 Ole Audun Werner Haabeth1†
Ole Audun Werner Haabeth1† Anders Aune Tveita1†
Anders Aune Tveita1† Marte Fauskanger1
Marte Fauskanger1 Fredrik Schjesvold1
Fredrik Schjesvold1 Kristina Berg Lorvik1
Kristina Berg Lorvik1 Peter O. Hofgaard2
Peter O. Hofgaard2 Hilde Omholt1Ludvig A. Munthe1
Hilde Omholt1Ludvig A. Munthe1 Zlatko Dembic3
Zlatko Dembic3 Alexandre Corthay1,4,5
Alexandre Corthay1,4,5 Bjarne Bogen1,2*
Bjarne Bogen1,2*- 1Department of Immunology, Centre for Immune Regulation, Oslo University Hospital, University of Oslo, Oslo, Norway
- 2KG Jebsen Centre for Research on Influenza Vaccines, Institute of Immunology, Oslo University Hospital, University of Oslo, Oslo, Norway
- 3Faculty of Dentistry, Molecular Genetics Laboratory, Department of Oral Biology, University of Oslo, Oslo, Norway
- 4Department of Biosciences, University of Oslo, Oslo, Norway
- 5Tumor Immunology Group, Department of Pathology, Oslo University Hospital, University of Oslo, Oslo, Norway
CD4+ T cells contribute to tumor eradication, even in the absence of CD8+ T cells. Cytotoxic CD4+ T cells can directly kill MHC class II positive tumor cells. More surprisingly, CD4+ T cells can indirectly eliminate tumor cells that lack MHC class II expression. Here, we review the mechanisms of direct and indirect CD4+ T cell-mediated elimination of tumor cells. An emphasis is put on T cell receptor (TCR) transgenic models, where anti-tumor responses of naïve CD4+ T cells of defined specificity can be tracked. Some generalizations can tentatively be made. For both MHCIIPOS and MHCIINEG tumors, presentation of tumor-specific antigen by host antigen-presenting cells (APCs) appears to be required for CD4+ T cell priming. This has been extensively studied in a myeloma model (MOPC315), where host APCs in tumor-draining lymph nodes are primed with secreted tumor antigen. Upon antigen recognition, naïve CD4+ T cells differentiate into Th1 cells and migrate to the tumor. At the tumor site, the mechanisms for elimination of MHCIIPOS and MHCIINEG tumor cells differ. In a TCR-transgenic B16 melanoma model, MHCIIPOS melanoma cells are directly killed by cytotoxic CD4+ T cells in a perforin/granzyme B-dependent manner. By contrast, MHCIINEG myeloma cells are killed by IFN-γ stimulated M1-like macrophages. In summary, while the priming phase of CD4+ T cells appears similar for MHCIIPOS and MHCIINEG tumors, the killing mechanisms are different. Unresolved issues and directions for future research are addressed.
Recent Advances in Tumor Immunology
The field of tumor immunology has come a long way since the formulation of the tumor immunosurveillance hypothesis by Thomas and Burnet (1–4). Although still debated, increasing evidence suggests that the immune system can detect and reject incipient tumors, and that CD4+ and CD8+ T cells play an important role as mediators of immunosurveillance (5). Furthermore, there is accumulating evidence that the immune system is not completely tolerant even to established tumors, based on the observation that tumor-infiltrating T cells, when expanded in vitro and injected back to lymphopenic patients, have a clinical effect in some patients (6). Further supporting the notion of ongoing immune responses to tumors, antibodies that block inhibitory molecules on T cells induce long-term remission in a subset of cancer patients (7). Finally, parameters that indicate immune activation in tumors are associated with improved prognosis (8).
CD4+ versus CD8+ T Cells in Tumor Immunology
Traditionally, CD8+ T cells have been thought to be the major mediators of effective anti-tumor T cell responses. Such a view is supported by the pronounced cytotoxic activity of CD8+ T cells in vitro, and the observation that tumors that escape CD8+ T cells onslaught may have altered or downregulated MHC class I antigen expression (9–11). Moreover, studies done in an MHC class I-restricted T cell receptor (TCR) transgenic mouse showed that CD8+ T cells, in the absence of CD4+ T cells, maintained their anti-tumor effect (12). Despite these observations, several studies indicate limited anti-tumor effects of CD8+ T cells alone (6, 13–16).
The helper function of tumor-reactive CD4+ T cells improves the efficacy of tumor-reactive CD8+ T cells (17–20). Similarly, treatment of a patient with metastatic melanoma with autologous CD4+ T cells specific for the tumor-associated antigen NY-ESO-1 resulted in sustained clinical remissions with evidence of endogenous immune responses against other tumor-derived antigens (21). In support of these findings, transfection of tumor cells with MHC class II genes resulted in increased protective immune responses against tumors (22, 23). Collectively, these results indicate an augmenting effect of CD4+ T cells on CD8+ T cell responses against tumors.
On the other hand, CD4+ T cells alone, in the absence of CD8+ T cells, have also been demonstrated to eliminate tumor cells. Thus, adoptive transfer experiments using primed CD4+ T cells generated by immunization with tumor cells conferred protection against a subsequent tumor challenge (24, 25). Moreover, naïve CD4+ T cells in TCR-transgenic mice conferred protection against tumor development upon subcutaneous (s.c.) injection of tumor cells (26, 27). Finally, using MHC class I-molecule and MHC class II-molecule restricted TCR-transgenic mice specific for the Dby H–Y antigen, CD4+ T cells were found to be more efficient at eradicating cancer cells than CD8+ T cells in a side-by-side comparison (28). Here, we will focus on the anti-tumor properties of CD4+ T cells in the absence of CD8+ T cells.
Pioneering Experiments on the Role of CD4+ T Cells in Eradication of Tumors
The role of CD4+ T cells was initially investigated in experiments where tumor-bearing mice were treated by adoptive transfer of T cells obtained from syngeneic mice immunized with irradiated tumor cells (25, 29), or with living tumor cells followed by surgical resection (24). It was shown that when T cells from tumor-immunized donors were purified prior to adoptive transfer, Lyt1+ 2− (CD4+) T cells had a superior ability to cure FBL-3 erythroleukemic tumors compared to Lyt1− 2+ (CD8+) T cells (29). Treatment with cyclophosphamide was required for the curative effect of CD4+ T cells to be observed. However, in the first reported experiments (29), a role of endogenous CD8+ T cells in the tumor-bearing host was not ruled out. In follow-up experiments, this possibility was formally excluded by the use of T cell deficient tumor-bearing recipients (25). Similar results were obtained using the X5563 plasmacytoma model (24), where transfer of purified Lyt1+ 2− (CD4+) T cells had superior therapeutic potential. In the following decades, experimental evidence supporting the anti-tumor properties of tumor-specific CD4+ T cells alone has accumulated (27, 28, 30–39).
TCR-Transgenic Models for CD4+ T Cell-Mediated Rejection of Tumors
The experiments referred to in the preceding section had features that prohibited detailed studies of the mechanisms of CD4+ T cell-mediated tumor protection. First, the CD4+ T cells were polyclonal. Second, CD4+ T cells were pre-primed cells obtained after immunization, making it impossible to study naïve CD4+ T cells in primary anti-tumor responses. Third, the relevant tumor-specific antigens were often not known.
The generation of TCR-transgenic mice that recognize tumor antigens presented on MHC class II molecules (Table 1) offered a novel approach to bypass these difficulties. In two models, these antigens are bona fide cancer antigens; the tumor-specific myeloma protein V region idiotype (Id) (26, 27) and the melanoma-associated tyrosinase-related protein 1 (Trp1) (35). In other TCR-transgenic models, the antigens are either minor histocompatibility antigen Dby (H-Y) (28), viral antigens such as the hemagglutinin (HA) (40–42), or xenogeneic proteins such as ovalbumin (OVA) (17, 43, 44). While the transgenic TCR specific for the mutated myeloma antigen was obtained after immunization of mice syngeneic to the tumor (45, 46), the transgenic TCR specific for the non-mutated antigen was obtained after immunization of Trp1-deficient mice. Thus, in the latter model, Trp1 represents a foreign antigen to which high-affinity TCRs are induced (due to a lack of T cell tolerance) (35).
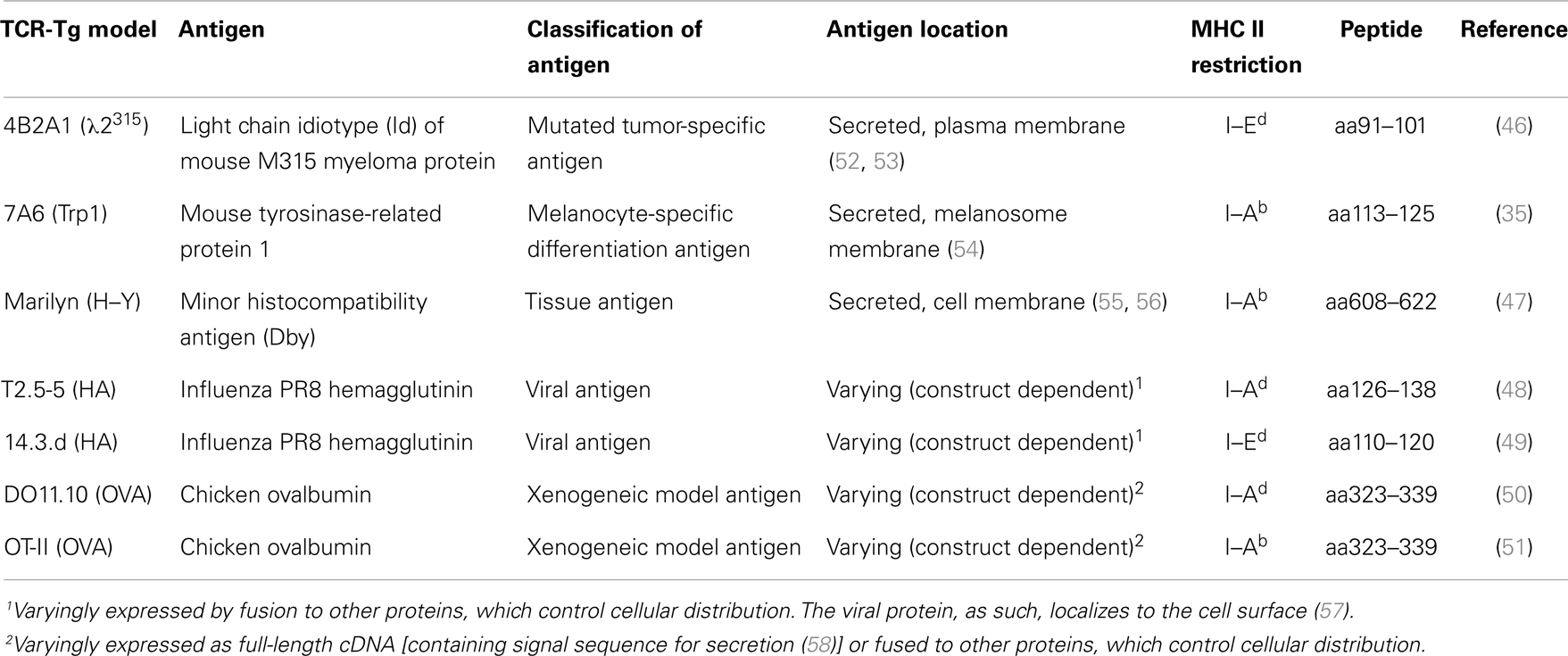
Table 1. TCR-transgenic models employed in studies of anti-tumor CD4+ T cell responses.
