 Kim L. Good-Jacobson
Kim L. Good-Jacobson- 1Immunology Division, Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, Australia
- 2Department of Medical Biology, University of Melbourne, Parkville, VIC, Australia
Understanding the regulation of antibody production and B-cell memory formation and function is core to finding new treatments for B-cell-derived cancers, antibody-mediated autoimmune disorders, and immunodeficiencies. Progression from a small number of antigen-specific B-cells to the production of a large number of antibody-secreting cells is tightly regulated. Although much progress has been made in revealing the transcriptional regulation of B-cell differentiation that occurs during humoral immune responses, there are still many questions that remain unanswered. Recent work on the expression and roles of histone modifiers in lymphocytes has begun to shed light on this additional level of regulation. This review will discuss the recent advancements in understanding how humoral immune responses, in particular germinal centers and memory cells, are modulated by histone modifiers.
Introduction
Pathogen clearance and formation of immunity requires the activation of B-cells and subsequent differentiation into antibody-secreting cells and memory cells. Humoral memory consists of both memory B-cells and long-lived plasma cells, the latter of which resides mainly in the bone marrow. Together, humoral memory cells are able to clear subsequent infections by the same pathogen more efficiently than responding B-cells during the initial response (1). The mechanisms underlying how memory is formed, and what controls its reactivation, are still unclear. In recent times, transcriptional regulation during B-cell differentiation (2–5) has been the focus of efforts to understand the intrinsic controls that regulate immune cell fates. In contrast, epigenetic regulation during a humoral immune response is relatively unknown. This review will discuss the limited information that is currently known about epigenetic regulators and their importance in the generation and maintenance of immune memory, focusing on the role of histone modifiers within the germinal center (GC).
Humoral Immune Responses and Germinal Centers
Humoral responses can be broadly categorized into either T-independent or T-dependent responses, with the production of high-affinity antibody and class-switched memory the main outcome of the latter (Figure 1). To this end, antigen-activated B-cells that receive T cell help and do not participate in the extrafollicular foci of low-affinity plasmablasts, or become early GC-independent memory (1, 6), can instead form GCs. GCs are divided into a light and dark zone. Within the dark zone, cells undergo multiple rounds of proliferation and adapt their antigen receptor to the immunizing antigen through the process of somatic hypermutation and class-switch recombination (7–11). B-cells then transition to the light zone, in which cells that have a high-affinity antigen receptor will be selected to continue to divide or to differentiate (12–14). In contrast, low-affinity cells and cells that have mutated their receptor to no longer be antigen-specific will die. High-affinity cells that are selected to survive may differentiate into plasma cells or memory cells.
