 Lillian Cohn
Lillian Cohn Lélia Delamarre
Lélia Delamarre- 1Laboratory of Molecular Immunology, Rockefeller University, New York, NY, USA
- 2Genentech, South San Francisco, CA, USA
Despite significant effort, the development of effective vaccines inducing strong and durable T-cell responses against intracellular pathogens and cancer cells has remained a challenge. The initiation of effector CD8+ T-cell responses requires the presentation of peptides derived from internalized antigen on class I major histocompatibility complex molecules by dendritic cells (DCs) in a process called cross-presentation. A current strategy to enhance the effectiveness of vaccination is to deliver antigens directly to DCs. This is done via selective targeting of antigen using monoclonal antibodies directed against endocytic receptors on the surface of the DCs. In this review, we will discuss considerations relevant to the design of such vaccines: the existence of DC subsets with specialized functions, the impact of the antigen intracellular trafficking on cross-presentation, and the influence of maturation signals received by DCs on the outcome of the immune response.
Introduction
Vaccination is the most effective way to prevent the spread of infectious diseases. We classify vaccines into two main types: preventative or therapeutic. Preventative vaccines typically elicit generation of specific antibodies and memory B cells. They are designed to block the spread of infection through these humoral immune responses (1). Alternatively, therapeutic vaccines are designed as a treatment to eradicate the cause of disease. Therapeutic vaccines are typically intended to activate or induce cytotoxic antigen-specific CD8+ T cells to eliminate virally infected cells or cancer cells. There are many conditions for which vaccination has diminished the devastating effects of disease, and the discovery of these vaccines has largely resulted from successful trial and error. However, there are many diseases for which no vaccine exists; e.g., human immunodeficiency virus, hepatitis C, malaria, and cancer. It is likely that cytotoxic CD8+ T-cell activity will be required to protect patients from these chronic conditions. For this reason, efforts are required to develop carefully designed therapeutic vaccines that will derive from our increasing understanding behind the mechanisms of the human immune system. Dendritic cells (DCs) are the antigen-presenting cells that initiate and direct adaptive immune responses, and thus are critically important in our consideration of vaccines designed to induce cellular immunity.
DCs induce and regulate immunity against pathogens, and tolerance against self-antigens and commensal microorganisms (2–4). In their immature state, DCs reside in the periphery where they are situated to recognize and capture antigens. Upon receiving an activating stimulus, DCs migrate to lymphoid organs whereby they present processed peptides derived from captured antigens to T cells in the context of major histocompatibility complex (MHC) class I or II (5). The immune response initiated by the DCs is dependent upon the context in which the antigen was captured. DCs induce tolerance under steady-state conditions, in the absence of infection or inflammation – generally in this case it is self-antigens that are processed and presented. The exact nature and state of tolerogenic DCs remain elusive. However, there is an increasing body of evidence suggesting that microenvironmental signals condition DCs to become tolerogenic (6). In this process, beta-catenin activation appears to play a central role (7–10), although other mechanisms also contribute to tolerance induction (8). In the presence of inflammatory signals, such as microbial products, proinflammatory cytokines, and other endogenous signals, DCs undergo a process called maturation. DC maturation is associated with dramatic functional and morphological changes that lead to an optimized ability to initiate T-cell immunity. It is characterized by an increase in cell surface expression of MHCI and MHCII molecules and accessory/costimulatory molecules, increased antigen processing, and induction of specific cytokine production (5). Maturation depends on both the nature of the stimuli and its extent and combination (11). Additionally, the DC compartment is diverse and contains different cell types with both conserved and unique functions and specialties. Indeed, different DC subsets possess different capacity for antigen presentation, cytokine production, and microbial sensing (12). Thus, it seems that different types of immune responses are initiated by specialized DC subsets.
The critical role of DCs to activate CD8+ T cells makes them an attractive target for vaccination against intracellular pathogens and diseases for which cellular immunity seems to be a crucial part of the immune response. One approach is cell-based immunotherapy with ex vivo generated DCs loaded with antigens (13). This approach however is laborious and expensive, and thus far clinical results have been limited. Another more promising approach to direct DCs involves selective targeting to DC-specific endocytic receptors by monoclonal antibody coupled or fused to a desired antigen. These complexes are internalized by the DCs, trafficked through the intracellular vesicular system, processed, and the antigenic peptides are loaded onto MHC and presented to T cells (14, 15). In mice, in the presence of adjuvant, these antigen–antibody conjugates induce robust immune responses (16). However, in the absence of adjuvant, these conjugates can promote a tolerogenic state (17). This in situ targeting strategy is in its infancy in human patients. The first clinical trials to evaluate this vaccine approach are in progress and their preliminary results are encouraging (18–20). Recent progress in understanding the biology of DCs should further help with optimization of a DC-targeted vaccine strategy: (1) identification of the human DC subsets with superior capacity at initiating CD8+ T-cell responses if any, (2) selection of the receptors based on expression pattern to target the desired DC subset(s), and also their ability to deliver antigen to intracellular compartments for processing and loading on MHC and (3) choice of the adjuvant(s) to induce the desired immune response. In this review, we will discuss the issues relevant to human vaccination through in vivo DC targeting: the existence of multiple DC subsets with specialized functions, how DCs handle external antigen for presentation on MHCI and the intracellular targeting that induces optimal immune responses, and finally the role of DC maturation signals in orchestrating the immune outcome.
Dendritic Cell Subsets
Increasingly it has become apparent that there exists a division of labor among DC subsets in both mice and in humans (12, 21, 22). The number of DC subsets identified, and the functional studies performed both in vivo in mice and in vitro using isolated DC subsets from humans yield evidence for specialization in T-cell priming and induction of immune responses, although the functions of the different DC subsets can partially overlap.
While the mouse DC network has been quite well characterized, until recently thorough studies with human blood DCs have been difficult due to their paucity in the blood and the difficulty to access human tissues. However recent genome-wide expression profiling studies helped identify the potential human counterparts to the mouse DC subsets (23, 24).
Human and mouse DCs can be divided in two main subsets: plasmacytoid DCs (pDCs) and conventional/myeloid DCs (mDCs) (Figure 1). pDCs play a crucial role against viral infection by producing vast amounts of type I interferon in response toll-like receptors (TLR) 7 and 9 and intracellular sensor triggering (25). pDCs have been shown to be rather poor at antigen presentation in comparison to mDCs (26–28), although recent studies suggest that efficient antigen delivery to pDCs via endocytic receptors can lead to robust presentation on both MHCI and MHCII (29–31). However, the influence of antigen presentation by pDCs in vivo has yet to be understood. Additionally, in mice there is evidence that suggest pDCs play a major role in the generation of tolerance (32, 33). Whether this is true for human pDCs is still unknown.
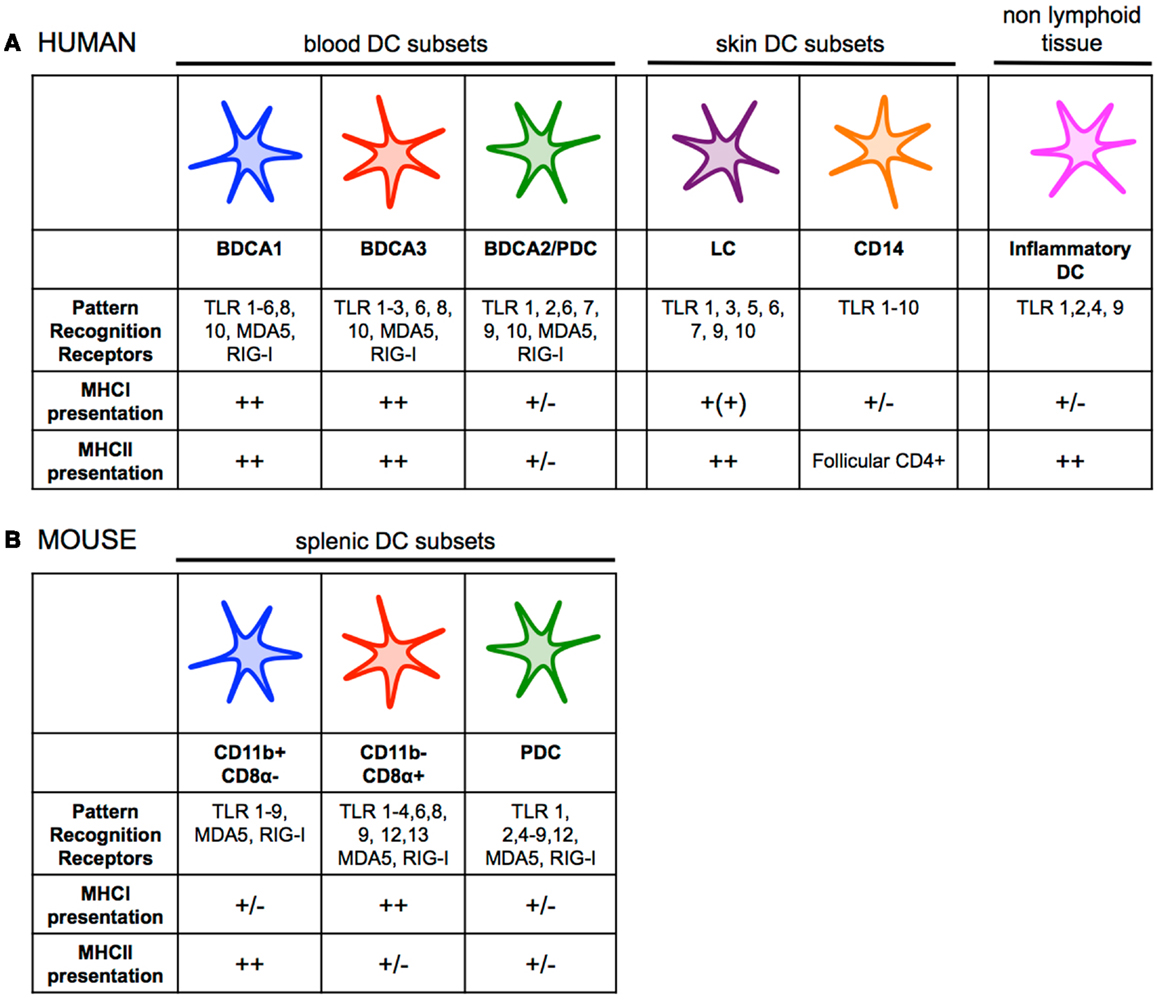
Figure 1. (A) Human dendritic cell subsets have overlapping functions and phenotypes, but also show some degree of specialization. BDCA1+ DCs and BDCA3+ DCs both efficiently present antigen on MHCI and MHCII. pDCs can present antigen to CD4+ and CD8+ T cells, but likely their primary role in the immune response is the production of type I interferon during viral infection. LCs seem to be specialized for cross-presentation on MHCI, while CD14+ dermal DCs prime naïve CD4+ T cells to generate follicular helper T cells. Inflammatory DCs are monocyte-derived, and are present at sites of inflammation. There is also partial overlap between expression of PRRs among DC subsets. (B) A clear division of labor exists among mouse splenic dendritic cell subsets. CD11b− CD8α+ DCs are far superior and essential at priming CD8+ T-cell responses, while CD11b+ DCs are specialized for presenting antigen on MHCII to stimulate helper T-cell immunity. pDCs can present antigen to CD4+ and CD8+ T cells, but likely their primary role in the immune response is the production of type I interferon during viral infection like their human counterparts. There is overlap between expression of PRRs among DC subsets, although CD11b− CD8α+ DCs express much higher levels of TLR3 while CD11b+ DCs uniquely express TLR5 and TLR7 (30, 35, 41, 58, 64, 147–151).
