 Nichol E. Holodick
Nichol E. Holodick Thomas L. Rothstein
Thomas L. Rothstein- Center for Oncology and Cell Biology, The Feinstein Institute for Medical Research, Manhasset, NY, USA
Peritoneal B-1a cells manifest unusual signaling characteristics that distinguish them from splenic B-2 cells. These include the failure of BCR engagement to trigger NF-κB activation and DNA replication. Despite extensive study, a clear explanation for these characteristics has not emerged. Here we aim to develop a unified paradigm based on previous reports and recent results, which proposes a central role for phosphatase activity. We hypothesize B-1a cells are unable to induce NF-κB or proliferate after BCR cross-linking due to increased phosphatase abundance or activity. This phosphatase abundance and/or activity may be the result of unique B-1a cell characteristics such as increased levels of HSP70 and/or constitutive secretion of IL-10. We speculate phosphatase activity cannot be overcome by BCR ligation alone due to insufficient Vav protein expression, which does not allow for proper production of reactive oxygen species, which inhibit phosphatases. Furthermore, constitutively active Lyn also plays a negative regulatory role in B-1a. We expect that a new focus on phosphatase activity and its suppression will be revealing for BCR signal transduction in B-1 cells.
B-1 Cell Overview
B-1 Cell Characteristics
B-1a cells are set apart from conventional B2 cells based on phenotypic and functional differences. B-1a cells are phenotypically characterized by the following cell surface markers: B220lo, CD5+, immunoglobulin (Ig) (sIg) Mhi, sIgDlo, Mac-1+, CD23−, and CD43+ (1, 2). In mice the largest proportion of B-1a cells are found in the peritoneal cavity with a small proportion but approximately equal sized population residing in the spleen (3, 4). The B-1a cell population originates during fetal life and persists throughout adult life by their ability to self-renew, meaning new B-1a cells are generated by mitosis of mature surface Ig-expressing B-1a cells. This process is regulated in a feedback fashion (5, 6). B-1a cell self-renewal is unlike development of B-2 cells, wherein mature cells derive from surface Ig-negative progenitors. Recently early appearing B-1a cells were shown to represent a separate lineage derived from a unique progenitor found both in the fetal liver and bone marrow that does not give rise to B-2 cells (7).
B-1a cells exhibit a number of functional characteristics different from conventional B-2 cells. B-1a cells spontaneously secrete IgM, which is often referred to as natural antibody and accumulates as the bulk of resting or non-immune IgM. Ig secreted by unstimulated B-1a cells varies less from germline than Ig secreted by B-2 cells, which is because B-1a immunoglobulin undergoes minimal if any somatic hypermutation and possesses little N-region addition (8–10). In addition, B-1a cells are repertoire skewed as evidenced by biased variable heavy chain (VH) gene usage in favor of VH11 and VH12 (9–13). This skewed, germline-like repertoire contains both antimicrobial and autoreactive specificities. B-1a cell-derived natural IgM has been shown to be essential for: (1) anti-microbial protection, through initial serological control of bacterial and viral infections (14–16), and (2) housekeeping homeostasis, by aiding in disposal of autoantigens through removal of apoptotic cell debris (17–19). In addition, housekeeping natural antibodies assist in elimination of toxic molecules such as oxidized low density lipoprotein (oxLDL), in particular by antibodies bearing the T15 idiotype, which helps control the inflammatory process leading to atherosclerotic plaques (20). These diverse functions may be facilitated by the characteristic polyreactivity of B-1a cell Ig.
Beyond spontaneous secretion of natural IgM antibody, B-1a cells express other distinct functions not shared by resting conventional B-2 cells. B-1a cells present antigen more potently than conventional B-2 cells, a property that has been attributed to constitutive expression of the co-stimulatory molecules B7.1 and B7.2 (21–23). Further, B-1a cells have been shown to induce pro-inflammatory Th17 cell differentiation and to generate immunosuppressive IL-10 (23, 24). Thus, in addition to antibody production, B-1a cells can influence other elements of the immune system in both positive and negative ways.
B-1a cells express unique signaling and proliferative characteristics, which seem in some ways hyperresponsive in comparison to B-2 cells but in other ways hyporesponsive. B-1a cells display constitutive expression of activated signaling mediators including ERK, NF-AT, and STAT3 (25, 26), which in B-2 cells require stimulation for activated expression (27). B-1a cells have also been shown to proliferate in response to treatment with phorbol ester as a single agent, in contrast to B-2 cells, which only respond to phorbol myristate acetate or phorbol dibutyrate in conjunction with a calcium ionophore (28). PMA responsiveness in B-1a cells is associated with rapid induction of cyclin D2 and activation of RB-phosphorylating cyclin D3-cdk4 complexes, neither of which occur in PMA-treated B-2 cells (29, 30). However, despite activated signaling mediators at rest and despite hyperresponsiveness to PMA, BCR signaling fails in B-1a cells – NF-κB is not induced nor is proliferation stimulated.
BCR Signaling in B-1a Cells
Despite the failure of BCR engagement to induce NF-κB activation in B-1a cells (31, 32), stimulation with LPS or PMA succeeds just as in B-2 cells (31), suggesting that key components involved in the pathway proximal to induction of this transcription factor are not lacking. Several previous studies have sought to determine why B-1a cells have an attenuated response to BCR cross-linking as compared to B-2 cells when the basic NF-κB machinery appears intact. The negative regulatory receptor CD5 and the tyrosine phosphatase SHP-1 were reported to play a role in the failure of B-1a cells to respond to BCR ligation. It was demonstrated B-1a cells from CD5 deficient mice could respond to BCR ligation, and SHP-1 was shown to be constitutively associated with the BCR in a CD5-dependent manner (33). The important regulatory role SHP-1 plays in B-1a cell development was presented in a separate study, which demonstrated an increase in B-1a cell number in the absence of SHP-1 and the negative regulation it provides; however, there was no change in the ability of SHP-1 deficient B-1a cells to proliferate in response to BCR crosslinking (34). Despite the initial clear results demonstrating CD5/SHP-1 negatively regulates BCR signaling in B-1a cells, it was later shown both B-1a (CD5+) and B-1b (CD5−) cells fail to respond to BCR ligation (35), raising questions about the role of CD5 and associated molecules. These results suggest some other element is responsible for the lack of B-1a cell responsiveness to BCR engagement, whereas the extent of CD5 involvement remains uncertain.
