 Bart Everts
Bart Everts Edward J. Pearce
Edward J. Pearce- 1Department of Pathology and Immunology, Washington University School of Medicine, St. Louis, MO, USA
- 2Department of Parasitology, Leiden University Medical Center, Leiden, Netherlands
Dendritic cells (DCs) are key regulators of both immunity and tolerance by controlling activation and polarization of effector T helper cell and regulatory T cell responses. Therefore, there is a major focus on developing approaches to manipulate DC function for immunotherapy. It is well known that changes in cellular activation are coupled to profound changes in cellular metabolism. Over the past decade there is a growing appreciation that these metabolic changes also underlie the capacity of immune cells to perform particular functions. This has led to the concept that the manipulation of cellular metabolism can be used to shape innate and adaptive immune responses. While most of our understanding in this area has been gained from studies with T cells and macrophages, evidence is emerging that the activation and function of DCs are also dictated by the type of metabolism these cells commit to. We here discuss these new insights and explore whether targeting of metabolic pathways in DCs could hold promise as a novel approach to manipulate the functional properties of DCs for clinical purposes.
Introduction
Dendritic cells (DCs) play a crucial role in the development of adaptive immune responses during infections and inflammatory diseases, as well as in the regulation of immune homeostasis during steady state, by governing the activation and maintenance of T cell responses. In response to many viral and bacterial infections, DCs promote the generation of effector CD4+ T helper 1 (Th1) and CD8+ T cell-dominated immune responses, while fungal and parasitic worm infections are predominantly associated with Th17 and Th2 responses, respectively. In addition to these effector responses, DCs can be instructed to become tolerogenic and promote regulatory T cells (Tregs), which regulate effector T cell responses, a process that is crucial for maintenance of immune homeostasis and control of autoimmune disorders and allergies. Because of the powerful immunoregulatory functions of DCs, there has been great interest in delineating the cellular processes that control the different properties of these cells, to ultimately identify ways to manipulate the function of DCs for the rational design of DC-based immune-interventions.
It has long been appreciated, especially in the cancer field, that changes in cellular activation coincide with, and are underpinned by, alterations in cellular metabolic state (1, 2). Importantly, over the last couple of years it is becoming increasingly clear that immune cell activation is also coupled to profound changes in cellular metabolism and that their fate and function are metabolically regulated (3). This has led to the idea that manipulation of cellular metabolism of immune cells can be used to shape innate and adaptive immune responses to our advantage. While most of our understanding in this area has been gained from studies with T cells (4–6) and macrophages (7, 8), evidence is emerging that metabolic processes also control the activation and immune-priming functions of DCs. In the current review, we will discuss these recent findings and explore whether targeting of metabolic pathways in DCs could hold promise as a novel approach to manipulate their functional properties for DC-based immunotherapy.
Role of Cellular Metabolism in DC function
Under non-inflammatory conditions, most DCs reside in peripheral tissues where they exist in a resting immature state. In this quiescent state, DCs are poorly immunogenic. However, upon triggering of a set of germline-encoded pattern recognition receptors, including Toll-like receptors (TLRs) by pathogen-derived products or inflammatory stimuli, DCs undergo a well-characterized process of cellular activation, termed DC maturation, which renders them highly immunogenic. This process involves an increase in capturing and processing of antigens for antigen presentation in context of major histocompatibility complex I (MHC-I) and MHC-II and the induction of expression of chemokine receptors, pro-inflammatory cytokines, and costimulatory molecules. This activation program endows DCs with the capacity to traffic, via tissue-draining lymphatics, to T cell zones of secondary lymphoid organs to efficiently prime and control effector T cell responses (9).
In T cells, catabolic metabolism centered around mitochondrial oxidative phosphorylation (OXPHOS) is associated with cellular longevity and quiescence, whereas cellular activation and proliferation are accompanied by a switch to glycolytic metabolism to support anabolic pathways needed for biosynthesis (4–6). Consistent with these observations, DCs when activated by TLR agonists, undergo a robust metabolic switch characterized by an increase in glycolysis and a concomitant progressive loss of OXPHOS (10–13). We have shown that in inflammatory DC subsets, such as murine GM-CSF-derived bone marrow DCs (14), this switch from OXPHOS to glycolysis is a direct consequence of TLR-induced inducible nitric oxide synthase (iNOS) expression that through the production of nitric oxide (NO) poisons the mitochondrial respiratory chain in an autocrine fashion (15). In this setting, in the absence of functional OXPHOS, TLR-agonist activated inflammatory DCs depend heavily on glycolysis as their sole source of ATP for survival both in vitro and in vivo (12). Consistent with this, in vitro and ex vivo TLR-activated Nos2−/− inflammatory DCs still have functional OXPHOS and as result do not display a long-term increase in glycolytic metabolism (12). Likewise, we did not observe a switch to glycolytic metabolism following TLR stimulation of conventional DCs (cDCs) ex vivo (12), which do not express iNOS in response to TLR stimulation. However, a more recent in vivo study showed that TLR-activated cDCs do display long-term diminished mitochondrial activity and enhanced glycolysis (13). They found that this metabolic shift is iNOS-independent and instead driven by TLR-induced autocrine type I interferon production. Despite the differences in mechanism underlying the metabolic switch, similar to inflammatory DCs, cDCs seem to rely on the glycolytic shift for ATP production for their survival (13) (Figure 1).
