 Maxine Bauzon
Maxine Bauzon Terry Hermiston
Terry Hermiston- Bayer HealthCare, US Innovation Center, Biologics Research, San Francisco, CA, USA
For the past 150 years cancer immunotherapy has been largely a theoretical hope that recently has begun to show potential as a highly impactful treatment for various cancers. In particular, the identification and targeting of immune checkpoints have given rise to exciting data suggesting that this strategy has the potential to activate sustained antitumor immunity. It is likely that this approach, like other anti-cancer strategies before it, will benefit from co-administration with an additional therapeutic and that it is this combination therapy that may generate the greatest clinical outcome for the patient. In this regard, oncolytic viruses are a therapeutic moiety that is well suited to deliver and augment these immune-modulating therapies in a highly targeted and economically advantageous way over current treatment. In this review, we discuss the blockade of immune checkpoints, how oncolytic viruses complement and extend these therapies, and speculate on how this combination will uniquely impact the future of cancer immunotherapy.
Introduction
Tumors are difficult to treat and in many instances lethal. The treatment challenge is not surprising as they are genetically unstable and complex biological systems with an ability to adapt to and thrive in often harsh and changing environments. Furthermore, this plasticity increases the probability that subpopulations will acquire resistance to any one therapy. Thus one could argue that a disease with such a complex etiology must be met with an equally complex therapeutic approach. Appropriately, oncologists have for some time combined chemotherapy, radiation and surgery and complemented these strategies with more targeted approaches such as tumor selective antibodies and/or small molecule kinase inhibitors (1). More recently, two alternative therapeutic approaches, cancer immunotherapy and oncolytic viruses, have begun to show promise that should further complement the oncologist’s repertoire of anti-cancer agents.
The area of cancer immunotherapy has had a long and complex history (2, 3). The idea that a patient’s own immune system could remove a tumor in much the same way it so efficiently removes invading microbes has been around for more than a century. Through the years, however, this concept of immunosurveillance has fallen in and out of favor perhaps appropriately given the complex and dynamic role, it is now believed to play in cancer, acting anywhere from anti to pro-tumorigenic (4–6). Research is beginning to elucidate the mechanisms by which tumors evade the immune system and in some instances how tumors use it to their advantage. From this research several promising immune-checkpoint inhibitor targets that are now translating into exciting clinical trial results have emerged (7–9).
Like cancer immunotherapy, the concept of oncolytic viruses is not new dating back to at least the beginning of the twentieth century when it was observed that on occasion tumor regression would follow a viral infection (10, 11). Although over 100 years have passed since these initial observations, the idea of using a replicating virus to selectively infect and kill tumor cells remains understandably appealing. Theoretically, either naturally or through genetic engineering, such an agent would spare normal neighboring cells while killing cancer cells by viral lysis. Furthermore, the progeny released from the lysed cancer cells would result in a self-perpetuating and amplifying therapy. Adding to their appeal is the ability of such agents to deliver exogenous genetic material whose product or products could augment the oncolytic viral treatment (12–14). Despite their theoretical promise, the reality is that oncolytic viruses have had limited clinical success as monotherapies perhaps due to an imbalanced focus on safety over potency. Recently however, there are several late-stage clinical trials showing promise which may eventually lead to clinical acceptance (15, 16).
Here, we suggest merging immune-checkpoint blockers with oncolytic viruses. We will discuss not only how these approaches could complement one another biologically for increased therapeutic benefit, but also how they may represent a unique opportunity to employ alternative biological formats not normally utilized commercially (e.g., Fabs, scFv) to increase both the safety and therapeutic profile of these agents. Finally we will touch upon how, together, these attributes might translate into a more economically appealing and clinically active therapy resulting in a truly new and disruptive treatment for malignancies.
Cancer Immunotherapy-Blockade of Immune Checkpoints
Immunotherapy works to direct the extensive repertoire of the host immune system to fight cancer. This approach strives to stimulate tumor suppression by (a) boosting the patient’s immune system, (b) decreasing the cancer-induced immunosuppression, and/or (c) increasing the immunogenicity of the tumor itself. If the immune system’s ability to rapidly respond to and clear invading microorganisms could be extended to malignant cells then a powerful therapeutic may be realized. Such an approach may hold greater potential than current treatment approaches as it may prove to be more potent, benefit many more cancer types, offer long-lasting protection against the disease, and come with fewer off-target effects. Advances in cellular and molecular immunology have provided enormous insight into the inter-play between tumors and immune cells and from this research have come strategies by which the immune system might be harnessed to fight cancer (7).
The blockade of immune checkpoints is a more recent approach taken to decrease cancer-induced immunosuppression. Immune checkpoints refer to a number of inhibitory pathways that play crucial roles in maintaining self-tolerance and immune homeostasis. Their function is to down-regulate T-cell signaling in order to prevent uncontrolled T-cell proliferation thereby protecting tissues from auto-immune damage while maintaining tolerance to self-antigens. It is becoming increasingly clear that tumors commandeer certain immune-checkpoint pathways particularly against T cells that are specific for tumor antigens. Preclinical and clinical data have demonstrated that this is a major mechanism utilized by the tumor to evade the immune system. If this could be reversed, the resulting amplification of T cells and their activity would be highly beneficial to the patient given the central role T cells play in cell-mediated immunity. The immune checkpoints are controlled by ligand–receptor interactions, which can be readily blocked by antibodies or disrupted by recombinant forms of ligands or receptors making them appealing therapeutic targets. For a list of immune-checkpoint targeting antibodies that are currently in clinical trial see Table 1.
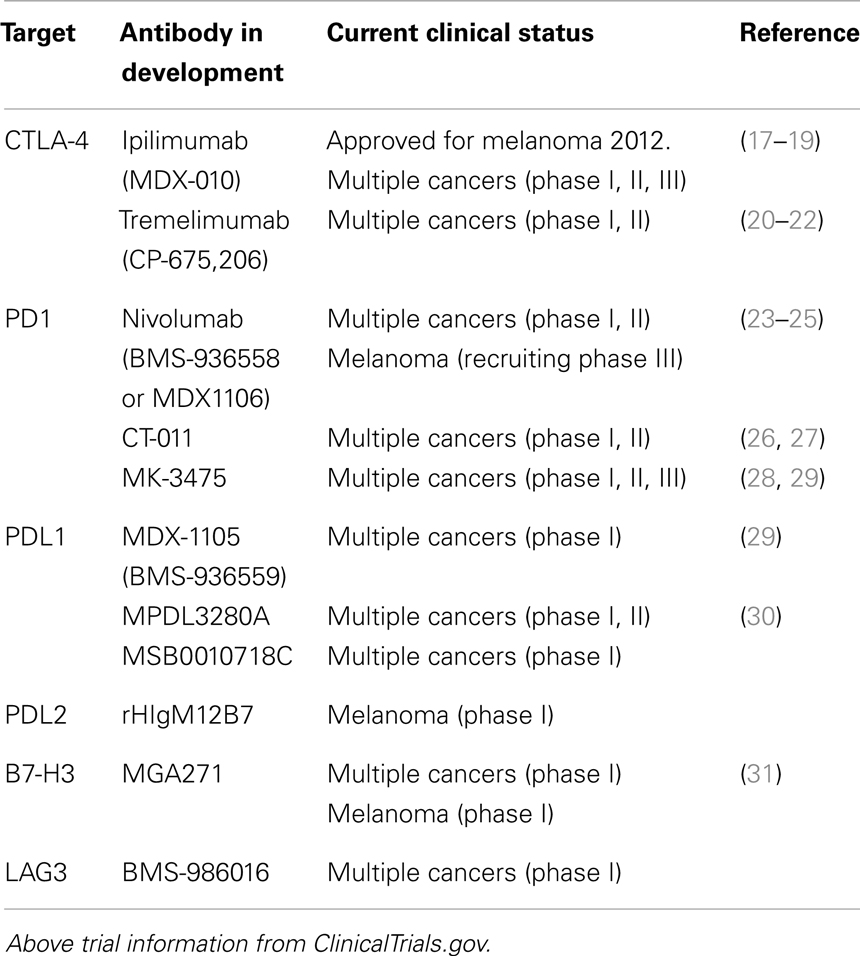
Table 1. The most advanced clinically evaluated immune-checkpoint blocking antibodies.