The src family kinase Lck, which characterizes T cells, was reported to be unexpectedly expressed in B-1a cells and responsible for defective NF-κB activation in response to BCR ligation (36). Dal Porto et al. reported peritoneal B-1a cells express Lck and are defective in BCR signaling whereas splenic B-1a cells do not express Lck and are not defective in BCR signaling (36). However, results published both before and after this study question the role of Lck in B-1 cells. An early investigation of kinase family members in peritoneal B-1a cells verified the presence of other src-kinases such as Lyn, Blk, Hck, and Syk, but not Lck (32). Subsequently Frances et al. re-examined Lck expression and found an absence of Lck in B-1a cells purified by various methods. However, despite the lack of Lck expression found in B-1a cells, defective BCR signaling was still observed (37), suggesting Lck expression does not correlate with B-1a cell hyporesponsiveness to BCR crosslinking. A few years later, it was shown by a separate group that splenic B-1a cells lacking Lck are, in fact, hyporesponsive to BCR signaling (38), unlike the finding by Dal Porto et al., which suggested splenic B-1a cells respond normally to BCR cross-linking (36). Together these studies do not support a role for Lck in the lack of NF-κB activation and proliferation by BCR-stimulated B-1 cells.
The inhibitory receptor Siglec-G has been shown to be highly expressed and functional in B cells (39). Siglec G plays an important role in B-1a cell signaling and over-expression inhibits Ca2+ signaling. As with SHP-1 deficiency, Siglec G deficiency enhances B-1a cell development and leads to an increase in B-1a cell number; these B-1a cells manifest enhanced signaling (39). Therefore, Siglec-G plays an important role in B-1 cell signaling and development but no clear or distinct role has been shown for Siglec G in inhibiting NF-κB activation and/or proliferation in response to BCR crosslinking.
It is important to recall the B-cell receptor complex does not function alone but is associated with additional signaling proteins, which include CD19, CD21, and CD81. These associated proteins are collectively termed the B-cell receptor co-complex, and greatly enhance the signal received after antigen binding to the BCR complex. When the BCR binds antigen coated with the complement component C3d, the complement receptor CD21 binds C3d resulting in activation of CD19 along with the BCR (40). Activation of the CD19/BCR co-complex enables B cells to respond to significantly less (10–100 fold less) antigen as compared to B cells lacking CD19 (41, 42).
CD19 is phosphorylated and activated by Lyn, a src family kinase; in turn CD19 amplifies Lyn activation and enhances activation of other src family kinase members. Subsequently Vav proteins are phosphorylated (43, 44). Lyn also serves an essential regulatory role by phosphorylation of cell surface receptors shown to negatively regulate the BCR response, such as CD22 (45). CD19 phosphorylation leads to activation of phosphatidylinositol 3-kinase (PI-3K), which phosphorylates inositol phospholipids leading to initiation of several signaling cascades via phospholipase C (PLC) and/or Ca2+ activation (46).
It is interesting to note B-1a cells express higher levels of CD19 than B-2 cells (47) and their development is greatly impaired in the absence of CD19 (48, 49). It has been shown BCR-induced CD19 signaling in B-1 cells is different from that of B-2 cells. In particular, following BCR engagement, B-1 cells experience shorter duration of CD19 phosphorylation and less PI-3K associated with phosphorylated CD19 (35). Both splenic and peritoneal B-1 cells overexpress Lyn and manifest impaired CD19 signaling; furthermore, Lyn inhibition allows B-1 cells to recover some responsiveness to BCR ligation (38). In addition, in Lynup/up mice that express constitutively active Lyn, splenic B-2 cells fail to proliferate in response to BCR ligation and thus acquire a signaling deficiency that parallels unmanipulated B-1 cells (50). Together these results suggest the unusual signaling characteristics of CD19 and Lyn may play an important role in the failure of B-1 cells to respond to BCR ligation. This may be exacerbated by the decreased levels of Vav1 and Vav2 in B-1 as compared to B-2 cells.
These studies over the past 20 years clearly demonstrate signaling differences between B-1a and conventional B-2 cells and point toward signaling in these two distinct subsets as being differentially regulated. However, a clear conclusion as to why B-1a cells do not activate NF-κB or proliferate in response to BCR cross-linking has not emerged. Here we synthesize a unifying hypothesis for the failure of B-1a cells to respond to BCR ligation. This hypothesis is placed in the context of the unique characteristics B-1a cells display.
B-1a Cell Signaling through the BCR is Not Defective
The loss of NF-κB activation and proliferation in response to BCR ligation suggests B-1 cells have complete or partial loss of certain signaling pathways (deletion model). However, signaling through the BCR of B-1a cells is not completely blocked. B-1a cells activate ERK, JNK, and NF-AT in response to BCR cross-linking (26). Moreover, BCR-induced activation has also been observed in more membrane proximal mediators. Recently we have shown intact functioning of key signaling mediators by demonstrating: (1) normal phosphorylation/activation of Syk and PLCγ2 (32) after BCR ligation, and (2) a src kinase requirement for BCR-induced Syk and PLCγ2 activation (51). Furthermore, we and others have observed calcium mobilization after BCR ligation in B-1a cells, which is comparable in degree to conventional B-2 cells [data not shown and (34, 52)]. Together these results demonstrate signaling in B-1a cells is functional and capable of activating membrane proximal mediators after BCR ligation. Despite seemingly normal functioning of membrane proximal mediators after BCR ligation, the fact remains that NF-κB is not activated in B-1a cells.
NF-κB activation occurs once NF-κB subunits are released from association with IκB, which allows the subunits to translocate into the nucleus (53). In previous reports the lack of NF-κB activation in response to BCR cross-linking in B-1a cells was demonstrated by an absence of nuclear expression of NF-κB components and a lack of κB-binding activity in nuclear extracts (31). However, the activation of mediators leading to NF-κB induction, such as IKKα/β phosphorylation or IκBα degradation have not been evaluated in B-1a cells after BCR ligation. Since membrane proximal signaling in B-1a cells has been shown to operate normally after BCR ligation, these more distal signaling events specific for NF-κB induction were assessed for activation status after BCR ligation. We found a large amount of phosphorylated IKKα/β protein detected at 30 min in conventional B-2 cells (Figure 1). Conversely, only a small amount of phosphorylated IKKα/β protein was detected at 30 min in B-1a cells. A dramatic decrease in IκBα protein occurs in B-2 cells after 90 min of anti-IgM stimulation (Figure 1). In contrast, only a small amount of IκBα degradation is observed in B-1a cells after 90 min of anti-IgM stimulation. These results show both phosphorylation of IKKα/β and the degradation of IκBα observed in B-1a cells is considerably less than that seen in B-2 cells stimulated with anti-IgM. Insufficient phosphorylation of IKKα/β and/or IκBα could prevent the translocation of NF-κB subunits into the nucleus. These results suggest the lack of NF-κB activation in B-1a cells after BCR ligation may originate with abnormal induction and/or regulation of phosphorylated IKKα/β and/or IκBα. If B-1a cells are still able to signal yet not able to activate NF-κB, it is possible NF-κB induction is being actively blocked, perhaps because proper phosphorylation of IKKα/β and/or IκBα is obstructed (inhibition model).
