 Martha-Lena Müller1
Martha-Lena Müller1 Samuel C. C. Chiang1†
Samuel C. C. Chiang1† Marie Meeths2,3†
Marie Meeths2,3† Bianca Tesi2,3
Bianca Tesi2,3 Miriam Entesarian2,3
Miriam Entesarian2,3 Daniel Nilsson3,4
Daniel Nilsson3,4 Stephanie M. Wood1Magnus Nordenskjöld3Jan-Inge Henter2Ahmed Naqvi5
Stephanie M. Wood1Magnus Nordenskjöld3Jan-Inge Henter2Ahmed Naqvi5 Yenan T. Bryceson1,6*
Yenan T. Bryceson1,6*- 1Centre for Infectious Medicine, Department of Medicine, Karolinska Institutet, Karolinska University Hospital Huddinge, Stockholm, Sweden
- 2Childhood Cancer Research Unit, Department of Women’s and Children’s Health, Karolinska Institutet, Karolinska University Hospital Solna, Stockholm, Sweden
- 3Clinical Genetics Unit, Department of Molecular Medicine and Surgery, Karolinska Institutet, Karolinska University Hospital Solna, Stockholm, Sweden
- 4Science for Life Laboratory, Department of Molecular Medicine and Surgery, Karolinska Institutet, Stockholm, Sweden
- 5Division of Hematology and Oncology, Hospital for Sick Children, Toronto, ON, Canada
- 6Broegelmann Research Laboratory, Institute of Clinical Sciences, University of Bergen, Bergen, Norway
Familial hemophagocytic lymphohistiocytosis (FHL) is an often-fatal hyperinflammatory disorder caused by autosomal recessive mutations in PRF1, UNC13D, STX11, and STXBP2. We identified a homozygous STX11 mutation, c.173T > C (p.L58P), in three patients presenting clinically with hemophagocytic lymphohistiocytosis from unrelated Pakistani families. The mutation yields an amino acid substitution in the N-terminal Habc domain of syntaxin-11 and resulted in defective natural killer cell degranulation. Notably, syntaxin-11 expression was decreased in patient cells. However, in an ectopic expression system, syntaxin-11 L58P was expressed at levels comparable to wild-type syntaxin-11, but did not bind Munc18-2. Moreover, another N-terminal syntaxin-11 mutant, R4A, also did not bind Munc18-2. Thus, we have identified a novel missense STX11 mutation causative of FHL type 4. The syntaxin-11 R4A and L58P mutations reveal that both the N-terminus and Habc domain of syntaxin-11 are required for binding to Munc18-2, implying similarity to the dynamic binary binding of neuronal syntaxin-1 to Munc18-1.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory disorder clinically diagnosed based on fulfillment of five out of eight criteria including fever, splenomegaly, bicytopenia, hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytosis, low/absent natural killer (NK) cell activity, hyperferritinemia, and high soluble interleukin (IL)-2 receptor levels (1). Familial hemophagocytic lymphohistiocytosis (FHL) typically presents in infancy (2, 3). The incidence of FHL has been estimated to 1 in 50,000 live births (4). Chemo- and immunotherapy succeeds in controlling the disease in the majority of patients, but persistent remission is rarely obtained. At present, hematopoietic stem cell transplantation (HSCT) is the only cure for FHL (5).
Familial hemophagocytic lymphohistiocytosis is associated with autosomal recessive mutations in genes including PRF1, UNC13D, STX11, and STXBP2 (6–10). In addition, Griscelli syndrome type 2 and Chediak Higashi syndrome, associated with autosomal recessive mutations RAB27A and LYST, respectively, may also present with HLH and are in addition characterized by hypopigmentation. These genes encode proteins required for cytotoxic granule biogenesis, secretion, and target cell death (11). STX11, associated with FHL type 4 (FHL4), has the shortest coding sequence among these genes and accounts for only a small fraction of FHL patients. Relative to other FHL subtypes, patients with STX11 non-sense mutations or Stx11-deficient mice typically display less severe disease (12, 13). Although syntaxin-11 (Stx11)-deficiency abrogates degranulation by both cytotoxic T cells and NK cells (14, 15). The exact molecular mechanisms are not clear. Stx11 has been shown to bind Munc18-2, as well as the SNARE domain-containing proteins SNAP-23 and Vti1b (9, 10, 16, 17). Missense mutations can be informative in elucidating how Stx11 acts to facilitate exocytosis. To date, only two STX11 missense mutations have been reported (18).
In this study, we report a novel STX11 missense mutation in three unrelated Pakistani families. The autosomal recessive mutation abrogated NK cell degranulation. Interestingly, biochemical analyses of this N-terminal mutation, in addition to another mutation at the conserved N-terminus of Stx11, revealed binding of the N-terminal Habc domain of Stx11 to Munc18-2, stabilizing Stx11 expression, and facilitating cytotoxic lymphocyte exocytosis.