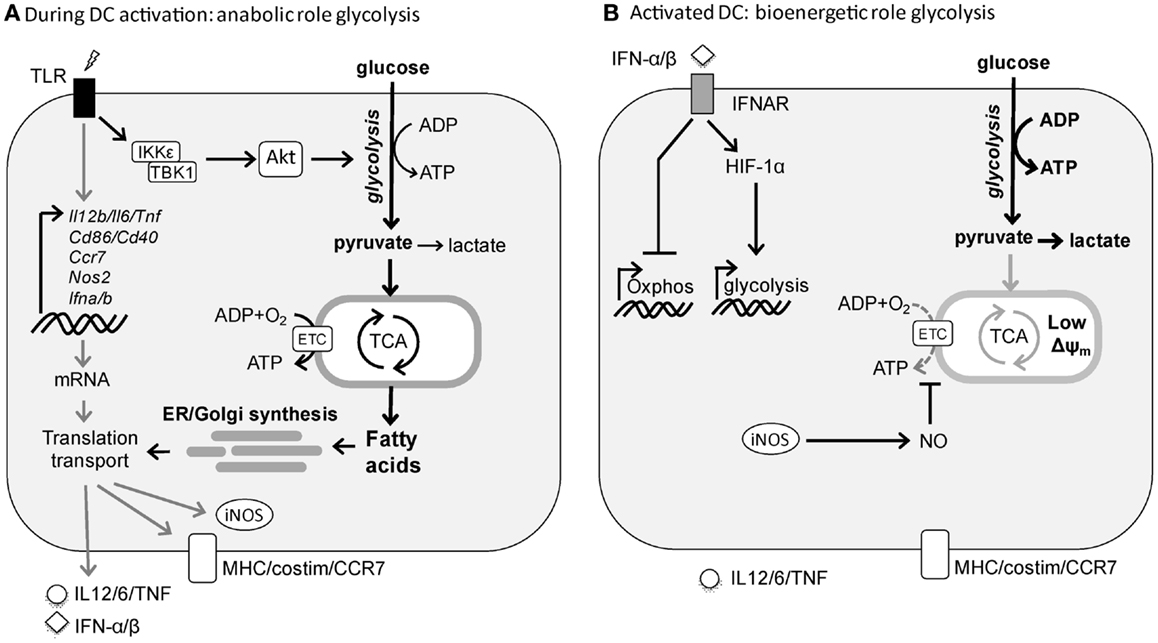
Figure 1. TLR-induced metabolic changes in dendritic cells. (A) Rapid induction of glycolysis in DCs by TLR stimulation serves an anabolic role in DC activation, by generating lipids for synthesis of additional membranes including ER and Golgi to support the increased demands of synthesis and transport of proteins required for DC maturation. (B) Following activation, DCs sustain high glycolytic rates to generate ATP to compensate for the loss of mitochondrial function. In cDCs this process appears to be driven by autocrine type I interferon, while in inflammatory DCs this is a direct consequence of iNOS-derived NO that blocks OXPHOS.
These studies suggest that the metabolic reprograming toward glycolytic metabolism is a consequence of TLR-driven DC activation, rather than a prerequisite for it. However, given the fact that TLR stimulation results in a rapid activation program in both cDCs and inflammatory DCs, we recently tested the hypothesis that rapid metabolic reprograming needs to occur in both types of DCs to meet the bioenergetic and anabolic needs of TLR-driven DC activation itself. Indeed, we observed that TLR stimulation in both cDCs and inflammatory DCs results within minutes in an increase in glycolytic rate that is maintained for several hours after which it returns to prestimulation levels in the absence of iNOS (16). Inhibition of this early metabolic reprograming blunts DC activation, migration, and T cell priming both in vitro and in vivo, illustrating its importance for DC biology. Functionally, as opposed to the long-term glycolytic commitment, the rapid increase in glycolysis appears not to be important as a rapid source of ATP, but rather to serve a central anabolic role by acting as a carbon source for both the pentose phosphate pathway (PPP) and the tricarboxylic (TCA) cycle to support the generation of NADPH and citrate, respectively, that are used for de novo fatty acid (FA) synthesis. Moreover, glycolysis-supported de novo FA synthesis plays a crucial role in DC activation and function at the posttranscriptional level, by allowing for the synthesis and expansion of membranes including Golgi and ER that are required for synthesis, transport, and secretion of proteins associated with TLR-driven DC activation (16). These findings share strong parallels with activated T cells that heavily rely on glycolysis as a carbon source for de novo FA synthesis to support the need for membrane synthesis required for cellular proliferation (17). However, in contrast to T cells, DCs do not proliferate and seem to use this pathway to expand the cellular machinery necessary for increased production and secretion of the mediators that are integral to DC activation (Figure 1). This is consistent with a recent study positively correlating lipid content with immunogenicity of DCs in the liver and showing that the immunogenicity of DCs with high lipid content is dependent on FA synthesis (18). Taken together, these studies illustrate that the induction of glycolysis plays a central role for DCs to acquire immunogenic properties as well as their survival following activation.