The inhibitory receptor, Cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), was the first checkpoint receptor to be extensively and successfully pursued as an anti-cancer target (32). The primary function of CTLA-4 is to regulate the magnitude of T-cell activation. It is expressed solely on T cells where it offsets the actions of CD28, a T-cell co-stimulatory receptor. Because CTLA-4 has a higher affinity for the CD28 ligands B7.1 and B7.2 it, in effect, out-competes CD28 for ligand binding resulting in an attenuated T-cell response (33–37). The lethal systemic immune hyperactivation phenotype of Ctla4-knockout mice clearly shows the importance of CTLA-4 and the need to keep T cells in check (38, 39). In 2011, an antibody against CTLA-4 (ipilimumab) was given FDA approval for the treatment of metastatic melanoma (20, 40–42). In a pivotal phase III randomized three-arm clinical trial, melanoma patients were treated with a glycoprotein 100 (gp100) peptide vaccine alone, ipilimumab alone, or the gp100 peptide and ipilimumab. Both ipilimumab groups demonstrated an increased survival of 3.5 months compared with the group receiving the gp100 peptide alone. Moreover, long-term survival was greatly increased with 18% of patients receiving ipilimumab surviving for greater than 2 years as compared with only 5% for the gp100 peptide alone cohort (17). Although ipilimumab treatment was relatively brief, spanning only 3 months, the finding of long-term progression-free survival supports the idea that immune-based therapies may actually result in a reprogramed immune system which can confer long-term antitumor immunity. Clinical trials are on-going evaluating the use of anti CTLA-4 antibodies in other cancer indications including lung, colorectal, renal, and ovarian (43).
The immune-checkpoint receptor, programed cell death 1 (PD1) and its ligands PDL1 and PLD2, are also emerging as promising targets. PD1 like CTLA-4 plays a role in regulating and maintaining the balance between T-cell activation and tolerance (44, 45). However, unlike CTLA-4, PD1 is more broadly expressed and can be found on other activated non-T-lymphocyte subsets including B cells and natural killer (NK) cells. Additionally while CTLA-4 primarily regulates T-cell activation, PD1 principally controls T-cell activity (46). The ligands PDL1 and PDL2 are commonly upregulated on the surface of many different human tumors with PDL1 being the predominant PD1 ligand on solid tumors. High expression levels of PDL1 have been shown on melanoma, lung, ovarian, and other human cancers (47, 48). PDL1 is also expressed on myeloid cells in the tumor microenvironment. Pdl, Pdl1, and Pdl2-knockout mice demonstrate a milder auto-immune phenotype than Ctla4-knockout mice (49–52). Preclinical studies have shown that blocking PD1 or its ligand PDL1 enhances immunity in vitro and mediates antitumor activity in preclinical models (53–55). Although the development of PD1 targeting antibodies is not as mature as that of CTLA-4 antibodies, preliminary clinical results look encouraging. In phase I trials of an anti-PD1 antibody (nivolumab), objective responses (complete or partial responses) were observed in those with non-small-cell lung cancer, melanoma, or renal-cell cancer with cumulative response rates ranging from 18 to 28%. Responses were durable with 20 of 31 responses lasting 1 year or more (56). In a separate phase I trial of patients with various advanced cancers, an anti-PDL1 antibody (MDX-1105) also induced durable tumor regression (objective response rate, 6–17%) and prolonged stabilization of disease (12–41% at 24 week) (57).
Beyond CTLA-4 and PD1, molecular immunology has begun to reveal additional receptors and ligands that serve an inhibitory immune function. These include B and T-lymphocyte attenuator (BTLA), T-cell membrane protein 3 (TIM3), Lymphocyte activation gene 3 (LAG3), adenosine A2a receptor (A2aR), and the B7 family of inhibitory ligands (58–66). Each has been associated with the inhibition of lymphocyte activity in preclinical models and consequently antibodies against a number of these targets are being actively pursued (58–66). Additionally, because multiple inhibitory ligands and receptors contribute to the tumor’s evasion of the immune system and appear to be non-redundant, there remains the possibility of further enhancing antitumor immunity by blocking multiple immune checkpoints. Currently several preclinical and clinical studies are on-going testing the effects of blocking a combination of immune checkpoints (Table 2) (67–73). In fact, a recently published phase I study in patients with melanoma that combined anti-CTLA-4 (ipilimumab) and anti-PD1(nivolumab) mAbs resulted in a rapid and deep tumor regression in a substantial proportion of patients (53% of patients had an objective response, all with tumor reduction of 80% or more) (74). These objective response rates exceeded the previously reported results with either mAb alone (17, 56).
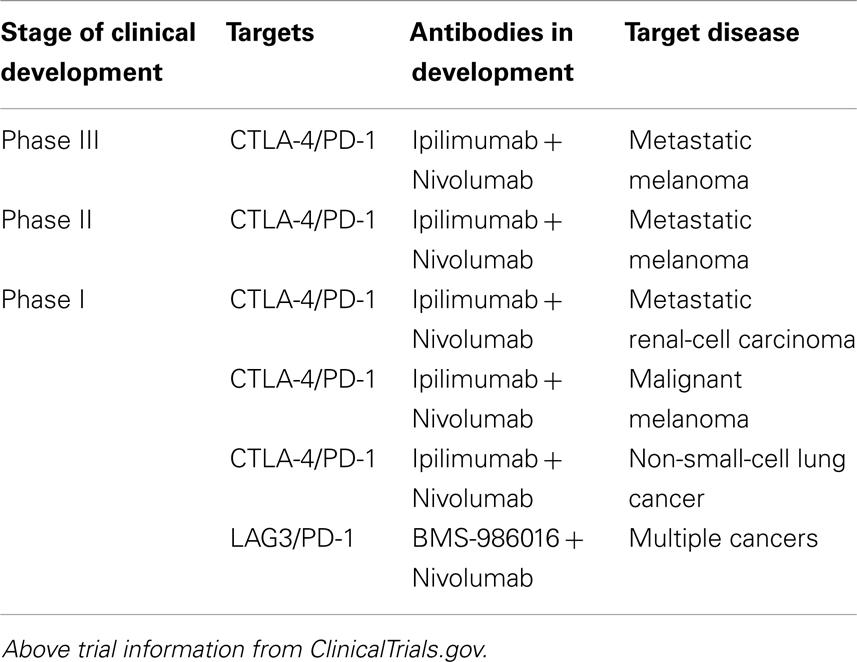
Table 2. The current clinical development of combined immune-checkpoint targeting agents.
Oncolytic Viruses as (Immuno)Therapies
Oncolytic viruses can be RNA or DNA based and derived from human (e.g., herpes simplex virus, adenovirus, measles virus) or animal [e.g., vesicular stomatitis virus (VSV), Newcastle disease virus, myxoma virus] viruses. By definition they selectively replicate in, and kill cancer cells. This selectivity can be a natural property of the virus or an engineered trait (75–81). Oncolytic viruses can also be genetically armed to improve or generate more tumor selective cell killing. For example, cell death can be induced by delivering tumor-suppressors (e.g., p53, p16), pro-apoptotic proteins (e.g., TRAIL, IL-24), or small hairpin RNA targeting cell survival or proliferation factors (e.g., hTERT, survivin) (82–87). Arming can also sensitize the tumor to chemo or radiotherapy (Prodrug enzymes, NIS) (88–90).
Although direct oncolysis was envisioned as the primary desired outcome of this therapeutic approach, research and clinical data is supporting the assertion that these productive tumor-specific infections can elicit additional antitumor effects. For example there is evidence that oncolytic viral therapy can induce tumor vasculature shutdown resulting in tumor necrosis (91, 92). Data also suggests that because oncolytic viruses result in highly pro-inflammatory and immunogenic events (tumor cell death and the release of tumor-specific antigens) (93–95) they can elicit a tumor-specific immune response (96). Additionally, viruses encode products that can be recognized by immune and non-immune cells as Pathogen-associated molecular patterns (PAMPs) and can also cause the release of Damage-associated molecular pattern molecules (DAMPs) (97). PAMPs are structural motifs which serve as “danger” signals to the host indicating the presence of virus that trigger host defenses. These danger signals can be structural proteins and glycolipids but are mainly nucleic acids including double-stranded RNA (dsRNA), viral single-stranded RNA, and CpG DNA (98, 99). DAMPs are host nuclear or cytosolic proteins with defined intracellular function that activate effector cells from the innate immune system when they are released outside the cell (100). Virus-induced changes such as an increase in pro-inflammatory cytokines and chemokines, a decrease in immunosuppressive cytokines, and the release of PAMPs and DAMPs at the site of the tumor may diminish or reverse the established immunosuppressive microenvironment and initiate antitumor immunity.