Materials and Methods
Patients and Controls
The studies were approved by the ethics committee at the Karolinska Institutet. Written consent was obtained from the patients’ families.
Cells and Antibodies
Peripheral blood mononuclear cells (PBMC) were isolated from peripheral blood by density gradient centrifugation (Lymphoprep, Axis-Shield) and maintained in complete medium (RPMI 1640 supplemented with 10% FBS and 2 mM l-glutamine; all Invitrogen). LAK cells were generated as previously described (19). The human erythroleukemia K562 and mouse mastocytoma P815 cell lines were maintained in complete medium. HEK-293T cells were maintained in DMEM (Invitrogen) supplemented with 10% FBS. Rabbit polyclonal anti-Stx11 and Munc18-2 (Proteintech Group) as well as mouse monoclonal anti-HA (clone 16B12, Covance) and anti-actin (C4, Fischer Scientific) antibodies were used for Western blotting. Mouse monoclonal anti-FLAG (M2, Sigma) was used for immunoprecipitation.
Functional Assays
For assessment of NK cell-mediated cytotoxicity, a standard 4-h 51Cr assay was used (14). Cytotoxic lymphocyte exocytosis was assessed by flow cytometry, as previously described (15). Samples were acquired on a Calibur instrument (BD Biosciences) and analyzed using Flowjo 9.4 software (Tree Star).
Plasmids and Sequence Analyses
Sequences encoding human Stx11 and Munc18-2 were cloned into a pDisplay vector backbone (Invitrogen) for expression on N-terminally tagged proteins. Stx11 mutations were generated by site-directed mutagenesis. Sequence analyses, alignments, and phylogenetic trees were performed and created with CLC Main Workbench software (v.6).
Biochemical Analyses
Patient and control PBMC or LAK cells were lysed in lysis buffer [20 mM Tris, pH 7.4, 2 mM EDTA, 1% Triton-X-100, 10% glycerol, 100 mM NaCl, protease inhibitors (Roche)]. The protein concentration in nuclei-depleted lysates was determined using Bradford assay (Thermo Scientific). Proteins were separated by SDS-PAGE (NuPAGE, Invitrogen), transferred to PVDF membranes (Millipore). The membranes were blocked with 5% skimmed milk, and blotted with specific antibodies. HEK-293T cells were transfected (Lipofectamine, Invitrogen) with plasmids encoding wild-type or mutated FLAG-tagged Stx11 (FLAG-Stx11) constructs, wild-type HA-tagged Munc18-2 (HA-Munc18-2, the empty vector, or combinations thereof). Twenty-four hours following transfection, the cells were lysed and the protein concentration was determined by Bradford assay (Thermo Scientific). For pull-down experiments, protein G-beads (Invitrogen) were pre-incubated with anti-FLAG mAb, washed in lysis buffer, and incubated with lysates from different FLAG-Stx11 transfected cells for 2 h at 4°C. Subsequently, FLAG-Stx11-loaded beads were washed and incubated with lysates from vector or HA-Munc18-2 transfected cells for 4 h at 4°C.
Results
Clinical and Immunological Characterization of Patients with a Homozygous STX11 Missense Mutation
Here, we describe two infants and one 5-year-old child born to unrelated Pakistani families that presented with HLH (Table 1). Patient A and B presented with a laboratory parameters consistent with a clinical diagnosis of HLH at the Aga Khan Hospital, Karachi. Patient C also presented with a hyperinflammatory syndrome and was later referred to the Aga Khan Hospital. For patient C, it has not been possible to retrieve laboratory parameters at initial presentation.