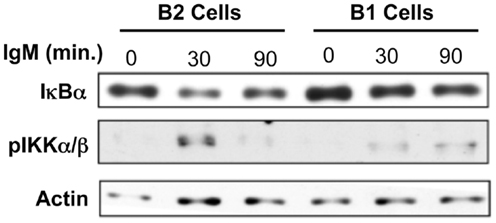
Figure 1. I-kappa-B-alpha degradation and IKKalpha/beta phosphorylation analysis in response to anti-IgM in B-1a cells. Sorted peritoneal B-1a and splenic B-2 cells were treated with anti-IgM (15 μg/ml) for the times indicated at 37°C. Afterward, the cells were washed, pelleted, lysed, and then used for western blot analysis. Results shown are representative of three independent experiments.
Regulation of BCR-Induced Signaling Leading to NF-κB Activation in B-1a Cells
Phosphatases
Our previous work emphasizes the role of phosphatases in regulating B-1a cell signaling. We examined the origin of constitutively phosphorylated ERK in B-1a cells. Despite phosphorylation of ERK, upstream signaling mediators such as Syk and PLCγ2 are not phosphorylated. However, we found after addition of tyrosine phosphatase inhibitors B-1a cells accumulated large amounts of phosphorylated Syk and PLCγ2, which did not occur with conventional B-2 cells. Moreover, this phosphorylation was blocked when B-1 cells were pre-treated with a src kinase inhibitor (51). These results suggest that signaling of upstream mediators by src kinases is taking place constitutively in B-1a cells, but rapid dephosphorylation prevents accumulation of phosphorylated intermediates. In other words, elevated phosphatase activity interferes with some, but not all, signaling events and raises the possibility that differential phosphatase expression and/or increased phosphatase activity could be playing a role in blocking anti-Ig-induced NF-κB induction in B-1a cells.
To test this we assessed IκBα degradation in anti-Ig-stimulated B-1a and B-2 cells pre-treated with the tyrosine phosphatase inhibitor sodium orthovanadate. Results are presented in Figure 2. These results show in the native state, as expected, IκBα is degraded in B-2 cells after anti-Ig stimulation for 30 and 90 min, whereas little to no IκBα degradation is seen in B-1a cells similarly stimulated by anti-Ig. However, in the presence of tyrosine phosphatase inhibition, IκBα is degraded in B-1a cells stimulated with anti-Ig, just as it is in unmanipulated B-2 cells. These results strongly suggest the failure of BCR-induced NF-κB activation in B-1a cells is the result of increased tyrosine phosphatase expression and/or activity, or is the result of insufficient phosphatase inactivation.
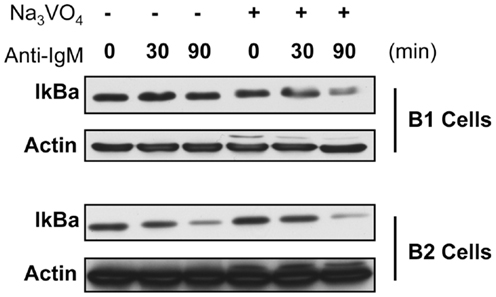
Figure 2. IκBa degradation in B-1a and B2 cells in the presence or absence of phosphatase inhibition. Sorted B-1a and B-2 cells were pre-treated for 1 h with cycloheximide (50 μM). Afterward, cells were either treated with or without anit-IgM (15 μg/ml) for 30 or 90 min, as indicated, or cells were cultured with 2 mM sodium orthovanadate (Na3VO4) for 15 min and then treated with anti-IgM for 30 or 90 min. Cells were collected, supernatants discarded, and pellets frozen at −20°C until lysed in NP-40 lysis buffer and used for western blot analysis of IκBα. Results shown are representative of two independent experiments.
Previous reports have attributed inhibition of NF-κB activation in other cell types to the activity of phosphatases, which is consistent with the effect of tyrosine phosphatase inhibition discussed above (Figure 2). It has been shown NF-κB activation can be affected by oxidation and/or reduction events (54, 55). Normally the cytosol is a reducing environment due to the presence of molecules such as glutathione (56), which is favorable for the activity of protein-tyrosine phosphatases (PTP). Oxidizing agents inactivate PTP by oxidizing the cysteine residue in the active site (57). Oxidizing agents, including reactive oxygen species (ROS) such as H2O2, have been shown to be produced inside lymphocytes where they regulate signaling by inhibiting PTP (55). Regardless of the stimulus, the cellular redox environment appears to play a role in the regulation of tyrosine phosphorylation events leading to activation of NF-κB (54).
The exact mechanism of how phosphatases regulate NF-κB activation is still not fully understood. It could be hypothesized that phosphatases regulate NF-κB activation by directly dephosphorylating IKKα/β thereby blocking phosphorylation of IκB proteins. Alternatively, phosphatases could act on upstream mediators specific to NF-κB activation such as NF-κB inducing kinase (NIK), which phosphorylates IKKα. An example of this type of regulation is provided in a study showing IL-1β-induced activation of NF-κB is dependent upon NIK activation, which requires ROS mediated inhibition of phosphatases for activation (58). Such studies demonstrate the role phosphatase activity and the redox environment can play in controlling NF-κB activation in response to various stimuli, which parallels the results shown here where phosphatase inhibitors cleared the way for anti-Ig-induced activation of NF-κB in B-1a cells (Figure 2). Further investigation into phosphatase expression and activity is likely to help unravel the mechanism which prevents NF-κB activation in B-1a cells in response to BCR ligation.
