity with those observed during canonical phagocytosis or macropinocytic events. Additionally, several molecules known to be molecular markers of membrane rafts, macropinocytosis, and phagocytosis have been demonstrated to be present at the invasion site. These events may or may not depend on the host cell lysosomes and cytoskeleton. In addition, after penetration, components of the host endosomal-lysosomal system, such as early endosomes, late endosomes, and lysosomes, participate in the formation of the nascent parasitophorous vacuole (PV). Dynamin, a molecule involved in vesicle formation, has been shown to be involved in the PV release from the host cell PM. This review focuses on the multiple pathways that T. cruzi can use to enter the host cells until complete PV formation. We will describe different endocytic processes, such as phagocytosis, macropinocytosis, and endocytosis using membrane microdomains and clathrin-dependent endocytosis and show results that are consistent with their use by this smart parasite. We will also discuss others mechanisms that have been described, such as active penetration and the process that takes advantage of cell membrane wound repair.
Introduction
Trypanosoma cruzi, the causative agent of Chagas disease, is an obligatory intracellular parasite that belongs to the Kinetoplastida order, and it is recognized by the WHO as one of the world’s 13 neglected tropical diseases, affecting 16 million people in Latin America. After the initial infection by the parasite, some patients can develop acute signs and symptoms, including fever, hepatosplenomegaly, and inflammatory reactions. These acute symptoms can be spontaneously resolved. However, the majority of patients are asymptomatic. After the acute phase, a symptomatic chronic form can develop 10–20 years after the initial infection, causing irreversible damage to the heart, esophagus, and colon, with severe disorders of nerve conduction in these organs. Therefore, Chagas disease is characterized as a chronic, systemic, and endemic disease affecting approximately 16 million in Latin America (1) and is considered the major parasitic disease burden of the American continent (2). This parasite presents a complex life cycle that occurs in both vertebrate and invertebrate hosts, where three major developmental stages are observed: epimastigotes, trypomastigotes, and amastigotes. The infective forms of T. cruzi (amastigotes and trypomastigotes) are able to infect a wide range of nucleated mammalian cells. The intracellular cycle can be divided into several steps and begins when the infective forms attach and are recognized by the host’s cell surface (3). Then, cell signaling processes lead to the internalization of the parasite in a process that involves the formation of an endocytic vacuole known as the PV. This review will focus on several processes that have been shown to be involved in the internalization of T. cruzi, such as phagocytosis, active entry, endocytosis dependent on membrane microdomains (flotillin- and caveolin-dependent), endocytosis mediated by clathrin and macropinocytosis (Figure 1).
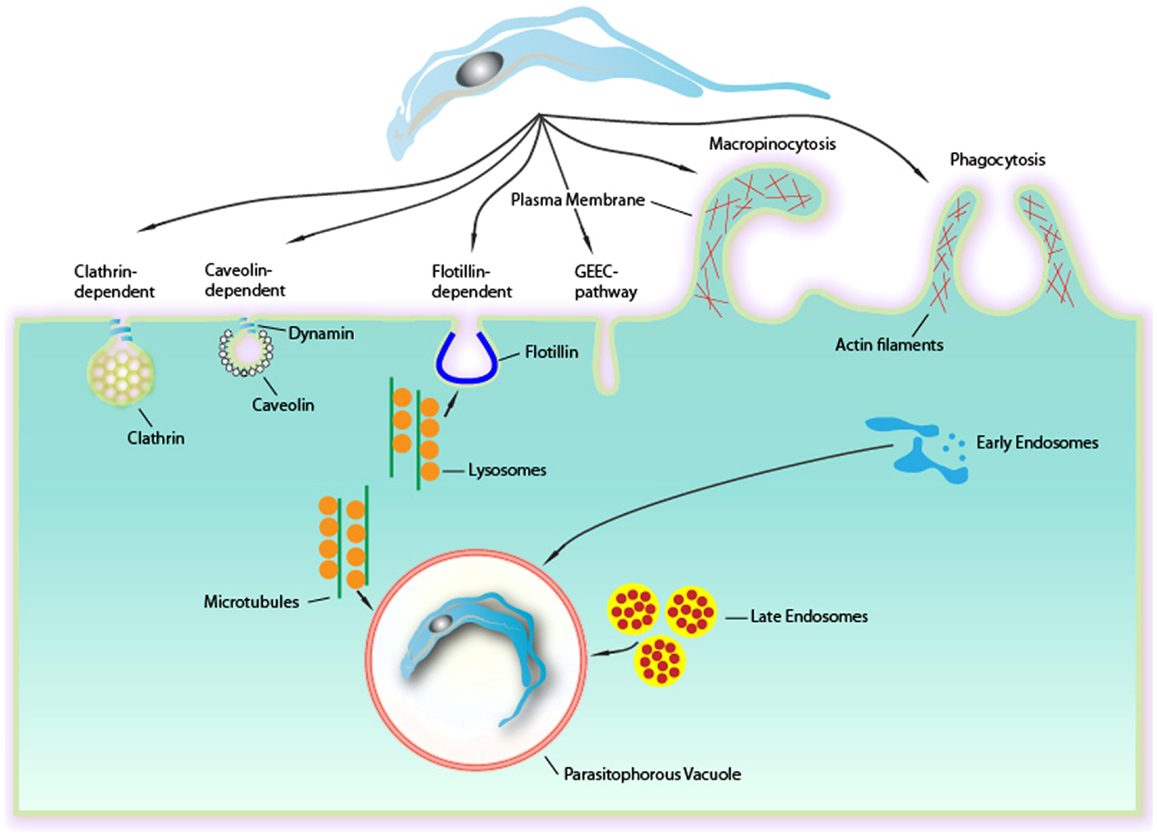
Figure 1. Endocytic mechanisms involved in Trypanosoma cruzi entry into mammalian cells can occur via several different mechanisms culminating in a formation of a PV. Although phagocytosis was the first endocytic mechanism described to be used by T. cruzi, others mechanisms as clathrin-mediated endocytosis, caveolar-dependent, and lipid raft-dependent endocytosis macropinocytosis seems to be involved. Formation of the PV is always depends on lysosomes. This fusion can occur at the site of entry of the parasite or after entry, with the PV preformed. The fusion of lysosomes in areas of entry-dependent flotillin was recently demonstrated, but it is believed that this can occur in other ways. The targeting of lysosomes to entrance region or the PV occurs via microtubules. Upon entry there is also the fusion of endocytic vesicles (endosomes and late initials) that together with the fusion of lysosomes leads to the maturation of the PV through their acidification. This allowed the destruction of this maturing vacuole of the parasite to escape later.
Recognition between Trypanosoma cruzi and the Mammalian Host Cell: A Mechanism Dependent on Receptors and Ligands
Classically, the interaction between host cells and T. cruzi has been divided into two different steps: adhesion (which includes recognition and signaling) and internalization (3). The internalization process is described as occurring through several pathways that resemble endocytic mechanisms. These two steps are easily distinguished because interactions performed at 4°C do not allow parasite internalization and the parasites remain attached to the host cell plasma membrane (PM), suggesting that the internalization process only occurs at higher temperatures (higher than 18°C) (4). Endocytic mechanisms control the lipid and protein composition of the PM, thereby regulating how cells interact with their environments (5). Endocytosis creates an essential interface between eukaryotic cells and their surroundings through the formation, budding, and maturation of PM-derived intermediates. That endocytosis comprises a sophisticated array of different pathways is now widely accepted (6). Mechanisms involved in cellular uptake are important for different processes in a wide variety of cell types. Classically, these mechanisms can be classified into a number of clathrin-independent pathways as well as clathrin-mediated endocytosis (CME), caveolae, phagocytosis, macropinocytosis, and circular dorsal ruffles (5). Additionally, pathogens often exploit endocytic routes to mediate their internalization into cells (7, 8). Although several studies have been conducted in the field of pathogen and host cell interactions, the molecular mechanisms, including the types of endocytic pathways and the proteins involved in cargo recruitment and internalization, are not completely clear (7). Actually, endocytic pathways start with the recognition between the molecules present and exposed on the cell surface and the product that will be internalized (7). Several T. cruzi molecules have been described as being involved in the process of invasion. One class of these molecules is the mucins, which are major T. cruzi surface glycoproteins (7). Many mucins have been reported as T. cruzi ligands because their sugar residues interact with mammalian host cells (9– 12). Other T. cruzi molecules involved in adhesion are trans-sialidases (active and inactive) and glycoproteins (gp82, gp80, gp35/50, and gp85) (13). With respect to the mammalian host cell, any class of molecules exposed on the host cell surface is believed to have the potential to be a T. cruzi receptor ligand. Most of the characterized receptor classes are carbohydrates that contain galactosyl, mannosyl, and sialyl residues (3, 14– 19) and lectin-like proteins, such as galectin 3 that bind to carbohydrate residues present on the parasite surface (20– 22). Some lectins, as mannose binding lectin, are involved in a humoral pattern-recognition molecule important for host defense. In the case of Chagas’ disease this lectin is involved in regulating host resistance and cardiac inflammation during infection (23). Other molecules that function as receptors is possibly involved in the pathogenesis of Chagas’ disease are endothelin 1 and bradikinin receptors. They are used by tripomastigotes to invade cardiovascular cells leading to a chagasic vasculopathy (24). Cytokeratin 18, fibronectin, laminin, and integrins are also receptor molecules because the Tc85 present on the trypomastigote surface has motifs that bind to these molecules, making a bridge between the parasite and the host cell (Figure 2) (25– 27). We will not describe all the putative molecules involved in T. cruzi-host cell interactions because this topic has been discussed in recent reviews (3, 28) and will be covered by other authors in this issue.