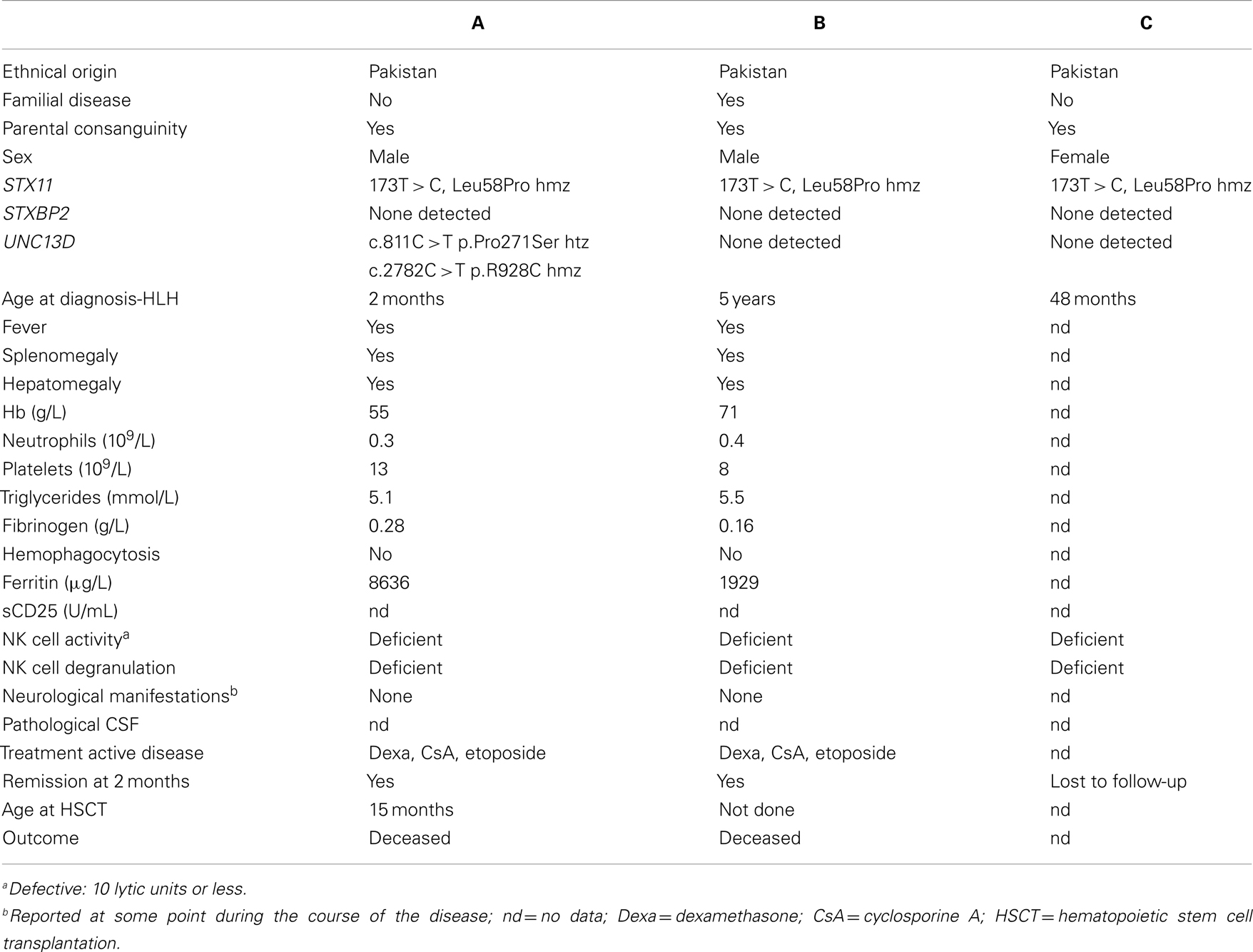
Table 1. Clinical, laboratory, and genetic findings in patients.
Due to suspicion of FHL, NK cell cytotoxicity, degranulation, and intracellular expression of granule constituents was assessed. All patients displayed defective lysis of K562 target cells and degranulation by NK cells in response to K562 target cells or engagement of the Fc receptor CD16 (Figures 1A,B). Notably, cytotoxicity and degranulation were partially restored by IL-2 stimulation (Figures 1C,D). Moreover, expression of cytotoxic granule constituents’ perforin, granzyme B, and CD107a was normal in patient NK cells, suggesting that granule integrity was not impaired (Figure 2). On the basis of these functional and phenotypic assessments, mutations in genes required for lymphocyte exocytosis and associated with FHL were suspected.
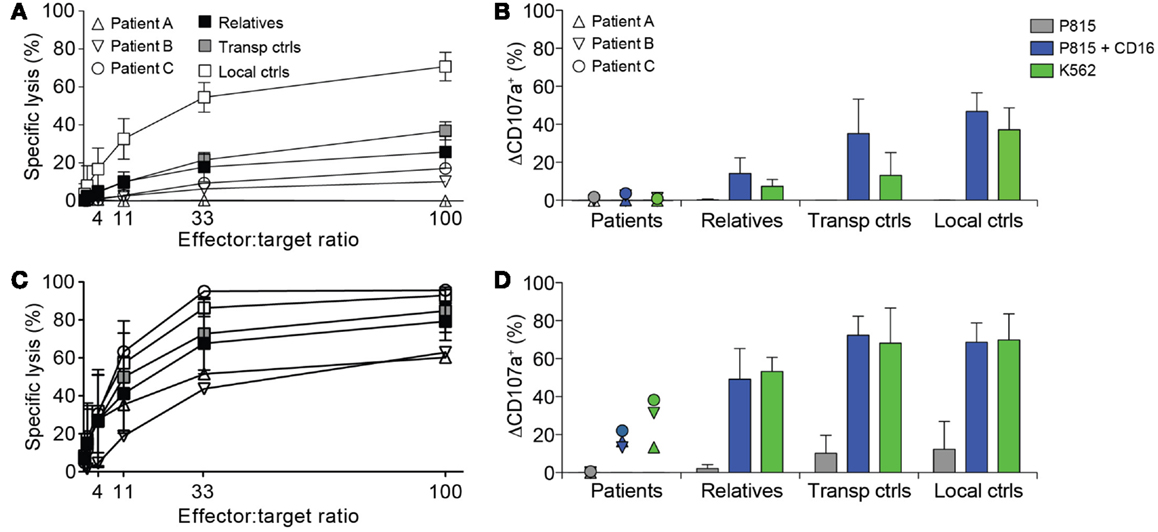
Figure 1. Defective NK cell degranulation in patients with homozygous STX11 c.173T > C mutations. (A–D) PBMC were isolated from three patients with homozygous STX11 c.173T > C, p.L58P mutations, relatives (n = 10, all six parents in addition to four siblings) as well as transport (n = 3) and local controls (n = 5). PBMC (A) freshly isolated or (C) stimulated with IL-2 overnight were mixed with 51Cr labeled K562 target cells. After 4 h, 51Cr-release was quantified in supernatants and specific lysis was calculated. Symbols indicate individual values for patients or mean values for relatives and controls. Error bars indicate SD. PBMC (B) freshly isolated or (D) stimulated with IL-2 overnight were mixed with target cells and antibodies, as indicated. The cells were stained with antibodies to lineage markers and CD107a. The frequency of cells expressing surface CD107a was determined by flow cytometry. Symbols indicate values for patients and bars mean for relatives and controls. Error bars indicate SD.