MHC Class II Status of Tumor Cells Used in Tumor Immunology Studies Focused on the Role of CD4+ T Cells
CD4+ T cells recognize peptides (about 13–17aa long) bound to the groove of MHC class II molecules (59) on professional antigen-presenting cells (APCs) (B cells, dendritic cells, macrophages, in addition to thymic epithelial cells) (60–62). However, in certain cells, MHC class II molecules may be induced by interferon gamma (IFN-γ) stimulation (63, 64). Thus, in CD4+ T cell immune responses to tumors, the MHC class II status of the tumor cells is of importance. The MHC II expression status of tumor cells used in studies with CD4+ TCR-transgenic mice is summarized in Table 2.
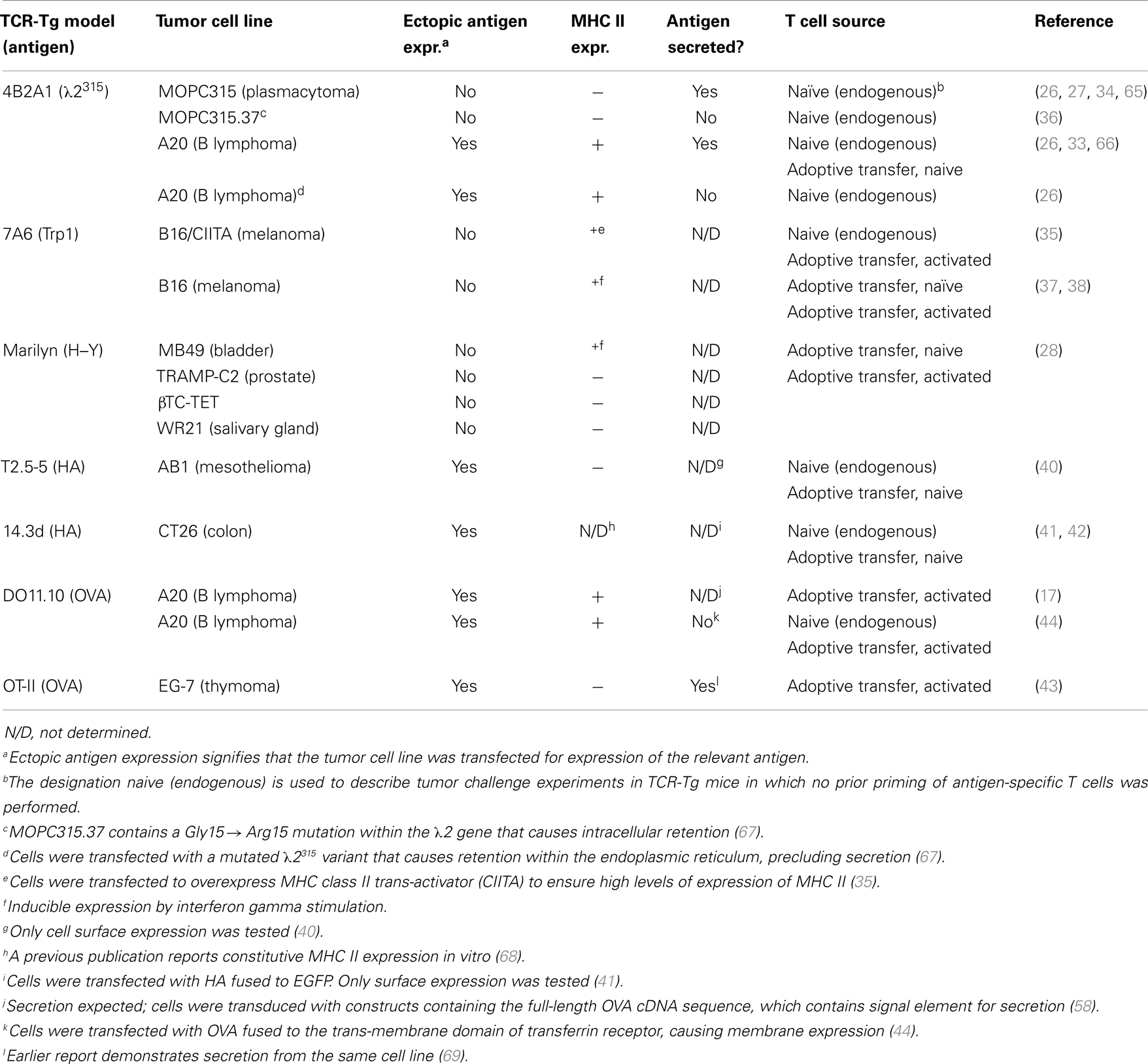
Table 2. Use of TCR-Tg models for studies of anti-tumor CD4+ T cell immune responses.
Direct and Indirect Killing of Tumor Cells by CD4+ T Cells
The antigen-specific interaction between CD4+ T cells and MHC IIPOS tumor cells is conceptually easy to grasp. On the other hand, the basis for antigen presentation and anti-tumor effector mechanisms are less obvious in the context of MHC IINEG tumors (25, 26, 31, 70) – simply because such cancer cells cannot directly stimulate MHC class II-restricted CD4+ T cells (Figure 1). In the following sections, we discuss mechanism of CD4+ T cell-mediated direct killing of MHC IIPOS tumor cells and indirect killing of MHC IINEG tumor cells. Emphasis is put on observations from TCR-transgenic models, where the T cell specificity is known and both naïve and primed CD4+ T cells are readily available.
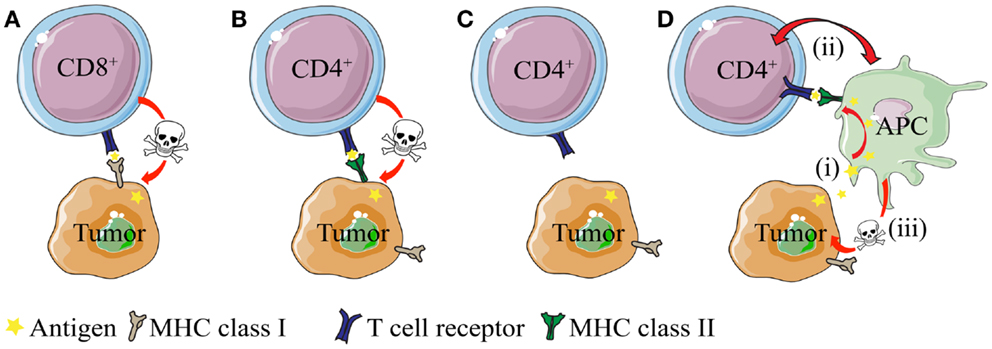
Figure 1. Direct and indirect killing of tumor cells by CD4+ T cells. (A) CD8+ T cells can directly kill tumor cells that express MHC class I molecules, whereas (B) cytotoxic CD4+ T cells can kill tumor cells that express MHC class II molecules. (C) While most tumor types express MHC class I molecules, they often lack expression of MHC class II. How do CD4+ T cells recognize and eliminate MHCIINEG tumor cells? (D) CD4+ T cells may kill MHC class II negative (MHC IINEG) tumors by a mechanism where (i) tumor antigen secreted by tumor cells is processed and presented by MHCIIPOS macrophages to CD4+ T cells. (ii) Bi-directional interaction/activation of macrophages and CD4+ T cells (iii) activates tumoricidal macrophages that in turn kill the tumor cells (In addition, activated CD4+ T cells themselves could possibly directly kill tumor cell in a TCR/MHC II-independent manner.).
Direct Killing of MHC Class IIPOS Tumor Cells
The existence of CD4+ T cells with cytotoxic properties has been increasingly recognized throughout the last three decades. Such cells are thought to function in a fashion analogous to cytotoxic CD8+ T cells, with antigen recognition triggering the release of cytotoxic mediators. CD4+ T cells displaying direct cytotoxicity in vitro toward MHC IIPOS targets, including tumor cells, have been described by several authors (37, 45, 70, 71). Correspondingly, efficient elimination of MHC IIPOS tumors by T cells with such properties is also observed in vivo (26, 28, 33, 35, 37, 38, 72).
Several effector mechanisms have been implicated for tumor-specific cytotoxic CD4+ T cells. In a model of Id-specific CD4+ T cell responses against an MHC IIPOS B lymphoma, in vitro cytotoxicity was shown to be dependent on signaling mediated by binding of Fas ligand (FasL) on CD4+ T cells to the death receptor Fas on tumor cells (66). Naïve T cells showed little killing activity, whereas Th1 differentiation greatly enhanced cytotoxicity. However, in vivo elimination of tumor cells was not affected in FasL-deficient (gld−/−) Id-specific TCR-Tg mice, suggesting that signaling through the Fas pathway is dispensable for tumor killing and that additional mechanisms are operational in vivo (66). Indeed, if the tumor antigen is secreted as is the case in the studies of Lundin et al. (33, 66), the indirect mechanism via Th1/M1 macrophages described below could also be active, and might play a prominent role in tumor rejection. In the Trp1-specific TCR-transgenic model, it was demonstrated that the rejection of B16 melanoma cells was abrogated in mice deficient for either granzyme B or perforin, indicating that these molecules are important for CD4+ T cell-mediated killing of MHC IIPOS tumor cells (37). In summary, different MHC IIPOS tumors may vary in susceptibility to various effector mechanisms of CD4+ T cells, as indicated by the observations addressed above.