While the metabolic features of immunogenic DCs are becoming more well characterized, there is still little known about the metabolism of tolerogenic DCs. Tolerogenic DCs, as opposed to immunogenic DCs, are generally characterized by the absence of traditional signs of activation, are maturation-resistant, and express increased levels of immunoregulatory factors, important for controlling Treg responses (19–21). Consistent with this immature, maturation-resistant phenotype, proteomic analysis of human DCs treated with dexamethasone and vitamin-D3, two well known immunosuppressive drugs that induce tolerogenic DCs, revealed increased expression of genes associated with mitochondrial metabolism and OXPHOS (22, 23). Furthermore, DCs conditioned by IL-4 acquire a phenotype highly reminiscent of alternatively activated (M2) macrophages and expression of M2-associated activation markers on DCs is required for optimal induction of IL-10-secreting T cells (24). The fact that M2 activation by IL-4 is dependent on increased fatty acid oxidation (FAO) and OXPHOS (25–27) makes it conceivable that there is a causal link between mitochondrial metabolism fueled by FAO and the acquisition of a tolerogenic phenotype by DCs. The observations that direct inhibition of glycolysis in TLR-activated DCs favors the induction of Foxp3-expressing Th cells at the expense of IFN-γ-producing Th1 cells (16), and that resveratrol and rosiglitazone, drugs known to promote FAO (28) and mitochondrial biogenesis (29), respectively, interfere with TLR-induced DC activation and can render them tolerogenic (30–33), would support this idea. However, these studies are mostly correlative and more work will be needed to elucidate whether there is a direct functional link between mitochondrial catabolic metabolism and the acquisition of tolerogenic properties of DCs.
Regulators of DC Metabolism
In recent years, major advances have been made in unraveling the signaling pathways in immune cells that regulate their metabolic state. The conserved kinase mammalian/mechanistic target of rapamycin (mTOR) and its upstream activators PI3K-Akt have been identified as central regulators of cellular activation and proliferation due to their ability to control glycolysis and anabolic metabolism (34–36). Consistent with a role for mTOR in regulating DC metabolism as well, cDCs isolated from mice with a DC-specific deletion of tuberous sclerosis 1 (Tsc1), a negative regulator of mTOR, display enhanced mTOR activity, an increase of expression of glycolytic and lipogenic genes, and of maturation markers at steady state (37). Also in response to TLR ligands inflammatory DCs depend on signaling through PI3K, Akt, and mTOR for their long-term commitment to glycolysis (10, 38). mTOR promotes anabolic pathways and glycolysis by driving expression and stabilization of transcription factors such as sterol-regulatory element binding protein (SREBP) (39, 40) and hypoxia-inducible factor (HIF)-1α (41), that control expression of genes involved in lipogenesis and glycolysis, respectively. While it remains unknown whether SREBP plays a role in DC metabolism, several studies have documented an important role for HIF-1α in supporting the long-term commitment in glycolytic metabolism of both inflammatory DCs and cDCs in response to TLR stimulation (11, 13). Moreover, consistent with its well-recognized role in regulating innate immune cell function under inflammatory conditions (42), TLR-induced DC activation and T cell priming appear to rely on HIF-1α (11, 13, 43). However, whether this is a consequence of the role of HIF-1α in promoting glycolytic metabolism and thereby cell survival, or a reflection of the direct control of expression of inflammatory cytokines independently from glycolytic regulation (44, 45), remains to be addressed. In addition to direct transcriptional regulation of glycolysis through HIF-1α, mTOR may regulate the TLR-induced commitment to glycolysis indirectly in inflammatory DCs, through induction of iNOS expression and NO production (46, 47) that forces these cells to switch to glycolysis in the absence of mitochondrial respiration (12).
In contrast to the clear role for the mTOR-HIF-1α axis in regulating TLR-induced long-term metabolic changes, recent evidence suggests that the early TLR-driven induction of glycolysis to support the anabolic demands of DC activation itself does not depend on mTOR or HIF-1α signaling (16, 48). Instead, there is a critical role for Akt in this response that directly enhances the enzymatic activity of rate-limiting glycolytic enzyme hexokinase-II (HK-II) by promoting its association with the mitochondria. Interestingly, Tank-Binding Kinase 1 (TBK1) and IκB kinase-(IKK) but not the canonical Akt activators PI3K or mTORC2 appear to be the crucial upstream regulators of Akt activation in this TLR-driven rapid induction of glycolysis (16). Taken together based on these recent findings a picture is emerging that TLR-signaling drives two functionally and temporally distinct waves in glycolytic metabolism in DCs that are controlled by largely separate signaling pathways (Figure 1).