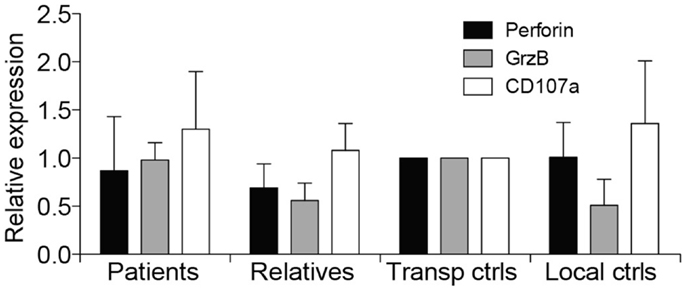
Figure 2. Intracellular expression of CD107a, perforin, and granzyme B in patients with homozygous STX11 c.173T > C mutations. Intracellular expression of perforin, granzyme B, and CD107a was examined in patient B and C, in addition to relatives (n = 8, all four parents in addition to four siblings) as well as transport (n = 2) and local controls (n = 4). Expression of granule constituents was normalized relative to that of the transport control. Bars indicate the mean value, Error bars indicate SD.
Sequencing of the coding regions and splice-sites of UNC13D, STX11, and STXBP2 revealed that all three patients were homozygous for a novel STX11 mutation, c.173T > C (p.L58P) (Table 1). The L58P localizes to an α-helical strand of the predicted Stx11 Habc domain. The parents were heterozygous for this STX11 mutation, but did not have any recorded history of inflammatory disease. In addition, patient A was heterozygous for a rare UNC13D c.811C > T (p.P271S; frequency 0.001 in a Caucasian population of 4294 individuals) variant inherited from the father and homozygous for an uncommon UNC13D c.2782C > T (p.R928C, frequency 0.01 in a Caucasian population of 4294 individuals) variant inherited from either parent. As no hypopigmentation was evident in the patients, RAB27A and LYST were not sequenced.
A Homozygous STX11 Missense Mutation Results in Selective Loss of SYNTAXIN-11 Expression in Patient NK Cells
To gain insights into how the Stx11 L58P missense mutation may cause disease, we analyzed Stx11 expression in PBMCs from patient C and controls. Stx11 levels were found to be greatly reduced in the patient (Figure 3A). PBMCs from the patient’s mother displayed low Munc18-2 expression as well as slightly decreased syntaxin-11 expression. Although Munc18-2 levels were comparable between the patient and controls, the loss of Stx11 expression may reflect differences in the distribution of immune cell subsets or the inflammatory state between the patient and controls. Thus, we generated LAK cells from patient C and controls. LAK cells from the patient also displayed a selective loss of Stx11 expression, whereas Munc18-2 expression was similar to that of control LAK cells (Figure 3B). LAK cells from the patient’s mother displayed syntaxin-11 and Munc18-2 levels similar to those of control LAK cells. Thus, Stx11 L58P might either be poorly expressed or be destabilized and degraded in the patient cells.
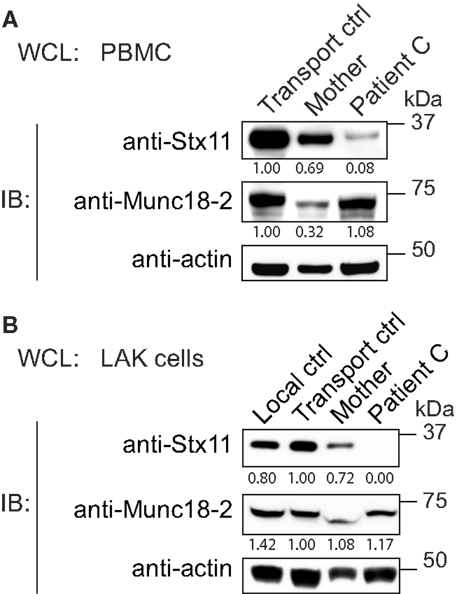
Figure 3. Low expression of syntaxin-11 L58P in patient lymphocytes. Whole cell lysates (WCL) prepared from (A) PBMC or (B) LAK cells from Stx11 L58P patient C and controls, as indicated, were analyzed by Western blotting for Stx11 and Munc18-2. β-actin was probed as a loading control. Densitometry values normalized to β-actin for each individual are indicated.
