 Wesley S. Bond1,2
Wesley S. Bond1,2 Tonia S. Rex1,2*
Tonia S. Rex1,2*- 1Vanderbilt Eye Institute, Vanderbilt University Medical Center, Nashville, TN, USA
- 2Vanderbilt Brain Institute, Vanderbilt University Medical Center, Nashville, TN, USA
Neuroinflammation is a normal and healthy response to neuronal damage. However, excessive or chronic neuroinflammation exacerbates neurodegeneration after trauma and in progressive diseases such as Alzheimer’s, Parkinson’s, age-related macular degeneration, and glaucoma. Therefore, molecules that modulate neuroinflammation are candidates as neuroprotective agents. Erythropoietin (EPO) is a known neuroprotective agent that indirectly attenuates neuroinflammation, in part, by inhibiting neuronal apoptosis. In this review, we provide evidence that EPO also modulates neuroinflammation upstream of apoptosis by acting directly on glia. Further, the signaling induced by EPO may differ depending on cell type and context possibly as a result of activation of different receptors. While significant progress has been made in our understanding of EPO signaling, this review also identifies areas for future study in terms of the role of EPO in modulating neuroinflammation.
Introduction
Inflammation is a physiologic response to injury and infection and is necessary for tissue healing. A similar process occurs in the central nervous system (CNS) in response to injury or disease and is termed neuroinflammation. In acute neuroinflammation, microglial cells become reactive, they phagocytose dying cells and release pro-inflammatory cytokines and chemokines to limit the area of injury [for review see Ref. (1)]. However, when neuroinflammation is severe or chronic, it can produce deleterious effects involving pro-inflammatory signaling pathways, increased oxidative stress, and death of nearby neurons. Neuroinflammation is a common mechanism influencing the severity and progression of neurodegenerative disease and injury and is, therefore, a potential target for neuroprotective therapies [for review see Ref. (2)].
Erythropoietin (EPO) was originally identified as a cytokine responsible for production of red blood cells by blocking apoptosis of progenitor cells [for reviews see Ref. (3–6)]. EPO is also produced at low levels in CNS tissue, and the EPO receptor (EpoR) homodimer is expressed on most CNS cell types, including neurons, astrocytes, and microglia [for review see Ref. (7)]. In the last 20 years, EPO has proven to be effective in preventing neuronal apoptosis in a wide-range of neurodegenerative conditions in the brain, retina, and spinal cord including acute, chronic, inherited, and induced degenerations. Briefly, EPO affects the regulators of apoptosis Bax, Bad, and Bcl-2/Bcl-xL by inhibiting formation of the Bax/Bcl complex and reducing activation of effector caspases. Comprehensive reviews on the anti-apoptotic effect of EPO in the CNS are available (4, 8, 9). EPO also blocks apoptosis in retinal neurons (10–23), showing that EPO acts similarly in all CNS tissue. Since the anti-apoptotic role of EPO is well-characterized it is not the focus of this review except to note that it is well accepted that EPO decreases neuroinflammation and its damaging effects in part by blocking apoptosis [Ref. (22, 23); Figure 1]. This review will discuss recent evidence that points to additional, apoptosis-independent, actions of EPO in modulating neuroinflammation including blocking reactive oxygen/nitrogen species (ROS/RNS) and glial reactivity. Accruing evidence that the signal transduction cascades activated by EPO may differ based on cell type will also be presented.
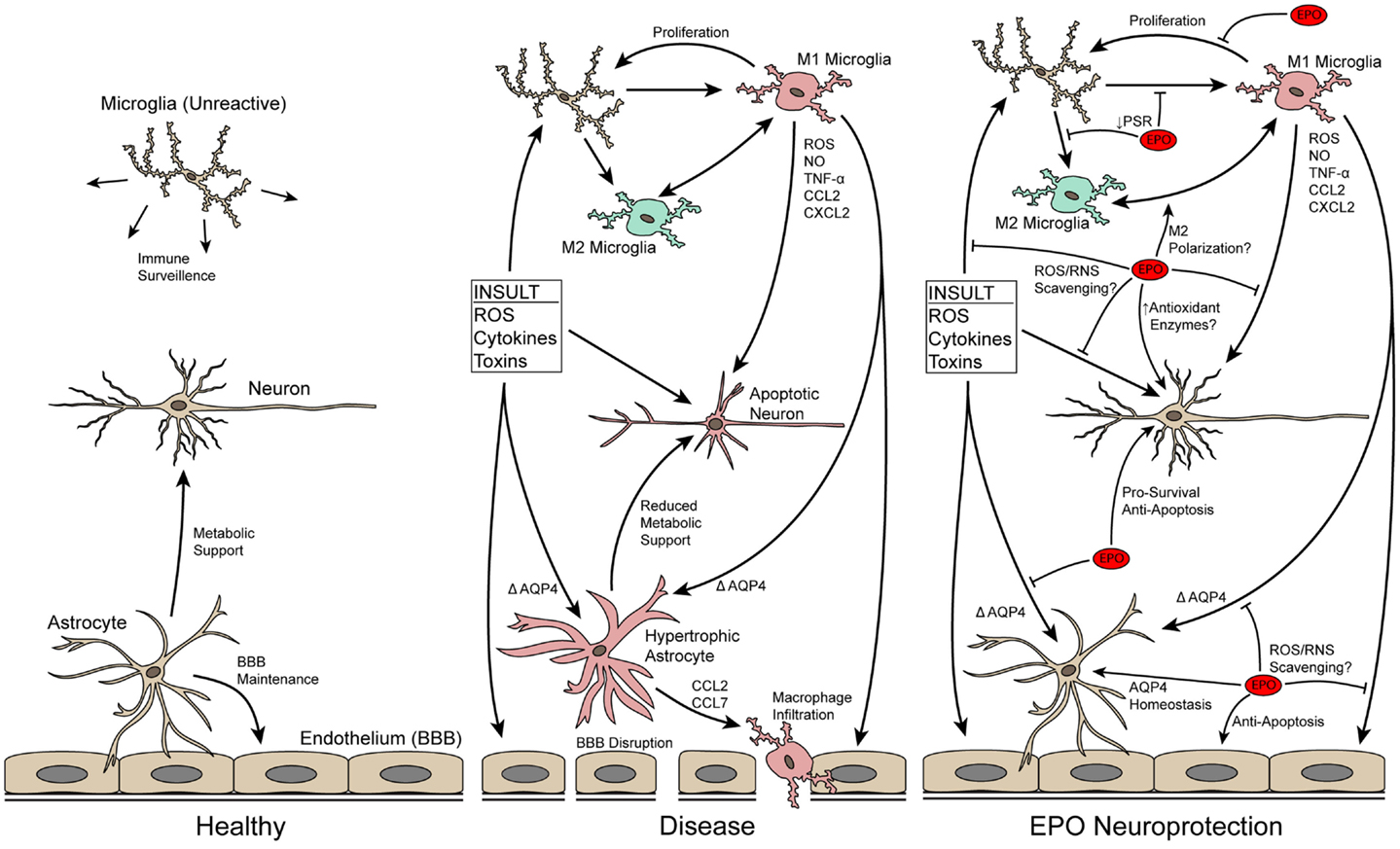
Figure 1. Schematic of neuron–glia interactions in healthy and diseased CNS tissue and how EPO may protect neurons by modulating neuroinflammation. In the normal CNS, microglia and astrocytes serve important support roles. In disease/trauma, the BBB breaks-down due to endothelial cell death and astrocyte hypertrophy, immune cells infiltrate into the CNS, microglia increase in number and become reactive, and neurons undergo apoptosis. EPO directly blocks apoptosis of neurons and preserves the BBB by blocking apoptosis of endothelial cells and decreasing astrocyte hypertrophy, thus, decreasing infiltration of immune cells. In addition, EPO may directly scavenge ROS/RNS to reduce local oxidative stress. EPO also has a direct effect on microglia, affecting their proliferative capability and possibly influencing their M1/M2 reactive state.