While the signaling pathways that promote the shift to glycolysis and anabolic metabolism required for TLR-induced activation and immunogenicity of DCs are starting to be characterized, much less is known about the signals in DCs that may antagonize these responses and that are potentially important for induction and function of tolerogenic DCs. In this respect, in T cells and in macrophages the metabolic sensor AMP Kinase (AMPK) is known to play a central role in antagonizing biosynthetic pathways, including lipogenesis, and has instead been shown to promote catabolic metabolism by, amongst other pathways, the activation of peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α that promotes mitochondrial biogenesis to increase mitochondrial OXPHOS (7, 35). Consistent with these observations, pharmacological activation of AMPK suppresses TLR-induced glucose consumption and activation of DCs, while knockdown of AMPK has the opposite effect (10, 49), suggesting an important role for AMPK signaling in the metabolic control of DC activation. Furthermore, systemic administration of drugs activating AMPK signaling to promote catabolic metabolism drives induction of tolerogenic immune responses in several inflammatory disease models (50–52). However, it remains to be determined whether these treatments exert their effects through direct induction of tolerogenic DCs. Moreover, resveratrol, a drug that has been linked to induction of tolerogenic DCs, is thought to favor catabolic metabolism through activation of the histone deacetylase Sirtuin 1, which is known to suppress HIF-1α function as well as enhance PGC-1α activity (29, 32, 53). In addition, DCs deficient for Nuclear factor-erythroid 2 p45-related factor-2 (NRF2) or PPAR-γ, downstream targets of PGC-1α, display increased maturation and T cell priming capacity (31, 54, 55). Hence, these studies may point toward an important role for the AMPK-PGC-1α axis in promoting mitochondria-centered catabolic metabolism in DCs, which may be crucial for the acquisition of a tolerogenic phenotype (Figure 2). However, how these signaling pathways are regulated under physiological conditions and to what extent the effects of these factors on DC biology can be attributed to direct regulation of DC metabolism are still unresolved questions.
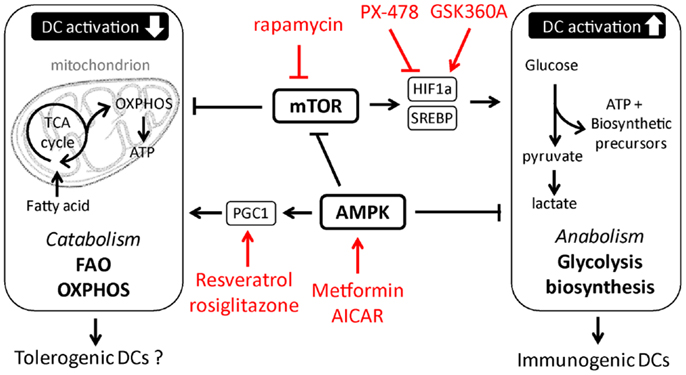
Figure 2. Putative metabolic pathways and upstream regulators in tolerogenic versus immunogenic dendritic cells. In red examples are depicted of pharmacological approaches currently tested or used in other therapeutic settings, that could be used to manipulate DC metabolism.
Manipulating DC Metabolism for Therapeutic Purposes?
There is a great interest in the use of DCs as targets for immune-intervention and for vaccine strategies, because of their powerful immune stimulatory as well as regulatory functions (56). The use of highly immunogenic DCs can be used to promote robust cellular and humoral immunity that is central for improving vaccination efficacy against a variety of infectious diseases and tumors, while the use of tolerogenic DCs will allow for induction of regulatory immune responses in settings where unwanted effector T cell responses need to be controlled, such as to prevent rejection following transplantation. It is of pivotal importance to identify and characterize the regulatory processes underpinning these different functions of DCs. It is becoming clear from the aforementioned studies that the activation and T cell-priming function of DCs is tightly regulated by their metabolic fate. What can we learn from these new metabolic insights in DC biology and would there be ways to use this knowledge in developing approaches to enhance DC-based immunotherapies? The idea of manipulating cellular metabolism for therapeutic purposes is not a new concept. In fact, in the cancer field there is great interest in the use of pharmacologicals that inhibit anabolic metabolism or glycolysis to reduce tumor growth (57–60). Likewise, studies in T cells have provided a clear proof of principle that targeting of cellular metabolism can provide a viable means for improving the efficacy of vaccinations (61, 62).
Based on the importance of anabolic metabolism and glycolysis in supporting DC activation and immunogenicity, and the possible role of catabolic metabolism in supporting tolerogenic DC function, it will be of great interest to assess whether promoting these types of metabolism in DCs can be used as a strategy to enhance the immunogenicity or tolerogenicity of DCs in therapeutic settings. It should be noted that some of the pharmacological approaches currently used to manipulate the immunogenicity of DCs, such as dexamethasone, Vitamin-D3, and rapamycin (63–66) that are known for their capacity to induce tolerogenic DCs, have been described to influence DC metabolism (22, 23, 38). Thus it is possible that direct targeting of metabolism of DCs as a single treatment may not be superior to some other already existing manipulations that also affect metabolism. It is therefore more conceivable that manipulation of metabolism of DCs for immunotherapy will be most effective when used in conjunction with existing approaches to complement and enhance their therapeutic efficacy. A second important advantage of direct enforcement of certain types of metabolism in DCs is that is it may render them more resistant to environmental metabolic manipulation. This is highly relevant since a key parameter that determines the efficacy of immunotherapies is how long targeted DCs retain their phenotype following their functional manipulation. The microenvironment, which DCs become exposed to in situ, may lead to the loss of immunogenicity or tolerogenicity and would significantly affect the outcome of the therapy. For instance, the immunostimulatory capacity of DCs is often suppressed in a tumor microenvironment (67). Given the important role for cellular metabolism in regulating DC function, many of the suppressive effects of tumors appear to be attributable to effects on DC metabolism. It has been shown that tumor-derived IL-10 can suppress glycolysis in DCs through down regulation of glycolytic enzyme pyruvate kinase (68). Additionally, yet unidentified tumor-derived factors can promote aberrant lipid accumulation in DCs, resulting in impaired T cell priming (69, 70). Moreover, immunogenic DCs are likely to be impaired in their function in a microenvironment where glucose will be scarce due to the high glycolytic rates of tumors themselves (71). Finally, caloric intake and mitochondrial activity are important determinants of organismal as well as cellular lifespan (72, 73). Therefore targeting DCs metabolism can also be used to manipulate DC longevity to affect their immunostimulatory potential. For example, mTOR inhibition has shown to increase the lifespan of TLR-activated DCs and enhance their capacity to induce protective tumor immunity (38).