Syntaxin-11 R4A and L58P Mutations Disrupt Binding to Munc18-2
Stx11 interacts with Munc18-2 and loss of Munc18-2 expression has previously been shown to result in loss of Stx11 expression as well, suggesting a requirement for Munc18-2 in stabilization of Stx11 (9, 10). With respect to N-terminal peptide sequences, human Stx19, Stx1A, Stx1B, Stx2, Stx3, and Stx4 represent close homologs to human Stx11 (Figure 4A). Remarkably, the N-peptide as well as the sequence surrounding the Stx11 L58 residue in the Habc are highly conserved between Stx11 and Stx1 isoforms, as well as other related Stxs (Figures 4B,C). Interestingly, studies of neuronally expressed Stx1A and Munc18-1 have revealed that the N-terminal residues (N-peptide) as well as the N-terminal Habc domains of Stx1 mediate interactions with Munc18-1 (20–22). As both the N-peptide and Habc domain of Stx1A are closely conserved to those of Stx11, we evaluated whether a Stx11 R4A mutation as well as the patient-derived Stx11 L58P mutation located to the Habc domain would interfere with binding of Munc18-2. Constructs for ectopic expression of FLAG-tagged wild-type and mutant Stx11 were transfected into HEK-293T cells. In transfection experiments, both Stx11 R4A and L58P mutants were expressed at levels comparable to Stx11 wild-type (Figure 4D). Notably, in pull-down experiments using beads loaded with FLAG-tagged Stx11 wild-type and Stx11 mutants neither FLAG-tagged Stx11 R4A nor L58P mutants bound HA-tagged Munc18-2 (Figure 4E). In contrast, the C-terminal Stx11 Q268X mutation previously associated with FHL4 (14), did not display impaired binding of Munc18-2 (Figure 4E). Together, the data show that mutations in the N-peptide or Habc domain of Stx11 can disrupt interactions with Munc18-2, demonstrating a critical role for both the N-peptide and Habc domain of Stx11 in binding of Munc18-2.
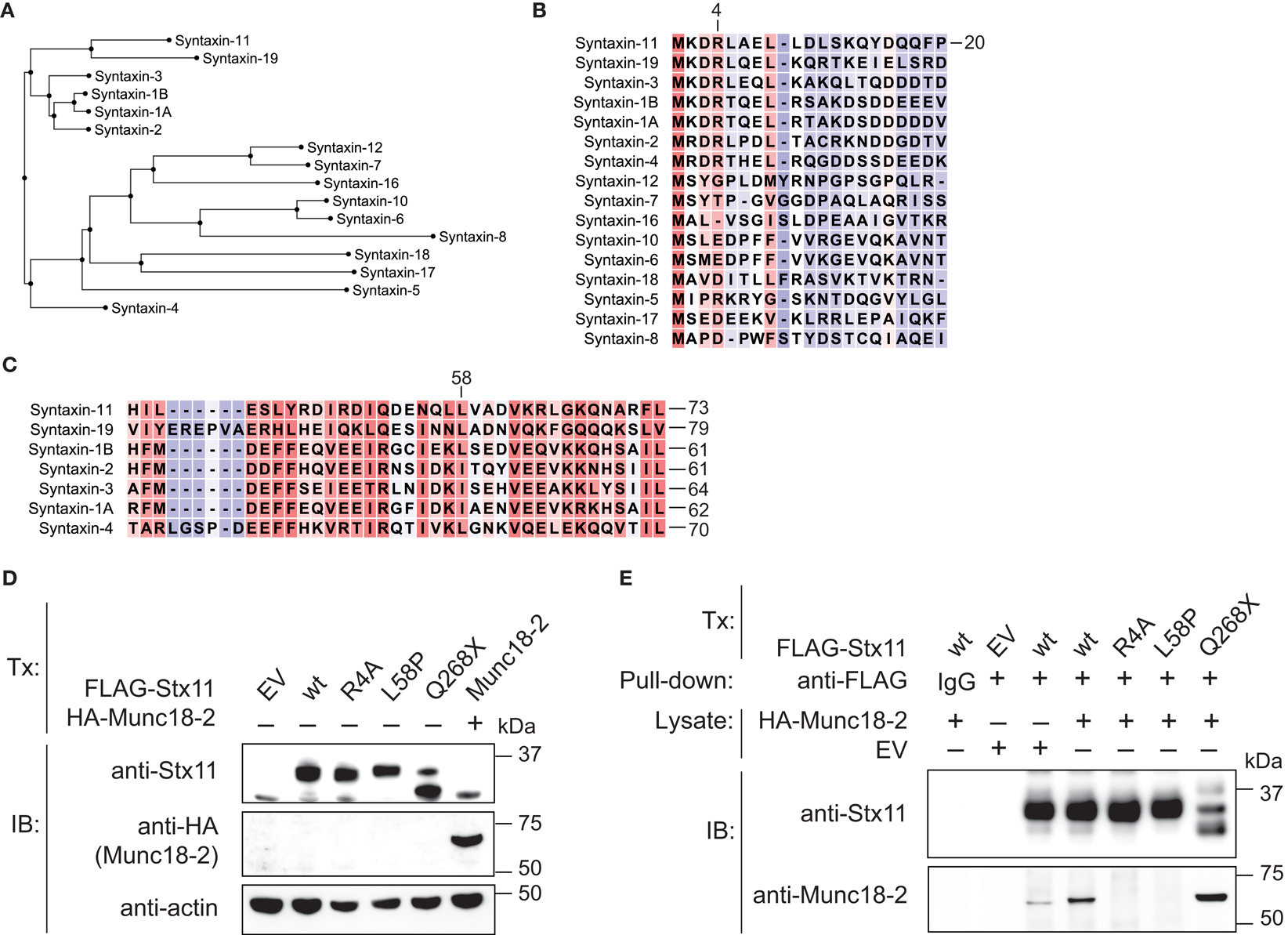
Figure 4. Abrogated binding of Munc18-2 to syntaxin-11 N-terminal R4A or Habc-domain L58P mutations. (A) Dendrogram comparing human syntaxins based on peptide sequence alignments of the N-termini (N-peptide and Habc domain; residues 1-167 of Stx11). (B) Amino acid alignments of human syntaxin (B) N-terminus and (C) Habc domain sequences. Red shade indicates a high degree of sequence homology, whereas