 Anita Schuch1,2
Anita Schuch1,2 Alexander Hoh
Alexander Hoh Robert Thimme
Robert Thimme- 1Department of Medicine II, University Hospital of Freiburg, Freiburg, Germany
- 2Faculty of Biology, University of Freiburg, Freiburg, Germany
- 3Spemann Graduate School of Biology and Medicine, University of Freiburg, Freiburg, Germany
Hepatitis B virus (HBV) infection is one of the main causes of chronic liver diseases that may progress to liver cirrhosis and hepatocellular carcinoma. Host immune responses are important factors that determine whether HBV infection is cleared or persists. Natural killer (NK) cells represent the main effector population of the innate immune system and are abundant in the human liver. Recently, it has been demonstrated that NK cells not only exhibit antiviral functions but may also regulate adaptive immune responses by deletion of HBV-specific CD8+ T cells. It is well-established that HBV-specific CD8+ T cells contribute to virus elimination. However, the mechanisms contributing to CD8+ T cell failure in chronic HBV infection are not well-understood. In this review, we will summarize the current knowledge about NK cells and CD8+ T cells and illustrate their contribution to viral clearance and persistence in HBV infection. Moreover, novel immunological in vitro model systems and techniques to analyze HBV-specific CD8+ T cells, which are barely detectable using current multimer staining methods, will be discussed.
Introduction
Hepatitis B virus (HBV) infection represents a major health care problem that affects around 350 million people worldwide, despite the availability of a prophylactic vaccine (1). The course of infection can be either acute or chronic, while persistence rate is considerably higher when HBV is acquired at birth or early infancy. Chronically infected patients are at risk of developing HBV-related diseases such as liver cirrhosis and hepatocellular carcinoma that account for 600,000 deaths annually (1). Although potent antiviral drugs such as nucleos(t)ide analogs and pegylated interferon-α (pegIFN-α) are available, treatment is rarely curative and patients often receive life-long therapy with the potential emergence of resistance and toxicity.
Natural killer (NK) cells, as part of the innate immune system, represent the first line of defense against viral infections. In addition to their antiviral effector functions, NK cells may also interact with and thereby negatively regulate HBV-specific CD8+ T cells (2). CD8+ T cells are thought to be the main effector cells since their experimental depletion delays the clearance of acute HBV infection in chimpanzees (3). Of note, persistent infection is characterized by impaired HBV-specific CD8+ T cell responses (4). The mechanisms responsible for this CD8+ T cell failure are less understood. Interestingly, different mechanisms that may lead to impaired and dysfunctional HBV-specific CD8+ T cell responses in chronically infected patients have been reported. Therefore, it is plausible that at least a part of immune-mediated liver damage is due to immune cells other than virus-specific T cells (5).
In the present review, we will focus on the role of NK cells and HBV-specific CD8+ T cells, which are thought to be responsible for both virus control and disease pathogenesis.
NK Cells
Natural killer cells represent the main effector cell population involved in innate immune responses against intracellular pathogens and abnormal cells (6). They are enriched in the liver (7) and account for one-third of the intrahepatic lymphocytes compared to 5–15% in the peripheral blood (8).
Natural killer cells do not express recombination-dependent antigen-specific receptors, therefore it is assumed that stimulation of NK cells is antigen-independent (9). However, NK cells have several traits in common with CD8+ T cells (10, 11): they share a common bipotential progenitor and exert similar killing mechanisms. Additionally, a number of cell surface molecules, referred to as “NK receptors” are also expressed on activated CD8+ T cells. Furthermore, murine NK cells have been described to mediate long-lived hapten-specific recall responses leading to the assumption of a NK cell memory (12).
Natural killer cell activation is regulated by the interplay of several activating and inhibitory receptors (10, 13) and cytokines such as type I IFN, interleukin (IL)-2, IL-12, IL-15, and IL-18 (14, 15). The best characterized activating receptor on the surface of NK cells is NKG2D. This receptor recognizes molecules, that are expressed at low levels on most cells but are upregulated upon infection or stress, such as MICA, MICB, and RAET1 proteins (10, 16, 17). Furthermore, NKp46 (18), NKp44 (19), and NKp30 (20) are important receptors involved in target cell recognition and killing (21). Another important NK cell stimulatory receptor is CD16, also known as the Fc receptor FcγRIII, which triggers antibody-dependent cell cytotoxicity (22). Moreover, NK cells are regulated by inhibitory receptors, which are known to mainly engage major histocompatibility complex (MHC) class I molecules expressed on the surface of target cells (23). Some of these inhibitory markers belong to a distinct family of receptors termed killer cell immunoglobulin-like receptors (KIRs), which include both activating and inhibitory molecules (22). Additionally, the CD94/NKG2A heterodimeric receptor is often used as an inhibitory marker (24).
Depending on the distribution of activating versus inhibitory signals and the prevalent cytokine milieu NK cells display at least two effector functions: they are able to produce a variety of antiviral active and immunoregulatory cytokines such as IFN-γ, tumor necrosis factor (TNF), granulocyte–macrophage colony stimulating factor (GM-CSF), and IL-10 (25, 26) and they can directly kill target cells through the release of perforin and granzymes at immunological synapses (27). Since hepatocytes are considered to be relatively resistant to cytotoxicity of NK cells via the perforin/granzyme pathway, tumor-necrosis-factor-related-apoptosis-inducing ligand (TRAIL) is likely to play a major role in hepatocellular damage (28).
Based on their expression of CD56, two NK cell subpopulations can be defined: CD56dim and CD56bright. CD56dim NK cells represent the major circulating subset and are regarded as developmentally mature. Furthermore, this subset is thought to exert mainly cytotoxic effector functions (29), although it has been shown that CD56dim NK cells are also able to produce large amounts of IFN-γ during the first hours after stimulation (30). By contrast, CD56bright NK cells represent an earlier stage of maturation. They comprise the minority in the peripheral blood and are considered as the main cytokine producers (29, 31–33). However, viral infections may alter these proportions, leading to a relative enrichment of the CD56bright population (34, 35).
Next to their antiviral function, NK cells have also been shown to regulate other immune cells thereby shaping both innate and adaptive immune responses. Indeed, various studies focused on the interaction between NK cells and innate immune cells such as monocyte-derived dendritic cells, plasmacytoid dendritic cells (pDCs), and macrophages. This crosstalk can modulate NK cell functions by direct cell-to-cell contact or the activity of soluble factors (36). For example, NK cell interaction with macrophages and DCs via CD40L/CD40 drives production of IL-12, which in turn not only induces NK cells to produce IFN-γ but also enhances NK cell cytotoxicity (37–39). Additionally, NK cell function can also be improved by type I IFN abundantly secreted by pDCs (40). Furthermore, NK cells can also interact with components of the adaptive immune system and may limit CD8+ T cell responses, as it has been shown in LCMV infection (41–43).