HSP70
The HSP70 family of proteins plays a role in facilitating protein folding and preventing aggregation in the ER (Grp78/Bip) and cytosol (HSC70 and HSP70) (59). High levels of protein within cells or stress may cause an increase in HSP expression as in the case of the unfolded protein response (60, 61). B-1a cells are constantly producing natural antibody and have nearly twice as much protein per cell than B-2 cells; in keeping with this, we found levels of HSP70 gene expression are higher in B-1a cells as compared with B-2 cells (Figure 3). Interestingly, HSPs have been shown to block the NF-κB pathway through inhibition of IKK or by increasing phosphatase activity leading to a decrease in IκBα phosphorylation (62). It is feasible there is enough overall decrease in IκBα degradation produced by the increased level of HSP70 or related proteins in B-1a cells to impair NF-κB translocation to the nucleus. Other chaperones such as HSP27 and clusterin have also been shown to inhibit NF-κB activation (59); however the expression levels of these chaperones have not been examined in B-1a and B-2 cells. In addition, the expression levels of cytosolic or ER-resident HSP70 family members have not been examined. Further analysis is required to determine the site of elevated HSP70 expression in B-1a cells, because only cytosolic HSP70 would result in inhibition of NF-κB. HSP70 remains an important candidate for modulation of B-1a cell NF-κB induction.
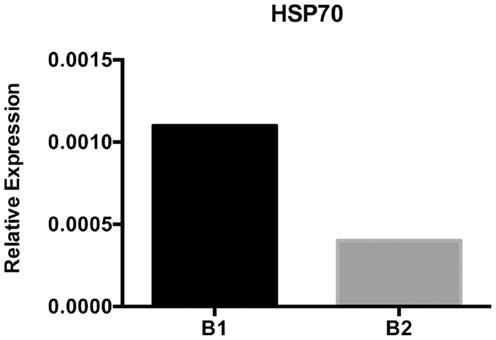
Figure 3. HSP70 expression analysis. Sorted naïve peritoneal B-1a and splenic B-2 cells were used for isolation of mRNA and subsequent cDNA synthesis after DNAse I treatment. HSP70 mRNA expression was assessed using semi-quantitative PCR. Results are representative of three independent experiments.
IL-10
B-1 cells have long been associated with IL-10 expression and secretion, and although B-1a and B-2 cells secrete IL-10 after antigenic stimulation (27), only B-1 cells secrete IL-10 spontaneously (24). Interestingly, it has been shown that IL-10 inhibits TNF-induced activation of NF-κB by preventing IKK activity and DNA binding of the NF-κB subunits (63). Further, IL-10 as well as IL-2 and IL-5 have been shown to regulate expression of certain phosphatases (64, 65). Therefore, it is possible that induced and/or constitutively secreted IL-10 in B-1 cells contributes to differences in phosphatase expression and/or activity in B-1a cells as compared to B-2 cells.
Discussion
BCR mediated signaling in B-1a cells functions normally in terms of phosphorylation of membrane proximal mediators such as PLCγ2 (51), Syk (51), and Ca2+ mobilization (34). While these findings illustrate functional membrane proximal BCR signaling, IKKαβ phosphorylation and IκBα degradation in response to BCR stimulation is impaired in B-1a cells (Figure 1). However, IκBα degradation is rescued if B-1a cells are pre-treated with the tyrosine phosphatase inhibitor, sodium orthovanadate (Figure 2). Collectively, these results suggest signaling in B-1a cells is differentially regulated by phosphatases, as compared to B-2 cells.
Phosphatase activity has been shown to play a role in regulating the activation of NF-κB (54, 58). Reduction/oxidation events within the cell, particularly generation of ROS, can regulate phosphatase activity (55, 57). If high levels of phosphatases are controlling BCR-induced signaling in B-1a cells and ROS play a role in controlling phosphatases, two questions arise: (1) what regulates expression of phosphatases in B-1a cells; and, (2) how do signals derived from LPS and CD40L generate enough ROS, or bypass the need for ROS, to allow NF-κB activation whereas BCR ligation fails to do so? The unique characteristics of B-1a cells (summarized in Figure 4) may relate to these issues.
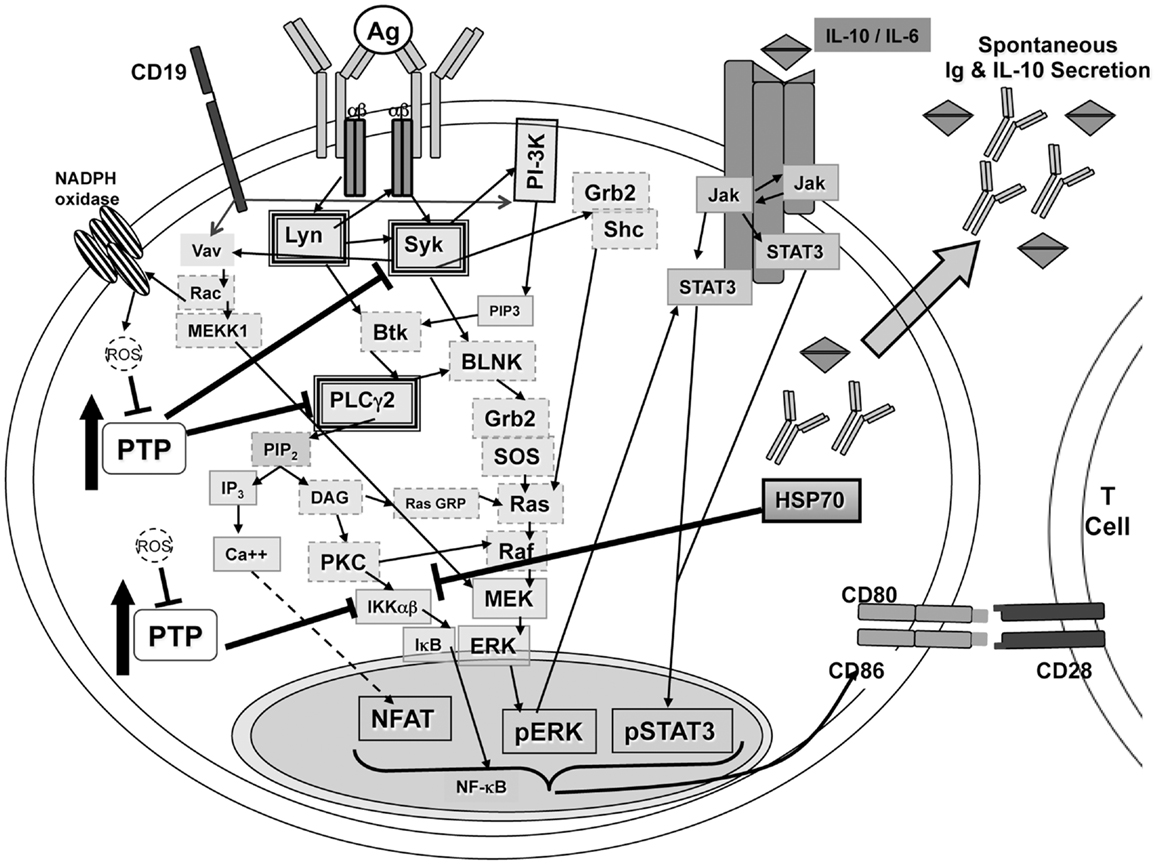
Figure 4. Signaling in B-1a cells. B-1a cells constitutively secrete IgM and IL-10 without prior stimulation. In addition, B-1a cells have constitutive levels of activated ERK, STAT3, and NF-AT, yet are not able to activate NF-κB in response to BCR ligation. It has been shown constitutive ERK activation in B-1 cells is the result of chronic signaling through the BCR (51). The constitutive ERK activation was shown to be dependent upon src kinases, PI-3K, Syk, and PLCγ2, which are heavily outlined in the illustration. Inhibition of PI-3K or Syk also blocked the constitutive levels of CD86 on B-1a cells, which is known to play an essential role during allogeneic stimulation of T cells (23, 51). All mediators outlined in a dashed gray line may play a role in signal transduction leading to constitutive ERK activation but have not been tested. Phosphatase activity, denoted as PTP, has been shown to control phosphorylation of Syk and PLCγ2 differentially in B-1a cells as compared to B2 cells (51). In addition, inhibition of phosphatase activity in B-1 cells was shown, Figure 2, to allow IκBα degradation in B-1a cells after BCR ligation. Therefore, PTP activity in B-1a cells inhibits NF-κB activation by an unknown mechanism. It is hypothesized that this mechanism involves HSP70 and IL-10, which were both shown to be expressed at a higher level in naive B-1a cells as compared to naive B2 cells, Figure 3 and Ref. (24). Furthermore, it has been shown that inhibition of constitutively active Lyn allowed for partial recovery of B-1a cells’ responsiveness to BCR ligation (38). In addition, suboptimal Vav levels in B-1a cells (35) may not be sufficient for the production of ROS, which are necessary to inhibit phosphatases to allow activation of NF-κB.