Indirect Killing of MHC Class IINEG Tumor Cells
In general, antibody-secreting plasma cells are MHC class II negative due to silencing of the MHC Class II trans-activator (CIITA) occurring during plasma cell differentiation (73, 74). Multiple myeloma (MM) is the malignant counterpart of plasma cells and usually express little if any MHC class II molecules. MHC class II negativity due to loss of CIITA expression appears to be a stable phenotype, although some studies have reported MHC II upregulation in MM cells exposed to retinoic acid (75) or IFN-γ (76, 77).
The work of our research group is based on experiments using the mineral oil-induced BALB/c plasmacytoma (MOPC)315 (52, 70). MOPC315 cells secrete a highly mutated and unique monoclonal IgA (myeloma protein). The λ2 light chain of the myeloma protein contains somatic mutations in positions 38, 50, 94, 95, and 96 that are unique to MOPC315 (78). Thus, the myeloma protein light chain is referred to as λ2315 (Figure 2A).
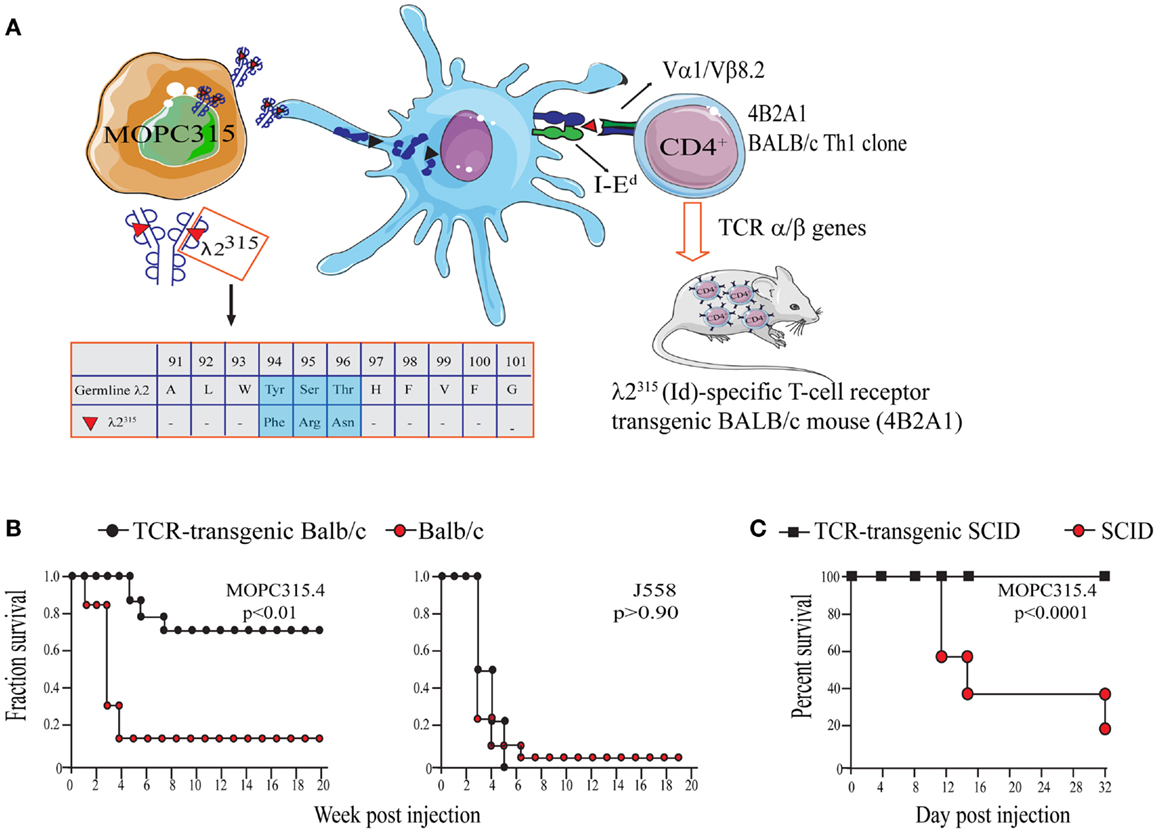
Figure 2. The MOPC315 myeloma model. Naïve tumor-specific CD4+ T cells protect against MHC IINEG tumor challenge in the absence of other T cells and B cells. (A) MOPC315 myeloma cells of BALB/c origin secrete an IgA M315 myeloma protein with a mutated λ2 light chain referred to as λ2315. M315 is endocytosed and processed by BALB/c APCs, and a CDR3 sequence that includes residues 91–101 of λ2315 is presented on the MHC class II molecule I-Ed to Id-specific CD4+ T cells. The peptide that is recognized by Id-specific CD4+ T cells contains somatic mutations in positions 94, 95, and 96 (45, 79, 80). Based on the αβ TCR of the Id-recognizing 4B2A1 clone, a TCR-transgenic mouse was generated (46). Most CD4+ T cells in this mouse express a transgenic TCR that can be tracked by a clonotype-specific mAb [Nomenclature: antigenic determinants in immunoglobulin variable (V) regions are called idiotopes (Id). The 91–101 peptide is thus an Id-peptide, and the CD4+ T cells that recognize this Id-peptide presented by I-Ed are called Id-specific]. (B) Id-specific TCR-transgenic mice on an immunosufficient background (BALB/c) are resistant to a challenge with IdPOS MOPC315 cells but succumb to IdNEG J558 myeloma cells [reproduced with permission from Proc Natl Acad Sci (26), Copyright 1994 National Academy of Sciences, U.S.A.]. (C). Id-specific TCR-transgenic mice on an immunodeficient background (SCID), lacking other T and B cells than Id-specific CD4+ T cells, are also resistant to MOPC315 tumor development [reproduced with permission from Immunity (34)]. Tumor resistance could be transferred with purified Id-specific CD4+ T cells to SCID mice (27).
By immunization of BALB/c mice with free λ2315 L chain, known from previous studies to stimulate T cells (81), I-Ed-restricted, Id-specific CD4+ T cell clones were generated (Figure 2A) (45). These clones recognize a unique Id-epitope, which depends on the somatic mutations in codons 94, 95, and 96 within the CDR3 loop of the λ2315 light chain (79). As would be expected, MOPC315 derived λ2315-immunoglobulin has to be endocytosed and processed by APCs prior to MHC class II presentation of the Id-peptide (80).
MOPC315 is found to be MHC class II negative by a number of criteria: (i) Negative staining with anti-MHC class II antibodies both in vitro, ex vivo (70), and in vivo (65). Lack of expression of MHC II molecules on MOPC315 was independently reported by others (82). (ii) Exposure to high amounts (500 ng/ml) of IFN-γ IL-4, or supernatant from activated Th1 cells, all failed to induce any detectable expression of MHC class II in vitro (70). (iii) Both in vitro-cultured (70) and ex vivo (65) MOPC315 cells failed to stimulate Id-specific MHC class II-restricted T cells in proliferation and cytokine secretion assays.
Idiotype-Specific CD4+ T Cell Clones Induce Killing of MHC Class II Negative Myeloma Cells in vitro – but Only in the Presence of MHC-Compatible APCs
A weak cytotoxicity that was greatly augmented by addition of high amounts of myeloma protein was observed when Id-specific CD4+ T cells were co-cultured with MHC-compatible spleen cells from BALB/c (H-2d) MHC IINEG MOPC315. Importantly, MHC II incompatible spleen cells from C57BL/6 failed to support cytotoxicity (70). Moreover, the cytotoxic effect could not be transferred by supernatants of activated T cells. It was suggested that some of the spleen cells, e.g., macrophages (Mϕ) stimulated by activated T cells, were important as cytotoxic effector cells in the in vitro cultures (70).
Naïve Id-Specific CD4+ T Cells in T Cell Receptor Transgenic Mice Protect Against Id+ Myeloma Cells in the Absence of CD8+ T Cells and B Cells
To facilitate studies of the role of Id-specific CD4+ T cells in tumor protection against MHC II negative MOPC315, an Id-specific TCR-transgenic mouse on syngeneic BALB/c background was established (46).
In initial experiments, naïve Id-specific T cells from TCR-transgenic mice did not respond to MOPC315 in vitro. Despite this, Id-specific TCR-transgenic mice were specifically protected against s.c. challenge with MOPC315 cells (26) (Figure 2B). Eradication of MOPC315 cells resulted in a change of T cell phenotype, since T cells of surviving TCR-transgenic mice had increased cytotoxicity against Id+ MHC IIPOS B lymphomas, and since they upon stimulation produced much IFNγ and some IL-4.
By breeding the TCR-Tg mice onto a SCID background, it was demonstrated that rejection of MOPC315 was independent of CD8+ T cells and B cells/antibodies (27, 34) (Figure 2C). Additionally, tumor protection could be transferred to SCID mice with adoptive transfer of purified Id-specific CD4+ T cells (27).
Id-Primed APC Can be Detected in Tumor Tissue of Large Established Myelomas
The finding that naïve CD4+ T cells could initiate rejection of a MHC II negative tumor indicated that host cells expressing MHC class II molecules were responsible for the presentation of Id to CD4+ T cells. In a subsequent study, it was demonstrated that s.c. MOPC315 tumors contained APCs that were able to stimulate Id-specific CD4+ T cells in vitro in an MHC-restricted manner (65). The great majority of MHC IIPOS tumor-infiltrating APCs were CD11b+CD11cLOWCD80+CD86+. These studies demonstrated that MHC class II negative MOPC315 tumors were infiltrated with Id-primed APCs with macrophage-like characteristics.