Below, we will summarize the current knowledge about of NK cells in acute and chronic HBV infection and discuss their role in regulating HBV-specific CD8+ T cell immunity.
NK Cells in Acute HBV Infection
Viral replication usually results in the activation of an innate immune response that is characterized by the rapid production of type I IFN. These cytokines induce the expression of interferon-stimulated genes (ISGs), which in turn exert several intracellular antiviral mechanisms to limit viral spread, including the upregulation of MHC I molecules on the surface of infected cells (44). However, HBV seems not to induce any detectable intrahepatic expression of ISGs in chimpanzees during the first weeks of infection and therefore has been postulated to be a “stealth virus” that does not activate the innate immune system (45). This assumption has been challenged in vitro by the finding that HBV replication elicits a strong and specific innate antiviral response in HepaRG cells with an upregulation of IFN-β and other ISGs resulting in a non-cytopathic clearance of HBV DNA (46). Furthermore, a significant reduction in HBV DNA has been reported in acutely infected chimpanzees long before the peak of T cell infiltration and liver damage, suggesting a contribution of non-cytopathic antiviral mechanisms to viral clearance (47). The influx of NK cells that recognize infected cells in the absence of MHC I expression has been suggested to contribute in this setting. Moreover, the induction of IFN-γ and TNF in the liver of chimpanzees during the described non-cytopathic pre-T cell phase of viral clearance supports this hypothesis, because these effector cytokines are produced not only by CD8+ T cells but also by NK cells.
Since the incubation period of acute HBV infection is predominantly asymptomatic and therefore difficult to study, only limited and partially contradicting information about the role of NK cells during the early stages of infection is available in humans. One of the leading studies was performed during the preclinical phase in two subjects with acute HBV infection characterized by persistently normal alanine aminotransferase (ALT) levels (48). NK cells were promptly activated before peak viremia occurred, as indicated by the early increase of NK cells expressing the activation markers CD69 and NKG2D. According to this, the highest number of circulating NK cells was found at an early stage in the incubation period of patients with acute HBV infection (49).
However, an impaired NK cell function in patients with acute hepatitis B has also been reported. Indeed, Dunn et al. showed that NK cell activation in acutely HBV-infected patients is significantly inhibited compared to healthy subjects, especially during the time of peak viremia (50). High viral load was also associated with a reduction of rather non-cytolytic than cytolytic NK cell effector functions. In addition, type I IFN, IFN-λ1, and IL-15, essential activators of NK cells, were barely detectable in these patients, supporting the dogma of HBV being a stealth virus. However, IL-10 levels increased early in the course of infection and the highest concentration was found at the time of peak viremia when NK cell IFN-γ production was severely reduced. This suggests a role for IL-10 in the inhibition of NK cell antiviral responses. The authors confirmed in vitro that addition of exogenous IL-10 to activated NK cells induces significant suppression of NK cell-derived IFN-γ, while blocking of IL-10 restored NK cell effector function (50).
However, it has also been reported that NK cells exert higher cytolytic activity and IFN-γ production during acute HBV infection. This was concomitant with the elevated expression of activating receptors such as NKp46, and lower levels of inhibitory markers, e.g., NKG2A (34). Furthermore, NK cell activation, measured by the expression of CD69, CD38, and HLA-DR, was correlated positively with ALT levels and negatively with viral load, suggesting a close association of activated NK cells with liver necroinflammation and HBV clearance in acute HBV infection. In addition to the altered phenotype, the frequency and subset distribution was also modified in patients with acute hepatitis B, showing a significant enrichment of CD56bright NK cells (34, 35).
The discrepancy between these different studies may arise from the fact that disease progression in the analyzed patients was either asymptomatic or symptomatic in concert with normal and elevated ALT levels, respectively. Overall, these results point to an important role of NK cells that are activated during acute HBV infection but might be functionally suppressed.
NK Cells in Chronic HBV Infection
Studies regarding phenotype and function of NK cells during chronic HBV infection have revealed, in part, conflicting results. Several reports conclude that NK cells exhibit selective defects in their antiviral function. This functional dichotomy features a conserved or enhanced cytolytic activity (51, 52) and a diminished cytokine production (51, 53) that may contribute to viral persistence and implicate a role for NK cells in disease pathogenesis. The mechanisms leading to this functional impairment are still not fully understood but thought to be heterogeneous.
Hepatitis B virus infection may alter the activation status and receptor expression patterns on the surface of NK cells. Indeed, the expression of inhibitory receptors such as NKG2A is elevated while activating receptors, CD16 and NKp30, are downregulated (53, 54) and this correlates with serum HBV DNA load. Interestingly, antiviral therapy partially restores NK cell phenotype and functionality (53). However, these findings are controversial, since Bonorio et al. showed decreased levels of NKG2A-expressing NK cells in chronic HBV infection (55) and it was also reported, that HBV infection does not alter NKG2A expression on NK cells (35). In addition to classical NK cell receptors other co-inhibitory molecules involved in immune responses may impair NK cell function. Of note, T cell immunoglobulin- and mucin-domain-containing molecule-3 (Tim-3) has been shown to be upregulated on NK cells during HBV infection and in vitro blockade was able to enhance NK cell cytotoxicity (56).
An impaired NK cell activation and function may also arise from modified expression patterns of ligands for inhibitory and activating NK cell receptors. Indeed, it has been shown that the decreased expression of NKG2D ligands, MICA/B, on HBV-infected hepatocytes inhibits NK cell lysis (57).
Furthermore, the immunosuppressive cytokine environment in chronic HBV infection, created through high levels of IL-10, may inhibit the ability of NK cells to produce IFN-γ (58), as has already been shown in acutely infected patients (50). This defect persists in patients with chronic HBV infection receiving antiviral therapy, but can be reversed in vitro by specific blockade of IL-10 and transforming growth factor (TGF)-β (58).
In addition, the interaction with other immune cells may alter the reactivity of NK cells during persistent viral infection. For example, several studies have revealed that HBV interferes with pDCs, thereby modulating pDC-NK cell crosstalk in vivo and in vitro (59–61). Although circulating and intrahepatic pDCs from patients with chronic HBV infection showed a more activated phenotype, their ability to respond to toll-like receptor (TLR) 9 stimulation was significantly impaired (60). Moreover, patient-derived mature pDCs were poor activators of NK cell cytotoxic function due to their impaired IFN-α secretion and reduced OX40L expression. HBV seems not only to directly inhibit pDC maturation in a TLR9-dependent manner, but also to abrogate the supporting function of monocytes regarding IFN-α production by pDCs (59).