Several agonist and antagonists of metabolic enzymes and upstream signaling pathways that could be used to manipulate DC metabolism have already been developed and tested for safety and efficacy in other systems (58–60, 74) (Figure 2). In addition to pharmacological approaches, genetic manipulation through introduction of small hairpin RNAs has shown to be a successful strategy to alter DC immunogenicity (75, 76) and could provide a feasible alternative to target DC metabolism. In recent years, there has been a major focus on manipulating the immunostimulatory properties ex vivo generated DCs for autologous DC vaccination. Some of these vaccines have made it to the clinic (77) or are currently in clinical trials (78–80). In addition, widespread enthusiasm has been generated by results from the in vivo use of nanoparticles, consisting of antibody covered micelles carrying antigens and potentially drugs or shRNA, that can be specifically targeted to DCs in situ (81, 82). Given the amenability of cellular metabolic intervention, it seems feasible that metabolism-targeted manipulations to DCs could be implemented in protocols for DC-based vaccinations.
Concluding Remarks
It is becoming increasingly clear that the metabolic phenotype of DCs dictates their activation and immunogenicity. However, many of the details and underlying mechanisms of how cellular metabolism controls the functional properties of DCs remain to be determined. For instance, the precise metabolic processes that underpin the function of tolerogenic DCs are still poorly defined. Moreover, do different in vivo DC subsets have different metabolic characteristics and are unique metabolic processes required for DCs to perform particular functions, such as cross presentation or the induction of Th1/2/17 cell responses? Addressing these and other questions will not only contribute to a better fundamental understanding of the biology of DCs, but will also aid in the rational design of metabolism-based approaches to enhance the efficacy of DC-based immunotherapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported by the National Institutes of Health grants to Edward J. Pearce (AI53825, CA164062), and by a Veni grant from Netherlands Organisation for Scientific Research to Bart Everts.
References
1. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
2. Vendelbo MH, Nair KS. Mitochondrial longevity pathways. Biochim Biophys Acta (2011) 1813(4):634–44. doi:10.1016/j.bbamcr.2011.01.029
3. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity (2013) 38(4):633–43. doi:10.1016/j.immuni.2013.04.005
4. Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol (2005) 5(11):844–52. doi:10.1038/nri1710
5. van der Windt GJ, Pearce EL. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev (2012) 249(1):27–42. doi:10.1111/j.1600-065X.2012.01150.x
6. Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol (2012) 33(4):168–73. doi:10.1016/j.it.2012.01.010
7. O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature (2013) 493(7432):346–55. doi:10.1038/nature11862
8. Shapiro H, Lutaty A, Ariel A. Macrophages, meta-inflammation, and immuno-metabolism. ScientificWorldJournal (2011) 11:2509–29. doi:10.1100/2011/397971
9. Joffre O, Nolte MA, Sporri R, Reis e Sousa C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev (2009) 227(1):234–47. doi:10.1111/j.1600-065X.2008.00718.x
10. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood (2010) 115(23):4742–9. doi:10.1182/blood-2009-10-249540
11. Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG, et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol (2008) 180(7):4697–705. doi:10.4049/jimmunol.180.7.4697
12. Everts B, Amiel E, van der Windt GJ, Freitas TC, Chott R, Yarasheski KE, et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood (2012) 120(7):1422–31. doi:10.1182/blood-2012-03-419747
13. Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, Longhi MP. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol (2014) 12(1):e1001759. doi:10.1371/journal.pbio.1001759
14. Dominguez PM, Ardavin C. Differentiation and function of mouse monocyte-derived dendritic cells in steady state and inflammation. Immunol Rev (2010) 234(1):90–104. doi:10.1111/j.0105-2896.2009.00876.x
15. Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett (1994) 345(1):50–4. doi:10.1016/0014-5793(94)00424-2
16. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol (2014) 15(4):323–32. doi:10.1038/ni.2833
17. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol (2013) 14(5):489–99. doi:10.1038/ni.2570
18. Ibrahim J, Nguyen AH, Rehman A, Ochi A, Jamal M, Graffeo CS, et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology (2012) 143(4):1061–72. doi:10.1053/j.gastro.2012.06.003
19. Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat Immunol (2010) 11(8):647–55. doi:10.1038/ni.1894
20. Svajger U, Obermajer N, Jeras M. Novel findings in drug-induced dendritic cell tolerogenicity. Int Rev Immunol (2010) 29(6):574–607. doi:10.3109/08830185.2010.522280
21. Maldonado RA, von Andrian UH. How tolerogenic dendritic cells induce regulatory T cells. Adv Immunol (2010) 108:111–65. doi:10.1016/B978-0-12-380995-7.00004-5
22. Ferreira GB, Kleijwegt FS, Waelkens E, Lage K, Nikolic T, Hansen DA, et al. Differential protein pathways in 1,25-dihydroxyvitamin d(3) and dexamethasone modulated tolerogenic human dendritic cells. J Proteome Res (2012) 11(2):941–71. doi:10.1021/pr200724e
23. Ferreira GB, van Etten E, Lage K, Hansen DA, Moreau Y, Workman CT, et al. Proteome analysis demonstrates profound alterations in human dendritic cell nature by TX527, an analogue of vitamin D. Proteomics (2009) 9(14):3752–64. doi:10.1002/pmic.200800848
24. Cook PC, Jones LH, Jenkins SJ, Wynn TA, Allen JE, MacDonald AS. Alternatively activated dendritic cells regulate CD4+ T-cell polarization in vitro and in vivo. Proc Natl Acad Sci U S A (2012) 109(25):9977–82. doi:10.1073/pnas.1121231109
25. Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity (2010) 33(5):699–712. doi:10.1016/j.immuni.2010.11.009
26. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab (2006) 4(1):13–24. doi:10.1016/j.cmet.2006.08.006
27. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature (2007) 447(7148):1116–20. doi:10.1038/nature05894
28. Szatmari I, Torocsik D, Agostini M, Nagy T, Gurnell M, Barta E, et al. PPARgamma regulates the function of human dendritic cells primarily by altering lipid metabolism. Blood (2007) 110(9):3271–80. doi:10.1182/blood-2007-06-096222
29. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell (2006) 127(6):1109–22. doi:10.1016/j.cell.2006.11.013
30. Iruretagoyena MI, Sepulveda SE, Lezana JP, Hermoso M, Bronfman M, Gutierrez MA, et al. Inhibition of nuclear factor-kappa B enhances the capacity of immature dendritic cells to induce antigen-specific tolerance in experimental autoimmune encephalomyelitis. J Pharmacol Exp Ther (2006) 318(1):59–67. doi:10.1124/jpet.106.103259
31. Klotz L, Dani I, Edenhofer F, Nolden L, Evert B, Paul B, et al. Peroxisome proliferator-activated receptor gamma control of dendritic cell function contributes to development of CD4+ T cell anergy. J Immunol (2007) 178(4):2122–31. doi:10.4049/jimmunol.178.4.2122
32. Svajger U, Obermajer N, Jeras M. Dendritic cells treated with resveratrol during differentiation from monocytes gain substantial tolerogenic properties upon activation. Immunology (2010) 129(4):525–35. doi:10.1111/j.1365-2567.2009.03205.x
33. Kim GY, Cho H, Ahn SC, Oh YH, Lee CM, Park YM. Resveratrol inhibits phenotypic and functional maturation of murine bone marrow-derived dendritic cells. Int Immunopharmacol (2004) 4(2):245–53. doi:10.1016/j.intimp.2003.12.009
34. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149(2):274–93. doi:10.1016/j.cell.2012.03.017
35. Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev (2012) 249(1):43–58. doi:10.1111/j.1600-065X.2012.01152.x
36. Xu X, Ye L, Araki K, Ahmed R. mTOR, linking metabolism and immunity. Semin Immunol (2012) 24(6):429–35. doi:10.1016/j.smim.2012.12.005
37. Wang Y, Huang G, Zeng H, Yang K, Lamb RF, Chi H. Tuberous sclerosis 1 (Tsc1)-dependent metabolic checkpoint controls development of dendritic cells. Proc Natl Acad Sci U S A (2013) 110(50):E4894–903. doi:10.1073/pnas.1308905110
38. Amiel E, Everts B, Freitas TC, King IL, Curtis JD, Pearce EL, et al. Inhibition of mechanistic target of rapamycin promotes dendritic cell activation and enhances therapeutic autologous vaccination in mice. J Immunol (2012) 189(5):2151–8. doi:10.4049/jimmunol.1103741
39. Bakan I, Laplante M. Connecting mTORC1 signaling to SREBP-1 activation. Curr Opin Lipidol (2012) 23(3):226–34. doi:10.1097/MOL.0b013e328352dd03
40. Im SS, Yousef L, Blaschitz C, Liu JZ, Edwards RA, Young SG, et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab (2011) 13(5):540–9. doi:10.1016/j.cmet.2011.04.001