Human mDCs can be divided into two main subsets based on the surface markers BDCA1/CD1c or BDCA3/CD141. A transcriptional comparison of mDCs has shown genetic similarity between human BDCA1+ DCs and BDCA3+ DCs from various tissues to murine CD11b+ and CD11b− DCs, respectively (23, 34–36). Human BDCA3+ DCs express a number of markers unique to mouse CD11b− CD8α+ and CD11b− CD103+ DCs including the lectin receptor Clec9A/DNGR1, the chemokine receptor XCR1, and Necl2 (37–39). Further, human BDCA3+ DCs and mouse CD11b− CD8α+ DCs share the expression of the transcription factors IRF8, BATF3 essential for their development (35, 40–43). Conversely, the transcriptional programing of mouse CD11b+ CD8α− DCs and human BDCA1+ is dependent on IRF4 (44, 45). Functional studies of the mouse and human mDCs revealed differences between the two species, however. A clear division of labor exists among the two mDC subsets in mice with CD11b− CD8α+ DCs and CD11b− CD103+ DCs being far superior and essential at priming CD8+ T-cell responses, while CD11b+ CD8α− DCs are specialized for presenting antigen on MHCII to stimulate helper T-cell immunity (12, 46, 47). This division of labor does not appear as clear between BDCA3+ DCs and BDCA1+ DCs at least in in vitro studies. Indeed both BDCA1+ DCs and BDCA3+ DCs can effectively cross-present antigens on MHCI (28, 31, 37, 38, 40, 41, 48–52). In addition, BDCA1+ DCs also produce high levels of IL-12 upon stimulation, a cytokine essential to inducing Th1 response and cross-priming of CD8+ T cells (28, 44, 48, 53, 54). BDCA3+ DCs and BDCA1+ DCs also exhibit a comparable capacity to present antigen on MHCII (28, 31). The skin contains two additional DC subsets that have been functionally characterized, the Langerhans cells (LCs) and the CD14+ DCs (36, 55). CD14+ DCs appear specialized in initiating humoral immune responses, while in vitro-derived LCs cross-present antigen on MHCI and prime CD8+ T cells of higher avidity as compared to CD14+ dermal DCs in vitro (26, 55). A side-by-side comparison of in vitro-derived LCs with CD14+ DCs suggests the two DC subset have similar capacity for cross-presentation (36). Importantly, LCs isolated from skin are incapable of cross-presentation of captured antigen, while they can present antigen on MHCII to CD4+ T cells (36, 56). Whether this deficiency is the result of the isolation procedure or a true characteristic of LCs remains to be confirmed.
Finally, the human equivalent of mouse inflammatory DCs was recently identified (57, 58). This DC subset is found in inflammatory microenvironments and can be divided into two main populations: CD16+ BDCA1+ DCs or CD16− BDCA1+ DCs. They have characteristic gene patterns similar to that of DCs and macrophages, and thus are likely derived from monocytes. Although there are limited data on the functional specialization of human inflammatory DCs, they appear highly plastic like their murine counterparts (57, 58).
One limitation of the studies aimed at characterizing the functional capacity of human DCs is that they are performed in vitro using T-cell lines or memory T cells. These assays permit to evaluate the DCs’ capacity for antigen presentation. However, other factors are also important for DC function in vivo and priming of immune responses. The enhanced capacity of LCs to prime CD8+ T-cell responses may at least partially result from their ability to express IL-15 upon maturation (59, 60). The costimulatory molecule CD70 also promotes the priming of CD8+ T-cell responses and the generation of CD8+ T-cell memory (61–63). CD70 has been found to be expressed on LCs and all three blood DCs subsets upon maturation [(64, 65); Delamarre, personal communication]. Finally, DC function may depend on environmental cues, resident BDCA3+ DCs constitutively produce IL-10, possibly in a vitamin D3-dependent manner, and thus mediate T-cell tolerance rather than immunity at steady-state (66). Granulocyte–macrophage colony stimulating factor (GMCSF) has recently been shown to enhance the cross-presentation capacity of mouse CD11b− CD8α+ DCs (67, 68).
Based on our current knowledge, there is no strong rational for the targeting of one DC subset over another to prime CD8+ T-cell responses in humans. Further in vivo studies are needed to identify the DC subsets if any that are specialized in cross-priming of CD8+ T cells. In this effort, it would be useful to better characterize DC subsets in non-human primates which appear to possess subpopulations of DCs that are similar to those present in humans (69) and therefore would be a more relevant model to humans than mice. Additionally, engagement of multiple DC subsets has been suggested to be important in generating a broad and potent T-cell response (70). For this reason, it may make sense to target a broad spectrum of DC subsets rather than a single DC subset.
Antigen Cross-Presentation Pathways
In the design of rational DC-targeted vaccines, there are important considerations related to the delivery of antigen to DCs and the downstream processing of antigen by DCs. Delivery of antigen to DCs is essential to generate strong and prolonged T-cell responses. DCs are able to non-specifically phagocytose and macropinocytose pathogen-associated antigen and can also uptake antigen more specifically via lectin receptors, Fcγ receptors, and scavenger receptors (5). It has been shown that antigens can be efficiently targeted to DCs using antibodies against these endocytic receptors (15, 71). This takes advantage of antibodies against DC-specific endocytic receptors either coupled or fused to antigen or attached to nanoparticles containing antigen. In mice, this delivery method is hundreds of times more efficient and potent than untargeted antigens and offers options for antigen presentation on both MHCI and MHCII to CD8+ and CD4+ T cells, respectively (72). In addition, this strategy can also extend antigen cross-presentation to pDCs, which display poor phagocytosis and macropinocytosis capacity, and thus could potentially further promote T-cell responses in vivo (28–31). Another benefit of employing this strategy for antigen delivery is that it can allow for delivery to both immature and mature DCs. Unlike the non-specific phagocytosis and macropinocytosis, endocytic receptor-driven uptake continues even after DC maturation (73, 74). It would be best to selectively target DCs to reduce the dose of antigen required, while additionally limiting cross-presentation by other cell types. Indeed B cells and other non-hematopoietic cells can cross-present exogenous antigens, albeit with less efficiency than DCs, and induce peripheral tolerance under steady-state conditions and could potentially negatively impact vaccination efficacy (75–78). In addition, the binding of a target receptor by non-DCs may trigger a signaling pathway and thus may potentially have unwanted side effects.
DC subsets express different pattern of endocytic receptors and therefore the choice of receptor will determine which DC subsets are delivered antigen (Table 1). The choice of receptor also matters for other reasons. Some receptors can trigger DC maturation and induce immune responses of various natures as further discussed in the next section. In addition, they determine antigen intracellular trafficking that impacts antigen fate (28, 79). Some antibodies may also differentially alter antigen cross-presentation by modulating receptor trafficking (80). Antigen processing and loading on MHCI and MHCII happens in distinct intracellular compartments. For presentation on MHCII, antigen processing and loading occurs in the endosomal compartments, and peptide–MHCII complexes are transported to the plasma membrane (5).