First, HSP70 and IL-10 can both play a role in influencing the expression of phosphatases (62–67). Here we demonstrated HSP70 is highly expressed in naïve B-1a cells as compared to B-2 cells (Figure 3). HSP70 has been shown to inhibit NF-κB signaling by inducing phosphatase activity and by directly interacting with IKKγ, thereby disrupting the IKKγ protein from binding to the IKKαβ complex, which renders it inactive (62, 66–68). It is possible one or both of these mechanisms operates in B-1a cells thereby blocking NF-κB activation after BCR ligation. However, the over-expression of HSP70 may merely reflect an increased need for chaperones in B-1a cells due to their continuous secretion of IgM. Furthermore, IL-10 is expressed in naïve B-1a cells but not in naïve B-2 cells (24). IL-10 has also been shown to inhibit NF-κB activation by preventing IKK activity (63) and has been shown to regulate the expression of certain phosphatases (64, 65). These are just two examples of how phosphatases might be differentially regulated in B-1a cells as compared to B-2 cells.
Secondly, the reason for lack of NF-κB activation in response to BCR ligation yet normal activation in response to CD40L or LPS may lie in the difference in signal strength delivered by these different stimuli. Perhaps a stronger signal is required, which may come from activation of receptors such as a toll-like receptor (TLR) or CD40. This type of additional signal requirement for activation of lymphocytes via inactivation of phosphatases has been previously suggested for B-2 cells (55). Further, it has been demonstrated B-1 cells proliferate in response to BCR ligation if the receptors are hyper-crosslinked (35). It may be that B-1a cells are relatively “exhausted” through tachyphylaxis as a result of chronic signaling (51) and thus require an unusually strong signal when delivered through the BCR as opposed to TLR or CD40. The high levels of constitutively active Lyn in B-1a cells may also be responsible for the requirement of an unusually strong signal through the BCR as the constitutively active Lyn might be activating inhibitory receptors such as SHP-1 and SHIP-1 (38).
We propose a certain level of phosphatase inactivation by ROS is required for full B-1 cell activation to occur. BCR cross-linking can cause an increase in ROS production, which can inactivate phosphatases thereby allowing signaling to propagate through the cell. After BCR ligation in B-1a cells NF-κB is not activated. Therefore, we hypothesize phosphatase activity must not be appropriately inhibited after BCR ligation to allow for NF-κB activation. Normally, ROS are generated by NADPH oxidase, which is activated by signal transduction via Rac or PKC (55, 69). Rac is a GTPase activated in B cells via the guanine nucleotide exchange factor Vav (70). Vav is recruited by CD19 and phosphorylated by Lyn (43). A few years ago it was reported the lack of proliferation to BCR cross-linking by B-1 cells is the result of defective CD19 signaling, which is due to a reduced level of Vav proteins in B-1 cells (35). The essential role of the Vav proteins is further demonstrated in B-2 cells deficient in Vav, which show no activation of NF-κB in response to BCR ligation (35). These results combined with the results presented here and Reth’s model of lymphocyte activation (55) suggest there is a lack of sufficient levels of Vav proteins in B-1 cells to activate Rac. Insufficient Rac activation would not allow for necessary ROS production through NADPH oxidase to overcome the phosphatase activity regulating NF-κB components. Therefore, a second signal or stronger signal is necessary in B-1 cells for phosphatase inactivation.
Hypothesis
In synthesizing previous reports and new data presented here, we suggest a new hypothesis for the lack of NF-κB activation and proliferation in B-1a cells after BCR ligation. We summarize this hypothesis in Figure 5.
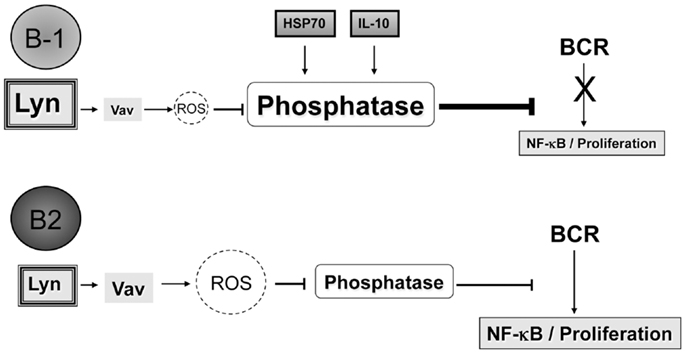
Figure 5. Differences in Vav proteins, IL-10, and/or HSP70 expression between B-1a and B2 cells lead to the inability of B-1a cells to activate NF-κB and proliferate after BCR ligation. It is hypothesized that B-1a cells have increased levels or differential expression of phosphatases as compared to B2 cells, which could be at least partially the result of the increased basal expression of IL-10 and/or HSP70 seen in B-1a cells but not B2 cells. Furthermore, suboptimal Vav levels in B-1 cells (35) may not be sufficient for the production of ROS, which are necessary to inhibit phosphatases to allow activation of NF-κB after BCR ligation. Over-expression of the src kinase Lyn may also play a role in the non-response of B-1a cells to BCR ligation, perhaps by phosphorylation of inhibitory receptors. The mechanism through which Lyn contributes to B-1a cell non-responsiveness to BCR ligation is not clear; however, it has been demonstrated blocking Lyn activity can allow B-1a cells to respond to respond to BCR crosslinking.