As mentioned above, NK cells may also exert regulatory functions (41–43). This is supported by a study in patients with chronic HBV infection, where in vitro depletion of NK cells increased HBV- but not CMV-specific CD8+ T cell responses (2). Elevated expression of TRAIL receptor 2 (TRAIL-R2) renders HBV-specific CD8+ T cells more susceptible to apoptosis by TRAIL-expressing NK cells. TRAIL-R2-expression patterns correlated with HBV DNA titer, thus the regulatory role of NK cells may be relevant during HBV flares. Longitudinal analysis of chronically HBV-infected patients already revealed a temporal correlation between ALT flares and TRAIL-expressing NK cells (62).
Taken together, these findings suggest that NK cells may exert a non-classical regulatory next to their classical antiviral function in HBV infection.
CD8+ T Cells
CD8+ T cells are a major component of cellular adaptive immunity. They normally mediate protection against intracellular pathogens and tumor cells. In order to be properly activated, CD8+ T cells require at least two signals: first, the recognition of their cognate antigen presented by MHC I molecules on antigen-presenting cells (APCs). This is mediated by the interaction of the antigen-specific T cell receptor (TCR) with peptide-MHC I complexes. Second, additional co-stimulatory signals have to be provided by the same APC to prevent anergy. Furthermore, different cytokine milieus may influence this activation process (63). Upon antigen-recognition, naïve CD8+ T cells undergo clonal expansion and differentiate into cytotoxic effector and memory T cells (64). According to their differentiation status, they are characterized by distinct expression patterns of surface markers such as CD45RA, CD27, CD28, and CCR7 (65). Similar to NK cells, the effector functions of CD8+ T cells comprise several mechanisms such as the secretion of cytokines (IFN-γ and TNF), the release of cytotoxic mediators (perforin/granzyme), and receptor-mediated induction of apoptosis (e.g., through TRAIL) (63). In the following, we will elaborate the current knowledge about phenotype and function of CD8+ T cells in the context of acute and chronic HBV infection.
CD8+ T Cells in Acute HBV Infection
Hepatitis B virus-specific CD8+ T cells play a major role in controlling and resolving HBV infection. Indeed, strong HBV-specific CD8+ T cell responses have been shown to correlate with viral clearance during acute infection (66). The antiviral role of CD8+ T cells has been further confirmed by depletion studies in experimentally infected chimpanzees, where in the absence of CD8+ T cells virus titer remained at high levels (3). Importantly, the re-appearance of CD8+ T cells in the circulation coincided with a decrease in viremia and the onset of liver disease. However, it has been shown that virus-specific CD8+ T cells are functionally impaired during the acute phase until infection is resolved (50, 66, 67). Furthermore, the mechanisms of CD8+ T cell-mediated antiviral control are still debated.
Indeed, studies in the transgenic mouse model revealed that HBV-specific CD8+ T cells are able to abolish viral replication in the liver while killing only a small fraction of hepatocytes (68). This was mediated by inflammatory cytokines such as IFN-γ and TNF. The contribution of non-cytolytic effector mechanisms has been further supported by findings in a cell culture model, where virus-specific CD8+ T cells were able to inhibit HBV replication in HepG2 2.2.15 cells with only minimal cell lysis (69). Of note, the authors could show that particularly IFN-γ and TNF are responsible for HBV inactivation in target cells, since blocking of these two cytokines abrogated the non-cytolytic inhibition of virus replication. Furthermore, in acutely infected chimpanzees most of HBV DNA elimination has been shown to occur before the peak of T cell infiltration and liver injury, also suggesting non-cytolytic effector mechanisms like the secretion of IFN-γ and TNF (47). Still, the contribution of cytolytic effects is supported by several studies (3, 70).
The development of cell culture models permits the investigation of the relative importance of cytolytic versus non-cytolytic effector functions and their impact on the suppression of HBV replication (71). Various cell lines transfected with the HBV genome have been established, e.g., HepG2.117 (72), HepG2.2.15 (73), or HepAD38 (74). Although the secreted virions are infectious, these cell lines are still quite artificial, since none of them is susceptible for HBV infection. Until the recent discovery of human sodium taurocholate cotransporting polypeptide (hNTCP) as HBV entry receptor (75, 76) in vitro infection could only be conducted in primary human hepatocytes, primary tupaia hepatocytes, and in differentiated HepaRG cells. The latter was the first hepatoma cell line being susceptible to HBV and supporting the full viral replication cycle after DMSO-dependent differentiation (77). However, cultures of differentiated HepaRG cells represent a mixture of biliary-like epithelial cells and hepatocyte-like cells and only the latter subset was shown to be susceptible for HBV infection with an infection efficacy below 20% (78). Hepatoma cell lines such as HuH7 and HepG2 cells lack hNTCP expression and are therefore non-permissive to HBV infection. Of note, infection rates with up to 70% can be achieved when HepG2 cells are transduced with hNTCP (76). These novel cell lines may be useful not only to study the molecular virology of HBV but also to improve our understanding of the relative contribution of cytolytic and non-cytolytic effector functions to viral clearance.
CD8+ T Cells in Chronic HBV Infection
In chronically HBV-infected individuals, virus-specific CD8+ T cell responses are rarely detectable (5, 79–82). The profiles of HBV-specific CD8+ T cell responses depend on the stage of disease and are highly influenced by the level of HBV replication. Indeed, circulating multispecific HBV-specific CD8+ T cell responses are predominantly detectable ex vivo in patients with low viral load. In individuals with a high level of HBV replication (>107 copies/ml) virus-specific CD8+ T cells were occasionally detectable only after in vitro expansion (81). However, the mechanisms responsible for the lack of functional CD8+ T cell responses are not completely understood. It might be that virus-specific CD8+ T cells are deleted. Indeed, the elevated intracellular expression of the pro-apoptotic protein Bcl2-interacting mediator (Bim) in HBV-specific CD8+ T cells of chronically infected patients supports this hypothesis (83).
Furthermore, HBV-specific CD8+ T cells may not or only insufficiently be primed by APCs and consequently may not expand upon antigen-encounter. Of note, several studies suggest a dysfunction of DCs in chronically HBV-infected patients, including reduced expression of co-stimulatory molecules, impaired cytokine secretion, and lower allostimulatory capacity compared to healthy subjects (84–87). According to this, HBV-specific CD8+ T cells would be expected to display a naïve phenotype, characterized by high expression levels of CD45RA, CD27, CD28, and CCR7 (65).
In addition, it is possible that they are not traceable since the frequencies of virus-specific CD8+ T cell responses are below the detection limit of conventional quantitative assays, as it has been shown in chronic HCV infection (Schmidt, unpublished data). New techniques for enumerating epitope-specific T cells from human peripheral blood based on the combination of tetramer staining, magnetic-bead enrichment, and multiparametric flow cytometry (88) may address at least the latter two possibilities.