blue shade indicates low homology. (D) HEK-293T cells were transfected with empty vector (EV), or constructs encoding FLAG-Stx11 wild-type (wt), R4A, L58P, Q268X, or HA-Munc18-2 wild-type. Twenty-four hours after transfection, WCL were prepared and analyzed for expression of Stx11, Munc18-2, and actin. (E) HEK-293T cells constructs as indicated. Twenty-four hours after transfection, WCL were prepared from Stx11 transfectants and Stx11 was immunoprecipitated to beads. The beads were washed extensively and thereafter incubated with WCL from EV or HA-Munc18-2 wt transfected HEK-293T cells. Beads were washed, proteins eluted, and analyzed by Western blotting with antibodies as indicated.
Discussion
We describe a novel autosomal recessive missense STX11 c.173T > C (p.L58P) mutation causative of FHL4 in three children from different Pakistani kindreds. Stx11 expression was absent in NK cells from a patient homozygous for this STX11 mutation. Consistent with previous studies, the STX11 mutation was associated with defective degranulation by resting NK cells (14, 23). Of note, whereas only STX11 mutations were detected in the other patients, patient A also carried UNC13D variants. By comparison, this patient presented at an earlier age than the patient B and C, and displayed less of a restoration of NK cell degranulation upon IL-2 stimulation. Thus, although an abrogation of NK cell degranulation is expected in Munc13-4 deficient patients, it is possible that the UNC13D variants contribute to the severity of disease presentation in patient A.
By sequence homology to Stx1A, the Stx11 L58P mutation is located in the first α-helix of the conserved Habc domain of Stx11 (20). Substitution to a proline residue at this position likely disrupts the conformation of the Stx11 Habc domain. Interestingly, biochemical analyses examining ectopically expressed, tagged Stx11 in a cell line revealed that the Stx11 L58P mutation disrupted interactions with tagged Munc18-2. Mutations in STXBP2 that lead to loss of Munc18-2 expression also cause loss of Stx11 expression in lymphocytes (9, 10). Thus, conversely, it is tenable that Stx11 mutations that disrupt Munc18-2 binding might reduce Stx11 expression through a similar mechanism, explaining the low expression of mutant Stx11 in patient cells.