Several oncolytic virus classes are currently in late-stage clinical trials (Table 3). The most advance of these, Talimogene laherparepvec (T-VEC, formerly OncoVex or JS1/ICP34.5-/ICP47-/GM-CSF; an HSV isolate selected for its potency over laboratory strains, it is deleted in both the ICP34.5 and ICP47 genes to further increase viral replication and tumor cell killing, it also expresses human GM-CSF for immune stimulation) has demonstrated some very promising clinical data. From recently announced results of a phase III trial in unresectable stage IIIB-IV melanoma receiving either T-VEC injected into the lesion or GM-CSF administered subcutaneously, the overall durable response rate (DRR) was 16.3% for T-VEC treated patients as compared to 2.1% for GM-CSF treated individuals (101). The objective overall response rate (ORR) was 26.4% for the T-VEC group (including 10.8% complete responders) compared to an ORR of 5.7% and a complete response rate of 0.7% in the GM-CSF alone group (101). Importantly, in a phase II trial, tumor shrinkage was noted in non-injected lesions, demonstrating that systemic immunity was induced (102). In addition, and across a number of viruses, studies have shown that both innate and adaptive immune responses are generated following viral tumor lysis (92, 103–111). This antitumor immunity is an important outcome of oncolytic viral therapy as it would lead to the destruction of tumor cells that escaped the initial viral lysis.
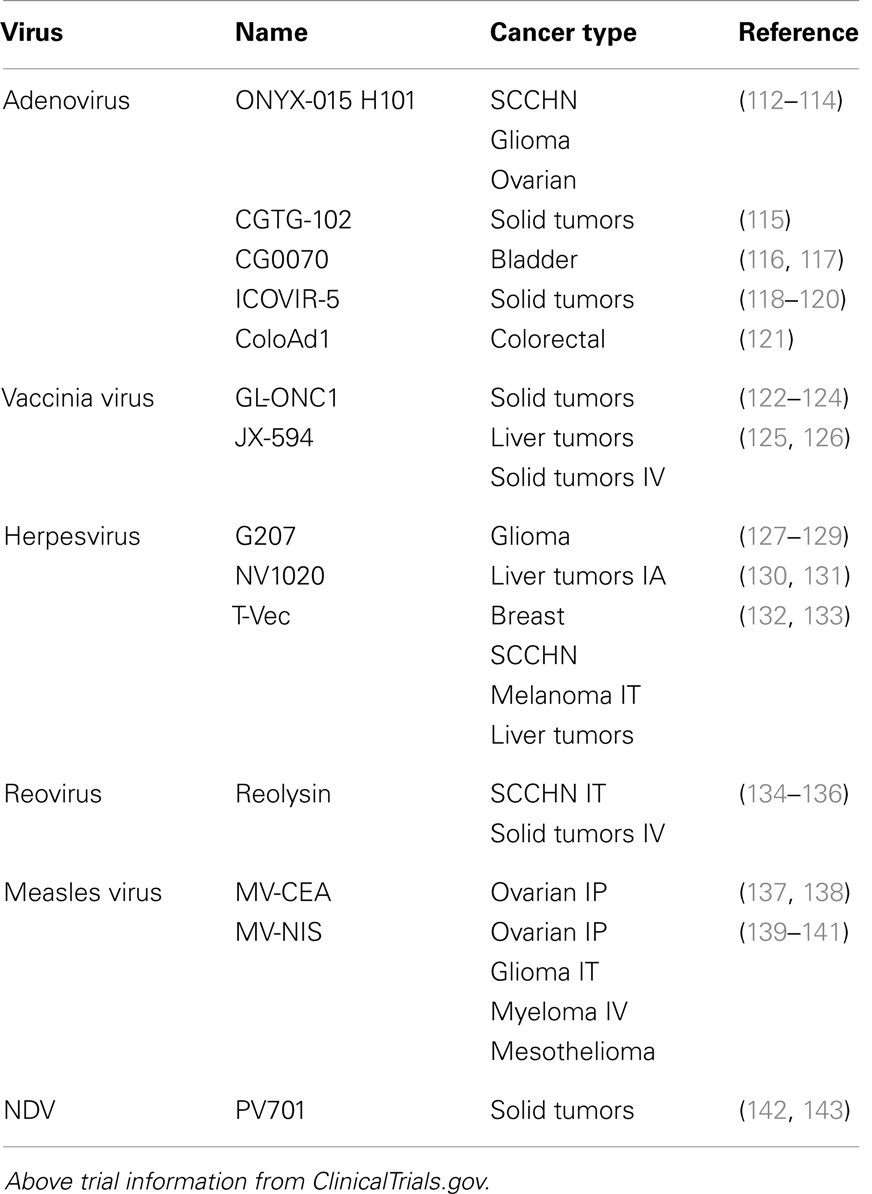
Table 3. The most advanced clinically evaluated oncolytic viruses.
Merging Oncolytic Viruses and Immune-Checkpoint Blocking
The realization that oncolytic viral therapy can itself be an immunotherapy has in many ways reinvigorated the field and expanded the possible approaches that can be taken to treat cancer. Similarly, the discovery and targeting of immune checkpoints has opened a new immuotherapeutic avenue generating very promising clinical results. The potential to combine oncolytic viruses with a blockade of immune checkpoints is a very exciting strategy that may be beneficial on many levels and help overcome current shortcomings associated with either approach alone. To date, there have been only a few preclinical studies combining oncolytic viruses and immune-checkpoint blockers (anti-CTLA-4 mAb) (144, 145). However, results have been promising with one study showing that replication competent VSV in combination with anti-CTLA-4 mAb resulted in the elimination of macroscopic tumor implants in the majority of test animals, an outcome that could not be achieved by either treatment alone (145). The study went on to show that the response was CD4 and CD8 T-cell mediated (145). When combining these two approaches, the exact virus/checkpoint combination will likely need to be determined empirically with many factors including indication and immune status of patient playing a role. However, in general an argument can be made that the greatest synergies between these strategies would be realized by delivering the immune-checkpoint therapy directly from the oncolytic virus (Table 4).

Table 4. The benefits of using an oncolytic virus to deliver immune-checkpoint blockers.
Increased Priming and Greater Immune Potency
Preclinical studies have shown that in mice bearing partially immunogenic tumors, treatment with CTLA-4 antibodies could elicit significant antitumor responses whereas poorly immunogenic tumors were refractory to anti-CTLA-4 administration (32, 146). However, these refractory tumors could be made more responsive by administering granulocyte-macrophage colony-stimulating factor (GM-CSF) in combination with the anti-CTLA-4 (146). These findings suggested that a CTLA-4 blockade enhances an already existing endogenous antitumor response resulting in tumor regression. But when the tumor is poorly immunogenic and does not induce a robust enough immune response the anti-immune checkpoint is not as efficacious. Similar results have been found in the clinic where analysis of pre-treatment tumors indicated that patients with high baseline expression levels of immune-related genes were more likely to respond favorably to ipilimumab (147). Just as the GM-CSF is used to help boost the initial innate immune response, oncolytic viruses could have a similar effect as it is clear that the oncolytic viral infection has pro-inflammatory properties, eliciting both an innate and adaptive immune response.
Enhanced Safety and Efficacy by Expressing Immune-Checkpoint Blockers from the Oncolytic Virus
The oncolytic virus and the immune-checkpoint blocker could be administered as two separate therapeutics but one of the most appealing aspects of the oncolytic viral approach is that it is localized to the tumor. This localization confers several advantages for both safety and potency. Clinical and preclinical data strongly suggest that a blockade of immune checkpoints is a very potent antitumor therapy. However, there are, in some cases, unwanted side effects. Given the importance of the immune checkpoints in maintaining immune homeostasis there is concern that a blockade of these receptors and/or ligands could lead to a break in immune self-tolerance resulting in autoimmune/autoinflammatory side effects (148). Blocking CTLA-4 as a therapy was initially questioned given its crucial role in the regulation of T-cell amplification. The phenotype of Ctla4-knockout mice also hinted at the possibility of a high number of unwanted immune-related effects. In the pivotal phase III trial of ipilimumab, Grade 3 or Grade 4 immune-related adverse events (including rash, colitis, hepatitis, and endocrinopathies) occurred in 10–15% of patients treated with the anti-CTLA-4 antibody as compared to 3% of those treated with gp100 alone. During this trial, there were 14 deaths related to ipilimumab (2.1%), 7 of which were due to immune-related adverse events (17). Delivering the immune-checkpoint blocker (Ab, Ab derivative or modified ligand or receptor) from the oncolytic virus would localize the treatment and mitigate the risks inherent in systemic delivery. In preclinical studies of a replication competent adenovirus armed with the coding region of a full length CTLA-4 antibody a 43-fold higher antibody concentration in the tumor as compared to the plasma was noted (144). Moreover, plasma levels in treated mice remained below the reported human safety threshold (144).