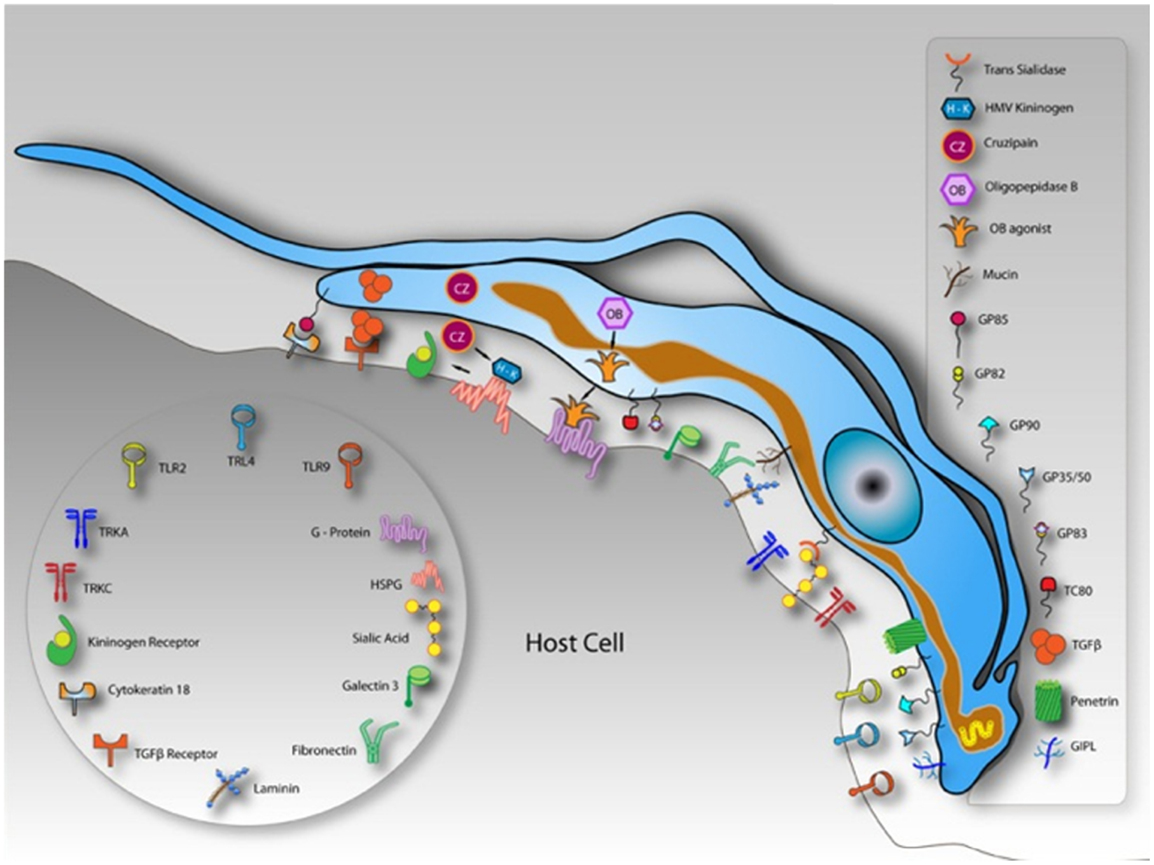
Figure 2. Schematic model demonstrating molecules involved on parasite-host cell interaction process and exposed on the surface of a hypothetical host cell and in trypomastigotes of Trypanosoma cruzi. After Ref. (3).
Phagocytosis
The process known as phagocytosis is a key mechanism of the innate immune response in which macrophages, dendritic cells, and other myeloid phagocytes internalize diverse microorganisms, dead or dying cells, and debris (29). Phagocytosis is an actin-dependent process that can be triggered by several types of ligands and receptors, leading to particle internalization (30). These receptors, called “pattern-recognition receptors” by Janneway (31) because of their capability to recognize pathogens, are present on the entire surface of phagocytic cells and are known as Fc receptors, complement receptors, scavenger receptors, mannose receptors, and receptors for extracellular matrix components (32). Accordingly, a classical zipper type of phagocytosis was described in addition to several unconventional phagocytic routes. In the classical zipper model, after the attachment of a pathogen to the receptor present on the host cell PM, bilateral protrusions extending from the host cell PM engulf the pathogen until a vacuole (completed sealed) is formed. Frequently, this type of phagocytosis occurs after some ligand binds to the Fc receptors or CR receptors (33). Unconventional methods of phagocytosis can be shared among three different groups according to the morphological features. The first is triggered phagocytosis, in which abundant membrane ruffles eventually enclose a spacious vacuole containing the microorganism to be ingested. This mechanism, frequently referred to as triggered macropinocytosis, is commonly driven by entero-invasive bacteria and requires a secretion of a type 3 bacteria protein complex that is responsible for translocating bacterial proteins into the host cells (34). Another unconventional mechanism is overlapping phagocytosis, which is morphologically described as forming pseudopods that do not fuse but slide past each other, resulting in pseudopod stacks to which lateral pseudopods are added. Coiling phagocytosis is characterized by the extension of unilateral pseudopods that rotate around the pathogens. Both overlapping and coiling phagocytosis are predominantly observed in professional phagocytic cells, indicating that this process is driven by the host cell (32). The signaling triggered by the pathogen varies depending on the nature of the receptors used. Basically, exposure to multivalent ligands induces clustering of these receptors in the plane of the membrane, initiating the phosphorylation of some tyrosine kinases. The remodeling of actin is unambiguously required for pseudopod extension, and in the case of FcγR, polymerization is driven by Rac1 and/or Rac2 and Cdc 42. Additionally, phosphoinositides provide an important contribution to actin remodeling during phagocytosis. Phosphatidylinositol-4,5-bisphosphate and phosphatidylinositol-3,4,5-participate in actin assembly, driving pseudopod formation. The conversion to phosphatidylinositol-3,4,5-trisphosphate is required for pseudopod extension and phagosomal closure. Phospholipases A and D have been considered essential to phagosome formation (35). With respect to the T. cruzi entry process, Nogueira and Cohn (36) were the first to propose that trypomastigotes enter peritoneal macrophages, L929, HeLa cell line and calf embryo fibroblasts by a phagocytic process because the treatment of these host cells with cytochalasin B (a drug that blocks the extension of actin filaments) inhibited the parasite internalization. Using cardiac muscle cells, Barbosa and Meirelles (37) demonstrated by transmission electron microscopy that trypomastigotes bind and induce a typical phagocytic process with host cell pseudopod extensions. These studies suggested the participation of endocytic mechanisms in both professional and non-professional phagocytes. In 1991, Hall et al. (38), using a macrophage cell line, described that the PV containing trypomastigotes presents CR3 receptors, β1 integrin, lysosomal membrane glycoproteins (lgp), and Fc receptors (the last only appears if trypomastigotes were previously opsonized). These results supported the hypothesis that T. cruzi can enter the host cell, mainly in macrophages, by phagocytosis. The recognition of Toll-like receptors 2 by trypomastigotes is also capable of inducing a phagocytic process (39) and initiating an inflammatory pathway. Additionally, several groups demonstrated the presence of PM components at the PV membrane, such as galactosyl and glycoconjugate residues (40) and sialoconjugates (41). Several signaling pathways are triggered by phagosome formation and are not different from those involved in the formation of the PV. In professional phagocytes, the activation of tyrosine kinase proteins during the initial contact with trypomastigotes was observed, followed by the recruitment of PI 3-kinase, which culminates in the polymerization of actin microfilaments and pseudopod extension. The participation of tyrosine kinases was demonstrated by Vieira et al. (42) using peritoneal macrophages treated with kinase inhibitors, such as genistein and staurosporine and this group suggested that the main process of trypomastigote entry was by phagocytosis. The participation of Rac1, Rho, and Cdc42 was also observed and will be discussed later. Currently, with new tools to study the endocytic types, the signaling pathways, and cellular components that are involved in different phagocytic mechanisms are being elucidated (macropinocytosis, CME, and participation of membrane microdomains) (5). In relation to amastigote the infection of mammalian cells seems be different when using and comparing different strains. While amastigotes from the T. cruzi I lineage (G strain) appears to induce phagocytosis by non-phagocytic cells (43, 44), amastigote from T. cruzi II as Y strain is largely phagocytized by macrophages, and occasionally by other cell types (43, 45). The amastigotes’ ability to induce phagocytosis was first demonstrated through cytochalasin D host cell’s treatment, where Procópio and colleagues (46) observed a drastic