EPO Limits Neuroinflammation and Cell Death by Decreasing ROS/RNS Levels
Oxidative/nitrosative stress refers to the undesirable modification of proteins, lipids, and DNA mainly thought to arise from mitochondrial dysfunction [for review see Ref. (24)]. Peroxynitrite, a by-product of superoxide and nitric oxide, can cause DNA damage and ultimately lead to necrosis, which in turn drives an inflammatory response that includes microglial reactivity. Reactive microglia are particularly effective at producing and releasing ROS/RNS [for review see Ref. (25)]. Oxidative/nitrosative stress and neuroinflammation have been implicated in a myriad of disease processes and has been shown to contribute to neuronal degeneration in Alzheimer’s, Parkinson’s, traumatic brain injury [for review see Ref. (26)], and glaucoma [for review see Ref. (27)].
Treatment with EPO decreases cellular damage caused by ROS/RNS, including lipid peroxidation (28–30), protein carbonylation (30), and protein nitrosylation (31) and significant progress has been made in elucidating how this is accomplished. EPO preserves mitochondrial membrane integrity in a β-amyloid model of Alzheimer’s disease (32). It also increases levels of antioxidant enzymes by increasing levels and/or translocation of nuclear factor erythroid 2-related factor 2 (Nrf-2) to the nucleus where it binds and activates the antioxidant response element (33–36). In neurons, the increased nuclear translocation of Nrf-2 appears to be mediated by PI-3K, ERK, and JNK, but not p38/MAPK (36). Some of the antioxidant enzymes increased by EPO in terms of both levels and activity include heme oxygenase (HO-1) (33, 36), peroxiredoxin (37), glutathione peroxidase, NAD(P)H:quinone oxidoreductase 1 (NQO1) (33–35), glutamate cysteine ligase, and glutathione S-transferase (33, 35). EPO also causes increases in the in vivo activity of the antioxidant proteins catalase (29), superoxide dismutase (30), and glutathione peroxidase (38). However, EPO’s positive effect on levels and activity of these enzymes is not consistently observed (39, 40). For example, EPO has no effect on expression of induced nitric oxide synthase (iNOS) in cultured activated microglia (41), even though it reduces total retinal levels of iNOS in a glaucoma model (42). These data suggest that EPO’s effect on antioxidant enzyme activity may contextual or cell-specific.
Two alternative methods for reduction of oxidative/nitrosative stress by EPO have been reported. First, there is emerging biochemical evidence that EPO is capable of directly scavenging ROS/RNS (43), including a study showing that EPO protects paraquat-treated astrocytes in a superoxide dismutase knockout mouse (44). Second, in some disease processes where iron accumulation is thought to be a key mediator of oxidative damage and degeneration, such as Parkinson’s disease [for review see Ref. (45)], EPO may indirectly promote an antioxidant effect through its increase in erythrocyte production and corresponding depletion of systemic iron. This is supported by the long-observed phenomenon of EPO treatment leading to systemic iron depletion [for review see Ref. (46)]. Therefore, EPO appears to act in a multi-pronged way to mitigate ROS/RNS levels thus preventing downstream damaging effects on cells that lead to apoptosis (Figure 1).
EPO Decreases Recruitment and Infiltration of Immune Cells
In neurodegenerative conditions, immune cells are recruited to the area of injury by the release of chemokines from the damaged neuronal tissue [for review see Ref. (47)]. Expression of two of these chemokines, CXCL2 and CCL7, is decreased by treatment with EPO in a stroke model (48). This suggests that EPO may limit recruitment of immune cells, which would in turn decrease release of pro-inflammatory cytokines into the CNS and migration of immune cells into the tissue. Migration of immune cells into the CNS occurs as a result of blood–brain/retina barrier (BBB/BRB) disruption after CNS trauma or in neurodegenerative diseases due to microvascular endothelial cell death (49), tight junction structural changes [for review see Ref. (50)], and astrocyte hypertrophy [Figure 1; for reviews see Ref. (51, 52)]. EPO preserves the BBB/BRB in multiple models (21, 53, 54) by blocking apoptosis of microvascular endothelial cells [for review see Ref. (9)] and astrocyte hypertrophy. Here, we will focus on the less well-characterized role of EPO in blocking astrocyte hypertrophy.
Dysregulation of aquaporin-4 is implicated in astrocyte swelling and disruption of the BBB/BRB in knockout models (55), though this effect is not consistently observed (56, 57). Astrocytes respond to EPO by activating JNK and p38-MAPK (53, 58), leading to modulation of aquaporin-4 levels (53, 58–60), decreased glial swelling (58), and reduced BBB permeability (54, 59). EPO also increases levels of tight junction proteins in these cells via activation of the MAPK cascade (53). Upregulation of the stress-induced intermediate filament protein, GFAP, is associated with glial hypertrophy and is also decreased by treatment with EPO (19, 60, 61). Interestingly, modulation of GFAP levels by EPO is independent of the MAPK pathway (60). Others have reported that EPO neither activates JAK2, Akt, ERK nor STAT in cultured astrocytes (53, 62), suggesting that these signaling molecules also might not be involved.
EPO Decreases Microglial Proliferation and Reactivity
Microglia are the key mediators of neuroinflammation. In the injured/degenerative CNS, innate microglia proliferate and convert into the M1 (pro-inflammatory) and M2 (alternative) reactive states in a manner similar to systemic macrophages [for review see Ref. (63)]. Reactive microglia can mediate both pro-inflammatory and anti-inflammatory states depending on their particular reactive state and the corresponding milieu of cytokines and chemokines released. The role of these cells is complex and current research suggests that while an overactive microglia response is deleterious, blocking it entirely can also be detrimental [for review see Ref. (64)]. In this review, we will focus on the damaging effects of chronically reactive microglia.