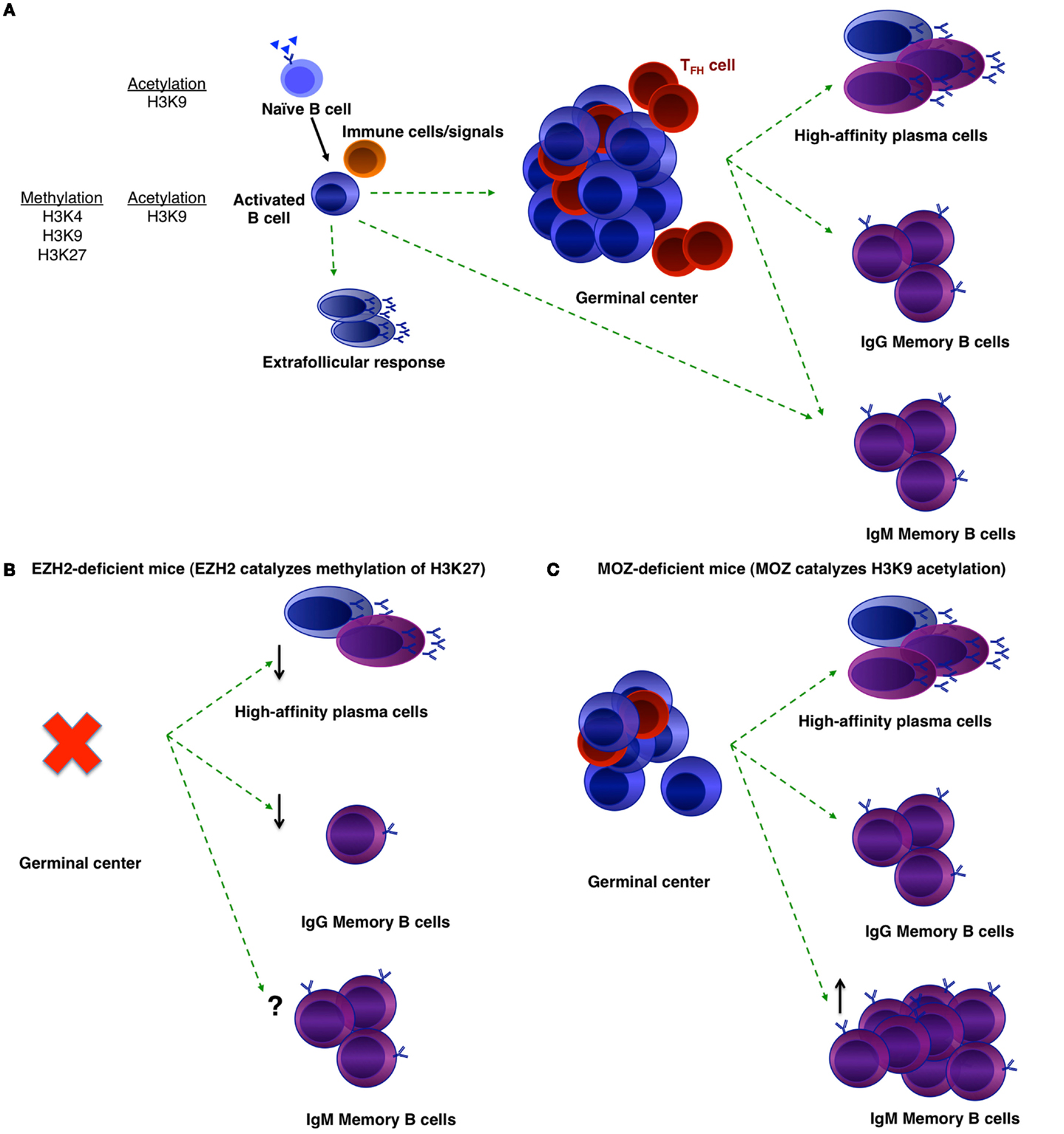
Figure 1. Histone modifications that may regulate B-cell differentiation. (A) B-cell differentiation during a T-dependent humoral response. Naïve B-cells that are specific to a foreign pathogen will become activated and receive help from accessory cells. They may become early memory B-cells or extrafollicular plasmablasts, or form a GC. GC B-cells then may either differentiate into long-lived plasma cells or memory B-cells. Within the memory population, there are IgM and IgG memory B-cells. Histone marks in naïve and activated B-cells are noted (15). (B,C) Phenotypes of EZH2-deficient (B) and MOZ-deficient (C) mice after immunization.
Long-lived plasma cells are generally high-affinity, sessile cells that reside in the bone marrow, relying on extrinsic factors from niche cells for their survival (16). These plasma cells continually secrete high-affinity antibody, resulting in lowering of the amount of an invading pathogen upon re-encounter. Together with memory B-cells, they contribute to maintaining immunity.
Regulation of Humoral Immune Responses
The activation, proliferation, and differentiation of antigen-activated B-cells during an immune response is orchestrated and regulated at both the cellular and molecular levels. During an immune response, B-cell behavior is regulated by both extrinsic and intrinsic mechanisms. B-cells respond to signals in the microenvironment, including cytokines, cell surface ligand/receptor pairings, and other soluble factors such as chemokines and cell survival molecules (17). For these signals to orchestrate cell behavior in a coordinated manner, cells integrate these signals, resulting in initiation or silencing of genes, which in turn directs cellular behavior.
Transcription factors are molecules that coordinate the expression of a number of genes, thus one transcription factor is often linked to the identity of a cell subset. Different B-cell subsets are associated with particular transcription factors. The transcriptional repressor B cell lymphoma 6 (BCL-6) is expressed in GC B-cells, regulating a program of genes required for GC function and as such is essential for the formation of GCs (18, 19). In contrast, the transcriptional repressor B lymphocyte-induced maturation protein 1 (Blimp-1) is expressed in plasma cells (20). BCL-6 and Blimp-1 were previously denoted as “master regulators” of B-cell differentiation, by reciprocally repressing each other (21). However, there are various lines of evidence demonstrating that, similar to the Th1/Th2 paradigm for T cells, the idea of master regulators was a useful concept but too simple to completely explain the genetic programs underlying B-cell differentiation (22). For example, in the case of memory B-cells, no master regulator transcription factor has been found, and both Bcl-6 and Prdm1, the gene encoding Blimp-1, are downregulated (2). Furthermore, plasma cell differentiation can be induced in the absence of Blimp-1 (23).