B-1a cells are unable to activate NF-κB or proliferate after BCR cross-linking due to increased phosphatase abundance and/or activity, which could be a result of basal expression of IL-10 and/or HSP70. The increased phosphatase abundance and/or activity is not able to be overcome by BCR ligation alone due to constitutively active Lyn and/or a lack of Vav protein expression in B-1 cells, which does not allow for proper production of ROS needed to inhibit the phosphatases present in B-1 cells.
Future Directions
A remaining question revolves around the nature of the phosphatase activity opposing BCR signaling in B-1a cells. Signaling deficiencies in antigen-experienced B cells, including germinal center B cells (71) and anergic B cells (72) have been attributed to SHP-1 and SHIP-1 respectively. Like anergic B cells (which express low levels of CD5) B-1a cells are thought to be antigen-experienced, so SHP-1 and SHIP-1 are candidates for BCR regulation here as well, although other possibilities, such as DUSP phosphatases, may be involved. Although elucidating the source of B-1a cell phosphatase activity is important, similar attention should focus on understanding the way in which phosphatase activity is normally overcome, a process that fails in B-1a cells.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol (2001) 19:595–621. doi: 10.1146/annurev.immunol.19.1.595
3. Hayakawa K, Hardy RR, Herzenberg LA. Peritoneal Ly-1 B cells: genetic control, autoantibody production, increased lambda light chain expression. Eur J Immunol (1986) 16:450–6. doi:10.1002/eji.1830160423
4. Hayakawa K, Hardy RR, Parks DR, Herzenberg LA. The “Ly-1 B” cell subpopulation in normal immunodefective, and autoimmune mice. J Exp Med (1983) 157:202–18. doi:10.1084/jem.157.1.202
5. Hayakawa K, Hardy RR, Stall AM, Herzenberg LA. Immunoglobulin-bearing B cells reconstitute and maintain the murine Ly-1 B cell lineage. Eur J Immunol (1986) 16:1313–6. doi:10.1002/eji.1830161021
6. Kantor AB, Stall AM, Adams S, Watanabe K, Herzenberg LA. De novo development and self-replenishment of B cells. Int Immunol (1995) 7:55–68. doi:10.1093/intimm/7.1.55
7. Montecino-Rodriguez E, Leathers H, Dorshkind K. Identification of a B-1 B cell-specified progenitor. Nat Immunol (2006) 7:293–301. doi:10.1038/ni1301
8. Feeney AJ. Lack of N regions in fetal and neonatal mouse immunoglobulin V-D-J junctional sequences. J Exp Med (1990) 172:1377–90. doi:10.1084/jem.172.5.1377
9. Pennell CA, Arnold LW, Haughton G, Clarke SH. Restricted Ig variable region gene expression among Ly-1+ B cell lymphomas. J Immunol (1988) 141:2788–96.
10. Tarlinton D, Stall AM, Herzenberg LA. Repetitive usage of immunoglobulin VH and D gene segments in CD5+ Ly-1 B clones of (NZB x NZW)F1 mice. EMBO J (1988) 7:3705–10.
11. Hardy RR, Carmack CE, Shinton SA, Riblet RJ, Hayakawa K. A single VH gene is utilized predominantly in anti-BrMRBC hybridomas derived from purified Ly-1 B cells. Definition of the VH11 family. J Immunol (1989) 142:3643–51.
12. Mercolino TJ, Locke AL, Afshari A, Sasser D, Travis WW, Arnold LW, et al. Restricted immunoglobulin variable region gene usage by normal Ly-1 (CD5+) B cells that recognize phosphatidyl choline. J Exp Med (1989) 169:1869–77. doi:10.1084/jem.169.6.1869
13. Wang H, Clarke SH. Positive selection focuses the VH12 B-cell repertoire towards a single B1 specificity with survival function. Immunol Rev (2004) 197:51–9. doi:10.1111/j.0105-2896.2004.0098.x
14. Baumgarth N, Herman OC, Jager GC, Brown LE, Herzenberg LA, Chen J. B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J Exp Med (2000) 192:271–80. doi:10.1084/jem.192.2.271
15. Ochsenbein AF, Fehr T, Lutz C, Suter M, Brombacher F, Hengartner H, et al. Control of early viral and bacterial distribution and disease by natural antibodies. Science (1999) 286:2156–9. doi:10.1126/science.286.5447.2156
16. Su SD, Ward MM, Apicella MA, Ward RE. The primary B cell response to the O/core region of bacterial lipopolysaccharide is restricted to the Ly-1 lineage. J Immunol (1991) 146:327–31.