Erythropoietin may directly influence the reactive state of the CNS microglia (Figure 1). One of the distinguishing features of a reactive microglia is its ability to phagocytose dying neurons. Apoptotic neurons increase levels of phosphatidylserine on the outer leaflet of the plasma membrane. Recognition of these residues is a key step in microglial phagocytosis. Reactive microglia express higher levels of the phosphatidylserine receptor [PSR; (65)]. EPO treatment decreases levels of the PSR on the microglial plasma membrane in vitro (66). This suggests that EPO treatment decreases the ability of microglial cells to phagocytose dying neurons. Active phagocytosis of apoptotic cells by microglia suppresses production of pro-inflammatory cytokines (65). Therefore, the decrease in PSR could suggest that EPO induces a pro-inflammatory state in the CNS. However, treatment with EPO leads to lower levels of pro-inflammatory cytokines in in vivo studies of neurodegenerative conditions [Ref. (22, 67–69); Figure 1]. The decrease in pro-inflammatory cytokines by EPO is not due to a direct block in production based on in vitro experiments (22, 41, 70). This suggests that the lower levels detected in the in vivo studies is likely an indirectly consequence of EPO limiting the number of reactive microglia present. In fact, fewer proliferating cellular nuclear antigen-positive primary microglial cells are detected after treatment with EPO (66). In summary, the data suggest that EPO redirects microglia back to or maintains microglia in a normal state and prevents microglial proliferation.
EPO Induces Signaling in Microglia and Macrophages
Microglia share similar characteristics with systemic macrophages and both can be found in the injured/degenerative CNS. Macrophages can infiltrate into the CNS where they take on a reactive microglial-like morphology. Like microglial cells, macrophages secrete neuroinflammatory cytokines and phagocytose dying cells. Therefore, they play an important role in chronic neuroinflammation. We will compare and contrast signaling by EPO in microglia and macrophages.
The EpoR is expressed on both macrophages and microglia (71–74). The investigations performed, to date, on microglial cells in culture have focused on the ability of EPO to block cell death rather than an effect on altering the reactive state of these cells. While the physiological relevance of microglial cell survival as opposed to proliferation or reactivity is unclear, these studies at a minimum demonstrate that EPO can activate signaling cascades in microglia. EPO-mediated protection of EOC-2 microglia-derived cells requires Wnt-1, PI-3K, and Akt, and also involves mTOR, and p70S6K [Ref. (71–73); Figure 2]. As a result of activation of the PI-3K/Akt pathway, the p65 subunit of NF-κB is translocated to the nucleus (73). Surprisingly, there is a concomitant increase in levels of Wnt-1 and de-activation of glycogen synthase kinase 3 (GSK-3α/β) by phosphorylation, which results in activation (phosphorylation) of β-catenin. Activated β-catenin translocates to the nucleus where it can sequester NF-κB (75), preventing it from activating gene expression (Figure 2). It is unclear why EPO would seemingly activate two conflicting pathways. One possibility is that EPO modulates each pathway in a context-dependent manner to modulate neuroinflammation, either promoting a neuroinflammatory state (activation of NF-κB) or decreasing it (sequestration of NF-κB by β-catenin). Additional studies to assess signaling pathways induced by EPO in primary microglia, particularly in the context of microglial proliferation and polarization are warranted.
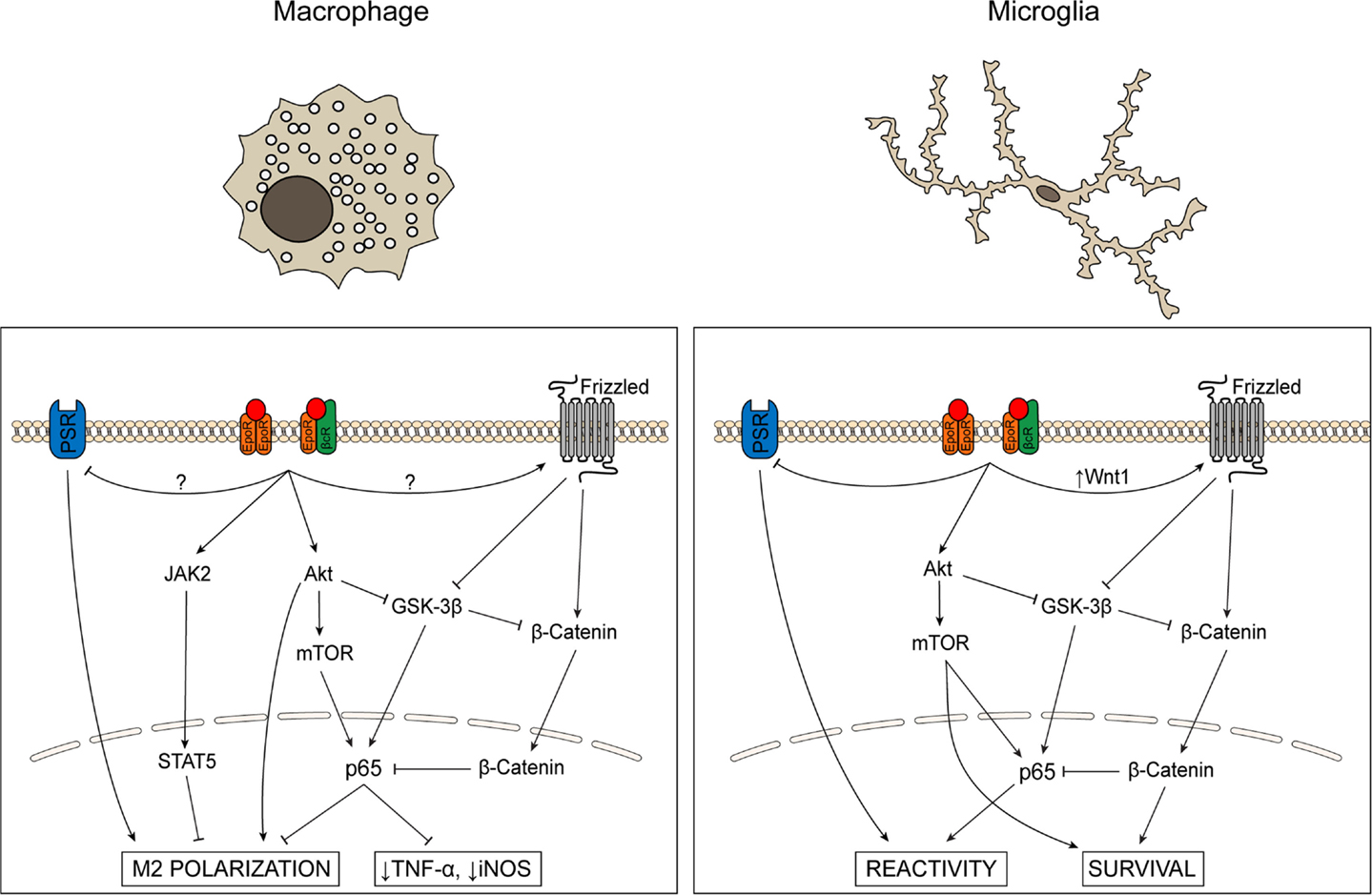
Figure 2. Schematic comparing signaling cascades activated by EPO in microglia and macrophages. The role of EPO in macrophages and microglia has primarily been evaluated in the context of inflammatory state and cell survival, respectively. In macrophages, EPO activates JAK/STAT and Akt signaling, inhibits GSK-3β activity, modulates NF-κB p65, decreases levels of pro-inflammatory cytokines and iNOS, and promotes phagocytosis and M2 activation state polarization. In microglia, EPO promotes cell survival, inhibits GSK-3β activity, and increases NF-κB p65 via PI-3K/Akt while simultaneously activating the Wnt-1/β-catenin pathway that results in sequestration of NF-κB in the nucleus. EPO also decreases PSR levels, suggesting decreased phagocytosis.