Memory B-cells are the centerpiece of the secondary response, in which foreign pathogens are cleared more quickly than a primary response (1). As such, resting memory B-cells have decreased expression of cell cycle inhibitors, correlated with their ability to enter division earlier than naïve B-cells (4). Transcriptionally, naïve and memory B-cells are actually quite similar (3, 4), despite the enhanced survival and proliferative capabilities. Therefore, it is likely there is an additional level of regulation that endows memory B-cells with the ability to respond more efficiently to pathogen infection than naïve B-cells.
Epigenetic Regulation
Genetic regulation also occurs via modification of histones. This is termed epigenetic regulation, i.e., stable inherited modifications of genetic material without altering the DNA sequence. The N-terminal tail of histones can be modified either to promote or inhibit transcription, via creating either an open chromatin structure (euchromatin) or a tightly packed structure (heterochromatin) (24). This is performed by histone modifiers, a group of enzymes such as methyltransferases, acetyltransferases, and histone deacetyltransferases (HDACs). Through these modifiers, histone structure and thus the ability of transcription to proceed is regulated (25, 26). DNA methylation is another form of epigenetic regulation, and recently it was demonstrated that inhibiting DNA methyltransferase 1 (DNMT1) can abrogate GC responses (27) (Table 1). However, due to space limitations, DNA methylation will not be discussed further here.
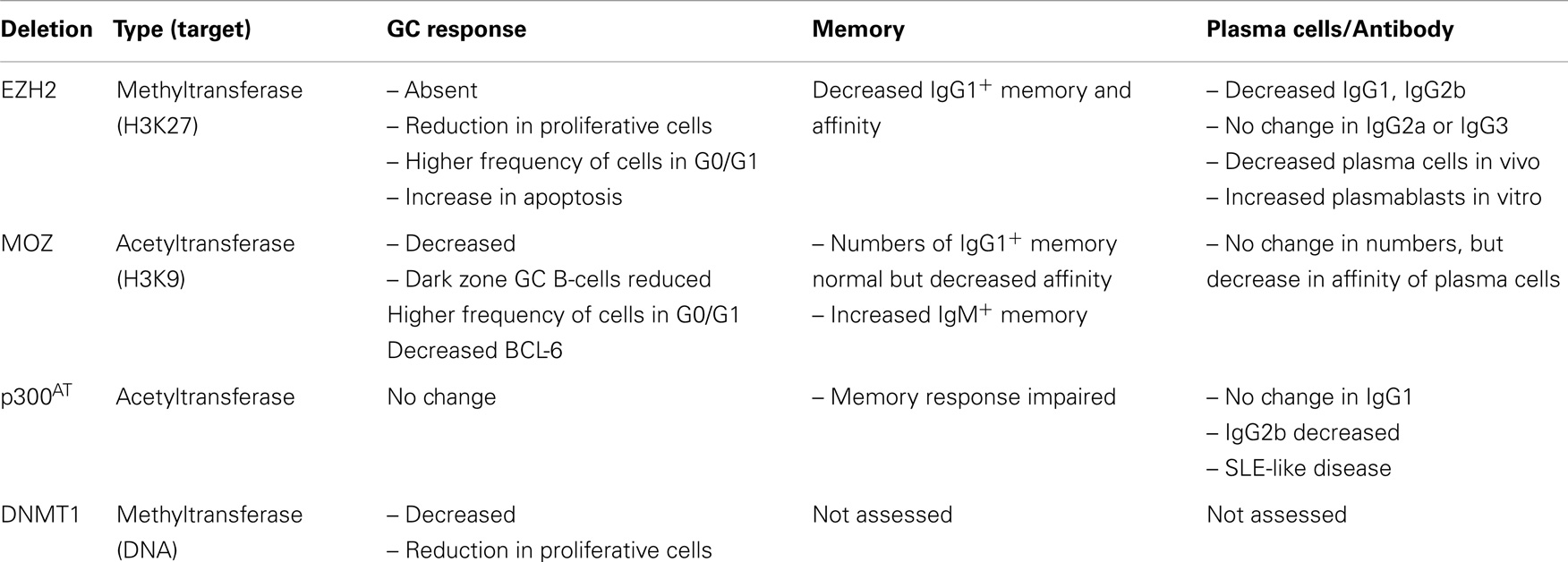
Table 1. Humoral responses in the absence of EZH2, MOZ, p300 (acetyltransferase activity), or DNMT1 [from Ref. (27–31)].
In recent years, epigenetic regulation of B-cell development – especially VDJ recombination (32) – has been revealed. However, much less is known about whether epigenetic modifiers can regulate B-cell differentiation during a humoral response. This mini-review will focus specifically on the current understanding of differential histone modifications during the formation of GC-dependent memory.