With respect to neuronal Stx1 binding of Munc18-1, the very N-terminal residues as well as the Habc domain of Stx1 mediate a binary interaction with Munc18-1 (20–22). Our data suggest that Stx11 binding to Munc18-2 has similar molecular requirements as either mutation of the conserved Stx11 N-peptide (R4A) or of the Habc domain (L58P) abrogated Munc18-2 binding. These observations are supported by recent publications demonstrating that mutations in the hydrophobic pocket of Munc18-2, which can bind the N-peptide of Stx11, abrogate Stx11 binding and mast cell degranulation (24). During preparation of this manuscript, the Munc18-2 crystal structure was reported (25). The crystal structure, as well as studies of how Munc18-2 mutations associated with FHL5 impact Stx11 binding, similarly suggest a requirement for the N-peptide and Habc domains of Stx11 for binding to Munc18-2 (25). With respect to neuronal exocytosis, it has recently been shown that mutations in the Habc domain of Stx1 abrogate interactions with Munc18-1, which usually keep Stx1 in a closed conformation, leading to reduced Mun18-1 expression (26). In contrast, mutations of the Stx1 N-peptide more specifically interfere with vesicle fusion (26). It will be interesting to further determine how the N-terminus and Habc domain of Stx11 regulate Stx11 conformation, trafficking, and granule exocytosis.
Stx11 has been shown to interact with additional proteins involved in vesicle exocytosis, including SNAP-23, VAMP2, and Vti1b (16, 17). In addition, the priming factor Munc13-4 has been shown to interact with different Stxs (27). These proteins interact with the Stx11 C-terminal SNARE domain, with preferential binding to the open conformation of Stx11. Speculatively, although other more direct mechanisms for degradation of Stx11 due to protein misfolding also may explain low Stx11 expression, disruption of the Stx11 – Munc18-2 interaction in patient NK cells may lead to Stx11 degradation based on a mechanism dependent on such facilitators and regulators of vesicle exocytosis. Thus, it is of interest to perform a more comprehensive screen of how different Stx11 mutations impact interactions with other proteins implicated in facilitating and regulating granule exocytosis.
In conclusion, we demonstrate that both the N-terminus and Habc domain of Stx11 are involved in binding to Munc18-2. In the patients homozygous for a Stx11 L58P mutation, it is quite possible that the abrogated interaction between Stx11 and Munc18-2 leads to destabilization of Stx11 expression. Further studies of Stx11 mutants may provide insights into mechanisms, specificity, and redundancy governing SNARE complex formation for lytic granule exocytosis by cytotoxic lymphocytes.
Author Contributions
Martha-Lena Müller designed research, performed biochemical experiments, analyzed and interpreted data, and drafted the manuscript; Samuel C. C. Chiang designed research, performed functional evaluations of lymphocytes, analyzed and interpreted data, and drafted the manuscript; Marie Meeths designed research, performed targeted sequencing of FHL genes, and drafted the manuscript; Bianca Tesi designed research and performed targeted sequencing of FHL genes; Miriam Entesarian, Daniel Nilsson, and Stephanie M. Wood designed experiments and interpreted data; Magnus Nordenskjöld and Jan-Inge Henter designed research and interpreted data; Ahmed Naqvi identified and cared for patients, collected clinical data, and drafted the manuscript; Yenan T. Bryceson designed research, interpreted data, and drafted the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement n. 311335, Swedish Research Council, Swedish Foundation for Strategic Research, Swedish Cancer Foundation, Swedish Children’s Cancer Foundation, Histiocytosis Association, Jeansson’s Foundation, Åke Wiberg’s Foundation, the Karolinska Institute Research Foundation, and the Stockholm County Council (ALF project).
References
1. Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer (2007) 48:124–31. doi: 10.1002/pbc.21039
2. Filipovich AH. The expanding spectrum of hemophagocytic lymphohistiocytosis. Curr Opin Allergy Clin Immunol (2011) 11:512–6. doi:10.1097/ACI.0b013e32834c22f5
3. Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med (2012) 63:233–46. doi:10.1146/annurev-med-041610-134208
4. Henter JI, Elinder G, Soder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand (1991) 80:428–35. doi:10.1111/j.1651-2227.1991.tb11878.x
5. Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr (2007) 166:95–109. doi:10.1007/s00431-006-0258-1
6. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science (1999) 286:1957–9. doi:10.1126/science.286.5446.1957
7. Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell (2003) 115:461–73. doi:10.1016/S0092-8674(03)00855-9
8. zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter JI, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet (2005) 14:827–34. doi:10.1093/hmg/ddi076
9. Cote M, Menager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest (2009) 119:3765–73. doi:10.1172/JCI40732
10. zur Stadt U, Rohr J, Seifert W, Koch F, Grieve S, Pagel J, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet (2009) 85:482–92. doi:10.1016/j.ajhg.2009.09.005
11. de Saint Basile G, Menasche G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol (2010) 10:568–79. doi:10.1038/nri2803
12. Kogl T, Muller J, Jessen B, Schmitt-Graeff A, Janka G, Ehl S, et al. Hemophagocytic lymphohistiocytosis in syntaxin-11-deficient mice: T-cell exhaustion limits fatal disease. Blood (2013) 121:604–13. doi:10.1182/blood-2012-07-441139
13. Sepulveda FE, Debeurme F, Menasche G, Kurowska M, Cote M, Pachlopnik Schmid J, et al. Distinct severity of HLH in both human and murine mutants with complete loss of cytotoxic effector PRF1, RAB27A, and STX11. Blood (2013) 121:595–603. doi:10.1182/blood-2012-07-440339
14. Bryceson YT, Rudd E, Zheng C, Edner J, Ma D, Wood SM, et al. Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood (2007) 110:1906–15. doi:10.1182/blood-2007-02-074468
15. Chiang SC, Theorell J, Entesarian M, Meeths M, Mastafa M, Al-Herz W, et al. Comparison of primary human cytotoxic T-cell and natural killer cell responses reveal similar molecular requirements for lytic granule exocytosis but differences in cytokine production. Blood (2013) 121:1345–56. doi:10.1182/blood-2012-07-442558
16. Valdez AC, Cabaniols JP, Brown MJ, Roche PA. Syntaxin 11 is associated with SNAP-23 on late endosomes and the trans-Golgi network. J Cell Sci (1999) 112(Pt 6):845–54.