In contrast, the EPO studies on cultured macrophages assess the activation state rather than survival of these cells. As in microglial cells, treatment with EPO activates the Akt/mTOR/NF-κB pathway. This pathway is implicated in shifting macrophage activation state polarization from M1 to M2 (76). For example, treatment of cultured macrophages with an EPO peptide caused a dose-dependent increase in phagocytosis and a corresponding decrease in TNF-α, suggesting that EPO shifts these cells from a pro-inflammatory to a phagocytic state but does not return them to a non-reactive state (77). Also, unlike in microglial cells, EPO inhibits NF-κB p65 in macrophages, leading to lower levels of TNF-α and NO (78). Further studies are needed to understand how EPO decreases NF-κB activity in macrophages while increasing it in microglia. It is feasible that the same signaling balance between the PI-3K/Akt and Wnt/β-catenin pathways are present in both microglial cells and macrophages, but that the balance is shifted to the Wnt/β-catenin pathway in activated macrophages, resulting in sequestration/inhibition of NF-κB. However, while activation of β-catenin by Frizzled has been demonstrated in macrophages (79), to date, no studies have investigated if EPO affects this pathway or if EPO has any effect on Wnt-1. Given the phenotypic similarity between systemic macrophages and resident microglia, as well as the identification of common signaling pathways affected by EPO in both cell types, the potential role of EPO in modulating additional pathways in microglia that have been identified in macrophages should be investigated.
EPO Signaling may be Cell Type and Context Dependent
The molecular pathways activated by EPO for neuroprotection are an area of active investigation. Many studies have reported changes in a myriad of signaling pathways in complex neuronal tissue in situ after a variety of insults and treatment with EPO (10, 21, 80–87). Since the EpoR is expressed in glia and endothelial cells in addition to neurons [for review see Ref. (7)], studies in primary cell cultures are helpful to parse out the role of EPO in these different cell types. These studies, when compared, suggest that EPO may activate different signal transduction cascades depending on cell type. For example, results from a combination of studies suggest that the p38-MAPK pathway is activated by EPO in astrocytes, but not in neurons. Whole tissue studies detected activation of p38-MAPK by EPO and, as mentioned above, EPO preserves astrocyte function at the BBB by activation of this pathway (53, 58). However, blocking activation of the p38-MAPK pathway in vivo had no effect on neuronal survival (87). Taken together, these observations suggest that functional effects of EPO via activation of p38-MAPK occur primarily in non-neuronal cells, including astrocytes.
Disparate signaling pathways may be initiated by EPO as a result of binding to different receptors. In hematopoietic cells, EPO activates the EpoR homodimer to induce downstream signaling and block apoptosis (88–92). In non-hematopoietic tissue, EPO may enact neuroprotection via an EpoR, interleukin beta common receptor (βcR) heterodimer (93). This is supported by evidence that the EpoR can associate with the βcR (94), and that binding of the EpoR to a dimerization-dependent, constitutively active βcR mutant initiates downstream signaling cascades (95). However, expression of the EpoR and the βcR does not appear to significantly overlap in the brain, despite an increase in microglial βcR expression following injury (96). This could imply that while a few cells or specific cell types may respond to EPO via an EpoR, βcR complex, the majority of cells may not. In fact, EPO could potentially act on an additional as of yet unidentified receptor. Neuronal survival was recently achieved by EPO therapy in mice that lack neuronal EpoR expression (97), thus suggesting that the EpoR is not necessary for blocking neuronal apoptosis. Further, forms of EPO that neither bind the EpoR nor initiate erythropoiesis are still neuroprotective (15, 98–101). Understanding whether EPO acts through canonical or non-canonical receptors, or independently of receptor interaction (i.e., direct ROS/RNS scavenging) is essential for developing new target-directed therapies and therefore should continue to be an area of active investigation.
In addition to direct binding of EPO to different receptors, activation of the EpoR can activate other surface receptors and channels. Signal transduction pathways downstream of EPO are significantly modulated by cross-talk with other surface receptors and cytosolic proteins in erythroid progenitors [for review see Ref. (5)]. We have already discussed the potential influence of cross-talk with the Wnt/β-catenin pathway in influencing downstream effects of EPO in microglial cells. Another example is the calcium channel, TRPC2 (102, 103), which facilitates calmodulin-dependent enhancement of EpoR-associated JAK2 signaling in erythroid progenitor cells (104). TRPC2 channels are also expressed in the CNS and expression may vary between cell types, which could also contribute to cell type specific responses to EPO (105, 106). This observation supports cell type specific signal transduction events downstream of even canonical EpoR signaling, dependent on cell type specific expression of these other proteins. Altogether these data suggest that EPO may be able to activate different receptors and that even signaling through the canonical EpoR homodimer may differ depending on cell type. Additional studies are needed to determine if these co-regulators of EPO signaling vary in expression among different CNS cell types and whether these factors play a role in differing responses to EPO.