17. Ehrenstein MR, Notley CA. The importance of natural IgM: scavenger, protector and regulator. Nat Rev Immunol (2010) 10:778–86. doi:10.1038/nri2849
18. Kaveri SV, Silverman GJ, Bayry J. Natural IgM in immune equilibrium and harnessing their therapeutic potential. J Immunol (2012) 188:939–45. doi:10.4049/jimmunol.1102107
19. Tsiantoulas D, Gruber S, Binder CJ. B-1 cell immunoglobulin directed against oxidation-specific epitopes. Front Immunol (2012) 3:415. doi:10.3389/fimmu.2012.00415
20. Weksler ME, Pawelec G, Franceschi C. Immune therapy for age-related diseases. Trends Immunol (2009) 30:344–50. doi:10.1016/j.it.2009.03.011
21. Sato T, Ishikawa S, Akadegawa K, Ito T, Yurino H, Kitabatake M, et al. Aberrant B1 cell migration into the thymus results in activation of CD4 T cells through its potent antigen-presenting activity in the development of murine lupus. Eur J Immunol (2004) 34:3346–58. doi:10.1002/eji.200425373
22. Vigna AF, Godoy LC, Rogerio de Almeida S, Mariano M, Lopes JD. Characterization of B-1b cells as antigen presenting cells in the immune response to gp43 from Paracoccidioides brasiliensis in vitro. Immunol Lett (2002) 83:61–6. doi:10.1016/S0165-2478(02)00070-6
23. Zhong X, Gao W, Degauque N, Bai C, Lu Y, Kenny J, et al. Reciprocal generation of Th1/Th17 and T(reg) cells by B1 and B2 B cells. Eur J Immunol (2007) 37:2400–4. doi:10.1002/eji.200737296
24. O’Garra A, Chang R, Go N, Hastings R, Haughton G, Howard M. Ly-1 B (B-1) cells are the main source of B cell-derived interleukin 10. Eur J Immunol (1992) 22:711–7. doi:10.1002/eji.1830220314
25. Karras JG, Wang Z, Huo L, Howard RG, Frank DA, Rothstein TL. Signal transducer and activator of transcription-3 (STAT3) is constitutively activated in normal, self-renewing B-1 cells but only inducibly expressed in conventional B lymphocytes. J Exp Med (1997) 185:1035–42. doi:10.1084/jem.185.6.1035
26. Wong SC, Chew WK, Tan JE, Melendez AJ, Francis F, Lam KP. Peritoneal CD5+ B-1 cells have signaling properties similar to tolerant B cells. J Biol Chem (2002) 277:30707–15. doi:10.1074/jbc.M202460200
27. Fan H, Rothstein TL. Lymphokine dependence of STAT3 activation produced by surface immunoglobulin cross-linking and by phorbol ester plus calcium ionophore treatment in B cells. Eur J Immunol (2001) 31:665–71. doi:10.1002/1521-4141(200102)31:2<665::AID-IMMU665>3.0.CO;2-1
28. Rothstein TL, Kolber DL. Peritoneal B cells respond to phorbol esters in the absence of co-mitogen. J Immunol (1988) 140:2880–5.
29. Tanguay DA, Colarusso TP, Doughty C, Pavlovic-Ewers S, Rothstein TL, Chiles TC. Cutting edge: differential signaling requirements for activation of assembled cyclin D3-cdk4 complexes in B-1 and B-2 lymphocyte subsets. J Immunol (2001) 166:4273–7.
30. Tanguay DA, Colarusso TP, Pavlovic S, Irigoyen M, Howard RG, Bartek J, et al. Early induction of cyclin D2 expression in phorbol ester-responsive B-1 lymphocytes. J Exp Med (1999) 189:1685–90. doi:10.1084/jem.189.11.1685
31. Morris DL, Rothstein TL. Abnormal transcription factor induction through the surface immunoglobulin M receptor of B-1 lymphocytes. J Exp Med (1993) 177:857–61. doi:10.1084/jem.177.3.857
32. Morris DL, Rothstein TL. Decreased surface IgM receptor-mediated activation of phospholipase C gamma 2 in B-1 lymphocytes. Int Immunol (1994) 6:1011–6. doi:10.1093/intimm/6.7.1011
33. Sen G, Bikah G, Venkataraman C, Bondada S. Negative regulation of antigen receptor-mediated signaling by constitutive association of CD5 with the SHP-1 protein tyrosine phosphatase in B-1 B cells. Eur J Immunol (1999) 29:3319–28. doi:10.1002/(SICI)1521-4141(199910)29:10<3319::AID-IMMU3319>3.3.CO;2-0
34. Pao LI, Lam KP, Henderson JM, Kutok JL, Alimzhanov M, Nitschke L, et al. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity (2007) 27:35–48. doi:10.1016/j.immuni.2007.04.016
35. Sen G, Wu HJ, Bikah G, Venkataraman C, Robertson DA, Snow EC, et al. Defective CD19-dependent signaling in B-1a and B-1b B lymphocyte subpopulations. Mol Immunol (2002) 39:57–68. doi:10.1016/S0161-5890(02)00047-0
36. Dal Porto JM, Burke K, Cambier JC. Regulation of BCR signal transduction in B-1 cells requires the expression of the Src family kinase Lck. Immunity (2004) 21:443–53. doi:10.1016/j.immuni.2004.07.018
37. Frances R, Tumang JR, Rothstein TL. B-1 cells are deficient in Lck: defective B cell receptor signal transduction in B-1 cells occurs in the absence of elevated Lck expression. J Immunol (2005) 175:27–31.
38. Dasu T, Sindhava V, Clarke SH, Bondada S. CD19 signaling is impaired in murine peritoneal and splenic B-1 B lymphocytes. Mol Immunol (2009) 46:2655–65. doi:10.1016/j.molimm.2009.04.015
39. Hoffmann A, Kerr S, Jellusova J, Zhang J, Weisel F, Wellmann U, et al. Siglec-G is a B1 cell-inhibitory receptor that controls expansion and calcium signaling of the B1 cell population. Nat Immunol (2007) 8:695–704. doi:10.1038/ni1480
40. Matsumoto AK, Kopicky-Burd J, Carter RH, Tuveson DA, Tedder TF, Fearon DT. Intersection of the complement and immune systems: a signal transduction complex of the B lymphocyte-containing complement receptor type 2 and CD19. J Exp Med (1991) 173:55–64. doi:10.1084/jem.173.1.55
41. Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science (1996) 271:348–50. doi:10.1126/science.271.5247.348
42. Matsumoto AK, Martin DR, Carter RH, Klickstein LB, Ahearn JM, Fearon DT. Functional dissection of the CD21/CD19/TAPA-1/Leu-13 complex of B lymphocytes. J Exp Med (1993) 178:1407–17. doi:10.1084/jem.178.4.1407
43. Fujimoto M, Fujimoto Y, Poe JC, Jansen PJ, Lowell CA, DeFranco AL, et al. CD19 regulates Src family protein tyrosine kinase activation in B lymphocytes through processive amplification. Immunity (2000) 13:47–57. doi:10.1016/S1074-7613(00)00007-8
44. Fujimoto M, Poe JC, Jansen PJ, Sato S, Tedder TF. CD19 amplifies B lymphocyte signal transduction by regulating Src-family protein tyrosine kinase activation. J Immunol (1999) 162:7088–94.
45. Smith KG, Tarlinton DM, Doody GM, Hibbs ML, Fearon DT. Inhibition of the B cell by CD22: a requirement for Lyn. J Exp Med (1998) 187:807–11. doi:10.1084/jem.187.5.807
46. Buhl AM, Pleiman CM, Rickert RC, Cambier JC. Qualitative regulation of B cell antigen receptor signaling by CD19: selective requirement for PI3-kinase activation, inositol-1,4,5-trisphosphate production and Ca2+ mobilization. J Exp Med (1997) 186:1897–910. doi:10.1084/jem.186.11.1897
47. Sato S, Ono N, Steeber DA, Pisetsky DS, Tedder TF. CD19 regulates B lymphocyte signaling thresholds critical for the development of B-1 lineage cells and autoimmunity. J Immunol (1996) 157:4371–8.