Id-Specific CD4+ T Cells are Present and Activated in Tumor Tissue
Given that Id-primed APC could be demonstrated in MOPC315 tumors, it was investigated if Id-specific CD4+ T cells were also present, and whether they were activated. In these experiments, a high amount of MOPC315 cells were injected in order to overcome the resistance of TCR-transgenic mice. A number of observations indicated that Id-specific CD4+ T cells were specifically activated in small s.c. MOPC315 tumors established in Id-specific TCR-transgenic mice: (i) The CD4+/CD8+ ratio was skewed toward CD4+ in tumor tissue. (ii) CD4+ blasts within the tumor were selectively enriched for cells expressing the Id-specific TCR. (iii) Id-specific CD4+ tumor-infiltrating lymphocytes (TIL) were activated (CD69+ CD25+), and proliferated (BrdU+) in clusters associated with MHC IIPOS tumor-infiltrating APC (65).
Secretion of Tumor-Specific Antigen is Required for CD4+ T Cell-Mediated Rejection of MHC IINEG Tumors
While it was clear that tumor-infiltrating APCs and lymph node cells take up the λ2315 antigen and display the Id-peptide on MHC class II molecules (34, 65), the precise source of the priming Id antigen was not established. To address this question, we used two secretory variants of MOPC315: one that secretes the complete M315 myeloma protein composed of α H chain and λ2315 L chain (MOPC315), and another that only secrets the free λ2315 L chain (MOPC315.26). In addition, we used two non-secretory variants: one where the free λ2315 L chain is retained intracellularly due to a point mutation (MOPC315.37) and another where no Ig is produced (MOPC315.36) (67, 83).
When Id-specific TCR-transgenic SCID mice were challenged with the four variants, protection was observed for the λ2315-secreting variants MOPC315 and MOPC315.26, while there was no protection against the antigen-negative MOPC315.36. Tumor take was significantly delayed, but still complete, in mice challenged with the MOPC315.37, which retains λ2315 intracellularly. This result was surprising since in MOPC315.37-containing Matrigels, macrophages were MHC IIHI, and Id-specific T cells were activated (CD69+). The only striking deficiency observed with MOPC315.37 in vivo was deficient T cell activation in draining lymph nodes, presumably due to poor local availability of the intracellularly retained tumor antigen. These results indicate that the extracellular concentration of secreted tumor-specific antigen is important for protection against an MHC IINEG tumor, most likely due to enhanced priming of APCs in draining lymph nodes as well as macrophages in tumors (36, 84).
Detection of Tumor-Specific CD4+ T Cells and Macrophages in Early Stages after Tumor Cell Challenge: The Matrigel Method
To study local events at the injection site at the early stages of the anti-tumor immune response, we injected the tumor cells suspended in a Matrigel solution (Figures 3 and 4). Matrigel is a liquid basement membrane preparation that jellifies rapidly at body temperature. Thus, a tumor bed of a defined size was generated that could be isolated and assayed to characterize infiltrating cells at any time point following tumor cell injection (Figure 3). Moreover, the defined volume of the gel plug allows quantitative assays of secreted factors within the tumor microenvironment (39). Initial experiments demonstrated that tumor cells embedded in Matrigel were rejected by TCR-transgenic SCID mice, although less efficiently than in the absence of Matrigel (34). Thus, events in the tumor cell-containing Matrigel most likely reflected those taking place during successful immunosurveillance of MHC II negative tumor cells by CD4+ T cells.
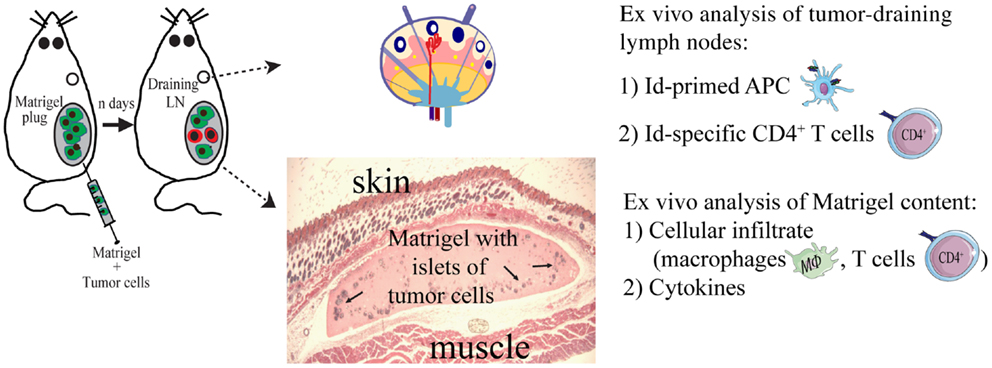
Figure 3. The Matrigel assay. A novel approach to unravel the dynamics of CD4+ T cell-mediated primary anti-tumor immune responses. At day 0, subcutaneous injections with MOPC315 tumor cells suspended in liquid Matrigel. When the Matrigel solution reaches body temperature, it jellifies and forms a plug containing the tumor cells. At various time points after injection (n days), the Matrigel plug and tumor-draining lymph nodes are dissected out and analyzed ex vivo for cellular content, function of cells, and cytokines (34, 36, 39, 85).
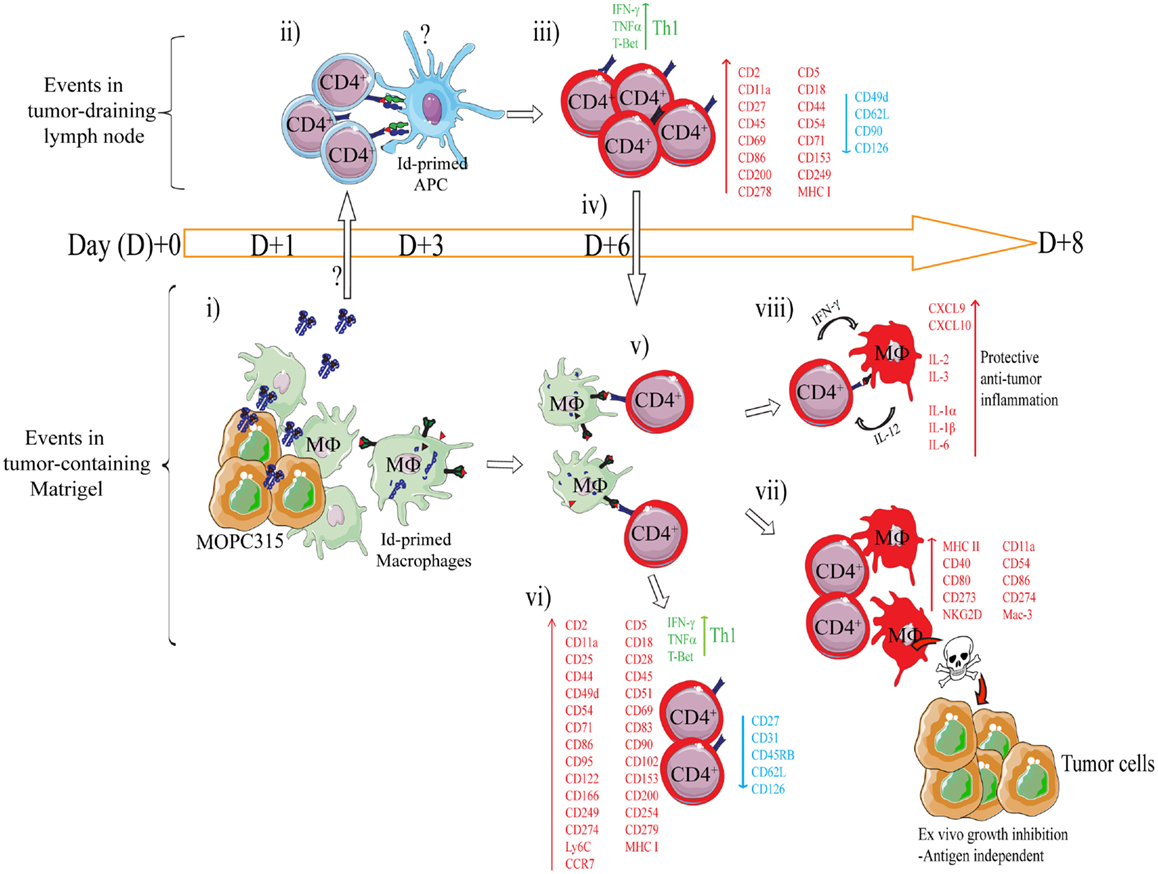
Figure 4. Mechanism of rejection of MHCIINEG myeloma cells by Id-specific CD4+ T cells. The following events are based on experiments where Id-secreting MOPC315 suspended in liquid Matrigel was injected subcutaneously in TCR-transgenic mice. (i–viii). (i) At the incipient tumor site, macrophages [CD11b+, CD11c−, CD80/CD86+ MHC IILO, F4/80+] start to infiltrate the tumor/Matrigel from day +1. Tumor-infiltrating macrophages become Id-primed by extracellular myeloma protein by the conventional MHC II presentation pathway (65). (ii) Extracellular Id+ myeloma protein (or possibly Id-primed tumor APCs) drain to lymph nodes where Id-primed APCs stimulate Id-specific CD4+ T cells. Uncertainties as to the mechanism of Id+ Ag draining and the identity of Id-primed APCs are indicated by a question mark (?). (iii) Id-specific CD4+ T cells become activated by day +3, are substantially expanded by day +6 (34), and polarize into Th1 cells by day +8 (39, 85). Upon activation in the tumor-draining lymph node, a number of molecules are significantly upregulated on the surface of the Id-specific CD4+ T cells, while some are consistently downregulated (85). (iv) Activated CD4+ T cells (CD62LLOW) leave the lymph node and accumulate at the tumor site from day +6 (34, 86). (v) At the incipient tumor site, infiltrating Id-specific CD4+ T cells are re-activated by Id-primed macrophages (34). (vi) Moreover, in addition to a sustained Th1 phenotype, the tumor-infiltrating CD4+ T cells dramatically change expression of a number of surface molecules (85). Several molecules are upregulated on both activated CD4+ T cells in the tumor-draining lymph node, and on tumor-infiltrating CD4+ T cells, although at higher levels in the latter population. (vii) IFN-γ produced by tumor-infiltrating Th1 cells activates macrophages that up-regulate MHC class II on the cell surface and show increased expression of M1-associated surface molecules (34, 39). IFN-γ-activated macrophages acquire a tumoricidal phenotype with the upregulation of cytotoxicity-associated markers including granzyme A/B, and NKG2D (39). In addition, purified activated macrophages can directly inhibit myeloma growth in vitro (34, 36, 39). The mechanisms underlying M1 macrophage-mediated growth inhibition is unknown, but once the macrophages are activated the growth inhibition is antigen independent (36). (viii) Analysis by gene expression profiling and Luminex multiplex cytokine analyses has revealed that the Id-specific CD4+ Th1-mediated anti-tumor immune response has a striking resemblance to the characteristics of acute inflammatory responses (39). Thus, we propose that Th1-mediated inflammatory responses may protect against cancer (87).