Conclusion
Erythropoietin is a pleiotropic protein, it influences erythropoiesis, BBB/BRB health, ROS/RNS levels, apoptosis, and glial reactivity seemingly simultaneously. EPO blocks apoptosis in a wide-range of cell types, including neurons, and it appears to be effective in a wide-range of neurodegenerative conditions. By preventing apoptosis, EPO indirectly decreases chronic neuroinflammation. In this review, we show evidence that EPO also modulates neuroinflammation by decreasing levels of ROS/RNS, limiting microglial infiltration by preserving the health of the microvascular endothelial cells and astrocytes at the BBB/BRB, and by acting directly on microglial cells to block proliferation and influence their reactive state. We propose that EPO may activate different signal transduction cascades in a context-dependent, cell type specific manner to enact its diverse functions. A caveat of this analysis is that not all signaling molecules have been assessed in all cell types. For example, Wnt-1 activation by EPO was reported in vascular endothelial cells (107) and in cultured microglia (71), but has not been assessed in astrocyte or neuronal cultures. There has been amazing progress in this field in the last 10 years, but there is still much to be understood about this complex, pleiotropic cytokine. Understanding the mechanism by which EPO modulates neuroinflammation may lead to novel therapeutic strategies for the treatment of neurodegenerative diseases and injuries.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Streit WJ, Mrak RE, Griffin WST. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation (2004) 1:14. doi: 10.1186/1742-2094-1-14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener (2009) 4:47. doi:10.1186/1750-1326-4-47
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Palis J. Primitive and definitive erythropoiesis in mammals. Front Physiol (2014) 5:3. doi:10.3389/fphys.2014.00003
4. Ghezzi P, Brines M. Erythropoietin as an antiapoptotic, tissue-protective cytokine. Cell Death Differ (2004) 11(Suppl 1):S37–44. doi:10.1038/sj.cdd.4401450
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Ingley E. Integrating novel signaling pathways involved in erythropoiesis. IUBMB Life (2012) 64:402–10. doi:10.1002/iub.1024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Broxmeyer HE. Erythropoietin: multiple targets, actions, and modifying influences for biological and clinical consideration. J Exp Med (2013) 210:205–8. doi:10.1084/jem.20122760
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Genc S, Koroglu TF, Genc K. Erythropoietin and the nervous system. Brain Res (2004) 1000:19–31. doi:10.1016/j.brainres.2003.12.037
8. Noguchi CT, Asavaritikrai P, Teng R, Jia Y. Role of erythropoietin in the brain. Crit Rev Oncol Hematol (2007) 64:159–71. doi:10.1016/j.critrevonc.2007.03.001
9. Maiese K, Chong ZZ, Shang YC, Wang S. Erythropoietin: new directions for the nervous system. Int J Mol Sci (2012) 13:11102–29. doi:10.3390/ijms130911102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Weishaupt JH, Rohde G, Pölking E, Siren A-L, Ehrenreich H, Bähr M. Effect of erythropoietin axotomy-induced apoptosis in rat retinal ganglion cells. Invest Ophthalmol Vis Sci (2004) 45:1514–22. doi:10.1167/iovs.03-1039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Tsai JC, Wu L, Worgul B, Forbes M, Cao J. Intravitreal administration of erythropoietin and preservation of retinal ganglion cells in an experimental rat model of glaucoma. Curr Eye Res (2005) 30:1025–31. doi:10.1080/02713680500320729
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Zhong L, Bradley J, Schubert W, Ahmed E, Adamis AP, Shima DT, et al. Erythropoietin promotes survival of retinal ganglion cells in DBA/2J glaucoma mice. Invest Ophthalmol Vis Sci (2007) 48:1212–8. doi:10.1167/iovs.06-0757
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Sullivan TA, Geisert EE, Hines-Beard J, Rex TS. Systemic adeno-associated virus-mediated gene therapy preserves retinal ganglion cells and visual function in DBA/2J glaucomatous mice. Hum Gene Ther (2011) 22:1191–200. doi:10.1089/hum.2011.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Sullivan T, Kodali K, Rex TS. Systemic gene delivery protects the photoreceptors in the retinal degeneration slow mouse. Neurochem Res (2011) 36:613–8. doi:10.1007/s11064-010-0272-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Sullivan TA, Geisert EE, Templeton JP, Rex TS. Dose-dependent treatment of optic nerve crush by exogenous systemic mutant erythropoietin. Exp Eye Res (2012) 96:36–41. doi:10.1016/j.exer.2012.01.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Grimm C, Wenzel A, Groszer M, Mayser H, Seeliger M, Samardzija M, et al. HIF-1-induced erythropoietin in the hypoxic retina protects against light-induced retinal degeneration. Nat Med (2002) 8:718–24. doi:10.1038/nm723
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Grimm C, Wenzel A, Stanescu D, Samardzija M, Hotop S, Groszer M, et al. Constitutive overexpression of human erythropoietin protects the mouse retina against induced but not inherited retinal degeneration. J Neurosci (2004) 24:5651–8. doi:10.1523/JNEUROSCI.1288-04.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Rex TS, Allocca M, Domenici L, Surace EM, Maguire AM, Lyubarsky A, et al. Systemic but not intraocular Epo gene transfer protects the retina from light-and genetic-induced degeneration. Mol Ther (2004) 10:855–61. doi:10.1016/j.ymthe.2004.07.027
19. Rex TS, Wong Y, Kodali K, Merry S. Neuroprotection of photoreceptors by direct delivery of erythropoietin to the retina of the retinal degeneration slow mouse. Exp Eye Res (2009) 89:735–40. doi:10.1016/j.exer.2009.06.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Shen J, Wu Y, Xu J-Y, Zhang J, Sinclair SH, Yanoff M, et al. ERK- and Akt-dependent neuroprotection by erythropoietin (EPO) against glyoxal-AGEs via modulation of Bcl-xL, Bax, and BAD. Invest Ophthalmol Vis Sci (2010) 51:35–46. doi:10.1167/iovs.09-3544
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Zhang J, Wu Y, Jin Y, Ji F, Sinclair SH, Luo Y, et al. Intravitreal injection of erythropoietin protects both retinal vascular and neuronal cells in early diabetes. Invest Ophthalmol Vis Sci (2008) 49:732–42. doi:10.1167/iovs.07-0721
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Villa P, Bigini P, Mennini T, Agnello D, Laragione T, Cagnotto A, et al. Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J Exp Med (2003) 198:971–5. doi:10.1084/jem.20021067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Yatsiv I, Grigoriadis N, Simeonidou C, Stahel PF, Schmidt OI, Alexandrovitch AG, et al. Erythropoietin is neuroprotective, improves functional recovery, and reduces neuronal apoptosis and inflammation in a rodent model of experimental closed head injury. FASEB J (2005) 19:1701–3. doi:10.1096/fj.05-3907fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Duchen MR. Roles of mitochondria in health and disease. Diabetes (2004) 53(Suppl 1):S96–102. doi:10.2337/diabetes.53.2007.S96
25. Block ML, Zecca L, Hong J-S. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci (2007) 8:57–69. doi:10.1038/nrn2038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci (2012) 322:254–62. doi:10.1016/j.jns.2012.05.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Chrysostomou V, Rezania F, Trounce IA, Crowston JG. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr Opin Pharmacol (2013) 13:12–5. doi:10.1016/j.coph.2012.09.008