Histone Modification Patterns in Different B-Cell Subsets
Germinal center B-cells and plasma cells have their own unique transcriptional program compared to naïve and memory B-cells (2, 18–20). A large number of gene expression changes occur during differentiation of a naïve B-cell to GC to memory or plasma cell. In addition, an antigen-activated B-cell has the ability to choose any one of those three fates during a response. Therefore, it is likely that regulation of heterochromatin or euchromatin states plays a large role in this adaptability. It could be hypothesized then that the pattern of histone marks is unique to different mature B-cell subsets. Indeed, assessment of H3K4me1, H3K4me3, H3Ac, H3K36me3, H3K27me3, and PolII demonstrated that human naïve and GC B-cells had different patterns of open chromatin (33). Thus, it appears that there is a change in the epigenetic landscape either upon B-cell activation or during the first couple of days during an immune response.
Changes to Histone Modifications upon Activation of B-Cells
The immediate epigenetic events that may occur upon activation of an antigen-specific B-cell are unknown. However, preliminary data have shown differences in histone marks between quiescent and activated B-cells (Figure 1). Methylation of various histone lysines was found to be decreased in resting cells compared to in vitro activated cells (15). For example, H3K4, H3K9, and H3K27 methylation increased after in vitro activation, whereas, H3K9 acetylation is present in both quiescent and activated cells (15). In contrast to H3K9 and H3K27 methylation, H3K4 methylation is a permissive mark. Although the authors suggest that histone lysine hypomethylation was a mechanism that endowed B-cells with reprograming potential (15), this has yet to be shown functionally.
Although it is clear that different B-cell subsets have different patterns of histone modifications, there is limited evidence on the role particular histone modifiers play during the early phase of humoral responses. For example, B-cells from a mouse engineered to have reduced acetyltransferase activity in p300 were still able to respond to T cell-derived stimuli such as anti-CD40, IL-4, and the T-independent stimuli LPS or CpG agonist (28). In contrast, there was a 50% reduction in the ability of these cells to respond to BCR stimulation (28). Because B-cell development is altered in these mice, it is not clear whether this defect is the result of a defect that occurred during B-cell development, as opposed to a direct role upon BcR engagement in the periphery.
An area of great interest currently is whether “bivalency,” i.e., the presence of both activating and repressive marks at the same loci, is important for lymphocyte plasticity in identity and function (22). Preliminary studies suggest that bivalency is an important regulator of gene expression during differentiation of naïve to GC B-cells. Enhancer of zeste homolog 2 (EZH2) is a histone methyltransferase and a polycomb group member that catalyzes methylation of H3K27 (34). A number of EZH2 target genes in centroblasts that were marked by H3K27me3 were also H3K27me3 marked in naïve B-cells, although likely not by EZH2 as its expression is very low in naïve B-cells (35). A study of bivalent genes in naïve and GC B-cells (with respect to the activating mark H3K4me3 and silencing mark H3K27me3) found that differentiation into GC B-cells resulted in ~1000 new bivalent domains (29). However, the vast majority of these promoters that had dual marks came from the acquisition of H3K27me3 (likely due to upregulation of EZH2) – i.e., already marked H3K4me3 promoters in naïve B-cells (29). As the transcriptional program in GCs is known to involve the large-scale repression of many genes, bivalency may allow GC B-cells to establish the transcriptional program required for the multiple rounds of proliferation and somatic hypermutation that occurs, while retaining the ability to differentiate into centrocytes and eventually plasma cells and memory B-cells. However, the likely complex roles of bivalent domains during B-cell differentiation are yet to be unraveled.
Regulation of GCs by EZH2 and MOZ
Polycomb group proteins are differentially expressed in the GC in human tonsils. BMI-1 and RING1 downregulation, and ENX and EED upregulation, occur upon differentiation into centroblasts (36). This was then reversed in centrocytes. EZH2 was also found to be upregulated in centroblasts (30, 35, 37). It has also been shown that while Ezh2 is expressed in plasmablasts, BMI-1 is expressed in plasma cells (38), correlating EZH2 expression with cycling cells in both the GC and in the plasmablast populations. The expression of Ezh2 is decreased, however, in PC and memory B-cell populations compared to GC B-cells (30).
To investigate the role of epigenetic regulation in B-cell differentiation during humoral responses, a number of groups have conditionally deleted histone modifiers (Table 1). Two such enzymes are EZH2 and the histone acetyltransferase monocytic leukemia zinc finger protein (MOZ) (Figure 1). EZH2 plays an important role during B-cell development by modulating Igh rearrangement (39), and has now been revealed to be essential for GCs (29, 30). The deletion of EZH2, by use of either Cγ1-Cre or CR2-Cre, dramatically reduced GC frequency, with the remaining GC cells EZH2+ (29, 30). Both research groups demonstrated the regulation of cell cycle genes by EZH2 (29, 30, 35), although GC B-cells were also found to undergo increased apoptosis in the absence of EZH2 (30).