Virus-specific CD8+ T cells isolated from the peripheral blood of chronically HBV-infected patients are functionally impaired and seem to have lost most of their ability to proliferate and to produce cytokines, like IFN-γ (82). The reported CD8+ T cell failure has been attributed to high levels of persisting viral antigens. Despite the constant presentation of viral peptides on MHC molecules, circulating sub-viral particles, comprised of soluble HBV surface antigen (HBsAg) along with HBeAg may drive chronic T cell stimulation. Particularly, the latter has been implicated in altering the reactivity of virus-specific CD8+ T cells (89). Moreover, accessory HBsAg seroconversion has been reported to induce a more potent restoration of CD8+ T cell responses than HBV viral load reduction alone (90).
CD8+ T cell dysfunction in chronic HBV infection follows a well-established pattern with elevated expression of inhibitory molecules such as programmed death-1 (PD-1) (82, 91), cytotoxic T lymphocyte antigen 4 (CTLA-4) (92), Tim-3 (93, 94), and 2B4 (CD244) (95) on T cells. Furthermore, the expression of corresponding ligands such as PD ligand (PD-L)1 has been shown to be increased on hepatocytes (96). According to this, highly viremic HBV-infected patients show a more severely impaired CD8+ T cell phenotype and T cell dysfunction is more profound in the liver than in the blood (91, 97). Blockade of these inhibitory pathways may at least partially restore HBV-specific CD8+ T cell functionality, as it has been shown in vitro (82, 91, 92, 94, 95). The potential relevance of blocking the PD-1 pathway was demonstrated in the HBV mouse model where HBV-transgenic mice were treated with blocking antibodies for PD-L1 prior to the adoptive transfer of HBV-specific cytotoxic T cells (98). This treatment resulted in an increased number of IFN-γ-producing CD8+ T cells in the liver and in a delayed suppression of these CD8+ T cells. Furthermore, a recent study could show that in vivo blockade of the PD-1/PD-L1 pathway, together with entecavir treatment and DNA vaccination, enhances virus-specific CD8+ T cell responses in the woodchuck model, leading to sustained immunological control of viral infection (99).
The combined modulation of these inhibitory pathways along with the activation of co-stimulatory pathways might be beneficial (92, 95, 100, 101). However, a more detailed insight into the relative contribution of individual inhibitory pathways to HBV-specific CD8+ T cell dysfunction and concomitant the impact of inhibitory receptor blockade on restoration of CD8+ T cell responses is necessary.
Moreover, the lack of CD4+ T cell help contributes to defective CD8+ T cell function (102). Increased regulatory T cell numbers (103–105), together with immunosuppressive cytokines such as IL-10 and TGF-β (50, 58, 106) impair virus-specific CD8+ T cell responses. It has also been reported that increased intrahepatic arginase levels (67, 107) and hence the lack of arginine lead to a functional silencing of CD8+ T cells due to the downregulation of the CD3ζ-chain (108).
Collectively, these studies demonstrate that several mechanisms may contribute to the diminished frequency and function of virus-specific CD8+ T cells in the chronic phase of HBV infection and that combined modulation of different pathways may lead to a restoration of HBV-specific T cell responses.
Conclusion
CD8+ T cell responses play an important role in HBV infection and contribute not only to viral clearance but also to liver injury. In the setting of chronic infection, several mechanisms of T cell dysfunction including expression of inhibitory molecules and pro-apoptotic proteins, as well as suppressive cell subsets and cytokines favor viral persistence (Figure 1). In addition, even antivirally active NK cells, which exert a unique influence in the early defense against HBV are supposed to control CD8+ T cells, particularly during hepatic flares.
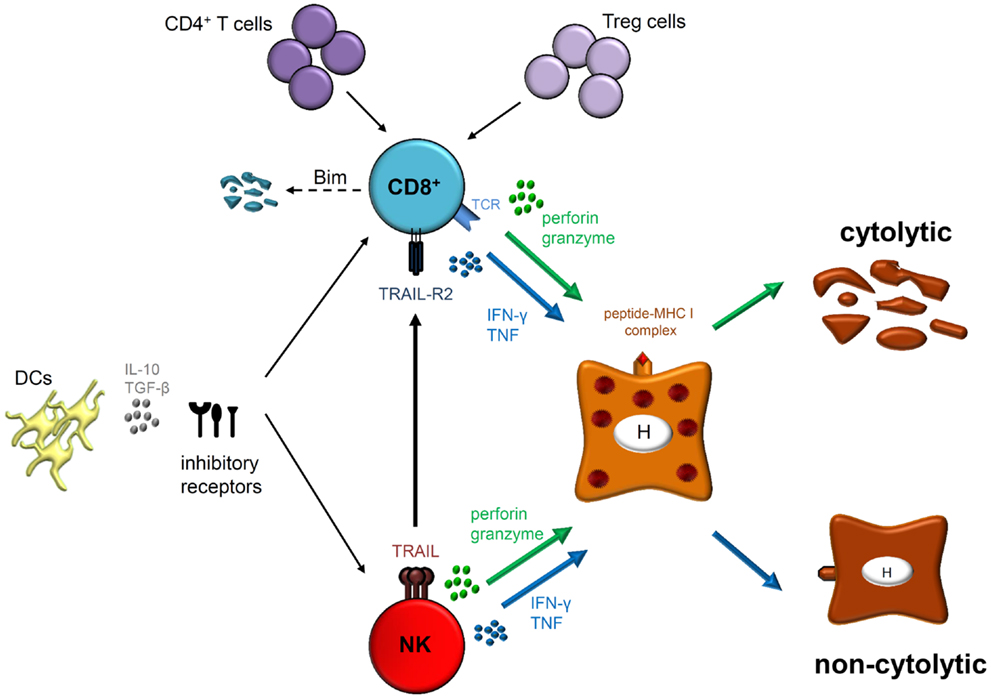
Figure 1. Natural killer cells as well as HBV-specific CD8+ T cells exhibit their antiviral functions by cytolytic (perforin and granzyme) and/or non-cytolytic (IFN-γ and TNF) mechanisms. However, CD8+ T cells express antigen-specific T cell receptors (TCRs) that interact with peptide-MHC I complexes on infected hepatocytes (H) whereas NK cell activation is thought to be antigen-independent. Effector functions and phenotype of both cell types are modulated during acute and chronic HBV infection. Indeed, different mechanisms play a role in regulating both effector populations, such as DCs, immunoregulatory cytokines (IL-10 and/or TGF-β) and expression of several inhibitory receptors. Furthermore, lack of CD4+ T cell help and interaction with regulatory T (Treg) cells may lead to CD8+ T cell dysfunction in chronically HBV-infected patients resulting in Bim-mediated apoptosis. Importantly, NK cells are also able to inhibit antiviral T cell responses by deleting HBV-specific CD8+ T cells in a TRAIL-dependent manner.