reduction of amastigotes penetration after actin polymerization inhibition. The analysis of the interaction type using these new approaches indicates that events initially described as phagocytosis may correspond to other endocytic pathways. The morphological analysis of the initial steps of T. cruzi invasion (trypomastigotes or amastigotes) using transmission and scanning electron microscopies revealed that this protozoan uses different mechanisms to invade host cells given that a wide type of morphological events can be observed when they are allowed to interact with the host cells. Using field emission scanning electron microscopy, we showed that even after a short interaction time, trypomastigotes, and amastigotes are ingested by peritoneal macrophages and by non-professional phagocytic cells (LLC-MK2). The macrophage PM can tightly recover T. cruzi, forming a funnel-like structure with bilateral projections of the host cell PM to internalize the parasites in a process described as a classical phagocytosis pathway, forming a long, large protrusion that recovers the parasite body, as characterized in the initial step of trigger phagocytosis (or macropinocytosis), or even forming a structure described as a coiled-coil phagosome in which the host cell PM forms coiled-coil projections (Figure 3) (47).
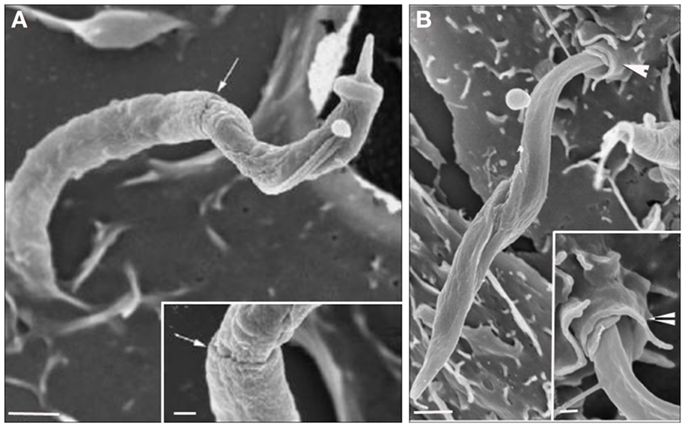
Figure 3. Field emission scanning electron microscopy of the interaction between peritoneal macrophages and T. cruzi. (A) T. cruzi was partially tightly recovered by the macrophage plasma membrane (PM) in a process described as phagocytosis. (B) T. cruzi flagella recovered by host cell PM in a process described as coiled phagocytosis. Bars = 1 μm [After Ref. (47)].
Autophagy as an Inductor of Autophagosome Formation
Phagosomes can also form inside cells in a process described as autophagy. Autophagy is a self-degradative process involved in developmental regulation, the response to nutrient stress, and the clearance of damaged proteins and organelles and plays an important role in balancing sources of energy at critical times in development and in response to nutrient stress (48). Autophagy also plays a housekeeping role in removing misfolded or aggregated proteins, clearing damaged organelles, and eliminating intracellular pathogens (48). Indeed, during autophagy, intracellular membranes engulf organelles and cytoplasmic debris, and this process can be used to engulf intracellular microorganisms into a phagosome (called an autophagosome in the case of autophagy). The intracellular machinery involved in this process is complex, involving several classes of proteins, including Atg proteins (proteins related to autophagy) (49). Currently, more than 32 genes for Atg proteins have been described in mammals (49). The formation of double membrane autophagosomes also requires the activation of the mTOR protein (mammalian target of rapamycin protein) and recruitment of microtubule-associated protein light chain 3 (LC3B) and lysosome (49). This mechanism can be induced by starvation or by the use of rapamycin (which activates the mTOR pathway). Romano and colleagues (50) demonstrated that both treatments are capable of reducing the internalization of T. cruzi into host cells and that the PV is labeled with LC3B, a molecular marker of the autophagy pathway. Martins et al. (51) showed that treating host cells with rapamycin impairs the binding of T. cruzi gp82 to the host cell. This surface molecule is required for adhesion and is one molecule described to be responsible for the exocytosis of lysosomes that can lead to trypomastigote internalization (51).
Membrane Rafts: Endocytosis Dependent on Caveolin or Flotillin
Due to their characteristic shape, caveolae have long been thought to be dynamic endocytic structures (52). In the case of mammalian cells, basically three different types of caveolin proteins are present: caveolin 1, caveolin 2, and caveolin 3 (52). Caveolin 1 and caveolin 2 are found in almost all cell types (excluding neurons and leukocytes, which do not present caveoles), and caveolin 3 mainly found in muscle cells (52). Each caveolae presents approximately 200 caveolin 1 molecules, and caveolae biogenesis is completely dependent on this protein (52). Based on this information, caveolin 1 is known to be the main caveolae marker. Caveolin 1 is also capable of binding to the GM1 ganglioside and to some GPI-anchored proteins. Cholesterol is another component of caveolae, and its depletion has been shown to promote the disorganization of the caveolar structure (53). The raft-associated proteins, flotillin 1 and flotillin 2, are also reported to play a role in endocytosis. Flotillin proteins show homology with caveolin 1, thus suggesting participation in lipid ordering (5, 54, 55). The domains that contain flotillins are morphologically distinct from caveolae because they display a flattened shape, whereas caveolae are spherical. However, both are enriched in cholesterol, GM1 and GPI-anchored proteins (55).
The host cell PM microdomains have been shown to be involved in T. cruzi entry in both non-phagocytic and phagocytic cells (56– 58). Fernandes et al. (56) and Barrias et al. (57) showed that cholesterol, the major component of membrane rafts, is involved in the T. cruzi entry process because the treatment of the host cells with drugs that remove or immobilize this component, such as beta cyclodextrin and filipin, impairs parasite internalization. We do not know yet if cholesterol is a direct participant in this recognition process or if the alterations caused by its removal or immobilization lead to membrane composition alterations that hide or remove receptors involved in this important process. Previously, Hissa et al. (58) showed that cholesterol depletion reduces T. cruzi penetration because lysosome exocytosis became unregulated after this treatment, impairing the release of acid sphingomyelinase from the lysosome, which induces endocytosis. During parasite internalization by the host cell, molecular markers of both types of membrane rafts, such as flotillin 1, caveolin 1, and GM1, were observed at the parasite-host cell PM interface (Figure 4) (57). These suggest the participation of microdomains in T. cruzi internalization by the host cells.