Although MOZ is a histone acetyltransferase, there were similarities between the phenotypes of MOZ-deficient and EZH2-deficient mice. Deletion of MOZ using Mb-1-Cre (in all B-cells) or Aicda-cre (specifically in activated B-cells) mice also resulted in a decrease in GC B-cells (31), although not to the extent of EZH2-deficiency (29, 30). This was found to be due to defective proliferation, correlating to a decrease specifically in dark zone B-cells (31). Thus, expression and/or function of EZH2 and MOZ can be localized to the dark zone of the GC. Somatic hypermutation and class-switch recombination is also known to be regulated epigenetically, however, this has been reviewed recently (32) and thus will not be discussed here. Given that a number of other histone modifiers are located either in the dark or light zone (36, 37), future investigations could assess whether these other modifiers regulate particular functions within the light zone.
B cell lymphoma 6 (BCL-6) is absolutely required for GC formation (18, 19). BCL-6 shares some common targets with EZH2 in GC B-cells. EZH2 binds approximately 1800 promoters in GC B-cells (35), and a portion of these were specific to GC B-cells. Within this GC-specific geneset, it appeared that EZH2 targets were involved in cellular proliferation and repression of differentiation (29, 30, 35). Interestingly, EZH2 targets that were not H3K27me3-marked in naïve B-cells were also bound by BCL-6 (35). Approximately half of the genes that were bound by both the polycomb repressor complex 2 and BCL-6 in wild-type GC B-cells were upregulated in EZH2 mutants (30). In contrast, EZH2-deficiency mostly did not affect the expression of BCL-6 targets that lack the H3K27me3 mark (30), and EZH2 does not modulate BCL-6 expression itself (30). In contrast, MOZ-deficient GC B-cells had decreased levels of BCL-6 (31), which may be associated with the gene expression program modulated by MOZ (31).
Regulation of Plasma Cells by Histone Modifiers
Conditional deletion of histone modifiers in B-cells has demonstrated that differentiation of GC cells into plasma cells is epigenetically regulated. In the case of MOZ, deficiency altered the affinity but not numbers of plasma cells, likely due to the reduction of dark zone B-cells (31). Similarly, the GC defect in EZH2-deficient mice resulted in a significant reduction in both numbers and affinity of plasma cells (30). However, when these authors stimulated EZH2-deficient cells in vitro, differentiation into plasmablasts was increased in the absence of EZH2. This was correlated to functional repression of the plasma cell genes Prdm1 and Irf4 by EZH2 (30), and the reduction of H3K27me3 marking at Irf4 and Prdm1 loci upon differentiation (30). In addition to Irf4 and Prdm1, EZH2 appears to regulate the genetic programs associated with differentiation of GC B-cells to plasma cells or memory B-cells (29, 30, 35). Thus, continued EZH2 expression is likely required to maintain the GC phenotype and prevent premature differentiation (35). It is known that EZH2 mutations are associated with malignant transformations (29, 30, 35, 40), but it is also possible that dysregulation of EZH2 may also play a role in antibody-mediated autoimmune disorders.
Lastly, it is likely that HDACs can also regulate plasma cell differentiation, although previous studies have had contrasting results on whether inhibiting HDACs inhibit or promote differentiation (41, 42). This will be important to determine as HDAC inhibitors are being used to treat lymphocyte malignancies (43–45). Dysregulation of gene expression during B-cell responses can lead to autoimmune diseases, and there is some evidence this could occur as a result of improper histone modifications. Mice lacking acetyltransferase activity in p300 specifically in B-cells develop a systemic lupus erythematosus-like disease (28). Thus, there is future potential to use epigenetic modifiers as treatment for antibody disorders.
Regulation of B-Cell Memory by EZH2 and MOZ
Immune memory is defined as the rapid and robust response that occurs upon secondary infections, clearing invading pathogens more quickly than the primary response. The memory B-cell population is phenotypically and functionally heterogeneous (1, 46, 47). Recently, a number of research groups have postulated that the heterogeneity evident within the memory population allows the pool to undergo specialized functions, i.e., differentiation into plasmablasts whilst being able to self-renew. IgM+ memory B-cells persist longer than IgG+ memory B-cells, and are able to initiate a response to secondary infections when IgG+ memory B-cells are present in low numbers (48, 49). In contrast, switched memory B-cells has been linked to the rapid production of antibody during secondary responses (48, 49). A number of genes expressed in IgM+ B-cells are silenced when those cells are engineered to signal through the cytoplasmic tail of IgG1 (50, 51). Therefore, regulation of gene transcription programs may be linked with the plasticity of the memory pool, allowing persistence in the presence of rapid activation and differentiation during re-infection.