41. Land SC, Tee AR. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem (2007) 282(28):20534–43. doi:10.1074/jbc.M611782200
42. Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell (2003) 112(5):645–57. doi:10.1016/S0092-8674(03)00154-5
43. Wobben R, Husecken Y, Lodewick C, Gibbert K, Fandrey J, Winning S. Role of hypoxia inducible factor-1alpha for interferon synthesis in mouse dendritic cells. Biol Chem (2013) 394(4):495–505. doi:10.1515/hsz-2012-0320
44. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature (2013) 496(7444):238–42. doi:10.1038/nature11986
45. Pawlus MR, Hu CJ. Enhanceosomes as integrators of hypoxia inducible factor (HIF) and other transcription factors in the hypoxic transcriptional response. Cell Signal (2013) 25(9):1895–903. doi:10.1016/j.cellsig.2013.05.018
46. Jin HK, Ahn SH, Yoon JW, Park JW, Lee EK, Yoo JS, et al. Rapamycin down-regulates inducible nitric oxide synthase by inducing proteasomal degradation. Biol Pharm Bull (2009) 32(6):988–92. doi:10.1248/bpb.32.988
47. Lisi L, Navarra P, Feinstein DL, Dello Russo C. The mTOR kinase inhibitor rapamycin decreases iNOS mRNA stability in astrocytes. J Neuroinflammation (2011) 8(1):1. doi:10.1186/1742-2094-8-1
48. Spirig R, Djafarzadeh S, Regueira T, Shaw SG, von Garnier C, Takala J, et al. Effects of TLR agonists on the hypoxia-regulated transcription factor HIF-1alpha and dendritic cell maturation under normoxic conditions. PLoS One (2010) 5(6):e0010983. doi:10.1371/journal.pone.0010983
49. Carroll KC, Viollet B, Suttles J. AMPKalpha1 deficiency amplifies proinflammatory myeloid APC activity and CD40 signaling. J Leukoc Biol (2013). 94(6):1113–21. doi:10.1189/jlb.0313157
50. Nath N, Giri S, Prasad R, Salem ML, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide ribonucleoside: a novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. J Immunol (2005) 175(1):566–74. doi:10.4049/jimmunol.175.1.566
51. Kim TB, Kim SY, Moon KA, Park CS, Jang MK, Yun ES, et al. Five-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside attenuates poly (I:C)-induced airway inflammation in a murine model of asthma. Clin Exp Allergy (2007) 37(11):1709–19. doi:10.1111/j.1365-2222.2007.02812.x
52. Kang KY, Kim YK, Yi H, Kim J, Jung HR, Kim IJ, et al. Metformin downregulates Th17 cells differentiation and attenuates murine autoimmune arthritis. Int Immunopharmacol (2013) 16(1):85–92. doi:10.1016/j.intimp.2013.03.020
53. Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol (2012) 13(4):225–38. doi:10.1038/nrm3293
54. Rangasamy T, Williams MA, Bauer S, Trush MA, Emo J, Georas SN, et al. Nuclear erythroid 2 p45-related factor 2 inhibits the maturation of murine dendritic cells by ragweed extract. Am J Respir Cell Mol Biol (2010) 43(3):276–85. doi:10.1165/rcmb.2008-0438OC
55. Aw Yeang HX, Hamdam JM, Al-Huseini LM, Sethu S, Djouhri L, Walsh J, et al. Loss of transcription factor nuclear factor-erythroid 2 (NF-E2) p45-related factor-2 (Nrf2) leads to dysregulation of immune functions, redox homeostasis, and intracellular signaling in dendritic cells. J Biol Chem (2012) 287(13):10556–64. doi:10.1074/jbc.M111.322420
56. Galluzzi L, Senovilla L, Vacchelli E, Eggermont A, Fridman WH, Galon J, et al. Trial watch: dendritic cell-based interventions for cancer therapy. Oncoimmunology (2012) 1(7):1111–34. doi:10.4161/onci.21494
57. Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis (2013) 4:e532. doi:10.1038/cddis.2013.60
58. Lee K, Kim HM. A novel approach to cancer therapy using PX-478 as a HIF-1alpha inhibitor. Arch Pharm Res (2011) 34(10):1583–5. doi:10.1007/s12272-011-1021-3
59. Dwarakanath BS, Singh D, Banerji AK, Sarin R, Venkataramana NK, Jalali R, et al. Clinical studies for improving radiotherapy with 2-deoxy-D-glucose: present status and future prospects. J Cancer Res Ther (2009) 5(Suppl 1):S21–6. doi:10.4103/0973-1482.55136
60. Choi YK, Park KG. Metabolic roles of AMPK and metformin in cancer cells. Mol Cells (2013) 36(4):279–87. doi:10.1007/s10059-013-0169-8
61. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460(7251):103–7. doi:10.1038/nature08097
62. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature (2009) 460(7251):108–12. doi:10.1038/nature08155
63. Haidinger M, Poglitsch M, Geyeregger R, Kasturi S, Zeyda M, Zlabinger GJ, et al. A versatile role of mammalian target of rapamycin in human dendritic cell function and differentiation. J Immunol (2010) 185(7):3919–31. doi:10.4049/jimmunol.1000296