28. Al-Qahtani JM, Abdel-Wahab BA, El-Aziz SMA. Long-term moderate dose exogenous erythropoietin treatment protects from intermittent hypoxia-induced spatial learning deficits and hippocampal oxidative stress in young rats. Neurochem Res (2014) 39:161–71. doi:10.1007/s11064-013-1201-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Yazihan N, Uzuner K, Salman B, Vural M, Koken T, Arslantas A. Erythropoietin improves oxidative stress following spinal cord trauma in rats. Injury (2008) 39:1408–13. doi:10.1016/j.injury.2008.03.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Barichello T, Simões LR, Generoso JS, Sangiogo G, Danielski LG, Florentino D, et al. Erythropoietin prevents cognitive impairment and oxidative parameters in Wistar rats subjected to pneumococcal meningitis. Transl Res (2014) 163:503–13. doi:10.1016/j.trsl.2013.12.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Lu M-J, Chen Y-S, Huang H-S, Ma M-C. Erythropoietin alleviates post-ischemic injury of rat hearts by attenuating nitrosative stress. Life Sci (2012) 90:776–84. doi:10.1016/j.lfs.2012.04.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Li G, Ma R, Huang C, Tang Q, Fu Q, Liu H, et al. Protective effect of erythropoietin on β-amyloid-induced PC12 cell death through antioxidant mechanisms. Neurosci Lett (2008) 442:143–7. doi:10.1016/j.neulet.2008.07.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Zhang J, Zhu Y, Zhou D, Wang Z, Chen G. Recombinant human erythropoietin (rhEPO) alleviates early brain injury following subarachnoid hemorrhage in rats: possible involvement of Nrf2-ARE pathway. Cytokine (2010) 52:252–7. doi:10.1016/j.cyto.2010.08.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Jin W, Wu J, Wang H, Kong J, Ni H, Liang W. Erythropoietin administration modulates pulmonary Nrf2 signaling pathway after traumatic brain injury in mice. J Trauma (2011) 71:680–6. doi:10.1097/TA.0b013e3181f6b984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Jin W, Ming X, Hou X, Zhu T, Yuan B, Wang J, et al. Protective effects of erythropoietin in traumatic spinal cord injury by inducing the Nrf2 signaling pathway activation. J Trauma Acute Care Surg (2014) 76:1228–34. doi:10.1097/TA.0000000000000211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Genc K, Egrilmez MY, Genc S. Erythropoietin induces nuclear translocation of Nrf2 and heme oxygenase-1 expression in SH-SY5Y cells. Cell Biochem Funct (2010) 28:197–201. doi:10.1002/cbf.1639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Meloni BP, Tilbrook PA, Boulos S, Arthur PG, Knuckey NW. Erythropoietin preconditioning in neuronal cultures: signaling, protection from in vitro ischemia, and proteomic analysis. J Neurosci Res (2006) 83:584–93. doi:10.1002/jnr.20755
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Kumral A, Gonenc S, Acikgoz O, Sonmez A, Genc K, Yilmaz O, et al. Erythropoietin increases glutathione peroxidase enzyme activity and decreases lipid peroxidation levels in hypoxic-ischemic brain injury in neonatal rats. Biol Neonate (2005) 87:15–8. doi:10.1159/000080490
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Kumral A, Tugyan K, Gonenc S, Genc K, Genc S, Sonmez U, et al. Protective effects of erythropoietin against ethanol-induced apoptotic neurodegeneration and oxidative stress in the developing C57BL/6 mouse brain. Brain Res Dev Brain Res (2005) 160:146–56. doi:10.1016/j.devbrainres.2005.08.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Comim CM, Cassol OJ Jr., Abreu I, Moraz T, Constantino LS, Vuolo F, et al. Erythropoietin reverts cognitive impairment and alters the oxidative parameters and energetic metabolism in sepsis animal model. J Neural Transm (2012) 119:1267–74. doi:10.1007/s00702-012-0774-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Wenker SD, Chamorro ME, Vittori DC, Nesse AB. Protective action of erythropoietin on neuronal damage induced by activated microglia. FEBS J (2013) 280:1630–42. doi:10.1111/febs.12172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Gui D-M, Yang Y, Li X, Gao D-W. Effect of erythropoietin on the expression of HIF-1 and iNOS in retina in chronic ocular hypertension rats. Int J Ophthalmol (2011) 4:40–3. doi:10.3980/j.issn.2222-3959.2011.01.09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Bailey DM, Lundby C, Berg RM, Taudorf S, Rahmouni H, Gutowski M, et al. On the antioxidant properties of erythropoietin and its association with the oxidative-nitrosative stress response to hypoxia in humans. Acta Physiol (2014) 212:175–87. doi:10.1111/apha.12313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Liu J, Narasimhan P, Song YS, Nishi T, Yu F, Lee Y-S, et al. Epo protects SOD2-deficient mouse astrocytes from damage by oxidative stress. Glia (2006) 53:360–5. doi:10.1002/glia.20289
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Weinreb O, Mandel S, Youdim MBH, Amit T. Targeting dysregulation of brain iron homeostasis in Parkinson’s disease by iron chelators. Free Radic Biol Med (2013) 62:52–64. doi:10.1016/j.freeradbiomed.2013.01.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Goodnough LT. Erythropoietin and iron-restricted erythropoiesis. Exp Hematol (2007) 35:167–72. doi:10.1016/j.exphem.2007.01.026
47. Rezai-Zadeh K, Gate D, Town T. CNS infiltration of peripheral immune cells: D-Day for neurodegenerative disease? J Neuroimmune Pharmacol (2009) 4:462–75. doi:10.1007/s11481-009-9166-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Mengozzi M, Cervellini I, Villa P, Erbayraktar Z, Gökmen N, Yilmaz O, et al. Erythropoietin-induced changes in brain gene expression reveal induction of synaptic plasticity genes in experimental stroke. Proc Natl Acad Sci U S A (2012) 109:9617–22. doi:10.1073/pnas.1200554109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Rizzo MT, Leaver HA. Brain endothelial cell death: modes, signaling pathways, and relevance to neural development, homeostasis, and disease. Mol Neurobiol (2010) 42:52–63. doi:10.1007/s12035-010-8132-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Luissint A-C, Artus C, Glacial F, Ganeshamoorthy K, Couraud P-O. Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation. Fluids Barriers CNS (2012) 9:23. doi:10.1186/2045-8118-9-23
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia (2013) 61:1939–58. doi:10.1002/glia.22575
52. Weiss N, Miller F, Cazaubon S, Couraud P-O. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta (2009) 1788:842–57. doi:10.1016/j.bbamem.2008.10.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Chu H, Ding H, Tang Y, Dong Q. Erythropoietin protects against hemorrhagic blood-brain barrier disruption through the effects of aquaporin-4. Lab Invest (2014) 94:1042–53. doi:10.1038/labinvest.2014.84
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Li Y, Ogle ME, Wallace GC, Lu ZY, Yu SP, Wei L. Erythropoietin attenuates intracerebral hemorrhage by diminishing matrix metalloproteinases and maintaining blood-brain barrier integrity in mice. Acta Neurochir Suppl (2008) 105:105–12. doi:10.1007/978-3-211-09469-3_22
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Zhou J, Kong H, Hua X, Xiao M, Ding J, Hu G. Altered blood-brain barrier integrity in adult aquaporin-4 knockout mice. Neuroreport (2008) 19:1–5. doi:10.1097/WNR.0b013e3282f2b4eb
56. Eilert-Olsen M, Haj-Yasein NN, Vindedal GF, Enger R, Gundersen GA, Hoddevik EH, et al. Deletion of aquaporin-4 changes the perivascular glial protein scaffold without disrupting the brain endothelial barrier. Glia (2012) 60:432–40. doi:10.1002/glia.22277
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Mitsuma T, Tani K, Hiroaki Y, Kamegawa A, Suzuki H, Hibino H, et al. Influence of the cytoplasmic domains of aquaporin-4 on water conduction and array formation. J Mol Biol (2010) 402:669–81. doi:10.1016/j.jmb.2010.07.060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Tang Z, Sun X, Huo G, Xie Y, Shi Q, Chen S, et al. Protective effects of erythropoietin on astrocytic swelling after oxygen-glucose deprivation and reoxygenation: mediation through AQP4 expression and MAPK pathway. Neuropharmacology (2013) 67:8–15. doi:10.1016/j.neuropharm.2012.10.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Gunnarson E, Song Y, Kowalewski JM, Brismar H, Brines M, Cerami A, et al. Erythropoietin modulation of astrocyte water permeability as a component of neuroprotection. Proc Natl Acad Sci U S A (2009) 106:1602–7. doi:10.1073/pnas.0812708106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Vitellaro-Zuccarello L, Mazzetti S, Madaschi L, Bosisio P, Fontana E, Gorio A, et al. Chronic erythropoietin-mediated effects on the expression of astrocyte markers in a rat model of contusive spinal cord injury. Neuroscience (2008) 151:452–66. doi:10.1016/j.neuroscience.2007.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Gonzalez FF, Larpthaveesarp A, McQuillen P, Derugin N, Wendland M, Spadafora R, et al. Erythropoietin increases neurogenesis and oligodendrogliosis of subventricular zone precursor cells after neonatal stroke. Stroke (2013) 44:753–8. doi:10.1161/STROKEAHA.111.000104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Diaz Z, Assaraf MI, Miller WH, Schipper HM. Astroglial cytoprotection by erythropoietin pre-conditioning: implications for ischemic and degenerative CNS disorders. J Neurochem (2005) 93:392–402. doi:10.1111/j.1471-4159.2005.03038.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol (2013) 39:3–18. doi:10.1111/nan.12011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia (2002) 40:133–9. doi:10.1002/glia.10154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. De SR, Ajmone-Cat MA, Nicolini A, Minghetti L. Expression of phosphatidylserine receptor and down-regulation of pro-inflammatory molecule production by its natural ligand in rat microglial cultures. J Neuropathol Exp Neurol (2002) 61:237–44.