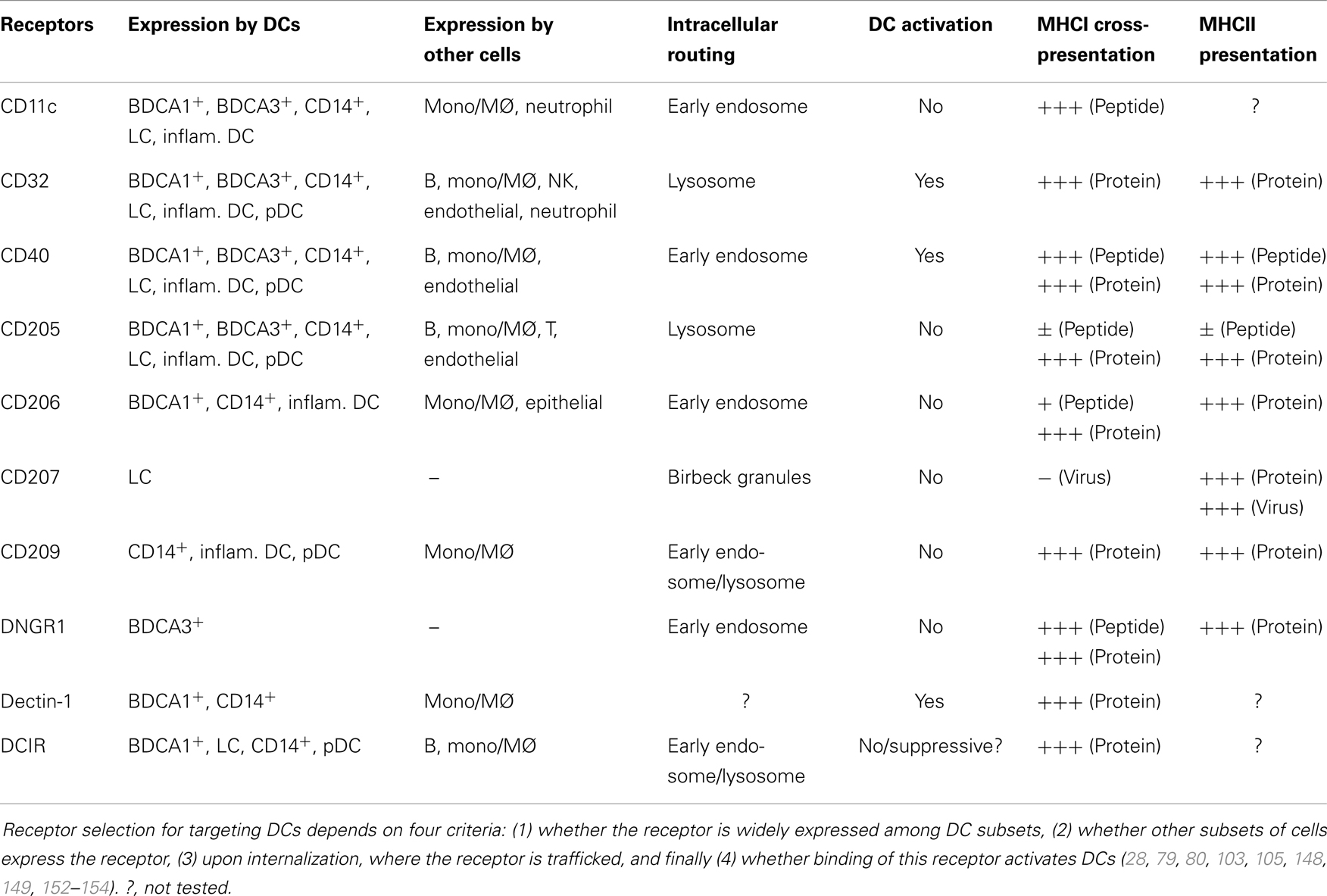
Table 1. Expression, intracellular localization, and ability to deliver antigen to MHCI and MHCII pathways of selected endocytic receptors and antigen.
Two main intracellular pathways for the cross-presentation of exogenous antigen on MHCI have been reported. They are referred to as the “cytosolic” and “vacuolar” pathways (Figure 2) (81, 82).
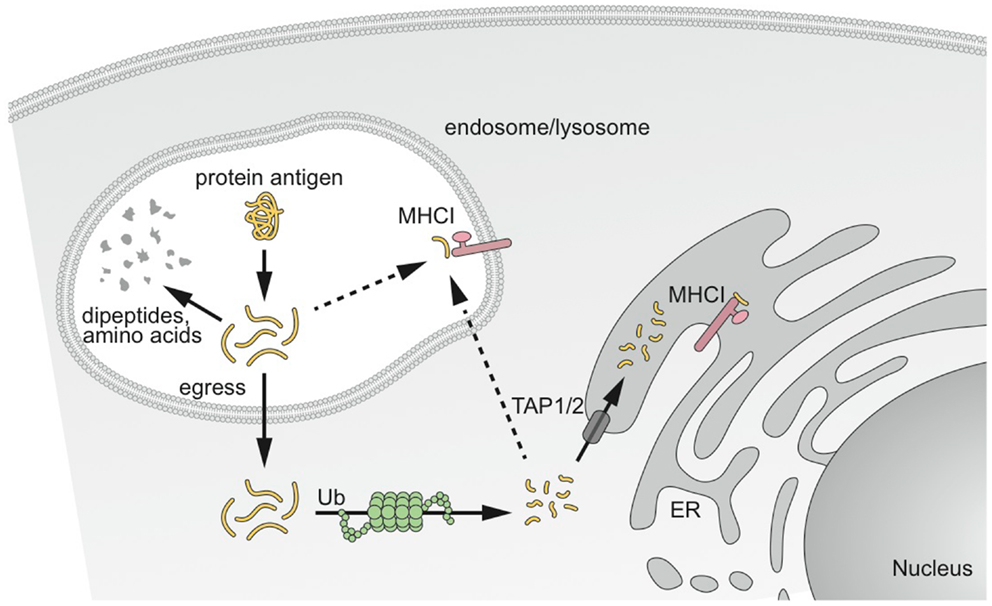
Figure 2. MHCI cross-presentation pathways of captured antigens. Antigen captured by DCs has different potential fates. Antigens destined for cross-presentation on MHCI have two different intracellular routes. Antigen can be transported from the endocytic vesicles to the cytosol to access the classical MHCI pathway involving proteasomal degradation and transport into the ER or back into the endosomal compartment for loading onto MHCI. The second pathway results in degradation and loading directly in endosomal compartments before peptide–MHCI complexes are transported to the plasma membrane. Modified from Delamarre and Mellman (14).
From extensive work with human and mouse DCs, the “cytosolic pathway” appears the most predominant pathway. It is proteasome-dependent, and therefore requires that internalized proteins escape the intracellular trafficking pathway and access the cytosol, where they are processed by the proteasome and transported into the ER and possibly in endocytic compartments by TAP1/2 transporters for loading onto MHCI (83–85). The molecular mechanism underlying transport of antigen from endocytic compartments to cytosol remains largely unknown. No specific transporter has been identified yet, despite substantial efforts from different laboratories. A role of the ER-associated degradation (ERAD) machinery has been suggested in antigen export to the cytosol (86, 87). Consistent with this finding, the recruitment of ER-resident proteins to the phagosomes, via the ER molecule Sec22b, is required for cross-presentation (88). Regardless of the exact mechanism, antigen transfer to the cytosol is rate-limiting to antigen access to the MHCI pathway. When the antigen actively gains access to the cytosol using listeriolysin O or a fusogenic virus, cross-presentation is 10-fold more efficient (28). ISCOMATRIX adjuvant, a saponin-based adjuvant, which disrupts lysosomal membranes and facilitates antigen translocation to the cytosol also enhances antigen cross-presentation (89).
The “vacuolar pathway” is dependent upon lysosomal proteolysis by cathepsins and IRAP (90, 91) and independent of the proteasome and TAP1/2 transporters. Exogenous antigens are degraded directly in endocytic compartments by lysosomal proteases and trimmed for loading onto MHCI.
The reason why certain antigens are cross-presented by one pathway rather than the other is unknown. The nature and the form of the antigen, and the ability of the proteolytic environment to generate MHCI epitopes are certainly contributing factors (90). Maybe counter intuitively, antigen intracellular targeting does not appear to influence the intracellular-processing pathway for cross-presentation in human blood DCs as cross-presentation of antigen required proteasomal processing independently of its intracellular targeting (79).
A feature essential to the ability of DCs to efficiently present antigens on MHCI and MHCII is their reduced ability for endosomal degradation. Although proteolysis is essential to the generation of MHC peptides, too much proteolytic activity leads to complete protein degradation into amino acids. Indeed, DCs are distinguished from other phagocytic cells (e.g., macrophages) by a remarkably low expression level of lysosomal proteases and a high lysosomal pH (92–94). The antigen susceptibility to degradation even by these reduced levels of proteases is a determinant factor to the efficiency at which MHCII–peptide complexes can be generated (95). Studies performed with murine DCs suggest that the MHCI pathway may be even more sensitive to lysosomal degradation. Indeed, inhibition of lysosomal proteases promotes antigen cross-presentation (96, 97). Murine CD11b− CD8α+ DCs, which exhibit an increased ability for cross-presentation in comparison to the CD11b+ CD8α− DCs, also generate high levels of reactive oxygen species in a NOX-2-dependent fashion so that their endocytic compartments stay at a more alkaline pH, thereby limiting antigen destruction (98). In addition, this phenomenon may also act to weaken or disrupt the vesicular membrane (99). As a result, antigen transport in the cytosol is increased. In addition, CD11b− CD8α+ DCs also have higher levels of lysosomal inhibitors and lower levels of lysosomal proteases than CD11b+ CD8α− DCs (46, 100). The constitutive activation of IRE-1α, a sensor of ER stress, is also a unique feature of CD11b− CD8α+ DCs and appears essential to antigen cross-presentation (101). The precise mechanism by which activated IRE-1α promotes the MHCI cross-presentation pathway remains to be elucidated. At least, some of the features of the murine CD11b− CD8α+ DCs are shared by human tonsil resident BDCA3+ DCs but also BDCA1+ DCs, both of which display similar cross-presentation capacity (51). Additionally, the three DC subsets efficiently export internalized proteins to the cytosol. However, another study found that blood BDCA3+ DCs superior at cross-presenting antigen delivered to lysosomes (28). Furthermore, blood BDCA3+ DCs express lower levels of lysosomal proteases than BDCA1+ DCs, suggesting that perhaps enhanced antigen release into the cytosol is favored by reduced lysosomal degradation. The lysosomal pH of blood DCs was not measured, and in the aforementioned study intracellular targeting of the antigen was not characterized. Further analysis will be needed to determine if different BDCA3+ DC subsets display different properties.