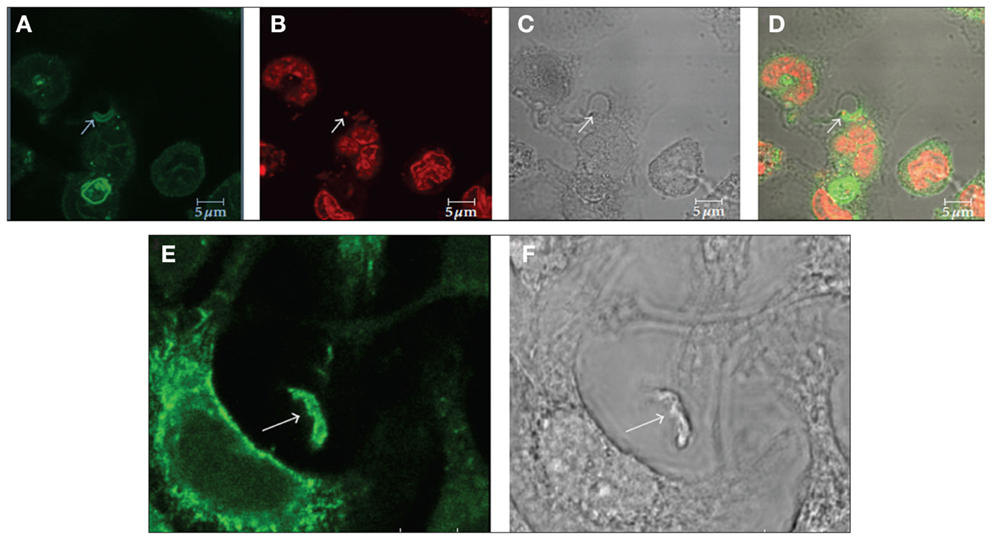
Figure 4. Immunofluorescence microscopy localization of GM1 (A–D) and flotillin 1 (E,F) during internalization of T. cruzi by macrophages suggests the participation of membrane microdomains in this process. (A–D) Co-localization of GM1, using cholera toxin subunit B (A) and an intracellular parasite (C: arrow). (B) Shows labeling of the nucleus and kinetoplast with propidium iodide. Corresponds to a DIC image; (D) is a merge image. (E,F) Co-localization of flotillin 1 (A), detected using a specific antibody, and trypomastigotes (B: arrows). Bars – 5 μm. After Ref. (57).
Macropinocytosis as Another Route to T. cruzi Penetration
Macropinocytosis represents a regulated form of endocytosis that mediates the non-selective uptake of solute molecules, nutrients, antigens, and some pathogens, such as viruses. This process of endocytosis was originally described as involving the assembly of large extensions of the PM (59). The molecular basis for the formation and maturation of macropinosomes has only recently begun to be defined. Macropinocytic events may begin with external stimuli that trigger the activation of tyrosine kinase receptors, inducing changes in the dynamics of actin filaments, which then leads to PM ruffling. The Ras GTPase superfamily plays an important function in the activation process (60). After activation of the tyrosine kinase receptor, three different signaling pathways are triggered, involving the proteins Rac1, Rabankyrin 5 (an effector of Rab5 protein), Arf6, PI3K, and p21-activated kinase Pak1 (activates Rac1) (61). Rabankyrin 5 has been used as a molecular marker to distinguish macropinosomes from other endocytic compartments (61). In addition, this mechanism is also characterized by the actin-dependent reorganization of the PM to form macropinosomes, which are morphologically heterogenic vesicles that lack coat structures. Na+/H+ exchangers have also been described to play an important role in the maintenance of a macropinocytic event. Indeed, drugs that inhibit these exchangers, such as amiloride and EIPA, are widely used to characterize macropinocytosis (60). Although PI3K, Rac, and Cdc 42 have already been described as proteins involved in T. cruzi entry into different cell types, Barrias and colleagues (62) recently showed, for the first time, the participation of this pathway in the internalization of trypomastigotes and amastigotes of T. cruzi into phagocytic and non-phagocytic cell types. The intense inhibition of the parasite internalization process occurred when the host cells were pre-treated with amiloride (an inhibitor of Na+/H+ exchangers) or with rottlerin (an inhibitor of PKC). Host cell treatment with PMA, a stimulator of macropinocytosis caused by PKC stimulation, promotes an increase in parasite internalization. The recruitment of phosphorylated proteins, actin, and Rabankyrin 5 to the site of parasite entry and the characteristic morphology of this process, as shown by fluorescence microscopy, support the view that macropinocytosis is another process used by T. cruzi to penetrate host cells (Figure 5) (62). Morphologically, the entry of trypomastigotes and amastigotes in peritoneal macrophages closely resembles the process described for macropinocytosis, where there are extensive unilateral extensions of the PM that result in a loose vacuole around the parasite (62).
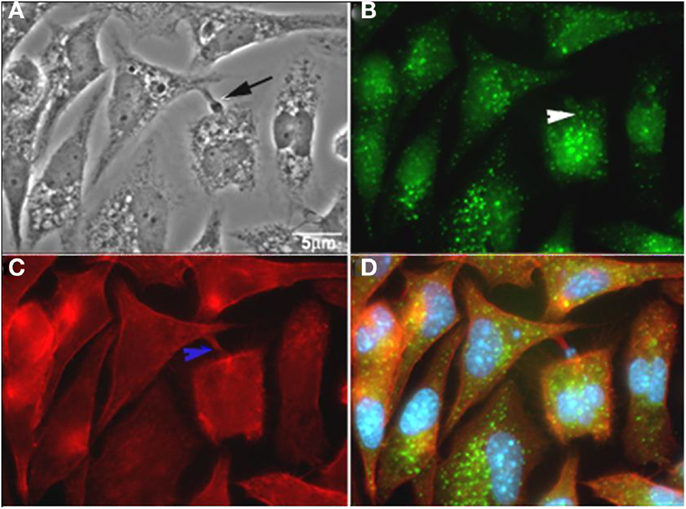
Figure 5. T. cruzi co-localizes with rabankyrin 5 and actin. (A) Phase contrast; (B) rabankyrin 5-Alexa 488; (C) phalloidin-Alexa 546; (D) merge (rabankyrin 5, phalloidin, and DAPI). Arrow indicate trypomastigotes, white arrowhead indicates rabankirin labeling around parasites and blue arrowhead indicates host cell actin around parasites. After Ref. (62).
Clathrin-Mediated Endocytosis
Clathrin-coated vesicles are formed during receptor-mediated endocytosis and organelle biogenesis at the trans-Golgi network (5). The clathrin coat itself is formed by the self-assembly of triskelion-shaped molecules composed of three clathrin heavy chains and associated clathrin light chain subunits (63). The diversity of the cargo and diversity of the adaptor and accessory proteins used to implement vesicle formation reflect the pathways’ adaptations to tools suited to the materials being packaged. Some well-known cargoes that use CME are tyrosine receptor kinase, GPCRs, transferrin receptor, LDL receptors, and anthrax toxin (64). Clathrin is also required for the internalization of large structures, such as bacteria (65), fungi hyphae (66), and large viruses (67), in a process that involves cooperation with actin. Recently, Nagajyotic and colleagues (68) demonstrated that the low-density lipoprotein receptor (LDLr) is important in the invasion and subsequent fusion of the PV containing T. cruzi with host cell lysosomes, thus suggesting the participation of clathrin-coated pits in parasite internalization because LDL receptors are concentrated in this vesicle. This demonstration was performed using an antibody against the clathrin light chain by immunofluorescence. Although this labeling was clearly observed around the vacuole, further studies should be conducted to demonstrate that the labeling is actually clathrin from the endocytic-coated vesicles and not from another cell site.