66. Chong ZZ, Kang J-Q, Maiese K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br J Pharmacol (2003) 138:1107–18. doi:10.1038/sj.bjp.0705161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Hellewell SC, Yan EB, Alwis DS, Bye N, Morganti-Kossmann MC. Erythropoietin improves motor and cognitive deficit, axonal pathology, and neuroinflammation in a combined model of diffuse traumatic brain injury and hypoxia, in association with upregulation of the erythropoietin receptor. J Neuroinflammation (2013) 10:156. doi:10.1186/1742-2094-10-156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Hagemeyer N, Boretius S, Ott C, Von Streitberg A, Welpinghus H, Sperling S, et al. Erythropoietin attenuates neurological and histological consequences of toxic demyelination in mice. Mol Med (2012) 18:628–35. doi:10.2119/molmed.2011.00457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Wang Y, Zhang ZG, Rhodes K, Renzi M, Zhang RL, Kapke A, et al. Post-ischemic treatment with erythropoietin or carbamylated erythropoietin reduces infarction and improves neurological outcome in a rat model of focal cerebral ischemia. Br J Pharmacol (2007) 151:1377–84. doi:10.1038/sj.bjp.0707285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Wilms H, Schwabedissen B, Sievers J, Lucius R. Erythropoietin does not attenuate cytokine production and inflammation in microglia – Implications for the neuroprotective effect of erythropoietin in neurological diseases. J Neuroimmunol (2009) 212:106–11. doi:10.1016/j.jneuroim.2009.04.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Shang YC, Chong ZZ, Wang S, Maiese K. Erythropoietin and Wnt1 govern pathways of mTOR, Apaf-1, and XIAP in inflammatory microglia. Curr Neurovasc Res (2011) 8:270–85. doi:10.2174/156720211798120990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Shang YC, Chong ZZ, Wang S, Maiese K. Prevention of β-amyloid degeneration of microglia by erythropoietin depends on Wnt1, the PI 3-K/mTOR pathway, Bad, and Bcl-xL. Aging (2012) 4:187–201.
73. Li F, Chong ZZ, Maiese K. Microglial integrity is maintained by erythropoietin through integration of Akt and its substrates of glycogen synthase kinase-3beta, beta-catenin, and nuclear factor-kappaB. Curr Neurovasc Res (2006) 3:187–201. doi:10.2174/156720206778018758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Lifshitz L, Tabak G, Gassmann M, Mittelman M, Neumann D. Macrophages as novel target cells for erythropoietin. Haematologica (2010) 95:1823–31. doi:10.3324/haematol.2010.025015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Deng J, Miller SA, Wang H-Y, Xia W, Wen Y, Zhou BP, et al. β-catenin interacts with and inhibits NF-κB in human colon and breast cancer. Cancer Cell (2002) 2:323–34. doi:10.1016/S1535-6108(02)00154-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Xu F, Kang Y, Zhang H, Piao Z, Yin H, Diao R, et al. Akt1-mediated regulation of macrophage polarization in a murine model of Staphylococcus aureus pulmonary infection. J Infect Dis (2013) 208:528–38. doi:10.1093/infdis/jit177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Liu Y, Luo B, Han F, Li X, Xiong J, Jiang M, et al. Erythropoietin-derived nonerythropoietic peptide ameliorates experimental autoimmune neuritis by inflammation suppression and tissue protection. PLoS One (2014) 9:e90942. doi:10.1371/journal.pone.0090942
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Nairz M, Schroll A, Moschen AR, Sonnweber T, Theurl M, Theurl I, et al. Erythropoietin contrastingly affects bacterial infection and experimental colitis by inhibiting nuclear factor-κB-inducible immune pathways. Immunity (2011) 34:61–74. doi:10.1016/j.immuni.2011.01.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Monick MM, Carter AB, Robeff PK, Flaherty DM, Peterson MW, Hunninghake GW. Lipopolysaccharide activates Akt in human alveolar macrophages resulting in nuclear accumulation and transcriptional activity of β-catenin. J Immunol (2001) 166:4713–20. doi:10.4049/jimmunol.166.7.4713
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Zhao J, Li G, Zhang Y, Su X, Hang C. The potential role of JAK2/STAT3 pathway on the anti-apoptotic effect of recombinant human erythropoietin (rhEPO) after experimental traumatic brain injury of rats. Cytokine (2011) 56:343–50. doi:10.1016/j.cyto.2011.07.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Wang G-B, Ni Y-L, Zhou X-P, Zhang W-F. The AKT/mTOR pathway mediates neuronal protective effects of erythropoietin in sepsis. Mol Cell Biochem (2014) 385:125–32. doi:10.1007/s11010-013-1821-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Zhou T-F, Yu J-G. Recombinant human erythropoietin attenuates neuronal apoptosis and cognitive defects via JAK2/STAT3 signaling in experimental endotoxemia. J Surg Res (2013) 183:304–12. doi:10.1016/j.jss.2012.11.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Kilic U, Kilic E, Soliz J, Bassetti CI, Gassmann M, Hermann DM. Erythropoietin protects from axotomy-induced degeneration of retinal ganglion cells by activating ERK-1/-2. FASEB J (2005) 19:249–51. doi:10.1096/fj.04-2493fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Kretz A, Happold CJ, Marticke JK, Isenmann S. Erythropoietin promotes regeneration of adult CNS neurons via Jak2/Stat3 and PI3K/AKT pathway activation. Mol Cell Neurosci (2005) 29:569–79. doi:10.1016/j.mcn.2005.04.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Xie Z, Chen F, Wu X, Zhuang C, Zhu J, Wang J, et al. Effects of supplemental erythropoietin on its receptor expression and signal transduction pathways in rat model of retinal detachment. Curr Eye Res (2012) 37:138–44. doi:10.3109/02713683.2011.647225
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Jia Y, Mo S-J, Feng Q-Q, Zhan M-L, Ouyang L-S, Chen J-C, et al. EPO-dependent activation of PI3K/Akt/FOXO3A signalling mediates neuroprotection in in vitro and in vivo models of Parkinson’s disease. J Mol Neurosci (2014) 53:117–24. doi:10.1007/s12031-013-0208-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Sättler MB, Merkler D, Maier K, Stadelmann C, Ehrenreich H, Bähr M, et al. Neuroprotective effects and intracellular signaling pathways of erythropoietin in a rat model of multiple sclerosis. Cell Death Differ (2004) 11(Suppl 2):S181–92. doi:10.1038/sj.cdd.4401504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Sawyer ST, Penta K. Association of JAK2 and STAT5 with erythropoietin receptors. Role of receptor phosphorylation in erythropoietin signal transduction. J Biol Chem (1996) 271:32430–7. doi:10.1074/jbc.271.50.32430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Chin H, Arai A, Wakao H, Kamiyama R, Miyasaka N, Miura O. Lyn physically associates with the erythropoietin receptor and may play a role in activation of the Stat5 pathway. Blood (1998) 91:3734–45.