Finally, recent studies from our group and others suggest that both early and late endosomal compartments are capable of serving as antigen portals for cytosolic entry and cross-presentation. However, early endosomal compartments appear to be far more efficient for some antigens. This is not dependent on internalization levels, but rather the low proteolytic activity of early endosomes (28, 79, 80, 97, 102). Surprisingly, there does not seem to be a direct correlation between the level of internalization and cross-presentation. CD40 and mannose receptor/CD206 both deliver antigen to early endosomes, but CD40, the receptor that is the least efficiently internalized, turns out to be the most efficient at promoting cross-presentation (79). Slow antigen internalization might preserve antigen and provide a continuous “time-release” pool of antigen that might be used over extended periods for the continuous formation of peptide–MHCI complexes. The importance of targeting antigen to compartments with low proteolytic activity most likely depends on the nature antigen and its stability. Chatterjee et al. used long peptides as antigen which are particularly susceptible to degradation and probably have reduced ability to survive long enough to escape into the cytosol. Protein antigens, however, may be inherently more resistant. This could explain why in some systems antigen delivered to lysosomes using DEC205 or FcγR, are efficiently cross-presented, with similar or better efficacy as antigen delivered to early endosomes via mannose receptor/CD206 (103–105).
Collectively, the data reviewed in this section indicate that targeting receptors for antigen delivery to DCs can promote CD8+ T-cell responses by increasing the amount of antigen delivered to the desired DC subset(s). It can also enhance antigen presentation by controlling its intracellular routing and degradation, and extend antigen cross-presentation to DCs that might not be optimally equipped.
Adjuvant
In absence of stimulation at steady-state DCs can induce tolerance. Antigen inoculation in absence of adjuvant leads to T-cell anergy or T-cell deletion (17, 72), and can induce regulatory T cells in the periphery (106–109). Hence, in vivo delivery of antigens to DCs in absence of adjuvant may also be a promising strategy to treat autoimmune disorders as reviewed elsewhere (110). But, to induce immunity rather than tolerance, it is essential to provide the DCs with an activation signal or “adjuvant” in addition to the vaccine antigen. Conserved components of microorganisms, or pathogen-associated molecular patterns (PAMPs) have been best characterized for their ability to activate DCs and their discovery offers the prospect of developing new vaccine adjuvants. PAMPs are recognized by pattern recognition receptors (PRRs) of the innate immune system. PRRs comprise a variety of receptors, including TLRs, cytosolic receptors [nucleotide-binding oligomerization domain-like (NOD-like) receptors (NLRs), RIG-I-like receptors (RLRs)], and C-type lectin receptors (111, 112). Activation of PPR signaling in DCs results in the enhancement of antigen presentation on MHCI and MHCII, cytokine production, and the upregulation of costimulatory molecules that are necessary for the induction of T-cell responses (5). Importantly, the nature of the adjuvant determines the type, the magnitude, the breadth, and the quality of the adaptive immune response. Differential patterns of expression of PRRs among DC subsets and different cytokine profiles induced by the triggering of distinct PRRs account for much of the diversity of phenotypes of the immune response (111, 113, 114) (Figure 1). Adding yet another level of complexity, adjuvants that trigger different pathways within a cell (115–117), or stimulate multiple cell types can cooperate to further enhance immune responses (70, 114, 118). In addition to PPRs, it was recently found that induction of stress response through sensing of amino acid starvation in DCs initiates autophagy and enhances MHCI cross-presentation (119). Stress sensors could therefore be possibly targeted to potentiate adjuvants.
The use of the mouse model to study and select adjuvants for human vaccine is limited because the pattern of expression of PRR can significantly differ between the two species. Because non-human primates express a similar repertoire of TLRs on immune cells to humans, they are a more relevant model to evaluate adjuvant effects (120, 121). While most adjuvants can induce antibody responses, generation of CD8+ T-cell immunity has proved particularly difficult (122). Immunization studies in non-human primates showed that Poly ICLC which stimulate multiple PPRs (TLR3, RIG-I, and MDA-5) and TLR7/8 agonists are currently the most potent known adjuvants for induction of T helper 1 and CD8+ T-cell responses (123–126). Poly ICLC and TLR7/8 agonist are the only TLR ligands capable of inducing both IL-12 and type I interferon, which are required for efficient cross-priming (53, 70, 114, 118). In mice, multiple cell types need to be stimulated for the production of IL-12 and type I interferon. IL-12 is produced by mDCs in response to Poly ICLC (through TLR3 triggering) and TLR7/8 agonist stimulation, whereas type I IFN is largely produced non-hematopoietic cells in response to Poly ICLC stimulation through MDA-5, and pDCs in response to TLR7/8 agonist, respectively. However, in mice reconstituted with a human immune system IL-12p70 and type I IFN production after TLR3 ligand stimulation resulted mainly from BDCA3+ DCs (53). Even more surprising is that those BDCA3+ DCs produce similar amounts of type I interferon as pDCs. These results are conflicting with those obtained after in vitro stimulation of BDCA3+ DCs isolated from human blood and human tissues which produce only limited amount type I interferon (28, 41). Further studies will be needed to confirm this observation. Another potential benefit of those TLRs is that they appear broadly expressed on human mDC subsets (Figure 1), and therefore they can engage multiple DC subsets, which has been shown to improve T-cell responses (70). Multiple clinical studies have been initiated to evaluate Poly ICLC and TLR7/8 agonists as vaccine adjuvants which will help establish their potency in humans (www.clinicaltrials.gov).
The co-delivery of adjuvant and antigen to DCs is critical for the priming of the immune response. Co-delivery has been realized by coupling antigen to adjuvant (127–129), fusing antigen to protein adjuvant, or co-encapsulation in particles (130–132), and has lead to significant increase in the magnitude of the immune responses and a better quality immune response (127). This enhanced T-cell priming may result from multiple effects: increased antigen uptake, altered intracellular routing, increased stability of the TLR agonist. The adjuvant effect may be even better achieved if the adjuvant and the antigen co-localize in the same endosomal compartments, as TLRs control MHCII presentation only in the compartments in which they are present (133, 134). Another benefit of coupled vaccines may be the local retention of the adjuvant at the site of injection, and thus the reduction of their toxicity. Indeed, free TLR agonists rapidly leave the site of injection and induce systemic innate responses resulting in high levels of serum cytokines (114). A more direct and controlled approach to reduce unwanted systemic effects of TLR agonists is to engineer their targeted delivery to DCs, although it might affect adjuvant effectiveness if activation of bystander cells contributes to the immune response (70, 118). Delivery of poly ICLC and TLR7/8 agonists through DEC205 or CD209 enhances DC activation and CD8+ T-cell response in mice. Moreover, potent CD8+ T-cell responses can be achieved with doses of adjuvant that do not induce toxic high serum cytokine levels (132).
Receptors other than TLRs have been shown to trigger DC activation. They are attractive due to their stimulatory capacity and their endocytic capacity that offer the potential of using a single molecule to deliver both antigen and activation signal to DCs. Dectin-1, a receptor involved in anti-fungal immunity, is a syk-coupled C-type lectin receptor that stimulate DC through its ITAM-like domain (112). Antigen delivery to human monocyte-derived DCs and BDCA1+ DCs through Dectin-1 leads to enhanced MHCI cross-presentation and cell activation in vitro (135, 136). However, mouse immunization studies suggest that Dectin-1 may be more potent at priming CD4+ T-cell responses than CD8+ T-cell responses (137). A more promising receptor may be the CD40 receptor, which is expressed by all DC subsets. Not only does it efficiently deliver antigen to the MHC presentation pathways in DCs (28, 79), but its ligation induces DC stimulation and promotes cross-presentation (138, 139). Immunization studies confirmed that anti-CD40 agonistic antibody/Ag conjugates can prime CD8+ T-cell responses in mice (140, 141). However, the use of agonist anti-CD40 antibodies in vaccine formulation may be limited by a narrow therapeutic window. CD40 is broadly expressed on B cells, monocytes, platelets, and endothelial cells, and CD40 ligation can induce high serum cytokine levels (142). It will be important to compare anti-CD40 antibodies with different agonistic function. Anti-CD40 with weaker agonistic function may be better tolerated and therefore allow higher antigen payload and vice versa for strong agonists. How this will impact the outcome of the immune response remains to be determined. CD32/FcγRII cross-linking also induces DC maturation and efficient antigen cross-presentation after immune complex internalization (73, 105, 143). Like CD40, it has the advantage of targeting most DCs, but could induce some toxicity because of its broad expression on other cells.