It is also possible to make expression of these immune-checkpoint blockers contingent upon a productive viral infection (i.e., selective replication that is restricted to the tumor cell) further increasing the safety of the therapeutic. This can be done by utilizing endogenous late viral promoters that are dependent upon the uptake and replication within the target tumor cell to express exogenous genes and has been described for human adenovirus (12, 13, 149). In the normal cell, this expression would be blocked as replication would not be achieved consequently confining expression to target cancer cells. Potency, like safety also benefits from this localized delivery, concentrating the therapeutic to the tumor and its microenvironment. Accumulation of virally delivered transgenes (including reporter genes, prodrug converting enzyme, anti-angiogenic factors, immunostimulatory factors) at the site of the infected tumor has been shown in numerous studies (97, 115, 132, 150–153). For example, PET imaging experiments have dramatically demonstrated the tumor localized expression of thymide kinase following infection with an oncolytic virus armed with the enzyme (154, 155). This accumulation was translated into efficacy upon administration of the prodrug Ganciclovir (154). Additionally, the self-perpetuating nature of an oncolytic infection results in sustained transgene expression (156) that will continue until tumor regression is complete and the virus is eliminated from the tumor site by the immune system (157). Therefore the amount of material produced would be directly related, in theory, to the tumor load, personalizing the respective dose to the individual and their tumor burden. It is also appealing to consider that this may eliminate peaks and valleys associated with the intravenous administration of the therapeutic as the virus expressed molecule would be generated on a more constant basis that might also benefit the patient.
Enablement of Alternative Therapeutics
Although viruses can be used to deliver an intact IgG, their focused delivery to the tumor site and their self-perpetuating nature allow for the use of alternative antibody formats such as diabodies, Fabs, and scFvs (144, 158). This could have a profound impact on any mAb-based antitumor therapeutic particulary immune-checkpoint blockers. From a safety standpoint, the use of these alternative Ab formats could be beneficial because IgGs, due to their size (150 kDa), have prolonged serum half-lives (>10 days) and are therefore more likely to have associated toxicities. If these alternative formats were to escape the tumor site their faster clearance reduces the risk for off-target events. For immune-checkpoint blockers, this could help to decrease the immune-related adverse events that have been associated with this therapeutic approach (148, 159). Additionally, smaller formats would potentially penetrate the tumor to a greater extent than a full length antibody. Studies have shown that an intact IgG molecule takes 54 h to move 1 mm into a solid tumor, whereas a Fab fragment travels the same distance in only 16 h (160). This enhanced penetration could increase overall efficacy. The diabodies in particular have been shown to provide rapid tissue penetration, high target retention, and rapid blood clearance presumably as a result of their multi-valent nature and intermediate size (55 kDa) (161). The use of alternative antibody formats also opens up the possibility of delivering multiple therapies from one oncolytic virus. This may have broad implications for the blockade of immune-checkpoint approach as studies are beginning to show that targeting multiple checkpoints may be more efficacious (67–71, 74). Without localized delivery, the use of these alternative formats would likely not be feasible as they would clear too rapidly (on the order of a few hours or minutes dependent upon the format) (162). This may necessitate the need for higher input doses or multiple injections of the Ab, which could potentially be cost prohibitive. Having localized delivery via the virus would avoid the need for full length Abs and make the smaller, faster-clearing formats viable therapies that are still capable of efficacious outcomes.
Economically Advantageous
Expression of immune-checkpoint blockers from an Oncolytic virus is economically appealing. If one assumes that the initial promising results seen with combination checkpoint blockers are maintained in larger phase II and III trials, the delivery of a combination of blockers from a virus would eliminate the need to commercially manufacture the molecules separately. This approach utilizes a single entity (the virus) to exploit the natural machinery of the virus and the tumor cell to continuously produce the therapeutic agents so long as the tumor cells continue to exist. Moreover, it has been demonstrated that multiple exogenous proteins can be delivered from a single virus (149). Due to their tumor selective localization, as mentioned previously, they would not need to express a full length antibody, making this approach potentially attractive and novel for delivering multiple-checkpoint inhibitors to the site. In addition, this therapy would have the potential added benefit of increased immunogenicity and/or direct tumor cell lysis offered by the oncolytic virus. Thus expressing a single biological agent with the ability to deliver multiple-checkpoint inhibitors that itself has anti-cancer activity is an interesting possibility. However, it should be kept in mind that the commercial manufacture of oncolytic viruses is behind that of antibodies and thus may be only a true economic advantage in the future with additional optimization.
Conclusion
In the fight against cancer, no single magic bullet has emerged. Despite several improvements in diagnostics and therapies nearly 7 million cancer-related deaths still occur every year worldwide (163). One reason is that cancer is complex and can evolve to thrive under harsh conditions and to evade the body’s natural defenses. Two promising therapeutic strategies have emerged; the blockade of immune checkpoints and oncolytic viruses and we believe that an argument can be made that the greatest potential for both of these therapies lies in the synergies that would be realized by delivering the immune-checkpoint therapy directly from the oncolytic virus (Table 5). We look forward to the continued evolution of these agents and to the exciting years ahead as we begin to see these agents come forward pre-clinically and clinically.
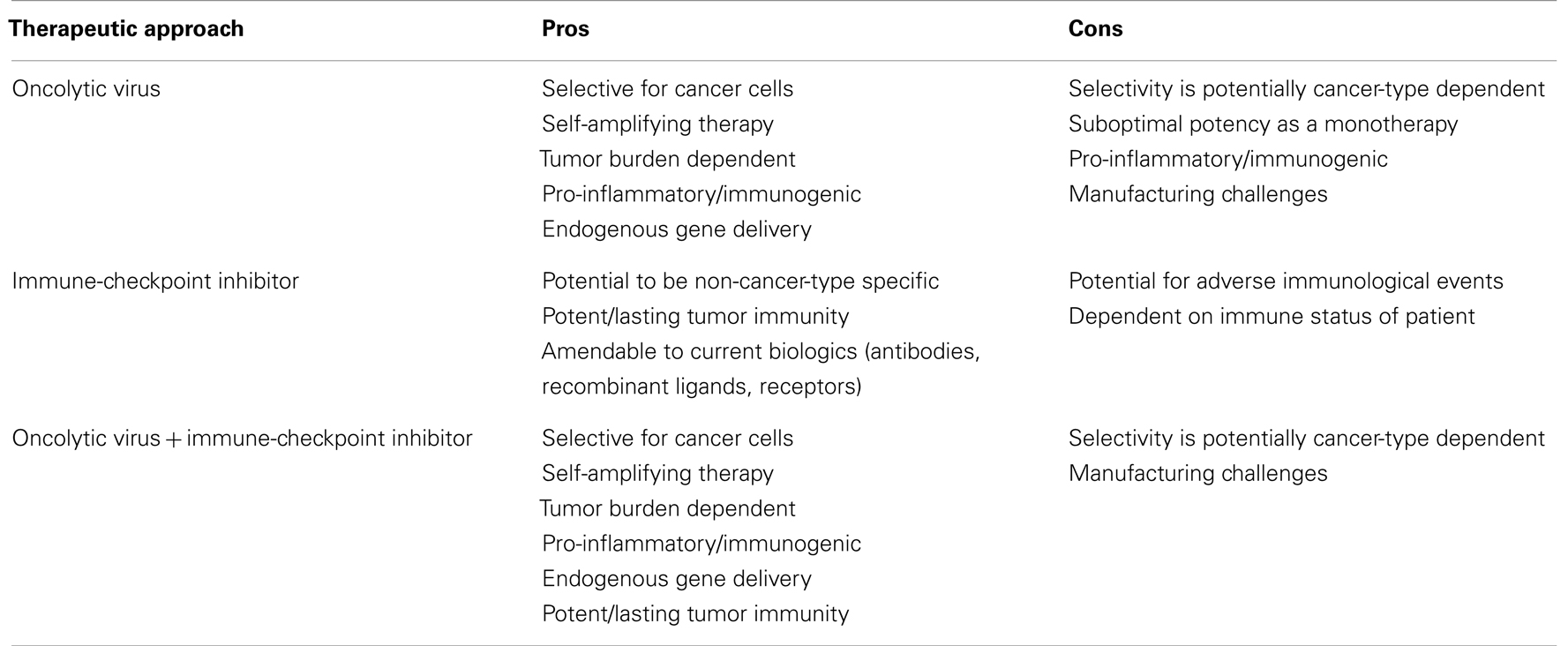
Table 5. The pros and cons of oncolytic viral, immune-checkpoint inhibition and combination therapy.