Endo-Lysosome Participation in T. cruzi Invasion
After the cargo binds to mammalian cell receptors and its internalization by different endocytic pathways culminating in the activation of many signaling events, the cargo is delivered to heterogeneous organelles known as early endosomes. These organelles are usually complex presenting long thin tubules connected to bulbous or vacuolar elements and pH 6.5–6.0. Early endosomes contain molecular markers, such as the Rab5 and EEA1 proteins (“early endosome antigen”), in their membranes. The tubules are responsible for molecular sorting and vesicle transport to the endoplasmic reticulum, PM, trans-Golgi network, and other destinations (69). This organelle loss tubular elements and matures, transforming in a late endosome. The maturation is marked by the switch of molecular marker Rab5 to Rab7 (70). The late endosome displays a vesicular appearance and moves through cellular microtubules in the minus direction, allowing it to occupy a perinuclear position. In addition to Rab7, late endosomes present Rab9, Cd63, and the mannose-6 phosphate receptor (70). The Lamp1 and Lamp2 proteins, which protect the organelle from acid hydrolases, are acquired during this maturation process through fusion with the lysosome in a coordinating system that culminates in an organelle containing many vesicles inside (multivesicular bodies) and with a low pH range (4.5–5.0). The participation of early and late endosomes in the T. cruzi-host cell interaction was first characterized by Wilkowsky and colleagues (71) when they demonstrated the recruitment of Rab5 and Rab7 to the PV containing the protozoan. Woolsey et al. (72), using a short interaction time between T. cruzi and non-professional phagocytic cells, showed that 50% or more of the invading T. cruzi trypomastigotes use the host cell PM during the PV formation. They suggested that this process was facilitated by host cell actin depolymerization and showed that this vacuole is enriched in products from PI 3-kinase and that it is negative for lysosomal markers. Approximately 20% of T. cruzi-containing vacuoles were positive for EEA1 and Rab 5, and approximately 20% were positive for Lamp1, a lysosome marker. Since 1994 (73), the exocytosis of lysosomes to the parasite site of entry has been described as playing an important role in parasite entry. Lysosomes are placed in the path of the host cell PM along microtubules in a kinesin-dependent method (74). They fuse with the PM in a Ca2+-dependent process (75). This process was described as the unique way the parasite used to enter and be kept inside the host cell. However, this process was subsequently shown to represent only approximately 20% of the parasite entrance, and a lysosome-independent process was described to account for approximately 50% of the parasite internalization process. In addition, in approximately 20% of the internalized parasite, there was participation of the early endosomes, as recognized by EEA1 labeling 10 min post-infection (72). They also described that the PV vacuole containing T. cruzi was observed labeled with the lysosome associated protein 1 (LAMP1) as well as with endocytic tracer from pre-labeled lysosomes. These results showed that the lysosome pathway was not the only one that presents fusion with lysosomes. Barr et al. (76) showed that an unusual 120-kDa alkaline peptidase (TSF) from a soluble fraction of T. cruzi induces repetitive calcium transients in primary isolated cardiac myocytes from dogs. Using thapsigargin, they also showed that Ca2+ depletion from intracellular stores, such as the sarcoplasmic reticulum, is able to inhibit Ca2+ transients and trypomastigote invasion. The authors also described that “the Ca2+ transients are dependent on release of Ca2+ from sarcoplasmic reticulum Ca2+ stores, but this release in not dependent on extracellular Ca2+ or on the classic model of Ca2+-induced Ca2+ release in cardiac myocytes.” In 1999, Meirelles et al. (77) also described that the sarcoplasmic reticulum Ca2+ ATPase (SERCA) participates in trypomastigote invasion into cardiomyocytes because thapsigargin inhibits 75% of this process. Recently, Fernandes et al. (78) showed that the entry of T. cruzi trypomastigotes into the host cell wounds the host cell PM by inducing a process of wound repair using Ca2+-dependent exocytosis of lysosomes. The lysosome exocytosis was triggered by an increase in calcium influx, derived from the extracellular space, which enters the host cell as soon as the PM is wounded. The wound repair of the host cell PM was performed with the lysosomal delivery of acid sphingomyelinase to the host PM and formation of endosomes enriched in ceramide, processes that facilitate parasite entry into the host cell (78). Besides, this mechanism may be involved with the tropism of T. cruzi for cardic cells since membrane repair is common in muscle cells, explaining part of the Chagas’ disease pathology (78).
Actin Cytoskeleton
The participation of the actin cytoskeleton during the initial step of the invasion has, until now, been under debate. The participation of the actin cytoskeleton in T. cruzi entry has been suggested since 1976 when Dvorak and colleagues (79) treated different host cells with cytochalasin B and demonstrated unequivocally that the internalization of trypomastigotes was impaired. Rosestolato et al. (80), using different host cells (professional and non-professional phagocytic cells) previously treated with cytochalasin D (CD) and then allowed to interact with the cell culture trypomastigote forms, also showed a drastic reduction of the parasites inside the host cells (81). Additionally, Barbosa and Meirelles (37), using heart muscle cells, clearly showed the evident participation of the actin cytoskeleton during T. cruzi invasion. In 2004, Woolsey and Burleigh (72) showed that actin depolymerization by cytochalasin D enhances parasite entry into the host cell at an early step and also blocks lysosome or early endosome fusion at the site of parasite entry. They also described, using NIH-3T3 fibroblasts expressing dominant-negative Rho, that after 15 min of infection, that there were three times more parasites inside than in the control cells but that the number of intracellular parasites drastically decreased until 1 h. They suggested that a cell with continuous actin cytoskeleton alterations was not able to retain the parasites inside the cell, showing the importance of actin polymerization and depolymerization on the interaction process. Our group showed (82) that cells overexpressing Rac 1 exhibited a higher internalization index for T. cruzi compared with normal cells. However, after 48 h, a reduced number of parasites were observed. Notably, these different results can be explained by different host cell treatments, whether the cells were washed after the incubation with cytochalasin, the interaction time after the drug treatment, the nature of the parasite strain, and other considerations. We also believe that despite the contradictory results, all these papers contribute to a better understanding of the complex process of the T. cruzi-host cell interaction and that it is not good scientific practice to neglect a thorough discussion of all published results, as frequently happens.
During the initial moments of the interaction process with T. cruzi trypomastigotes, the host cell transient calcium increase has been reported (73– 76). Host cells treated with thapsigargin, an inhibitor of endoplasmic reticulum Ca2+-ATPase (83) that reduces parasite entry into the host cell (84), showed the participation of the intracellular calcium store in this process and its involvement in lysosome exocytosis.
Parasitophorous Vacuole’ Closure
In mammalian cells, several molecules that selectively regulate the assembly of an endocytic vacuole have been identified. Among them, dynamin has been shown to play a major role in processes such as CME, synaptic vesicle recycling, phagocytosis, transport from the trans-Golgi network, and ligand uptake through caveolae (85). Dynamin is a GTPase family comprising three isoforms: dynamins 1, 2, and 3 (86). One protein class that interacts with dynamin is phosphatidylinositol-3-kinase (PI3K) (87). Dynamin interacts with the p85 regulatory subunit of PI3K, and this interaction stimulates the GTPase activity of dynamin. Gold and colleagues (88) reported that the inhibition of PI3K prevents the recruitment of dynamin 2 to the site of particle binding, suggesting that in phagocytosis, the activation of PI3K is upstream of dynamin. According to some models, dynamin is a mechanochemical enzyme that is directly responsible for pinching off the vesicle (86). Other authors consider that dynamin is a regulatory protein that recruits the downstream partner, which, in turn, drives the fission step (87, 89). Using dominant-negative dynamin (K44A) HeLa cells, Wilkowsky and colleagues (71) showed that dynamin is involved in the invasion of T. cruzi in non-phagocytic host cells. Subsequently, Barrias et al. (62) showed that the GTPase activity of this protein is important for the fission of PVs in both phagocytic and non-phagocytic cell lines through the use of dynasore, which has the ability to block the GTPase activity of dynamin, acting as a potent inhibitor of endocytic pathways by blocking coated vesicle formation within seconds of its addition.