Using this system, a longitudinal characterization of the immune response within the tumor microenvironment and draining lymph nodes was undertaken (34, 36, 39, 85). The findings are summarized in Figure 4. Briefly, secreted myeloma protein is presented by APC in tumor-draining lymph nodes to Id-specific CD4+ T cells. Upon recognition, T cells are activated, polarize into Th1 cells, and migrate to the Matrigel/tumor. In the Matrigel/tumor, Th1 cells become re-activated by tumor-infiltrating macrophages that has endocytosed and processed myeloma protein. Th1 derived IFN-γ activates macrophages into tumoricidal M1 macrophages (34, 36, 39, 65).
Unresolved Issues and Directions for Future Research
Use of MHC Class II Negative Tumor Cell Lines in Tumor Immunology
While MHC class II positivity in tumor cells is generally to be trusted, MHC class II negativity should, for obvious reasons, be viewed with a healthy skepticism. In the case of MOPC315, many attempts by others and us have consistently failed to detect expression of MHC class II molecules in vitro as well as in vivo, even when MOPC315 cells were exposed to IFN-γ (34, 70, 82, 88). In several other models, such as the use of the erythroleukemia cell line FBL-3 (25), the UV-induced fibrosarcoma 6132A-PRO (31), and the methylcholanthrene-induced Mc51.9 (32); no MHC class II was detected on tumor cells even after IFN-γ exposure, similar to MOPC315. In the B16 melanoma model, Quezada et al. showed that the cell line used in their experiments expressed MHC class II, but only when the tumor-bearing hosts were subjected to a combination of irradiation and adoptive transfer of Trp1-specific CD4+ T cells together with anti-CTLA mAb (37). Xie et al. also reported that B16 cells express MHC class II by immunofluorescence staining of tumor biopsies, but the identity of the MHC class IIPOS cells within the sections was not further characterized, complicating interpretation (38). In contrast, Hung et al. reports the use of B16 tumor cells that were described to be MHC IINEG (30).
Peres-Diez et al. (28) reported that expression of MHC class II molecules on tumor cells was not required for rejection mediated by CD4+ cells. In note, they found that: H-2k H-Y+ tumor cells were rejected by I–Ab-restricted, H–Y-specific CD4+ T cells in an immunodeficient H-2b mouse. An alternative approach to ensure the absence of the relevant MHC class II molecule in a tumor cell line would be to delete the corresponding MHC class II molecule genes from the tumor cells.
Is Secretion of Tumor-Specific Antigen by MHC IINEG Tumors Required?
The tumor-specific antigen used in our own studies, the MOPC315 myeloma protein, is a highly secreted antigen, with serum levels reaching milligrams per milliliter levels. Concentrations of myeloma protein in tumor tissues would be expected to be even higher. Surprisingly, a non-secreting myeloma variant that only expresses an intracellularly retained mutated Id+ L chain, but in high amounts, was not rejected (36). In the absence of sufficient tumor antigen secretion, it might be expected that either spontaneous necrosis or apoptosis of tumor cells containing such high amounts of intracellular tumor antigen could prime tumor-infiltrating APC with tumor-specific antigen. This is apparently not the case for the non-secreting variant of MOPC315. It remains to be seen whether cytotoxic drug treatment of mice with tumors caused by this particular MOPC315 variant could enhance Id priming of APCs via uptake of necrotic or apoptotic cells.
In other MHC IINEG models where tumor cells is reported to be rejected by CD4+ T cells (28, 31, 32), there is scarce information as to whether tumor-specific antigen is secreted or not (Table 2). In the case of H-Y antigen, which clearly must be transferred from the tumor cells to host APC for MHC II presentation (28), there is little information about the extent of secretion of the antigen. In yet other cases, the tumor-specific antigen is simply not known (25), precluding any analysis of secretion status. It should further be noted that in some experiments [e.g., Ref. (24, 25, 37)] it has not been rigorously excluded that non-malignant normal cells could also produce the “tumor-specific” antigen. This possibility is virtually excluded in the MOPC315 model since CD4+ T cells recognize a somatically mutated tumor-specific antigen unique to MOPC315 myeloma cells. By and large, it appears that secretion of tumor-specific antigen facilitates priming of host APC and stimulation of CD4+ T cells. However, it is possible that the requirement of secretion could vary for distinct tumors and tumor-specific antigens, perhaps related to differences in susceptibility for cross-presentation of antigen associated with either necrotic or apoptotic tumor cells, or secreted vesicles such as exosomes.
What about MHC IIPOS tumors – do they also require secretion of tumor-specific antigen? For MHC IIPOS B lymphoma, a transfectant that secretes λ2315 was rejected, while another transfectant expressing a mutated intracellularly retained λ2315 was not (26). Similarly, A20 cells expressing HA, which apparently was negligibly secreted since HA was not found in serum, was not rejected (89). The Dby minor histocompatibility antigen (H–Y) (28) and Trp1 (35, 37, 38) have both been reported to be secreted by tumor cells. A strategy to test the hypothesis that secretion of tumor-specific antigen is required for rejection of MHC IIPOS tumors would be to transfect MOPC315.37 with CIIITA so that the tumor cells become MHC IIPOS. If this transfectant is rejected in Id-specific TCR-transgenic mice, this would weaken the hypothesis.
By Which Pathway is Tumor Antigen Presented by APC in Draining Lymph Nodes?
In the tumor models where it has been tested, be they MHC IINEG (28, 34, 65) or MHC IIPOS (37, 38), there was an apparent need for tumor-specific antigen to be presented by host APC to stimulate naïve (but not memory) CD4+ T cells. Thus, in the case of the B16 MHC IIPOS model, no rejection by naïve Trp1-specific CD4+ T cells was obtained in hosts that lacked MHC class II molecules. By contrast, transfer of CD4+ T cells that first had been primed in vitro could readily reject B16 tumors (37, 38). These findings indicate that MHC IIPOS tumor cells themselves are incapable of stimulating naïve Trp1-specific CD4+ T cells, and that priming by professional host APC is required. In addition, experiments reported by Xie et al. (38) using Trp1-deficient mice indicate that Trp1 derived from host tissue is redundant for priming APC and that Trp1 derived from B16 tumor cells suffice, at least for stimulation of memory CD4+ T cells. It is still, however, unclear how the Trp1 antigen is transferred from tumors to host APC, and in which anatomical compartment priming of CD4+ T cells take place.
The conclusions of the above experiments are supported by previous observations in the MOPC315 model, which directly demonstrate activation of Id-specific CD4+ T cells in draining lymph nodes (34, 36, 85). Moreover, treatment with the sphingosine phosphate receptor modulator fingolimod that abrogates egress of T cells from lymph nodes led to a decreased number of Id-specific CD4+ T cells within the tumor, resulting in failure of tumor rejection (86). Consistent with these findings, the non-secreting MOPC315.37 variant caused little activation of CD4+ T cells in draining lymph nodes, and tumor rejection did not occur.
Idiotype-primed APCs are readily found in lymph nodes that drain MOPC315 tumors (Dembic and Bogen, unpublished experiments). It should therefore be possible by cell purifications and characterizations to reveal the identity of these Id-primed APCs in lymph nodes. Information from such experiments could help to define the mechanisms by which APC get primed by secreted tumor antigen. For example, if the predominant features of Id-primed APCs are that of a residential dendritic cell, this may signify priming by soluble antigen arriving to the lymph node via afferent lymphatic vessels.
Elimination of MHC IINEG Tumor Cells
It is well documented that Th1/IFN-γ-activated M1 macrophages isolated from tumors under conditions of tumor rejection can directly inhibit the growth of MHC IINEG myeloma cells in vitro (34, 36, 39). However, the molecular mechanisms mediating the inhibition of tumor cell growth remain to be established. Possibly, reactive oxygen species could be of importance, since resistance against B16 cells [although in later work reported to be MHC class IIPOS under conditions of rejection (37)] was reduced in iNOS−/− and NOX2−/− mice (30). Results of Perez-Diez et al. indicate that under some circumstances, NK cells activated by CD4+ T cells are important, but the effector mechanisms employed by such NK cells have not been addressed (28).
It is also possible that CD4+ T cells could themselves directly kill tumor cells, e.g., through FasL/Fas interactions, similar to what has been described for killing of MHC IIPOS B lymphoma cells (33), or a perforin/granzyme B-dependent mechanism as described for killing of the MHC IIPOS B16 cells (37). The efficacy of killing mechanisms of CD4+ T cells could also differ for different tumors. Thus, even though Th1 cells efficiently killed transfected A20 cells in vitro by a FasL-dependent mechanism, the same cells could not kill MOPC315 (26, 66). Finally, it has been reported that IFN-γ produced by tumor-specific Th1 cells mediate tumor rejection by means of angiostatic effects, thus causing starvation of the tumor (32).
Do CD4+ T Cell-Mediated Immune Responses Against MHC IINEG Tumor Cells Convey Bystander Killing of Tumor Cells That have Lost Expression of Antigen?
In theory, macrophage-mediated killing of MHC IINEG tumors could be expected to indiscriminately kill surrounding cells, including tumor cells that have lost expression of antigen (“bystander killing”). If true, this would be a clinically important asset of Th1/M1 macrophage-mediated killing of tumor cells (34, 36, 39). The previously described angiostatic properties of Th1 derived IFNγ (32) would also be expected to cause bystander killing. On the other hand, direct killing of MHC IIPOS tumor cells by cytotoxic CD4+ T cells was demonstrated not to induce bystander killing (37).
What CD4+ T Cell Phenotypes Support Anti-Tumor Immunity?