Conflict of Interest Statement
Dr. Terry Hermiston serves on the following boards: Scientific Advisory Board for Psioxus Therapeutics and Board of Directors for Jennerex Biotherapeutics. The other co-author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol (2012) 30(7):679–92. doi: 10.1038/nbt.2284
2. Parish CR. Cancer immunotherapy: the past, the present and the future. Immunol Cell Biol (2003) 81(2):106–13. doi:10.1046/j.0818-9641.2003.01151.x
3. Waldmann TA. Immunotherapy: past, present and future. Nat Med (2003) 9(3):269–77. doi:10.1038/nm0303-269
4. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer (2006) 6(1):24–37. doi:10.1038/nrc1782
6. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol (2011) 29:235–71. doi:10.1146/annurev-immunol-031210-101324
7. Davis ID, Jefford M, Parente P, Cebon J. Rational approaches to human cancer immunotherapy. J Leukoc Biol (2003) 73(1):3–29. doi:10.1189/jlb.0502261
8. Pardoll DM. Spinning molecular immunology into successful immunotherapy. Nat Rev Immunol (2002) 2(4):227–38. doi:10.1038/nri774
9. Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. J Clin Oncol (2011) 29(36):4828–36. doi:10.1200/JCO.2011.38.0899
10. Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther (2007) 15(4):651–9. doi:10.1038/sj.mt.6300108
11. Moore AE. The destructive effect of the virus of Russian Far East encephalitis on the transplantable mouse sarcoma 180. Cancer (1949) 2(3):525–34. doi:10.1002/1097-0142(194905)2:3<525::AID-CNCR2820020317>3.0.CO;2-O
12. Hermiston T. Gene delivery from replication-selective viruses: arming guided missiles in the war against cancer. J Clin Invest (2000) 105(9):1169–72. doi:10.1172/JCI9973
13. Hermiston T. Fighting fire with fire: attacking the complexity of human tumors with armed therapeutic viruses. Curr Opin Mol Ther (2002) 4(4):334–42.
14. Jeyaretna DS, Kuroda T. Recent advances in the development of oncolytic HSV-1 vectors: ‘arming’ of HSV-1 vectors and application of bacterial artificial chromosome technology for their construction. Curr Opin Mol Ther (2007) 9(5):447–66.
15. Patel MR, Kratzke RA. Oncolytic virus therapy for cancer: the first wave of translational clinical trials. Transl Res (2013) 161(4):355–64. doi:10.1016/j.trsl.2012.12.010
16. Rowan K. Oncolytic viruses move forward in clinical trials. J Natl Cancer Inst (2010) 102(9):590–5. doi:10.1093/jnci/djq165
17. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
18. Acharya UH, Jeter JM. Use of ipilimumab in the treatment of melanoma. Clin Pharmacol (2013) 5(Suppl 1):21–7.
19. McDermott D, Haanen J, Chen TT, Lorigan P, O’Day S. Efficacy and safety of ipilimumab in metastatic melanoma patients surviving more than 2 years following treatment in a phase III trial (MDX010-20). Ann Oncol (2013) 24(10):2694–8. doi:10.1093/annonc/mdt291
20. Ribas A. Clinical development of the anti-CTLA-4 antibody tremelimumab. Semin Oncol (2010) 37(5):450–4. doi:10.1053/j.seminoncol.2010.09.010
21. Calabro L, Morra A, Fonsatti E, Cutaia O, Amato G, Giannarelli D, et al. Tremelimumab for patients with chemotherapy-resistant advanced malignant mesothelioma: an open-label, single-arm, phase 2 trial. Lancet Oncol (2013) 14(11):1104–11. doi:10.1016/S1470-2045(13)70381-4
22. Tarhini AA. Tremelimumab: a review of development to date in solid tumors. Immunotherapy (2013) 5(3):215–29. doi:10.2217/imt.13.9
23. McDermott DF, Atkins MB. PD-1 as a potential target in cancer therapy. Cancer Med (2013) 2(5):662–73.
24. Robert C, Soria JC, Eggermont AM. Drug of the year: programmed death-1 receptor/programmed death-1 ligand-1 receptor monoclonal antibodies. Eur J Cancer (2013) 49(14):2968–71. doi:10.1016/j.ejca.2013.07.001
25. Weber JS, Kudchadkar RR, Yu B, Gallenstein D, Horak CE, Inzunza HD, et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol (2013) 31(34):4311–8. doi:10.1200/JCO.2013.51.4802
26. Benson DM Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood (2010) 116(13):2286–94. doi:10.1182/blood-2010-02-271874
27. Rosenblatt J, Glotzbecker B, Mills H, Vasir B, Tzachanis D, Levine JD, et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J Immunother (2011) 34(5):409–18. doi:10.1097/CJI.0b013e31821ca6ce
28. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med (2013) 369(2):134–44. doi:10.1056/NEJMoa1305133
29. Tang PA, Heng DY. Programmed death 1 pathway inhibition in metastatic renal cell cancer and prostate cancer. Curr Oncol Rep (2013) 15(2):98–104. doi:10.1007/s11912-012-0284-2
30. Forde PM, Reiss KA, Zeidan AM, Brahmer JR. What lies within: novel strategies in immunotherapy for non-small cell lung cancer. Oncologist (2013) 18(11):1203–13. doi:10.1634/theoncologist.2013-0171
31. Loo D, Alderson RF, Chen FZ, Huang L, Zhang W, Gorlatov S, et al. Development of an Fc-enhanced anti-B7-H3 monoclonal antibody with potent antitumor activity. Clin Cancer Res (2012) 18(14):3834–45. doi:10.1158/1078-0432.CCR-12-0715
32. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1996) 271(5256):1734–6. doi:10.1126/science.271.5256.1734
33. Azuma M, Ito D, Yagita H, Okumura K, Phillips JH, Lanier LL, et al. B70 antigen is a second ligand for CTLA-4 and CD28. Nature (1993) 366(6450):76–9. doi:10.1038/366076a0
34. Freeman GJ, Gribben JG, Boussiotis VA, Ng JW, Restivo VA Jr, Lombard LA, et al. Cloning of B7-2: a CTLA-4 counter-receptor that costimulates human T cell proliferation. Science (1993) 262(5135):909–11. doi:10.1126/science.7694363
35. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med (1991) 174(3):561–9. doi:10.1084/jem.174.3.561
36. Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity (1994) 1(9):793–801. doi:10.1016/S1074-7613(94)80021-9
37. Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, et al. Reversal of the TCR stop signal by CTLA-4. Science (2006) 313(5795):1972–5. doi:10.1126/science.1131078
38. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3(5):541–7. doi:10.1016/1074-7613(95)90125-6
39. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science (1995) 270(5238):985–8. doi:10.1126/science.270.5238.985
40. Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci U S A (2003) 100(8):4712–7. doi:10.1073/pnas.0830997100
41. Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A (2003) 100(14):8372–7. doi:10.1073/pnas.1533209100
42. Ribas A, Camacho LH, Lopez-Berestein G, Pavlov D, Bulanhagui CA, Millham R, et al. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J Clin Oncol (2005) 23(35):8968–77. doi:10.1200/JCO.2005.01.109
43. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi:10.1038/nrc3239
44. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192(7):1027–34. doi:10.1084/jem.192.7.1027
45. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi:10.1146/annurev.immunol.26.021607.090331
46. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25(21):9543–53. doi:10.1128/MCB.25.21.9543-9553.2005
47. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8(8):793–800. doi:10.1038/nm0902-1039c
48. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med (2007) 13(1):84–8. doi:10.1038/nm1517
49. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11(2):141–51. doi:10.1016/S1074-7613(00)80089-8
50. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science (2001) 291(5502):319–22. doi:10.1126/science.291.5502.319
51. Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, et al. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A (2004) 101(29):10691–6. doi:10.1073/pnas.0307252101
52. Zhang Y, Chung Y, Bishop C, Daugherty B, Chute H, Holst P, et al. Regulation of T cell activation and tolerance by PDL2. Proc Natl Acad Sci U S A (2006) 103(31):11695–700. doi:10.1073/pnas.0601347103
53. Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol (2009) 10(11):1185–92. doi:10.1038/ni.1790
54. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206(13):3015–29. doi:10.1084/jem.20090847
55. Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood (2009) 114(8):1545–52. doi:10.1182/blood-2009-03-206672
56. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
57. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi:10.1056/NEJMoa1200694
58. Goldberg MV, Drake CG. LAG-3 in cancer immunotherapy. Curr Top Microbiol Immunol (2011) 344:269–78. doi:10.1007/82_2010_114
59. He C, Qiao H, Jiang H, Sun X. The inhibitory role of b7-h4 in antitumor immunity: association with cancer progression and survival. Clin Dev Immunol (2011) 2011:695834. doi:10.1155/2011/695834
60. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21(4):503–13. doi:10.1016/j.immuni.2004.08.010
61. Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res (2011) 71(10):3540–51. doi:10.1158/0008-5472.CAN-11-0096
62. Waickman AT, Alme A, Senaldi L, Zarek PE, Horton M, Powell JD. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol Immunother (2012) 61(6):917–26. doi:10.1007/s00262-011-1155-7
63. Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol (2003) 4(7):670–9. doi:10.1038/ni944
64. Yi KH, Chen L. Fine tuning the immune response through B7-H3 and B7-H4. Immunol Rev (2009) 229(1):145–51. doi:10.1111/j.1600-065X.2009.00768.x
65. Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, et al. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood (2008) 111(1):251–9. doi:10.1182/blood-2007-03-081646
66. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol (2005) 6(12):1245–52. doi:10.1038/ni1271
67. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med (2010) 207(10):2175–86. doi:10.1084/jem.20100637
68. Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, et al. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res (2012) 72(4):887–96. doi:10.1158/0008-5472.CAN-11-2637
69. Grosso JF, Goldberg MV, Getnet D, Bruno TC, Yen HR, Pyle KJ, et al. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J Immunol (2009) 182(11):6659–69. doi:10.4049/jimmunol.0804211
70. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207(10):2187–94. doi:10.1084/jem.20100643
71. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res (2012) 72(4):917–27. doi:10.1158/0008-5472.CAN-11-1620
72. Baitsch L, Legat A, Barba L, Fuertes Marraco SA, Rivals JP, Baumgaertner P, et al. Extended co-expression of inhibitory receptors by human CD8 T-cells depending on differentiation, antigen-specificity and anatomical localization. PLoS One (2012) 7(2):e30852. doi:10.1371/journal.pone.0030852
73. Nirschl CJ, Drake CG. Molecular pathways: co-expression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res (2013) 19:4917–24. doi:10.1158/1078-0432.CCR-12-1972
74. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369(2):122–33. doi:10.1056/NEJMoa1302369
75. Grandi P, Fernandez J, Szentirmai O, Carter R, Gianni D, Sena-Esteves M, et al. Targeting HSV-1 virions for specific binding to epidermal growth factor receptor-vIII-bearing tumor cells. Cancer Gene Ther (2010) 17(9):655–63. doi:10.1038/cgt.2010.22
76. Kelly K, Nawrocki S, Mita A, Coffey M, Giles FJ, Mita M. Reovirus-based therapy for cancer. Expert Opin Biol Ther (2009) 9(7):817–30. doi:10.1517/14712590903002039
77. Kinoh H, Inoue M, Komaru A, Ueda Y, Hasegawa M, Yonemitsu Y. Generation of optimized and urokinase-targeted oncolytic Sendai virus vectors applicable for various human malignancies. Gene Ther (2009) 16(3):392–403. doi:10.1038/gt.2008.167
78. Kolodkin-Gal D, Zamir G, Edden Y, Pikarsky E, Pikarsky A, Haim H, et al. Herpes simplex virus type 1 preferentially targets human colon carcinoma: role of extracellular matrix. J Virol (2008) 82(2):999–1010. doi:10.1128/JVI.01769-07
79. Mansour M, Palese P, Zamarin D. Oncolytic specificity of Newcastle disease virus is mediated by selectivity for apoptosis-resistant cells. J Virol (2011) 85(12):6015–23. doi:10.1128/JVI.01537-10
80. Menotti L, Nicoletti G, Gatta V, Croci S, Landuzzi L, De Giovanni C, et al. Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells. Proc Natl Acad Sci U S A (2009) 106(22):9039–44. doi:10.1073/pnas.0812268106
81. Stanford MM, McFadden G. Myxoma virus and oncolytic virotherapy: a new biologic weapon in the war against cancer. Expert Opin Biol Ther (2007) 7(9):1415–25. doi:10.1517/14712598.7.9.1415
82. Chen L, Chen D, Gong M, Na M, Li L, Wu H, et al. Concomitant use of Ad5/35 chimeric oncolytic adenovirus with TRAIL gene and taxol produces synergistic cytotoxicity in gastric cancer cells. Cancer Lett (2009) 284(2):141–8. doi:10.1016/j.canlet.2009.04.026
83. Ma J, He X, Wang W, Huang Y, Chen L, Cong W, et al. E2F promoter-regulated oncolytic adenovirus with p16 gene induces cell apoptosis and exerts antitumor effect on gastric cancer. Dig Dis Sci (2009) 54(7):1425–31. doi:10.1007/s10620-008-0543-0
84. Shen W, Wang CY, Wang XH, Fu ZX. Oncolytic adenovirus mediated Survivin knockdown by RNA interference suppresses human colorectal carcinoma growth in vitro and in vivo. J Exp Clin Cancer Res (2009) 28:81. doi:10.1186/1756-9966-28-81
85. van Beusechem VW, van den Doel PB, Grill J, Pinedo HM, Gerritsen WR. Conditionally replicative adenovirus expressing p53 exhibits enhanced oncolytic potency. Cancer Res (2002) 62(21):6165–71.
86. Zhao L, Dong A, Gu J, Liu Z, Zhang Y, Zhang W, et al. The antitumor activity of TRAIL and IL-24 with replicating oncolytic adenovirus in colorectal cancer. Cancer Gene Ther (2006) 13(11):1011–22. doi:10.1038/sj.cgt.7700969
87. Zheng JN, Pei DS, Sun FH, Zhang BF, Liu XY, Gu JF, et al. Inhibition of renal cancer cell growth by oncolytic adenovirus armed short hairpin RNA targeting hTERT gene. Cancer Biol Ther (2009) 8(1):84–91. doi:10.4161/cbt.8.1.7204
88. Dingli D, Bergert ER, Bajzer Z, O’Connor MK, Russell SJ, Morris JC. Dynamic iodide trapping by tumor cells expressing the thyroidal sodium iodide symporter. Biochem Biophys Res Commun (2004) 325(1):157–66. doi:10.1016/j.bbrc.2004.09.219
89. Dingli D, Peng KW, Harvey ME, Greipp PR, O’Connor MK, Cattaneo R, et al. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood (2004) 103(5):1641–6. doi:10.1182/blood-2003-07-2233
90. Zheng FQ, Xu Y, Yang RJ, Wu B, Tan XH, Qin YD, et al. Combination effect of oncolytic adenovirus therapy and herpes simplex virus thymidine kinase/ganciclovir in hepatic carcinoma animal models. Acta Pharmacol Sin (2009) 30(5):617–27. doi:10.1038/aps.2009.33
91. Breitbach CJ, Arulanandam R, De Silva N, Thorne SH, Patt R, Daneshmand M, et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res (2013) 73(4):1265–75. doi:10.1158/0008-5472.CAN-12-2687
92. Liu TC, Hwang T, Park BH, Bell J, Kirn DH. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther (2008) 16(9):1637–42. doi:10.1038/mt.2008.143
93. Benencia F, Courreges MC, Conejo-Garcia JR, Mohamed-Hadley A, Zhang L, Buckanovich RJ, et al. HSV oncolytic therapy upregulates interferon-inducible chemokines and recruits immune effector cells in ovarian cancer. Mol Ther (2005) 12(5):789–802. doi:10.1016/j.ymthe.2005.03.026
94. Endo Y, Sakai R, Ouchi M, Onimatsu H, Hioki M, Kagawa S, et al. Virus-mediated oncolysis induces danger signal and stimulates cytotoxic T-lymphocyte activity via proteasome activator upregulation. Oncogene (2008) 27(17):2375–81. doi:10.1038/sj.onc.1210884
95. Steele L, Errington F, Prestwich R, Ilett E, Harrington K, Pandha H, et al. Pro-inflammatory cytokine/chemokine production by reovirus treated melanoma cells is PKR/NF-kappaB mediated and supports innate and adaptive anti-tumour immune priming. Mol Cancer (2011) 10:20. doi:10.1186/1476-4598-10-20
96. Tong AW, Senzer N, Cerullo V, Templeton NS, Hemminki A, Nemunaitis J. Oncolytic viruses for induction of anti-tumor immunity. Curr Pharm Biotechnol (2012) 13(9):1750–60. doi:10.2174/138920112800958913
97. Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer (2009) 9(1):64–71. doi:10.1038/nrc2545
98. Prestwich RJ, Errington F, Diaz RM, Pandha HS, Harrington KJ, Melcher AA, et al. The case of oncolytic viruses versus the immune system: waiting on the judgment of Solomon. Hum Gene Ther (2009) 20(10):1119–32. doi:10.1089/hum.2009.135
99. Thompson AJ, Locarnini SA. Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunol Cell Biol (2007) 85(6):435–45. doi:10.1038/sj.icb.7100100
100. Rubartelli A, Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol (2007) 28(10):429–36. doi:10.1016/j.it.2007.08.004
101. Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y, et al. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer (2013) 12(1):103. doi:10.1186/1476-4598-12-103
102. Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G, et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol (2009) 27(34):5763–71. doi:10.1200/JCO.2009.24.3675
103. Bhat R, Dempe S, Dinsart C, Rommelaere J. Enhancement of NK cell antitumor responses using an oncolytic parvovirus. Int J Cancer (2011) 128(4):908–19. doi:10.1002/ijc.25415
104. Errington F, Steele L, Prestwich R, Harrington KJ, Pandha HS, Vidal L, et al. Reovirus activates human dendritic cells to promote innate antitumor immunity. J Immunol (2008) 180(9):6018–26.