Concluding Remarks
More than 100 years after Carlos Chagas’ discovery about T. cruzi and Chagas’ disease, we still have many important gaps in the knowledge of the basic aspects of the protozoan biology and its interaction with host cells. It is now clear that the parasite uses several surface-associated molecules to interact with a not yet completely defined set of macromolecules exposed on the host cell surface. We also now know that several internalization processes are triggered following the parasite ligand-host cell receptor interactions. However, we still do not know which ligand-receptor complex triggers each of the types of internalization. New methods and approaches are necessary to better understand the parasite-host cell interaction process. The use of parasite molecules to recover latex beads and their use to interact with host cells may provide new information as to how different parasite molecules could act in the parasite-host cell junction. Unfortunately, neither gene knock-out nor gene silencing is effective with T. cruzi. The use of other methodologies, such as high-throughput technology, gene knock-out of host cell molecules by RNAi and microarray platforms, can provide new insights into this fascinating field of research. Altogether, it is clear that now we have much more information on the process of interaction of T. cruzi with host cells, especially the various mechanisms the parasite uses to penetrate into host cells. It is now important to identify the key molecules involved on each process and develop drugs able to inhibit the infection of the cells by the parasite, opening a new approach to the treatment of the acute phase of Chagas disease, where amplification of the infection through successive invasion of the cells plays a fundamental role.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rassi A Jr, Rassi A, Marcondes de Rezende J. American trypanosomiasis (Chagas disease). Infect Dis Clin North Am (2012) 26(2):275–91. doi:10.1016/j.idc.2012.03.002
2. Rassi A Jr, Rassi A, Marin-Neto JA. Chagas disease. Lancet (2010) 375(9723):1388–402. doi:10.1016/S0140-6736(10)60061-X
3. De Souza W, de Carvalho TM, Barrias ES. Review on Trypanosoma cruzi: host cell interaction. Int J Cell Biol (2010) 2010:295394. doi:10.1155/2010/295394
4. De Meirelles MNL, Araújo Jorge TC, De Souza W. Interaction of Trypanosoma cruzi with macrophages in vitro: dissociation of the attachment and internalization phases by low temperature and cytochalasin B. Z Parasitenkd (1982) 68(1):7–14. doi:10.1007/BF00926652
5. Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem (2009) 78:857–902. doi:10.1146/annurev.biochem.78.081307.110540
6. Howes MT, Mayor S, Parton RG. Molecules, mechanisms, and cellular roles of clathrin-independent endocytosis. Curr Opin Cell Biol (2010) 22(4):519–27. doi:10.1016/j.ceb.2010.04.001
7. Sibley LD, Andrews NW. Cell invasion by un-palatable parasites. Traffic (2000) 1(2):100–6. doi:10.1034/j.1600-0854.2000.010202.x
8. Coombes JL, Robey EA. Dynamic imaging of host-pathogen interactions in vivo. Nat Rev Immunol (2010) 10(5):353–64. doi:10.1038/nri2746
9. Villalta F, Kierszenbaum F. Host cell invasion by Trypanosoma cruzi: role of cell surface galactose residues. Biochem Biophys Res Commun (1984) 119(1):228–35. doi:10.1016/0006-291X(84)91642-5
10. Yoshida N, Mortara RA, Araguth MF, Gonzalez JC, Russo M. Metacyclic neutralizing effect of monoclonal antibody 10D8 directed to the 35- and 50-kilodalton surface glycoconjugates of Trypanosoma cruzi. Infect Immun (1989) 57(6):1663–7.
11. Di Noia JM, Sanchez DO, Frasch ACC. The protozoan Trypanosoma cruzi has a family of genes resembling the mucin genes of mammalian cells. J Biol Chem (1995) 270(41):24146–9. doi:10.1074/jbc.270.41.24146
12. Buscaglia CA, Campo VA, Frasch AC, DiNoia JM. Trypanosoma cruzi surface mucins: host-dependent coat diversity. Nat Rev Microb (2006) 4(3):229–36. doi:10.1038/nrmicro1351
13. Schenkman S, Diaz C, Nussenzweig V. Attachment of Trypanosoma cruzi trypomastigotes to receptors at restricted cell surface domains. Exp Parasitol (1991) 72(1):76–86. doi:10.1016/0014-4894(91)90123-E
14. Alves MJ, Colli W. Role of the gp85/trans-sialidase superfamily of glycoproteins in the interaction of Trypanosoma cruzi with host structures. Subcell Biochem (2008) 47:58–69. doi:10.1007/978-0-387-78267-6_4
15. Abuin G, Colli W, De Souza W, Alves MJM. A surface antigen of Trypanosoma cruzi involved in cell invasion (Tc-85) is heterogeneous in expression and molecular constitution. Mol Biochem Parasitol (1989) 35(3):229–37. doi:10.1016/0166-6851(89)90209-0
16. Andrews NW, Katzin AM, Colli W. Mapping of surface glycoproteins of Trypanosoma cruzi by two dimensional electrophoresis. A correlation with the cell invasion capacity. Eur J Biochem (1984) 140(3):599–604. doi:10.1111/j.1432-1033.1984.tb08144.x
17. Araya JE, Cano MI, Yoshida N, Da Silveira JF. Cloning and characterization of a gene for the stage-specific 82-kDa surface antigen of metacyclic trypomastigotes of Trypanosoma cruzi. Mol Biochem Parasitol (1984) 65(1):161–9. doi:10.1016/0166-6851(94)90124-4
18. Previato JO, Andrade AFB, Pessolani MCV, Mendonça-Previato L. Incorporation of sialic acid into Trypanosoma cruzi macromolecules. Mol Biochem Parasitol (1985) 16(1):85–96. doi:10.1016/0166-6851(85)90051-9
19. Tomlinson S, Pontes De Carvalho LC, Vandekerckhove F, Nussenzweig V. Role of sialic acid in the resistance of Trypanosoma cruzi trypomastigotes to complement. J Immunol (1994) 153(7):3141–7.
20. Moody TN, Ochieng J, Villalta F. Novel mechanism that Trypanosoma cruzi uses to adhere to the extracellular matrix mediated by human galectin-3. FEBS Lett (2000) 470(3):305–8. doi:10.1016/S0014-5793(00)01347-8
21. Vray B, Camby I, Vercruysse V, Mijatovic T, Bovin NV, Ricciardi-Castagnoli P, et al. Up-regulation of galectin-3 and its ligands by Trypanosoma cruzi infection with modulation of adhesion and migration of murine dendritic cells. Glycobiology (2004) 14(7):647–57. doi:10.1093/glycob/cwh068
22. Kleshchenko YY, Moody TN, Furtak VA, Ochieng J, Lima MF, Villalta F. Human galectin-3 promotes Trypanosoma cruzi adhesion to human coronary artery smooth muscle cells. Infect Immun (2004) 72(11):6717–21. doi:10.1128/IAI.72.11.6717-6721.2004
23. Rothfuchs AG, Roffê E, Gibson A, Cheever AW, Ezekowitz AB, Takahashi K, et al. Mannose-binding lectin regulates host resistance and pathology during experimental infection with Trypanosoma cruzi. PLoS ONE (2012) 7(11):e47835. doi:10.1371/journal.pone.0047835
24. Andrade D, Serra R, Svensjö E, Lima APC, Ramos Junior ES, Fortes FS, et al. Trypanosoma cruzi invades host cells through the activation of endothelin and bradykinin receptors: a converging pathway leading to chagasic vasculopathy. Br J Pharmacol (2012) 165(5):1333–47. doi:10.1111/j.1476-5381.2011.01609.x
25. Barbosa HS, Meirelles MN. Ultrastructural detection in vitro of WGA-, RCA I-, and Con A-binding sites involved in the invasion of heart muscle cells by Trypanosoma cruzi. Parasitol Res (1992) 78(5):404–9. doi:10.1007/BF00931696
26. Magdesian MN, Giordano R, Juliano MA, Juliano L, Schumacher RI, Colli W, et al. Infection by Trypanosoma cruzi: identification of a parasite ligand and its host cell receptor. J Biol Chem (2001) 276(22):19382–9. doi:10.1074/jbc.M011474200
27. Claser C, Curcio M, Mello SM, Silveira EV, Monteiro HP, Rodrigues MM. Silencing cytokeratin 18 gene inhibits intracellular replication of Trypanosoma cruzi in HeLa cells but not binding and invasion of trypanosomes. BMC Cell Biol (2008) 9:68. doi:10.1186/1471-2121-9-68
28. Villalta F, Scharfstein J, Ashton AW, Tyler KM, Guan F, Mukherjee S, et al. Perspectives on the Trypanosoma cruzi-host cell receptor interactions. Parasitol Res (2009) 104(6):1251–60. doi:10.1007/s00436-009-1383-3
29. Stuart LM, Ezekowitz RA. Phagocytosis: elegant complexity. Immunity (2005) 22(5):539–50. doi:10.1016/j.immuni.2005.05.002
30. Haglund CM, Welch MD. Pathogens and polymers: microbe-host interactions illuminate the cytoskeleton. J Cell Biol (2011) 195(1):7–17. doi:10.1083/jcb.201103148
31. Janeway CA. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol (1989) 54:1–13. doi:10.1101/SQB.1989.054.01.003
32. Rittig MG, Schröppel K, Seack KH, Sander U, N’Diaye EN, Maridonneau-Parini I, et al. Coiling phagocytosis of trypanosomatids and fungal cells. Infect Immun (1998) 66(9):4331–9.