Conclusion
Recent advances in DC biology and the mechanisms controlling adaptive immune responses have offered new insights for the rational design of novel vaccines. Immunization studies in mice indicate that there is a clear benefit to the targeting of antigens to DCs. A major challenge, however, remains to translate this approach developed in mice to humans. The preliminary data obtained from the first clinical trials testing vaccines targeting DEC205 (CDX-1401, Celldex) and mannose receptor/CD206 (CDX-1307, Celldex) indicate that this strategy can elicit immune responses (18–20), but maybe not as strong as one could have expected based on the mouse data. One explanation is that immunologist’s favorite model antigen for mouse studies is ovalbumin, which is exceptionally immunogenic, and may lead to overestimating vaccine efficacy. Mouse and human immune systems have also significant differences that make translation difficult (144). Although the intracellular mechanisms involved in antigen cross-presentation pathway and the DC lineage appear conserved between the two species, the specialization of the DC subsets may not be conserved. In addition, the pattern of expression of endocytic receptors for antigen delivery and TLRs for DC activation are different between mice and humans. Clearly, using a different model such as mice with a reconstituted immune system or non-human primates, which have a human immune system more similar to the human immune system is essential to optimize these vaccines. Additionally, analysis of the immune response to successful human viral vaccines that induce potent CD8+ T-cell responses could help further determine the mechanisms that control immune responses to vaccination and identify predictors of vaccine efficacy (145).
Another challenge specific to the therapeutic treatment of cancer and maybe persistent viral infection is that they developed mechanisms to evade immune clearance by impairing T-cell function (146). The presence of these suppressive factors may limit vaccine efficacy, and combination of a vaccine with immunomodulatory molecules to neutralize inhibitory signals may be necessary to produce effective T-cell immune response.
In spite of these challenges, we view the present as an exciting time to study vaccine development and foresee that continuing to design DC-based therapies will allow us to prevent and treat many of the major illnesses for which no vaccine currently exists.
Conflict of Interest Statement
Lélia Delamarre is an employee of Genentech, and hence declares a competing financial interest. Lillian Cohn declares no conflict of interest.
Acknowledgments
The authors thank Allison Bruce (Genentech) for excellent assistance with artwork.
References
1. Pulendran B, Ahmed R. Immunological mechanisms of vaccination. Nat Immunol (2011) 12:509–17. doi: 10.1038/ni.2039
2. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392:245–52. doi:10.1038/32588
3. Lanzavecchia A, Sallusto F. Regulation of T cell immunity by dendritic cells. Cell (2001) 106:263–6. doi:10.1016/S0092-8674(01)00455-X
4. Scott CL, Aumeunier AM, Mowat AM. Intestinal CD103+ dendritic cells: master regulators of tolerance? Trends Immunol (2011) 32:412–9. doi:10.1016/j.it.2011.06.003
5. Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol (2005) 23:975–1028. doi:10.1146/annurev.immunol.22.012703.104538
6. Manicassamy S, Pulendran B. Dendritic cell control of tolerogenic responses. Immunol Rev (2011) 241:206–27. doi:10.1111/j.1600-065X.2011.01015.x
7. Jiang A, Bloom O, Ono S, Cui W, Unternaehrer J, Jiang S, et al. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity (2007) 27:610–24. doi:10.1016/j.immuni.2007.08.015
8. Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, Wang YC, et al. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science (2010) 329:849–53. doi:10.1126/science.1188510
9. Liang X, Fu C, Cui W, Ober-Blobaum JL, Zahner SP, Shrikant PA, et al. beta-Catenin mediates tumor-induced immunosuppression by inhibiting cross-priming of CD8+ T cells. J Leukoc Biol (2014) 95:179–90. doi:10.1189/jlb.0613330
10. Shan M, Gentile M, Yeiser JR, Walland AC, Bornstein VU, Chen K, et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science (2013) 342:447–53. doi:10.1126/science.1237910
11. Macagno A, Napolitani G, Lanzavecchia A, Sallusto F. Duration, combination and timing: the signal integration model of dendritic cell activation. Trends Immunol (2007) 28:227–33. doi:10.1016/j.it.2007.03.008
12. Heath WR, Carbone FR. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat Immunol (2009) 10:1237–44. doi:10.1038/ni.1822
13. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer (2012) 12:265–77. doi:10.1038/nrc3258
14. Delamarre L, Mellman I. Harnessing dendritic cells for immunotherapy. Semin Immunol (2011) 23:2–11. doi:10.1016/j.smim.2011.02.001
15. Tacken PJ, Figdor CG. Targeted antigen delivery and activation of dendritic cells in vivo: steps towards cost effective vaccines. Semin Immunol (2011) 23:12–20. doi:10.1016/j.smim.2011.01.001
16. Trumpfheller C, Longhi MP, Caskey M, Idoyaga J, Bozzacco L, Keler T, et al. Dendritic cell-targeted protein vaccines: a novel approach to induce T-cell immunity. J Intern Med (2012) 271:183–92. doi:10.1111/j.1365-2796.2011.02496.x
17. Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med (2001) 194:769–79. doi:10.1084/jem.194.6.769
18. Morse MA, Chapman R, Powderly J, Blackwell K, Keler T, Green J, et al. Phase I study utilizing a novel antigen-presenting cell-targeted vaccine with Toll-like receptor stimulation to induce immunity to self-antigens in cancer patients. Clin Cancer Res (2011) 17:4844–53. doi:10.1158/1078-0432.CCR-11-0891
19. Morse MA, Bradley DA, Keler T, Laliberte RJ, Green JA, Davis TA, et al. CDX-1307: a novel vaccine under study as treatment for muscle-invasive bladder cancer. Expert Rev Vaccines (2011) 10:733–42. doi:10.1586/erv.11.20
20. Riedmann EM. CDX-1401 combined with TLR agonist: positive phase 1 results. Hum Vaccin Immunother (2012) 8:1742. doi:10.4161/hv.23373
21. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol (2013) 31:563–604. doi:10.1146/annurev-immunol-020711-074950
22. Haniffa M, Collin M, Ginhoux F. Ontogeny and functional specialization of dendritic cells in human and mouse. Adv Immunol (2013) 120:1–49. doi:10.1016/B978-0-12-417028-5.00001-6
23. Crozat K, Guiton R, Guilliams M, Henri S, Baranek T, Schwartz-Cornil I, et al. Comparative genomics as a tool to reveal functional equivalences between human and mouse dendritic cell subsets. Immunol Rev (2010) 234:177–98. doi:10.1111/j.0105-2896.2009.00868.x
24. Haniffa M, Collin M, Ginhoux F. Identification of human tissue cross-presenting dendritic cells: a new target for cancer vaccines. Oncoimmunology (2013) 2:e23140. doi:10.4161/onci.23140
25. Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol (2011) 29:163–83. doi:10.1146/annurev-immunol-031210-101345
26. Klechevsky E, Flamar AL, Cao Y, Blanck JP, Liu M, O’Bar A, et al. Cross-priming CD8+ T cells by targeting antigens to human dendritic cells through DCIR. Blood (2010) 116:1685–97. doi:10.1182/blood-2010-01-264960
27. Smed-Sorensen A, Chalouni C, Chatterjee B, Cohn L, Blattmann P, Nakamura N, et al. Influenza A virus infection of human primary dendritic cells impairs their ability to cross-present antigen to CD8 T cells. PLoS Pathog (2012) 8:e1002572. doi:10.1371/journal.ppat.1002572
28. Cohn L, Chatterjee B, Esselborn F, Smed-Sorensen A, Nakamura N, Chalouni C, et al. Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3+ dendritic cells at cross presentation. J Exp Med (2013) 210:1049–63. doi:10.1084/jem.20121251
29. Tel J, Benitez-Ribas D, Hoosemans S, Cambi A, Adema GJ, Figdor CG, et al. DEC-205 mediates antigen uptake and presentation by both resting and activated human plasmacytoid dendritic cells. Eur J Immunol (2011) 41:1014–23. doi:10.1002/eji.201040790
30. Tel J, Sittig SP, Blom RA, Cruz LJ, Schreibelt G, Figdor CG, et al. Targeting uptake receptors on human plasmacytoid dendritic cells triggers antigen cross-presentation and robust type I IFN secretion. J Immunol (2013) 191:5005–12. doi:10.4049/jimmunol.1300787
31. Tel J, Schreibelt G, Sittig SP, Mathan TS, Buschow SI, Cruz LJ, et al. Human plasmacytoid dendritic cells efficiently cross-present exogenous Ags to CD8+ T cells despite lower Ag uptake than myeloid dendritic cell subsets. Blood (2013) 121:459–67. doi:10.1182/blood-2012-06-435644
32. de Heer HJ, Hammad H, Soullie T, Hijdra D, Vos N, Willart MA, et al. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J Exp Med (2004) 200:89–98. doi:10.1084/jem.20040035