90. Okutani Y, Kitanaka A, Tanaka T, Kamano H, Ohnishi H, Kubota Y, et al. Src directly tyrosine-phosphorylates STAT5 on its activation site and is involved in erythropoietin-induced signaling pathway. Oncogene (2001) 20:6643–50. doi:10.1038/sj.onc.1204807
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Myklebust JH, Blomhoff HK, Rusten LS, Stokke T, Smeland EB. Activation of phosphatidylinositol 3-kinase is important for erythropoietin-induced erythropoiesis from CD34(+) hematopoietic progenitor cells. Exp Hematol (2002) 30:990–1000. doi:10.1016/S0301-472X(02)00868-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Chen C, Sytkowski AJ. Erythropoietin regulation of Raf-1 and MEK: evidence for a Ras-independent mechanism. Blood (2004) 104:73–80. doi:10.1182/blood-2003-04-1340
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Brines M, Grasso G, Fiordaliso F, Sfacteria A, Ghezzi P, Fratelli M, et al. Erythropoietin mediates tissue protection through an erythropoietin and common β-subunit heteroreceptor. Proc Natl Acad Sci U S A (2004) 101:14907–12. doi:10.1073/pnas.0406491101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Jubinsky PT, Krijanovski OI, Nathan DG, Tavernier J, Sieff CA. The beta chain of the interleukin-3 receptor functionally associates with the erythropoietin receptor. Blood (1997) 90:1867–73.
95. Blake TJ, Jenkins BJ, D’Andrea RJ, Gonda TJ. Functional cross-talk between cytokine receptors revealed by activating mutations in the extracellular domain of the beta-subunit of the GM-CSF receptor. J Leukoc Biol (2002) 72:1246–55.
96. Nadam J, Navarro F, Sanchez P, Moulin C, Georges B, Laglaine A, et al. Neuroprotective effects of erythropoietin in the rat hippocampus after pilocarpine-induced status epilepticus. Neurobiol Dis (2007) 25:412–26. doi:10.1016/j.nbd.2006.10.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Xiong Y, Mahmood A, Qu C, Kazmi H, Zhang ZG, Noguchi CT, et al. Erythropoietin improves histological and functional outcomes after traumatic brain injury in mice in the absence of the neural erythropoietin receptor. J Neurotrauma (2010) 27:205–15. doi:10.1089/neu.2009.1001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Leist M, Ghezzi P, Grasso G, Bianchi R, Villa P, Fratelli M, et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science (2004) 305:239–42. doi:10.1126/science.1098313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Hines-Beard J, Desai S, Haag R, Esumi N, D’Surney L, Parker S, et al. Identification of a therapeutic dose of continuously delivered erythropoietin in the eye using an inducible promoter system. Curr Gene Ther (2013) 13:275–81. doi:10.2174/15665232113139990024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Mennini T, De Paola M, Bigini P, Mastrotto C, Fumagalli E, Barbera S, et al. Nonhematopoietic erythropoietin derivatives prevent motoneuron degeneration in vitro and in vivo. Mol Med (2006) 12:153–60. doi:10.2119/2006-00045.Mennini
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Colella P, Iodice C, Di Vicino U, Annunziata I, Surace EM, Auricchio A. Non-erythropoietic erythropoietin derivatives protect from light-induced and genetic photoreceptor degeneration. Hum Mol Genet (2011) 20:2251–62. doi:10.1093/hmg/ddr115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Chu X, Cheung JY, Barber DL, Birnbaumer L, Rothblum LI, Conrad K, et al. Erythropoietin modulates calcium influx through TRPC2. J Biol Chem (2002) 277:34375–82. doi:10.1074/jbc.M205541200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Tong Q, Hirschler-Laszkiewicz I, Zhang W, Conrad K, Neagley DW, Barber DL, et al. TRPC3 is the erythropoietin-regulated calcium channel in human erythroid cells. J Biol Chem (2008) 283:10385–95. doi:10.1074/jbc.M710231200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Kakihana K, Yamamoto M, Iiyama M, Miura O. Calmodulin physically interacts with the erythropoietin receptor and enhances Jak2-mediated signaling. Biochem Biophys Res Commun (2005) 335:424–31. doi:10.1016/j.bbrc.2005.07.095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Fusco FR, Martorana A, Giampà C, De March Z, Vacca F, Tozzi A, et al. Cellular localization of TRPC3 channel in rat brain: preferential distribution to oligodendrocytes. Neurosci Lett (2004) 365:137–42. doi:10.1016/j.neulet.2004.04.070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Chung YH, Sun Ahn H, Kim D, Hoon Shin D, Su Kim S, Yong Kim K, et al. Immunohistochemical study on the distribution of TRPC channels in the rat hippocampus. Brain Res (2006) 1085:132–7. doi:10.1016/j.brainres.2006.02.087
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Chong ZZ, Hou J, Shang YC, Wang S, Maiese K. EPO relies upon novel signaling of Wnt1 that requires Akt1, FoxO3a, GSK-3β, and β-catenin to foster vascular integrity during experimental diabetes. Curr Neurovasc Res (2011) 8:103–20. doi:10.2174/156720211795495402
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: erythropoietin, neuroinflammation, microglia, astrocytes, signaling pathways
Citation: Bond WS and Rex TS (2014) Evidence that erythropoietin modulates neuroinflammation through differential action on neurons, astrocytes, and microglia. Front. Immunol. 5:523. doi: 10.3389/fimmu.2014.00523
Received: 05 August 2014; Accepted: 06 October 2014;
Published online: 22 October 2014.
Edited by:
Pietro Ghezzi, Brighton and Sussex Medical School, UKReviewed by:
Ilaria Cervellini, Brighton and Sussex Medical School, UKPaolo Bigini, Mario Negri Institute, Italy
Copyright: © 2014 Bond and Rex. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tonia S. Rex, Department of Ophthalmology and Visual Sciences, Vanderbilt University, 11425 Langford MRB-IV, 2213 Garland Avenue, Nashville, TN 37232 USA e-mail:dG9uaWEucmV4QHZhbmRlcmJpbHQuZWR1