In the absence of EZH2, GC-derived IgG1+ memory B-cells and antibody produced in a secondary response were significantly reduced (30). It is likely that the reduction in memory formation and function is a result of the absence of functional GCs (29, 30). High affinity IgG1+ memory B-cells were also reduced in the absence of MOZ (31). The latter study also investigated the IgM+ memory B-cell subset, which has been linked to longevity of the memory population (48, 49). In the absence of MOZ, the make-up of the memory B-cell population was altered such that IgM+ memory B-cell numbers were increased. It is likely that as a result, secondary GC formation was increased in these mice, whereas, secondary plasmablast formation was decreased (31). Thus, MOZ regulated the composition and functional outcome of the memory compartment. More work is now needed to investigate in detail the role of epigenetic regulation in memory B-cell formation and function.
Concluding Remarks
Histone modifications are an important component of gene expression regulation. Specifically, in both T and B-cells, during development and during differentiation in the periphery, patterns of histone modifications are unique to different lymphocyte subsets. These modifications likely allow adaptability of cells – i.e., for the ability of an antigen-activated B-cell to undergo differentiation into either a memory B-cell, GC or plasmablast.
The enzymes that catalyze modifications of histones, such as EZH2 and MOZ, have recently been shown to play important roles in formation, maintenance and modulation of B-cell populations. Thus, these new studies demonstrate that programing of B-cell subsets by epigenetic changes influence differentiation decisions during immune responses. However, it is only the beginning for these types of studies. A better understanding of epigenetic regulation of humoral responses will be attained as the targets for each modifier in B-cell subsets, factors involved in facilitating modifications, and interactions between known regulatory complexes are revealed. It will be important to use an integrated approach to identify histone modifications important for B cell generation and function, and the transcriptional networks they regulate. Thus, in addition to ChIP-sequencing and gene-targeted mice, it will be essential to use new methods that can systematically initiate histone marks during B cell responses to unravel the role of particular modifications during memory formation and secondary responses.
Revealing the roles of other histone modifiers has the potential to reveal the molecular mechanisms underlying the production of a memory population that is able to persist in the absence of antigen whilst being poised to respond to subsequent infections. This not only has implications for vaccines and immunodeficiencies that are unable to produce memory cells, it will also result in a wider understanding of how epigenetic regulation controls gene expression during programs of cell differentiation. Understanding these fundamental cellular processes are applicable not only to B-cell and hematopoietic development, but also more generally for developmental processes. It is noteworthy that it is precisely these transcriptional networks that are predictive in disease, particularly autoimmune diseases and cancers.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors’ work is made possible by a National Health and Medical Research Council (NHMRC) Australia project grant (1022458), and through Victorian State Government Operational Infrastructure Support and the Australian Government NHMRC IRIISS.
References
1. Tarlinton D, Good-Jacobson K. Diversity among memory B cells: origin, consequences, and utility. Science (2013) 341(6151):1205–11. doi: 10.1126/science.1241146
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Klein U, Tu Y, Stolovitzky GA, Keller JL, Haddad J Jr, Miljkovic V, et al. Transcriptional analysis of the B cell germinal center reaction. Proc Natl Acad Sci U S A (2003) 100(5):2639–44. doi:10.1073/pnas.0437996100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Good KL, Avery DT, Tangye SG. Resting human memory B cells are intrinsically programmed for enhanced survival and responsiveness to diverse stimuli compared to naive B cells. J Immunol (2009) 182(2):890–901. doi:10.4049/jimmunol.182.2.890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Good KL, Tangye SG. Decreased expression of Kruppel-like factors in memory B cells induces the rapid response typical of secondary antibody responses. Proc Natl Acad Sci U S A (2007) 104(33):13420–5. doi:10.1073/pnas.0703872104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Tomayko MM, Anderson SM, Brayton CE, Sadanand S, Steinel NC, Behrens TW, et al. Systematic comparison of gene expression between murine memory and naive B cells demonstrates that memory B cells have unique signaling capabilities. J Immunol (2008) 181(1):27–38. doi:10.4049/jimmunol.181.1.27
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Taylor JJ, Pape KA, Jenkins MK. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med (2012) 209(3):597–606. doi:10.1084/jem.20111696
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Rajewsky K. Clonal selection and learning in the antibody system. Nature (1996) 381(6585):751–8. doi:10.1038/381751a0
8. Takahashi Y, Cerasoli DM, Dal Porto JM, Shimoda M, Freund R, Fang W, et al. Relaxed negative selection in germinal centers and impaired affinity maturation in bcl-xL transgenic mice. J Exp Med (1999) 190(3):399–410. doi:10.1084/jem.190.3.399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Takahashi Y, Dutta PR, Cerasoli DM, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V affinity maturation develops in two stages of clonal selection. J Exp Med (1998) 187(6):885–95. doi:10.1084/jem.187.6.885
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Han S, Hathcock K, Zheng B, Kepler TB, Hodes R, Kelsoe G. Cellular interaction in germinal centers. Roles of CD40 ligand and B7-2 in established germinal centers. J Immunol (1995) 155(2):556–67.
11. Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature (1991) 354(6352):389–92. doi:10.1038/354389a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol (2012) 30:429–57. doi:10.1146/annurev-immunol-020711-075032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Meyer-Hermann M, Mohr E, Pelletier N, Zhang Y, Victora GD, Toellner KM. A theory of germinal center B cell selection, division, and exit. Cell Rep (2012) 2(1):162–74. doi:10.1016/j.celrep.2012.05.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell (2010) 143(4):592–605. doi:10.1016/j.cell.2010.10.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Baxter J, Sauer S, Peters A, John R, Williams R, Caparros ML, et al. Histone hypomethylation is an indicator of epigenetic plasticity in quiescent lymphocytes. EMBO J (2004) 23(22):4462–72. doi:10.1038/sj.emboj.7600414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Peperzak V, Vikstrom I, Walker J, Glaser SP, Lepage M, Coquery CM, et al. Mcl-1 is essential for the survival of plasma cells. Nat Immunol (2013) 14(3):290–7. doi:10.1038/ni.2527
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Good-Jacobson KL, Shlomchik MJ. Plasticity and heterogeneity in the generation of memory B cells and long-lived plasma cells: the influence of germinal center interactions and dynamics. J Immunol (2010) 185(6):3117–25. doi:10.4049/jimmunol.1001155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Shaffer AL, Yu X, He Y, Boldrick J, Chan EP, Staudt LM. BCL-6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity (2000) 13(2):199–212. doi:10.1016/S1074-7613(00)00020-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science (1997) 276(5312):589–92. doi:10.1126/science.276.5312.589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity (2002) 17(1):51–62. doi:10.1016/S1074-7613(02)00335-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Lin KI, Tunyaplin C, Calame K. Transcriptional regulatory cascades controlling plasma cell differentiation. Immunol Rev (2003) 194:19–28. doi:10.1034/j.1600-065X.2003.00040.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Murphy KM, Stockinger B. Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol (2010) 11(8):674–80. doi:10.1038/ni.1899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Kallies A, Hasbold J, Fairfax K, Pridans C, Emslie D, McKenzie BS, et al. Initiation of plasma-cell differentiation is independent of the transcription factor Blimp-1. Immunity (2007) 26(5):555–66. doi:10.1016/j.immuni.2007.04.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Tarakhovsky A. Tools and landscapes of epigenetics. Nat Immunol (2010) 11(7):565–8. doi:10.1038/ni0710-565
25. Strahl BD, Allis CD. The language of covalent histone modifications. Nature (2000) 403(6765):41–5. doi:10.1038/47412
26. Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol (2007) 8(4):284–95. doi:10.1038/nrm2145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Shaknovich R, Cerchietti L, Tsikitas L, Kormaksson M, De S, Figueroa ME, et al. DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood (2011) 118(13):3559–69. doi:10.1182/blood-2011-06-357996
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Forster N, Gallinat S, Jablonska J, Weiss S, Elsasser HP, Lutz W. p300 protein acetyltransferase activity suppresses systemic lupus erythematosus-like autoimmune disease in mice. J Immunol (2007) 178(11):6941–8. doi:10.4049/jimmunol.178.11.6941
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell (2013) 23(5):677–92. doi:10.1016/j.ccr.2013.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest (2013) 123(12):5009–22. doi:10.1172/JCI70626
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Good-Jacobson KL, Chen Y, Voss AK, Smyth GK, Thomas T, Tarlinton D. Regulation of germinal center responses and B-cell memory by the chromatin modifier MOZ. Proc Natl Acad Sci U S A (2014) 111(26):9585–90. doi:10.1073/pnas.1402485111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Li G, Zan H, Xu Z, Casali P. Epigenetics of the antibody response. Trends Immunol (2013) 34(9):460–70. doi:10.1016/j.it.2013.03.006
33. Zhang J, Jima D, Moffitt AB, Liu Q, Czader M, Hsi ED, et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood (2014) 123(19):2988–96. doi:10.1182/blood-2013-07-517177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature (2011) 469(7330):343–9. doi:10.1038/nature09784