Cell culture models taking advantage of hepatoma cell lines that are transduced with the recently identified HBV entry receptor, hNTCP, may allow novel insights into HBV immunobiology and pathogenesis, revealing the relative contribution of cytolytic and non-cytolytic mechanisms to viral clearance. Furthermore, enrichment techniques could uncover whether HBV-specific CD8+ T cells are actually deleted in chronically HBV-infected patients and could also elucidate the phenotype of the detectable virus-specific CD8+ T cells. A better understanding of the mechanisms leading to viral persistence may result in new therapeutic treatment strategies that aim to remedy the T cell defects described, thereby augmenting functional responses and decreasing antigen-unspecific liver damage.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Honer Zu Siederdissen C, Cornberg M. The role of HBsAg levels in the current management of chronic HBV infection. Ann Gastroenterol (2014) 27:105–12.
2. Peppa D, Gill US, Reynolds G, Easom NJ, Pallett LJ, Schurich A, et al. Up-regulation of a death receptor renders antiviral T cells susceptible to NK cell-mediated deletion. J Exp Med (2013) 210:99–114. doi:10.1084/jem.20121172
3. Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, et al. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol (2003) 77:68–76. doi:10.1128/JVI.77.1.68-76.2003
4. Bertoletti A, Ferrari C. Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Gut (2012) 61:1754–64. doi:10.1136/gutjnl-2011-301073
5. Maini MK, Boni C, Lee CK, Larrubia JR, Reignat S, Ogg GS, et al. The role of virus-specific CD8(+) cells in liver damage and viral control during persistent hepatitis B virus infection. J Exp Med (2000) 191:1269–80. doi:10.1084/jem.191.8.1269
6. Doherty DG, O’Farrelly C. Innate and adaptive lymphoid cells in the human liver. Immunol Rev (2000) 174:5–20. doi:10.1034/j.1600-0528.2002.017416.x
7. Gregoire C, Chasson L, Luci C, Tomasello E, Geissmann F, Vivier E, et al. The trafficking of natural killer cells. Immunol Rev (2007) 220:169–82. doi:10.1111/j.1600-065X.2007.00563.x
8. Mackay IR. Hepatoimmunology: a perspective. Immunol Cell Biol (2002) 80:36–44. doi:10.1046/j.1440-1711.2002.01063.x
9. Chen Y, Wei H, Gao B, Hu Z, Zheng S, Tian Z. Activation and function of hepatic NK cells in hepatitis B infection: an underinvestigated innate immune response. J Viral Hepat (2005) 12:38–45. doi:10.1111/j.1365-2893.2005.00543.x
10. Lanier LL. NK cell recognition. Annu Rev Immunol (2005) 23:225–74. doi:10.1146/annurev.immunol.23.021704.115526
11. Lanier LL. Back to the future – defining NK cells and T cells. Eur J Immunol (2007) 37:1424–6. doi:10.1002/eji.200737418
12. O’Leary JG, Goodarzi M, Drayton DL, Von Andrian UH. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat Immunol (2006) 7:507–16. doi:10.1038/ni1332
13. Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol (2008) 9:495–502. doi:10.1038/ni1581
14. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol (2001) 22:633–40. doi:10.1016/S1471-4906(01)02060-9
15. Kimura K, Kakimi K, Wieland S, Guidotti LG, Chisari FV. Interleukin-18 inhibits hepatitis B virus replication in the livers of transgenic mice. J Virol (2002) 76:10702–7. doi:10.1128/JVI.76.21.10702-10707.2002
16. Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol (2003) 3:781–90. doi:10.1038/nri1199
17. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol (2013) 31:413–41. doi:10.1146/annurev-immunol-032712-095951
18. Sivori S, Vitale M, Morelli L, Sanseverino L, Augugliaro R, Bottino C, et al. p46, a novel natural killer cell-specific surface molecule that mediates cell activation. J Exp Med (1997) 186:1129–36. doi:10.1084/jem.186.7.1129
19. Vitale M, Bottino C, Sivori S, Sanseverino L, Castriconi R, Marcenaro E, et al. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J Exp Med (1998) 187:2065–72. doi:10.1084/jem.187.12.2065
20. Pende D, Parolini S, Pessino A, Sivori S, Augugliaro R, Morelli L, et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J Exp Med (1999) 190:1505–16. doi:10.1084/jem.190.10.1505
21. Moretta L, Pietra G, Montaldo E, Vacca P, Pende D, Falco M, et al. Human NK cells: from surface receptors to the therapy of leukemias and solid tumors. Front Immunol (2014) 5:87. doi:10.3389/fimmu.2014.00087
22. Campbell KS, Hasegawa J. Natural killer cell biology: an update and future directions. J Allergy Clin Immunol (2013) 132:536–44. doi:10.1016/j.jaci.2013.07.006
23. Moretta A, Bottino C, Vitale M, Pende D, Biassoni R, Mingari MC, et al. Receptors for HLA class-I molecules in human natural killer cells. Annu Rev Immunol (1996) 14:619–48. doi:10.1146/annurev.immunol.14.1.619
24. Lazetic S, Chang C, Houchins JP, Lanier LL, Phillips JH. Human natural killer cell receptors involved in MHC class I recognition are disulfide-linked heterodimers of CD94 and NKG2 subunits. J Immunol (1996) 157:4741–5.
25. Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol (1999) 17:189–220. doi:10.1146/annurev.immunol.17.1.189
26. Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol (2001) 19:65–91. doi:10.1146/annurev.immunol.19.1.65
27. Tang H, Li C, Wang L, Zhang H, Fan Z. Granzyme H of cytotoxic lymphocytes is required for clearance of the hepatitis B virus through cleavage of the hepatitis B virus X protein. J Immunol (2012) 188:824–31. doi:10.4049/jimmunol.1102205
28. Mundt B, Kuhnel F, Zender L, Paul Y, Tillmann H, Trautwein C, et al. Involvement of TRAIL and its receptors in viral hepatitis. FASEB J (2003) 17:94–6. doi:10.1096/fj.02-0537fje
29. Inngjerdingen M, Kveberg L, Naper C, Vaage JT. Natural killer cell subsets in man and rodents. Tissue Antigens (2011) 78:81–8. doi:10.1111/j.1399-0039.2011.01714.x
30. De Maria A, Bozzano F, Cantoni C, Moretta L. Revisiting human natural killer cell subset function revealed cytolytic CD56(dim)CD16+ NK cells as rapid producers of abundant IFN-gamma on activation. Proc Natl Acad Sci U S A (2011) 108:728–32. doi:10.1073/pnas.1012356108
31. Chan A, Hong DL, Atzberger A, Kollnberger S, Filer AD, Buckley CD, et al. CD56bright human NK cells differentiate into CD56dim cells: role of contact with peripheral fibroblasts. J Immunol (2007) 179:89–94. doi:10.4049/jimmunol.179.1.89
32. Ouyang Q, Baerlocher G, Vulto I, Lansdorp PM. Telomere length in human natural killer cell subsets. Ann N Y Acad Sci (2007) 1106:240–52. doi:10.1196/annals.1392.001
33. Romagnani C, Juelke K, Falco M, Morandi B, D’Agostino A, Costa R, et al. CD56brightCD16− killer Ig-like receptor-NK cells display longer telomeres and acquire features of CD56dim NK cells upon activation. J Immunol (2007) 178:4947–55. doi:10.4049/jimmunol.178.8.4947
34. Zhao J, Li Y, Jin L, Zhang S, Fan R, Sun Y, et al. Natural killer cells are characterized by the concomitantly increased interferon-gamma and cytotoxicity in acute resolved hepatitis B patients. PLoS One (2012) 7:e49135. doi:10.1371/journal.pone.0049135
35. Lunemann S, Malone DF, Hengst J, Port K, Grabowski J, Deterding K, et al. Compromised function of natural killer cells in acute and chronic viral hepatitis. J Infect Dis (2013) 209(9):1362–73. doi:10.1093/infdis/jit561
36. Moretta A, Marcenaro E, Parolini S, Ferlazzo G, Moretta L. NK cells at the interface between innate and adaptive immunity. Cell Death Differ (2008) 15:226–33. doi:10.1038/sj.cdd.4402170
37. Carbone E, Ruggiero G, Terrazzano G, Palomba C, Manzo C, Fontana S, et al. A new mechanism of NK cell cytotoxicity activation: the CD40-CD40 ligand interaction. J Exp Med (1997) 185:2053–60. doi:10.1084/jem.185.12.2053
38. Nakajima A, Kodama T, Morimoto S, Azuma M, Takeda K, Oshima H, et al. Antitumor effect of CD40 ligand: elicitation of local and systemic antitumor responses by IL-12 and B7. J Immunol (1998) 161:1901–7.
39. Rakhmilevich AL, Alderson KL, Sondel PM. T-cell-independent antitumor effects of CD40 ligation. Int Rev Immunol (2012) 31:267–78. doi:10.3109/08830185.2012.698337
40. Della Chiesa M, Romagnani C, Thiel A, Moretta L, Moretta A. Multidirectional interactions are bridging human NK cells with plasmacytoid and monocyte-derived dendritic cells during innate immune responses. Blood (2006) 108:3851–8. doi:10.1182/blood-2006-02-004028
41. Lang PA, Lang KS, Xu HC, Grusdat M, Parish IA, Recher M, et al. Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc Natl Acad Sci U S A (2012) 109:1210–5. doi:10.1073/pnas.1118834109
42. Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature (2012) 481:394–8. doi:10.1038/nature10624
43. Cook KD, Whitmire JK. The depletion of NK cells prevents T cell exhaustion to efficiently control disseminating virus infection. J Immunol (2013) 190:641–9. doi:10.4049/jimmunol.1202448
44. Chisari FV, Isogawa M, Wieland SF. Pathogenesis of hepatitis B virus infection. Pathol Biol (Paris) (2010) 58:258–66. doi:10.1016/j.patbio.2009.11.001
45. Wieland S, Thimme R, Purcell RH, Chisari FV. Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci U S A (2004) 101:6669–74. doi:10.1073/pnas.0401771101
46. Lucifora J, Durantel D, Testoni B, Hantz O, Levrero M, Zoulim F. Control of hepatitis B virus replication by innate response of HepaRG cells. Hepatology (2010) 51:63–72. doi:10.1002/hep.23230
47. Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV. Viral clearance without destruction of infected cells during acute HBV infection. Science (1999) 284:825–9. doi:10.1126/science.284.5415.825
48. Fisicaro P, Valdatta C, Boni C, Massari M, Mori C, Zerbini A, et al. Early kinetics of innate and adaptive immune responses during hepatitis B virus infection. Gut (2009) 58:974–82. doi:10.1136/gut.2008.163600
49. Webster GJ, Reignat S, Maini MK, Whalley SA, Ogg GS, King A, et al. Incubation phase of acute hepatitis B in man: dynamic of cellular immune mechanisms. Hepatology (2000) 32:1117–24. doi:10.1053/jhep.2000.19324
50. Dunn C, Peppa D, Khanna P, Nebbia G, Jones M, Brendish N, et al. Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology (2009) 137:1289–300. doi:10.1053/j.gastro.2009.06.054
51. Oliviero B, Varchetta S, Paudice E, Michelone G, Zaramella M, Mavilio D, et al. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology (2009) 137:1151–60. doi:10.1053/j.gastro.2009.05.047
52. Zhang Z, Zhang S, Zou Z, Shi J, Zhao J, Fan R, et al. Hypercytolytic activity of hepatic natural killer cells correlates with liver injury in chronic hepatitis B patients. Hepatology (2011) 53:73–85. doi:10.1002/hep.23977
53. Tjwa ET, Van Oord GW, Hegmans JP, Janssen HL, Woltman AM. Viral load reduction improves activation and function of natural killer cells in patients with chronic hepatitis B. J Hepatol (2011) 54:209–18. doi:10.1016/j.jhep.2010.07.009
54. Nattermann J, Feldmann G, Ahlenstiel G, Langhans B, Sauerbruch T, Spengler U. Surface expression and cytolytic function of natural killer cell receptors is altered in chronic hepatitis C. Gut (2006) 55:869–77. doi:10.1136/gut.2005.076463