105. Errington F, White CL, Twigger KR, Rose A, Scott K, Steele L, et al. Inflammatory tumour cell killing by oncolytic reovirus for the treatment of melanoma. Gene Ther (2008) 15(18):1257–70. doi:10.1038/gt.2008.58
106. Li H, Dutuor A, Fu X, Zhang X. Induction of strong antitumor immunity by an HSV-2-based oncolytic virus in a murine mammary tumor model. J Gene Med (2007) 9(3):161–9. doi:10.1002/jgm.1005
107. Nakamori M, Fu X, Rousseau R, Chen SY, Zhang X. Destruction of nonimmunogenic mammary tumor cells by a fusogenic oncolytic herpes simplex virus induces potent antitumor immunity. Mol Ther (2004) 9(5):658–65. doi:10.1016/j.ymthe.2004.02.019
108. Prestwich RJ, Errington F, Ilett EJ, Morgan RS, Scott KJ, Kottke T, et al. Tumor infection by oncolytic reovirus primes adaptive antitumor immunity. Clin Cancer Res (2008) 14(22):7358–66. doi:10.1158/1078-0432.CCR-08-0831
109. Prestwich RJ, Harrington KJ, Vile RG, Melcher AA. Immunotherapeutic potential of oncolytic virotherapy. Lancet Oncol (2008) 9(7):610–2. doi:10.1016/S1470-2045(08)70163-3
110. Prestwich RJ, Ilett EJ, Steele L, Errington F. The importance of the immune system in reovirus therapy. Clin Oncol (R Coll Radiol) (2008) 20(10):769. doi:10.1016/j.clon.2008.08.004
111. Sobol PT, Boudreau JE, Stephenson K, Wan Y, Lichty BD, Mossman KL. Adaptive antiviral immunity is a determinant of the therapeutic success of oncolytic virotherapy. Mol Ther (2011) 19(2):335–44. doi:10.1038/mt.2010.264
112. Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science (1996) 274(5286):373–6. doi:10.1126/science.274.5286.373
113. Cassel WA, Murray DR, Phillips HS. A phase II study on the postsurgical management of stage II malignant melanoma with a Newcastle disease virus oncolysate. Cancer (1983) 52(5):856–60. doi:10.1002/1097-0142(19830901)52:5<856::AID-CNCR2820520519>3.0.CO;2-4
114. Wallack MK, Sivanandham M, Balch CM, Urist MM, Bland KI, Murray D, et al. Surgical adjuvant active specific immunotherapy for patients with stage III melanoma: the final analysis of data from a phase III, randomized, double-blind, multicenter vaccinia melanoma oncolysate trial. J Am Coll Surg (1998) 187(1):69–77. discussion 9, doi:10.1016/S1072-7515(98)00097-0
115. Kanerva A, Nokisalmi P, Diaconu I, Koski A, Cerullo V, Liikanen I, et al. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin Cancer Res (2013) 19(10):2734–44. doi:10.1158/1078-0432.CCR-12-2546
116. Burke JM, Lamm DL, Meng MV, Nemunaitis JJ, Stephenson JJ, Arseneau JC, et al. A first in human phase 1 study of CG0070, a GM-CSF expressing oncolytic adenovirus, for the treatment of nonmuscle invasive bladder cancer. J Urol (2012) 188(6):2391–7. doi:10.1016/j.juro.2012.07.097
117. Ramesh N, Ge Y, Ennist DL, Zhu M, Mina M, Ganesh S, et al. CG0070, a conditionally replicating granulocyte macrophage colony-stimulating factor – armed oncolytic adenovirus for the treatment of bladder cancer. Clin Cancer Res (2006) 12(1):305–13. doi:10.1158/1078-0432.CCR-05-1059
118. Alonso MM, Cascallo M, Gomez-Manzano C, Jiang H, Bekele BN, Perez-Gimenez A, et al. ICOVIR-5 shows E2F1 addiction and potent antiglioma effect in vivo. Cancer Res (2007) 67(17):8255–63. doi:10.1158/0008-5472.CAN-06-4675
119. Alonso MM, Gomez-Manzano C, Jiang H, Bekele NB, Piao Y, Yung WK, et al. Combination of the oncolytic adenovirus ICOVIR-5 with chemotherapy provides enhanced anti-glioma effect in vivo. Cancer Gene Ther (2007) 14(8):756–61. doi:10.1038/sj.cgt.7701067
120. Cascallo M, Alonso MM, Rojas JJ, Perez-Gimenez A, Fueyo J, Alemany R. Systemic toxicity-efficacy profile of ICOVIR-5, a potent and selective oncolytic adenovirus based on the pRB pathway. Mol Ther (2007) 15(9):1607–15. doi:10.1038/sj.mt.6300239
121. Kuhn I, Harden P, Bauzon M, Chartier C, Nye J, Thorne S, et al. Directed evolution generates a novel oncolytic adenovirus for the treatment of colon cancer. PLoS One (2008) 3(6):e2409. doi:10.1371/journal.pone.0002409
122. Ascierto ML, Worschech A, Yu Z, Adams S, Reinboth J, Chen NG, et al. Permissivity of the NCI-60 cancer cell lines to oncolytic Vaccinia Virus GLV-1h68. BMC Cancer (2011) 11:451. doi:10.1186/1471-2407-11-451
123. Chen NG, Yu YA, Zhang Q, Szalay AA. Replication efficiency of oncolytic vaccinia virus in cell cultures prognosticates the virulence and antitumor efficacy in mice. J Transl Med (2011) 9:164. doi:10.1186/1479-5876-9-164
124. Ehrig K, Kilinc MO, Chen NG, Stritzker J, Buckel L, Zhang Q, et al. Growth inhibition of different human colorectal cancer xenografts after a single intravenous injection of oncolytic vaccinia virus GLV-1h68. J Transl Med (2013) 11:79. doi:10.1186/1479-5876-11-79
125. Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med (2013) 19(3):329–36. doi:10.1038/nm.3089
126. Kim JH, Oh JY, Park BH, Lee DE, Kim JS, Park HE, et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol Ther (2006) 14(3):361–70. doi:10.1016/j.ymthe.2006.05.008
127. Aghi MK, Chiocca EA. Phase Ib trial of oncolytic herpes virus G207 shows safety of multiple injections and documents viral replication. Mol Ther (2009) 17(1):8–9. doi:10.1038/mt.2008.275
128. Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther (2000) 7(10):867–74. doi:10.1038/sj.gt.3301205
129. Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med (1995) 1(9):938–43. doi:10.1038/nm0995-938
130. Geevarghese SK, Geller DA, de Haan HA, Horer M, Knoll AE, Mescheder A, et al. Phase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liver. Hum Gene Ther (2010) 21(9):1119–28. doi:10.1089/hum.2010.020
131. Kelly KJ, Wong J, Fong Y. Herpes simplex virus NV1020 as a novel and promising therapy for hepatic malignancy. Expert Opin Investig Drugs (2008) 17(7):1105–13. doi:10.1517/13543784.17.7.1105
132. Kaufman HL, Bines SD. OPTIM trial: a Phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future Oncol (2010) 6(6):941–9. doi:10.2217/fon.10.66
133. Sivendran S, Pan M, Kaufman HL, Saenger Y. Herpes simplex virus oncolytic vaccine therapy in melanoma. Expert Opin Biol Ther (2010) 10(7):1145–53. doi:10.1517/14712598.2010.495383
134. Galanis E, Markovic SN, Suman VJ, Nuovo GJ, Vile RG, Kottke TJ, et al. Phase II trial of intravenous administration of Reolysin(®) (Reovirus Serotype-3-dearing Strain) in patients with metastatic melanoma. Mol Ther (2012) 20(10):1998–2003. doi:10.1038/mt.2012.146
135. Morris DG, Feng X, DiFrancesco LM, Fonseca K, Forsyth PA, Paterson AH, et al. REO-001: a phase I trial of percutaneous intralesional administration of reovirus type 3 dearing (Reolysin®) in patients with advanced solid tumors. Invest New Drugs (2013) 31(3):696–706. doi:10.1007/s10637-012-9865-z
136. Stoeckel J, Hay JG. Drug evaluation: reolysin – wild-type reovirus as a cancer therapeutic. Curr Opin Mol Ther (2006) 8(3):249–60.