64. Ohtani M, Nagai S, Kondo S, Mizuno S, Nakamura K, Tanabe M, et al. Mammalian target of rapamycin and glycogen synthase kinase 3 differentially regulate lipopolysaccharide-induced interleukin-12 production in dendritic cells. Blood (2008) 112(3):635–43. doi:10.1182/blood-2008-02-137430
65. Macedo C, Turquist H, Metes D, Thomson AW. Immunoregulatory properties of rapamycin-conditioned monocyte-derived dendritic cells and their role in transplantation. Transplant Res (2012) 1(1):16. doi:10.1186/2047-1440-1-16
66. Fischer R, Turnquist HR, Taner T, Thomson AW. Use of rapamycin in the induction of tolerogenic dendritic cells. Handb Exp Pharmacol (2009) 188:215–32. doi:10.1007/978-3-540-71029-5_10
67. Bluth MJ, Zaba LC, Moussai D, Suarez-Farinas M, Kaporis H, Fan L, et al. Myeloid dendritic cells from human cutaneous squamous cell carcinoma are poor stimulators of T-cell proliferation. J Invest Dermatol (2009) 129(10):2451–62. doi:10.1038/jid.2009.96
68. Zhang Z, Liu Q, Che Y, Yuan X, Dai L, Zeng B, et al. Antigen presentation by dendritic cells in tumors is disrupted by altered metabolism that involves pyruvate kinase M2 and its interaction with SOCS3. Cancer Res (2010) 70(1):89–98. doi:10.1158/0008-5472.CAN-09-2970
69. Cao W, Ramakrishnan R, Tuyrin VA, Veglia F, Condamine T, Amoscato A, et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J Immunol (2014) 192(6):2920–31. doi:10.4049/jimmunol.1302801
70. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med (2010) 16(8):880–6. doi:10.1038/nm.2172
71. Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol (2006) 18(6):598–608. doi:10.1016/j.ceb.2006.10.005
72. Poulsen MM, Jorgensen JO, Jessen N, Richelsen B, Pedersen SB. Resveratrol in metabolic health: an overview of the current evidence and perspectives. Ann N Y Acad Sci (2013) 1290:74–82. doi:10.1111/nyas.12141
73. Guarente L. Calorie restriction and sirtuins revisited. Genes Dev (2013) 27(19):2072–85. doi:10.1101/gad.227439.113
74. Beuck S, Schanzer W, Thevis M. Hypoxia-inducible factor stabilizers and other small-molecule erythropoiesis-stimulating agents in current and preventive doping analysis. Drug Test Anal (2012) 4(11):830–45. doi:10.1002/dta.390
75. Sioud M, Saeboe-Larssen S, Hetland TE, Kaern J, Mobergslien A, Kvalheim G. Silencing of indoleamine 2,3-dioxygenase enhances dendritic cell immunogenicity and antitumour immunity in cancer patients. Int J Oncol (2013) 43(1):280–8. doi:10.3892/ijo.2013.1922
76. Cathelin D, Met O, Svane IM. Silencing of the glucocorticoid-induced leucine zipper improves the immunogenicity of clinical-grade dendritic cells. Cytotherapy (2013) 15(6):740–9. doi:10.1016/j.jcyt.2013.02.005
77. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med (2010) 363(5):411–22. doi:10.1056/NEJMoa1001294
78. Bregy A, Wong TM, Shah AH, Goldberg JM, Komotar RJ. Active immunotherapy using dendritic cells in the treatment of glioblastoma multiforme. Cancer Treat Rev (2013) 39(8):891–907. doi:10.1016/j.ctrv.2013.05.007
79. Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care (2011) 34(9):2026–32. doi:10.2337/dc11-0472
80. Hilkens CM, Isaacs JD. Tolerogenic dendritic cell therapy for rheumatoid arthritis: where are we now? Clin Exp Immunol (2013) 172(2):148–57. doi:10.1111/cei.12038
81. Unger WW, van Beelen AJ, Bruijns SC, Joshi M, Fehres CM, van Bloois L, et al. Glycan-modified liposomes boost CD4+ and CD8+ T-cell responses by targeting DC-SIGN on dendritic cells. J Control Release (2012) 160(1):88–95. doi:10.1016/j.jconrel.2012.02.007
Keywords: metabolism, oxidative phosphorylation, mitochondria, glycolysis, TLR signaling, immunogenic dendritic cells, tolerogenic dendritic cells, immunotherapy
Citation: Everts B and Pearce EJ (2014) Metabolic control of dendritic cell activation and function: recent advances and clinical implications. Front. Immunol. 5:203. doi: 10.3389/fimmu.2014.00203
Received: 01 February 2014; Paper pending published: 02 April 2014;
Accepted: 24 April 2014; Published online: 08 May 2014.
Edited by:
Marianne Boes, University Medical Centre Utrecht, NetherlandsReviewed by:
Amir Ghaemmaghami, The University of Nottingham, UKCurdin Conrad, Centre Hospitalier Universitaire Vaudois, Switzerland
Copyright: © 2014 Everts and Pearce. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edward J. Pearce, Department of Pathology and Immunology, Washington University School of Medicine, Campus Box 8118, 660 S. Euclid Avenue, St. Louis, MO 63110, USA e-mail:ZWR3YXJkcGVhcmNlQHBhdGgud3VzdGwuZWR1