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Velichutina I, Shaknovich R, Geng H, Johnson NA, Gascoyne RD, Melnick AM, et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood (2010) 116(24):5247–55. doi:10.1182/blood-2010-04-280149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Raaphorst FM, van Kemenade FJ, Fieret E, Hamer KM, Satijn DP, Otte AP, et al. Cutting edge: polycomb gene expression patterns reflect distinct B cell differentiation stages in human germinal centers. J Immunol (2000) 164(1):1–4. doi:10.4049/jimmunol.164.1.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. van Galen JC, Dukers DF, Giroth C, Sewalt RG, Otte AP, Meijer CJ, et al. Distinct expression patterns of polycomb oncoproteins and their binding partners during the germinal center reaction. Eur J Immunol (2004) 34(7):1870–81. doi:10.1002/eji.200424985
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Tarte K, Zhan F, De Vos J, Klein B, Shaughnessy J Jr. Gene expression profiling of plasma cells and plasmablasts: toward a better understanding of the late stages of B-cell differentiation. Blood (2003) 102(2):592–600. doi:10.1182/blood-2002-10-3161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Su IH, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT, et al. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol (2003) 4(2):124–31. doi:10.1038/ni876
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet (2010) 42(2):181–5. doi:10.1038/ng.518
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Lee SC, Bottaro A, Insel RA. Activation of terminal B cell differentiation by inhibition of histone deacetylation. Mol Immunol (2003) 39(15):923–32. doi:10.1016/S0161-5890(03)00029-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Kienzler AK, Rizzi M, Reith M, Nutt SL, Eibel H. Inhibition of human B-cell development into plasmablasts by histone deacetylase inhibitor valproic acid. J Allergy Clin Immunol (2013) 131(6):1695–9. doi:10.1016/j.jaci.2013.01.018
43. Ramalingam SS, Kummar S, Sarantopoulos J, Shibata S, LoRusso P, Yerk M, et al. Phase I study of vorinostat in patients with advanced solid tumors and hepatic dysfunction: a National Cancer Institute Organ Dysfunction Working Group study. J Clin Oncol (2010) 28(29):4507–12. doi:10.1200/JCO.2010.30.2307
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Zain J, Kaminetzky D, O’Connor OA. Emerging role of epigenetic therapies in cutaneous T-cell lymphomas. Expert Rev Hematol (2010) 3(2):187–203. doi:10.1586/ehm.10.9
45. Lemoine M, Younes A. Histone deacetylase inhibitors in the treatment of lymphoma. Discov Med (2010) 10(54):462–70.
46. Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, et al. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol (2014) 15(7):631–7. doi:10.1038/ni.2914
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Tangye SG, Good KL. Human IgM+CD27+ B cells: memory B cells or “memory” B cells? J Immunol (2007) 179(1):13–9. doi:10.4049/jimmunol.179.1.13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science (2011) 331(6021):1203–7. doi:10.1126/science.1201730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Dogan I, Bertocci B, Vilmont V, Delbos F, Megret J, Storck S, et al. Multiple layers of B cell memory with different effector functions. Nat Immunol (2009) 10(12):1292–9. doi:10.1038/ni.1814
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Horikawa K, Martin SW, Pogue SL, Silver K, Peng K, Takatsu K, et al. Enhancement and suppression of signaling by the conserved tail of IgG memory-type B cell antigen receptors. J Exp Med (2007) 204(4):759–69. doi:10.1084/jem.20061923
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Martin SW, Goodnow CC. Burst-enhancing role of the IgG membrane tail as a molecular determinant of memory.[comment]. Nat Immunol (2002) 3(2):182–8. doi:10.1038/ni752
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: humoral memory, B-cells, germinal centers, epigenetics, EZH2, MOZ, histone modifiers
Citation: Good-Jacobson KL (2014) Regulation of germinal center, B-cell memory, and plasma cell formation by histone modifiers. Front. Immunol. 5:596. doi: 10.3389/fimmu.2014.00596
Received: 01 September 2014; Accepted: 06 November 2014;
Published online: 19 November 2014.
Edited by:
Paolo Casali, University of Texas School of Medicine – Health Science Center SA, USACopyright: © 2014 Good-Jacobson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kim L. Good-Jacobson, Immunology Division, Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, VIC 3052, Australia e-mail:amFjb2Jzb25Ad2VoaS5lZHUuYXU=