137. Galanis E, Hartmann LC, Cliby WA, Long HJ, Peethambaram PP, Barrette BA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res (2010) 70(3):875–82. doi:10.1158/0008-5472.CAN-09-2762
138. Msaouel P, Dispenzieri A, Galanis E. Clinical testing of engineered oncolytic measles virus strains in the treatment of cancer: an overview. Curr Opin Mol Ther (2009) 11(1):43–53.
139. Allen C, Opyrchal M, Aderca I, Schroeder MA, Sarkaria JN, Domingo E, et al. Oncolytic measles virus strains have significant antitumor activity against glioma stem cells. Gene Ther (2013) 20(4):444–9. doi:10.1038/gt.2012.62
140. Li H, Peng KW, Russell SJ. Oncolytic measles virus encoding thyroidal sodium iodide symporter for squamous cell cancer of the head and neck radiovirotherapy. Hum Gene Ther (2012) 23(3):295–301. doi:10.1089/hum.2011.128
141. Touchefeu Y, Khan AA, Borst G, Zaidi SH, McLaughlin M, Roulstone V, et al. Optimising measles virus-guided radiovirotherapy with external beam radiotherapy and specific checkpoint kinase 1 inhibition. Radiother Oncol (2013) 108(1):24–31. doi:10.1016/j.radonc.2013.05.036
142. Hotte SJ, Lorence RM, Hirte HW, Polawski SR, Bamat MK, O’Neil JD, et al. An optimized clinical regimen for the oncolytic virus PV701. Clin Cancer Res (2007) 13(3):977–85. doi:10.1158/1078-0432.CCR-06-1817
143. Lorence RM, Roberts MS, O’Neil JD, Groene WS, Miller JA, Mueller SN, et al. Phase 1 clinical experience using intravenous administration of PV701, an oncolytic Newcastle disease virus. Curr Cancer Drug Targets (2007) 7(2):157–67. doi:10.2174/156800907780058853
144. Dias JD, Hemminki O, Diaconu I, Hirvinen M, Bonetti A, Guse K, et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther (2012) 19(10):988–98. doi:10.1038/gt.2011.176
145. Gao Y, Whitaker-Dowling P, Griffin JA, Barmada MA, Bergman I. Recombinant vesicular stomatitis virus targeted to Her2/neu combined with anti-CTLA4 antibody eliminates implanted mammary tumors. Cancer Gene Ther (2009) 16(1):44–52. doi:10.1038/cgt.2008.55
146. van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med (1999) 190(3):355–66.
147. Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother (2012) 61(7):1019–31. doi:10.1007/s00262-011-1172-6
148. Corsello SM, Barnabei A, Marchetti P, De Vecchis L, Salvatori R, Torino F. Endocrine side effects induced by immune checkpoint inhibitors. J Clin Endocrinol Metab (2013) 98(4):1361–75. doi:10.1210/jc.2012-4075
149. Bauzon M, Castro D, Karr M, Hawkins LK, Hermiston TW. Multigene expression from a replicating adenovirus using native viral promoters. Mol Ther (2003) 7(4):526–34. doi:10.1016/S1525-0016(03)00023-6
150. Lampe J, Bossow S, Weiland T, Smirnow I, Lehmann R, Neubert W, et al. An armed oncolytic measles vaccine virus eliminates human hepatoma cells independently of apoptosis. Gene Ther (2013) 20:1033–41. doi:10.1038/gt.2013.28
151. Lee JH, Roh MS, Lee YK, Kim MK, Han JY, Park BH, et al. Oncolytic and immunostimulatory efficacy of a targeted oncolytic poxvirus expressing human GM-CSF following intravenous administration in a rabbit tumor model. Cancer Gene Ther (2010) 17(2):73–9. doi:10.1038/cgt.2009.50
152. Todo T. “Armed” oncolytic herpes simplex viruses for brain tumor therapy. Cell Adh Migr (2008) 2(3):208–13. doi:10.4161/cam.2.3.6353
153. Tysome JR, Lemoine NR, Wang Y. Update on oncolytic viral therapy – targeting angiogenesis. Onco Targets Ther (2013) 6:1031–40. doi:10.2147/OTT.S46974
154. Abate-Daga D, Andreu N, Camacho-Sanchez J, Alemany R, Herance R, Millan O, et al. Oncolytic adenoviruses armed with thymidine kinase can be traced by PET imaging and show potent antitumoural effects by ganciclovir dosing. PLoS One (2011) 6(10):e26142. doi:10.1371/journal.pone.0026142
155. Jacobs A, Tjuvajev JG, Dubrovin M, Akhurst T, Balatoni J, Beattie B, et al. Positron emission tomography-based imaging of transgene expression mediated by replication-conditional, oncolytic herpes simplex virus type 1 mutant vectors in vivo. Cancer Res (2001) 61(7):2983–95.
156. Ichikawa T, Chiocca EA. Comparative analyses of transgene delivery and expression in tumors inoculated with a replication-conditional or -defective viral vector. Cancer Res (2001) 61(14):5336–9.
157. Zhang Q, Yu YA, Wang E, Chen N, Danner RL, Munson PJ, et al. Eradication of solid human breast tumors in nude mice with an intravenously injected light-emitting oncolytic vaccinia virus. Cancer Res (2007) 67(20):10038–46. doi:10.1158/0008-5472.CAN-07-0146
158. Frentzen A, Yu YA, Chen N, Zhang Q, Weibel S, Raab V, et al. Anti-VEGF single-chain antibody GLAF-1 encoded by oncolytic vaccinia virus significantly enhances antitumor therapy. Proc Natl Acad Sci U S A (2009) 106(31):12915–20. doi:10.1073/pnas.0900660106
159. Beck KE, Blansfield JA, Tran KQ, Feldman AL, Hughes MS, Royal RE, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol (2006) 24(15):2283–9. doi:10.1200/JCO.2005.04.5716
160. Jain RK, Baxter LT. Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: significance of elevated interstitial pressure. Cancer Res (1988) 48(24 Pt 1):7022–32.
161. Robinson MK, Doss M, Shaller C, Narayanan D, Marks JD, Adler LP, et al. Quantitative immuno-positron emission tomography imaging of HER2-positive tumor xenografts with an iodine-124 labeled anti-HER2 diabody. Cancer Res (2005) 65(4):1471–8. doi:10.1158/0008-5472.CAN-04-2008
162. Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol (2005) 23(9):1126–36. doi:10.1038/nbt1142
Keywords: oncolytic virus, cancer immunotherapy, immune-checkpoint inhibitors, CTLA-4, PD1, PDL1, PDL2, blockade of checkpoint inhibitors
Citation: Bauzon M and Hermiston T (2014) Armed therapeutic viruses – a disruptive therapy on the horizon of cancer immunotherapy. Front. Immunol. 5:74. doi: 10.3389/fimmu.2014.00074
Received: 13 December 2013; Paper pending published: 25 January 2014;
Accepted: 11 February 2014; Published online: 24 February 2014.
Edited by:
Volker Schirrmacher, German Cancer Research Center, GermanyCopyright: © 2014 Bauzon and Hermiston. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Terry Hermiston, Bayer HealthCare, US Innovation Center, Biologics Research, 455 Mission Bay Blvd. South, San Francisco, CA 94158, USA e-mail:dGVycnkuaGVybWlzdG9uQGJheWVyLmNvbQ==