33. Goubier A, Dubois B, Gheit H, Joubert G, Villard-Truc F, Asselin-Paturel C, et al. Plasmacytoid dendritic cells mediate oral tolerance. Immunity (2008) 29:464–75. doi:10.1016/j.immuni.2008.06.017
34. MacDonald KP, Munster DJ, Clark GJ, Dzionek A, Schmitz J, Hart DN. Characterization of human blood dendritic cell subsets. Blood (2002) 100:4512–20. doi:10.1182/blood-2001-11-0097
35. Robbins SH, Walzer T, Dembele D, Thibault C, Defays A, Bessou G, et al. Novel insights into the relationships between dendritic cell subsets in human and mouse revealed by genome-wide expression profiling. Genome Biol (2008) 9:R17. doi:10.1186/gb-2008-9-1-r17
36. Haniffa M, Shin A, Bigley V, Mcgovern N, Teo P, See P, et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity (2012) 37:60–73. doi:10.1016/j.immuni.2012.04.012
37. Bachem A, Guttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med (2010) 207:1273–81. doi:10.1084/jem.20100348
38. Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med (2010) 207:1283–92. doi:10.1084/jem.20100223
39. Crozat K, Tamoutounour S, vu Manh TP, Fossum E, Luche H, Ardouin L, et al. Cutting edge: expression of XCR1 defines mouse lymphoid-tissue resident and migratory dendritic cells of the CD8alpha+ type. J Immunol (2011) 187:4411–5. doi:10.4049/jimmunol.1101717
40. Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med (2010) 207:1261–71. doi:10.1084/jem.20092618
41. Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med (2010) 207:1247–60. doi:10.1084/jem.20092140
42. Jaiswal H, Kaushik M, Sougrat R, Gupta M, Dey A, Verma R, et al. Batf3 and Id2 have a synergistic effect on Irf8-directed classical CD8alpha+ dendritic cell development. J Immunol (2013) 191:5993–6001. doi:10.4049/jimmunol.1203541
43. Poulin LF, Reyal Y, Uronen-Hansson H, Schraml B, Sancho D, Murphy KM, et al. DNGR-1 is a specific and universal marker of mouse and human Batf3-dependent dendritic cells in lymphoid and non-lymphoid tissues. Blood (2012) 119:6052–62. doi:10.1182/blood-2012-01-406967
44. Schlitzer A, Mcgovern N, Teo P, Zelante T, Atarashi K, Low D, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity (2013) 38:970–83. doi:10.1016/j.immuni.2013.04.011
45. van der Lugt B, Khan AA, Hackney JA, Agrawal S, Lesch J, Zhou M, et al. Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat Immunol (2014) 15:161–7. doi:10.1038/ni.2795
46. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science (2007) 315:107–11. doi:10.1126/science.1136080
47. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science (2008) 322:1097–100. doi:10.1126/science.1164206
48. Mittag D, Proietto AI, Loudovaris T, Mannering SI, Vremec D, Shortman K, et al. Human dendritic cell subsets from spleen and blood are similar in phenotype and function but modified by donor health status. J Immunol (2011) 186:6207–17. doi:10.4049/jimmunol.1002632
49. Segura E, Valladeau-Guilemond J, Donnadieu MH, Sastre-Garau X, Soumelis V, Amigorena S. Characterization of resident and migratory dendritic cells in human lymph nodes. J Exp Med (2012) 209:653–60. doi:10.1084/jem.20111457
50. van de Ven R, van den Hout MF, Lindenberg JJ, Sluijter BJ, van Leeuwen PA, Lougheed SM, et al. Characterization of four conventional dendritic cell subsets in human skin-draining lymph nodes in relation to T-cell activation. Blood (2012) 118:2502–10. doi:10.1182/blood-2011-03-344838
51. Segura E, Durand M, Amigorena S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J Exp Med (2013) 210:1035–47. doi:10.1084/jem.20121103
52. Yu CI, Becker C, Wang Y, Marches F, Helft J, Leboeuf M, et al. Human CD1c+ dendritic cells drive the differentiation of CD103+ CD8+ mucosal effector T cells via the cytokine TGF-beta. Immunity (2013) 38:818–30. doi:10.1016/j.immuni.2013.03.004
53. Meixlsperger S, Leung CS, Ramer PC, Pack M, Vanoaica LD, Breton G, et al. CD141+ dendritic cells produce prominent amounts of IFN-alpha after dsRNA recognition and can be targeted via DEC-205 in humanized mice. Blood (2013) 121:5034–44. doi:10.1182/blood-2012-12-473413
54. Nizzoli G, Krietsch J, Weick A, Steinfelder S, Facciotti F, Gruarin P, et al. Human CD1c+ dendritic cells secrete high levels of IL-12 and potently prime cytotoxic T-cell responses. Blood (2013) 122:932–42. doi:10.1182/blood-2013-04-495424
55. Klechevsky E, Morita R, Liu M, Cao Y, Coquery S, Thompson-Snipes L, et al. Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity (2008) 29:497–510. doi:10.1016/j.immuni.2008.07.013
56. van der Vlist M, de Witte L, de Vries RD, Litjens M, de Jong MA, Fluitsma D. Human Langerhans cells capture measles virus through Langerin and present viral antigens to CD4+ T cells but are incapable of cross-presentation. Eur J Immunol (2011) 41:2619–31. doi:10.1002/eji.201041305
57. Segura E, Amigorena S. Inflammatory dendritic cells in mice and humans. Trends Immunol (2013) 34:440–5. doi:10.1016/j.it.2013.06.001
58. Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, et al. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity (2013) 38:336–48. doi:10.1016/j.immuni.2012.10.018
59. Banchereau J, Thompson-Snipes L, Zurawski S, Blanck JP, Cao Y, Clayton S, et al. The differential production of cytokines by human Langerhans cells and dermal CD14(+) DCs controls CTL priming. Blood (2012) 119:5742–9. doi:10.1182/blood-2011-08-371245
60. Romano E, Cotari JW, Barreira da Silva R, Betts BC, Chung DJ, Avogadri F, et al. Human Langerhans cells use an IL-15R-alpha/IL-15/pSTAT5-dependent mechanism to break T-cell tolerance against the self-differentiation tumor antigen WT1. Blood (2012) 119:5182–90. doi:10.1182/blood-2011-09-382200
61. Hendriks J, Gravestein LA, Tesselaar K, van Lier RA, Schumacher TN, Borst J. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol (2000) 1:433–40. doi:10.1038/80877
62. Soares H, Waechter H, Glaichenhaus N, Mougneau E, Yagita H, Mizenina O, et al. A subset of dendritic cells induces CD4+ T cells to produce IFN-gamma by an IL-12-independent but CD70-dependent mechanism in vivo. J Exp Med (2007) 204:1095–106. doi:10.1084/jem.20070176
63. Ballesteros-Tato A, Leon B, Lund FE, Randall TD. Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8(+) T cell responses to influenza. Nat Immunol (2010) 11:216–24. doi:10.1038/ni.1838
64. van der Aar AM, de Groot R, Sanchez-Hernandez M, Taanman EW, van Lier RA, Teunissen MB, et al. Cutting edge: virus selectively primes human Langerhans cells for CD70 expression promoting CD8+ T cell responses. J Immunol (2011) 187:3488–92. doi:10.4049/jimmunol.1101105
65. Polak ME, Newell L, Taraban VY, Pickard C, Healy E, Friedmann PS, et al. CD70-CD27 interaction augments CD8+ T-cell activation by human epidermal Langerhans cells. J Invest Dermatol (2012) 132:1636–44. doi:10.1038/jid.2012.26
66. Chu CC, Ali N, Karagiannis P, DI Meglio P, Skowera A, Napolitano L, et al. Resident CD141 (BDCA3)+ dendritic cells in human skin produce IL-10 and induce regulatory T cells that suppress skin inflammation. J Exp Med (2012) 209:935–45. doi:10.1084/jem.20112583
67. Sathe P, Pooley J, Vremec D, Mintern J, Jin JO, Wu L, et al. The acquisition of antigen cross-presentation function by newly formed dendritic cells. J Immunol (2011) 186:5184–92. doi:10.4049/jimmunol.1002683
68. Zhan Y, Vega-Ramos J, Carrington EM, Villadangos JA, Lew AM, Xu Y. The inflammatory cytokine, GM-Csf, alters the developmental outcome of murine dendritic cells. Eur J Immunol (2012) 42:2889–900. doi:10.1002/eji.201242477
69. Lore K. Isolation and immunophenotyping of human and rhesus macaque dendritic cells. Methods Cell Biol (2004) 75:623–42. doi:10.1016/S0091-679X(04)75026-8
70. Kastenmuller K, Wille-Reece U, Lindsay RW, Trager LR, Darrah PA, Flynn BJ, et al. Protective T cell immunity in mice following protein-TLR7/8 agonist-conjugate immunization requires aggregation, type I IFN, and multiple DC subsets. J Clin Invest (2011) 121:1782–96. doi:10.1172/JCI45416
71. Caminschi I, Lahoud MH, Shortman K. Enhancing immune responses by targeting antigen to DC. Eur J Immunol (2009) 39:931–8. doi:10.1002/eji.200839035
72. Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med (2002) 196:1627–38. doi:10.1084/jem.20021598
73. Platt CD, Ma JK, Chalouni C, Ebersold M, BOU-Reslan H, Carano RA, et al. Mature dendritic cells use endocytic receptors to capture and present antigens. Proc Natl Acad Sci U S A (2010) 107:4287–92. doi:10.1073/pnas.0910609107
74. Drutman SB, Trombetta ES. Dendritic cells continue to capture and present antigens after maturation in vivo. J Immunol (2010) 185:2140–6. doi:10.4049/jimmunol.1000642