48. Engel P, Zhou LJ, Ord DC, Sato S, Koller B, Tedder TF. Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity (1995) 3:39–50. doi:10.1016/1074-7613(95)90157-4
49. Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature (1995) 376:352–5. doi:10.1038/376352a0
50. Hibbs ML, Harder KW, Armes J, Kountouri N, Quilici C, Casagranda F, et al. Sustained activation of Lyn tyrosine kinase in vivo leads to autoimmunity. J Exp Med (2002) 196:1593–604. doi:10.1084/jem.20020515
51. Holodick NE, Tumang JR, Rothstein TL. Continual signaling is responsible for constitutive ERK phosphorylation in B-1a cells. Mol Immunol (2009) 46:3029–36. doi:10.1016/j.molimm.2009.06.011
52. Cohen DP, Rothstein TL. Elevated levels of protein kinase C activity and alpha-isoenzyme expression in murine peritoneal B cells. J Immunol (1991) 146:2921–7.
53. Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev (2004) 18:2195–224. doi:10.1101/gad.1228704
54. Anderson MT, Staal FJ, Gitler C, Herzenberg LA. Separation of oxidant-initiated and redox-regulated steps in the NF-kappa B signal transduction pathway. Proc Natl Acad Sci U S A (1994) 91:11527–31. doi:10.1073/pnas.91.24.11527
55. Reth M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat Immunol (2002) 3:1129–34. doi:10.1038/ni1202-1129
56. Nordberg J, Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med (2001) 31:1287–312. doi:10.1016/S0891-5849(01)00724-9
57. van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature (2003) 423:773–7. doi:10.1038/nature01681
58. Li Q, Engelhardt JF. Interleukin-1beta induction of NFkappaB is partially regulated by H2O2-mediated activation of NFkappaB-inducing kinase. J Biol Chem (2006) 281:1495–505. doi:10.1074/jbc.M511153200
59. Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett (2007) 581:3702–10. doi:10.1016/j.febslet.2007.05.039
60. Liu CY, Kaufman RJ. The unfolded protein response. J Cell Sci (2003) 116:1861–2. doi:10.1242/jcs.00408
61. Salminen A, Paimela T, Suuronen T, Kaarniranta K. Innate immunity meets with cellular stress at the IKK complex: regulation of the IKK complex by HSP70 and HSP90. Immunol Lett (2008) 117:9–15. doi:10.1016/j.imlet.2007.12.017
62. Malhotra V, Wong HR. Interactions between the heat shock response and the nuclear factor-kappa B signaling pathway. Crit Care Med (2002) 30:S89–95. doi:10.1097/00003246-200201001-00012
63. Schottelius AJ, Mayo MW, Sartor RB, Baldwin AS Jr. Interleukin-10 signaling blocks inhibitor of kappaB kinase activity and nuclear factor kappaB DNA binding. J Biol Chem (1999) 274:31868–74. doi:10.1074/jbc.274.45.31868
64. Hammer M, Mages J, Dietrich H, Schmitz F, Striebel F, Murray PJ, et al. Control of dual-specificity phosphatase-1 expression in activated macrophages by IL-10. Eur J Immunol (2005) 35:2991–3001. doi:10.1002/eji.200526192
65. Rui L, Healy JI, Blasioli J, Goodnow CC. ERK signaling is a molecular switch integrating opposing inputs from B cell receptor and T cell cytokines to control TLR4-driven plasma cell differentiation. J Immunol (2006) 177:5337–46.
66. Ding XZ, Tsokos GC, Kiang JG. Overexpression of HSP-70 inhibits the phosphorylation of HSF1 by activating protein phosphatase and inhibiting protein kinase C activity. FASEB J (1998) 12:451–9.
67. Lee KH, Lee CT, Kim YW, Han SK, Shim YS, Yoo CG. Heat shock protein 70 negatively regulates the heat-shock-induced suppression of the IkappaB/NF-kappaB cascade by facilitating IkappaB kinase renaturation and blocking its further denaturation. Exp Cell Res (2005) 307:276–84. doi:10.1016/j.yexcr.2005.03.014
68. Lee KH, Lee CT, Kim YW, Han SK, Shim YS, Yoo CG. Preheating accelerates mitogen-activated protein (MAP) kinase inactivation post-heat shock via a heat shock protein 70-mediated increase in phosphorylated MAP kinase phosphatase-1. J Biol Chem (2005) 280:13179–86. doi:10.1074/jbc.M410059200
69. Mizrahi A, Molshanski-Mor S, Weinbaum C, Zheng Y, Hirshberg M, Pick E. Activation of the phagocyte NADPH oxidase by Rac Guanine nucleotide exchange factors in conjunction with ATP and nucleoside diphosphate kinase. J Biol Chem (2005) 280:3802–11. doi:10.1074/jbc.M410257200
70. O’Rourke LM, Tooze R, Turner M, Sandoval DM, Carter RH, Tybulewicz VL, et al. CD19 as a membrane-anchored adaptor protein of B lymphocytes: costimulation of lipid and protein kinases by recruitment of Vav. Immunity (1998) 8:635–45. doi:10.1016/S1074-7613(00)80568-3
71. Khalil AM, Cambier JC, Shlomchik MJ. B cell receptor signal transduction in the GC is short-circuited by high phosphatase activity. Science (2012) 336:1178–81. doi:10.1126/science.1213368
Keywords: B cells, signal transduction, protein kinases/phosphatases, rodent
Citation: Holodick NE and Rothstein TL (2013) Atypical response of B-1 cells to BCR ligation: a speculative model. Front. Immunol. 4:457. doi: 10.3389/fimmu.2013.00457
Received: 29 August 2013; Accepted: 02 December 2013;
Published online: 16 December 2013.
Edited by:
Barbara L. Kee, University of Chicago, USAReviewed by:
Shiv Pillai, Massachusetts General Hospital, USAAaron James Marshall, University of Manitoba, Canada
Copyright: © 2013 Holodick and Rothstein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nichol E. Holodick, Center for Oncology and Cell Biology, The Feinstein Institute for Medical Research, 350 Community Drive, Manhasset, NY 11030, USA e-mail:bmhvbG9kaWNrQG5zaHMuZWR1