33. Rittig MG, Burmester GR, Krause A. Coiling phagocytosis: when the zipper jams, the cup is deformed. Trends Microbiol (1998) 6(10):384–8. doi:10.1016/S0966-842X(98)01343-2
34. Francis CL, Ryan TA, Jones BD, Smith SJ, Falkow S. Ruffles induced by Salmonella and other stimuli direct macropinocytosis of bacteria. Nature (1993) 364:639–42. doi:10.1038/364639a0
35. Grinstein S. Imaging signal transduction during phagocytosis: phospholipids, surface charge and eletrostatic interaction. Am J Physiol Cell Physiol (2011) 299:876–81. doi:10.1152/ajpcell.00342.2010
36. Nogueira N, Cohn Z. Trypanosoma cruzi: mechanism of entry and intracellular fate in mammalian cells. J Exp Med (1976) 143(6):1402–20. doi:10.1084/jem.143.6.1402
37. Barbosa HS, Meirelles MNL. Evidence of participation of cytoskeleton of heart muscle cells during the invasion of Trypanosoma cruzi. Cell Struct Funct (1995) 20(4):275–84. doi:10.1247/csf.20.275
38. Hall BF, Furtado GC, Joiner KA. Characterization of host cell-derived membrane proteins of the vacuole surrounding different intracellular forms of Trypanosoma cruzi in J774 cells: evidence for phagocyte receptor sorting during the early stages of parasite entry. J Immunol (1991) 147(12):4313–21.
39. Maganto-Garcia E, Punzon C, Terhorst C, Fresno M. Rab5 activation by toll-like receptor 2 is required for Trypanosoma cruzi internalization and replication in macrophages. Traffic (2008) 9(8):1299–315. doi:10.1111/j.1600-0854.2008.00760.x
40. De Meirelles MNL, De Araújo Jorge TC, De Souza W. Interaction of Trypanosoma cruzi with macrophages in vitro: dissociation of the attachment and internalization phases by low temperature and cytochalasin B. Z Parasitenkd (1982) 68(1):7–14. doi:10.1007/BF00926652
41. Carvalho TMU, De Souza W, Coimbra E. Internalization of components of the host cell plasma membrane during infection by Trypanosoma cruzi. Mem Inst Oswaldo Cruz (1999) 94(1):143–7. doi:10.1590/S0074-02761999000700016
42. Vieira MCF, De Carvalho TU, De Souza W. Effect of protein kinase inhibitors on the invasion process of macrophages by Trypanosoma cruzi. Biochem Biophys Res Commun (1994) 203(2):967–71. doi:10.1006/bbrc.1994.2276
43. Mortara RA, Andreoli WK, Taniwaki NN. Mammalian cell invasion and intracellular trafficking by Trypanosoma cruzi infective forms. An Acad Bras Cienc (2005) 77:77–94. doi:10.1590/S0001-37652005000100006
44. Mortara RA, Andreoli WK, Fernandes MC, Silva CV, Fernandes AB, L’Abbate C, et al. Host cell actin remodelling in response to Trypanosoma cruzi: trypomastigote versus amastigote entry. In: B. Burleigh, D. Soldati-Favre, editors. Molecular Mechanism of Parasite Invasion. Chapter 8. Austin, TX: Landes Bioscience and Springer Science+Business Media (2008). p. 101–8. [Subcellular Biochemistry 47:101–9]. doi:10.1007/978-0-387-78267-6_8
45. Ley V, Andrews NW, Robbins ES, Nussenzweig V. Amastigotes of Trypanosoma cruzi sustain an infective cycle in mammalian cells. J Exp Med (1988) 168:649–59. doi:10.1084/jem.168.2.649
46. Procópio DO, Silva S, Cunningan CC, Mortara RA. Trypanosoma cruzi: effect of protein kinase inhibitors and cytoskeletal protein organization and expression on host cell invasion by amastigotes and metacyclic trypomastigotes. Exp Parasitol (1998) 90:1–13. doi:10.1006/expr.1998.4314
47. Barrias ES, Reignault LC, De Souza W, Carvalho TM. Dynasore, a dynamin inhibitor, inhibits Trypanosoma cruzi entry into peritoneal macrophages. PLoS ONE (2010) 25(1):e7764. doi:10.1371/journal.pone.0007764
48. Glick D, Barth S, MacLeod K. Autophagy: cellular and molecular mechanisms. J Pathol (2010) 221:3–12. doi:10.1002/path.2697
49. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol (2011) 10(27):107–32. doi:10.1146/annurev-cellbio-092910-154005
50. Romano PS, Arboit MA, Vázquez CL, Colombo MI. The autophagic pathway is a key component in the lysosomal dependent entry of Trypanosoma cruzi into the host cell. Autophagy (2009) 5(1):6–18. doi:10.4161/auto.5.1.7160
51. Martins RM, Alves RM, Macedo S, Yoshida N. Starvation and rapamycin differentially regulate host cell lysosome exocytosis and invasion by Trypanosoma cruzi metacyclic forms. Cell Microbiol (2011) 13(7):943–54. doi:10.1111/j.1462-5822.2011.01590.x
52. Hansen CG, Nichols BJ. Exploring the caves: cavins, caveolins and caveolae. Trends Cell Biol (2010) 20(4):177–86. doi:10.1016/j.tcb.2010.01.005
53. Westermann M, Steiniger F, Richter W. Belt-like localisation of caveolin in deep caveolae and its re-distribution after cholesterol depletion. Histochem Cell Biol (2005) 123:613–20. doi:10.1007/s00418-004-0750-5
54. Glebov OO, Bright NA, Nichols BJ. Flotillin-1 defines a clathrin-independent endocytic pathway in mammalian cells. Nat Cell Biol (2006) 8:46–54. doi:10.1038/ncb1342
55. Frick M, Bright NA, Riento K, Bray A, Merrified C, Nichols BJ. Coassembly of flotillins induces formation of membrane microdomains, membrane curvature, and vesicle budding. Curr Biol (2007) 17:1151–6. doi:10.1016/j.cub.2007.05.078
56. Fernandes MC, Cortez M, Geraldo Yoneyama KA, Straus AH, Yoshida N, Mortara RA. Novel strategy in Trypanosoma cruzi cell invasion: implication of cholesterol and host cell microdomains. Inter J Parasitol (2007) 37(13):1431–41. doi:10.1016/j.ijpara.2007.04.025
57. Barrias ES, Dutra JMF, De Souza W, Carvalho TMU. Participation of macrophage membrane rafts in Trypanosoma cruzi invasion process. Biochem Biophys Res Commun (2007) 363(3):828–34. doi:10.1016/j.bbrc.2007.09.068
58. Hissa B, Duarte JG, Kelles LF, Santos FP, del Puerto HL, Gazzinelli-Guimarães PH, et al. Membrane cholesterol regulates lysosome-plasma membrane fusion events and modulates Trypanosoma cruzi invasion of host cells. PLoS Negl Trop Dis (2012) 6(3):e1583. doi:10.1371/journal.pntd.0001583
59. Kerr MC, Teasdale RD. Defining Macropinocytosis. Traffic (2009) 10:364–71. doi:10.1111/j.1600-0854.2009.00878.x
60. Lim JP, Gleeson PA. Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol Cell Biol (2011) 89(8):836–43. doi:10.1038/icb.2011.20
61. Schnatwinkel C, Christoforidis S, Lindsay MR, Uttenweiler-Joseph S, Wilm M, Parton RG, et al. The Rab5 effector Rabankyrin-5 regulates and coordinates different endocytic mechanisms. PLoS Biol (2004) 2(9):E261. doi:10.1371/journal.pbio.0020261
62. Barrias ES, Reignault LC, De Souza W, Carvalho TM. Trypanosoma cruzi uses macropinocytosis as an additional entry pathway into mammalian host cell. Microbes Infect (2012) 14(14):1340–51. doi:10.1016/j.micinf.2012.08.003
63. Mooren OL, Galletta BJ, Cooper JA. Roles for actin assembly in endocytosis. Annu Rev Biochem (2012) 81:661–86. doi:10.1146/annurev-biochem-060910-094416
64. Andersson ER. The role of endocytosis in activating and regulating signal transduction. Cell Mol Life Sci (2012) 69(11):1755–71. doi:10.1007/s00018-011-0877-1
65. Law HT, Lin AE, Kim Y, Quach B, Nano FE, Guttman JA. Francisella tularensis uses cholesterol and clathrin-based endocytic mechanisms to invade hepatocytes. Sci Rep (2011) 1:192. doi:10.1038/srep00192
66. Moreno-Ruiz E, Galán-Díez M, Zhu W, Fernández-Ruiz E, d’Enfert C, Filler SG, et al. Candida albicans internalization by host cells is mediated by a clathrin-dependent mechanism. Cell Microbiol (2009) 11(8):1179–89. doi:10.1111/j.1462-5822.2009.01319.x
67. Mercer J, Schelhaas M, Helenius A. Virus entry by endocytosis. Annu Rev Biochem (2009) 79:803–33. doi:10.1146/annurev-biochem-060208-104626
68. Nagajyothi F, Weiss LM, Silver DL, Desruisseaux MS, Scherer PE, Herz J, et al. Trypanosoma cruzi utilizes the host low density lipoprotein receptor in invasion. PLoS Negl Trop Dis (2011) 5(2):e953. doi:10.1371/journal.pntd.0000953
69. Jovic M, Sharma M, Rahajeng J, Caplan S. The early endosome: a busy sorting station for proteins at the crossroads. Histol Histopathol (2010) 25(1):99–112.