75. Werner-Klein M, Dresch C, Marconi P, Brocker T. Transcriptional targeting of B cells for induction of peripheral CD8 T cell tolerance. J Immunol (2007) 178:7738–46. doi:10.4049/jimmunol.178.12.7738
76. Reynoso ED, Turley SJ. Unconventional antigen-presenting cells in the induction of peripheral CD8(+) T cell tolerance. J Leukoc Biol (2009) 86:795–801. doi:10.1189/jlb.0509362
77. Morlacchi S, Soldani C, Viola A, Sarukhan A. Self-antigen presentation by mouse B cells results in regulatory T-cell induction rather than anergy or clonal deletion. Blood (2011) 118:984–91. doi:10.1182/blood-2011-02-336115
78. Murray SE, Toren KG, Parker DC. Peripheral CD4(+) T-cell tolerance is induced in vivo by rare antigen-bearing B cells in follicular, marginal zone, and B-1 subsets. Eur J Immunol (2013) 43:1818–27. doi:10.1002/eji.201242784
79. Chatterjee B, SMED-Sorensen A, Cohn L, Chalouni C, Vandlen R, Lee BC, et al. Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood (2012) 120:2011–20. doi:10.1182/blood-2012-01-402370
80. Tacken PJ, Ginter W, Berod L, Cruz LJ, Joosten B, Sparwasser T, et al. Targeting DC-SIGN via its neck region leads to prolonged antigen residence in early endosomes, delayed lysosomal degradation, and cross-presentation. Blood (2011) 118:4111–9. doi:10.1182/blood-2011-04-346957
81. Amigorena S, Savina A. Intracellular mechanisms of antigen cross presentation in dendritic cells. Curr Opin Immunol (2010) 22:109–17. doi:10.1016/j.coi.2010.01.022
82. Segura E, Amigorena S. Cross-presentation by human dendritic cell subsets. Immunol Lett (2013) 158:73–8. doi:10.1016/j.imlet.2013.12.001
83. Houde M, Bertholet S, Gagnon E, Brunet S, Goyette G, Laplante A, et al. Phagosomes are competent organelles for antigen cross-presentation. Nature (2003) 425:402–6. doi:10.1038/nature01912
84. Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, van Endert P, Amigorena S. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature (2003) 425:397–402. doi:10.1038/nature01911
85. Ackerman AL, Kyritsis C, Tampe R, Cresswell P. Access of soluble antigens to the endoplasmic reticulum can explain cross-presentation by dendritic cells. Nat Immunol (2005) 6:107–13. doi:10.1038/ni1147
86. Ackerman AL, Giodini A, Cresswell P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity (2006) 25:607–17. doi:10.1016/j.immuni.2006.08.017
87. Giodini A, Rahner C, Cresswell P. Receptor-mediated phagocytosis elicits cross-presentation in nonprofessional antigen-presenting cells. Proc Natl Acad Sci U S A (2009) 106:3324–9. doi:10.1073/pnas.0813305106
88. Cebrian I, Visentin G, Blanchard N, Jouve M, Bobard A, Moita C, et al. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell (2011) 147:1355–68. doi:10.1016/j.cell.2011.11.021
89. Schnurr M, Orban M, Robson NC, Shin A, Braley H, Airey D, et al. ISCOMATRIX adjuvant induces efficient cross-presentation of tumor antigen by dendritic cells via rapid cytosolic antigen delivery and processing via tripeptidyl peptidase II. J Immunol (2009) 182:1253–9. doi:10.4049/jimmunol.0990022
90. Shen L, Sigal LJ, Boes M, Rock KL. Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity (2004) 21:155–65. doi:10.1016/j.immuni.2004.07.004
91. Saveanu L, Carroll O, Weimershaus M, Guermonprez P, Firat E, Lindo V, et al. IRAP identifies an endosomal compartment required for MHC class I cross-presentation. Science (2009) 325:213–7. doi:10.1126/science.1172845
92. Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I. Activation of lysosomal function during dendritic cell maturation. Science (2003) 299:1400–3. doi:10.1126/science.1080106
93. Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science (2005) 307:1630–4. doi:10.1126/science.1108003
94. McCurley N, Mellman I. Monocyte-derived dendritic cells exhibit increased levels of lysosomal proteolysis as compared to other human dendritic cell populations. PLoS One (2010) 5:e11949. doi:10.1371/journal.pone.0011949
95. Delamarre L, Couture R, Mellman I, Trombetta ES. Enhancing immunogenicity by limiting susceptibility to lysosomal proteolysis. J Exp Med (2006) 203:2049–55. doi:10.1084/jem.20052442
96. Accapezzato D, Visco V, Francavilla V, Molette C, Donato T, Paroli M, et al. Chloroquine enhances human CD8+ T cell responses against soluble antigens in vivo. J Exp Med (2005) 202:817–28. doi:10.1084/jem.20051106
97. Belizaire R, Unanue ER. Targeting proteins to distinct subcellular compartments reveals unique requirements for MHC class I and II presentation. Proc Natl Acad Sci U S A (2009) 106:17463–8. doi:10.1073/pnas.0908583106
98. Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell (2006) 126:205–18. doi:10.1016/j.cell.2006.05.035
99. Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene (2008) 27:6434–51. doi:10.1038/onc.2008.310
100. Savina A, Peres A, Cebrian I, Carmo N, Moita C, Hacohen N, et al. The small GTPase Rac2 controls phagosomal alkalinization and antigen crosspresentation selectively in CD8(+) dendritic cells. Immunity (2009) 30:544–55. doi:10.1016/j.immuni.2009.01.013
101. Osorio F, Tavernier SJ, Hoffmann E, Saeys Y, Martens L, Vetters J, et al. The unfolded-protein-response sensor IRE-1alpha regulates the function of CD8alpha dendritic cells. Nat Immunol (2014) 15:248–57. doi:10.1038/ni.2808
102. Burgdorf S, Kautz A, Bohnert V, Knolle PA, Kurts C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science (2007) 316:612–6. doi:10.1126/science.1137971
103. Bozzacco L, Trumpfheller C, Huang Y, Longhi MP, Shimeliovich I, Schauer JD, et al. HIV gag protein is efficiently cross-presented when targeted with an antibody towards the DEC-205 receptor in Flt3 ligand-mobilized murine DC. Eur J Immunol (2010) 40:36–46. doi:10.1002/eji.200939748
104. Tsuji T, Matsuzaki J, Kelly MP, Ramakrishna V, Vitale L, He LZ, et al. Antibody-targeted NY-ESO-1 to mannose receptor or DEC-205 in vitro elicits dual human CD8+ and CD4+ T cell responses with broad antigen specificity. J Immunol (2011) 186:1218–27. doi:10.4049/jimmunol.1000808
105. Flinsenberg TW, Compeer EB, Koning D, Klein M, Amelung FJ, van Baarle D, et al. Fcgamma receptor antigen targeting potentiates cross-presentation by human blood and lymphoid tissue BDCA-3+ dendritic cells. Blood (2012) 120:5163–72. doi:10.1182/blood-2012-06-434498
106. Yamazaki S, Iyoda T, Tarbell K, Olson K, Velinzon K, Inaba K, et al. Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J Exp Med (2003) 198:235–47. doi:10.1084/jem.20030422
107. Yamazaki S, Dudziak D, Heidkamp GF, Fiorese C, Bonito AJ, Inaba K, et al. CD8+ CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J Immunol (2008) 181:6923–33. doi:10.4049/jimmunol.181.10.6923
108. Loschko J, Heink S, Hackl D, Dudziak D, Reindl W, Korn T, et al. Antigen targeting to plasmacytoid dendritic cells via Siglec-H inhibits Th cell-dependent autoimmunity. J Immunol (2011) 187:6346–56. doi:10.4049/jimmunol.1102307
109. Idoyaga J, Fiorese C, Zbytnuik L, Lubkin A, Miller J, Malissen B, et al. Specialized role of migratory dendritic cells in peripheral tolerance induction. J Clin Invest (2013) 123:844–54. doi:10.1172/JCI65260
110. Romani N, Thurnher M, Idoyaga J, Steinman RM, Flacher V. Targeting of antigens to skin dendritic cells: possibilities to enhance vaccine efficacy. Immunol Cell Biol (2010) 88:424–30. doi:10.1038/icb.2010.39
111. Schenten D, Medzhitov R. The control of adaptive immune responses by the innate immune system. Adv Immunol (2011) 109:87–124. doi:10.1016/B978-0-12-387664-5.00003-0
112. Kerrigan AM, Brown GD. Syk-coupled C-type lectins in immunity. Trends Immunol (2011) 32:151–6. doi:10.1016/j.it.2011.01.002
113. Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity (2010) 33:492–503. doi:10.1016/j.immuni.2010.10.002
114. Kwissa M, Nakaya HI, Oluoch H, Pulendran B. Distinct TLR adjuvants differentially stimulate systemic and local innate immune responses in nonhuman primates. Blood (2012) 119:2044–55. doi:10.1182/blood-2011-10-388579
115. Ahonen CL, Doxsee CL, McGurran SM, Riter TR, Wade WF, Barth RJ, et al. Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J Exp Med (2004) 199:775–84. doi:10.1084/jem.20031591
116. Ahonen CL, Wasiuk A, Fuse S, Turk MJ, Ernstoff MS, Suriawinata AA, et al. Enhanced efficacy and reduced toxicity of multifactorial adjuvants compared with unitary adjuvants as cancer vaccines. Blood (2008) 111:3116–25. doi:10.1182/blood-2007-09-114371
117. Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, et al. Programming the magnitude and persistence of antibody responses with innate immunity. Nature (2011) 470:543–7. doi:10.1038/nature09737