Naïve CD4+ T cells in Id-specific TCR-transgenic mice, which eradicate injected MHC IINEG tumor cells, develop into IFNγ-secreting Th1 TILs that induce macrophage polarization into tumoricidal M1 macrophages (33, 34, 39). Transfer of naïve Id-specific CD4+ T cells could cure established MHC IIPOS tumors (33). In the Trp1-specific TCR-transgenic model, naïve (37, 38), Th1 (35), and Th17 (35) cells have been demonstrated to eradicate MHC IIPOS tumors. Collectively, these results indicate that the primary anti-tumor response of naïve CD4+ T cells is followed by T cell differentiation into Th1 (or possibly Th17) cells that confer anti-tumor immunity irrespective of MHC class II expression on tumor cells. While Th1 cells are clearly associated with anti-tumor immunity, variable effects have been observed with other CD4+ T cell subsets, reviewed in Ref. (90). Moreover, recent studies suggest that effector CD4+ T cells retain some degree of functional plasticity (91, 92). The plasticity of effector Th populations may explain the differential effects of the various Th cell populations in tumor immunity. In addition, exploiting the plasticity of Th cell subsets may be utilized in immune therapy.
Tolerance Induction of Tumor-Specific CD4+ T Cells
Use of TCR-transgenic mice offers the possibility of studying tolerance development by following the fate and function of tumor-reactive CD4+ T cells. When Id-specific TCR-transgenic mice failed to reject high amounts of injected MHC IINEG MOPC315 cells, CD4+ T cells in peripheral lymphoid organs and in the tumor became deleted (93). The extent of deletion became more profound as tumor size increased. The deletion of peripheral tumor-specific CD4+ T cells seen in this model for a highly secreted tumor antigen resembles that of exhaustion observed in chronic viral diseases. In addition to peripheral deletion of Id-specific CD4+ T cells, progressive MOPC315 tumors also caused thymocyte deletion. It was demonstrated that circulatory myeloma protein gained access to the thymus and was presented in an MHC class II context by thymic APCs, thus causing negative selection of thymocytes (94).
In a recent paper, T cell characteristics in Trp1-specific TCR-transgenic mice developing B16 tumor recurrence following adoptive therapy were studied. Recurrence was associated with increased FoxP3+ Treg cell numbers, and increased expression of inhibitory ligands, including PD-1 and CTLA-4 inhibitory receptors on both Treg and effector CD4+ cells (95). Tumor recurrence could be prevented by concomitant depletion of Tregs and administration of checkpoint blockade antibodies. Collectively, these results indicate that CD4+ T cells must eliminate tumor antigen-secreting tumor cells efficiently within a short timeframe. If the elimination is incomplete, T cell tolerance is induced by multiple mechanisms.
It has been shown that MHC IIPOS A20 cells, are not rejected after i.v. injection in HA-specific TCR-transgenic mice, but induce anergy in CD4+ T cells via priming of bone marrow derived APCs (89, 96). Interestingly, when presentation by bone marrow derived APCs was prevented by the use of bone marrow chimeras, anergy did not occur, and tumor cells were rejected (72). Thus, it might seem that tumor cells that poorly secrete tumor antigen could favor anergy development by induction tolerogenic APCs. The above results are consistent with previous observations that A20 cells expressing a non-secreted λ2315 were not rejected in Id-specific TCR-transgenic mice (26) (although it was not tested if anergy was induced). These results, obtained with non-secreting MHC IIPOS A20 transfectants in two different TCR-transgenic models, are in support of the notion that tumor-specific antigen, perhaps via presentation of apoptotic or necrotic tumor cells by a special type of APC, favor induction of T cell anergy. In contrast, secretion of tumor-specific antigen and presentation (perhaps by another type of host APCs) in lymph nodes, may favor induction of potent primary anti-tumor CD4+ T cell responses.
Dichotomous Role of Th Cells in B Cell Cancers
This review paper has focused on CD4+ T cell-mediated eradication of tumor cells. However, CD4+ T cells may also induce tumors. This dichotomy may especially apply to B cell tumors since B cells are known to proliferate in response to help from CD4+ T cells. Extensive and prolonged B cell proliferation could indeed predispose to genetic instability and malignant transformation. In fact, B lymphoma development has been associated with continuous antigenic exposure in chronic infectious diseases caused by Helicobacter pylori, EBV, and hepatitis C. Moreover, chronic immune responses to self antigens in autoimmune diseases such as systemic lupus erythematosus, Sjögren’s syndrome and rheumatoid arthritis have also been linked to development of B cell lymphomas, reviewed in Ref. (97, 98). Further supporting a role for chronic antigen stimulation, diffuse large B cell lymphomas (98, 99) and follicular B cell lymphomas (98, 100, 101) are frequently infiltrated with T cells. In Ig- and TCR-transgenic mice, chronic stimulation of Id+ B cells by Id-specific CD4+ Th2 cells results in the induction of Id+ B lymphomas (102). Moreover, two separate studies have shown that proliferation of B lymphomas (103) and MM (104) was augmented by the presence of CD4+ T cells.
The MOPC315 model, reviewed herein, was used in the experiments were Id+ lymphomas were induced. Interestingly, when such induced lymphoma cells were injected s.c into naïve Id-specific TCR-transgenic mice, the lymphoma cells were promptly rejected (102). Thus, Id+ B lymphoma cells were eliminated by mice having naïve CD4+ T cells with an identical Id-specific TCR to that of the B lymphoma-inducing Th2 cells. If naïve T cells in the protected mice differentiated into tumor-eliminating Th1 cells was not investigated. However, analogous experiments indicate that Th1 is the primary response to subcutaneously inoculated B lymphomas (34, 39). These results suggest that B lymphoma cells induced by Th2 cells are rejected by Th1 cells expressing an identical TCR. The finding has obvious implications for T cell therapy: if a B cell tumor is initiated by Th2 cells, it may be treated by Th1 cells of the same specificity (and possibly vice versa). The same may apply to other combinations of Th cells such as Th17/Th1 etc. Thus, re-education of T cell phenotype may become part of the tumor immunotherapy armamentarium. Given the plasticity of CD4+ subsets (91, 92), such re-education may become a real possibility.
Concluding Remarks
How Disparate are the Mechanisms for Rejection of MHC IIPOS and MHC IINEG Tumors?
The data reviewed herein suggest that the difference between direct and indirect killing of tumors relates predominantly to the effector stage of tumor cell killing. Thus, CD4+ T cells can kill MHC IIPOS cells directly, while killing of MHC IINEG occurs indirectly via macrophages or possibly NK cells, angiostatic effects, or all of these. In contrast, the primary activation of naïve tumor-specific CD4+ T cells appears to be similar for the direct and indirect mechanisms, in that presentation of tumor-specific antigen by host APC seems to be required. However, the evidence for this in the context of MHC IIPOS tumors is largely circumstantial. In an MHC IINEG myeloma model, secretion of tumor-specific myeloma protein clearly facilitates priming of APC in lymph nodes and stimulation of naive CD4+ T cells that subsequently infiltrate the tumor site. Thus, the nature of the antigen, by virtue of its cellular localization and accessibility to APCs, might determine the ability of the antigen to serve as an efficient tumor-specific antigen in CD4+ T cell responses. A more in-depth analysis of such factors might be of value in reconciling observations made in the various TCR-transgenic models.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank everyone in Bogen lab who, over the years, have contributed to the work described in this review. The work has been generously supported by the Norwegian Cancer Society, The Multiple Myeloma Research Foundation, The Research Council of Norway, The South-Eastern Norway Regional Health Authority, and numerous funds and legacies mentioned in the original research reports. The authors apologize for any omissions of publications that could not be included due to space constraints.
Abbreviations
APC, antigen-presenting cell; CIITA, class II trans-activator; FasL, Fas ligand; HA, hemagglutinin; Id, idiotype; IFN-γ, interferon gamma; Ig, immunoglobulin; MHC, major histocompatibility complex; MM, multiple myeloma; MOPC, mineral oil-induced plasmacytoma; OVA, ovalbumin; s.c., subcutaneous; TCR-Tg, T cell receptor transgenic; TIL, tumor-infiltrating lymphocyte; Trp1, tyrosinase-related protein 1; SCID, severe combined immunodeficiency.
References
1. Burnet M. Cancer; a biological approach. I. The processes of control. Br Med J (1957) 1:779–86. doi: 10.1136/bmj.1.5022.779
2. Thomas L. Discussion in Cellular and Humoral Aspects of the Hypersensitive State. New York: Hoebner-Harper (1959). p. 529–32.
5. Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature (2007) 450:903–7. doi:10.1038/nature06309
6. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science (2002) 298:850–4. doi:10.1126/science.1076514
7. Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu Rev Med (2013). doi:10.1146/annurev-med-092012-112807
8. Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity (2013) 39:11–26. doi:10.1016/j.immuni.2013.07.008
9. Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol (2002) 3:999–1005. doi:10.1038/ni1102-999
10. Garrido F, Cabrera T, Aptsiauri N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: implications for immunotherapy. Int J Cancer (2010) 127:249–56. doi:10.1002/ijc.25270
11. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature (2012) 482:400–4. doi:10.1038/nature10755
12. Hanson HL, Donermeyer DL, Ikeda H, White JM, Shankaran V, Old LJ, et al. Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity (2000) 13:265–76. doi:10.1016/S1074-7613(00)00026-1
13. Lee KH, Wang E, Nielsen MB, Wunderlich J, Migueles S, Connors M, et al. Increased vaccine-specific T cell frequency after peptide-based vaccination correlates with increased susceptibility to in vitro stimulation but does not lead to tumor regression. J Immunol (1999) 163:6292–300.
14. Dudley ME, Wunderlich JR, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother (2002) 25:243–51. doi:10.1097/00002371-200205000-00007
15. Peterson AC, Harlin H, Gajewski TF. Immunization with melan-A peptide-pulsed peripheral blood mononuclear cells plus recombinant human interleukin-12 induces clinical activity and T-cell responses in advanced melanoma. J Clin Oncol (2003) 21:2342–8. doi:10.1200/JCO.2003.12.144
16. Boon T, Coulie PG, Van Den Eynde BJ, Van Der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol (2006) 24:175–208. doi:10.1146/annurev.immunol.24.021605.090733
17. Nishimura T, Iwakabe K, Sekimoto M, Ohmi Y, Yahata T, Nakui M, et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J Exp Med (1999) 190:617–27. doi:10.1084/jem.190.5.617
18. Gao FG, Khammanivong V, Liu WJ, Leggatt GR, Frazer IH, Fernando GJ. Antigen-specific CD4+ T-cell help is required to activate a memory CD8+ T cell to a fully functional tumor killer cell. Cancer Res (2002) 62:6438–41.