55. Bonorino P, Ramzan M, Camous X, Dufeu-Duchesne T, Thelu MA, Sturm N, et al. Fine characterization of intrahepatic NK cells expressing natural killer receptors in chronic hepatitis B and C. J Hepatol (2009) 51:458–67. doi:10.1016/j.jhep.2009.05.030
56. Ju Y, Hou N, Meng J, Wang X, Zhang X, Zhao D, et al. T cell immunoglobulin- and mucin-domain-containing molecule-3 (Tim-3) mediates natural killer cell suppression in chronic hepatitis B. J Hepatol (2010) 52:322–9. doi:10.1016/j.jhep.2009.12.005
57. Tang KF, Chen M, Xie J, Song GB, Shi YS, Liu Q, et al. Inhibition of hepatitis B virus replication by small interference RNA induces expression of MICA in HepG2.2.15 cells. Med Microbiol Immunol (2009) 198:27–32. doi:10.1007/s00430-008-0101-6
58. Peppa D, Micco L, Javaid A, Kennedy PT, Schurich A, Dunn C, et al. Blockade of immunosuppressive cytokines restores NK cell antiviral function in chronic hepatitis B virus infection. PLoS Pathog (2010) 6:e1001227. doi:10.1371/journal.ppat.1001227
59. Woltman AM, Op Den Brouw ML, Biesta PJ, Shi CC, Janssen HL. Hepatitis B virus lacks immune activating capacity, but actively inhibits plasmacytoid dendritic cell function. PLoS One (2011) 6:e15324. doi:10.1371/journal.pone.0015324
60. Martinet J, Dufeu-Duchesne T, Bruder Costa J, Larrat S, Marlu A, Leroy V, et al. Altered functions of plasmacytoid dendritic cells and reduced cytolytic activity of natural killer cells in patients with chronic HBV infection. Gastroenterology (2012) 143:1586–1596e1588. doi:10.1053/j.gastro.2012.08.046
61. Shi CC, Tjwa ET, Biesta PJ, Boonstra A, Xie Q, Janssen HL, et al. Hepatitis B virus suppresses the functional interaction between natural killer cells and plasmacytoid dendritic cells. J Viral Hepat (2012) 19:e26–33. doi:10.1111/j.1365-2893.2011.01496.x
62. Dunn C, Brunetto M, Reynolds G, Christophides T, Kennedy PT, Lampertico P, et al. Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell-mediated liver damage. J Exp Med (2007) 204:667–80. doi:10.1084/jem.20061287
63. Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity (2011) 35:161–8. doi:10.1016/j.immuni.2011.07.010
64. Youngblood B, Hale JS, Ahmed R. T-cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunology (2013) 139:277–84. doi:10.1111/imm.12074
65. Appay V, Van Lier RA, Sallusto F, Roederer M. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A (2008) 73:975–83. doi:10.1002/cyto.a.20643
66. Maini MK, Boni C, Ogg GS, King AS, Reignat S, Lee CK, et al. Direct ex vivo analysis of hepatitis B virus-specific CD8(+) T cells associated with the control of infection. Gastroenterology (1999) 117:1386–96. doi:10.1016/S0016-5085(99)70289-1
67. Sandalova E, Laccabue D, Boni C, Watanabe T, Tan A, Zong HZ, et al. Increased levels of arginase in patients with acute hepatitis B suppress antiviral T cells. Gastroenterology (2012) 143:78–87e73. doi:10.1053/j.gastro.2012.03.041
68. Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity (1996) 4:25–36. doi:10.1016/S1074-7613(00)80295-2
69. Phillips S, Chokshi S, Riva A, Evans A, Williams R, Naoumov NV. CD8(+) T cell control of hepatitis B virus replication: direct comparison between cytolytic and noncytolytic functions. J Immunol (2010) 184:287–95. doi:10.4049/jimmunol.0902761
70. Ando K, Guidotti LG, Wirth S, Ishikawa T, Missale G, Moriyama T, et al. Class I-restricted cytotoxic T lymphocytes are directly cytopathic for their target cells in vivo. J Immunol (1994) 152:3245–53.
71. Dandri M, Lutgehetmann M, Petersen J. Experimental models and therapeutic approaches for HBV. Semin Immunopathol (2013) 35:7–21. doi:10.1007/s00281-012-0335-7
72. Sun D, Nassal M. Stable HepG2- and Huh7-based human hepatoma cell lines for efficient regulated expression of infectious hepatitis B virus. J Hepatol (2006) 45:636–45. doi:10.1016/j.jhep.2006.05.019
73. Sells MA, Chen ML, Acs G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci U S A (1987) 84:1005–9. doi:10.1073/pnas.84.4.1005
74. Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, et al. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother (1997) 41:1715–20.
75. Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife (2012) 1:e00049. doi:10.7554/eLife.00049
76. Ni Y, Lempp FA, Mehrle S, Nkongolo S, Kaufman C, Falth M, et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology (2014) 146:1070–1083e1076. doi:10.1053/j.gastro.2013.12.024
77. Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, et al. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci U S A (2002) 99:15655–60. doi:10.1073/pnas.232137699
78. Hantz O, Parent R, Durantel D, Gripon P, Guguen-Guillouzo C, Zoulim F. Persistence of the hepatitis B virus covalently closed circular DNA in HepaRG human hepatocyte-like cells. J Gen Virol (2009) 90:127–35. doi:10.1099/vir.0.004861-0
79. Ferrari C, Penna A, Bertoletti A, Valli A, Antoni AD, Giuberti T, et al. Cellular immune response to hepatitis B virus-encoded antigens in acute and chronic hepatitis B virus infection. J Immunol (1990) 145:3442–9.
80. Bertoletti A, Costanzo A, Chisari FV, Levrero M, Artini M, Sette A, et al. Cytotoxic T lymphocyte response to a wild type hepatitis B virus epitope in patients chronically infected by variant viruses carrying substitutions within the epitope. J Exp Med (1994) 180:933–43. doi:10.1084/jem.180.3.933
81. Webster GJ, Reignat S, Brown D, Ogg GS, Jones L, Seneviratne SL, et al. Longitudinal analysis of CD8+ T cells specific for structural and nonstructural hepatitis B virus proteins in patients with chronic hepatitis B: implications for immunotherapy. J Virol (2004) 78:5707–19. doi:10.1128/JVI.78.11.5707-5719.2004
82. Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T, et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol (2007) 81:4215–25. doi:10.1128/JVI.02844-06
83. Lopes AR, Kellam P, Das A, Dunn C, Kwan A, Turner J, et al. Bim-mediated deletion of antigen-specific CD8 T cells in patients unable to control HBV infection. J Clin Invest (2008) 118:1835–45. doi:10.1172/JCI33402
84. Wang FS, Xing LH, Liu MX, Zhu CL, Liu HG, Wang HF, et al. Dysfunction of peripheral blood dendritic cells from patients with chronic hepatitis B virus infection. World J Gastroenterol (2001) 7:537–41.