17. Offenhauser C, Lei N, Roy S, Collins BM, Stow JL, Murray RZ. Syntaxin 11 binds Vti1b and regulates late endosome to lysosome fusion in macrophages. Traffic (2011) 12:762–73. doi:10.1111/j.1600-0854.2011.01189.x
18. Marsh RA, Satake N, Biroschak J, Jacobs T, Johnson J, Jordan MB, et al. STX11 mutations and clinical phenotypes of familial hemophagocytic lymphohistiocytosis in North America. Pediatr Blood Cancer (2010) 55:134–40. doi:10.1002/pbc.22499
19. Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood (2006) 107:159–66. doi:10.1182/blood-2005-04-1351
20. Fernandez I, Ubach J, Dulubova I, Zhang X, Sudhof TC, Rizo J. Three-dimensional structure of an evolutionarily conserved N-terminal domain of syntaxin 1A. Cell (1998) 94:841–9. doi:10.1016/S0092-8674(00)81742-0
21. Dulubova I, Khvotchev M, Liu S, Huryeva I, Sudhof TC, Rizo J. Munc18-1 binds directly to the neuronal SNARE complex. Proc Natl Acad Sci U S A (2007) 104:2697–702. doi:10.1073/pnas.0611318104
22. Rickman C, Medine CN, Bergmann A, Duncan RR. Functionally and spatially distinct modes of munc18-syntaxin 1 interaction. J Biol Chem (2007) 282:12097–103. doi:10.1074/jbc.M700227200
23. Bryceson YT, Pende D, Maul-Pavicic A, Gilmour KC, Ufheil H, Vraetz T, et al. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood (2012) 119:2754–63. doi:10.1182/blood-2011-08-374199
24. Bin NR, Jung CH, Piggott C, Sugita S. Crucial role of the hydrophobic pocket region of Munc18 protein in mast cell degranulation. Proc Natl Acad Sci U S A (2013) 110:4610–5. doi:10.1073/pnas.1214887110
25. Hackmann Y, Graham SC, Ehl S, Honing S, Lehmberg K, Arico M, et al. Syntaxin binding mechanism and disease-causing mutations in Munc18-2. Proc Natl Acad Sci U S A (2013) 110:E4482–91. doi:10.1073/pnas.1313474110
26. Zhou P, Pang ZP, Yang X, Zhang Y, Rosenmund C, Bacaj T, et al. Syntaxin-1 N-peptide and Habc-domain perform distinct essential functions in synaptic vesicle fusion. EMBO J (2013) 32:159–71. doi:10.1038/emboj.2012.307
Keywords: familial hemophagocytic lymphohistiocytosis, syntaxin-11, Munc18-2, N-peptide
Citation: Müller M-L, Chiang SCC, Meeths M, Tesi B, Entesarian M, Nilsson D, Wood SM, Nordenskjöld M, Henter J-I, Naqvi A and Bryceson YT (2014) An N-terminal missense mutation in STX11 causative of FHL4 abrogates syntaxin-11 binding to Munc18-2. Front. Immunol. 4:515. doi: 10.3389/fimmu.2013.00515
Received: 13 November 2013; Paper pending published: 27 November 2013;
Accepted: 27 December 2013; Published online: 14 January 2014.
Edited by:
Konrad Krzewski, National Institutes of Health, USAReviewed by:
Peter Van Der Sluijs, Universitair Medisch Centrum Utrecht, NetherlandsClaudio Guillermo Giraudo, University of Pennsylvania, USA
Copyright: © 2014 Müller, Chiang, Meeths, Tesi, Entesarian, Nilsson, Wood, Nordenskjöld, Henter, Naqvi and Bryceson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yenan T. Bryceson, Department of Medicine, Center for Infectious Medicine, F59, Karolinska Institutet, Karolinska University Hospital Huddinge, S-141 86 Stockholm, Sweden e-mail:eWVuYW4uYnJ5Y2Vzb25Aa2kuc2U=
†Samuel C. C. Chiang and Marie Meeths have contributed equally to this work.