19. Janssen EM, Lemmens EE, Wolfe T, Christen U, Von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature (2003) 421:852–6. doi:10.1038/nature01441
20. Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol (2005) 174:2591–601.
21. Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med (2008) 358:2698–703. doi:10.1056/NEJMoa0800251
22. Ostrand-Rosenberg S, Thakur A, Clements V. Rejection of mouse sarcoma cells after transfection of MHC class II genes. J Immunol (1990) 144:4068–71.
23. Ostrand-Rosenberg S, Roby CA, Clements VK. Abrogation of tumorigenicity by MHC class II antigen expression requires the cytoplasmic domain of the class II molecule. J Immunol (1991) 147:2419–22.
24. Fujiwara H, Fukuzawa M, Yoshioka T, Nakajima H, Hamaoka T. The role of tumor-specific Lyt-1+2– T cells in eradicating tumor cells in vivo. I. Lyt-1+2– T cells do not necessarily require recruitment of host’s cytotoxic T cell precursors for implementation of in vivo immunity. J Immunol (1984) 133:1671–6.
25. Greenberg PD, Kern DE, Cheever MA. Therapy of disseminated murine leukemia with cyclophosphamide and immune Lyt-1+,2– T cells. Tumor eradication does not require participation of cytotoxic T cells. J Exp Med (1985) 161:1122–34. doi:10.1084/jem.161.5.1122
26. Lauritzsen GF, Weiss S, Dembic Z, Bogen B. Naive idiotype-specific CD4+ T cells and immunosurveillance of B-cell tumors. Proc Natl Acad Sci U S A (1994) 91:5700–4. doi:10.1073/pnas.91.12.5700
27. Bogen B, Munthe L, Sollien A, Hofgaard P, Omholt H, Dagnaes F, et al. Naive CD4+ T cells confer idiotype-specific tumor resistance in the absence of antibodies. Eur J Immunol (1995) 25:3079–86. doi:10.1002/eji.1830251114
28. Perez-Diez A, Joncker NT, Choi K, Chan WFN, Anderson CC, Lantz O, et al. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood (2007) 109:5346–54. doi:10.1182/blood-2006-10-051318
29. Greenberg PD, Cheever MA, Fefer A. Eradication of disseminated murine leukemia by chemoimmunotherapy with cyclophosphamide and adoptively transferred immune syngeneic Lyt-1+2– lymphocytes. J Exp Med (1981) 154:952–63. doi:10.1084/jem.154.3.952
30. Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med (1998) 188:2357–68. doi:10.1084/jem.188.12.2357
31. Mumberg D, Monach PA, Wanderling S, Philip M, Toledano AY, Schreiber RD, et al. CD4(+) T cells eliminate MHC class II-negative cancer cells in vivo by indirect effects of IFN-gamma. Proc Natl Acad Sci U S A (1999) 96:8633–8. doi:10.1073/pnas.96.15.8633
32. Qin Z, Blankenstein T. CD4+ T cell – mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity (2000) 12:677–86. doi:10.1016/S1074-7613(00)80218-6
33. Lundin KU, Hofgaard PO, Omholt H, Munthe LA, Corthay A, Bogen B. Therapeutic effect of idiotype-specific CD4+ T cells against B-cell lymphoma in the absence of anti-idiotypic antibodies. Blood (2003) 102:605. doi:10.1182/blood-2002-11-3381
34. Corthay A, Skovseth DK, Lundin KU, Rosjo E, Omholt H, Hofgaard PO, et al. Primary antitumor immune response mediated by CD4+ T cells. Immunity (2005) 22:371–83. doi:10.1016/j.immuni.2005.02.003
35. Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood (2008) 112:362–73. doi:10.1182/blood-2007-11-120998
36. Corthay A, Lundin KU, Lorvik KB, Hofgaard PO, Bogen B. Secretion of tumor-specific antigen by myeloma cells is required for cancer immunosurveillance by CD4+ T cells. Cancer Res (2009) 69:5901–7. doi:10.1158/0008-5472.CAN-08-4816
37. Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med (2010) 207:637–50. doi:10.1084/jem.20091918
38. Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EKM, et al. Naive tumor-specific CD4+ T cells differentiated in vivo eradicate established melanoma. (2010) 207:651–67. doi:10.1084/jem.20091921
39. Haabeth OA, Lorvik KB, Hammarstrom C, Donaldson IM, Haraldsen G, Bogen B, et al. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat Commun (2011) 2:240. doi:10.1038/ncomms1239
40. Marzo AL, Lake RA, Robinson BW, Scott B. T-cell receptor transgenic analysis of tumor-specific CD8 and CD4 responses in the eradication of solid tumors. Cancer Res (1999) 59:1071–9.
41. Klein L, Trautman L, Psarras S, Schnell S, Siermann A, Liblau R, et al. Visualizing the course of antigen-specific CD8 and CD4 T cell responses to a growing tumor. Eur J Immunol (2003) 33:806–14. doi:10.1002/eji.200323800
42. Zhou G, Drake CG, Levitsky HI. Amplification of tumor-specific regulatory T cells following therapeutic cancer vaccines. Blood (2006) 107:628–36. doi:10.1182/blood-2005-07-2737
43. Chamoto K, Wakita D, Narita Y, Zhang Y, Noguchi D, Ohnishi H, et al. An essential role of antigen-presenting cell/T-helper type 1 cell-cell interactions in draining lymph node during complete eradication of class II-negative tumor tissue by T-helper type 1 cell therapy. Cancer Res (2006) 66:1809–17. doi:10.1158/0008-5472.CAN-05-2246
44. Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest (2013) 123:2447–63. doi:10.1172/JCI64859
45. Bogen B, Malissen B, Haas W. Idiotope-specific T cell clones that recognize syngeneic immunoglobulin fragments in the context of class II molecules. Eur J Immunol (1986) 16:1373–8. doi:10.1002/eji.1830161110
46. Bogen B, Gleditsch L, Weiss S, Dembic Z. Weak positive selection of transgenic T cell receptor-bearing thymocytes: importance of major histocompatibility complex class II, T cell receptor and CD4 surface molecule densities. Eur J Immunol (1992) 22:703–9. doi:10.1002/eji.1830220313
47. Lantz O, Grandjean I, Matzinger P, Di Santo JP. Gamma chain required for naive CD4+ T cell survival but not for antigen proliferation. Nat Immunol (2000) 1(1):54–8. doi:10.1038/76917
48. Scott B, Liblau R, Degermann S, Marconi LA, Ogata L, Caton AJ, et al. A role for non-MHC genetic polymorphism in susceptibility to spontaneous autoimmunity. Immunity (1994) 1(1):73–83. doi:10.1016/1074-7613(94)90011-6
49. Kirberg J, Baron A, Jakob S, Rolink A, Karjalainen K, von Boehmer H. Thymic selection of CD8+ single positive cells with a class II major histocompatibility complex-restricted receptor. J Exp Med (1994) 180(1):25–34. doi:10.1084/jem.180.1.25
50. Murphy KM, Heimberger AB, Loh DY. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science (1990) 250(4988):1720–23.
51. Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol (1998) 76(1):34–40. doi:10.1046/j.1440-1711.1998.00709.x
52. Eisen HN, Simms ES, Potter M. Mouse myeloma proteins with antihapten antibody activity. The protein produced by plasma cell tumor MOPC-315. Biochemistry (1968) 7:4126–34. doi:10.1021/bi00851a048
53. Hannestad K, Kao MS, Eisen HN. Cell-bound myeloma proteins on the surface of myeloma cells: potential targets for the immune system. Proc Natl Acad Sci U S A (1972) 69(8):2295–9.
54. Xu Y, Setaluri V, Takechi Y, Houghton AN. Sorting and secretion of a melanosome membrane protein, gp75/TRP1. J Invest Dermatol (1997) 109(6):788–95. doi:10.1111/1523-1747.ep12340971
55. Zenzes MT, Müller U, Aschmoneit I, Wolf U. Studies on H-Y antigen in different cell fractions of the testis during pubescence: immature germ cells are H-Y antigen negative. Hum Genet (1978) 45(3):297–303. doi:10.1007/BF00278727
57. Copeland CS, Doms RW, Bolzau EM, Webster RG, Helenius A. Assembly of influenza hemagglutinin trimers and its role in intracellular transport. J Cell Biol (1986) 103(4):1179–91. doi:10.1083/jcb.103.4.1179
58. Meek RL, Walsh KA, Palmiter RD. The signal sequence of ovalbumin is located near the NH2 terminus. J Biol Chem (1982) 257(20):12245–51.
59. Rudensky A, Preston-Hurlburt P, Hong SC, Barlow A, Janeway CA Jr. Sequence analysis of peptides bound to MHC class II molecules. Nature (1991) 353:622–7. doi:10.1038/353622a0
60. Benoist C, Mathis D. Regulation of major histocompatibility complex class-II genes: X, Y and other letters of the alphabet. Annu Rev Immunol (1990) 8:681–715. doi:10.1146/annurev.immunol.8.1.681
61. Glimcher LH, Kara CJ. Sequences and factors: a guide to MHC class-II transcription. Annu Rev Immunol (1992) 10:13–49. doi:10.1146/annurev.iy.10.040192.000305
62. Ting JP, Baldwin AS. Regulation of MHC gene expression. Curr Opin Immunol (1993) 5:8–16. doi:10.1016/0952-7915(93)90074-3
63. Steimle V, Siegrist CA, Mottet A, Lisowska-Grospierre B, Mach B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science (1994) 265:106–9. doi:10.1126/science.8016643
64. Mach B, Steimle V, Martinez-Soria E, Reith W. Regulation of MHC class II genes: lessons from a disease. Annu Rev Immunol (1996) 14:301–31. doi:10.1146/annurev.immunol.14.1.301
65. Dembic Z, Schenck K, Bogen B. Dendritic cells purified from myeloma are primed with tumor-specific antigen (idiotype) and activate CD4+ T cells. Proc Natl Acad Sci U S A (2000) 97:2697–702. doi:10.1073/pnas.050579897
66. Lundin KU, Screpanti V, Omholt H, Hofgaard PO, Yagita H, Grandien A, et al. CD4+ T cells kill Id+ B-lymphoma cells: FasLigand-Fas interaction is dominant in vitro but is redundant in vivo. Cancer Immunol Immunother (2004) 53:1135–45. doi:10.1007/s00262-004-0538-4
67. Wu GE, Hozumi N, Murialdo H. Secretion of a lambda 2 immunoglobulin chain is prevented by a single amino acid substitution in its variable region. Cell (1983) 33:77–83. doi:10.1016/0092-8674(83)90336-7
68. Esumi N, Hunt B, Itaya T, Frost P. Reduced tumorigenicity of murine tumor cells secreting gamma-interferon is due to nonspecific host responses and is unrelated to class I major histocompatibility complex expression. Cancer Res (1991) 51(4):1185–9.