85. Beckebaum S, Cicinnati VR, Dworacki G, Muller-Berghaus J, Stolz D, Harnaha J, et al. Reduction in the circulating pDC1/pDC2 ratio and impaired function of ex vivo-generated DC1 in chronic hepatitis B infection. Clin Immunol (2002) 104:138–50. doi:10.1006/clim.2002.5245
86. van der Molen RG, Sprengers D, Binda RS, De Jong EC, Niesters HG, Kusters JG, et al. Functional impairment of myeloid and plasmacytoid dendritic cells of patients with chronic hepatitis B. Hepatology (2004) 40:738–46. doi:10.1002/hep.20366
87. Duan XZ, Zhuang H, Wang M, Li HW, Liu JC, Wang FS. Decreased numbers and impaired function of circulating dendritic cell subsets in patients with chronic hepatitis B infection (R2). J Gastroenterol Hepatol (2005) 20:234–42. doi:10.1111/j.1440-1746.2004.03529.x
88. Alanio C, Lemaitre F, Law HK, Hasan M, Albert ML. Enumeration of human antigen-specific naive CD8+ T cells reveals conserved precursor frequencies. Blood (2010) 115:3718–25. doi:10.1182/blood-2009-10-251124
89. Reignat S, Webster GJ, Brown D, Ogg GS, King A, Seneviratne SL, et al. Escaping high viral load exhaustion: CD8 cells with altered tetramer binding in chronic hepatitis B virus infection. J Exp Med (2002) 195:1089–101. doi:10.1084/jem.20011723
90. Boni C, Laccabue D, Lampertico P, Giuberti T, Vigano M, Schivazappa S, et al. Restored function of HBV-specific T cells after long-term effective therapy with nucleos(t)ide analogues. Gastroenterology (2012) 143:963–73. doi:10.1053/j.gastro.2012.07.014
91. Fisicaro P, Valdatta C, Massari M, Loggi E, Biasini E, Sacchelli L, et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology (2010) 138:682–93. doi:10.1053/j.gastro.2009.09.052
92. Schurich A, Khanna P, Lopes AR, Han KJ, Peppa D, Micco L, et al. Role of the coinhibitory receptor cytotoxic T lymphocyte antigen-4 on apoptosis-prone CD8 T cells in persistent hepatitis B virus infection. Hepatology (2011) 53:1494–503. doi:10.1002/hep.24249
93. Nebbia G, Peppa D, Schurich A, Khanna P, Singh HD, Cheng Y, et al. Upregulation of the Tim-3/galectin-9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS One (2012) 7:e47648. doi:10.1371/journal.pone.0047648
94. Wu W, Shi Y, Li S, Zhang Y, Liu Y, Wu Y, et al. Blockade of Tim-3 signaling restores the virus-specific CD8(+) T-cell response in patients with chronic hepatitis B. Eur J Immunol (2012) 42:1180–91. doi:10.1002/eji.201141852
95. Raziorrouh B, Schraut W, Gerlach T, Nowack D, Gruner NH, Ulsenheimer A, et al. The immunoregulatory role of CD244 in chronic hepatitis B infection and its inhibitory potential on virus-specific CD8+ T-cell function. Hepatology (2010) 52:1934–47. doi:10.1002/hep.23936
96. Muhlbauer M, Fleck M, Schutz C, Weiss T, Froh M, Blank C, et al. PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. J Hepatol (2006) 45:520–8. doi:10.1016/j.jhep.2006.05.007
97. Knolle PA, Thimme R. Hepatic immune regulation and its involvement in viral hepatitis infection. Gastroenterology (2014) 146(5):1193–207. doi:10.1053/j.gastro.2013.12.036
98. Maier H, Isogawa M, Freeman GJ, Chisari FV. PD-1:PD-L1 interactions contribute to the functional suppression of virus-specific CD8+ T lymphocytes in the liver. J Immunol (2007) 178:2714–20. doi:10.4049/jimmunol.178.5.2714
99. Liu J, Zhang E, Ma Z, Wu W, Kosinska A, Zhang X, et al. Enhancing virus-specific immunity in vivo by combining therapeutic vaccination and PD-L1 blockade in chronic hepadnaviral infection. PLoS Pathog (2014) 10:e1003856. doi:10.1371/journal.ppat.1003856
100. Fisicaro P, Valdatta C, Massari M, Loggi E, Ravanetti L, Urbani S, et al. Combined blockade of programmed death-1 and activation of CD137 increase responses of human liver T cells against HBV, but not HCV. Gastroenterology (2012) 143:1576–1585e1574. doi:10.1053/j.gastro.2012.08.041
101. Schurich A, Pallett LJ, Lubowiecki M, Singh HD, Gill US, Kennedy PT, et al. The third signal cytokine IL-12 rescues the anti-viral function of exhausted HBV-specific CD8 T cells. PLoS Pathog (2013) 9:e1003208. doi:10.1371/journal.ppat.1003208
102. Yang PL, Althage A, Chung J, Maier H, Wieland S, Isogawa M, et al. Immune effectors required for hepatitis B virus clearance. Proc Natl Acad Sci U S A (2010) 107:798–802. doi:10.1073/pnas.0913498107
103. Stoop JN, van der Molen RG, Baan CC, van der Laan LJ, Kuipers EJ, Kusters JG, et al. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis B virus infection. Hepatology (2005) 41:771–8. doi:10.1002/hep.20649
104. Xu D, Fu J, Jin L, Zhang H, Zhou C, Zou Z, et al. Circulating and liver resident CD4+CD25+ regulatory T cells actively influence the antiviral immune response and disease progression in patients with hepatitis B. J Immunol (2006) 177:739–47. doi:10.4049/jimmunol.177.1.739
105. Yang G, Liu A, Xie Q, Guo TB, Wan B, Zhou B, et al. Association of CD4+CD25+Foxp3+ regulatory T cells with chronic activity and viral clearance in patients with hepatitis B. Int Immunol (2007) 19:133–40. doi:10.1093/intimm/dxl130
106. Das A, Ellis G, Pallant C, Lopes AR, Khanna P, Peppa D, et al. IL-10-producing regulatory B cells in the pathogenesis of chronic hepatitis B virus infection. J Immunol (2012) 189:3925–35. doi:10.4049/jimmunol.1103139
107. Chisari FV. Regulation of human lymphocyte function by a soluble extract from normal human liver. J Immunol (1978) 121:1279–86.
Keywords: natural killer cells, CD8+ T cells, hepatitis B virus, functional dichotomy, T cell failure
Citation: Schuch A, Hoh A and Thimme R (2014) The role of natural killer cells and CD8+ T cells in hepatitis B virus infection. Front. Immunol. 5:258. doi: 10.3389/fimmu.2014.00258
Received: 09 April 2014; Paper pending published: 25 April 2014;
Accepted: 19 May 2014; Published online: 03 June 2014.
Edited by:
Lynn B. Dustin, University of Oxford, UKReviewed by:
John J. Miles, Queensland Institute of Medical Research, AustraliaMala K. Maini, University College London, UK
Copyright: © 2014 Schuch, Hoh and Thimme. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Thimme, Department of Medicine II, University Hospital of Freiburg, Hugstetter Street 55, Freiburg 79106, Germany e-mail:cm9iZXJ0LnRoaW1tZUB1bmlrbGluaWstZnJlaWJ1cmcuZGU=