71. Wilkowsky SE, Barbieri MA, Stahl PD, Isola EL. Regulation of Trypanosoma cruzi invasion of nonphagocytic cells by the endocytically active GTPases dynamin, Rab5, and Rab7. Biochem Biophys Res Commun (2002) 291(3):516–21. doi:10.1006/bbrc.2002.6474
72. Woolsey AM, Burleigh BA. Host cell actin polymerization is required for cellular retention of Trypanosoma cruzi and early association with endosomal/lysosomal compartments. Cell Microbiol (2003) 6(9):829–38. doi:10.1111/j.1462-5822.2004.00405.x
73. Tardieux I, Nathanson MH, Andrews NW. Role in host cell invasion of Trypanosoma cruzi-induced cytosolic free Ca2+ transients. J Exp Med (1994) 179(3):1017–22. doi:10.1084/jem.179.3.1017
74. Rodríguez A, Samoff E, Rioult MG, Chung A, Andrews NW. Host cell invasion by trypanosomes requires lysosomes and microtubule/kinesin-mediated transport. J Cell Biol (1996) 134(2):349–62. doi:10.1083/jcb.134.2.349
75. Rodríguez A, Martinez I, Chung A, Berlot CH, Andrews NW. cAMP regulates Ca2+-dependent exocytosis of lysosomes and lysosome-mediated cell invasion by trypanosomes. J Biol Chem (1999) 274(24):16754–9. doi:10.1074/jbc.274.24.16754
76. Barr SC, Han W, Andrews NW, Lopez JW, Ball BA, Pannabecker TL, et al. A factor from Trypanosoma cruzi induces repetitive cytosolic free Ca2+ transients in isolated primary canine cardiac myocytes. Infect Immun (1996) 64(5):1770–7.
77. Meirelles MN, Pereira MC, Singer RH, Soeiro MN, Garzoni LR, Silva DT, et al. Trypanosoma cruzi-cardiomyocytes: new contributions regarding a better understanding of this interaction. Mem Inst Oswaldo Cruz (1999) 94(1):149–52. doi:10.1590/S0074-02761999000700017
78. Fernandes MC, Cortez M, Flannery AR, Tam C, Mortara RA, Andrews NW. Trypanosoma membrane repair pathway for cell invasion. J Exp Med (2011) 208(5):909–21. doi:10.1084/jem.20102518
79. Dvorak JA, Howe CL. The attraction of Trypanosoma cruzi to vertebrate cells in vitro. J Protozool (1976) 23(4):534–7.
80. Rosestolato CT, Da Matta Furniel Dutra J, De Souza W, De Carvalho TM. Participation of host cell actin filaments during interaction of trypomastigote forms of Trypanosoma cruzi with host cells. Cell Struct Funct (2002) 27(2):91–8. doi:10.1247/csf.27.91
81. Meirelles MN, Araújo Jorge TC, De Souza W. Interaction of Trypanosoma cruzi with macrophages in vitro: dissociation of the attachment and internalization phases by low temperature and cytochalasin B. Z Parasitenkd (1982) 68(1):7–14. doi:10.1007/BF00926652
82. Dutra JM, Bonilha VL, De Souza W, Carvalho TM. Role of small GTPases in Trypanosoma cruzi invasion in MDCK cell lines. Parasitol Res (2005) 96(3):171–7. doi:10.1007/s00436-005-1333-7
83. Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci U S A (1990) 87:2466–70. doi:10.1073/pnas.87.7.2466
84. Yoshida N, Favoreto S Jr, Ferreira AT, Manque PM. Signal transduction induced in Trypanosoma cruzi metacyclic trypomastigotes during the invasion of mammalian cells. Braz J Med Biol Res (2000) 33(3):269–78. doi:10.1590/S0100-879X2000000300003
85. Schmid SL, McNiven MA, De Camilli P. Dynamin and its partners: a progress report. Curr Opin Cell Biol (1998) 10(4):504–12. doi:10.1016/S0955-0674(98)80066-5
86. Praefcke GJ, McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol (2004) 2:133–47. doi:10.1038/nrm1313
87. Otsuka A, Abe T, Watanabe M, Yagisawa H, Takei K, Yamada H. Dynamin 2 is required for actin assembly in phagocytosis in Sertoli cells. Biochem Biophys Res Commun (2008) 378(3):478–82. doi:10.1016/j.bbrc.2008.11.066
88. Gold ES, Underhil DM, Morrissette NS, Guo J, McNiven MA, Anderem A. Dynamin 2 is required for phagocytosis in macrophages. J Exp Med (1999) 190(12):1849–56. doi:10.1084/jem.190.12.1849
Keywords: Trypanosoma cruzi, mammalian cell, endocytosis, phagocytosis, active penetration, host cell, interaction
Citation: Barrias ES, de Carvalho TMU and De Souza W (2013) Trypanosoma cruzi: entry into mammalian host cells and parasitophorous vacuole formation. Front. Immunol. 4:186. doi: 10.3389/fimmu.2013.00186
Received: 17 October 2012; Accepted: 25 June 2013;
Published online: 01 August 2013.
Edited by:
Abhay Satoskar, The Ohio State University, USAReviewed by:
Emilio Luis Malchiodi, University of Buenos Aires, ArgentinaRavi Durvasula, University of New Mexico School of Medicine, USA
Copyright: © 2013 Barrias, de Carvalho and De Souza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emile Santos Barrias, Laboratório de Biologia, Instituto Nacional de Metrologia, Qualidade e Tecnologia, Avenida Nossa Senhora das Graças, 50 Duque de Caxias, Rio de Janeiro 25250-020, Brazil e-mail:ZXNiYXJyaWFzQGlubWV0cm8uZ292LmJy;
Tecia Maria Ulisses de Carvalho, Laboratório de Ultratestrutura Celular Hertha Meyer, Instituto de Biofı́sica Carlos Chagas Filho, Avenida Carlos Chagas Filho, Cidade Universitária, Rio de Janeiro 21941-902, Brazil e-mail:dGVjaWFAYmlvZi51ZnJqLmJy