69. Moore MW, Carbone FR, Bevan MJ. Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell (1988) 54(6):777–85.
70. Lauritzsen GF, Bogen B. The role of idiotype-specific, CD4+ T cells in tumor resistance against major histocompatibility complex class II molecule negative plasmacytoma cells. Cell Immunol (1993) 148:177–88. doi:10.1006/cimm.1993.1100
71. Tite JP, Janeway CA Jr. Cloned helper T cells can kill B lymphoma cells in the presence of specific antigen: Ia restriction and cognate vs. noncognate interactions in cytolysis. Eur J Immunol (1984) 14:878–86. doi:10.1002/eji.1830141004
72. Horna P, Cuenca A, Cheng F, Brayer J, Wang HW, Borrello I, et al. In vivo disruption of tolerogenic cross-presentation mechanisms uncovers an effective T-cell activation by B-cell lymphomas leading to antitumor immunity. Blood (2006) 107:2871–8. doi:10.1182/blood-2005-07-3014
73. Silacci P, Mottet A, Steimle V, Reith W, Mach B. Developmental extinction of major histocompatibility complex class II gene expression in plasmocytes is mediated by silencing of the transactivator gene CIITA. J Exp Med (1994) 180:1329–36. doi:10.1084/jem.180.4.1329
74. Reith W, Mach B. The bare lymphocyte syndrome and the regulation of MHC expression. Annu Rev Immunol (2001) 19:331–73. doi:10.1146/annurev.immunol.19.1.331
75. Sanda T, Iida S, Kayukawa S, Ueda R. Induction of class II major histocompatibility complex expression in human multiple myeloma cells by retinoid. Haematologica (2007) 92:115–20. doi:10.3324/haematol.10300
76. Yi Q, Dabadghao S, Osterborg A, Bergenbrant S, Holm G. Myeloma bone marrow plasma cells: evidence for their capacity as antigen-presenting cells. Blood (1997) 90:1960–7.
77. Zhao M, Flynt FL, Hong M, Chen H, Gilbert CA, Briley NT, et al. MHC class II transactivator (CIITA) expression is upregulated in multiple myeloma cells by IFN-gamma. Mol Immunol (2007) 44:2923–32. doi:10.1016/j.molimm.2007.01.009
78. Bothwell AL, Paskind M, Reth M, Imanishi-Kari T, Rajewsky K, Baltimore D. Somatic variants of murine immunoglobulin lambda light chains. Nature (1982) 298:380–2. doi:10.1038/298380a0
79. Bogen B, Lambris JD. Minimum length of an idiotypic peptide and a model for its binding to a major histocompatibility complex class II molecule. EMBO J (1989) 8:1947–52.
80. Weiss S, Bogen B. B-lymphoma cells process and present their endogenous immunoglobulin to major histocompatibility complex-restricted T cells. Proc Natl Acad Sci U S A (1989) 86:282–6. doi:10.1073/pnas.86.1.282
81. Jorgensen T, Hannestad K. Helper T cell recognition of the variable domains of a mouse myeloma protein (315). Effect of the major histocompatibility complex and domain conformation. J Exp Med (1982) 155:1587–96. doi:10.1084/jem.155.6.1587
82. Foy TM, Lynch RG, Waldschmidt TJ. Ontogeny and distribution of the murine B cell Fc gamma RII. J Immunol (1992) 149:1516–23.
83. Winberry L, Marks A, Baumal R. Immunoglobulin production and secretion by variant clones of the MOPC 315 mouse myeloma cell line. J Immunol (1980) 124:1174–82.
84. Corthay A, Lorvik KB, Bogen B. Is secretion of tumour-specific antigen important for cancer eradication by CD4(+) T cells? – Implications for cancer immunotherapy by adoptive T cell transfer. Scand J Immunol (2011) 73:527–30. doi:10.1111/j.1365-3083.2011.02558.x
85. Lorvik KB, Haabeth OA, Clancy T, Bogen B, Corthay A. Molecular profiling of tumor-specific T1 cells activated in vivo. Oncoimmunology (2013) 2:e24383. doi:10.4161/onci.24383
86. Lorvik KB, Bogen B, Corthay A. Fingolimod blocks immunosurveillance of myeloma and B-cell lymphoma resulting in cancer development in mice. Blood (2012) 119:2176–7. doi:10.1182/blood-2011-10-388892
87. Haabeth OA, Bogen B, Corthay A. A model for cancer-suppressive inflammation. Oncoimmunology (2012) 1:1146–55. doi:10.4161/onci.21542
88. Bogen B, Schenck K, Munthe LA, Dembic Z. Deletion of idiotype (Id)-specific T cells in multiple myeloma. Acta Oncol (2000) 39:783–8. doi:10.1080/028418600750063505
89. Staveley-O’Carroll K, Sotomayor E, Montgomery J, Borrello I, Hwang L, Fein S, et al. Induction of antigen-specific T cell anergy: an early event in the course of tumor progression. Proc Natl Acad Sci U S A (1998) 95:1178–83. doi:10.1073/pnas.95.3.1178
90. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer (2012) 12:298–306. doi:10.1038/nrc3245
91. Murphy KM, Stockinger B. Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol (2010) 11:674–80. doi:10.1038/ni.1899
92. O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science (2010) 327:1098–102. doi:10.1126/science.1178334
93. Bogen B. Peripheral T cell tolerance as a tumor escape mechanism: deletion of CD4+ T cells specific for a monoclonal immunoglobulin idiotype secreted by a plasmacytoma. Eur J Immunol (1996) 26:2671–9. doi:10.1002/eji.1830261119
94. Lauritzsen GF, Hofgaard PO, Schenck K, Bogen B. Clonal deletion of thymocytes as a tumor escape mechanism. Int J Cancer (1998) 78:216–22. doi:10.1002/(SICI)1097-0215(19981005)78:2<216::AID-IJC16>3.0
95. Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A, et al. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol (2013) 190:4899–909. doi:10.4049/jimmunol.1300271
96. Sotomayor EM, Borrello I, Rattis FM, Cuenca AG, Abrams J, Staveley-O’carroll K, et al. Cross-presentation of tumor antigens by bone marrow-derived antigen-presenting cells is the dominant mechanism in the induction of T-cell tolerance during B-cell lymphoma progression. Blood (2001) 98:1070–7. doi:10.1182/blood.V98.4.1070
97. Morse HC III, Kearney JF, Isaacson PG, Carroll M, Fredrickson TN, Jaffe ES. Cells of the marginal zone – origins, function and neoplasia. Leuk Res (2001) 25:169–78. doi:10.1016/S0145-2126(00)00107-7
98. Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer (2005) 5:251–62. doi:10.1038/nrc1589
99. Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood (2005) 105:1851–61. doi:10.1182/blood-2004-07-2947
100. Ansell SM, Stenson M, Habermann TM, Jelinek DF, Witzig TE. CD4+ T-cell immune response to large B-cell non-Hodgkin’s lymphoma predicts patient outcome. J Clin Oncol (2001) 19:720–6.
101. Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med (2004) 351:2159–69. doi:10.1056/NEJMoa041869
102. Zangani MM, Froyland M, Qiu GY, Meza-Zepeda LA, Kutok JL, Thompson KM, et al. Lymphomas can develop from B cells chronically helped by idiotype-specific T cells. J Exp Med (2007) 204:1181–91. doi:10.1084/jem.20061220
103. Os A, Burgler S, Ribes AP, Funderud A, Wang D, Thompson KM, et al. Chronic lymphocytic leukemia cells are activated and proliferate in response to specific T helper cells. Cell Rep (2013) 4:566–77. doi:10.1016/j.celrep.2013.07.011
Keywords: tumor immunology, CD4+ T cells, MHC class II, T cell receptor transgenic, transgenic mouse models, tumor antigen, T helper 1, multiple myeloma
Citation: Haabeth OAW, Tveita AA, Fauskanger M, Schjesvold F, Lorvik KB, Hofgaard PO, Omholt H, Munthe LA, Dembic Z, Corthay A and Bogen B (2014) How do CD4+ T cells detect and eliminate tumor cells that either lack or express MHC class II molecules? Front. Immunol. 5:174. doi: 10.3389/fimmu.2014.00174
Received: 17 February 2014; Paper pending published: 07 March 2014;
Accepted: 02 April 2014; Published online: 15 April 2014.
Edited by:
Fang-Ping Huang, Imperial College London, UKReviewed by:
Luca Gattinoni, National Cancer Institute, USAJunko Matsuzaki, Roswell Park Cancer Institute, USA
William J. Magner, Roswell Park Cancer Institute, USA
Copyright: © 2014 Haabeth, Tveita, Fauskanger, Schjesvold, Lorvik, Hofgaard, Omholt, Munthe, Dembic, Corthay and Bogen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bjarne Bogen, Institute of Immunology, University of Oslo, OUS Rikshospitalet HF, A2/A3, Sognsvannsveien 20, Oslo N-0027, Norway e-mail:Ymphcm5lLmJvZ2VuQG1lZGlzaW4udWlvLm5v
†Ole Audun Werner Haabeth and Anders Aune Tveita have contributed equally to this work.