118. Longhi MP, Trumpfheller C, Idoyaga J, Caskey M, Matos I, Kluger C, et al. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med (2009) 206:1589–602. doi:10.1084/jem.20090247
119. Ravindran R, Khan N, Nakaya HI, Li S, Loebbermann J, Maddur MS, et al. Vaccine activation of the nutrient sensor GCN2 in dendritic cells enhances antigen presentation. Science (2014) 343:313–7. doi:10.1126/science.1246829
120. Ketloy C, Engering A, Srichairatanakul U, Limsalakpetch A, Yongvanitchit K, Pichyangkul S, et al. Expression and function of Toll-like receptors on dendritic cells and other antigen presenting cells from non-human primates. Vet Immunol Immunopathol (2008) 125:18–30. doi:10.1016/j.vetimm.2008.05.001
121. Gujer C, Sundling C, Seder RA, Karlsson Hedestam GB, Lore K. Human and rhesus plasmacytoid dendritic cell and B-cell responses to Toll-like receptor stimulation. Immunology (2011) 134:257–69. doi:10.1111/j.1365-2567.2011.03484.x
122. Koup RA, Douek DC. Vaccine design for CD8 T lymphocyte responses. Cold Spring Harb Perspect Med (2011) 1:a007252. doi:10.1101/cshperspect.a007252
123. Wille-Reece U, Flynn BJ, Lore K, Koup RA, Miles AP, Saul A, et al. Toll-like receptor agonists influence the magnitude and quality of memory T cell responses after prime-boost immunization in nonhuman primates. J Exp Med (2006) 203:1249–58. doi:10.1084/jem.20052433
124. Stahl-Hennig C, Eisenblatter M, Jasny E, Rzehak T, Tenner-Racz K, Trumpfheller C, et al. Synthetic double-stranded RNAs are adjuvants for the induction of T helper 1 and humoral immune responses to human papillomavirus in rhesus macaques. PLoS Pathog (2009) 5:e1000373. doi:10.1371/journal.ppat.1000373
125. Park H, Adamson L, Ha T, Mullen K, Hagen SI, Nogueron A, et al. Polyinosinic-polycytidylic acid is the most effective TLR adjuvant for SIV Gag protein-induced T cell responses in nonhuman primates. J Immunol (2013) 190:4103–15. doi:10.4049/jimmunol.1202958
126. Tewari K, Flynn BJ, Boscardin SB, Kastenmueller K, Salazar AM, Anderson CA, et al. Poly(I:C) is an effective adjuvant for antibody and multi-functional CD4+ T cell responses to Plasmodium falciparum circumsporozoite protein (CSP) and alphaDEC-CSP in non human primates. Vaccine (2010) 28:7256–66. doi:10.1016/j.vaccine.2010.08.098
127. Wille-Reece U, Wu CY, Flynn BJ, Kedl RM, Seder RA. Immunization with HIV-1 Gag protein conjugated to a TLR7/8 agonist results in the generation of HIV-1 Gag-specific Th1 and CD8+ T cell responses. J Immunol (2005) 174:7676–83. doi:10.4049/jimmunol.174.12.7676
128. Khan S, Bijker MS, Weterings JJ, Tanke HJ, Adema GJ, van Hall T, et al. Distinct uptake mechanisms but similar intracellular processing of two different toll-like receptor ligand-peptide conjugates in dendritic cells. J Biol Chem (2007) 282:21145–59. doi:10.1074/jbc.M701705200
129. Oh JZ, Kedl RM. The capacity to induce cross-presentation dictates the success of a TLR7 agonist-conjugate vaccine for eliciting cellular immunity. J Immunol (2010) 185:4602–8. doi:10.4049/jimmunol.1001892
130. Kaiser-Schulz G, Heit A, Quintanilla-Martinez L, Hammerschmidt F, Hess S, Jennen L, et al. Polylactide-coglycolide microspheres co-encapsulating recombinant tandem prion protein with CpG-oligonucleotide break self-tolerance to prion protein in wild-type mice and induce CD4 and CD8 T cell responses. J Immunol (2007) 179:2797–807. doi:10.4049/jimmunol.179.5.2797
131. Schlosser E, Mueller M, Fischer S, Basta S, Busch DH, Gander B, et al. TLR ligands and antigen need to be coencapsulated into the same biodegradable microsphere for the generation of potent cytotoxic T lymphocyte responses. Vaccine (2008) 26:1626–37. doi:10.1016/j.vaccine.2008.01.030
132. Tacken PJ, ter Huurne M, Torensma R, Figdor CG. Antibodies and carbohydrate ligands binding to DC-SIGN differentially modulate receptor trafficking. Eur J Immunol (2012) 42:1989–98. doi:10.1002/eji.201142258
133. Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature (2006) 440:808–12. doi:10.1038/nature04596
134. Hoffmann E, Kotsias F, Visentin G, Bruhns P, Savina A, Amigorena S. Autonomous phagosomal degradation and antigen presentation in dendritic cells. Proc Natl Acad Sci U S A (2012) 109:14556–61. doi:10.1073/pnas.1203912109
135. Weck MM, Appel S, Werth D, Sinzger C, Bringmann A, Grunebach F, et al. Dectin-1 is involved in uptake and cross-presentation of cellular antigens. Blood (2008) 111:4264–72. doi:10.1182/blood-2006-10-051375
136. Ni L, Gayet I, Zurawski S, Duluc D, Flamar AL, Li XH, et al. Concomitant activation and antigen uptake via human dectin-1 results in potent antigen-specific CD8+ T cell responses. J Immunol (2010) 185:3504–13. doi:10.4049/jimmunol.1000999
137. Carter RW, Thompson C, Reid DM, Wong SY, Tough DF. Preferential induction of CD4+ T cell responses through in vivo targeting of antigen to dendritic cell-associated C-type lectin-1. J Immunol (2006) 177:2276–84. doi:10.4049/jimmunol.177.4.2276
138. Delamarre L, Holcombe H, Mellman I. Presentation of exogenous antigens on major histocompatibility complex (MHC) class I and MHC class II molecules is differentially regulated during dendritic cell maturation. J Exp Med (2003) 198:111–22. doi:10.1084/jem.20021542
139. Flamar AL, Xue Y, Zurawski SM, Montes M, King B, Sloan L, et al. Targeting concatenated HIV antigens to human CD40 expands a broad repertoire of multifunctional CD4+ and CD8+ T cells. AIDS (2013) 27:2041–51. doi:10.1097/QAD.0b013e3283624305
140. Barr TA, McCormick AL, Carlring J, Heath AW. A potent adjuvant effect of CD40 antibody attached to antigen. Immunology (2003) 109:87–92. doi:10.1046/j.1365-2567.2003.01634.x
141. Schjetne KW, Fredriksen AB, Bogen B. Delivery of antigen to CD40 induces protective immune responses against tumors. J Immunol (2007) 178:4169–76. doi:10.4049/jimmunol.178.7.4169
142. Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res (2013) 19:1035–43. doi:10.1158/1078-0432.CCR-12-2064
143. Regnault A, Lankar D, Lacabanne V, Rodriguez A, Thery C, Rescigno M, et al. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med (1999) 189:371–80. doi:10.1084/jem.189.2.371
144. Davis MM. Immunology taught by humans. Sci Transl Med (2012) 4:117fs2. doi:10.1126/scitranslmed.3003385
145. Pulendran B, Oh JZ, Nakaya HI, Ravindran R, Kazmin DA. Immunity to viruses: learning from successful human vaccines. Immunol Rev (2013) 255:243–55. doi:10.1111/imr.12099
146. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39:1–10. doi:10.1016/j.immuni.2013.07.012
147. Reis e Sousa C. Toll-like receptors and dendritic cells: for whom the bug tolls. Semin Immunol (2004) 16:27–34. doi:10.1016/j.smim.2003.10.004
148. Lindstedt M, Lundberg K, Borrebaeck CA. Gene family clustering identifies functionally associated subsets of human in vivo blood and tonsillar dendritic cells. J Immunol (2005) 175:4839–46. doi:10.4049/jimmunol.175.8.4839
149. van der Aar AM, Sylva-Steenland RM, Bos JD, Kapsenberg ML, DE Jong EC, Teunissen MB. Loss of TLR2, TLR4, and TLR5 on Langerhans cells abolishes bacterial recognition. J Immunol (2007) 178:1986–90. doi:10.4049/jimmunol.178.4.1986
150. Klechevsky E, Liu M, Morita R, Banchereau R, Thompson-Snipes L, Palucka AK, et al. Understanding human myeloid dendritic cell subsets for the rational design of novel vaccines. Hum Immunol (2009) 70:281–8. doi:10.1016/j.humimm.2009.02.004
151. Hespel C, Moser M. Role of inflammatory dendritic cells in innate and adaptive immunity. Eur J Immunol (2012) 42:2535–43. doi:10.1002/eji.201242480
152. Engering A, Geijtenbeek TB, van Vliet SJ, Wijers M, van Liempt E, Demaurex N, et al. The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for presentation to T cells. J Immunol (2002) 168:2118–26. doi:10.4049/jimmunol.168.5.2118
153. Amigorena S. Fcgamma receptors and cross-presentation in dendritic cells. J Exp Med (2002) 195:F1–3. doi:10.1084/jem.20011925
Keywords: dendritic cells, MHC class I, CD8+ T cells, vaccination, adjuvants, immunologic
Citation: Cohn L and Delamarre L (2014) Dendritic cell-targeted vaccines. Front. Immunol. 5:255. doi: 10.3389/fimmu.2014.00255
Received: 05 February 2014; Paper pending published: 16 April 2014;
Accepted: 15 May 2014; Published online: 30 May 2014.
Edited by:
Marianne Boes, University Medical Centre Utrecht, NetherlandsReviewed by:
Masaaki Murakami, Osaka University, JapanJoke M. M. Den Haan, VU University Medical Center, Netherlands
Copyright: © 2014 Cohn and Delamarre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lélia Delamarre, Genentech, MS#231, 1 DNA way, South San Francisco, CA 94080, USA e-mail:ZGVsYW1hcnJlLmxlbGlhQGdlbmUuY29t